

Abstract
O-Sulfation is an important chemical code widely existing in bioactive molecules, but the scalable and facile synthesis of complex bioactive molecules carrying O-sulfation remains challenging. Herein, we report a general approach to O-sulfation via the Sulfur (VI) Fluoride Exchange (SuFEx) reaction between aryl fluorosulfates and silylated hydroxyl groups. Efficient sulfate diester formation was achieved through systematic optimization of the electronic properties of aryl fluorosulfates. The versatility of this O-sulfation strategy was demonstrated in the scalable syntheses of a variety of complex molecules carrying sulfate diesters at various positions, including monosaccharides, disaccharides, amino acid, and steroid. Selective hydrolytic and hydrogenolytic removal of the aryl masking groups from sulfate diesters yielded the corresponding O-sulfated products in excellent yields. This strategy provides a powerful tool for the synthesis of O-sulfated bioactive compounds.
Keywords: O-sulfation, SuFEx, early-stage sulfation, glycosylation, carbohydrate chemistry
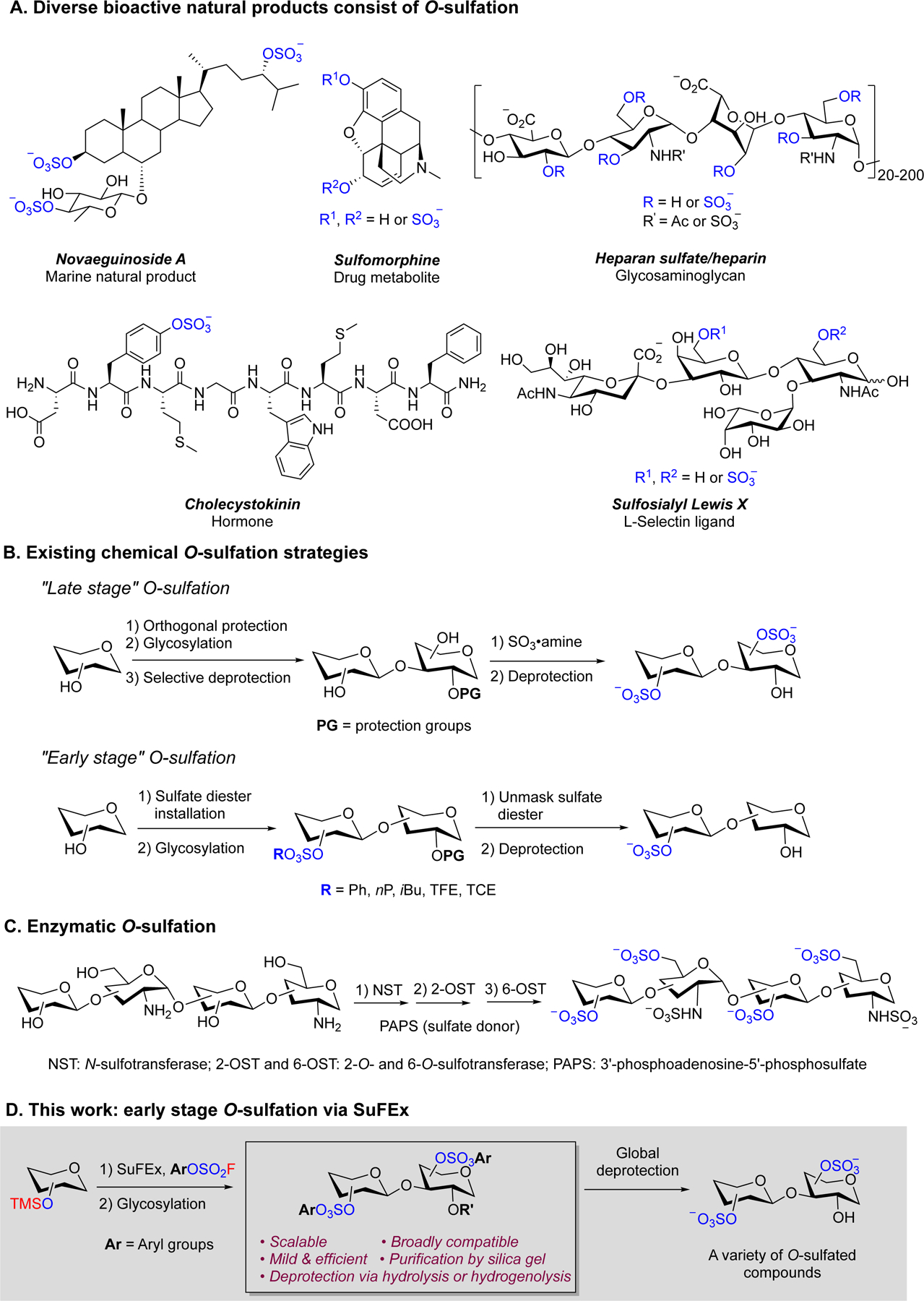
O-Sulfation widely exists in polysaccharides, liposaccharides, peptides, proteins, marine natural products, and drug metabolites in nature (Figure 1A). The spatiotemporal distribution of the O-sulfation in these molecules plays important roles in a variety of biological activities such as telomerase inhibition,[1] cell signaling,[2] anticoagulation,[3] drug detoxification,[4] and cancer metastasis.[5] However, the lack of synthetic tools for efficient and scalable O-sulfation imposes a significant constraint on our abilities to access bioactive molecules carrying complex sulfation patterns and use them to study the structure-function relationships of the sulfate modifications in biology.
Figure 1.
Significance of O-sulfation and approaches to O-sulfation in carbohydrate and other substrates.
To date, sulfur trioxide-nitrogen base remains the most common chemical reagent to introduce O-sulfation to a variety of substrates (Figure 1B).[6] O-Sulfation using this reagent can only be performed at the late stage of synthesis due to the challenging purification of the highly polar O-sulfated products and their chemical instability. These challenges are frequently exacerbated in the synthesis of polysulfated compounds of which O-sulfation at multiple residues are targeted.[7] Efforts to address these deficiencies have led to the development of the so-called “early-stage” sulfation strategies, in which targeted hydroxyl groups were converted to sulfate diesters at the early stage of synthesis that were deprotected to generate O-sulfate in the final step (Figure 1B). Penney and Perlin first reported the phenyl sulfate diester as a precursor of O-sulfation,[8] but their method necessitated chemically labile reagents, harsh reaction conditions, and long reaction time. Further optimization of the reactivity and stability of the phenyl sulfate diester was not explored, either. Since this seminal work, other sulfate diesters were employed in early-stage O-sulfation approaches, including those with neopentyl (nP),[9] isobutyl (iBu),[9] trifluoroethylene (TFE),[10] and 2, 2, 2-trichloroethylene (TCE)[11] groups. However, incompatibility with common reaction conditions and the deactivating effect on the modified carbohydrate substrates in glycosylation limited their utility in the synthesis of O-sulfated complex carbohydrates.[12] Recently, enzymatic O-sulfation by sulfotransferases emerged as a promising strategy with excellent yields and regioselectivity (Figure 1C).[13] However, the stringent specificity of sulfotransferase enzymes has caused the inflexible reaction sequence in polysulfation[13b] and limited substrate scope. The highly polar O-sulfated carbohydrates from enzymatic reactions also required high-resolution purification techniques.[13d]
Reported by Sharpless and coworkers in 2014, the SuFEx reaction has been employed to prepare a myriad of sulfate diesters.[14] We envisioned that the SuFEx reaction between an aliphatic or aromatic silylether and an aryl fluorosulfate could serve as a general early-stage O-sulfation approach to site-specifically install sulfate diesters onto carbohydrate and non-carbohydrate substrates (Figure 1D). Subsequent selective deprotection of the aryl sulfate monoester would lead to the desired O-sulfated compounds. Compared to the existing chemical and enzymatic O-sulfation strategies, advantages of this approach include the ability to balance stability and reactivity to accommodate different substrates and achieve efficient deprotection.
We first investigated the model reaction between trimethylsilyl (TMS)-protected galactopyranose (1)[15] and substituted aryl fluorosulfates (Figure 2A). Aryl fluorosulfates 2a-j carrying variable substitutions ranging from strongly electron-withdrawing groups to electron-donating groups were readily prepared from corresponding phenols (Table S1).[14c,d] A systematic screening identified that 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) efficiently catalyzed the coupling of 1 with 2a-f carrying electron-withdrawing substitutions, providing 3a-f in excellent yields (Figure 2B and Table S2). In contrast, the coupling of 1 with 2g-j carrying electron-neutral or electron-donating substitutions were sluggish, affording 3g-j in modest to low yields from 76% to 29%. These results suggested that the reaction rates positively correlated with the electron deficiency of the aryl fluorosulfates. The SuFEx reactivity of the electron-rich aryl fluorosulfates could be improved when 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD)[16] was used as the catalyst (e.g., 2i in Figure 2B and Table S3).
Figure 2.

Synthesis of aryl sulfate diesters as protected O-sulfation.
We then sought to generate the desired 6-O-sulfated galactose (4) through the selective deprotection of the aryl groups in 3a-j. Previous studies have shown that the fission rate of the two S–O bonds in a sulfate diester is determined by the pKa difference between the two leaving groups.[17] Consistently, we observed that 4 was efficiently generated as the sole product when 3a-d carrying strongly electron-withdrawing groups were treated with 5 M sodium methoxide in methanol at room temperature (Figure 2B and Table S4). In contrast, the hydrolysis of 3e-j carrying weak electron-withdrawing or electron-neutral groups was inefficient (Figure 2B and Figures S1–S2. See Figure S3 for mechanistic analysis).
Concerns over undesired side reactions caused by strong base treatment[18] prompted us to investigate an alternative deprotection strategy. Penney and Perlin first reported hydrogenolysis of phenyl sulfate monoester using PtO2 and hydrogen gas (H2) in modest yields.[8] Inspired by this work, we studied the selective hydrogenolysis of the aryl sulfate monoester in glycosyl aryl sulfate diesters by Pd(OH)2/C/H2, and observed high deprotection efficiency in an acetonitrile/methanol/water solution of phosphate salts (Table S5). Notably, this condition is insensitive to the electronic properties of the aryl substitution, yielding 4 from all substrates 3a-j in excellent yields at room temperature (Figure 2B). Gas chromatography-mass spectrometry (GC-MS) analysis of the hydrogenolysis reaction revealed a mechanism of oxidative addition of palladium into the aryl C–O bond followed by the reductive elimination of arenes (Figures S4–S6), as opposed to the previously proposed mechanism of hydrogenation of the aryl group followed by S–O bond fission to release cyclohexanol.[8] Our observation that hydrogen gas can be replaced by ammonium formate, a far less potent hydrogenation agent, without affecting the hydrogenolysis efficiency, further proved the oxidative addition-reductive elimination mechanism (Figure S7).[19]
Our subsequent investigation was focused on the feasibility of this strategy for a variety of substrates (Figure 2C). Initial attempts to install p-nitrophenyl sulfate diester onto the O-6 position of methyl 2,3,4-tri-O-benzyl-α-D-glucopyranoside by reacting with 2a generated an ether byproduct methyl 2,3,4-tri-O-benzyl-6-O-(p-nitrophenyl)-α-D-glucopyranoside along with the targeted sulfate diester. We attributed this side reaction to the susceptibility of the electron-deficient sulfate diesters to nucleophiles. Indeed, O-6 substitution using 2e provided sulfate diesters 5 and 6 in 70% and 78% yields, respectively, without ether formation. Furthermore, O-6 substitution of the glucosyl thiol donor required 2j carrying the strongly electron-donating p-methoxy group, affording 7 in 72% yield. In these reactions, we also discovered that aryl fluorosulfates remained stable in the presence of silylethers before the introduction of organobase catalysts, making it possible for a one-pot procedure combining in situ hydroxyl silylation by hexamethyldisilazane (HMDS)[20] and the subsequent SuFEx reaction. Following the one-pot procedure, sulfate diesters 8 was prepared in 77% yield. No major difference in reaction efficiency was found between the stepwise and one-pot procedures, as shown in the preparation of 9a and 14a. In addition to simplifying the operation, the one-pot procedure could also circumvent the challenge to isolate TMS silylethers that are often unstable on silica gel chromatography. Other silylether protecting groups, such as tert-butyldimethylsilyl (TBS) in 9a and tert-butyldiphenylsilyl (TBDPS) in 9b, remained stable in both stepwise and one-pot procedures. Sulfate diesters at different positions like O-3 and O-4 of glucose were also successfully obtained, giving products 10, 11, 12a and 12b in 67%, 94%, 60%, and 67% yields, respectively.[21] Moreover, 13 and 14a, monosaccharides carrying two sulfate diesters were prepared successfully in 73% and 71% yields, respectively. The efficiency and scale of disubstitution could be further improved to 95% in multi-gram quantities when switching from 2a to 2e. Disubstituted disaccharide 15 was also synthesized in gram scale from the corresponding dual-TMS-protected substrate, with 2j being found to enable the highest SuFEx efficiency of all aryl fluorosulfates tested at 67% yield. Besides carbohydrates, the SuFEx reaction was further applied to install sulfate diesters on non-carbohydrate substrates. Estrone sulfate diester 16 and tyrosine sulfate diester 17 were efficiently prepared in 89% and 94% yields, respectively. It is noteworthy that all compounds mentioned above were prepared in readily scalable procedures and purified by silica gel chromatography. Taken together, these examples highlight the broad substrate scope of our method and its versatility in tuning the electronic properties of the aryl fluorosulfates to optimize their reactivities.
Next, the compatibility of our approach with frequently used reagents and reaction conditions in carbohydrate and peptide chemistries was examined (Figure 3). First, the aryl sulfate diesters as the protected sulfates could overcome the acid sensitivity of the nonprotected sulfates (Figure 3A). The anomeric O-methyl group of 5 was converted to the O-acetyl group in 18 by sulfuric acid in 85% yield. Disulfated 1,6-anhydroidose 14b was readily converted into the ring-opening product 19 in 81% yield by strong Lewis acid scandium triflate.[22] Basic conditions were tolerated (Figure 3B). From 9c and 21, removal of the TBS and acetyl protecting groups by tetrabutylammonium fluoride (TBAF) and sodium methoxide afforded 20 in excellent yields. Notably, TBAF compatibility was a challenge for the state-of-the-art TCE sulfate diesters due to the tendency of the undesired fluorination.[12a] While tyrosine p-nitrophenylsulfate diester demonstrated limited stability in 20% piperidine in DMF (Table S6), the m-nitrophenyl sulfate diester 17 was perfectly stable under the same condition. Oxidizing conditions were also well tolerated (Figure 3C). The anomeric p-methoxyphenyl group of 6 was removed to form 22 in 92% yield by cerium ammonium nitrate (CAN). Oxidation of the thiol donor 7 by m-chloroperoxybenzoic acid (m-CPBA) afforded sulfoxide 23 in 79% yield. Oxidative debenzylation of 14a by sodium bromate and sodium dithionite quantitatively yielded glycosyl acceptors 24. Reducing conditions were tolerated (Figure 3D). Acid-promoted reductive benzylidene opening of 8 exposed O-6 or O-4 positions as free hydroxyls to afford 25 and 26 in quantitative and 95% yields, respectively. Zinc-catalyzed azide reduction of 15 afforded the free amine 27 in 78% yield. Finally, the sulfate diesters are also compatible with the sulfur trioxide-nitrogen base reagent (Figure 3E). N-Sulfation of 27 by SO3∙pyridine generated 28 in 72% yield. Overall, these results indicated excellent compatibilities of the aryl sulfate diesters with acidic, basic, oxidizing, and reducing conditions commonly used in carbohydrate and peptide chemistries.
Figure 3.

Compatibility of aryl sulfate diesters to common reagents used in carbohydrate and peptide chemistries.
We then examined the glycosylation reactions of both donors and acceptors modified by glycosyl aryl sulfate diesters (Figure 4). First, the disubstituted acceptor 24 was coupled with imidate donor 29, successfully yielding disaccharides 30 in 65% yield (Figure 4A). Next, glycosylation of acceptor 33 using O-6 sulfate diester-substituted glucosyl donors including the bromide donor (in situ generated from 1-O-acetyl-glucopyranoside 18), fluoride donor (31), and the phosphate donor (32) all proceeded efficiently, forming disaccharide 34 in excellent (73–92%) yields (Figure 4B). Because sulfate diester modifications could deactivate carbohydrate building blocks in glycosylation reactions, coupling the glycosyl donor and acceptor became challenging when both were modified by sulfate diesters in previous early stage sulfation strategies.[10c,12a] Our initial attempt to couple the bromide donor and the 1,6-anhydroidosyl acceptor 20 generated the disaccharide product 35 in 51% yield (Figure 4C). The yield of the disaccharide was improved to 79% when thiol donor 7 was employed (Figure 4C). However, it was found that 7 was partially decomposed after three days at 4 °C, likely attributed to the intramolecular nucleophilic attack at the O-6 sulfate diester by the anomeric thiol group. This challenge was overcome by oxidizing 7 into the sulfoxide donor 23, which can be stored in long term without any signs of decomposition. Glycosylation efficiency was further improved when the sulfoxide donor 23 was employed, affording 36 in 95% yield even when both the donor and the acceptor carried sulfate diester substitutions (Figure 4C).
Figure 4.

Preparation of O-sulfated complex molecules.
Because hydrogenolysis was milder and less prone to side reactions than hydrolysis, it became the method of choice for sulfate diester deprotection (Figure 4D). Indeed, global deprotection of multigram amounts of 14b by Pd(OH)2/C/H2 successfully yielded O-2, O-4 disulfated idose 37 in 95% yield. Furthermore, disulfated 38, trisulfated 39, and disulfated 40 were also efficiently prepared in quantitative, 83%, and 90% yields, respectively. The deprotection of non-carbohydrate sulfate diesters was also efficient. Hydrogenolysis of 16 and 17 exclusively furnished sulfoestrone 41 and sulfotyrosine 42 in high yields, without any desulfated byproducts being observed. The excellent selectivity of hydrogenolysis is attributed to the strong electron-withdrawing p- or m-NO2 substitution that weakens the nearby aryl C–O bond and makes it susceptible to oxidative addition by palladium.
In summary, a facile and scalable approach for early stage O-sulfation via SuFEx reaction was developed for both carbohydrates and non-carbohydrate compounds. The SuFEx coupling reactions were optimized to efficiently generate sulfate diesters. The sulfate diesters demonstrated excellent compatibility with a broad range of reaction conditions and can be efficiently deprotected to yield O-sulfated products. This strategy provides a powerful tool for the synthesis of complex O-sulfated small molecules and macromolecules including carbohydrates, glycomimetic polymers, and peptides.
Supplementary Material
Acknowledgements
We thank Marek Domin for assistance in mass spectrometry characterizations and Professor James Morken for helpful comments. The research is supported by a startup fund from Boston College to J.N. and the NIH Director’s New Innovator Award (1DP2HG011027-01) to J.N.
Footnotes
Conflict of interest
Authors through Boston College have filed a patent application on the O-sulfation approach.
References
- [1].Warabi K, Hamada T, Nakao Y, Matsunaga S, Hirota H, van Soest RW, Fusetani N, J. Am. Chem. Soc 2005, 127, 13262–13270. [DOI] [PubMed] [Google Scholar]
- [2].Bowman KG, Bertozzi CR, Chem. Biol 1999, 6, R9–R22. [DOI] [PubMed] [Google Scholar]
- [3].McLean J, Am. J. Physiol 1916, 41, 250–257. [Google Scholar]
- [4].Ratzka A, Vogel H, Kliebenstein DJ, Mitchell-Olds T, Kroymann J, Proc. Natl. Acad. Sci. U.S.A 2002, 99, 11223–11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sasisekharan R, Shriver Z, Venkataraman G, Narayanasami U, Nat. Rev. Cancer 2002, 2, 521–528. [DOI] [PubMed] [Google Scholar]
- [6].a) Lee J-C, Lu X-A, Kulkarni SS, Wen Y-S, Hung S-C, J. Am. Chem. Soc 2004, 126, 476–477; [DOI] [PubMed] [Google Scholar]; b) Tully SE, Mabon R, Gama CI, Tsai SM, Liu X, Hsieh-Wilson LC, J. Am. Chem. Soc 2004, 126, 7736–7737. [DOI] [PubMed] [Google Scholar]
- [7].Al-Horani RA, Desai UR, Tetrahedron 2010, 66, 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Penney CL, Perlin AS, Carbohydr. Res 1981, 93, 241–246. [Google Scholar]
- [9].Simpson LS, Widlanski TS, J. Am. Chem. Soc 2006, 128, 1605–1610. [DOI] [PubMed] [Google Scholar]
- [10].a) Proud AD, Prodger JC, Flitsch SL, Tetrahedron Lett 1997, 38, 7243–7246; [Google Scholar]; b) Karst NA, Islam TF, Linhardt RJ, Org. Lett 2003, 5, 4839–4842; [DOI] [PubMed] [Google Scholar]; c) Karst NA, Islam TF, Avci FY, Linhardt RJ, Tetrahedron Lett 2004, 45, 6433–6437. [Google Scholar]
- [11].a) Liu Y, Lien I-FF, Ruttgaizer S, Dove P, Taylor SD, Org. Lett 2004, 6, 209–212; [DOI] [PubMed] [Google Scholar]; b) Ingram LJ, Taylor SD, Angew. Chem. Int. Ed 2006, 45, 3503–3506; [DOI] [PubMed] [Google Scholar]; c) Ingram LJ, Desoky A, Ali AM, Taylor SD, J. Org. Chem 2009, 74, 6479–6485; [DOI] [PubMed] [Google Scholar]; d) Desoky AY, Taylor SD, J. Org. Chem 2009, 74, 9406–9412. [DOI] [PubMed] [Google Scholar]
- [12].a) Tiruchinapally G, Yin Z, El‐Dakdouki M, Wang Z, Huang X, Chem. Eur. J 2011, 17, 10106–10112; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Matsushita K, Sato Y, Funamoto S, Tamura J.-i., Carbohydr. Res 2014, 396, 14–24. [DOI] [PubMed] [Google Scholar]
- [13].a) Xu Y, Masuko S, Takieddin M, Xu H, Liu R, Jing J, Mousa SA, Linhardt RJ, Liu J, Science 2011, 334, 498–501; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen Y, Li Y, Yu H, Sugiarto G, Thon V, Hwang J, Ding L, Hie L, Chen X, Angew. Chem. Int. Ed 2013, 52, 11852–11856; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu J, Linhardt RJ, Nat. Prod. Rep 2014, 31, 1676–1685; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang X, Pagadala V, Jester HM, Lim AM, Pham TQ, Goulas AMP, Liu J, Linhardt RJ, Chem. Sci 2017, 8, 7932–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Dong J, Krasnova L, Finn M, Sharpless KB, Angew. Chem. Int. Ed 2014, 53, 9430–9448; [DOI] [PubMed] [Google Scholar]; b) Dong J, Sharpless KB, Kwisnek L, Oakdale JS, Fokin VV, Angew. Chem. Int. Ed 2014, 53, 9466–9470; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Gao B, Zhang L, Zheng Q, Zhou F, Klivansky LM, Lu J, Liu Y, Dong J, Wu P, Sharpless KB, Nat. Chem 2017, 9, 1083; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yang C, Flynn JP, Niu J, Angew. Chem. Int. Ed 2018, 57, 16194–16199. [DOI] [PubMed] [Google Scholar]
- [15].Gembus V, Marsais F, Levacher V, Synlett 2008, 1463–1466. [Google Scholar]
- [16].Yatvin J, Brooks K, Locklin J, Angew. Chem. Int. Ed 2015, 54, 13370–13373. [DOI] [PubMed] [Google Scholar]
- [17].Younker JM, Hengge AC, J. Org. Chem 2004, 69, 9043–9048. [DOI] [PubMed] [Google Scholar]
- [18].a) Herczeg M, Lázár L, Mándi A, Borbás A, Komáromi I, Lipták A, Antus S, Carbohydr. Res 2011, 346, 1827–1836; [DOI] [PubMed] [Google Scholar]; b) Lázár L, Mező E, Herczeg M, Lipták A, Antus S, Borbás A, Tetrahedron 2012, 68, 7386–7399. [Google Scholar]
- [19].a) Guan B-T, Lu X-Y, Zheng Y, Yu D-G, Wu T, Li K-L, Li B-J, Shi Z-J, Org. Lett 2010, 12, 396–399; [DOI] [PubMed] [Google Scholar]; b) Liang Q, Xing P, Huang Z, Dong J, Sharpless KB, Li X, Jiang B, Org. Lett 2015, 17, 1942–1945; [DOI] [PubMed] [Google Scholar]; c) Ram S, Ehrenkaufer RE, Synthesis 1988, 91–95. [Google Scholar]
- [20].Joseph AA, Verma VP, Liu XY, Wu CH, Dhurandhare VM, Wang CC, Eur. J. Org. Chem 2012, 2012, 744–753. [Google Scholar]
- [21].The major isolated byproducts were desilylated carbohydrate substrates. For more information, please see Figure S9.
- [22].a) Zulueta MML, Lin S-Y, Lin Y-T, Huang C-J, Wang C-C, Ku C-C, Shi Z, Chyan C-L, Irene D, Lim L-H, J. Am. Chem. Soc 2012, 134, 8988–8995; [DOI] [PubMed] [Google Scholar]; b) Lee J-C, Tai C-A, Hung S-C, Tetrahedron Lett 2002, 43, 851–855. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.