

Abstract
Objective.
This study sought to conduct a comprehensive search for genetic risk of cognitive decline in the context of geriatric depression.
Design.
A genomewide association study (GWAS) analysis in the Neurocognitive Outcomes of Depression in the Elderly (NCODE) study.
Setting.
Longitudinal, naturalistic follow-up study
Participants.
Older depressed adults, both outpatients and inpatients, receiving care at an academic medical center.
Measurements.
The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) neuropsychological battery was administered to the study participants at baseline and a minimum of twice within a subsequent three-year period in order to measure cognitive decline. A GWAS analysis was conducted to identify genetic variation that is associated with baseline and change in the CERAD Total Score (CERAD-TS) in NCODE.
Results.
The GWAS of baseline CERAD-TS revealed a significant association with an intergenic SNP on chromosome 6, rs17662598, that surpassed adjustment for multiple testing (p=3.7x10-7, FDR q=0.0371). For each additional G allele, average baseline CERAD-TS decreased by 8.656 points. The most significant SNP that lies within a gene was rs11666579 in SLC27A1 (p=1.1x10-5). Each additional copy of the G allele was associated with an average decrease of baseline CERAD-TS of 4.829 points. SLC27A1 is involved with processing docosahexaenoic acid (DHA), an endogenous neuroprotective compound in the brain. Decreased levels of DHA have been associated with the development of Alzheimer’s disease (AD). The most significant SNP associated with CERAD-TS decline over time was rs73240021 in GRXCR1 (p=1.1x10-6), a gene previously linked with deafness. However, none of the associations within genes survived adjustment for multiple testing.
Conclusions.
Our GWAS of cognitive function and decline among individuals with LLD has identified promising candidate genes that, upon replication in other cohorts of LLD, may be potential biomarkers for cognitive decline and suggests DHA supplementation as a possible therapy of interest.
Introduction
The relationship between depression and cognitive function is complex. Depression, especially when occurring in later life, has long been associated with executive impairment, attentional problems, and slowed speed of information processing (Butters et al., 2004; Koenig et al., 2015). Other studies have identified memory impairment as a concern among older depressed patients (Lee et al., 2007). Cognitive impairments in late-life depression may persist despite adequate treatment of mood symptoms (Lee et al., 2007; Mackin et al., 2014); for instance, we previously reported two-year outcomes among older cognitively impaired, non-demented depressives that included both normal cognition and cognitive decline, the latter consisting of various forms of cognitive impairment as well as dementia (Steffens et al., 2009). This is consistent with over 30 years of epidemiological research linking depression in mid- and late-life to later development of Alzheimer’s disease (AD) (Devanand et al., 1996; Jorm et al., 1991; Kokmen et al., 1991; Speck et al., 1995; Steffens et al., 1997; Saczynski et al., 2010). Other studies have found an association between late-life depression and development of vascular dementia (Alexopoulos et al., 1993; Diniz et al., 2013). The heterogeneity of cognitive trajectories of late-life depression presents a challenge for early detection and treatment of what may be both distinct and overlapping etiologies of disease. Given the rapid aging of populations, there is a pressing scientific need for approaches that help elucidate unique and shared variance in trajectories of cognitive decline associated with late-life depression.
Recent studies suggest that the variance in the presentations of cognitive impairment and late-life depression may be explained by genetic polymorphisms (Brzezinska et al., 2020). Large-scale genetic studies to identify loci or genes associated with increased risk of cognitive decline or dementia in the context of depression have been limited. One strategy employed has been to examine genes and alleles known to increase AD risk, including the epsilon-4 allele of Apolipoprotein E gene (APOE ε4) (Saunders et al., 1993). Another candidate gene approach has been to examine genes associated with risk for depression where there is a plausible scientific basis supporting a link with AD risk. For example, single nucleotide polymorphisms (SNPs) of genes encoding cholinergic muscarinic receptors, which have been related to depression, (Chee and Cumming, 2018). Other studies have found genetic loci common to depression and AD that were related to inflammatory, serotonergic, neurotrophic, and immune pathways (Kang et al., 2015; Kitzlerova et al., 2018; Lutz et al., 2020), and the angiotensin-converting enzyme (ACE) gene (Zettergren et al., 2017). Genetic polymorphisms of brain-derived neurotrophic factor (BDNF), interleukin 1-beta (IL1B), and methylenetetrahydrofolate reductase (MTHFR) confer increased risk to both late-life depression and AD (Ye et al., 2016). Despite these findings, some have suggested that a common genetic predisposition for depression and AD may be unlikely (Herbert and Lucassen, 2016).
In comparison to candidate gene approaches, which are hypothesis driven, genome-wide association studies (GWAS) may help identify putative genes that increase the risk for cognitive decline and dementia among depressed individuals in an unbiased manner. GWAS analyses that sought to identify genetic loci linking depression and cognitive change have implicated genes related to cerebrovascular disease (Rutten-Jacobs et al., 2018), presynaptic function (White et al., 2017), and the complement pathway (Hamilton et al., 2012). However, one GWAS study found no evidence to support a common polygenic structure for AD and MDD (Gibson et al., 2017).
To date, there has not been a GWAS of cognitive decline in the context of geriatric depression, which may be due to a lack of a consensus on how to conceptualize “cognitive decline” as a phenotypic target. One approach is to define cognitive decline clinically based on the established diagnostic criteria that characterize it. An example of this is a consensus diagnostic approach that has been used in many population-based studies (Plassman et al., 2006; Plassman et al., 2007). However, because diagnoses of cognitive impairment share common neuropsychological deficits with late-life depression (Zihl et al., 2010), an alternative approach is to track decline on an objective index of cognitive function. To address the former issue, we examine cognitive decline as a clinical diagnosis based on expert consensus; to address the latter issue, we examine change on a validated neuropsychological battery, such as the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) instrument (Morris et al., 1989). The CERAD Total Score has been shown to be a valid global measure of AD progression and of annualized change between AD and control groups (Rossetti et al., 2010). As such, change in the CERAD Total Score (CERAD-TS) may be a useful phenotypic target for GWAS. We hypothesize that cognitive decline within a geriatric depressed cohort may represent distinct underlying genetic risks and pathways than simply geriatric depression alone. Moreover, these genetic risk factors may lay the path for subsequent neurodegenerative disorders in the same individuals.
We undertook a GWAS analysis in Neurocognitive Outcomes of Depression in the Elderly (NCODE), a longitudinal study of older depressed adults that characterized the incidence of cognitive decline and development of cognitive disorders including AD. We examined both by clinical diagnosis, as well as by a neuropsychological phenotype. We hypothesized that this approach would identify genetic markers that might be candidates for future genetic studies of cognitive impairment and cognitive decline in LLD.
Methods
The Sample
The methods of the NCODE study, including a description of the sample, have been previously described (Steffens et al., 2004). In brief, the NCODE sample consists of participants originally enrolled in the Conte Center for the Study of Depression in the Elderly, a National Institute for Mental Health (NIMH)-supported study of depressed and non-depressed older adults (age 60 and above) at Duke University Medical Center. Some individuals were enrolled beginning in 1995 into the NIMH-supported Clinical Research Center at Duke and have subsequently agreed to continue participating in the longitudinal study associated with the Conte Center, spanning a study period from 1995 to 2011. As part of the enrollment evaluation, a geriatric psychiatrist interviewed each depressed participant and assessed depression symptoms with several standardized clinical assessments (Steffens et al., 2004). Depressed participants met Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) criteria for major depressive episode. Depressed participants entering the study with a Mini-Mental State Exam (MMSE) less than 25 were followed clinically to assess cognition and determine whether a diagnosis of baseline dementia warranted exclusion. In the present analysis, based on the clinical judgment of the study geriatric psychiatrist following established study protocol, clinically evident dementia was excluded at or close to baseline in all participants.
Participants with psychotic depression were included, as were those with comorbid anxiety disorders, as long as major depression was deemed by the treating geriatric psychiatrist on the study to be the primary psychiatric disorder.
The sample for the current study initially consisted of 271 individuals meeting criteria for major depressive episode on NCODE study entry who were referred to a series of Consensus Diagnostic Conferences (CDCs, see below). The study was approved by the Institutional Review Board at Duke University Medical Center, and the study procedures were explained to all participants, who then provided written informed consent to participate.
Clinical Follow-up of Depressed Participants
The NCODE study operates in a naturalistic treatment milieu using treatment guidelines established by the Duke Affective Disorders Program (Steffens et al., 2002a). Treatment modalities available included antidepressant medications, electroconvulsive therapy, and individual and group cognitive-behavioral psychotherapy. Treatment was monitored to ensure that clinical guidelines were followed appropriately. Patients were evaluated when clinically indicated and at least every 3 months for the duration of study participation. The protocol recommends that participants receive continuation treatment for at least 1 to 2 years (some indefinitely) once they achieve remission. Each participant was thus assured to receive the most appropriate care we were able to provide.
Referral of Participants with Cognitive Impairment
Participants had the option of referral to the Memory Disorders Clinic at Duke University Medical Center when (1) they self-reported cognitive complaints, (2) family members reported cognitive concerns to the study geriatric psychiatrist, or (3) the psychiatrist had a clinical suspicion of cognitive impairment or dementia. The study sought to obtain copies of medical records from these referrals when they occurred.
Neuropsychological Battery
The neuropsychological test battery was administered to depressed participants at baseline while still symptomatic and then annually regardless of depression status. A trained psychometric technician supervised by a licensed clinical neuropsychologist administered testing. The full battery is described elsewhere (Steffens et al., 2004), while the current study focuses on the tests in the battery that constitute the CERAD-TS. The CERAD TS was computed based on the original publication by Chandler et al. (Chandler et al., 2005), and includes score ranges from Animal Naming (0-24); 15-item Boston Naming Test (0-15); Constructional Praxis (0-11); and Word List Learning (0-30), Delayed Recall (-10), and Recognition Memory Discriminability (true positives – false positives: 0-10). The CERAD TS is the sum of these individual tests and ranges from 0-100. Longitudinal CERAD-TS was utilized to assess cognitive change. Participants with baseline CERAD-TS and two or more CERAD TS over the first three annual follow-up evaluations were included (N=145).
Consensus Diagnostic Conference
Clinical diagnoses were made by a consensus panel of experts in dementia, based on a model developed in several epidemiological studies of dementia (Plassman et al., 2006; Plassman et al., 2007). The panel consisted of a core group of experts, including 3 to 4 geriatric psychiatrists, a cognitive neuroscientist, 1 to 2 neuropsychologists specializing in memory disorders, and a neurologist specializing in memory disorders. Panel members reviewed the following information for each participant presented: (1) initial and most recent clinical depression study notes, (2) neuropsychological testing profiles and provisional diagnoses for all participants who underwent testing, and (3) neurological consultations when available. The treating study psychiatrist briefly presented the case, and a neuropsychologist summarized the neuropsychological findings to the group. Discussion among the panel members would ensue until a consensus clinical diagnosis was assigned. Panel members chose among several clinical diagnoses (Steffens et al., 2004). Dementia diagnoses were based on criteria from the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition. (American Psychiatric Association, 1994). For AD diagnoses featured in the present study, we used published criteria for diagnoses of probable and possible AD (McKhann et al., 1984); diagnoses of other types of dementia were based on currently accepted criteria (McKeith et al., 1996; Roman et al., 1993; The Lund and Manchester Groups, 1994). Individuals with cognitive impairment not meeting criteria for dementia, were included a broad category of cognitive impairment, no dementia (CIND). Diagnosis of CIND was based on prior work (Plassman et al., 2000; Plassman et al., 2006), defined as mild cognitive or functional impairment that does not meet criteria for dementia, such as performance on neuropsychological measures that was below expectation based on the individual’s premorbid history, and scores at least 1.5 standard deviations (SDs) below published norms on any test. Finally, individuals with no cognitive impairment were diagnosed as cognitively normal. As mentioned previously, all participants in this study met criteria for major depression at the time of study enrollment. For the purposes of the current study, we used the following diagnostic groups at the time of censure, which was 5 years from the time of study enrollment: 1) cognitive impairment (CI), which encompasses diagnoses of CIND, AD, and non-AD dementias; 2) Alzheimer’s disease only (AD), and 3) cognitively normal (CN), which reflects with no diagnoses of cognitive impairment during the study period.
Genotyping
DNA was extracted from whole blood using the Puregene system (Gentra Systems, Minneapolis, MN). A total of 576 samples were genotyped with the Infinium PsychArray-24 v1.3 BeadChip (Illumina, San Diego, CA), which included 552 study samples (271 depressed and 381 non-depressed), 12 replicates, and 12 internal quality control (QC) samples. Resultant genotype data was analyzed using the GenomeStudio software (Illumina, San Diego, CA) in order to call individual genotypes. Samples with whole genome amplified DNA were removed (n = 18). Several quality control (QC) methods were employed including call rate > 98% (n=4 samples excluded) and exclusion of gender discrepancies (n=6 samples excluded). Cryptic relatedness was performed using PLINK (Purcell et al., 2007), which resulted in the exclusion of one duplicate and two first degree relatives of other study samples. Identity by descent estimates for all replicates and their matched study sample were 1, as expected. Principal components analysis (PCA) was run using the smartpca program from the software package EIGENSOFT (Patterson et al., 2006) in order to identify remaining outliers (n=0 excluded) and calculate eigenvectors to use as covariates in the statistical analysis. Finally, we required probes to have a call rate > 97% and display no deviation from Hardy-Weinberg Equilibrium (HWE) in the control samples (p-values > 10−6). In total, 521 samples and 398,317 probes passed genotyping QC checks.
Imputation
To increase genomic coverage, we imputed missing genotypes using a global reference panel from the 1000 Genomes Project (www.1000genomes.org). Samples were pre-phased using SHAPEIT (Delaneau et al., 2011) and genotypes imputed using IMPUTE2 (Howie et al., 2009). Imputed probes with certainty < 90% were zeroed out for specific individuals and were subsequently removed from the entire data set if the call rate was < 97% in all samples. Imputed probes were also removed if HWE p-values were < 10−6 in controls or if the minor allele frequency (MAF) was < 5%. A subset of genotyped calls were masked and imputed to determine the average imputation accuracy. The concordance between imputed and true genotype was 98.3%. After all quality control steps, 3,730,665 autosomal probes were available for statistical analysis.
Statistical Analysis
After removing participants with missing clinical data, 271 depressed participants were analyzed. To reduce genetic heterogeneity, the primary analyses were conducted in 222 depressed participants of Caucasian ancestry. Potential covariates were examined with respect to two diagnoses: Alzheimer’s disease (AD) and the broader definition of any cognitive impairment (CI); each of these groups were separately compared to the reference group of individuals with the cognitively normal diagnosis (CN) using chi-square tests for categorical covariates and t-tests for continuous covariates in SAS v9.4 (SAS Institute, Cary, NC). Trajectories of CERAD decline were obtained from beta estimates of CERAD-TS regressed on time (years) for each participant. To assess how well the beta estimate fit the longitudinal data, we examined the distribution of root-mean-square error (RMSE). Three participants were excluded from this analysis due to RMSE values more than three SDs from the mean, indicating the trajectory of CERAD decline was not linear for those participants. Genome-wide SNPs were assessed for association with AD and CI compared to (CN) participants using logistic regression with an additive genetic model implemented in PLINK. In addition to dichotomous outcomes, linear regression models were used to investigate the associations between genome-wide SNPs and baseline CERAD-TS or CERAD decline scores. Several relevant variables were considered for inclusion as covariates in the regression models: age, sex, race, years of education, and the cumulative illness rating scale (CIRS) total score. Due to significant confounding among several pairs of these variables, only age, sex, and two genome-wide principal components were included as covariates. Additionally, baseline CERAD-TS was covaried in the models of CERAD decline. In an effort to reduce genomic redundancy, LD-clumping was performed on the Caucasian subset in PLINK using previously reported thresholds (p1=1, p2=1, r2=0.25, 500kb window) (Ripke et al., 2014). False discovery rate (FDR) q-values were calculated and quantile-quantile (QQ) plots were generated using the R packages qvalue and qqman, respectively.
Results
Among the 271 depressed NCODE participants, 123 experienced cognitive decline over time; 31 (14.76%) were assigned a diagnosis of AD and 92 (33.95%) were assigned diagnoses related to CI, including those with AD and other dementias. As shown in Table 1, compared with CN participants, those with AD were older at time of enrollment and completed fewer years of education. There was no difference in the proportion of females or Caucasian ancestry or in mean CIRS total score between AD and CN participants. As expected, those with AD had significantly lower average CERAD-TS at baseline compared with CN participants. Of interest, CERAD-TS for depressed AD participants declined at a faster rate compared with depressed CN participants. Results for CI participants compared to CN participants yielded similar results (Table 1).
Table 1.
Participant characteristics
| AD (N=12) | CI (N=31) | CN (N=111) | p-value (AD vs. CN) |
p-value (CI vs. CN) |
|
|---|---|---|---|---|---|
| % female | 67.74% | 65.22% | 61.45% | 0.5045 | 0.5439 |
| age at enrollment (mean, SD) | 73.35 (7.09) | 71.97 (6.75) | 66.82 (5.93) | <0.0001 | <0.0001 |
| % Caucasian | 86.67% | 80.22% | 82.68% | 0.5885 | 0.6198 |
| years education (mean, SD) | 13.45 (3.25) | 13.39 (3.22) | 14.78 (2.28) | 0.0058 | <0.0001 |
| CIRS total score (mean, SD) | 4.24 (2.10) | 5.00 (2.84) | 4.87 (3.2) | 0.3106 | 0.7456 |
| CERAD total score (mean, SD) | 60.77 (13.37) | 63.20 (10.66) | 79.29 (8.46) | <0.0001 | <0.0001 |
| 3 year CERAD decline score (mean, SD) | −1.73 (4.94) | −0.09 (3.97) | 0.87 (2.41) | 0.0022 | 0.0935 |
AD=Alzheimer’s disease, CI=cognitively impaired, CN=cognitively normal, CIRS=cumulative illness rating scale, CERAD=Consortium to Establish a Registry for Alzheimer’s Disease)
Comparisons for AD vs.CN and CI vs CN groups used chi-square tests for categorical covariates and t-tests for continuous covariates
Many potential covariates were correlated with each other and therefore they were not all included in the subsequent GWAS analyses. Younger participants completed more years of education (p=0.0048) and had a lower CIRS total score (p=0.0016). Males and those of Caucasian ancestry completed more years of education (p=0.0005 and 0.0006, respectively). 90% of males were of Caucasian ancestry, while 77% of females were Caucasian (p=0.0019). Because 82% of the participants were Caucasian, we limited the GWAS analysis to Caucasians (N = 222).
Clinical diagnosis
Among the depressed individuals, we were interested in identifying genetic variants that significantly predicted AD vs. CN (Figure 1a) and CI vs CN (Figure 1b). None of these analyses resulted in a genome-wide significant result. Again, we note that CI is a broad construct that incorporates dementia including AD.
Figure 1.
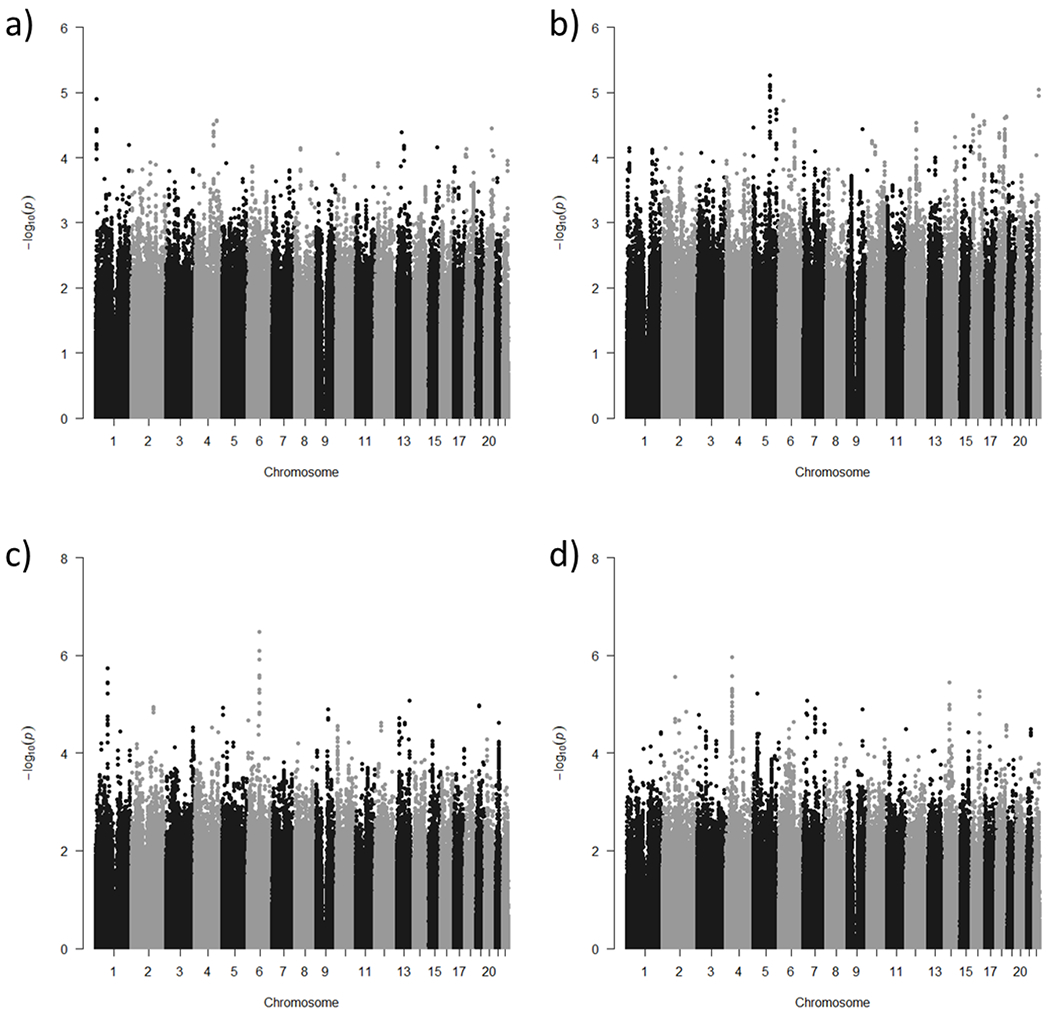
Manhattan plots of GWAS results a) AD vs. CN b) CI vs. CN c) baseline CERAD total score d) CERAD decline score
AD vs.CN. The most significant SNP in the analysis of AD compared to CN was rs754804 with a minor allele frequency of 0.06, located in an intergenic region of chromosome 1 (p=1.25x10−5). Individuals with the T allele were 32 times more likely to have AD than be CN. The most significant SNP in a gene was rs17851751, which is a nonsynonymous variant in ZMAT4 on chromosome 8 (p=7.1x10−5). Individuals with the C allele were 6.5 times more likely to have AD than be CN.
CI vs. CN. The most significant SNP when comparing CI to CN participants was rs79966641 located in an intron of DMXL1 on chromosome 5 (p=5.4x10−6). Individuals with the A allele were 6.3 times more likely to be CI than CN.
Neuropsychological phenotype
We explored whether there were genetic variants influencing CERAD score, both at baseline and decline.
Baseline cognitive analyses.
The GWAS of baseline CERAD-TS revealed a significant intergenic SNP on chromosome 6, rs17662598, that surpassed adjustment for multiple testing (p=3.7x10−7, FDR q=0.0371). For each additional G allele, average baseline CERAD-TS was 8.656 points lower compared to those with the AA genotype. The most significant SNP that lies within a gene was rs11666579 in SLC27A1 (p=1.1x10−5). Each additional copy of the G allele was associated with an average CERAD baseline score 4.829 points lower than those with the TT genotype.
Longitudinal cognitive analyses.
The most significant SNP associated with CERAD decline over time was rs73240021 in GRXCR1 (p=1.1x10−6). However, this association did not survive adjustment for multiple testing.
Discussion
This study represents, to our knowledge, the first GWAS of cognitive decline in late-life depression. We compared those patients who subsequently developed AD to those who remained cognitively intact, as well as those with cognitive impairment to those who remained cognitively intact. We hypothesized that a quantitative measure of cognitive decline might provide more statistical power for this analysis. Thus, we also examined GWAS of CERAD baseline cognitive performance, as well as cognitive decline, as measured by change in CERAD-TS over at least three annual time points including baseline. Analyses related to AD vs CN and CI vs CN did not reach genome-wide statistical significance, with the most significant SNPs being located in an intergenic region on chromosome 1 (rs754804, p=1.25x10−5) and within ZMAT4 (rs17851751, p=7.1x10−5) for AD vs CN analyses; and in an intron of DMXL1 (rs79966641, p=5.4x10−6) for CI vs CN analyses. Analyses of baseline CERAD-TS revealed a genome-wide significant association with a SNP on chromosome 6, rs17662598 (p=3.7x10−7, FDR q=0.0371). We also identified a SNP lying within SLC27A1 (rs11666579), that did not reach genome-wide significance (p=1.1x10−5). Our analyses of CERAD-TS decline revealed a SNP in GRXCR1 (rs73240021) that did not reach genome-wide significance (p=1.1x10−6).
The most compelling association that we detected was in the GWAS of baseline CERAD-TS, which identified rs17662598, an intergenic SNP that remains significant after adjusting for multiple comparisons. This SNP has been identified as an expression QTL (eQTL) in Genotype Tissue Expression (GTEx) database, but only in testis. There are no known candidate regulatory elements (cREs) directly overlapping rs17662598 according to the Encyclopedia of DNA Elements (ENCODE) database, but there are three cREs within 2kb of the associated SNP (http://screen.encodeproject.org/search/?q=rs17662598&assembly=hg19&uuid=0). Additional research will be necessary to understand how this highly statistically significant association reflects underlying biology of cognitive function. A SNP in SLC27A1 was also nominally associated (p=1.05x10−5), though it did not reach genome-wide significance (q=0.2053). SLC27A1 is the fatty acid transport protein 1 (FATP1), which has docosahexaenoic acid (DHA) as a substrate. DHA is an endogenous neuroprotective compound, and decreased levels of DHA in the brain are associated with the development of AD (Ochiai et al., 2019). The GWAS of CERAD decline identified a SNP in GRXCR1, a gene associated with autosomal-recessive nonsyndromic hearing impairment (Schraders et al., 2010). This is notable, as hearing impairment has been associated with cognitive decline and depression in late life (Rutherford et al., 2018).
The intergenic SNP rs754804, found in the AD vs CN GWAS, is 10kb from the gene SLC45A1. This gene has been previously associated with intellectual disability with neuropsychiatric features (Srour et al., 2017). It is possible that variation in rs754804 is regulating expression of SLC45A1, however the GTEx database shows no significant eQTLs in any tissue. Looking in the ENCODE database, there are no directly overlapping cRE, but there are five cREs within 2kb of this SNP (http://screen.encodeproject.org/search/?q=rs754804&uuid=0&assembly=hg19). Thus, it is possible that the association with this SNP is driven by these other regulatory elements. The most significant SNP that fell in a gene was a nonsynonymous SNP in ZMAT4 (rs17851751) associated with AD. Interestingly, ZMAT4 has previously been associated with refractive error (Fan et al., 2014), which has in turn been associated with cognitive function (Ong et al., 2013).
For CI vs. CN analyses, DMXL1, lying in a region on chromosome 5, has been associated with astrocytomas (van den Boom et al., 2006), and a link has been hypothesized between AD and astrocytomas (Lehrer, 2018). DMXL1 has also been associated with primary open-angle glaucoma (Davis et al., 2011).
The strengths of our study include the careful clinical assessment and the novelty of our approach. The diagnosis of MDD was assigned by a geriatric psychiatrist based on a comprehensive standardized assessment, and participants received ongoing care. Participants completed neuropsychological testing annually, and cognitive diagnoses were assigned by an expert consensus panel based on current clinical histories. The diagnostic process using consensus diagnoses has been shown to be reliable and valid (Breitner et al., 1995). In addition, this study represents the first GWAS of cognitive decline in the context of geriatric depression, which was examined across both clinical diagnosis and neuropsychological phenotype.
Despite the strengths, we also acknowledge that the study has several limitations. The most significant limitation is the sample size. Notably, most GWAS analyses are conducted in samples of several thousand individuals and our study had only a few hundred individuals. This certainly impacted the statistical power to identify associations. However, despite the small sample, we did identify one genomewide significant association, and several other plausible candidate genes that were nominally significant. Additionally, our approach to conceptualize cognitive decline quantitatively by using annual CERAD assessments was quite novel. Nonetheless, having more CERAD assessments over a longer period of time could provide a more informative construct of cognitive decline. While our results are intriguing, they are simply a first step in understanding the genetic architecture of cognitive decline in geriatric depression. As such, we have refrained from reporting effect sizes. Future work should build upon these findings and ideally include much larger samples.
The advantage of the GWAS approach over previous candidate gene approaches is the potential to identify new genes and pathways related to the development of a particular disorder or condition. While the chip we used in this study did not include APOE variants, we note that we failed to find an association between APOE genotype and incident dementia in our prior NCODE study (Steffens et al., 2007), highlighting the importance of research that seeks to discover new genetic paths linking depression, cognitive decline and dementia. In the present study, the results related to cognitive performance and cognitive decline are particularly intriguing and point towards DHA biology and hearing impairment as being related to baseline and longitudinal cognition in depression.
Acknowledgements
This study was supported by R01 MH108578 from the U.S. National Institute of Mental Health and by the Leo and Anne Albert Charitable Trust.
Footnotes
Conflict of interest
None.
References
- Alexopoulos GS, Meyers BS, Young RC, Mattis S and Kakuma T (1993). The course of geriatric depression with “reversible dementia”: a controlled study. American Journal of Psychiatry, 150, 1693–1699. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (1994). Diagnostic and Statistical Manual of Mental Disorders, 4th ed. Washington, DC. [Google Scholar]
- Breitner JC, et al. (1995). Alzheimer’s disease in the National Academy of Sciences-National Research Council Registry of Aging Twin Veterans. III. Detection of cases, longitudinal results, and observations on twin concordance. Arch Neurol, 52, 763–771. [DOI] [PubMed] [Google Scholar]
- Brzezinska A, Bourke J, Rivera-Hernandez R, Tsolaki M, Wozniak J and Kazmierski J (2020). Depression in Dementia or Dementia in Depression? Systematic Review of Studies and Hypotheses. Curr Alzheimer Res, 17, 16–28. [DOI] [PubMed] [Google Scholar]
- Burke SL, Maramaldi P, Cadet T and Kukull W (2016). Associations between depression, sleep disturbance, and apolipoprotein E in the development of Alzheimer’s disease: dementia. Int Psychogeriatr, 28, 1409–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butters MA, et al. (2004). The nature and determinants of neuropsychological functioning in late-life depression. Arch Gen Psychiatry, 61, 587–595. [DOI] [PubMed] [Google Scholar]
- Chandler MJ, et al. (2005). A total score for the CERAD neuropsychological battery. Neurology, 65, 102–106. [DOI] [PubMed] [Google Scholar]
- Chee LY and Cumming A (2018). Polymorphisms in the Cholinergic Receptors Muscarinic (CHRM2 and CHRM3) Genes and Alzheimer’s Disease. Avicenna J Med Biotechnol, 10, 196–199. [PMC free article] [PubMed] [Google Scholar]
- Davis LK, et al. (2011). Copy number variations and primary open-angle glaucoma. Invest Ophthalmol Vis Sci, 52, 7122–7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaneau O, Marchini J and Zagury JF (2011). A linear complexity phasing method for thousands of genomes. Nat Methods, 9, 179–181. [DOI] [PubMed] [Google Scholar]
- Devanand DP, et al. (1996). Depressed mood and the incidence of Alzheimer’s disease in the elderly living in the community. Archives of General Psychiatry, 53, 175–182. [DOI] [PubMed] [Google Scholar]
- Diniz BS, Butters MA, Albert SM, Dew MA, Reyholds CF 3rd. (2013) Late-life depression and risk of vascular dementia and Alzheimer’s disease: systematic reviwe and meta-analysis of community-based cohort studies. Br J Psychiatry, 202, 329–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q, et al. (2014). Education influences the association between genetic variants and refractive error: a meta-analysis of five Singapore studies. Hum Mol Genet, 23, 546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson J, et al. (2017). Assessing the presence of shared genetic architecture between Alzheimer’s disease and major depressive disorder using genome-wide association data. Transl Psychiatry, 7, e1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton G, et al. (2012). Alzheimer’s disease risk factor complement receptor 1 is associated with depression. Neurosci Lett, 10, 6–9. [DOI] [PubMed] [Google Scholar]
- Herbert J and Lucassen PJ (2016). Depression as a risk factor for Alzheimer’s disease: Genes, steroids, cytokines and neurogenesis - What do we need to know? Front Neuroendocrinol, 41, 153–171. [DOI] [PubMed] [Google Scholar]
- Howie BN, Donnelly P and Marchini J (2009). A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet, 5, e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorm AF, et al. (1991). Psychiatric history and related exposures as risk factors for Alzheimer’s disease: A collaborative re-analysis of case-control studies. International Journal of Epidemiology, 20(suppl 2), S43–S47. [DOI] [PubMed] [Google Scholar]
- Kang HJ, et al. (2015). Associations of cytokine genes with Alzheimer’s disease and depression in an elderly Korean population. J Neurol Neurosurg Psychiatry, 86, 1002–1007. [DOI] [PubMed] [Google Scholar]
- Kitzlerova E, et al. (2018). Interactions Among Polymorphisms of Susceptibility Loci for Alzheimer’s Disease or Depressive Disorder. Med Sci Monit, 24, 2599–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig AM, et al. (2015). Neuropsychological functioning in the acute and remitted States of late-life depression. J Alzheimers Dis, 45, 175–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokmen E, Beard CM, Chandra V, Offord KP, Schoenberg BS and Ballard DJ (1991). Clinical risk factors for Alzheimer’s disease: A population-based case-control study. Neurology, 41, 1393–1397. [DOI] [PubMed] [Google Scholar]
- Lee JS, Potter GG, Wagner HR, Welsh-Bohmer KA and Steffens DC (2007). Persistent mild cognitive impairment in geriatric depression. Int Psychogeriatr, 19, 125–135. [DOI] [PubMed] [Google Scholar]
- Lehrer S (2018). Glioma and Alzheimer’s Disease. J Alzheimers Dis Rep, 2, 213–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz MW, Sprague D, Barrera J, Chiba-Falek O (2020). Shared Genetic Etiology Underlying Alzheimer’s Disease and Major Depressive Disorder. Transl Psychiatry, 10, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackin RS, et al. (2014). Cognitive outcomes after psychotherapeutic interventions for major depression in older adults with executive dysfunction. Am J Geriatr Psychiatry, 22, 1496–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeith IG, et al. (1996). Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology, 47, 1113–1124. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D and Stadlan EM (1984). Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology, 34, 939–944. [DOI] [PubMed] [Google Scholar]
- Morris JC, et al. (1989). The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology, 39, 1159–1165. [DOI] [PubMed] [Google Scholar]
- Ochiai Y, Uchida Y, Tachikawa M, Couraud PO and Terasaki T (2019). Amyloid beta25-35 impairs docosahexaenoic acid efflux by down-regulating fatty acid transport protein 1 (FATP1/SLC27A1) protein expression in human brain capillary endothelial cells. J Neurochem, 150, 385–401. [DOI] [PubMed] [Google Scholar]
- Ong SY, et al. (2013). Myopia and cognitive dysfunction: the singapore malay eye study. Invest Ophthalmol Vis Sci, 54, 799–803. [DOI] [PubMed] [Google Scholar]
- Patterson N, Price AL and Reich D (2006). Population structure and eigenanalysis. PLoS Genet, 2, e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plassman BL, et al. (2000). Documented head injury in early adulthood increases risk of Alzheimer’s disease and other dementias 50 years later. Neurology, 55, 1158–1166. [DOI] [PubMed] [Google Scholar]
- Plassman BL, et al. (2006). Duke Twins Study of Memory in Aging in the NAS-NRC Twin Registry. Twin Res Hum Genet, 9, 950–957. [DOI] [PubMed] [Google Scholar]
- Plassman BL, et al. (2007). Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology, 29, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet, 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH and Holmans PA (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature, 511, 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roman GC, et al. (1993). Vascular dementia: diagnostic criteria for research studies. Report of the NINDS-AIREN International Workshop. Neurology, 43, 250–260. [DOI] [PubMed] [Google Scholar]
- Rossetti HC, Munro Cullum C, Hynan LS and Lacritz LH (2010). The CERAD Neuropsychologic Battery Total Score and the progression of Alzheimer disease. Alzheimer Dis Assoc Disord, 24, 138–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutherford BR, Brewster K, Golub JS, Kim AH and Roose SP (2018). Sensation and Psychiatry: Linking Age-Related Hearing Loss to Late-Life Depression and Cognitive Decline. Am J Psychiatry, 175, 215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Older Adults With Mild Cognitive Impairment. Am J Geriatr Psychiatry. [Google Scholar]
- Rutten-Jacobs LCA, et al. (2018). Genetic Study of White Matter Integrity in UK Biobank (N=8448) and the Overlap With Stroke, Depression, and Dementia. Stroke, 49, 1340–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saczynski JS, Beiser A, Seshadri S, Auerbach S, Wolf PA and Au R (2010). Depressive symptoms and risk of dementia: the Framingham Heart Study. Neurology, 75, 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos LE, Beckman D and Ferreira ST (2016). Microglial dysfunction connects depression and Alzheimer’s disease. Brain Behav Immun, 55, 151–165. [DOI] [PubMed] [Google Scholar]
- Saunders AM, et al. (1993). Association of apolipoprotein E allele E4 with late-onset familial and sporadic Alzheimer’s disease. Neurology, 43, 1467–1472. [DOI] [PubMed] [Google Scholar]
- Schraders M, et al. (2010). Homozygosity mapping reveals mutations of GRXCR1 as a cause of autosomal-recessive nonsyndromic hearing impairment. Am J Hum Genet, 86, 138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speck CE, et al. (1995). History of depression as a risk factor for Alzheimer’s disease. Epidemiology, 6, 366–369. [DOI] [PubMed] [Google Scholar]
- Srour M, et al. (2017). Dysfunction of the cerebral glucose transporter SLC45A1 in individuals with intellectual disability and epilepsy. Am J Hum Genet, 100, 824–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens DC, McQuoid DR and Krishnan KR (2002a). The Duke Somatic Treatment Algorithm for Geriatric Depression (STAGED) approach. Psychopharmacol Bull, 36, 58–68. [PubMed] [Google Scholar]
- Steffens DC, McQuoid DR and Potter GG (2009). Outcomes of older cognitively impaired individuals with current and past depression in the NCODE study. J Geriatr Psychiatry Neurol, 22, 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steffens DC, Plassman BL, Helms MJ, Welsh-Bohmer KA, Saunders AM and Breitner JC (1997). A twin study of late-onset depression and apolipoprotein E epsilon 4 as risk factors for Alzheimer’s disease. Biological Psychiatry, 41, 851–856. [DOI] [PubMed] [Google Scholar]
- Steffens DC, et al. (2007). Longitudinal magnetic resonance imaging vascular changes, apolipoprotein E genotype, and development of dementia in the neurocognitive outcomes of depression in the elderly study. Am J Geriatr Psychiatry, 15, 839–849. [DOI] [PubMed] [Google Scholar]
- Steffens DC, et al. (2004). Methodology and preliminary results from the Neurocognitive Outcomes of Depression in the Elderly study. Journal of Geriatric Psychiatry and Neurology, 17, 202–211. [DOI] [PubMed] [Google Scholar]
- The Lund and Manchester Groups (1994). Clinical and neuropathological criteria for frontotemporal dementia. J Neurol Neurosurg Psychiatry, 57, 416–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J, Wolter M, Blaschke B, Knobbe CB and Reifenberger G (2006). Identification of novel genes associated with astrocytoma progression using suppression subtractive hybridization and real-time reverse transcription-polymerase chain reaction. Int J Cancer, 119, 2330–2338. [DOI] [PubMed] [Google Scholar]
- White CC, et al. (2017). Identification of genes associated with dissociation of cognitive performance and neuropathological burden: Multistep analysis of genetic, epigenetic, and transcriptional data. PLoS Med, 14, e1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Q, Bai F and Zhang Z (2016). Shared Genetic Risk Factors for Late-Life Depression and Alzheimer’s Disease. J Alzheimers Dis, 52, 1–15. [DOI] [PubMed] [Google Scholar]
- Zettergren A, et al. (2017). The ACE Gene Is Associated with Late-Life Major Depression and Age at Dementia Onset in a Population-Based Cohort. Am J Geriatr Psychiatry, 25, 170–177. [DOI] [PubMed] [Google Scholar]
- Zihl J, Reppermund S, Thum S and Unger K (2010). Neuropsychological profiles in MCI and in depression: Differential cognitive dysfunction patterns or similar final common pathway disorder? J Psychiatr Res, 44, 647–654. [DOI] [PubMed] [Google Scholar]