

ABSTRACT
The phylum Chlamydiae constitutes a group of obligate intracellular bacteria that infect a remarkably diverse range of host species. Some representatives are significant pathogens of clinical or veterinary importance. For instance, Chlamydia trachomatis is the leading infectious cause of blindness and the most common bacterial agent of sexually transmitted diseases. Chlamydiae are exceptionally dependent on their eukaryotic host cells as a consequence of their developmental biology. At the same time, host cell death is an integral part of the chlamydial infection cycle. It is therefore not surprising that the bacteria have evolved exquisite and versatile strategies to modulate host cell survival and death programs to their advantage. The recent introduction of tools for genetic modification of Chlamydia spp., in combination with our increasing awareness of the complexity of regulated cell death in eukaryotic cells, and in particular of its connections to cell-intrinsic immunity, has revived the interest in this virulence trait. However, recent advances also challenged long-standing assumptions and highlighted major knowledge gaps. This review summarizes current knowledge in the field and discusses possible directions for future research, which could lead us to a deeper understanding of Chlamydia’s virulence strategies and may even inspire novel therapeutic approaches.
Keywords: intracellular bacteria, virulence strategies, regulated cell death, bacterial exit, bacterial toxicity, cell-autonomous immunity
A comprehensive review that highlights recent advances and major knowledge gaps in our understanding of the diverse mechanisms and roles of pathogen-mediated host cell death modulation during infection with the obligate intracellular pathogen Chlamydia trachomatis and its relatives.
INTRODUCTION
The phylum Chlamydiae is a group of obligate intracellular bacteria that infect a wide range of host species, including members of all major groups of vertebrates, as well as invertebrates, and even unicellular eukaryotes (Horn 2008). From a medical perspective, the most important representatives are species in the genus Chlamydia. For instance, Chlamydia trachomatis (serovars A–C) is the causative agent of trachoma, an ocular disease that is the leading infectious cause of blindness (Taylor et al. 2014). Chlamydia trachomatis (serovars D–K) is also the most frequent bacterial agent of sexually transmitted diseases and as such a significant cause of infertility and adverse pregnancy outcomes (Newman et al. 2015). Moreover, certain sexually transmitted strains of C. trachomatis (serovars L1–L3) can cause lymphogranuloma venereum (LGV) (Ceovic and Gulin 2015). Chlamydia pneumoniae, the second major human pathogenic Chlamydia species, primarily causes respiratory tract infections, yet was also proposed to be associated with atherosclerosis and neurological disorders (Burillo and Bouza 2010). Other Chlamydia species, such as Chlamydia psittaci, infect primarily animals, but can occasionally cause severe zoonotic infections (Longbottom and Coulter 2003). Moreover, some animal pathogenic species, such as the mouse pathogen Chlamydia muridarum and the guinea pig pathogen Chlamydia caviae, are frequently used as models in research, due to the availability of convenient in vivo infection models (Rank 2012). Chlamydial species within other genera and families in the phylum Chlamydiae are often collectively referred to as environmental chlamydiae (Horn 2008). While recent studies uncovered an astonishing diversity in this group, their biology and impact are less well explored (Collingro, Köstlbacher and Horn 2020). It is clear that a better understanding of these microbes could provide valuable insights into the evolution of virulence strategies in Chlamydiae and their role in host adaptation (Horn 2008). Although this review will primarily focus on species in the genus Chlamydia, it will therefore also summarize available data on cultured environmental species.
All cultured Chlamydiae share a unique lifestyle that is characterized by a strict dependence on a eukaryotic host cell and a biphasic developmental cycle (Ward 1988) (Fig. 1). The infective developmental form, called the elementary body (EB), invades the host cell in a process that leads to the generation of a membrane-bound pathogen-containing vacuole, named the inclusion. Within the inclusion, the EB differentiates into the replicative form, the reticulate body (RB). Due to their adaptation to the intracellular growth niche, Chlamydiae have highly reduced metabolic capacities (Stephens et al. 1998; Collingro et al. 2011; Omsland et al. 2014). The bacteria evolved to redirect nutrients and lipids from host cell compartments to the inclusion (Saka and Valdivia 2010). RBs are also energy parasites that can directly feed on the host cell's pool of adenosine triphosphate (ATP) (Hatch, Al-Hossainy and Silverman 1982), although the bacteria also have the capacity to generate ATP on their own (Omsland et al. 2012). Chlamydiae actively modulate host cellular functions, in part by exploiting their type III secretion system to deliver effector proteins into the host cell cytosol (Peters et al. 2007). A special class of these effectors, the inclusion membrane proteins (Incs), is inserted into the membrane of the inclusion (Rockey and Alzhanov 2006). After several rounds of division, RBs asynchronously re-differentiate into EBs, which are eventually released from the host cell to infect neighboring cells (Hybiske and Stephens 2007). The length of this developmental cycle can be variable, but typically ranges from 36 to 96 h for Chlamydia spp. in cultured cells (Miyairi et al. 2006).
Figure 1.
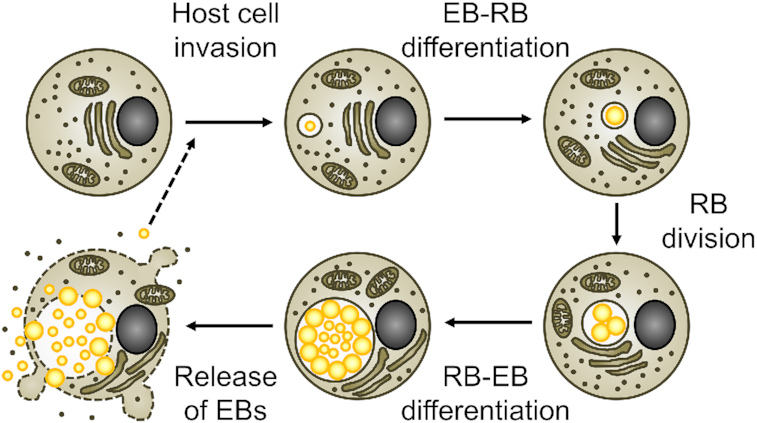
The chlamydial developmental cycle. Schematic representation of the different stages of the Chlamydia developmental cycle. Bacteria are indicated as yellow spheres. Small and large spheres represent EBs and RBs, respectively.
As a consequence of this unique lifestyle, Chlamydiae are absolutely dependent on the integrity of their host cell until they have completed their developmental cycle, i.e. until a significant number of bacteria have re-differentiated into mature EBs. During the past decades, the interaction of pathogenic Chlamydia spp. with their host cells’ death and survival pathways has been the subject of extensive investigations. While these studies revealed a complex picture in which Chlamydia spp. both induced and blocked host cell death, they particularly highlighted the pronounced anti-apoptotic trait of the pathogen (Ying et al. 2007; Sharma and Rudel 2009). However, recent findings challenged the effectiveness of Chlamydia’s anti-apoptotic activities as means to preserve host cell viability (Sixt et al. 2018). At the same time, advances in our ability to genetically modify Chlamydia spp. (Sixt and Valdivia 2016) revealed cell death as a host cellular defense response that could be effective, but is actively suppressed by the pathogen (Sixt et al. 2017; Weber et al. 2017; Giebel et al. 2019). A profound understanding of this complex interplay between host and pathogen is critical for our understanding of Chlamydia diseases and anti-chlamydial host defenses. In the future, it may even enable us to exploit Chlamydia’s interference with cell death as target for novel anti-chlamydial treatment strategies.
This review aims to provide a comprehensive and structured overview of current knowledge, with focus on recent advances, to establish links between distinct facets of Chlamydia-mediated cell death modulation, and to highlight knowledge gaps. Its objective is to stimulate cross-disciplinary discussions and to inspire future research.
MODES OF CELL DEATH
Accidental and regulated forms of cell death
Extreme physical, mechanical or chemical insults on eukaryotic cells, for example high temperatures, shear stress or extreme pH variations, can cause the instantaneous death of the cell. Such accidental cell death (ACD) does not depend on the activity of cellular regulators (Galluzzi et al. 2018). As counter pole to ACD, the term programmed cell death (PCD) was first introduced in 1964 to describe the observation that in multicellular organisms certain cells seemed to be programmed to die at specific stages during development (Lockshin 1964). However, genetically encoded cell-intrinsic death programs, which govern PCD, act in not only such developmentally programmed events but also cell death that is induced in response to severe non-compensable perturbations of the cellular homeostasis. Therefore, the more general term regulated cell death (RCD) was introduced more recently to encompass all forms of cell death that result from the activation of molecular death machineries (Galluzzi et al. 2018).
Morphology of cell death
Morphologically, ACD is a form of necrosis, characterized by cell swelling and a sudden rupture of the plasma membrane (Proskuryakov, Konoplyannikov and Gabai 2003). Based on the characteristics of cells dying from PCD, two major forms of cell death distinct from necrosis were described: cell death involving autophagy, characterized by extensive cytoplasmic vacuolization, and apoptosis, involving cell shrinkage, membrane blebbing, nuclear condensation and cell fragmentation (Kerr, Wyllie and Currie 1972; Schweichel and Merker 1973). However, the increasing focus on molecular determinants for the classification of cell death, as opposed to morphological traits, and the developing awareness that RCD serves functions beyond development have led to the recognition of several modes of regulated necrosis, such as necroptosis (Degterev et al. 2005) and pyroptosis (Boise and Collins 2001; Cookson and Brennan 2001), that are executed by a molecular suicide machinery, but share morphological characteristics with ACD (Fig. 2).
Figure 2.
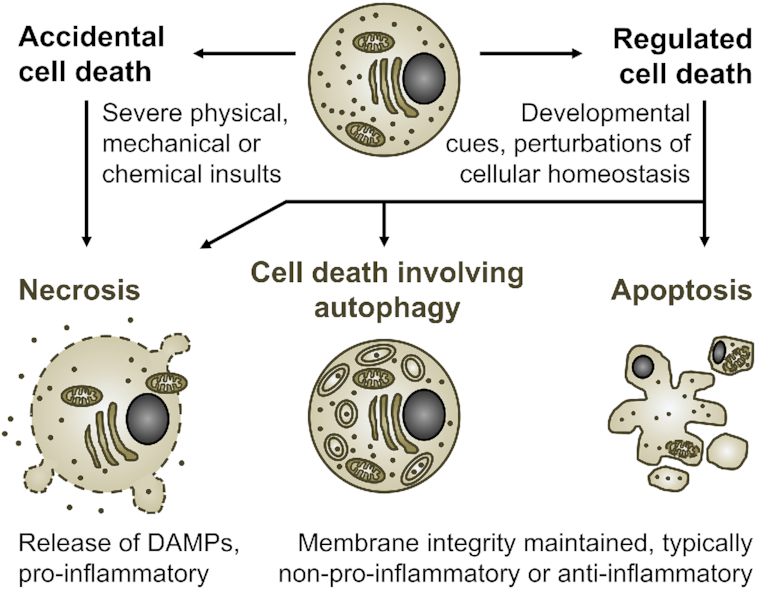
Inducers, cell morphologic presentation, and inflammatory potential of accidental and regulated cell death. Note that immunogenic forms of apoptosis, which are accompanied by release of DAMPs, have also been described. Moreover, under conditions in which phagocytic removal of dying cells is insufficient, even cells that initiate non-necrotic forms of cell death will eventually become secondary necrotic.
Inflammatory potential and immunogenicity of cell death
The way in which a cell dies has a strong impact on how it is perceived by surrounding cells and by the immune system (Galluzzi et al. 2017). Necrotic forms of cell death lead to the release of cellular content into the surroundings of the dying cell. Among the molecules released are those that act as danger-associated molecular patterns (DAMPs), for instance ATP and high mobility group protein B1 (HMGB1) (Kono and Rock 2008). The pro-inflammatory environment that is thereby generated supports the development of immune responses against antigens present in the dying cells (Galluzzi et al. 2017). In contrast, under physiological conditions, the plasma membrane integrity of cells dying by apoptosis or cell death involving autophagy is maintained until the cells are phagocytosed and degraded (Kerr, Wyllie and Currie 1972; Schweichel and Merker 1973). This enables a safe and usually immunologically silent removal. However, immunogenic forms of apoptosis, which are accompanied by release of DAMPs, have been described (Galluzzi et al. 2017). Moreover, under conditions in which phagocytic removal of dying cells is insufficient, apoptotic cells will eventually become secondary necrotic (Silva, do Vale and dos Santos 2008).
Molecular basis of selected death programs
Apoptosis
Two major pathways of apoptotic signaling can be distinguished: intrinsic and extrinsic apoptosis (Elmore 2007) (Fig. 3A). Intrinsic apoptosis can result from a variety of microenvironmental perturbations, such as growth factor withdrawal, DNA damage or mitotic defects (Galluzzi et al. 2018). A central step in the intrinsic pathway is the mitochondrial outer membrane permeabilization (MOMP) (Galluzzi, Kepp and Kroemer 2016). MOMP causes the release of mitochondrial cytochrome c (CYC), followed by the formation of a cytosolic signaling complex that leads to the activation of the initiator caspase caspase-9 (CASP9) (Bao and Shi 2007). CASP9 in turn mediates the activation of effector caspases [caspase-3 (CASP3) and caspase-7 (CASP7)], whose proteolytic action initiates cell demolition (Kumar 1999). MOMP is mediated by the activity of the pro-apoptotic B-cell lymphoma 2 (BCL-2) family proteins BCL-2-associated X protein (BAX) and BCL-2-antagonist killer (BAK), which is tightly regulated by pro-apoptotic BH3-only proteins and anti-apoptotic BCL-2 family proteins (Czabotar et al. 2014). In the extrinsic pathway, engagement of death receptors, such as Fas receptor (CD95/FAS) or tumor necrosis factor receptor 1 (TNFR1), can lead to the formation of a signaling complex that activates the initiator caspase caspase-8 (CASP8) (Guicciardi and Gores 2009). Depending on the cell type, the proteolytic activity of CASP8 may be sufficient to directly activate effector caspases resulting in cell death (type I cells) or induction of cell death may require an amplification of pro-death signaling via CASP8-mediated cleavage of BH3-interacting domain death agonist (BID) and subsequent induction of MOMP (type II cells) (Kantari and Walczak 2011).
Figure 3.
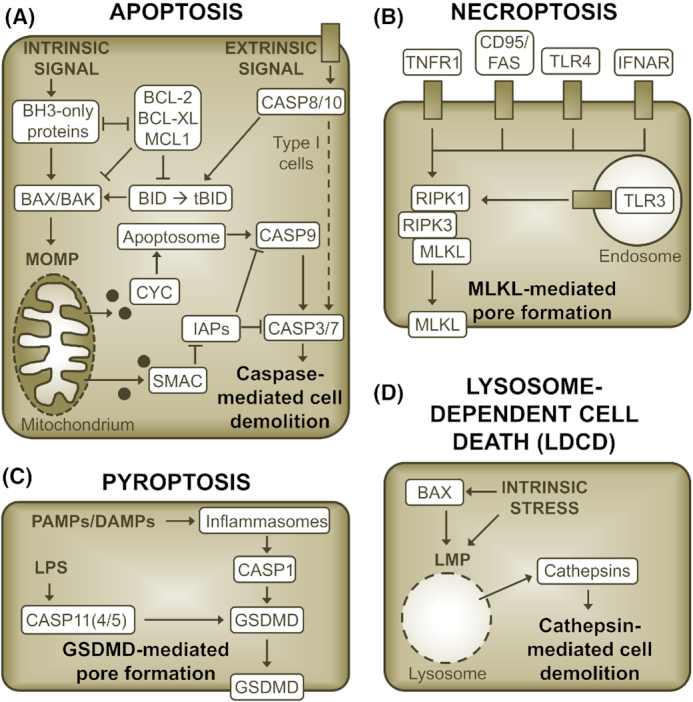
The molecular basis of selected cell death programs. Illustration of key events in apoptosis (A), necroptosis (B), pyroptosis (C) and lysosome-dependent cell death (D). The representations are simplified. Cross-talks between pathways and links to inflammatory signaling are not depicted. Moreover, in (B), the signaling events between the receptors and the effectors of necroptosis are not shown. RIPK1 is dispensable for some modes of necroptosis.
Necroptosis
Necroptosis is a necrotic mode of RCD that can be activated downstream of certain death receptors, such as CD95/FAS and TNFR1, and pathogen recognition receptors, such as Toll-like receptor 3 (TLR3) and Toll-like receptor 4 (TLR4) (Degterev et al. 2005; Pasparakis and Vandenabeele 2015) (Fig. 3B). In the context of TNFR1 signaling, necroptosis is considered a back-up for apoptosis, because it is only activated under circumstances in which CASP8 is absent or its activity blocked (Degterev et al. 2005; Tummers and Green 2017). Mechanistically, necroptosis induction depends on the activation of receptor-interacting serine/threonine-protein kinase 3 (RIPK3), which phosphorylates mixed lineage kinase domain-like protein (MLKL), resulting in the formation of MLKL oligomers that induce plasma membrane permeabilization (Sun et al. 2012b; Cai et al. 2014). During tumor necrosis factor alpha (TNFα)-induced necroptosis, RIPK3 activation depends on receptor-interacting serine/threonine-protein kinase 1 (RIPK1) and can hence be blocked by inhibitors of RIPK1 (Degterev et al. 2008). However, RIPK1 is not involved in all types of necroptotic signaling (Pasparakis and Vandenabeele 2015).
Pyroptosis
Pyroptosis is a form of regulated necrosis that depends on the activation of one or more inflammatory caspases (Cookson and Brennan 2001; Jimenez Fernandez and Lamkanfi 2015) (Fig. 3C). These enzymes are activated in response to pathogen-associated molecular patterns (PAMPs) or DAMPs (Jimenez Fernandez and Lamkanfi 2015). While murine caspase-11 (CASP11) [and human caspase-4 (CASP4) and caspase-5 (CASP5)] can directly respond to bacterial lipopolysaccharide (LPS) (Shi et al. 2014), caspase-1 (CASP1) is activated indirectly via inflammasomes (Martinon, Burns and Tschopp 2002). Once activated beyond a critical threshold, inflammatory caspases catalyze the proteolytic cleavage of gasdermin D (GSDMD) (Kovacs and Miao 2017). The N-terminal fragment of GSDMD can then oligomerize at the plasma membrane, resulting in pore formation and cell death (Kovacs and Miao 2017). Because of the involvement of inflammatory caspases, pyroptosis is usually accompanied by secretion of interleukin 1 beta (IL-1β) and interleukin 18 (IL-18) and hence mediates robust pro-inflammatory effects (Jimenez Fernandez and Lamkanfi 2015).
Lysosome-dependent cell death
Soon after their discovery in the 1950s, lysosomes were dubbed cellular 'suicide bags', because it was suggested that lysosomal rupture could be a major mechanism of RCD (De Duve 1959). While the role of lysosome-dependent cell death (LDCD) may not be as fundamental as initially suggested, examples exist in which intracellular perturbations, such as oxidative stress or disruption of the cytoskeleton, can cause lysosomal membrane permeabilization (LMP) (Aits et al. 2015; Wang, Gomez-Sintes and Boya 2018) (Fig. 3D). LMP can also be mediated by BAX, which in some instances may precede BAX-mediated MOMP (Kagedal et al. 2005; Bove et al. 2014; Guan et al. 2015). The proteolytic enzymes of the cathepsin family, which are released from lysosomes during LMP, are considered main executors of LDCD, because inhibition of cathepsin activity can in many instances ameliorate LDCD (Wang, Gomez-Sintes and Boya 2018). However, the mode of cell death induced by LMP also depends on its extent. While massive LMP appears to induce rapid necrotic death, partial LMP may for instance induce MOMP and apoptosis or inflammasome activation and pyroptosis (Wang, Gomez-Sintes and Boya 2018). Moreover, in some instances, LMP does not initiate cell death by itself, but accompanies and amplifies other death signals, such as during certain instances of apoptosis (Galluzzi et al. 2018).
Roles of cell death during infection
It is well known that the encounter of eukaryotic cells with extracellular pathogens or the invasion by intracellular pathogens can eventually result in the death of the cell. Different scenarios of infection-associated cell death can be distinguished (Fig. 4). First, cell death may be mediated by a pathogen-driven process, such as by the action of toxins or cytolytic enzymes, for instance to enable release of intracellular pathogens, to facilitate pathogen dissemination or to mediate depletion of immune cells (Fig. 4A). Pathogen-driven host cell death depends on virulence factors and may or may not rely on the (partial) induction of host RCD programs. Second, cell death may be induced by host cell-intrinsic mechanisms in response to microenvironmental perturbations caused by the presence of the pathogen, such as nutrient deprivation, DNA damage or oxidative stress (Fig. 4B). Third, cell death may be triggered by the host cell as a defense response, for example to restrict intracellular pathogen replication by removing the replicative niche, to entrap the pathogen to limit its dissemination, to alert neighboring cells and immune cells, and/or to shape the subsequent immune response against the pathogen (Fig. 4C). Both forms of host cell-driven cell death, stress-induced and defensive, may be actively counteracted by the pathogen. Immune mediators, such as cytotoxic cytokines and cell-mediated killing mechanisms, can promote host cell death either by inducing cellular stress or by supporting death-inducing cell-intrinsic defense responses. Given these complex implications of host cell death, it is not surprising that a pathogen that is as dependent on its host cell as Chlamydia has evolved both pro- and anti-death activities, which can be variably active under distinct circumstances.
Figure 4.
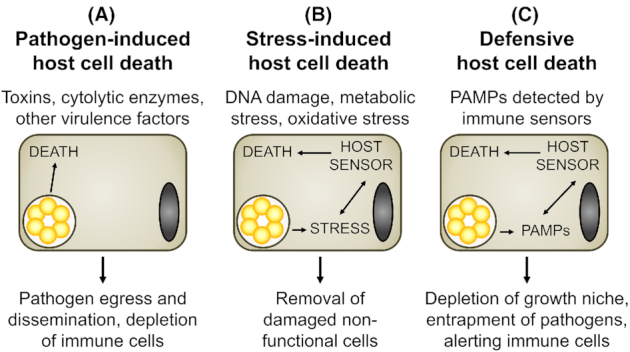
The various roles of infection-associated cell death. (A) Host cell death can be triggered by the pathogen, for instance to mediate pathogen release, spread to deeper tissue layers, or immune cell depletion. (B) Host cell death may be triggered by the host cell itself as response to non-compensable infection-induced stress, such as DNA damage, oxidative stress or metabolic stress. (C) Upon detection of an invading pathogen by immune sensors, host cells may trigger cell death as defense response. Note that immune mediators, such as cytotoxic cytokines and immune cell-mediated killing mechanisms, can promote host cell death either by inducing cellular stress (B) or by supporting death-inducing host cell-intrinsic defense responses (C).
PRO-DEATH ACTIVITIES OF CHLAMYDIAE
Host cell death as exit strategy
Cell death is an integral part of the Chlamydia infection cycle
Host cell death has long been recognized as the final stage of the Chlamydia infection cycle, enabling the release of EBs and spread of infection. The Chlamydia developmental cycle was first described for C. psittaci in 1932 based on light microscopic observations (Bedson and Bland 1932). Subsequently, electron microscopic analyses gave deeper insights into the morphological differences between EBs and RBs and their relation to each other (Swain 1955; Tajima, Nomura and Kubota 1957). In the 1960s, studies in cell culture revealed that infectivity was lost after the invading EBs had differentiated into RBs and reappeared once RBs had re-differentiated into EBs (Bernkopf, Mashiah and Becker 1962; Higashi, Tamura and Iwanaga 1962). Moreover, host cell death was observed at late stages and was accompanied by an increase of infectivity in culture supernatants (Higashi, Tamura and Iwanaga 1962; Friis 1972; Todd and Storz 1975). It can be expected that the mode by which host cells release EBs greatly impacts the magnitude of tissue damage and inflammation at the infection site. It will also shape the nature of the subsequent immune response and will affect the viability and spreading potential of the released bacteria. Astonishingly, our knowledge of the molecular events that govern Chlamydia egress is still scarce.
The role of lysosomes in Chlamydia exit
Several studies conducted in the 1970s suggested that Chlamydia exit could be preceded by LMP (Kordova, Wilt and Sadiq 1971; Kordova and Wilt 1972; Kordova, Hoogstraten and Wilt 1973; Todd and Storz 1975). Cell fractionation experiments and ultrastructural protein localization studies demonstrated that in bovine cells infected with Chlamydia pecorum, enzymes were released from lysosomes prior to host cell death (Todd and Storz 1975). LMP also appeared to precede cell death in murine fibroblasts and macrophages infected with C. psittaci (Kordova, Wilt and Sadiq 1971; Kordova and Wilt 1972; Kordova, Hoogstraten and Wilt 1973). Interestingly, these observations were not followed up upon in more recent studies. A critical reevaluation of a potential role of LMP in Chlamydia exit using more advanced tools would be timely.
The role of the apoptotic program in Chlamydia exit
In 1998, Ojcius and colleagues reported that cell death induced by C. caviae in human epithelial (HeLa) cells and murine macrophages was accompanied by features that were characteristic for apoptosis (Ojcius et al. 1998). One of these features was DNA fragmentation, which was detected by various techniques, such as the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, which enables microscopic detection of DNA strand breaks, the DNA ladder assay, which enables the display of nucleosomal DNA fragmentation on agarose gels, and flow cytometry of propidium iodide-stained permeabilized cells, which enables the detection of cells with sub-diploid DNA content (Ojcius et al. 1998). Furthermore, ultrastructural changes indicative for apoptosis, such as cell shrinkage and chromatin condensation, were observed (Ojcius et al. 1998). DNA fragmentation and nuclear condensation were also detected in cells infected with C. psittaci, C. muridarum or C. trachomatis L2 (Gibellini, Panaya and Rumpianesi 1998; Perfettini et al. 2000; Perfettini et al. 2002b; Ying et al. 2006). Moreover, TUNEL-positive cells were detected in the genital tract tissue of mice infected with C. muridarum (Perfettini et al. 2000). Finally, the surface of mouse embryonic fibroblasts (MEFs) that died after infection with C. muridarum or C. trachomatis L2 could be stained with fluorescent Annexin V (ANX5) (Perfettini et al. 2003a; Jungas et al. 2004). This is a marker for apoptosis, because ANX5 binds phosphatidylserine, a lipid that typically resides in the inner leaflet of the plasma membrane, but in apoptotic cells is translocated to the outer leaflet to trigger phagocytic removal of the dying cell (Koopman et al. 1994). In some studies, the use of microscopic assays also confirmed that the above-mentioned apoptosis-like features occurred indeed in cells that contained Chlamydia inclusions (Gibellini, Panaya and Rumpianesi 1998; Ojcius et al. 1998; Perfettini et al. 2000; Ying et al. 2006).
Although the features described above are generally considered hallmarks of caspase-dependent apoptosis, these studies also reported that cell death induced by C. caviae or C. trachomatis L2 could not be blocked by a specific inhibitor of CASP3 nor by the pan caspase inhibitor Z-VAD-FMK, suggesting that it was a caspase-independent form of cell death (Ojcius et al. 1998; Perfettini et al. 2002b; Ying et al. 2006). Indeed, no activation of apoptotic effector caspases could be observed in late stage infected cells (Perfettini et al. 2002b; Ying et al. 2006). The reliability of ANX5 staining for detection of apoptosis in Chlamydia-infected cells is also questionable, because Chlamydia infection itself can induce phosphatidylserine externalization in the absence of an activation of the apoptotic program (Galle et al. 2019). Moreover, DNA ladders induced by C. trachomatis L2 were atypical and it was therefore proposed that the mechanism of DNA fragmentation in infected cells was different from that that operates during apoptosis (Ying et al. 2006). In disagreement with the above-mentioned findings, a recent study reported that cell death induced at late stage of infection with C. trachomatis (L2 or D) can be blocked or delayed by Z-VAD-FMK and is accompanied by effector caspase activity (Foschi et al. 2019). In this study, cell death was monitored at the cell population level and the occurrence of apoptotic features was not directly demonstrated in inclusion-containing cells.
Interestingly, BAX activation was detected in HeLa cells infected with C. caviae or C. muridarum, but not after infection with C. trachomatis L2, and in the former cells activated BAX appeared to co-localize with mitochondria (Perfettini et al. 2002b; Jungas et al. 2004). Furthermore, overexpression of the BAX inhibitor 1 (BI-1) or the anti-apoptotic protein BCL-2 significantly reduced the occurrence of apoptotic nuclear morphology in C. caviae-infected cells (Perfettini et al. 2002b). A reduction in 'apoptotic' cell death, inferred from reduced emergence of cells with sub-diploid DNA content or nuclear condensation, was also observed during C. muridarum or C. trachomatis L2 infection in BAX- or BAK-deficient MEFs when compared with wild-type cells (Perfettini et al. 2003a; Ying et al. 2006). However, no release of mitochondrial CYC was observed in infected wild-type MEFs (Ying et al. 2006), which together with the caspase-independent nature of Chlamydia-induced cell death indicated that the proposed pro-death role of these proteins during infection must differ from their usual role in apoptotic signaling. It is possible that in the context of infection, BAX and BAK may contribute to cell death by inducing LMP instead of MOMP. But further work is needed to test this hypothesis experimentally.
In cultures of BAX-deficient MEFs, spread of C. muridarum infection to formerly uninfected cells appeared to be impeded (Perfettini et al. 2003a). Moreover, in absence of BAX, infected MEFs appeared to die more frequently by necrosis (Perfettini et al. 2003a). Consistently, in BAX-deficient mice, infection was cleared faster, but signs of enhanced inflammation and tissue damage were observed (Perfettini et al. 2003a). Based on these findings, it was suggested that BAX-mediated host cell death, compared with host cell necrosis, provides a more efficient and silent mode of spread by promoting uptake of dying infected cells through phagocytosis and by avoiding release of DAMPs (Perfettini et al. 2003a).
It is important to mention that the above-described 'apoptotic' features were not observed in all instances of late stage Chlamydia-mediated cell death. Depending on the combination of host cell type and Chlamydia strain studied, the default mode could also be necrotic. For instance, Jungas et al reported that in contrast to their observations made in MEFs, in HeLa cells, C. trachomatis L2 and C. muridarum induced a necrotic mode of host cell death that resulted in the release of HMGB1 (Jungas et al. 2004). Native egress of C. trachomatis L2 from HeLa cells was also not affected by BAX/BAK-deficiency (Kerr et al. 2017).
The variable nature of the findings described above, suggests that the question if components of the apoptotic machinery take part in Chlamydia exit may not have a simple answer. Because some of the observations made may represent unnatural behavior of immortalized cell lines or may have been influenced by the use of cells from non-matched host species, future studies, including mechanistic studies such as those described below, should preferentially focus on more natural infection systems, for example by using primary cells or tissues derived from the natural host species and infection site. Moreover, the possibility that distinct Chlamydia species use different exit strategies should not be neglected.
Late stage host cell death is a pathogen-triggered process
In 2007, Hybiske and Stephens developed an elegant approach to monitor the fate of individual infected HeLa cells by live cell microcopy (Hybiske and Stephens 2007). More specifically, the use of a cell line that expressed cytosolic green fluorescent protein (GFP) enabled the authors to detect host cell lysis as efflux of GFP from the host cell into the medium. Moreover, inclusion rupture was detectable as influx of GFP into the inclusion lumen. Using this approach, the authors demonstrated that late stage host cell death was a sequential inside-out process that was initiated by the rupture of the inclusion membrane, followed by the rupture of other intracellular structures, such as the nuclear envelope, and completed with the rupture of the host plasma membrane (Hybiske and Stephens 2007). The entire process was completed within ∼20 min. Morphological features characteristic for apoptosis were not observed (Hybiske and Stephens 2007). Protease inhibitors, in particular the cysteine protease inhibitor E64, blocked inclusion rupture and significantly delayed host cell death. In contrast, host plasma membrane rupture could be blocked by inhibition of calcium signaling (Hybiske and Stephens 2007).
The observed order of membrane rupture events led Hybiske and Stephens suggest that at least the initial step, the inclusion rupture, was triggered by the bacteria (Hybiske and Stephens 2007). Further support for an active contribution of the bacteria came from a recent study that demonstrated that the lytic exit of C. trachomatis L2 from HeLa cells was blocked or delayed when chloramphenicol, an inhibitor of bacterial protein synthesis, was added at a late stage of infection (Yang et al. 2015). Moreover, experiments with C. trachomatis L2 mutants that were deficient for the protease Chlamydia proteasome-like activity factor (CPAF) or for plasmid-encoded PGP4, suggested that these virulence factors contribute to host cell lysis (Yang et al. 2015). The underlying mechanisms are unclear, but it was suggested that PGP4, likely non-directly, promotes inclusion rupture by destabilizing the actin cage that provides the inclusion with mechanical support (Yang et al. 2015). While CPAF is known to cleave the intermediate filament protein vimentin, which could further destabilize the cytoskeletal support of the inclusion (Kumar and Valdivia 2008), recent work demonstrated that CPAF-dependent vimentin cleavage in infected cells occurs only post-inclusion lysis and that inclusion lysis was observed also during infection with a CPAF-deficient mutant (Snavely et al. 2014). Moreover, CPAF cannot represent the protease that Hybiske and Stephens found to contribute to inclusion rupture (Hybiske and Stephens 2007), because CPAF is a serine protease that is insensitive to the E64 protease inhibitor (Paschen et al. 2008).
Another Chlamydia factor that was proposed to contribute to inclusion and/or host plasma membrane lysis is the C. trachomatis protein CT153, which contains a membrane attack complex/perforin (MACPF) domain and was proposed to have pore-forming activity (Taylor et al. 2010; Taylor and Nelson 2014). The absence of functional CT153 orthologs from genomes of multiple Chlamydia strains, including strains of C. pneumoniae, C. caviae and C. abortus (Taylor and Nelson 2014), precludes a universal role of the MACPF domain-containing protein in Chlamydia exit. However, the recent generation of MACPF domain-containing protein deficient transposon insertion mutants, both in C. trachomatis and C. muridarum (LaBrie et al. 2019; Wang et al. 2019), now provides the opportunity for experimental clarification of its role in these species.
Late stage host cell death partially represents a host cell-driven process
To study exit of C. trachomatis L2 from infected HeLa cells, Kerr and colleagues developed an approach that was similar to that described before by Hybiske and Stephens and led to comparable observations, although nuclear disruption was not observed (Kerr et al. 2017). The authors moreover used multiphoton ablation to selectively rupture chlamydial inclusions inside infected cells (Kerr et al. 2017). This procedure revealed that inclusion rupture at any studied time point during infection resulted in subsequent host plasma membrane rupture, which, like native egress (Hybiske and Stephens 2007), was dependent on intracellular calcium signaling (Kerr et al. 2017). Laser-assisted inclusion rupture at earlier time points resulted in a slower progression to plasma membrane rupture than late stage inclusion disruption, suggesting that higher bacterial load may accelerate the process. Because chloramphenicol treatment and inhibition of CPAF did not affect ablation-triggered cell death, the authors suggested that the second step of host cell lysis, the rupture of the plasma membrane, was a host cell-driven process (Kerr et al. 2017). Moreover, the authors suggested that host calpains might contribute to the first step of host cell lysis, inclusion rupture, because inclusion rupture was delayed in presence of calpain inhibitors (Kerr et al. 2017). A requirement for host factors in lytic exit was further supported by the observation that cycloheximide (CHX), an inhibitor of host protein synthesis, could block or delay the natural egress of C. psittaci and C. trachomatis L2 from epithelial cells (Gibellini, Panaya and Rumpianesi 1998; Yang et al. 2015). However, another study reported that CHX had no significant effect on late stage cell death induced by C. caviae (Ojcius et al. 1998).
Cell death induced by laser ablation of inclusions was not accompanied by the activation of apoptotic caspases and could not be blocked by BAX/BAK-deficiency or Z-VAD-FMK (Kerr et al. 2017). Cell death was also unaffected by the CASP1 inhibitor VX765, RIPK1-deficiency or the RIPK1 inhibitor necrostatin-1 (Kerr et al. 2017). However, because necroptosis can proceed in absence of RIPK1 (Pasparakis and Vandenabeele 2015) and pyroptosis can be induced by other inflammatory caspases not all of which are efficiently blocked by Z-VAD-FMK (Chauvier et al. 2007; Jimenez Fernandez and Lamkanfi 2015), further experimentation is needed to clearly exclude an involvement of the necroptotic and pyroptotic pathways. It also remains an intriguing idea that Chlamydia spp. may co-opt pre-existing host cellular defense programs to trigger their exit from their host cells, as discussed below.
Alternative host cell fates
Infection with Chlamydia spp. not always results in the death of the host cell (Fig. 5). For instance, infected cells, in particular macrophages, may clear infection by mediating pathogen degradation in phagolysosomes or via autophagy (Yong, Chi and Kuo 1987; Sun et al. 2012a) (Fig. 5A). Host cell death does also not occur during persistent infection (Fig. 5B). The term persistent infection in this context refers to the observation that in cell culture certain unfavorable conditions, such as nutrient deprivation or exposure to penicillin or interferon gamma (IFN-γ), can temporarily interrupt the developmental cycle of Chlamydia (Matsumoto and Manire 1970; Beatty, Byrne and Morrison 1993; Coles et al. 1993). Under these conditions, the bacteria can survive inside their host cell for extended periods of time without undergoing division, differentiating into EBs, or causing host cell lysis (Matsumoto and Manire 1970; Beatty, Byrne and Morrison 1993; Coles et al. 1993; Perfettini et al. 2002a; Foschi et al. 2020). Persistent bacteria often appear as abnormally enlarged RBs, also known as aberrant bodies (ABs) (Weiss 1950; Beatty, Byrne and Morrison 1993; Coles et al. 1993). When conditions improve, persistent bacteria can eventually resume cell division and progression through the development cycle (Matsumoto and Manire 1970; Beatty, Byrne and Morrison 1993; Coles et al. 1993), which also includes induction of host cell lysis (Perfettini et al. 2002a; Skilton et al. 2009; Foschi et al. 2020). In vivo, this stress program may contribute to long-term persistence of Chlamydia in the infected host and to recurrent infections (Beatty, Morrison and Byrne 1994; Schuchardt and Rupp 2016).
Figure 5.
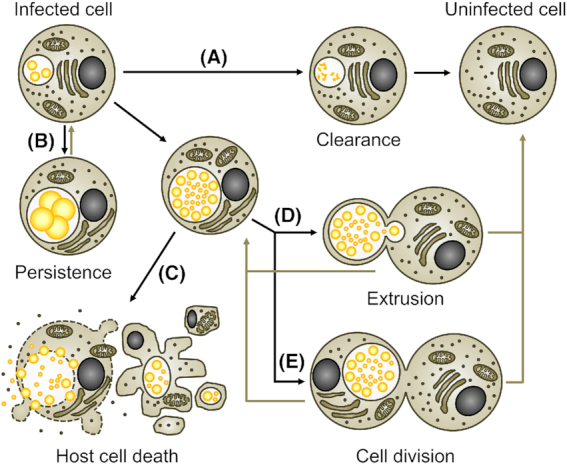
Alternative fates of Chlamydia-infected cells. The encounter with Chlamydia does not always lead to the death of the infected cell. (A) Under certain conditions, in particular after invasion of a professional phagocyte, Chlamydia infection can be cleared by the host cell, for instance by phagolysosomal destruction of the pathogen. (B) Under unfavorable growth conditions, Chlamydia’s progress through the developmental cycle is blocked and the bacteria can persist inside the viable host cell for prolonged periods of time without inducing host cell death. Under favorable growth conditions in permissive host cells, Chlamydia can complete its developmental cycle and form infectious EBs that are released from host cells through either induction of host cell death (C) or extrusion (D). During extrusion, host cells remain viable. Complete extrusion can lead to the formation of inclusion-free cells. (E) In some instances, host cell mitosis can give rise to one infected and one inclusion-free daughter cell, while in other instances two infected daughter cells can arise.
Besides bacterial egress involving cell death (Fig. 5C), Chlamydia can also be released from host cells by a mechanism that maintains host cell viability (Fig. 5D). Early studies described this process as exocytotic release, extrusion of Chlamydia vacuoles or liberation of bacteria in cytoplasmic fragments surrounded by cell membranes (Doughri, Storz and Altera 1972; de la Maza and Peterson 1982; Todd and Caldwell 1985). This extrusion process was studied in greater detail by Hybiske and Stephens (Hybiske and Stephens 2007). Extrusions are bacteria-filled vesicles that pinch off from infected cells. They are bound by membrane derived from the host plasma membrane and contain bacteria encased in an intact inclusion surrounded by a layer of host cell-derived cytoplasm (Hybiske and Stephens 2007). During extrusion, the infected cell may release the entire load of intracellular bacteria or may retain a smaller inclusion (Hybiske and Stephens 2007). While host cells survive the process, it is possible that cells that retain parts of the inclusion will proceed to host cell lysis later. Similarly, released extrusions will eventually lyse to release EBs so that these can infect new host cells. Extrusions also display phosphatidylserine at their surface, which was shown to facilitate uptake of extrusions by professional phagocytes in a manner analogous to the clearance of apoptotic bodies (Zuck et al. 2017). While this process enabled enhanced pathogen survival in the phagocytes, likely by preventing phagocyte activation, and was suggested to enable the bacteria to exploit these cells as vehicles for dissemination, it did only infrequently result in productive infection of the phagocytes (Sherrid and Hybiske 2017; Zuck et al. 2017). Like the above-mentioned clearance of infection by intracellular degradation of the bacteria, complete extrusion represents another incident in which infected cells can give rise to inclusion-free cells. A third event that can lead to this outcome is the mitotic division of an infected cell, which often gives rise to one uninfected daughter cell (Campbell, Richmond and Yates 1989) (Fig. 5E).
While the majority of the cultured environmental chlamydiae also cause host cell death at the end of their developmental cycle (Kahane et al. 1999; Greub and Raoult 2002; Goy, Croxatto and Greub 2008), examples of delayed cell death or long-term co-existence with the host cell exist. For example, in cultures infected with Simkania negevensis, the process of bacterial replication and EB formation was completed within ∼3 days of infection, yet release of EBs did not occur before day 12 (Kahane et al. 1999; Kahane, Kimmel and Friedman 2002). It is possible that this species lacks a bacterial factor involved in the induction of host cell death. One candidate may be CPAF, as the genome of S. negevensis does not encode a CPAF homolog (Collingro et al. 2011). Further experimentation is needed to test this idea. In the case of the Parachlamydiaceae, which naturally infect free-living amoebae, the growth of the bacteria may be synchronized with the replication of their host (Horn 2008). For instance, depending on the host strain and growth temperature, infection of Acanthamoeba spp. with Protochlamydia amoebophila or Parachlamydia acanthamoebae either resulted in host cell lysis or enabled the establishment of stable co-cultures (Fritsche, Sobek and Gautom 1998; Greub, La Scola and Raoult 2003b).
Overall, the impact of these alternative host cell fates and lifestyles on bacterial replication and survival, host adaptation, pathogenesis and immune responses is only insufficiently understood.
Replication-independent cytotoxicity of Chlamydia spp.
Chlamydia toxicity in mice
Chlamydia spp. also have a cytotoxic potential that is independent of their ability to establish infection and that has historically been discussed in relation to their in vivo toxicity. In the early 1940s, Rake and co-workers demonstrated that intravenous injection of Chlamydia (C. trachomatis LGV, C. muridarum or C. psittaci) caused rapid death in mice (Rake and Jones 1943; Rake and Jones 1944). A major proportion of the mice showed signs of toxemia and died within 4–24 h post-inoculation (Rake and Jones 1944). Chlamydia muridarum and C. psittaci, but not C. trachomatis, caused a biphasic curve of death, as some mice that survived the early period after inoculation died up to few weeks later. Yet, in contrast to the deaths that occurred rapidly, these delayed deaths were preceded by typical signs of infection, such as ruffled fur, hunched back and loss of weight (Rake and Jones 1944). Toxicity for mice upon intravenous inoculation was later confirmed by others for various strains of Chlamydia (Manire and Meyer 1950; Bell, Snyder and Murray 1959; Wang and Grayston 1963; Taverne, Blyth and Reeve 1964).
The above-mentioned observations led to the idea that Chlamydia spp. produce a toxin (Rake and Jones 1944). This hypothetical toxin was suggested to be of low potency, because killing of mice required injection of very high infection doses (Rake and Jones 1944). The 'toxin' could not be separated from the bacteria and any procedure that affected the infectivity of the inoculum, for example formalin treatment or prolonged extracellular incubation, also diminished its toxicity (Rake and Jones 1944). Experiments with purified EBs, RBs and EB cell walls of C. psittaci indicated that only the EB form was toxic (Christoffersen and Manire 1969). Moreover, antisera generated against formalin-killed bacteria or during infection with sublethal doses could inactivate the 'toxin' and protect mice from death when they were administered together with toxic doses of intravenously injected bacteria or when they were used to pretreat bacterial suspensions before injection into mice (Rake and Jones 1944; Bell, Snyder and Murray 1959; Wang and Grayston 1963).
Immediate cytotoxicity of high multiplicities of infection
In 1976, Moulder and colleagues proposed that the above-described rapid killing of mice can be explained by direct physical damaging of cells resulting from the ingestion of high numbers of bacteria (Moulder et al. 1976). The authors showed that high doses of C. psittaci caused rapid death of cultured L cells (murine fibroblasts), a phenomenon that was named immediate cytotoxicity (Moulder et al. 1976). Indeed, when cells were treated with 500–1000 ID50 per cell, where ID50 is the dose required to establish infection in 50% of the cells, morphological changes such as rounding were observed as early as 30 min after infection (Moulder et al. 1976). Cell monolayers were completely destroyed at 24 h post-infection (hpi) (Moulder et al. 1976) and the cells released inorganic ions, an indicator of necrotic death (Chang and Moulder 1978). Immediate cytotoxicity of necrotic nature and variable strength was also observed after infection with high doses of C. trachomatis or C. muridarum, and in other cell types, such as in HeLa cells and murine macrophages (Moulder et al. 1976; Kuo 1978; Wyrick, Brownridge and Ivins 1978).
Chloramphenicol and rifampin, inhibitors of bacterial protein synthesis and transcription, respectively, did not diminish toxicity of high doses of C. psittaci for L cells (Moulder et al. 1976). Moreover, while UV-inactivated bacteria could invade host cells and cause toxicity, treatments that prevented bacterial entry, such as heat-inactivation of the bacteria or low temperature during inoculation, blocked immediate toxicity of Chlamydia (Moulder et al. 1976; Kuo 1978). Pretreatment of C. psittaci with antisera that had been raised against C. psittaci could also block both its infectivity and immediate toxicity (Moulder et al. 1976). It thus seemed that immediate cytotoxicity of C. psittaci, like its rapid toxicity for mice, was independent of bacterial replication, yet it was dependent on host cell invasion.
When L cells were infected with lower doses of C. psittaci (10–100 ID50 per cell), both multiplication-independent and multiplication-dependent toxicity were observed, while at doses below 10 ID50 per cell, multiplication-independent toxicity disappeared and induction of host cell death was dependent both on entry and intracellular bacterial replication (Kellogg, Horoschak and Moulder 1977; Chang and Moulder 1978). Moreover, at these low doses, host cell damage was only apparent at ∼48–72 hpi, reflecting late stage host cell death (Kellogg, Horoschak and Moulder 1977; Chang and Moulder 1978).
The idea that immediate cytotoxicity was solely a consequence of physical damage caused by ingestion of bacteria was challenged by the finding that different chlamydial species and strains displayed a distinct toxic potential (Belland et al. 2001). Moreover, while heat-inactivation of C. psittaci abolished its ability to induce immediate toxicity in murine macrophages, the treatment rather enhanced than prevented uptake of the bacteria by these cells and phagocytosis of equivalent numbers of latex beads was non-toxic to the cells (Wyrick, Brownridge and Ivins).
Role of LPS in replication-independent toxicity of Chlamydia
Like in other gram negative bacteria, the outer membrane of Chlamydia spp. contains a form of LPS (Nurminen et al. 1983). LPS is an endotoxin that after release into the bloodstream can cause severe systemic inflammatory reactions, leading to fever, endotoxin shock, tissue injury and death (Galanos and Freudenberg 1993). In this context, LPS mainly acts indirectly through activation of immune cells, which for instance also stimulates secretion of the cytotoxic cytokine TNFα (Galanos and Freudenberg 1993). Chlamydial LPS was shown to possess comparably low endotoxic activity, which was proposed to be attributed to its structural characteristics, such as the higher hydrophobicity of its lipid A moiety (Brade et al. 1986; Ingalls et al. 1995; Heine et al. 2003; Yang et al. 2019).
A potential involvement of LPS in Chlamydia’s toxicity for mice and in immediate cytotoxicity was studied by Ivins and Wyrick (Ivins and Wyrick 1978). After injection of high doses of C. psittaci, similar levels of mortality were observed in the endotoxin-resistant mouse strain C3H/HeJ compared with endotoxin-sensitive C3H/HeN mice. When macrophages derived from these mice were challenged with C. psittaci, only a slight reduction in immediate toxicity was observed in cells derived from C3H/HeJ mice (Ivins and Wyrick 1978). The authors further argued that a major role for LPS in Chlamydia toxicity appeared unlikely, because LPS is highly heat-stable (Magalhaes et al. 2007), whereas the toxic potential of Chlamydia was heat-labile (Rake and Jones 1944; Moulder et al. 1976; Wyrick, Brownridge and Ivins 1978). A more recent study also showed that intraperitoneal injection of purified C. trachomatis E LPS was non-toxic to mice, even when used at 100 times higher amounts than toxic doses of E.coli LPS (Yang et al. 2019).
Interestingly, C. trachomatis LPS was shown to affect the viability of human sperm with 500 times higher potency than E.coli LPS (Galdiero et al. 1994; Hosseinzadeh, Pacey and Eley 2003). Sperm death occurred rapidly, was accompanied by apoptotic effector caspase activation, and could be partially blocked by Z-VAD-FMK (Eley et al. 2005). However, while the toxicity of LPS purified from C. trachomatis E or C. trachomatis LGV was comparable in strength, purified EBs of C. trachomatis E were significantly more toxic than EBs of C. trachomatis LGV (Hosseinzadeh, Pacey and Eley 2003). It was thus suggested that also the rapid toxicity for sperm cells could not be explained by LPS alone.
Role of bacterial proteins in replication-independent toxicity of Chlamydia
Chlamydia spp. produce several proteins that are cytotoxic for human cells upon contact or when ectopically expressed inside the cells. For instance, exposure to the C. trachomatis heat shock proteins HSP60 and HSP10 induced rapid death in human fibroblasts and in epithelial cells (Equils et al. 2006; Jha et al. 2011). This cell death appeared to be of apoptotic nature (Equils et al. 2006; Jha et al. 2011). Chlamydia genomes also encode a protein named Chlamydia protein associated with death domains (CADD), which has a domain that is homologous to death domains found in members of the mammalian TNF receptor family (Stenner-Liewen et al. 2002). CADD interacted in vitro with various death receptors and co-localized with CD95/FAS during infection. However, while ectopic expression of CADD in uninfected cells caused caspase-dependent apoptosis, infected cells were resistant (Stenner-Liewen et al. 2002). It is unknown whether the above-mentioned proteins could contribute to the immediate toxicity of Chlamydia spp., yet it is unlikely, because immediate cytotoxicity caused by high doses of bacteria appears to be a non-apoptotic form of cell death. Moreover, although the Chlamydia protease CPAF induced non-apoptotic cell death when expressed in active form ectopically in the cytosol of uninfected cells (Paschen et al. 2008), and may play a role in Chlamydia exit (Yang et al. 2015), a role in immediate cytotoxicity is less likely, because the protease, or at least the bulk of the protease present in infected cells, was shown to enter the host cell cytosol only at late stages of infection (Snavely et al. 2014).
The analysis of the C. muridarum genome revealed the presence of three genes (TC0437, TC0438, TC0439) that encode proteins with significant homology to the large cytotoxins A and B of Clostridium difficile (Belland et al. 2001; Carlson et al. 2004). These clostridial toxins act as UDP-glucosyltransferases and interfere with the activity of RHO family GTPases, causing disruption of the actin cytoskeleton, cell rounding and eventually cell death (Carter, Rood and Lyras 2010). The amino acid residues that mediate UDP-glucose binding and glucosyltransferase activity are well conserved in the C. muridarum toxin homologs (Belland et al. 2001; Carlson et al. 2004). Interestingly, the cytotoxin locus differs significantly between Chlamydia spp. (Belland et al. 2001; Carlson et al. 2004). In C. trachomatis, only fragmented and/or truncated homologs of TC0438 could be found and homologs of TC0437 or TC0439 were absent. Significant variability could also be seen between distinct strains and potential correlations between toxin genotypes and disease groups were observed (Belland et al. 2001; Carlson et al. 2004). Genitotropic strains appeared to carry a gene (CT166 in C. trachomatis D) that encodes the intact N-terminal part of the toxin, including both the UDP-glucose binding and glycosyltransferase domains. In contrast, ocular strains were found to encode a protein that contains only the UDP-glucose binding domain. Finally, in genomes of LGV strains both domains were absent (Belland et al. 2001; Carlson et al. 2004).
A possible connection between toxin genes and immediate cytotoxicity was proposed based on the observation that the morphological and cytoskeletal changes induced by high doses of Chlamydia were similar than those induced by the clostridial toxins (Belland et al. 2001). In addition, a correlation between strength of immediate toxicity and the presence of intact toxin genes was observed (Belland et al. 2001). Chlamydia muridarum mutants that have nonsense mutations in the toxin genes TC0437 or TC0439 also displayed a slightly reduced immediate toxicity and induced less pathology in a murine genital tract infection model (Rajaram et al. 2015). While limited conclusions could be drawn from this study, due to the presence of additional mutations in these strains, these data demonstrated that neither of these toxins alone was sufficient to explain the observed cytotoxicity. Moreover, the fact that LGV strains of C. trachomatis typically do not contain an intact toxin gene, but still induce some degree of immediate cytotoxicity (Moulder et al. 1976), and cause death in mice (Rake and Jones 1944), suggests that the multiplication-independent toxicity of high doses of Chlamydia could be caused by a combination of different factors. It should be noted that the toxin genotype of LGV strains may also be variable. For instance, a recent study reported the isolation of a LGV strain that appeared to be a recombinant between C. trachomatis L2 and D strains, carried an intact toxin gene (CT166) and was more cytotoxic towards cultured cells than C. trachomatis L2 (Somboonna et al. 2011).
An important question that remains is whether the multiplication-independent toxicity of Chlamydia is significant for disease manifestation. For instance, it is possible that the products of the Chlamydia toxin genes, when present at low amounts, are non-toxic, but rather have specific roles in modulating host cellular processes in favor of bacterial entry, survival and replication. Consistent with this idea, it was shown that CT166 from C. trachomatis D acts on the small GTPases RAC, RHOA and RAS and thereby modifies various cellular processes, such as cell proliferation and cell migration (Thalmann et al. 2010; Bothe et al. 2015). However, cells may encounter high doses of Chlamydia when they are exposed to neighboring infected cells or extrusions that lyse and release their entire load of bacteria. In this context, immediate toxicity may contribute to tissue damage and inflammation and potentially pathogen dissemination. The molecular nature of cell death induced by high doses of Chlamydia and the potential involvement of host RCD and defense programs also needs to be further explored.
Induction of cell death in bystander cells
Apart from multiplication-dependent and -independent effects on the viability of infected cells, Chlamydia spp. can also affect the fate of uninfected cells. Dual staining for Chlamydia inclusions and hallmarks of apoptosis in epithelial cell cultures infected with various Chlamydia species and strains indicated enhanced levels of apoptosis in inclusion-free bystander cells (Ojcius et al. 1998; Schöier et al. 2001; Greene et al. 2004). Augmented levels of apoptosis in inclusion-free cells were also observed in the genital tract of mice infected with C. muridarum (Perfettini et al. 2000). Furthermore, co-culture with infected macrophages induced apoptosis in uninfected T cells (Jendro et al. 2000; Jendro et al. 2004; Sessa et al. 2009). Several studies suggested that apoptosis in bystander cells is induced by soluble factors, such as TNFα, interferon alpha (IFN-α) and interferon beta (IFN-β), which are secreted by infected cells. For instance, cell-free supernatants of infected macrophage cultures were sufficient to induce apoptosis in T cells and this T cell apoptosis could be blocked by TNFα depletion (Jendro et al. 2004; Sessa et al. 2009). Moreover, depletion of TNFα in C. muridarum-infected mice reduced the incidence of apoptosis in uninfected cells at the site of infection (Perfettini et al. 2000). TNFα depletion in mice and guinea pigs also increased the numbers of inflammatory cells in infected tissues, likely by preventing their death (Darville, Andrews and Rank 2000). Similarly, reduced levels of macrophage apoptosis were observed in lung tissues of C. muridarum-infected interferon-α/β receptor (IFNAR)-deficient mice compared with wild-type mice (Qiu et al. 2008). Besides possible direct contributions to tissue damage, inflammation and post-infection sequelae, Chlamydia-induced cell death in bystander cells may therefore also impair anti-chlamydial immune responses via depletion of immune cells.
ANTI-DEATH ACTIVITIES OF CHLAMYDIAE
Chlamydia-mediated inhibition of apoptosis
Premature host cell death disrupts Chlamydia development
The reports that late stage host cell death induced by Chlamydia spp. proceeds in the absence of caspase activation are in good agreement with the observation that Chlamydia spp. inhibit the apoptotic machinery in infected cells (Fig. 6). This phenomenon was first described in 1998 (Fan et al. 1998), attracted a lot of attention and was the subject of intensive investigation (Perfettini et al. 2003b; Byrne and Ojcius 2004; Häcker, Kirschnek and Fischer 2006; Ying et al. 2007; Sharma and Rudel 2009). While newer findings challenged this idea, as discussed below, the anti-apoptotic trait was proposed to enable the pathogen to maintain host cell viability in situations of cellular stress. Premature host cell death can be detrimental for Chlamydia spp., because the bacteria depend on a host cell that provides a suitable replicative niche. Moreover, EBs are only formed during the late stage of the developmental cycle and RBs are non-infectious and fragile (Tamura, Matsumoto and Higashi 1967). Consistently, it was shown that experimental induction of apoptosis at mid-stage of infection could abrogate formation of infectious bacteria (Ying et al. 2008b). In this system, apoptosis was triggered by inducible overexpression of the pro-apoptotic protein BCL-2-like protein 11 (BIM) in HeLa cells infected with C. trachomatis L2 (Ying et al. 2008b). While Z-VAD-FMK blocked the morphological signs of apoptosis in these cells, it failed to restore normal production of infectious EBs (Ying et al. 2008b). This suggested that even in the absence of caspase-mediated cell demolition, partial induction of the apoptotic pathway, causing MOMP and hence mitochondrial dysfunction, can also impair chlamydial development (Ying et al. 2008b).
Figure 6.
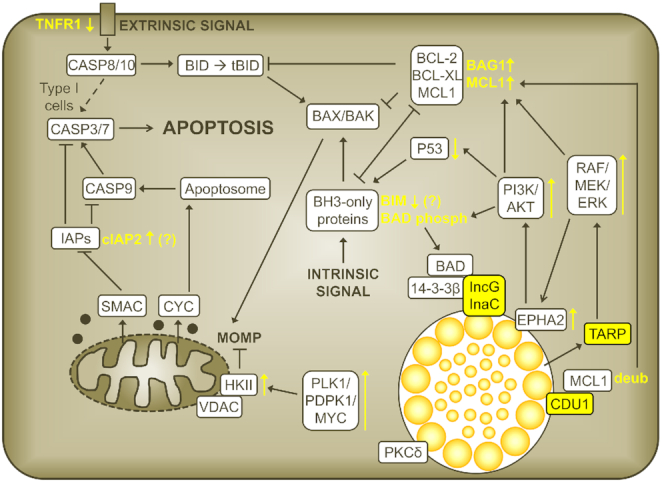
Anti-apoptotic activities of C. trachomatis. The illustration gives an overview of the main anti-apoptotic activities described for C. trachomatis. Chlamydia factors and Chlamydia-mediated activities are indicated in yellow. Upward facing arrows next to factors or pathways indicate upregulation or activation; downward facing arrows indicate downregulation. Note that the anti-apoptotic activities of Chlamydia spp. were shown to be species specific. For instance, NFκB activation was reported to play a role in apoptosis inhibition during infection with C. pneumoniae, but not during infection with C. trachomatis. It is therefore not shown.
Overview of chlamydial anti-apoptotic activities
Cells infected with C. trachomatis or C. pneumoniae were shown to be profoundly protected against apoptosis induced by various experimental stimuli. These include conditions that cause intracellular stress and are known to induce the intrinsic pathway of apoptosis in uninfected cells [for instance, UV-irradiation, the DNA-damaging drug etoposide and the kinase inhibitor staurosporine (STS)] (Fan et al. 1998; Dean and Powers 2001; Fischer et al. 2001; Rajalingam et al. 2001; Airenne et al. 2002; Fischer et al. 2004b; Greene et al. 2004). Furthermore, infected cells were also protected against immunological mediators of cell death that activate the extrinsic pathway of apoptosis, such as TNFα and CD95L/FASL (Fan et al. 1998; Fischer et al. 2001; Rajalingam et al. 2001; Airenne et al. 2002; Fischer et al. 2004a). Resistance to granzyme B/perforin (GRB/PRF)-mediated killing and to poly(I:C)-induced apoptosis was also reported (Fan et al. 1998; Böhme et al. 2009). Protection against pro-apoptotic stimuli was also observed during infection with other Chlamydia spp., including for example C. psittaci, C. caviae and C. muridarum, albeit the degree of protection appeared to vary (Fan et al. 1998; Greene et al. 2004; Zhong et al. 2006; Messinger et al. 2015). Furthermore, apoptosis inhibition by Chlamydia spp. was observed in a variety of cell lines and primary cells from diverse origins, including epithelial cells, fibroblasts, endothelial cells, monocytes and lymphoid cells (Fan et al. 1998; Dean and Powers 2001; Fischer et al. 2001; Rajalingam et al. 2001; Airenne et al. 2002; Fischer et al. 2004a,b; Greene et al. 2004), and not only during active but also during persistent infection (Dean and Powers 2001; Airenne et al. 2002; Paland et al. 2006; Li et al. 2018).
When Chlamydia-infected cells were exposed to the above-mentioned stimuli, they failed to develop typical characteristics of apoptotic cells, such as nuclear condensation and/or fragmentation, DNA double-strand breaks, nucleosomal DNA degradation, proteolytic activation of effector CASP3 and effector caspase activity (Fan et al. 1998; Dean and Powers 2001; Fischer et al. 2001; Rajalingam et al. 2001; Airenne et al. 2002; Fischer et al. 2004a,b; Greene et al. 2004). In infected cultures, protection was restricted to cells carrying considerable inclusions, while inclusion-free cells and cells containing very small inclusions were still susceptible to apoptosis induction (Fan et al. 1998; Rajalingam et al. 2001; Fischer et al. 2004b; Xiao et al. 2004; Xiao et al. 2005; Zhong et al. 2006). When infections were carried out at low multiplicities of infection (MOI), strong resistance to apoptosis-inducing conditions was usually established until ∼24 hpi and was maintained until the end of the infection cycle (Fan et al. 1998; Dean and Powers 2001; Rajalingam et al. 2001). However, infections at higher MOIs established the same level of resistance earlier (Fan et al. 1998; Rajalingam et al. 2001). Exposure to heat- or UV-inactivated bacteria failed to mediate apoptosis resistance (Geng et al. 2000; Airenne et al. 2002; Fischer et al. 2004a). Several studies also showed that the anti-apoptotic state could not be established in presence of rifampin or chloramphenicol (Fan et al. 1998; Fischer et al. 2001; Böhme et al. 2009). While some authors interpreted this finding as an indication that apoptosis inhibition depends on bacterial protein synthesis, it should be noted that early addition of these antibiotics would not only inhibit the synthesis of virulence factors, but also entirely prevent the formation of inclusions. Indeed, Fischer et al showed that rifampin was highly effective in blocking C. pneumoniae-mediated protection against apoptosis (induced at 72 hpi) when added at the time of infection, but already significantly less effective when added at 6 hpi and ineffective when added at 24 hpi (Fischer et al. 2001). Furthermore, chloramphenicol could sensitize cells infected with C. trachomatis L2 to poly(I:C)-induced apoptosis (induced at 20 hpi) when added at 2.5 hpi, but not when added at 20 hpi (Böhme et al. 2009). LPC-011, which blocks the synthesis of LPS by inhibiting the enzyme UDP-3-O-(R-3-hydroxymyristoyl)-N-acetylglucosamine deacetylase (LPXC), also sensitized cells infected with C. trachomatis to STS-induced apoptosis (Wang, Rockey and Dolan 2020). Yet the inhibitor is also known to induce abnormal bacterial development and to block inclusion formation when used at high concentrations (Nguyen et al. 2011).
Inhibition of apoptosis at the pre-mitochondrial and at the post-mitochondrial levels
Chlamydia spp. block the apoptosis machinery at various levels. When infected cells were exposed to stimuli that activate the intrinsic pathway, apoptosis was inhibited at a level upstream of MOMP. Activation of BAX and BAK did not occur in infected cells (Fischer et al. 2004b; Xiao et al. 2004; Paland et al. 2006; Zhong et al. 2006). Consequently, there was no release of CYC from mitochondria (Fan et al. 1998; Dean and Powers 2001; Fischer et al. 2001; Rajalingam et al. 2001; Airenne et al. 2002; Fischer et al. 2004a,b; Greene et al. 2004; Xiao et al. 2004) and no activation of CASP9 (Fischer et al. 2001, 2004a; Xiao et al. 2005) or CASP3 (Fan et al. 1998; Fischer et al. 2001; Rajalingam et al. 2001; Xiao et al. 2004). However, addition of CYC to cell extracts derived from C. pneumoniae-infected cells also failed to induce CASP9/3 activation, suggesting that infection also blocked apoptosis at a level downstream of the mitochondria (Fischer et al. 2001).
In the context of the extrinsic pathway, most studies suggested that Chlamydia spp. fail to block early apoptotic signaling events activated by death receptors (Fischer et al. 2004a; Paland et al. 2006; Rajalingam et al. 2006; Sixt et al. 2018). These events include the activation of CASP8 and the cleavage of BID, which connects the extrinsic pathway with the intrinsic pathway. Indeed, Chlamydia failed to protect type I cells, in which CASP8 activation is sufficient to activate the apoptotic effector caspases (Kantari and Walczak 2011), from CD95L/FASL-mediated apoptosis (Fischer et al. 2004a). However, Chlamydia infection could protect type II cells (Fischer et al. 2004a), in which a mitochondrial amplification of the apoptotic signal by tBID-mediated activation of BAX/BAK and induction of MOMP is required (Kantari and Walczak 2011). In these cells, exposure to CD95L/FASL (anti-FAS antibody) or TNFα/CHX did not result in BAX/BAK activation, release of mitochondrial CYC or activation of CASP9/3 (Fan et al. 1998; Rajalingam et al. 2001; Fischer et al. 2004a; Xiao et al. 2005; Paland et al. 2006).
Induction of survival signaling pathways
Chlamydia trachomatis activates several survival signaling pathways in infected cells. These include for instance the phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) pathway and the RAF/MEK/ERK mitogen-activated protein kinase (MAPK) pathway (Verbeke et al. 2006; Paland et al. 2008; Rajalingam et al. 2008; Du et al. 2011; Kun et al. 2013; Siegl et al. 2014). Importantly, depletion of AKT using specific siRNAs or pharmacologic inhibition of PI3K with LY294002 sensitized infected cells to STS- and GRB-induced apoptosis (Verbeke et al. 2006; Rajalingam et al. 2008). In the presence of LY294002, STS also induced the release of mitochondrial CYC in infected cells, suggesting that the induction of the PI3K/AKT pathway contributes to the apoptotic block at the pre-mitochondrial level (Verbeke et al. 2006). The MEK inhibitor U0126 and the RAF inhibitor GW5074 also sensitized infected cells to STS- and GRB- induced apoptosis (Rajalingam et al. 2008; Du et al. 2011; Kun et al. 2013). Moreover, C. trachomatis L2 also induces the polo-like kinase 1/3-phosphoinositide-dependent protein kinase 1/Myc proto-oncogene (PLK1/PDPK1/MYC) signaling pathway, which was shown to contribute to protection against TNFα/CHX-induced apoptosis (Al-Zeer et al. 2017).
Chlamydia trachomatis induces survival pathways by various means. For instance, a recent study indicated that the host surface receptor Ephrin A2 (EPHA2) acts as receptor for Chlamydia EBs during the early stage of infection, but is subsequently internalized, remains associated with the Chlamydia inclusion and mediates lasting PI3K activation (Subbarayal et al. 2015). The ERK pathway was involved in infection-induced upregulation of EPHA2 and infection-induced AKT activation was found to depend on EPHA2 signaling during mid-phase of infection (Subbarayal et al. 2015). Depletion of EPHA2 sensitized cells to TNFα/CHX-induced apoptosis at 16 hpi (Subbarayal et al. 2015). Moreover, one study linked early ERK activation to the secreted Chlamydia effector protein translocated actin recruiting phosphoprotein (TARP), which phosphorylates Src homology 2 domain-containing-transforming protein C1 (SHC1), which in turn activates the MEK/ERK pathway (Mehlitz et al. 2010). Depletion of SHC1 blocked early ERK activation and sensitized C. trachomatis-infected cells to TNFα/CHX-induced apoptosis at 6 hpi (Mehlitz et al. 2010). However, it should be noted that a very high MOI of 50 had to be used in this experiment to establish protection against apoptosis in control cells at this early time point during infection. Recent studies also suggested that the plasmid-encoded secreted Chlamydia protein PGP3 may contribute to apoptosis inhibition (He et al. 2019), likely via induction of the ERK signaling pathway (Luo et al. 2019).
During infection with other Chlamydia spp., alternative signaling pathways have been implicated in promoting host cell survival. For instance, PI3K inhibition did not sensitize C. pneumoniae-infected HeLa cells to STS-induced apoptosis (Verbeke et al. 2006). Instead, infection with C. pneumoniae induced nuclear factor kappa B (NFκB) activation in human epithelial cells (Paland et al. 2006). CAPE, an inhibitor of NFκB nuclear translocation, as well as siRNA-mediated depletion of the P65 subunit of NFκB, sensitized infected cells to TNFα/CHX- and STS-induced apoptosis (Paland et al. 2006). Chlamydia pneumoniae also induced NFκB activation in the human monocytic cell line Mono Mac 6 (Wahl et al. 2001, 2003). Because inhibition of NFκB activation induced apoptosis in these cells, the authors suggested that NFκB activation during infection was important for maintenance of host cell survival (Wahl et al. 2001, 2003). In contrast to these findings, no activation of NFκB, assessed by monitoring of NFκB nuclear translocation and IκB degradation, was observed in human epithelial cells infected with C. trachomatis (Xiao et al. 2005). Furthermore, inhibition of NFκB activation in these cells did not result in sensitization to STS- or TNFα/CHX-induced apoptosis, and P65-deficient C. trachomatis-infected MEFs maintained protection against TNFα/CHX-induced apoptosis (Xiao et al. 2005). In the case of C. psittaci, little is known about the contribution of survival pathways to anti-apoptosis. Yet, induction of the janus kinase /signal transducer and activator of transcription protein 3 (JAK/STAT3) pathway was proposed to contribute to apoptosis resistance during infection with this species (Sun et al. 2017). Moreover, persistent infection of HeLa cells with C. psittaci also activated the ERK pathway and U0126 sensitized infected cells to STS-induced apoptosis (Li et al. 2018).
It should be mentioned that the use of pharmacologic inhibitors for the testing of the contribution of specific signaling pathways to Chlamydia anti-apoptosis is complicated by the fact that certain inhibitors, at least at high concentrations and when added early during infection, affect chlamydial growth. Because cells that contain only small inclusions are not well protected from apoptosis (Rajalingam et al. 2001; Zhong et al. 2006), it is plausible that each condition that blocks inclusion establishment or growth may also interfere with Chlamydia anti-apoptosis, independently of whether the drug target is involved in apoptosis inhibition or not. While some authors included respective controls, this issue was not accounted for in all studies.
Interference with pro-apoptotic BCL-2 family proteins
Various explanations for the profound Chlamydia-mediated block of the apoptotic machinery upstream of MOMP have been proposed. The majority of these suggest that Chlamydia interferes with the balance between pro-apoptotic BH3-only proteins and anti-apoptotic BCL-2 family proteins, which controls the activation state of BAX and BAK and thus regulates MOMP. Initially, based on the analysis of host protein levels in lysates of infected cells, it was proposed that Chlamydia spp. cause the degradation of BH3-only proteins (Fischer et al. 2004b; Dong et al. 2005; Ying et al. 2005). Degradation of BIM appeared to start at ∼14–16 hpi and to be complete at ∼24–26 hpi (Fischer et al. 2004b), a time point that correlated well with the time at which potent protection against apoptosis was established in infected cells. Similar observations were made for other BH3-only proteins, including BCL-2-binding component 3 (PUMA), BCL-2-modifying factor (BMF), Phorbol-12-myristate-13-acetate-induced protein 1 (NOXA), BCL-2-interacting killer (BIK) and tBID (Fischer et al. 2004b; Dong et al. 2005; Ying et al. 2005). BCL-2-associated agonist of cell death (BAD) was reported to be degraded in some studies (Fischer et al. 2004b; Ying et al. 2005; Verbeke et al. 2006), but not in others (Dong et al. 2005). The loss of BH3-only proteins was explained by proteolytic activity, because it could be prevented by addition of proteasome inhibitors and because mRNA levels appeared to be unaffected (Fischer et al. 2004b; Ying et al. 2005). However, other authors reported that the levels of BH3-only proteins remained unchanged during infection (Rajalingam et al. 2008). Today, it is clear that the occasionally observed degradation was caused by the Chlamydia protease CPAF, yet only post-lysis in sample buffer when conditions used for the generation of protein samples were inappropriate for blocking residual CPAF activity (Chen et al. 2012).
While the broad degradation of BH3-only proteins was revealed as an experimental artifact, there is still a debate concerning BIM, which one group reported to be degraded even when samples were prepared in a way that prevented CPAF post-lysis activity (Dille et al. 2015). Furthermore, Chlamydia infection was also proposed to interfere with BH3-only proteins in other ways. For instance, infection with C. trachomatis L2 caused phosphorylation of BAD, which promotes its association with the host protein 14-3-3β, which in turn was recruited to the Chlamydia inclusion (Verbeke et al. 2006). Significantly, inhibition of PI3K with LY294002, which sensitized cells to STS-induced apoptosis, blocked BAD phosphorylation and recruitment (Verbeke et al. 2006). The authors proposed that BAD sequestration at the inclusion interferes with its pro-apoptotic function. Recruitment of 14-3-3β was proposed to be mediated by the Inc protein IncG (Scidmore and Hackstadt 2001). Consistent with this idea, C. pneumoniae, a species that lacks an IncG homolog, did not recruit 14-3-3β (Scidmore and Hackstadt 2001) and protected infected cells from STS-induced apoptosis in a PI3K-independent manner (Verbeke et al. 2006). However, genetic evidence from experiments with a C. trachomatis IncG mutant still awaits to be collected. It is possible that other bacterial factors could also be involved. For instance, reduced recruitment of 14-3-3β to inclusions was observed during infection with a C. trachomatis mutant that was deficient for an Inc protein named inclusion membrane protein for actin assembly (InaC) (Kokes et al. 2015).
Interference with anti-apoptotic BCL-2 family proteins
Infection with C. trachomatis L2 did not affect the expression or stability of the anti-apoptotic BCL-2 family proteins BCL-2 or B-cell lymphoma-extra-large (BCL-XL) (Dong et al. 2005; Ying et al. 2005). However, levels of induced myeloid leukemia cell differentiation protein (MCL1), another anti-apoptotic member of the BCL-2 family, were strongly upregulated in infected human epithelial cells, at both the mRNA and the protein level (Hess et al. 2001; Rajalingam et al. 2008). The MEK inhibitor U0126 reduced MCL1 mRNA and protein levels and the PI3K inhibitor LY294002 reduced MCL1 protein levels, suggesting a RAF/MEK/ERK-pathway-dependent transcriptional upregulation of MCL1 and a PI3K-dependent stabilization of MCL1 protein levels in infected cells (Rajalingam et al. 2008). Stabilization of MCL1 in C. trachomatis-infected cells was also shown to be a consequence of reduced MCL1 ubiquitination and hence reduced proteosomal degradation (Fischer et al. 2017). This appeared to be mediated by the Chlamydia deubiquitinating enzyme 1 (CDU1), which localizes to the inclusion membrane (Fischer et al. 2017). Co-immunoprecipitation experiments and in vitro binding assays indicated that CDU1 and MCL1 directly interact with each other and an in vitro deubiquitination assay confirmed that MCL1 is a substrate for CDU1 (Fischer et al. 2017). While MCL1 levels were reduced during infection with a CDU1-deficient mutant of C. trachomatis, compared with infection with wild-type bacteria, MCL1 levels were still increased compared with uninfected cells and cells infected with the mutant were not sensitized to TNFα/CHX-induced apoptosis (Fischer et al. 2017). This finding can likely be explained by Chlamydia using redundant mechanisms to maintain elevated MCL1 levels. Indeed, depletion of MCL1 using siRNAs or shRNAs could sensitize C. trachomatis-infected cells to apoptosis induced by STS, TNFα/CHX or GRB (Rajalingam et al. 2008), at least at the mid-stage of infection (24 hpi). No sensitization was seen at later stages of infection (48 hpi) or in cells containing large inclusions (Rajalingam et al. 2008). Contrasting with the results reported for human cells, in MEFs, MCL1 deficiency only caused a mild sensitization to STS-induced apoptosis (Ying et al. 2008a). However, enhanced levels of MCL1 were also seen in HeLa cells that were persistently infected with C. psittaci and in human neutrophils that were infected with C. pneumoniae (Sarkar et al. 2015; Li et al. 2018).
Besides MCL1, other anti-apoptotic proteins may be upregulated during infection and may contribute to maintenance of host cell survival. For example, one study showed that C. trachomatis (and C. muridarum) infection increased the levels of BCL-2-associated athanogene 1 (BAG1), a BCL-2 binding protein, in a manner dependent on the RAF/MEK/ERK signaling pathway (Kun et al. 2013). Depletion of BAG1 sensitized infected cells to STS- and TNFα/CHX-induced apoptosis (Kun et al. 2013). An importance of de novo host protein synthesis for Chlamydia anti-apoptosis appears to disagree with the observation that CHX did not generally sensitize infected cells to apoptosis (Fan et al. 1998; Airenne et al. 2002; Fischer et al. 2004b). It is possible that Chlamydia infection primarily affects levels of anti-apoptotic proteins by protein stabilization, rather than by modulating their expression, or that redundant anti-apoptotic strategies can compensate for effects mediated by CHX.
Inhibition of apoptosis at a post-mitochondrial stage by IAPs
The inhibitors of apoptosis proteins (IAPs) constitute a group of proteins that have a baculovirus IAP repeat (BIR) domain that allows for direct interaction with caspases (Salvesen and Duckett 2002). IAPs regulate caspase activity, for instance by mediating caspase ubiquitination, which promotes proteosomal degradation (Vaux and Silke 2005). During MOMP, the IAP-inhibiting protein second mitochondria-derived activator of caspases (SMAC) is released from mitochondria together with CYC to promote apoptosis (Du et al. 2000; Verhagen et al. 2000). A role for IAPs in Chlamydia-mediated apoptosis inhibition may therefore depend on Chlamydia’s simultaneous capacity to prevent or reduce MOMP. Moreover, conflicting reports on the involvement of IAPs in Chlamydia anti-apoptosis have been published. Infection of human epithelial cells with C. trachomatis L2 was reported to cause transcriptional upregulation of cellular inhibitor of apoptosis protein 2 (cIAP2), but not cellular inhibitor of apoptosis protein 1 (cIAP1) or X-linked inhibitor of apoptosis (XIAP) (Rajalingam et al. 2006). In this study, depletion of cIAP2 using specific siRNAs sensitized infected cells, except such that contained very large inclusions, to TNFα/CHX-induced apoptosis (Rajalingam et al. 2006). Interestingly, sensitization was also observed in cells depleted for cIAP1 or XIAP, an observation that the authors explained with the finding that IAPs act in complexes and can affect the stability of each other (Rajalingam et al. 2006). However, another study reported that neither depletion of IAPs nor addition of SMAC mimetics (IAP inhibitors) sensitized C. trachomatis-infected HeLa cells to TNFα/CHX-induced apoptosis (Waguia Kontchou et al. 2016). Moreover, deficiency for XIAP, cIAP1, cIAP2, or cIAP1 and cIAP2 did not sensitize C. trachomatis-infected MEFs to TNFα/CHX-induced apoptosis (Ying et al. 2008a). In contrast, enhanced levels of cIAP2 mRNA and protein were also observed in human cells infected with C. pneumoniae (Wahl et al. 2003; Paland et al. 2006) and depletion of cIAP2 or cIAP1 sensitized these cells to TNFα/CHX- and STS-induced apoptosis (Paland et al. 2006). In these cells, upregulation of cIAP2 could be blocked by CAPE, an inhibitor of NFκB nuclear translocation, and MG132, a proteasome inhibitor that blocks IκB degradation (Wahl et al. 2003; Paland et al. 2006). It was therefore proposed to be NFκB-dependent in the context of C. pneumoniae infection.
Interference with CASP8 activation
Two studies reported conflicting observations in respect to direct effects of C. trachomatis L2 infection on TNFR1 signaling. One study reported that infection caused a reduction of surface-exposed TNFR1 (Paland et al. 2008). Interestingly, total TNFR1 expression was increased in infected cells, yet more TNFR1 was shed into culture supernatants or was internalized (Paland et al. 2008). These changes were dependent on the induction of the RAF/MEK/ERK signaling pathway (Paland et al. 2008). In contrast, another study showed that C. trachomatis blocked TNFR1 internalization, an event that is required for TNFR1-mediated pro-apoptotic signaling (Waguia Kontchou et al. 2016). Consistent with this idea, but in conflict with reports from other groups (Paland et al. 2006; Rajalingam et al. 2006; Sixt et al. 2018), the authors found that infection blocked TNFα/CHX-induced processing of CASP8 and BID (Waguia Kontchou et al. 2016). A role for cellular FLICE-like inhibitory protein (cFLIP), a regulatory protein that can prevent recruitment and activation of CASP8 (Irmler et al. 1997), in apoptosis inhibition was excluded in this context, because cFLIP depletion did not sensitize infected cells to TNFα/CHX-induced apoptosis (Waguia Kontchou et al. 2016). In contrast, cFLIP was implicated in C. trachomatis’ ability to protect HeLa cells from poly(I:C)-induced apoptosis (Böhme et al. 2009). In this system, infection blocked poly(I:C)-induced activation of CASP8, CASP9 and CASP3, as well as BID cleavage (Böhme et al. 2009). While cFLIP protein levels were not affected by infection, depletion of cFLIP sensitized infected cells to poly(I:C)-induced CASP8 activation (Böhme et al. 2009).
Anti-apoptotic (side) effects of metabolic reprogramming
Chlamydia spp. modulate host cell metabolism to their benefit and do so in part by mechanisms that can modify host cell resistance to apoptosis. For instance, activation of MYC in C. trachomatis-infected cells via the PLK1/PDPK1/MYC signaling pathway was shown to result in upregulation of hexokinase-II (HKII) and enhanced association of HKII with the mitochondrial voltage-dependent anion channel (VDAC) (Al-Zeer et al. 2017). While this can promote glycolysis and oxidative phosphorylation, HKII can thereby also compete with pro-apoptotic signals that could otherwise act on VDAC (Pastorino, Shulga and Hoek 2002; Majewski et al. 2004). Indeed, dissociation of HKII from VDAC using clotrimazole or N-HKII (a competitive peptide), sensitized C. trachomatis-infected cells to TNFα/CHX-induced apoptosis (Al-Zeer et al. 2017).
Chlamydia trachomatis infection was also shown to cause a pronounced reduction in tumor suppressor P53 protein levels in infected cells (Gonzalez et al. 2014; Siegl et al. 2014). This seems to be the result of several redundant mechanisms. For instance, infection induced PI3K/AKT-dependent activation of the E3 ubiquitin-protein ligase MDM2 (also called HDM2 in human cells), a protein that mediates ubiquitination and proteosomal degradation of P53 (Gonzalez et al. 2014; Siegl et al. 2014). Indeed, LY294002, proteasome inhibitors, inhibitors of P53-MDM2 interaction (nutlin3a and RITA), and depletion of MDM2 rescued P53 levels in infected cells (Gonzalez et al. 2014; Siegl et al. 2014). Another study reported that C. trachomatis induced upregulation of miR-30c-5p, a miRNA that is known to downregulate expression of P53 (Chowdhury et al. 2017). Interference with miR-30c-5p function resulted in enhanced levels of P53 in infected cells (Chowdhury et al. 2017). Infection with other Chlamydia spp., such as C. pneumoniae, C. psittaci and C. muridarum, also triggered a drop in P53 levels (Gonzalez et al. 2014; Siegl et al. 2014). However, while a reduction in P53 levels was observed in various human cells, including HeLa cells, HUVECs and primary cells from fallopian tubes, it was not observed in MEFs (Gonzalez et al. 2014; Siegl et al. 2014). Importantly, P53 is a pro-apoptotic protein and nutlin3a partially re-sensitized C. trachomatis-infected HeLa cells to TNFα/CHX-induced apoptosis (Gonzalez et al. 2014). However, experimental interference with P53 downregulation by itself did not induce massive apoptosis in infected cells, but disrupted Chlamydia development (Gonzalez et al. 2014; Siegl et al. 2014). Reduced P53 levels may therefore benefit Chlamydia spp. primarily by contributing to the metabolic reprogramming of the host cell (Siegl et al. 2014).
Another study suggested that C. trachomatis interferes with the pro-apoptotic function of protein kinase C delta (PKCδ) by sequestering the kinase at the Chlamydia inclusion (Tse et al. 2005). Expression of a PKCδ variant that is catalytically active, but that lacks its diacylglycerol binding domain and can thus not be recruited to the inclusion membrane, induced apoptosis in infected cells (Tse et al. 2005). However, in uninfected cells, PKCδ typically localizes to the Golgi apparatus and the expression of the mutant variant that failed to bind lipids induced apoptosis in these cells as well (Tse et al. 2005). It is possible that the sequestration of PKCδ at the Chlamydia inclusion reflects the acquisition of Golgi-derived host lipids by the Chlamydia inclusion, rather than a bona fide anti-apoptotic virulence strategy.
Inhibition of apoptosis by environmental chlamydiae
The anti-apoptotic capacities of environmental chlamydiae are not well explored, yet available literature indicates species-specific differences. The Parachlamydiaceae may lack anti-apoptotic activities and appear to induce apoptosis instead (Greub, Mege and Raoult 2003a; Ito et al. 2012; Sixt et al. 2012; Matsuo et al. 2013; Brokatzky, Kretz and Häcker 2020), as discussed in more detail below. A single study reported that S. negevensis could protect HeLa cells from STS- and TNFα/CHX-induced apoptosis (Karunakaran, Mehlitz and Rudel 2011). Apoptotic signaling was blocked at a pre-mitochondrial step. Indeed, infection prevented activation of BAX and BAK, release of mitochondrial CYC, and activation of CASP9 and CASP3, while CASP8 was still activated in presence of TNFα/CHX (Karunakaran, Mehlitz and Rudel 2011). The authors did not observe changes in the levels of the BH3-only proteins or the anti-apoptotic proteins BCL-2 and MCL1, yet cIAP1 and cIAP2 were upregulated in infected cells (Karunakaran, Mehlitz and Rudel 2011). Furthermore, depletion of cIAP1 and/or cIAP2, as well as inhibition of PI3K sensitized infected cells to TNFα/CHX-induced apoptosis (Karunakaran, Mehlitz and Rudel 2011). Rhabdochlamydia porcellionis, a species that naturally infects arthropod hosts, was shown to inhibit actinomycin D- and UV-induced apoptosis in infected insect cells (Sixt et al. 2013). In contrast, Waddlia chondrophila, a species that has been isolated from aborted bovine fetuses, failed to block STS-induced apoptosis in HeLa cells (Dille et al. 2015).
Significance of Chlamydia-mediated apoptosis inhibition
The profound inhibition of the apoptotic machinery in Chlamydia-infected cells has commonly been explained as an attempt of the bacteria to protect host cells from infection-induced stress and to maintain host cell viability under adverse growth conditions or when the cell is under attack by immune mediators (Ying et al. 2007; Sharma and Rudel 2009). Indeed, a recent study described that in spite of C. trachomatis’ anti-apoptotic trait, low levels of MOMP (known as minority MOMP), were induced in infected cells in a BAX/BAK-dependent manner (Brokatzky et al. 2019). The finding was interpreted as evidence for the presence of pro-apoptotic signals in infected cells, explaining the need for potent bacteria-mediated suppression (Brokatzky et al. 2019).
However, while interference with Chlamydia’s anti-apoptotic strategies could re-sensitize host cells to pro-apoptotic conditions, as described above, it did not cause massive spontaneous cell death in infected cells. Moreover, a recent study in which my co-workers and I monitored the effect of pro-apoptotic conditions on the fate of human epithelial cells infected with C. trachomatis L2 by live cell imaging demonstrated that infected cells were not protected from cell death (Sixt et al. 2018). While apoptosis inducers, such as STS and TNFα/CHX, failed to induce typical hallmarks of apoptosis in infected cells, these cells died by a necrotic type of death accompanied by early loss of plasma membrane integrity. This necrotic death was also not significantly delayed compared with apoptotic death induced by the same conditions in uninfected cells (Sixt et al. 2018). Mechanistically, TNFα/CHX-induced necrosis of infected cells was independent of the necroptotic signaling pathway, but was dependent on CASP8, whose activation was not blocked by infection (Sixt et al. 2018). Consistent with the inability of C. trachomatis to protect its growth niche under the tested pro-apoptotic conditions, pro-apoptotic stimulation abolished formation of infectious progeny. Yet, in the presence of TNFα/CHX, progeny formation could be rescued by inhibition of CASP8 or CASP8 deficiency (Sixt et al. 2018).
While it may seem at first glance puzzling that these observations have not been reported before, it is plausible that the phenomenon has been overlooked, because previous studies primarily focused on the detection of specific hallmarks of apoptosis and used very short periods of incubation with apoptosis-inducing conditions. Moreover, consistent with the findings of our recent study (Sixt et al. 2018), Jungas et al. reported that STS failed to induce apoptosis in Chlamydia-infected cells, but still caused a reduction in cell numbers in cultures of infected adherent cells (Jungas et al. 2004). Further experimentation will be required to test whether similar observations can be made during infection with other Chlamydia spp. and in primary cells. Moreover, a better understanding of the nature of the pro-apoptotic signals that arise during infection is required.
It may also seem counterintuitive that Chlamydia spp. use so many redundant mechanisms to block apoptosis, if these strategies are not sufficient to maintain cell viability. However, it is possible that Chlamydia anti-apoptosis, resulting in preferential host cell death by necrosis, is either primarily a side effect of the metabolic reprogramming of the host cell, or that it has other benefits for the bacteria in vivo. For instance, apoptotic death of infected cells, in contrast to necrotic death, could be expected to lead to preferential uptake by phagocytes, which are not the preferred host cells for Chlamydia, have a high capacity to restrict Chlamydia growth, and can stimulate immune responses for example by antigen (cross)-presentation. Moreover, while the uptake of bacteria encased within apoptotic bodies may forestall phagocyte activation, it may reduce the bacteria's capacity to interact with their new host cell and to modulate its biology in a similar way as free bacteria could do. In the future, it will therefore be important to clarify in which ways distinct modes of host cell death can affect survival and spread of the bacteria, disease progression and the immune response.
Chlamydia-mediated prolongation of the life span of short-lived cells
In contrast to the above-mentioned observations in C. trachomatis-infected epithelial cells, in which Chlamydia-mediated inhibition of apoptosis did not achieve prolonged maintenance of host cell viability (Sixt et al. 2018), it is well established that infection with C. pneumoniae can prolong the life span of certain inherently short-lived cells, such as neutrophils, by delaying apoptotic death (van Zandbergen et al. 2004; Rupp et al. 2009; Sarkar et al. 2015). The mechanism behind this pro-survival effect in neutrophils appears to be distinct from Chlamydia-mediated anti-apoptosis in epithelial cells, as it may at least in part represent a non-specific response of neutrophils to infection conditions. Indeed, exposure to Chlamydia LPS, heat-killed bacteria or recombinant interleukin 8 (IL-8), a cytokine secreted by infected cells, also enhanced the life span of neutrophils (van Zandbergen et al. 2004; Sarkar et al. 2015). It was also shown that C. pneumoniae caused a PI3K-dependent activation of NFκB in infected neutrophils (Sarkar et al. 2015). Inhibitors of PI3K or NFκB activation could reduce infection-induced IL-8 release and could partially re-sensitize infected neutrophils to spontaneous apoptosis (Sarkar et al. 2015). The MEK inhibitor U0126 could also re-sensitize infected neutrophils to spontaneous apoptosis (Sarkar et al. 2015).
Importantly, the above-mentioned studies showed that apoptosis induction in C. pneumoniae-infected neutrophils was only delayed, not entirely blocked. Indeed, it was suggested that apoptotic neutrophils may serve as Trojan Horses enabling the bacteria to be taken up into macrophages in a manner that forestalls macrophage activation and allows prolonged intracellular bacterial survival in the phagocyte, which could then serve as vehicle for pathogen dissemination (Rupp et al. 2009). As discussed above, how infection can be established in a macrophage after uptake of bacteria entrapped within a host cell corpse, and if this is a species-specific trait, needs to be further explored.
Chlamydia-mediated inhibition of defensive host cell death
Host cell death as a cell-autonomous defense response
Cell-autonomous immunity is the capacity of eukaryotic cells, including non-immune cells of metazoan origin, to detect invading pathogens and to respond to the threat by inducing cellular defense responses (Randow, MacMicking and James 2013). Host cell death is one of these responses, because the controlled demise of infected cells can limit the replication and spread of intracellular pathogens (Jorgensen, Rayamajhi and Miao 2017). As will be discussed below, current knowledge suggests that Chlamydia spp. can trigger several cell death-inducing defense responses that have the potential to effectively disrupt Chlamydia development and replication. However, the bacteria also have evolved powerful counterstrategies, most notable such that shield the bacteria from the host cell's innate immune sensors.
Apoptosis restricts growth of Parachlamydiaceae in animal cells
In certain host-pathogen systems, apoptosis is induced as a defense mechanism that can block pathogen replication, if not actively counteracted by the pathogen. This has been well illustrated by Clem and co-workers, who demonstrated the importance of anti-apoptotic virulence factors for the growth of baculovirus in insect cells (Clem and Miller 1993; Clarke and Clem 2003). Studies with environmental chlamydiae, in particular studies with the naturally amoeba-infecting Parachlamydiaceae, suggested that apoptosis might have a similar protective potential against members of the phylum Chlamydiae. Certain animal-derived cells, including insect cells and human cells (epithelial cells and macrophages), were shown to induce rapid (or at least premature) cell death in response to infection with P. amoebophila and/or Pa. acanthamoebae (Greub, Mege and Raoult 2003a; Ito et al. 2012; Sixt et al. 2012; Matsuo et al. 2013; Brokatzky, Kretz and Häcker 2020). This cell death was confirmed to be of apoptotic nature, as it was accompanied by typical apoptotic changes in nuclear morphology, nucleosomal DNA fragmentation, DNA double-strand breaks, externalization of phosphatidylserine and activation of apoptotic effector caspases (Greub, Mege and Raoult 2003a; Ito et al. 2012; Sixt et al. 2012; Matsuo et al. 2013; Brokatzky, Kretz and Häcker 2020). Induction of apoptosis was dependent on the viability of the bacteria and required direct contact between the bacteria and the host cells (Ito et al. 2012; Sixt et al. 2012; Brokatzky, Kretz and Häcker 2020). Host cell apoptosis was blocked in presence of the pan caspase inhibitor Z-VAD-FMK (Ito et al. 2012; Sixt et al. 2012; Matsuo et al. 2013). BAX/BAK-deficiency or overexpression of BCL-XL could diminish cell death in infected HeLa cells (Brokatzky, Kretz and Häcker 2020). Interestingly, in the insect cell model, inhibition of apoptosis during infection with Pa. acanthamoebae was sufficient to maintain host cell viability and to enable effective replication and formation of infectious EBs (Sixt et al. 2012). Beneficial effects of apoptosis inhibition on Pa. acanthamoebae survival in human host cells were also described more recently (Brokatzky, Kretz and Häcker 2020). It is currently unknown how host cell death is induced during infection with Parachlamydiaceae. Moreover, cell death was not observed in each host/pathogen system studied. For instance, apoptosis induction was not observed in primary peripheral blood mononuclear cells (PBMCs) that were infected with P. amoebophila or in HEp2 cells that were infected with Pa. acanthamoebae (Ito et al. 2012).
Chlamydia spp. actively counteract premature host cell death
The findings discussed above could in principle be explained by an inability of Parachlamydiaceae to protect their host cells against apoptosis caused by infection-induced cellular stress, due to a lack of anti-apoptotic virulence strategies. However, given the considerations described above, which challenge the existence of such strong pro-death signals and the potency of Chlamydia’s anti-apoptotic trait, it is also plausible that cell death induced during infection with Parachlamydiaceae is a host defense response. That means more critical than the bacteria's inability to target the cell death machineries per se, may be an inability to block the defense pathways that act upstream of their induction.
Indeed, recent studies that exploited new genetic tools for Chlamydia spp. demonstrated that C. trachomatis actively counteracts cellular defense pathways that activate premature host cell death (Sixt et al. 2017; Weber et al. 2017). More precisely, it was shown that C. trachomatis mutants that were deficient for certain Incs, such as Chlamydia promoter of survival (CpoS)/CT229, CT383 or IncC/CT233, induced premature host cell death, which impeded formation of infectious EBs (Sixt et al. 2017; Weber et al. 2017). This host cell death was at least partially dependent on the intracellular innate immune sensor Stimulator of Interferon Genes (STING) (Sixt et al. 2017; Weber et al. 2017), giving support to the idea that this cell death was induced as part of a host cell-intrinsic defense response. Cell death induced during infection with these mutants was partially apoptotic and partially necrotic (Sixt et al. 2017; Weber et al. 2017), yet the exact nature of the death pathway(s) activated remains to be uncovered. Moreover, it is not understood how the above-mentioned Inc proteins can prevent cell death induction. It was proposed that their absence causes inclusion membrane instability, which could enhance the release of PAMPs from the inclusion into the host cell cytosol, where they could be sensed, for instance by STING. Furthermore, because CpoS/CT229 interacts with host Rab GTPases, lack of CpoS/CT229 may influence inclusion membrane stability by affecting Chlamydia’s ability to subvert host cellular trafficking processes (Sixt et al. 2017; Faris et al. 2019). It is possible that cell death induced as consequence of premature inclusion lysis could be mechanistically related to late stage host cell death, which is also preceded by inclusion rupture (Hybiske and Stephens 2007; Kerr et al. 2017) (Fig. 7).
Figure 7.
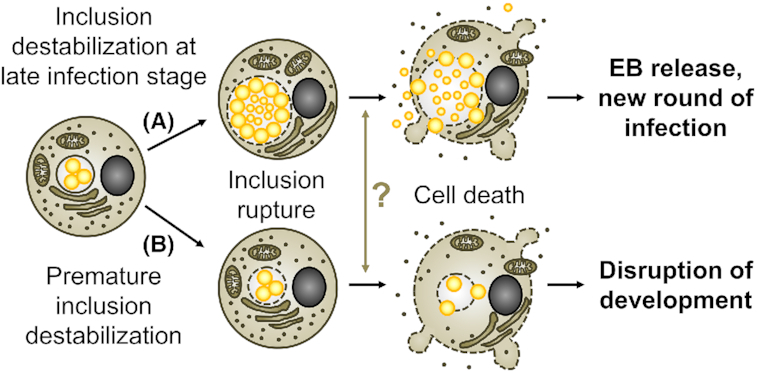
Host cell death as consequence of inclusion instability. (A) Host cell lysis at late stages of infection with C. trachomatis is preceded by inclusion rupture. (B) Conditions that cause premature inclusion rupture, such as laser-mediated disruption of inclusions and potentially lack of certain Inc proteins, result in premature host cell death. The molecular machineries that execute cell death in these distinct situations are not well understood. Yet, it is conceivable that Chlamydia co-opts pre-existing host cellular defense responses to mediate its exit from its host cell.
More recently, a genetic screen in C. muridarum uncovered several mutants that were more sensitive to IFN-γ-mediated restriction of bacterial growth in mouse cells (Giebel et al. 2019). A closer analysis of one mutant, which carried a specific point mutation in the putative Inc protein TC0574, revealed premature inclusion lysis, followed by induction of host cell death (Giebel et al. 2019). Moreover, levels of inclusion lysis and cell death were strongly enhanced when IFN-γ or IFN-β were added to the culture (Giebel et al. 2019). In contrast to the cell death observed during infection with the above-mentioned C. trachomatis Inc mutants (Sixt et al. 2017; Weber et al. 2017), this inclusion lysis could be blocked by the pan caspase inhibitor Z-VAD-FMK (Giebel et al. 2019). Interestingly, inclusion lysis could also be inhibited partially by specific inhibitors of CASP3, CASP8 and CASP9, while exposure to pro-apoptotic stimuli, such as STS or TNFα/CHX, enhanced inclusion lysis in cells infected with the TC0574 mutant (Giebel et al. 2019). The mechanism of inclusion lysis and the nature of the cell death program induced during infection with the mutant remain to be determined.
The C. trachomatis and C. muridarum Inc mutants mentioned above were significantly attenuated in mouse models of genital infection (Sixt et al. 2017; Weber et al. 2017; Giebel et al. 2019). These observations highlight the protective potential of premature host cell death, as well as the importance for Chlamydia spp. to have evolved strategies to block this host defense mechanism.
Interactions of Chlamydia spp. with necroptotic and the pyroptotic pathways
While necroptosis can be induced downstream of cytokine receptors and immune sensor proteins (Pasparakis and Vandenabeele 2015), a potential role for necroptosis as a defense mechanism during infection with Chlamydia spp. is not well explored. It appears that in contrast to its potent anti-apoptotic activities, C. trachomatis L2 does not cause a general block in the necroptotic pathway. Indeed, infection failed to block RIPK3 and MLKL activation in response to the necroptosis inducer TSZ (a mixture of TNFα, SMAC mimetic BV6 and Z-VAD-FMK) and even appeared to sensitize host cells to TSZ-induced necrotic death (Sixt et al. 2018).
Pyroptosis, a pro-inflammatory form of necrosis activated by inflammatory caspases, is another prominent example of a cell-autonomous defense response that triggers the death of the infected cell (Cookson and Brennan 2001). Numerous studies showed that Chlamydia spp. can, typically within few hours after infection, induce activation of canonical inflammasomes, more precisely activation of the NACHT, LRR and PYD domains-containing protein 3 (NLRP3) and absent in melanoma 2 (AIM2) inflammasomes (Abdul-Sater et al. 2009, 2010; He et al. 2010; Shimada et al. 2011; Nagarajan et al. 2012; Itoh et al. 2014; Finethy et al. 2015; Webster et al. 2017). Consequently, infection can lead to the activation of CASP1 and secretion of the pro-inflammatory cytokines IL-1β and IL-18 (Ojcius et al. 1998; Lu, Shen and Brunham 2000; Cheng et al. 2008; Abdul-Sater et al. 2009; Prantner et al. 2009; Abdul-Sater et al. 2010; He et al. 2010; Shimada et al. 2011; Nagarajan et al. 2012; Itoh et al. 2014; Finethy et al. 2015; Chen et al. 2017; Webster et al. 2017). Most of these studies investigated the interaction of Chlamydia spp. with human or murine monocytes or macrophages (Ojcius et al. 1998; Cheng et al. 2008; Prantner et al. 2009; Abdul-Sater et al. 2010; He et al. 2010; Shimada et al. 2011; Nagarajan et al. 2012; Itoh et al. 2014; Finethy et al. 2015; Chen et al. 2017; Webster et al. 2017). Few studies demonstrated that inflammasome activation also occurs in epithelial cells, such as in HeLa cells, although with slower kinetics (Lu, Shen and Brunham 2000; Cheng et al. 2008; Abdul-Sater et al. 2009). Chlamydia pneumoniae was a more potent inducer of inflammasome and CASP1 activation when compared with C. trachomatis (D or L2), C. caviae and C. muridarum (He et al. 2010; Itoh et al. 2014). A subset of the above-mentioned studies also reported induction of pyroptosis (Finethy et al. 2015; Chen et al. 2017; Webster et al. 2017). Moreover, there is evidence that C. trachomatis and C. muridarum also induce the non-canonical inflammasome pathway and that both CASP1 and CASP11 contribute to pyroptotic death in infected murine bone marrow-derived macrophages (BMDMs) (Finethy et al. 2015; Webster et al. 2017).
While Chlamydia infection appeared to be self-sufficient in providing both signals required for inflammasome activation, priming of macrophages with extracellular ATP, LPS or IFN-γ enhanced or accelerated the response (Prantner et al. 2009; He et al. 2010; Finethy et al. 2015). Moreover, STING-dependent induction of type I interferon (IFN) production and autocrine IFN signaling were also implicated in enhancing inflammasome activation and pyroptosis in Chlamydia-infected cells (Webster et al. 2017). This suggests that Chlamydia virulence strategies that aim at blocking STING activation, or at dampening the IFN response by other means, may also help the pathogen to limit pyroptosis. Moreover, a recent study provided evidence that the unique structure of chlamydial LPS reduces its potential to activate the non-canonical inflammasome pathway (Yang et al. 2019). More precisely, it was shown that in contrast to E.coli LPS, LPS from C. trachomatis had a very low ability to induce activation of CASP1 and IL-1β release (through CASP11-mediated inflammasome activation) and pyroptosis, when transfected into murine BMDMs (Yang et al. 2019). The low toxicity of C. trachomatis L2 LPS upon transfection was confirmed in a more recent study (Wang, Rockey and Dolan 2020).
The role of pyroptosis in controlling Chlamydia infection is not well understood and its investigation is complicated by the fact that inflammasome activation has multiple additional effects on the immune response, such as via the secretion of pro-inflammatory cytokines. Furthermore, some studies suggested that CASP1-deficiency or inhibition in infected host cells can negatively affect intracellular chlamydial growth (Abdul-Sater et al. 2009; Christian et al. 2011; Itoh et al. 2014).
It should be noted that the recent observation that C. trachomatis can induce minority MOMP in infected cells clearly shows that the role of the apoptotic machinery in immunity against Chlamydia can also not be reduced to its pro-death function. Indeed, it was shown that minority MOMP can cause DNA damage and mitochondrial damage, which in turn may act as danger signals boosting cytokine secretion and cell-autonomous immunity (Brokatzky et al. 2019).
Chlamydia-mediated inhibition of NETosis
NETosis is a special form of regulated cell death that occurs in neutrophils as part of the host defense. It enables the cells to release so-called neutrophil extracellular traps (NETs), which are large extracellular structures that are composed of proteins and decondensed chromatin (Brinkmann et al. 2004). NETs can trap pathogens and block their dissemination. Moreover, NETs contain antimicrobials that can kill pathogens directly (Papayannopoulos 2018). The role of NETs in anti-chlamydial immunity is not well explored. A recent study provided evidence that C. trachomatis L2 can block neutrophil activation and hence also NETosis (Rajeeve et al. 2018). Mechanistically, this was linked to the Chlamydia protease CPAF, which was shown to cleave formyl peptide receptor 2 (FPR2), a surface receptor required for neutrophil activation (Rajeeve et al. 2018). The CPAF-deficient mutant was attenuated in a mouse model of infection and this attenuation was not observed in a FPR2-deficient mouse model nor in neutropenic mice (Rajeeve et al. 2018). While these data clearly demonstrated the role of neutrophils in anti-chlamydial immunity and the significance of CPAF as virulence factor, further studies are required to clarify the relative role of NETosis compared with other antibacterial neutrophil responses.
CONCLUSIONS AND PERSPECTIVES
In summary, the interaction of Chlamydia spp. (and their relatives) with host cellular survival and death pathways is highly complex and our understanding of the underlying mechanisms and their significance remains scarce despite extensive research progress.
While it is clear that late stage host cell death is an exit strategy that promotes Chlamydia spread to formerly uninfected cells, the molecular mechanisms that regulate this important event in Chlamydia’s infection cycle remain obscure. It will be important to identify the virulence factors that regulate the execution of bacterial egress and its timing during the development cycle, as well as to clarify the potential roles of host RCD programs. Furthermore, our understanding of the relevance of alternative exit strategies, including their influence on bacterial viability and infectivity, pathogen dissemination, inflammation and tissue damage, and the nature of the host immune response is very limited. Potential effects of phenomena such as bystander cell death and multiplication-independent cytotoxicity should also not be neglected.
Similarly, as we learn more and more about the mechanisms of Chlamydia anti-apoptosis, our knowledge about anti-apoptotic Chlamydia virulence factors remains scarce and it becomes clear that the function of this virulence trait is not well understood. The number of anti-apoptotic virulence strategies described for Chlamydia spp. is vast, yet the relative importance of these strategies is unknown. Moreover, to evaluate the significance of apoptosis inhibition in infected cells, we would need to have a better understanding of the nature and strength of pro-apoptotic signals that infected cells typically encounter in vivo. Finally, while recent advances demonstrated the potential of host cell death to serve as host defense response in pathogen restriction, we know little about how these defense programs are trigged and executed by the host or how they are blocked by the pathogen.
Fortunately, it can be predicted that the recently established genetic tools for Chlamydia spp. will empower and inspire researchers to apply novel approaches to tackle these questions. For instance, once the molecular basis of bacterial egress is better defined, these tools may be used to directly modify the relative frequency of the distinct exit strategies, such as extrusion vs host cell death, or apoptosis-like vs necrotic host cell death, and to study the consequences for the bacteria and the host in cell culture and in vivo. In fact, a recent study reported that a C. trachomatis mutant deficient for the Inc CT228 displayed enhanced rates of extrusion and infection with the strain was cleared slower in a murine genital tract infection model (Shaw et al. 2018). However, whether these phenotypes are mechanistically linked still needs to be explored.
Interestingly, past research on Chlamydia exit and anti-apoptosis has led to partially conflicting findings. It is possible that some reported observations reflect unnatural behavior of immortalized cell lines. Hence, it will be an important task for future research to reassess findings made in cell lines in more natural infection systems, as well as to confirm that similar mechanisms operate in more recent isolates of the bacteria. However, we should also be open for the possibility that some of these discrepancies may reflect true biological differences. For instance, as described above, there is clear evidence that C. trachomatis and C. pneumoniae use different molecular mechanisms to block host cell apoptosis and it is plausible that similar differences may exist between other species and in the context of exit strategies. Indeed, it is an intriguing possibility that these differences could directly contribute to the distinct disease spectra and tropisms seen among Chlamydia spp.
In conclusion, the interaction of Chlamydia spp. with host cell death and survival pathways remains an active and stimulating field of research (Fig. 8). A deeper knowledge in this area will be critical for our understanding of Chlamydia diseases and anti-chlamydial immunity. Moreover, it is tempting to speculate that it may even inspire novel anti-virulence or host-directed therapeutic strategies. For instance, it may well be possible to fight infectious agents by modulation of pathogen exit strategies or by exploiting host cell-intrinsic defense responses that act by inducing cell death in infected cells.
Figure 8.

Important areas for future research. This figure gives an overview of highly relevant questions that need to be addressed in depth in the future.
ACKNOWLEDGMENTS
I would like to thank the research community for their invaluable contributions to our knowledge, for inspiring discussions and for continuous support and collaboration. I would also like to apologize to all colleagues whose work was not discussed in this review.
FUNDING
This work was supported by the Swedish Research Council (Vetenskapsrådet) [2018-02286 to BSS and 2016-06598 to the Laboratory for Molecular Infection Medicine Sweden (MIMS)].
Conflict of interest
The author declares no conflict of interest.
REFERENCES
- Abdul-Sater AA, Koo E, Hacker Get al. Inflammasome-dependent caspase-1 activation in cervical epithelial cells stimulates growth of the intracellular pathogen Chlamydia trachomatis. J Biol Chem. 2009;284:26789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdul-Sater AA, Said-Sadier N, Padilla EVet al. Chlamydial infection of monocytes stimulates IL-1beta secretion through activation of the NLRP3 inflammasome. Microbes Infect. 2010;12:652–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Airenne S, Surcel HM, Tuukkanen Jet al. Chlamydia pneumoniae inhibits apoptosis in human epithelial and monocyte cell lines. Scand J Immunol. 2002;55:390–8. [DOI] [PubMed] [Google Scholar]
- Aits S, Kricker J, Liu Bet al. Sensitive detection of lysosomal membrane permeabilization by lysosomal galectin puncta assay. Autophagy. 2015;11:1408–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Zeer MA, Xavier A, Lubad MAet al. Chlamydia trachomatis prevents apoptosis via activation of PDPK1-MYC and enhanced mitochondrial binding of hexokinase II. EBioMedicine. 2017;23:100–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Q, Shi Y. Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ. 2007;14:56–65. [DOI] [PubMed] [Google Scholar]
- Beatty WL, Byrne GI, Morrison RP. Morphologic and antigenic characterization of interferon gamma-mediated persistent Chlamydia trachomatis infection in vitro. Proc Natl Acad Sci USA. 1993;90:3998–4002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty WL, Morrison RP, Byrne GI. Persistent chlamydiae: from cell culture to a paradigm for chlamydial pathogenesis. Microbiol Rev. 1994;58:686–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedson SP, Bland JOW.. A morphological study of psittacosis virus, with the description of a developmental cycle. Br J Exp Pathol. 1932;13:461–6. [Google Scholar]
- Belland RJ, Scidmore MA, Crane DDet al. Chlamydia trachomatis cytotoxicity associated with complete and partial cytotoxin genes. Proc Natl Acad Sci USA. 2001;98:13984–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell SD Jr, Snyder JC, Murray ES. Immunization of mice against toxic doses of homologous elementary bodies of trachoma. Science. 1959;130:626–7. [DOI] [PubMed] [Google Scholar]
- Bernkopf H, Mashiah P, Becker Y. Correlation between morphological and biochemical changes and the appearance of infectivity in FL cell cultures infected with trachoma agent. Ann N Y Acad Sci. 1962;98:62–81. [DOI] [PubMed] [Google Scholar]
- Boise LH, Collins CM. Salmonella-induced cell death: apoptosis, necrosis or programmed cell death?. Trends Microbiol. 2001;9:64–7. [DOI] [PubMed] [Google Scholar]
- Bothe M, Dutow P, Pich Aet al. DXD motif-dependent and -independent effects of the Chlamydia trachomatis cytotoxin CT166. Toxins (Basel). 2015;7:621–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove J, Martinez-Vicente M, Dehay Bet al. BAX channel activity mediates lysosomal disruption linked to Parkinson disease. Autophagy. 2014;10:889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brade L, Schramek S, Schade Uet al. Chemical, biological, and immunochemical properties of the Chlamydia psittaci lipopolysaccharide. Infect Immun. 1986;54:568–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkmann V, Reichard U, Goosmann Cet al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–5. [DOI] [PubMed] [Google Scholar]
- Brokatzky D, Dorflinger B, Haimovici Aet al. A non-death function of the mitochondrial apoptosis apparatus in immunity. EMBO J. 2019;38:e100907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brokatzky D, Kretz O, Häcker G. Apoptosis functions in defense against infection of mammalian cells with environmental chlamydiae. Infect Immun. 2020;88:e00851–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burillo A, Bouza E. Chlamydophila pneumoniae. Infect Dis Clin North Am. 2010;24:61–71. [DOI] [PubMed] [Google Scholar]
- Byrne GI, Ojcius DM. Chlamydia and apoptosis: life and death decisions of an intracellular pathogen. Nat Rev Microbiol. 2004;2:802–8. [DOI] [PubMed] [Google Scholar]
- Böhme L, Albrecht M, Riede Oet al. Chlamydia trachomatis-infected host cells resist dsRNA-induced apoptosis. Cell Microbiol. 2009;12:1340–51. [DOI] [PubMed] [Google Scholar]
- Cai Z, Jitkaew S, Zhao Jet al. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat Cell Biol. 2014;16:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell S, Richmond SJ, Yates P. The development of Chlamydia trachomatis inclusions within the host eukaryotic cell during interphase and mitosis. J Gen Microbiol. 1989;135:1153–65. [DOI] [PubMed] [Google Scholar]
- Carlson JH, Hughes S, Hogan Det al. Polymorphisms in the Chlamydia trachomatis cytotoxin locus associated with ocular and genital isolates. Infect Immun. 2004;72:7063–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter GP, Rood JI, Lyras D. The role of toxin A and toxin B in Clostridium difficile-associated disease: past and present perspectives. Gut Microbes. 2010;1:58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceovic R, Gulin SJ. Lymphogranuloma venereum: diagnostic and treatment challenges. Infect Drug Resist. 2015;8:39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GT, Moulder JW. Loss of inorganic ions from host cells infected with Chlamydia psittaci. Infect Immun. 1978;19:827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauvier D, Ankri S, Charriaut-Marlangue Cet al. Broad-spectrum caspase inhibitors: from myth to reality? Cell Death Differ. 2007;14:387–91. [DOI] [PubMed] [Google Scholar]
- Chen AL, Johnson KA, Lee JKet al. CPAF: a Chlamydial protease in search of an authentic substrate. PLoS Pathog. 2012;8:e1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng W, Shivshankar P, Li Zet al. Caspase-1 contributes to Chlamydia trachomatis-induced upper urogenital tract inflammatory pathologies without affecting the course of infection. Infect Immun. 2008;76:515–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Liu X, Yu Xet al. Chlamydia muridarum infection of macrophages stimulates IL-1beta secretion and cell death via activation of caspase-1 in an RIP3-independent manner. Biomed Res Int. 2017;2017:1592365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury SR, Reimer A, Sharan Met al. Chlamydia preserves the mitochondrial network necessary for replication via microRNA-dependent inhibition of fission. J Cell Biol. 2017;216:1071–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian JG, Heymann J, Paschen SAet al. Targeting of a chlamydial protease impedes intracellular bacterial growth. PLoS Pathog. 2011;7:e1002283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen G, Manire GP. The toxicity of meningopneumonitis organisms (Chlamydia psittaci) at different stages of development. J Immunol. 1969;103:1085–8. [PubMed] [Google Scholar]
- Clarke TE, Clem RJ. Insect defenses against virus infection: the role of apoptosis. Int Rev Immunol. 2003;22:401–24. [DOI] [PubMed] [Google Scholar]
- Clem RJ, Miller LK. Apoptosis reduces both the in vitro replication and the in vivo infectivity of a baculovirus. J Virol. 1993;67:3730–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles AM, Reynolds DJ, Harper Aet al. Low-nutrient induction of abnormal chlamydial development: a novel component of chlamydial pathogenesis? FEMS Microbiol Lett. 1993;106:193–200. [DOI] [PubMed] [Google Scholar]
- Collingro A, Köstlbacher S, Horn M. Chlamydiae in the environment. Trends Microbiol. 2020, 10.1016/j.tim.2020.05.020. [DOI] [PubMed] [Google Scholar]
- Collingro A, Tischler P, Weinmaier Tet al. Unity in variety – the pan-genome of the Chlamydiae. Mol Biol Evol. 2011;28:3253–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–4. [DOI] [PubMed] [Google Scholar]
- Czabotar PE, Lessene G, Strasser Aet al. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014;15:49–63. [DOI] [PubMed] [Google Scholar]
- Darville T, Andrews CW Jr, Rank RG. Does inhibition of tumor necrosis factor alpha affect chlamydial genital tract infection in mice and guinea pigs? Infect Immun. 2000;68:5299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean D, Powers VC. Persistent Chlamydia trachomatis infections resist apoptotic stimuli. Infect Immun. 2001;69:2442–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Duve C. Lysosomes, a new group of cytoplasmic particles. In: Hayashi T (ed.). Subcellular Particles. New York: The Ronald Press Co., 1959, 128–59. [Google Scholar]
- Degterev A, Hitomi J, Germscheid Met al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A, Huang Z, Boyce Met al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9. [DOI] [PubMed] [Google Scholar]
- de la Maza LM, Peterson EM. Scanning electron microscopy of McCoy cells infected with Chlamydia trachomatis. Exp Mol Pathol. 1982;36:217–26. [DOI] [PubMed] [Google Scholar]
- Dille S, Kleinschnitz EM, Kontchou CWet al. In contrast to Chlamydia trachomatis, Waddlia chondrophila grows in human cells without inhibiting apoptosis, fragmenting the Golgi apparatus, or diverting post-Golgi sphingomyelin transport. Infect Immun. 2015;83:3268–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong F, Pirbhai M, Xiao Yet al. Degradation of the proapoptotic proteins Bik, Puma, and Bim with Bcl-2 domain 3 homology in Chlamydia trachomatis-infected cells. Infect Immun. 2005;73:1861–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughri AM, Storz J, Altera KP. Mode of entry and release of chlamydiae in infections of intestinal epithelial cells. J Infect Dis. 1972;126:652–7. [DOI] [PubMed] [Google Scholar]
- Du C, Fang M, Li Yet al. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. [DOI] [PubMed] [Google Scholar]
- Du K, Zheng Q, Zhou Met al. Chlamydial antiapoptotic activity involves activation of the Raf/MEK/ERK survival pathway. Curr Microbiol. 2011;63:341–6. [DOI] [PubMed] [Google Scholar]
- Eley A, Hosseinzadeh S, Hakimi Het al. Apoptosis of ejaculated human sperm is induced by co-incubation with Chlamydia trachomatis lipopolysaccharide. Hum Reprod. 2005;20:2601–7. [DOI] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Equils O, Lu D, Gatter Met al. Chlamydia heat shock protein 60 induces trophoblast apoptosis through TLR4. J Immunol. 2006;177:1257–63. [DOI] [PubMed] [Google Scholar]
- Fan T, Lu H, Hu Het al. Inhibition of apoptosis in Chlamydia-infected cells: blockade of mitochondrial cytochrome c release and caspase activation. J Exp Med. 1998;187:487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faris R, Merling M, Andersen SEet al. Chlamydia trachomatis CT229 subverts Rab GTPase-dependent CCV trafficking pathways to promote chlamydial infection. Cell Rep. 2019;26:3380–90.e5. [DOI] [PubMed] [Google Scholar]
- Finethy R, Jorgensen I, Haldar AKet al. Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in Chlamydia-infected macrophages. Infect Immun. 2015;83:4740–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Harrison KS, Ramirez Yet al. Chlamydia trachomatis-containing vacuole serves as deubiquitination platform to stabilize Mcl-1 and to interfere with host defense. eLife. 2017;6:e21465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SF, Harlander T, Vier Jet al. Protection against CD95-induced apoptosis by chlamydial infection at a mitochondrial step. Infect Immun. 2004a;72:1107–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SF, Schwarz C, Vier Jet al. Characterization of antiapoptotic activities of Chlamydia pneumoniae in human cells. Infect Immun. 2001;69:7121–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SF, Vier J, Kirschnek Set al. Chlamydia inhibit host cell apoptosis by degradation of proapoptotic BH3-only proteins. J Exp Med. 2004b;200:905–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foschi C, Bortolotti M, Marziali Get al. Survival and death of intestinal cells infected by Chlamydia trachomatis. PLoS One. 2019;14:e0215956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foschi C, Bortolotti M, Polito Let al. Insights into penicillin-induced Chlamydia trachomatis persistence. Microb Pathog. 2020;142:104035. [DOI] [PubMed] [Google Scholar]
- Friis RR. Interaction of L cells and Chlamydia psittaci: entry of the parasite and host responses to its development. J Bacteriol. 1972;110:706–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche TR, Sobek D, Gautom RK. Enhancement of in vitro cytopathogenicity by Acanthamoeba spp. following acquisition of bacterial endosymbionts. FEMS Microbiol Lett. 1998;166:231–6. [DOI] [PubMed] [Google Scholar]
- Galanos C, Freudenberg MA. Mechanisms of endotoxin shock and endotoxin hypersensitivity. Immunobiology. 1993;187:346–56. [DOI] [PubMed] [Google Scholar]
- Galdiero F, Sommese L, Gorga Fet al. Toxic effect on human spermatozoa by Chlamydia trachomatis purified lipopolysaccharide. FEMS Microbiol Lett. 1994;115:197–200. [DOI] [PubMed] [Google Scholar]
- Galle JN, Fechtner T, Eierhoff Tet al. A Chlamydia pneumoniae adhesin induces phosphatidylserine exposure on host cells. Nat Commun. 2019;10:4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Buque A, Kepp Oet al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. [DOI] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Kroemer G. Mitochondrial regulation of cell death: a phylogenetically conserved control. Microbial Cell. 2016;3:101–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SAet al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Shane RB, Berencsi Ket al. Chlamydia pneumoniae inhibits apoptosis in human peripheral blood mononuclear cells through induction of IL-10. J Immunol. 2000;164:5522–9. [DOI] [PubMed] [Google Scholar]
- Gibellini D, Panaya R, Rumpianesi F. Induction of apoptosis by Chlamydia psittaci and Chlamydia trachomatis infection in tissue culture cells. Zentralbl Bakteriol. 1998;288:35–43. [DOI] [PubMed] [Google Scholar]
- Giebel AM, Hu S, Rajaram Ket al. Genetic screen in Chlamydia muridarum reveals role for an interferon-induced host cell death program in antimicrobial inclusion rupture. mBio. 2019;10:e00385–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez E, Rother M, Kerr MCet al. Chlamydia infection depends on a functional MDM2-p53 axis. Nat Commun. 2014;5:5201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goy G, Croxatto A, Greub G. Waddlia chondrophila enters and multiplies within human macrophages. Microbes Infect. 2008;10:556–62. [DOI] [PubMed] [Google Scholar]
- Greene W, Xiao Y, Huang Yet al. Chlamydia-infected cells continue to undergo mitosis and resist induction of apoptosis. Infect Immun. 2004;72:451–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, La Scola B, Raoult D. Parachlamydia acanthamoebae is endosymbiotic or lytic for Acanthamoeba polyphaga depending on the incubation temperature. Ann N Y Acad Sci. 2003b;990:628–34. [DOI] [PubMed] [Google Scholar]
- Greub G, Mege J-L, Raoult D. Parachlamydia acanthamoebae enters and multiplies within human macrophages and induces their apoptosis. Infect Immun. 2003a;71:5979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greub G, Raoult D.. Crescent bodies of Parachlamydia acanthamoebae and its life cycle within Acanthamoeba polyphaga: an electron micrograph study. Appl Environ Microbiol. 2002;68:3076–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JJ, Zhang XD, Sun Wet al. DRAM1 regulates apoptosis through increasing protein levels and lysosomal localization of BAX. Cell Death Dis. 2015;6:e1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guicciardi ME, Gores GJ.. Life and death by death receptors. FASEB J. 2009;23:1625–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch TP, Al-Hossainy E, Silverman JA. Adenine nucleotide and lysine transport in Chlamydia psittaci. J Bacteriol. 1982;150:662–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Zhao Y, Yang Xet al. Chlamydia trachomatis pORF5 plasmid-encoded protein regulates autophagy and apoptosis of HeLa cells. Biotechnol Biotec Eq. 2019;33:1269–79. [Google Scholar]
- Heine H, Muller-Loennies S, Brade Let al. Endotoxic activity and chemical structure of lipopolysaccharides from Chlamydia trachomatis serotypes E and L2 and Chlamydophila psittaci 6BC. Eur J Biochem. 2003;270:440–50. [DOI] [PubMed] [Google Scholar]
- Hess S, Rheinheimer C, Tidow Fet al. The reprogrammed host: Chlamydia trachomatis-induced up-regulation of glycoprotein 130 cytokines, transcription factors, and antiapoptotic genes. Arthritis Rheum. 2001;44:2392–401. [DOI] [PubMed] [Google Scholar]
- He X, Mekasha S, Mavrogiorgos Net al. Inflammation and fibrosis during Chlamydia pneumoniae infection is regulated by IL-1 and the NLRP3/ASC inflammasome. J Immunol. 2010;184:5743–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi N, Tamura A, Iwanaga M. Developmental cycle and reproductive mechanism of the meningopneumonitis virus in strain L cells. Ann N Y Acad Sci. 1962;98:100–21. [DOI] [PubMed] [Google Scholar]
- Horn M. Chlamydiae as symbionts in eukaryotes. Annu Rev Microbiol. 2008;62:113–31. [DOI] [PubMed] [Google Scholar]
- Hosseinzadeh S, Pacey AA, Eley A. Chlamydia trachomatis-induced death of human spermatozoa is caused primarily by lipopolysaccharide. J Med Microbiol. 2003;52:193–200. [DOI] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS.. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. Proc Natl Acad Sci USA. 2007;104:11430–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häcker G, Kirschnek S, Fischer SF. Apoptosis in infectious disease: how bacteria interfere with the apoptotic apparatus. Med Microbiol Immunol. 2006;195:11–9. [DOI] [PubMed] [Google Scholar]
- Ingalls RR, Rice PA, Qureshi Net al. The inflammatory cytokine response to Chlamydia trachomatis infection is endotoxin mediated. Infect Immun. 1995;63:3125–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmler M, Thome M, Hahne Met al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–5. [DOI] [PubMed] [Google Scholar]
- Ito A, Matsuo J, Nakamura Set al. Amoebal endosymbiont Protochlamydia induces apoptosis to human immortal HEp-2 cells. PLoS One. 2012;7:e30270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh R, Murakami I, Chou Bet al. Chlamydia pneumoniae harness host NLRP3 inflammasome-mediated caspase-1 activation for optimal intracellular growth in murine macrophages. Biochem Biophys Res Commun. 2014;452:689–94. [DOI] [PubMed] [Google Scholar]
- Ivins BE, Wyrick PB. Response of C3H/HeJ and C3H/HeN mice and their peritoneal macrophages to the toxicity of Chlamydia psittaci elementary bodies. Infect Immun. 1978;22:620–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendro MC, Deutsch T, Korber Bet al. Infection of human monocyte-derived macrophages with Chlamydia trachomatis induces apoptosis of T cells: a potential mechanism for persistent infection. Infect Immun. 2000;68:6704–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jendro MC, Fingerle F, Deutsch Tet al. Chlamydia trachomatis-infected macrophages induce apoptosis of activated T cells by secretion of tumor necrosis factor-alpha in vitro. Med Microbiol Immunol. 2004;193:45–52. [DOI] [PubMed] [Google Scholar]
- Jha R, Vardhan H, Bas Set al. Chlamydia trachomatis heat shock proteins 60 and 10 induce apoptosis in endocervical epithelial cells. Inflamm Res. 2011;60:69–78. [DOI] [PubMed] [Google Scholar]
- Jimenez Fernandez D, Lamkanfi M. Inflammatory caspases: key regulators of inflammation and cell death. Biol Chem. 2015;396:193–203. [DOI] [PubMed] [Google Scholar]
- Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17:151–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungas T, Verbeke P, Darville Tet al. Cell death, BAX activation, and HMGB1 release during infection with Chlamydia. Microbes Infect. 2004;6:1145–55. [DOI] [PubMed] [Google Scholar]
- Kagedal K, Johansson AC, Johansson Uet al. Lysosomal membrane permeabilization during apoptosis–involvement of Bax?. Int J Exp Pathol. 2005;86:309–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahane S, Everett KD, Kimmel Net al. Simkania negevensis strain ZT: growth, antigenic and genome characteristics. Int J Syst Bacteriol. 1999;49 Pt 2:815–20. [DOI] [PubMed] [Google Scholar]
- Kahane S, Kimmel N, Friedman MG. The growth cycle of Simkania negevensis. Microbiology. 2002;148:735–42. [DOI] [PubMed] [Google Scholar]
- Kantari C, Walczak H. Caspase-8 and bid: caught in the act between death receptors and mitochondria. Biochim Biophys Acta. 2011;1813:558–63. [DOI] [PubMed] [Google Scholar]
- Karunakaran K, Mehlitz A, Rudel T. Evolutionary conservation of infection-induced cell death inhibition among Chlamydiales. PLoS One. 2011;6:e22528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellogg KR, Horoschak KD, Moulder JW. Toxicity of low and moderate multiplicities of Chlamydia psittaci for mouse fibroblasts (L cells). Infect Immun. 1977;18:531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr MC, Gomez GA, Ferguson Cet al. Laser-mediated rupture of chlamydial inclusions triggers pathogen egress and host cell necrosis. Nat Commun. 2017;8:14729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokes M, Dunn JD, Granek JAet al. Integrating chemical mutagenesis and whole-genome sequencing as a platform for forward and reverse genetic analysis of Chlamydia. Cell Host Microbe. 2015;17:716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H, Rock KL.. How dying cells alert the immune system to danger. Nat Rev Immunol. 2008;8:279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman G, Reutelingsperger CP, Kuijten GAet al. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–20. [PubMed] [Google Scholar]
- Kordova N, Hoogstraten J, Wilt JC. Lysosomes and the “toxicity” of Rickettsiales. IV. Ultrastructural studies of macrophages infected with a cytopathic L cell-grown C. psittaci 6BC strain. Can J Microbiol. 1973;19:315–20. [DOI] [PubMed] [Google Scholar]
- Kordova N, Wilt JC, Sadiq M. Lysosomes in L cells infected with Chlamydia psittaci 6BC strain. Can J Microbiol. 1971;17:955–9. [DOI] [PubMed] [Google Scholar]
- Kordova N, Wilt JC.. Lysosomes and the “toxicity” of Rickettsiales. I. Cytochemical studies of macrophages inoculated in vitro with C. psittaci 6BC. Can J Microbiol. 1972;18:457–64. [DOI] [PubMed] [Google Scholar]
- Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27:673–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S Mechanisms mediating caspase activation in cell death. Cell Death Differ. 1999;6:1060–6. [DOI] [PubMed] [Google Scholar]
- Kumar Y, Valdivia RH.. Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe. 2008;4:159–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kun D, Xiang-Lin C, Ming Zet al. Chlamydia inhibit host cell apoptosis by inducing Bag-1 via the MAPK/ERK survival pathway. Apoptosis. 2013;18:1083–92. [DOI] [PubMed] [Google Scholar]
- Kuo CC. Immediate cytotoxicity of Chlamydia trachomatis for mouse peritoneal macrophages. Infect Immun. 1978;20:613–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBrie SD, Dimond ZE, Harrison KSet al. Transposon mutagenesis in Chlamydia trachomatis identifies CT339 as a ComEC homolog important for DNA uptake and lateral gene transfer. mBio. 2019;10:e01343–019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang C, Wen Yet al. ERK1/2 and the Bcl-2 family proteins Mcl-1, tBid, and Bim are involved in inhibition of apoptosis during persistent Chlamydia psittaci infection. Inflammation. 2018;41:1372–83. [DOI] [PubMed] [Google Scholar]
- Lockshin RA. Programmed cell death—II. Endocrine potentiation of the breakdown of the intersegmental muscles of silkmoths. J Insect Physiol. 1964;10:643–9. [Google Scholar]
- Longbottom D, Coulter LJ. Animal chlamydioses and zoonotic implications. J Comp Pathol. 2003;128:217–44. [DOI] [PubMed] [Google Scholar]
- Lu H, Shen C, Brunham RC. Chlamydia trachomatis infection of epithelial cells induces the activation of caspase-1 and release of mature IL-18. J Immunol. 2000;165:1463–9. [DOI] [PubMed] [Google Scholar]
- Luo F, Shu M, Gong Set al. Antiapoptotic activity of Chlamydia trachomatis Pgp3 protein involves activation of the ERK1/2 pathway mediated by upregulation of DJ-1 protein. Pathog Dis. 2019;77:ftaa003. [DOI] [PubMed] [Google Scholar]
- Magalhaes PO, Lopes AM, Mazzola PGet al. Methods of endotoxin removal from biological preparations: a review. J Pharm Pharm Sci. 2007;10:388–404. [PubMed] [Google Scholar]
- Majewski N, Nogueira V, Bhaskar Pet al. Hexokinase-mitochondria interaction mediated by Akt is required to inhibit apoptosis in the presence or absence of Bax and Bak. Mol Cell. 2004;16:819–30. [DOI] [PubMed] [Google Scholar]
- Manire GP, Meyer KF.. The toxins of the psittacosis-lymphogranuloma group of agents; the toxicity of various members of the psittacosis-lymphogranuloma venereum group. J Infect Dis. 1950;86:226–32. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Manire GP.. Electron microscopic observations on the effects of penicillin on the morphology of Chlamydia psittaci. J Bacteriol. 1970;101:278–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuo J, Nakamura S, Ito Aet al. Protochlamydia induces apoptosis of human HEp-2 cells through mitochondrial dysfunction mediated by chlamydial protease-like activity factor. PLoS One. 2013;8:e56005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlitz A, Banhart S, Maurer APet al. Tarp regulates early Chlamydia-induced host cell survival through interactions with the human adaptor protein SHC1. J Cell Biol. 2010;190:143–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messinger JE, Nelton E, Feeney Cet al. Chlamydia infection across host species boundaries promotes distinct sets of transcribed anti-apoptotic factors. Front Cell Infect Microbiol. 2015;5:96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyairi I, Mahdi OS, Ouellette SPet al. Different growth rates of Chlamydia trachomatis biovars reflect pathotype. J Infect Dis. 2006;194:350–7. [DOI] [PubMed] [Google Scholar]
- Moulder JW, Hatch TP, Byrne GIet al. Immediate toxicity of high multiplicities of Chlamydia psittaci for mouse fibroblasts (L cells). Infect Immun. 1976;14:277–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarajan UM, Sikes JD, Yeruva Let al. Significant role of IL-1 signaling, but limited role of inflammasome activation, in oviduct pathology during Chlamydia muridarum genital infection. J Immunol. 2012;188:2866–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman L, Rowley J, Vander Hoorn Set al. Global estimates of the prevalence and incidence of four curable sexually transmitted infections in 2012 based on systematic review and global reporting. PLoS One. 2015;10:e0143304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen BD, Cunningham D, Liang Xet al. Lipooligosaccharide is required for the generation of infectious elementary bodies in Chlamydia trachomatis. Proc Natl Acad Sci USA. 2011;108:10284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurminen M, Leinonen M, Saikku P. et al. The genus-specific antigen of Chlamydia: resemblance to the lipopolysaccharide of enteric bacteria. Science. 1983;220:1279–81. [DOI] [PubMed] [Google Scholar]
- Ojcius DM, Souque P, Perfettini JLet al. Apoptosis of epithelial cells and macrophages due to infection with the obligate intracellular pathogen Chlamydia psittaci. J Immunol. 1998;161:4220–6. [PubMed] [Google Scholar]
- Omsland A, Sager J, Nair Vet al. Developmental stage-specific metabolic and transcriptional activity of Chlamydia trachomatis in an axenic medium. Proc Natl Acad Sci USA. 2012;109:19781–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omsland A, Sixt BS, Horn Met al. Chlamydial metabolism revisited: interspecies metabolic variability and developmental stage-specific physiologic activities. FEMS Microbiol Rev. 2014;38:779–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paland N, Bohme L, Gurumurthy RKet al. Reduced display of tumor necrosis factor receptor I at the host cell surface supports infection with Chlamydia trachomatis. J Biol Chem. 2008;283:6438–48. [DOI] [PubMed] [Google Scholar]
- Paland N, Rajalingam K, Machuy Net al. NF-κB and inhibitor of apoptosis proteins are required for apoptosis resistance of epithelial cells persistently infected with Chlamydophila pneumoniae. Cell Microbiol. 2006;8:1643–55. [DOI] [PubMed] [Google Scholar]
- Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. 2018;18:134–47. [DOI] [PubMed] [Google Scholar]
- Paschen SA, Christian JG, Vier Jet al. Cytopathicity of Chlamydia is largely reproduced by expression of a single chlamydial protease. J Cell Biol. 2008;182:117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasparakis M, Vandenabeele P.. Necroptosis and its role in inflammation. Nature. 2015;517:311–20. [DOI] [PubMed] [Google Scholar]
- Pastorino JG, Shulga N, Hoek JB. Mitochondrial binding of hexokinase II inhibits Bax-induced cytochrome c release and apoptosis. J Biol Chem. 2002;277:7610–8. [DOI] [PubMed] [Google Scholar]
- Perfettini JL, Darville T, Dautry-Varsat Aet al. Inhibition of apoptosis by gamma interferon in cells and mice infected with Chlamydia muridarum (the mouse pneumonitis strain of Chlamydia trachomatis). Infect Immun. 2002a;70:2559–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfettini JL, Darville T, Gachelin Get al. Effect of Chlamydia trachomatis infection and subsequent tumor necrosis factor alpha secretion on apoptosis in the murine genital tract. Infect Immun. 2000;68:2237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perfettini JL, Hospital V, Stahl Let al. Cell death and inflammation during infection with the obligate intracellular pathogen, Chlamydia. Biochimie. 2003b;85:763–9. [DOI] [PubMed] [Google Scholar]
- Perfettini JL, Ojcius DM, Andrews CW Jret al. Role of proapoptotic BAX in propagation of Chlamydia muridarum (the mouse pneumonitis strain of Chlamydia trachomatis) and the host inflammatory response. J Biol Chem. 2003a;278:9496–502. [DOI] [PubMed] [Google Scholar]
- Perfettini JL, Reed JC, Israel Net al. Role of Bcl-2 family members in caspase-independent apoptosis during Chlamydia infection. Infect Immun. 2002b;70:55–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J, Wilson DP, Myers Get al. Type III secretion à la Chlamydia. Trends Microbiol. 2007;15:241–51. [DOI] [PubMed] [Google Scholar]
- Prantner D, Darville T, Sikes JDet al. Critical role for interleukin-1beta (IL-1beta) during Chlamydia muridarum genital infection and bacterial replication-independent secretion of IL-1beta in mouse macrophages. Infect Immun. 2009;77:5334–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proskuryakov SY, Konoplyannikov AG, Gabai VL. Necrosis: a specific form of programmed cell death? Exp Cell Res. 2003;283:1–16. [DOI] [PubMed] [Google Scholar]
- Qiu H, Fan Y, Joyee AGet al. Type I IFNs enhance susceptibility to Chlamydia muridarum lung infection by enhancing apoptosis of local macrophages. J Immunol. 2008;181:2092–102. [DOI] [PubMed] [Google Scholar]
- Rajalingam K, Al-Younes H, Muller Aet al. Epithelial cells infected with Chlamydophila pneumoniae (Chlamydia pneumoniae) are resistant to apoptosis. Infect Immun. 2001;69:7880–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalingam K, Sharma M, Lohmann Cet al. Mcl-1 is a key regulator of apoptosis resistance in Chlamydia trachomatis-infected cells. PLoS One. 2008;3:e3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajalingam K, Sharma M, Paland Net al. IAP-IAP complexes required for apoptosis resistance of C. trachomatis-infected cells. PLoS Pathog. 2006;2:e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajaram K, Giebel AM, Toh Eet al. Mutational analysis of the Chlamydia muridarum plasticity zone. Infect Immun. 2015;83:2870–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajeeve K, Das S, Prusty BKet al. Chlamydia trachomatis paralyses neutrophils to evade the host innate immune response. Nat Microbiol. 2018;3:824–35. [DOI] [PubMed] [Google Scholar]
- Rake G, Jones HP. A toxic factor associated with the agent of lymphogranuloma venereum. Proc Soc Exp Biol Med. 1943;53:86–8. [Google Scholar]
- Rake G, Jones HP.. Studies on lymphogranuloma venereum. II. The association of specific toxins with agents of the lymphogranuloma-psittacosis group. J Exp Med. 1944;79:463–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randow F, MacMicking JD, James LC. Cellular self-defense: how cell-autonomous immunity protects against pathogens. Science. 2013;340:701–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rank RG. In Vivo Chlamydial Infection. Intracellular Pathogens I: Chlamydiales. In: Tan M, Bavoil P (eds.). Washington, DC: ASM Press, 2012, 285–310. [Google Scholar]
- Rockey DD, Alzhanov DT. Proteins in the Chlamydial Inclusion Membrane. Chlamydia Genomics and Pathogenesis. In: Bavoil P, Wyrick PB (eds.). Norfolk, UK: Horizon Bioscience, 2006, 235–54. [Google Scholar]
- Rupp J, Pfleiderer L, Jugert Cet al. Chlamydia pneumoniae hides inside apoptotic neutrophils to silently infect and propagate in macrophages. PLoS One. 2009;4:e6020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saka HA, Valdivia RH.. Acquisition of nutrients by Chlamydiae: unique challenges of living in an intracellular compartment. Curr Opin Microbiol. 2010;13:4–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvesen GS, Duckett CS. IAP proteins: blocking the road to death's door. Nat Rev Mol Cell Biol. 2002;3:401–10. [DOI] [PubMed] [Google Scholar]
- Sarkar A, Moller S, Bhattacharyya Aet al. Mechanisms of apoptosis inhibition in Chlamydia pneumoniae-infected neutrophils. Int J Med Microbiol. 2015;305:493–500. [DOI] [PubMed] [Google Scholar]
- Schuchardt L, Rupp J.. Chlamydia trachomatis as the cause of infectious infertility: acute, repetitive or persistent long-term infection?. In: Häcker G (ed.). Biology of Chlamydia. Cham: Springer, 2016, 159–82. [DOI] [PubMed] [Google Scholar]
- Schweichel JU, Merker HJ.. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–66. [DOI] [PubMed] [Google Scholar]
- Schöier J, Öllinger K, Kvarnström Met al. Chlamydia trachomatis-induced apoptosis occurs in uninfected McCoy cells late in the developmental cycle and is regulated by the intracellular redox state. Microb Pathog. 2001;31:173–84. [DOI] [PubMed] [Google Scholar]
- Scidmore MA, Hackstadt T.. Mammalian 14-3-3beta associates with the Chlamydia trachomatis inclusion membrane via its interaction with IncG. Mol Microbiol. 2001;39:1638–50. [DOI] [PubMed] [Google Scholar]
- Sessa R, Di Pietro M, Schiavoni Get al. Chlamydia pneumoniae induces T cell apoptosis through glutathione redox imbalance and secretion of TNF-α. Int J Immunopathol Pharmacol. 2009;22:659–68. [DOI] [PubMed] [Google Scholar]
- Sharma M, Rudel T.. Apoptosis resistance in Chlamydia-infected cells: a fate worse than death?. FEMS Immunol Med Microbiol. 2009;55:154–61. [DOI] [PubMed] [Google Scholar]
- Shaw JH, Key CE, Snider TAet al. Genetic inactivation of Chlamydia trachomatis inclusion membrane protein CT228 alters MYPT1 recruitment, extrusion production, and longevity of infection. Front Cell Infect Microbiol. 2018;8:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrid AM, Hybiske K.. Chlamydia trachomatis cellular exit alters interactions with host dendritic cells. Infect Immun. 2017;85:e00046–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Zhao Y, Wang Yet al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92. [DOI] [PubMed] [Google Scholar]
- Shimada K, Crother TR, Karlin Jet al. Caspase-1 dependent IL-1beta secretion is critical for host defense in a mouse model of Chlamydia pneumoniae lung infection. PLoS One. 2011;6:e21477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegl C, Prusty BK, Karunakaran Ket al. Tumor suppressor p53 alters host cell metabolism to limit Chlamydia trachomatis infection. Cell Rep. 2014;9:918–29. [DOI] [PubMed] [Google Scholar]
- Silva MT, do Vale A, dos Santos NM. Secondary necrosis in multicellular animals: an outcome of apoptosis with pathogenic implications. Apoptosis. 2008;13:463–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt BS, Bastidas RJ, Finethy Ret al. The Chlamydia trachomatis inclusion membrane protein CpoS counteracts STING-mediated cellular surveillance and suicide programs. Cell Host Microbe. 2017;21:113–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt BS, Hiess B, König Let al. Lack of effective anti-apoptotic activities restricts growth of Parachlamydiaceae in insect cells. PLoS One. 2012;7:e29565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt BS, Kostanjsek R, Mustedanagic Aet al. Developmental cycle and host interaction of Rhabdochlamydia porcellionis, an intracellular parasite of terrestrial isopods. Environ Microbiol. 2013;15:2980–93. [DOI] [PubMed] [Google Scholar]
- Sixt BS, Nunez-Otero C, Kepp Oet al. Chlamydia trachomatis fails to protect its growth niche against pro-apoptotic insults. Cell Death Differ. 2018;26:1485–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sixt BS, Valdivia RH.. Molecular genetic analysis of Chlamydia species. Annu Rev Microbiol. 2016;70:179–98. [DOI] [PubMed] [Google Scholar]
- Skilton RJ, Cutcliffen LT, Barlow Det al. Penicillin induced persistence in Chlamydia trachomatis: high quality time lapse video analysis of the developmental cycle. PLoS One. 2009;4:e7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snavely EA, Kokes M, Dunn JDet al. Reassessing the role of the secreted protease CPAF in Chlamydia trachomatis infection through genetic approaches. Pathog Dis. 2014;71:336–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somboonna N, Wan R, Ojcius DMet al. Hypervirulent Chlamydia trachomatis clinical strain is a recombinant between lymphogranuloma venereum (L(2)) and D lineages. mBio. 2011;2:e00045–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenner-Liewen F, Liewen H, Zapata JMet al. CADD, a Chlamydia protein that interacts with death receptors. J Biol Chem. 2002;277:9633–6. [DOI] [PubMed] [Google Scholar]
- Stephens RS, Kalman S, Lammel Cet al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–9. [DOI] [PubMed] [Google Scholar]
- Subbarayal P, Karunakaran K, Winkler ACet al. EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog. 2015;11:e1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HS, Eng EW, Jeganathan Set al. Chlamydia trachomatis vacuole maturation in infected macrophages. J Leukoc Biol. 2012a;92:815–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wang H, Wang Zet al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012b;148:213–27. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhou P, Chen Set al. The JAK/STAT3 signaling pathway mediates inhibition of host cell apoptosis by Chlamydia psittaci infection. Pathog Dis. 2017;75:ftx088. [DOI] [PubMed] [Google Scholar]
- Swain RH. A microscopical study of the reproduction of psittacosis virus. Br J Exp Pathol. 1955;36:507–14. [PMC free article] [PubMed] [Google Scholar]
- Tajima M, Nomura Y, Kubota Y. Structure and development of viruses of the psittacosis-lymphogranuloma group observed in the electron microscope. J Bacteriol. 1957;74:605–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura A, Matsumoto A, Higashi N. Purification and chemical composition of reticulate bodies of the meningopneumonitis organisms. J Bacteriol. 1967;93:2003–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverne J, Blyth WA, Reeve P. Toxicity of the agents of trachoma and inclusion conjunctivitis. J Gen Microbiol. 1964;37:271–5. [DOI] [PubMed] [Google Scholar]
- Taylor HR, Burton MJ, Haddad Det al. Trachoma. Lancet. 2014;384:2142–52. [DOI] [PubMed] [Google Scholar]
- Taylor LD, Nelson, DE. Chlamydial MACPF protein CT153. Subcell Biochem. 2014;80:255–69. [DOI] [PubMed] [Google Scholar]
- Taylor LD, Nelson DE, Dorward DWet al. Biological characterization of Chlamydia trachomatis plasticity zone MACPF domain family protein CT153. Infect Immun. 2010;78:2691–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalmann J, Janik K, May Met al. Actin re-organization induced by Chlamydia trachomatis serovar D – evidence for a critical role of the effector protein CT166 targeting Rac. PLoS One. 2010;5:e9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd WJ, Caldwell HD.. The interaction of Chlamydia trachomatis with host cells: ultrastructural studies of the mechanism of release of a biovar II strain from HeLa 229 cells. J Infect Dis. 1985;151:1037–44. [DOI] [PubMed] [Google Scholar]
- Todd WJ, Storz J.. Ultrastructural cytochemical evidence for the activation of lysosomes in the cytocidal effect of Chlamydia psittaci. Infect Immun. 1975;12:638–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse SM, Mason D, Botelho RJet al. Accumulation of diacylglycerol in the Chlamydia inclusion vacuole: possible role in the inhibition of host cell apoptosis. J Biol Chem. 2005;280:25210–5. [DOI] [PubMed] [Google Scholar]
- Tummers B, Green DR. Caspase-8: regulating life and death. Immunol Rev. 2017;277:76–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Zandbergen G, Gieffers J, Kothe Het al. Chlamydia pneumoniae multiply in neutrophil granulocytes and delay their spontaneous apoptosis. J Immunol. 2004;172:1768–76. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Silke J. IAPs, RINGs and ubiquitylation. Nat Rev Mol Cell Biol. 2005;6:287–97. [DOI] [PubMed] [Google Scholar]
- Verbeke P, Welter-Stahl L, Ying Set al. Recruitment of BAD by the Chlamydia trachomatis vacuole correlates with host-cell survival. PLoS Pathog. 2006;2:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen AM, Ekert PG, Pakusch Met al. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. [DOI] [PubMed] [Google Scholar]
- Waguia Kontchou C, Tzivelekidis T, Gentle IEet al. Infection of epithelial cells with Chlamydia trachomatis inhibits TNF-induced apoptosis at the level of receptor internalization while leaving non-apoptotic TNF-signalling intact. Cell Microbiol. 2016;18:1583–95. [DOI] [PubMed] [Google Scholar]
- Wahl C, Maier S, Marre Ret al. Chlamydia pneumoniae induces the expression of inhibitor of apoptosis 2 (c-IAP2) in a human monocytic cell line by an NF-κB-dependent pathway. Int J Med Microbiol. 2003;293:377–81. [DOI] [PubMed] [Google Scholar]
- Wahl C, Oswald F, Simnacher Uet al. Survival of Chlamydia pneumoniae-infected Mono Mac 6 cells is dependent on NF-κB binding activity. Infect Immun. 2001;69:7039–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Gomez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19:918–31. [DOI] [PubMed] [Google Scholar]
- Wang SP, Grayston JT.. Classification of trachoma virus strains by protection of mice from toxic death. J Immunol. 1963;90:849–56. [PubMed] [Google Scholar]
- Wang X, Rockey DD, Dolan BP. Chlamydia lipooligosaccharide has varied direct and indirect roles in evading both innate and adaptive host-immune responses. Infect Immun. 2020;88:e00198–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, LaBrie SD, Carrell SJet al. Development of transposon mutagenesis for Chlamydia muridarum. J Bacteriol. 2019;201:e00366–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward ME. The chlamydial developmental cycle. Micobiology of Chlamydia. In: Barron AL (ed.). Boca Raton, FL: CRC Press, 1988, 71–95. [Google Scholar]
- Weber MM, Lam JL, Dooley CAet al. Absence of specific Chlamydia trachomatis inclusion membrane proteins triggers premature inclusion membrane lysis and host cell death. Cell Rep. 2017;19:1406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SJ, Brode S, Ellis Let al. Detection of a microbial metabolite by STING regulates inflammasome activation in response to Chlamydia trachomatis infection. PLoS Pathog. 2017;13:e1006383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss E. The effect of antibiotics on agents of the psittacosis-lymphogranuloma group. I. The effect of penicillin. J Infect Dis. 1950;87:249–63. [DOI] [PubMed] [Google Scholar]
- Wyrick PB, Brownridge EA, Ivins BE. Interaction of Chlamydia psittaci with mouse peritoneal macrophages. Infect Immun. 1978;19:1061–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Zhong Y, Greene Wet al. Chlamydia trachomatis infection inhibits both Bax and Bak activation induced by staurosporine. Infect Immun. 2004;72:5470–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Zhong Y, Su Het al. NF-κB activation is not required for Chlamydia trachomatis inhibition of host epithelial cell apoptosis. J Immunol. 2005;174:1701–8. [DOI] [PubMed] [Google Scholar]
- Yang C, Briones M, Chiou Jet al. Chlamydia trachomatis lipopolysaccharide evades the canonical and noncanonical inflammatory pathways to subvert innate immunity. mBio. 2019;10:e00595–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Starr T, Song Let al. Chlamydial lytic exit from host cells is plasmid regulated. mBio. 2015;6:e01648–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Christian JG, Paschen SAet al. Chlamydia trachomatis can protect host cells against apoptosis in the absence of cellular inhibitor of apoptosis proteins and Mcl-1. Microbes Infect. 2008a;10:97–101. [DOI] [PubMed] [Google Scholar]
- Ying S, Fischer SF, Pettengill Met al. Characterization of host cell death induced by Chlamydia trachomatis. Infect Immun. 2006;74:6057–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Pettengill M, Latham ERet al. Premature apoptosis of Chlamydia-infected cells disrupts chlamydial development. J Infect Dis. 2008b;198:1536–44. [DOI] [PubMed] [Google Scholar]
- Ying S, Pettengill M, Ojcius DMet al. Host-cell survival and death during Chlamydia infection. Curr Immunol Rev. 2007;3:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying S, Seiffert BM, Hacker Get al. Broad degradation of proapoptotic proteins with the conserved Bcl-2 homology domain 3 during infection with Chlamydia trachomatis. Infect Immun. 2005;73:1399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong EC, Chi EY, Kuo CC. Differential antimicrobial activity of human mononuclear phagocytes against the human biovars of Chlamydia trachomatis. J Immunol. 1987;139:1297–1302. [PubMed] [Google Scholar]
- Zhong Y, Weininger M, Pirbhai Met al. Inhibition of staurosporine-induced activation of the proapoptotic multidomain Bcl-2 proteins Bax and Bak by three invasive chlamydial species. J Infect. 2006;53:408–14. [DOI] [PubMed] [Google Scholar]
- Zuck M, Ellis T, Venida Aet al. Extrusions are phagocytosed and promote Chlamydia survival within macrophages. Cell Microbiol. 2017;19:e12683. [DOI] [PubMed] [Google Scholar]