

Abstract
The U.S. Food and Drug Administration recently approved two poly‐adenosine diphosphate‐ribose polymerase (PARP) inhibitors, olaparib and rucaparib, for treatment of biomarker‐positive metastatic castrate resistant prostate cancer. The benefits of PARP inhibition have been well characterized in patients who have BRCA1 and BRCA2 mutations in several forms of cancer. BRCA1 and BRCA2 occupy key roles in DNA damage repair, which is comprised of several different pathways with numerous participants. Patients with mutations in other key genes within the DNA damage repair pathway may also respond to treatment with PARP inhibitors, and identification of these alterations could significantly increase the percentage of patients that may benefit from PARP inhibition. This review focuses on the potential for synthetically lethal interactions between PARP inhibitors and non‐BRCA DNA damage repair genes.
Implications for Practice
The treatment potential of PARP inhibition has been well characterized in patients with BRCA1 and BRCA2 mutations, but there is compelling evidence for expanding the use of PARP inhibitors to mutations of other non‐BRCA DNA damage repair (DDR) genes. This could increase the percentage of patients that may benefit from treatment with PARP inhibitors alone or in combination with other therapies. Understanding the significance of PARP inhibitor‐sensitizing alterations in other common non‐BRCA DDR genes will help guide clinical decisions to provide targeted treatment options to a wider population of patients.
Keywords: Poly(ADP‐ribose) polymerase inhibitor, Prostate cancer, Olaparib, Rucaparib, BRCA, DNA damage repair, Homologous recombination repair, Mismatch repair, Mutation, Biomarker
Short abstract
The FDA recently approved two poly‐ADP‐ribose polymerase (PARP) inhibitors for the treatment of biomarker‐positive metastatic castrate resistant prostate cancer. This review highlights non‐BRCA DNA damage repair gene alterations that may increase patient sensitivity to PARP inhibition alone or in combination with other therapies.
Introduction
Until the U.S. Food and Drug Administration's (FDA's) recent approval of two poly‐adenosine diphosphate (ADP) ribose polymerase (PARP) inhibitors, rucaparib and olaparib, the treatment armamentarium for men with metastatic‐castration resistant prostate cancer (mCRPC) included first‐generation androgen receptor (AR) axis‐targeting agents (flutamide, bicalutamide, and nilutamide), next‐generation AR axis‐targeting agents (abiraterone, enzalutamide, apalutamide, darolutamide), taxane‐based chemotherapies (docetaxel, cabazitaxel), and the radiopharmaceutical radium‐223 (for bone metastases), as well as immunotherapy with Sipuleucel‐T [1, 2]. Although these therapies have considerably improved outcomes over the past decade, prostate cancer remains the second most common cause of cancer‐related mortality in American men [3]. The approval of pembrolizumab for solid tumors designated as microsatellite instability‐high (MSI‐H) or those with mismatch repair deficiencies served as the first tumor‐agnostic treatment regimen that ushered in the era of precision medicine for mCRPC. PARP inhibitors, which take advantage of DNA damage repair (DDR) germline and somatic mutations, introduce a new genetically stratified approach to treating prostate cancer and have previously been approved for treatment of certain forms of breast and ovarian cancers [4, 5]. PARP inhibitors are most often associated with pathogenic alterations of the DDR genes BRCA1 and BRCA2, but there is compelling evidence for expanding the use of PARP inhibitors beyond BRCA to mutations of other non‐BRCA DDR genes, which could increase the percentage of patients that may benefit from treatment with PARP inhibitors. There is also increasing evidence to suggest that certain DDR mutations do not confer sensitivity to PARP inhibition, and understanding when to not prescribe PARP inhibitors is also critical for physicians as they make clinical decisions. This review will highlight non‐BRCA DDR gene alterations that may increase patient sensitivity to PARP inhibition alone or in combination with other therapies.
DNA Damage Repair and PARP Inhibition
PARPs form a large class of 16 enzymes. PARP1 and PARP2 respond to DNA damage and facilitate the cell's DDR response [6]. In healthy cells, PARP1 recognizes and binds to DNA at the site of single‐strand breaks (SSBs) and double‐strand breaks (DSBs). A series of structural allosteric changes of PARP1 follow, which allows for the recruitment of acceptor proteins and production of a negatively charged poly(ADP‐ribose) branched polymer composed of NAD+ that links the damaged site to surrounding chromatin in a process known as PARylation [7, 8]. The negative charge repulses PARP1 from the complex and acts as a target for DDR proteins and polymerases, including XRCC1, POLβ, and LIG3, for continuation of the damage repair response [8]. Should the SSB repair process fail, an accumulation of SSBs leads to replication arrest, followed by potentially lethal DSB formations at collapsed replication forks. DSBs can also occur independently from failed SSB repair, as they are commonly the result of harmful ionizing radiation damage to the cell. Healthy cells have the ability to recognize and repair DSBs via one of the four main DSB repair pathways: homologous recombination (HR), nonhomologous end joining (NHEJ), alternative NHEJ, or single‐strand annealing [9]. In addition to SSBs, PARP1 can recognize DSBs and competes with the Ku protein complex and the MRN complex for localization at free DNA ends. Depending on the phase of the cell cycle, either the Ku protein complex or PARP1 will carry out NHEJ or alt‐NHEJ, respectively [10].
Classic NHEJ requires that the Ku protein complex activates and recruits the catalytic subunit of DNA‐PK (DNA‐dependent protein kinase) to the site of damage, thereby forming the DNA‐PK complex, which can be phosphorylated by a number of factors, including ATM, a protein linked to the HR DSB repair pathway [11]. XCCR4 stabilizes the complex, whereas Artemis and other repair and ligation factors are recruited to complete repair. PARP1 has been implicated in the stabilization of the Ku protein complex in some cases [10]. In the absence of the Ku protein complex, PARP1 can induce alt‐NHEJ, which operates similarly to SSB repair, with XRCC1 and LIG3 filling key roles for repair [12, 13].
Recognition of DSB damage by the MRN complex, composed of MRE11, RAD50, and NBS1, initiates the HR repair pathway, which provides a homology‐directed, high‐fidelity repair of DSBs through various subpathways, depending on cell cycle stage and molecular competition [14]. Following recognition by the MRN complex, ATM is recruited to the site of damage to activate the MRN‐ATM signaling axis. Together with ATR, the MRN‐ATM signaling axis recruits and activates a number of downstream targets, including BRCA1, BRCA2, PALB2, CHEK1, CHEK2, RAD51, and P53 [12, 13, 14, 15, 16, 17, 18, 19, 20].
Another form of DNA damage repair concerns the resolution of mismatched nucleotides that arise from replication errors, repair errors, or chemical damage. Mismatch repair (MMR) is initiated following recognition of an error by either MutSα (MSH2/MSH6) or MutSβ (MSH2/MSH3), depending on the size of the error: MutSα preferentially repairs smaller indels, whereas MutSβ localizes to larger tracts of base errors. Depletions in MutSα are more deleterious; however, as larger errors can be repaired through other pathways, should the function of MutSβ be hindered [21, 22]. Following recognition and localization by either MutSα or MutSβ, MutLα (MLH1/PMS2) is recruited to form a tetrameric complex that forms DNA incisions [23]. Exo1, and a number of other polymerases and ligation proteins, are recruited to complete the repair [10, 24]. These DNA repair mechanisms are summarized in Figure 1.
Figure 1.
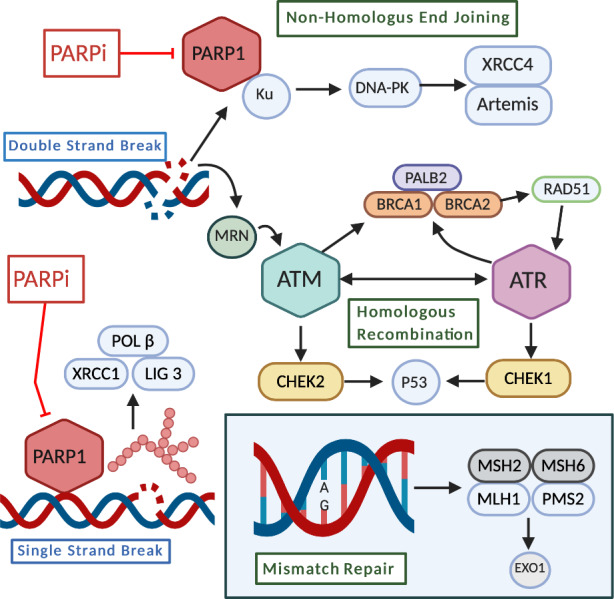
DNA damage repair mechanisms. Single‐strand break (SSB) repair: PARP1 detects single‐strand DNA breaks and facilitates the formation of a negatively charged, branched polymer that recruits XRCC1, LIG 3, and POL β to the site of damage for ligation and repair. Inhibition of PARP1 at this stage leads to an accumulation of SSBs that ultimately results in DSBs [7, 8]. Double‐strand break (DSB) repair: nonhomologous end joining–DNA ends are bound by Ku proteins, which are stabilized by PARP1, and form the DNA‐PK (DNA‐dependent protein kinase) complex following recruitment of DNA‐PKcs (catalytic subunit of DNA‐PK). XCCR4 and Artemis are recruited, which stabilize and recruit other repair factors to the site of damage [9, 10, 11]. Homologous recombination: damage is detected by the MRN complex, which recruits and activates ATM. ATM can activate the PALB2, BRCA1, and BRCA2 complex, ATR, or CHEK2, depending on cell cycle phase. RAD51 is activated and conducts a search for a homologous template used for repair, which activates other factors necessary for repair. Ultimately, P53 is stabilized by either CHEK1 or CHEK2 and proofs the repair [12, 13, 14, 15, 16, 17, 18, 19, 20]. Mismatch repair: a MSH2/MSH6 heterodimer recognizes and localizes mismatched base pair errors and forms a complex with MLH1 and PMS2. This complex recruits the exonuclease EXO1 as well as other repair and ligation factors [21, 22, 23, 24].Figure was created using BioRender.com.Abbreviations: PARP, poly(ADP‐ribose) polymerase; PARPi, PARP inhibitor.
Due to the significant roles that PARP1 fulfills in mediating the cell's DDR response, PARP inhibitors emerged as a potential therapy to sensitize tumor cells to conventional DNA damage‐causing cancer treatments. Nicotinamide analogs were shown to inhibit poly(ADP‐ribose) polymer formation and to increase tumor cell sensitivity to dimethyl sulphate [25], and further drug development led to the first‐generation clinical introduction of veliparib, rucaparib, olaparib, niraparib, and second‐generation talazoparib. There is some variation in size and structure among available PARP inhibitors, but generally, the inhibitors bind to active sites of PARP1, preventing PARylation and the ability of bound PARP1 to release from DNA‐chromatin structures. This process has been described as “PARP trapping,” and available PARP inhibitors vary in how effectively PARP can be trapped [26]. Approved prostate cancer (PCa) PARP inhibitors, rucaparib and olaparib, are relatively similar in their ability to trap PARP. Talazoparib has been shown to bind chromatin, DNA, and PARP to around a 100‐fold greater degree than olaparib and rucaparib, but this cannot be directly correlated to clinical efficacy [27]. Studies investigating head‐to‐head PARP inhibitor efficacy, target, and toxicity comparisons are ongoing [25, 28, 29, 30, 31, 32].
In 2005, PARP inhibitors were implicated as a possible means of treatment for patients with cancer with germline BRCA1 and BRCA2 mutations [33, 34]. Patients possessing germline mutations in DNA damage repair genes have a higher risk of developing certain cancers, including prostate, breast, and ovarian cancers [35, 36, 37]. There is also evidence suggesting germline mutations in DNA damage repair genes are more likely to be associated with aggressive disease types. Cells harboring these heterozygous mutations often lose the wild‐type functioning allele during tumorigenesis and are rendered unable to repair DSBs via the HR repair pathway, thus driving carcinogenesis and the emergence of a tumor that is genetically distinct from the normal tissues around it. Treating patients possessing these DDR genetic mutations with PARP inhibitors offers a combinatory approach that introduces errors in DNA damage detection and takes advantage of the preexisting malfunctioning HR pathway within the tumor microenvironment, likely resulting in tumor cell death. This exploitative approach is known as “synthetic lethality,” in which two normally nonlethal events synergistically produce a fatal effect [30]. The capability to induce lethal events confined to tumor tissue harboring loss‐of‐function mutations in essential DNA damage repair genes poses great therapeutic potential across many types of cancers [33, 38]. In 2014, the FDA approved olaparib as a monotherapy treatment of advanced ovarian cancer in patients with deleterious germline BRCA mutations, with approvals of other PARP inhibitors in other cancers following [39]. FDA approvals of PARP inhibitors are listed in Tables 1 and 2.
Table 1.
FDA approvals for poly(ADP‐ribose) polymerase inhibitors in all cancers
| Disease and drug | Treatment | Approval date |
|---|---|---|
| Ovarian cancer | ||
| Olaparib | Advanced ovarian cancer | 12/19/2014 |
| Ovarian cancer maintenance therapy | 08/17/2017 | |
| First‐line maintenance therapy in BRCA‐mutated advanced ovarian cancer | 12/19/2018 | |
| First‐line maintenance therapy with bevacizumab for HRD‐positive advanced ovarian cancer | 05/08/2020 | |
| Rucaparib | Advanced ovarian cancer | 12/19/2016 |
| Maintenance therapy of recurrent ovarian cancer | 09/16/2018 | |
| Niraparib | Recurrent ovarian cancer | 03/27/2017 |
| Late‐line treatment of recurrent ovarian cancer | 10/23/2019 | |
| First‐line monotherapy for platinum‐responsive advanced ovarian cancer regardless of biomarker status | 04/29/2020 | |
| Breast cancer | ||
| Olaparib | Germline BRCA‐mutated metastatic breast cancer | 01/12/2018 |
| Talazoparib | Germline BRCA‐mutated HER2‐negative locally advanced or metastatic breast cancer | 10/16/2018 |
| Pancreatic cancer | ||
| Olaparib | First‐line maintenance therapy in BRCA‐mutated metastatic pancreatic cancer | 12/30/2019 |
| Prostate cancer | ||
| Rucaparib | Monotherapy for BRCA1/2 mutant mCRPC | 05/15/2020 |
| Olaparib | HR repair gene mutated mCRPC | 05/20/2020 |
Abbreviations: FDA, U.S. Food and Drug Administration; HR, homologous recombination; HRD, homologous recombination deficient mCRPC, metastatic castration‐resistant prostate cancer.
Table 2.
FDA approvals for poly(ADP‐ribose) polymerase inhibitors in prostate cancer
| Drug | Approved dose | Treatment group | Side effects | Clinical trials contributing to approval |
|---|---|---|---|---|
| Rucaparib | 600 mg orally twice daily, with or without food | Men with deleterious BRCA germline or somatic mutations and mCRPC who have previously been treated with AR‐directed therapy and a taxane‐based chemotherapy | Fatigue (including asthenia), nausea, anemia, ALT/AST increased, decreased appetite, rash, constipation, thrombocytopenia, vomiting, diarrhea | TRITON2: NCT02952534 |
| Olaparib | 300 mg orally twice daily with or without food | Men with deleterious, or suspected deleterious, germline or somatic HR repair mutated mCRPC following disease progression after enzalutamide or abiraterone treatments | Nausea, fatigue (including asthenia), anemia, vomiting, diarrhea, decreased appetite, headache, neutropenia, dysgeusia, cough, dyspnea, dizziness, dyspepsia, leukopenia, thrombocytopenia, and abdominal pain | PROfound: NCT02987543 |
Abbreviations: FDA, U.S. Food and Drug Administration; ALT, alanine aminotransferase; AR, androgen receptor; AST, aspartate aminotransferase; HR, homologous recombination; mCRPC, metastatic castration‐resistant prostate cancer.
DDR Mutations and Prostate Cancer
A number of studies have reported the frequencies of somatic and germline mutations in DDR genes at several disease stages of PCa, but whether or not patient mutation status (germline or somatic) indicates clinical benefit has yet to be seen [40]. In 2015, Robinson et al. evaluated 150 cases of mCRPC and found that 22.7% of tumors harbored deleterious DDR germline or somatic mutations in BRCA1, BRCA2, ATM, CDK12, FANCA, RAD51B, and RAD51C [41]. Pritchard et al. found that 11.8% of screened patients with mCRPC had at least one germline mutation in a DDR gene [42], and Abida et al., in 2017, found that 27% of screened patients across all stages of PCa possessed germline or somatic alterations in either BRCA1/2, ATM, and CHEK2 [43]. The recent PROfound trial screened 4,425 patients with mCRPC for 15 genes with direct or indirect roles in HR. A total of 2,792 patients were successfully sequenced, and qualifying alterations were found in 778 of 2,792 (28%) patients [44]. These reported frequencies in sequenced patients have been corroborated by several other studies in mCRPC [45, 46, 47, 48, 49, 50, 51], as seen in Table 3.
Table 3.
Frequencies of germline vs. somatic mutations in DNA damage repair genes and evidence‐based clinical applications in prostate cancer
| Gene | Frequency of somatic mutation, a % | Frequency of germline mutation, b % | Evidence‐based clinical application c |
|---|---|---|---|
| ATM | 3.7–5 | 1.6 | Modest activity of PARPi as monotherapy (TRITON 2, TOPARP‐B, TALAPRO‐1). Consideration of PARPi + ATR inhibitor combination. |
| BRCA1 | 1 | 0.9 | Clear benefit of PARPi (PROfound, TRITON 2, TOPARP‐B, TALAPRO‐1, GALAHAD). |
| BRCA2 | 6–7 | 5.4 | Clear benefit of PARPi (PROfound, TRITON 2, TOPARP‐B, TALAPRO‐1, GALAHAD). |
| BRIP1 | 0.5 | 0.2 | Limited data of potential activity (TRITON 2). More studies recommended. |
| CDK12 | 2.8–10 | Modest activity of PARPi as monotherapy (TRITON 2). Consideration of PARPi + PD‐1/PD‐L1 inhibitor combination. | |
| CHEK2 | 1–2 | 1.9 | Limited data of potential activity (TRITON 2; TOPARP‐B). More studies recommended including PARPi + ATR inhibitor combinations. |
| FANCA | 0.1–3 | 0.1 | Limited data of potential activity (TRITON 2). More studies recommended. |
| NBN | 0.5–1 | 0.3 | Modest activity in limited data. More studies recommended. |
| PALB2 | 0.5–2 | 0.4 | Potential benefit of PARPi (PROfound, TRITON 2, TOPARP‐B, TALAPRO‐1). More studies warranted. |
| RAD51B | 3 | Limited data available (TRITON 2). More studies recommended. | |
| RAD51D | 2.7 | 0.4 | Limited data available. More studies recommended. |
Ongoing clinical trials measuring PARP inhibitor response (classified by select DNA damage repair genes) are listed in supplemental online Table 1.
Abbreviations: PARPi, PARP inhibitor; PD‐1, programmed cell death protein‐1; PD‐L1, programmed death ligand‐1.
Due to the benefits seen in breast and ovarian cancers treated with PARP inhibitors, coupled with the frequency of DDR mutations seen in PCa, a number of clinical trials have arisen to evaluate the effects of PARP inhibition when used to treat PCa. The canonical use of PARP inhibitors in PCa offers a molecular‐stratified approach that is novel to the PCa treatment regimen, which has thus far lacked targeted treatment options and the associated biomarkers. Accompanying the recent approval of rucaparib and olaparib by the FDA for the treatment of mCRPC, several trials are examining the effects of other PARP inhibitors when used to treat PCa at different disease stages, some in combination with current standard of care drugs. Summaries of completed and ongoing trials are listed in Table 4.
Table 4.
Ongoing clinical trials of poly(ADP‐ribose) polymerase inhibitors
| Drug and trial no. | Status | Phase | Treatment | Patient population | Primary endpoint | Biomarkers included in eligibility criteria |
|---|---|---|---|---|---|---|
| Olaparib | ||||||
| NCT03434158 (IMANOL) | Active | II | Olaparib, maintenance | Patients with mCRPC after docetaxel treatment reaching partial or stable response | Progression‐free survival | BRCA1, BRCA2, ATM, FANC genes, CHEK2, MLH1, MSH2, MSH6, PMS2, PALB2, RAD51C, MRE11 |
| NCT03432897 | Active | II | Olaparib, neoadjuvant | Patients with locally advanced Pca and defects in DNA repair genes | PSA response rate | BRCA1, BRCA 2, ATM, CHEK1, CHEK2, FANCONIS ANEMIA (FANCL), HDAC2, PALB2, BARD1, BRIP1, CDK12, PPP2R2A, RAD51B, RAD51C, RAD51D, or RAD54L |
| NCT03810105 | Active | II | Olaparib + durvalumab | Patients with castration sensitive, biochemically recurrent, nonmetastatic PCa and DDR mutations | Undetectable PSA | BRCA1, BRCA2, ATM, CHEK2, FANCA, RAD51C, RAD51D, PALB2, BRIP1, BARD1, or CDK12 |
| NCT03012321 | Active | II | Abiraterone/prednisone, olaparib vs. abiraterone/prednisone + olaparib | Patients with mCRPC and DDR defects | Objective progression‐free survival | ATM, BRCA1, BRCA2, FANCA, PALB2, RAD51, ERCC3, MRE11, NBN, MLH3, CDK12, CHEK2, HDAC2, ATR, PMS2, GEN1, MSH2, MSH6, BRIP1, FAM175A |
| NCT03570476 | Suspended (COVID‐19) | II | Olaparib, neoadjuvant | Patients with localized PCa and DNA repair deficiencies | Pathological complete response rate | BRCA1, BRCA2, ATM, PALB2 (germline) or BRCA1, BRCA2, PALB2, FANCA, ATM (somatic) |
| NCT02987543 (PROfound) | Active, not recruiting | III | Olaparib vs. enzalutamide or abiraterone | Patients with mCRPC who have failed prior treatment with a new hormonal agent and have qualifying tumor mutation in an HR gene | Radiographic progression‐free survival | BRCA1, BRCA2, ATM, BRIP1, BARD1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, and RAD54L |
| NCT03516812 | Active, not yet recruiting | II | Olaparib + durvalumab | Nonmetastatic patients predicted to have a high neoantigen load that have received local therapy | Undetectable PSA | CDK12, mismatch repair deficiencies, or HR repair deficiencies |
| NCT03263650 | Active | II | Olaparib following cabazitaxel | Patients with aggressive variant Pca | Progression‐free survival | None |
| NCT03317392 | Active | I/II | Olaparib + radium 223 | Patients with mCRPC that has spread to the bone | Maximum tolerated dose, progression‐free survival | None |
| NCT03787680 (TRAP) | Active | II | Olaparib + AZD6738 | Patients with mCRPC with or without DDR mutations | Complete or partial response in DNA repair proficient patients | General DNA repair deficiency |
| NCT03047135 | Active | II | Olaparib only | Patients with high‐risk biochemically recurrent PCa following radical prostatectomy | PSA repsonse rate | None |
| NCT02893917 | Active, not recruiting | II | Olaparib with or without cediranib | Patients with mCRPC | Progression‐free survival | None |
| NCT03732820 (PROpel) | Active | II | Olaparib + abiraterone vs. placebo + abiraterone | Patients with mCRPC who have received no prior cytotoxic chemotherapy or new hormonal agents | Radiological progression‐free survival | None |
| NCT01972217 | Active, not recruiting | II | Olaparib + abiraterone vs. placebo + abiraterone | Patients with mCRPC |
Part A: adverse events, dose limiting toxicities; part B: radiological progression‐free survival, progression or death |
None |
| Rucaparib | ||||||
| NCT03413995 (TRIUMPH) | Active | II | Rucaparib only | Patients with mCRPC who have not yet been treated with ADT and germline mutations in DNA damage genes | PSA response rate | BRCA1, BRCA2, ATM, CHEK2, NBN, RAD50, RAD51C, RAD51D, PALB2, MRE11, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM |
| NCT03442556 | Active | II | Carboplatin and docetaxel followed by maintenance rucaparib | Patients with mCRPC and HR repair deficiency | Radiographic progression‐free survival | ATM, BRCA1, BRCA2 |
| NCT03533946 (ROAR) | Active | II | Rucaparib only | Patients with castration sensitive PCa demonstrating "BRCAness" | PSA response rate | ATM, ATR, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, ERCC3, FAM175A, FANCA, FANCL, GEN1, HDAC2, MLH1, MRE11, NBN, PALB2, PPP2R2A, RAD51, RAD54L |
| NCT04253262 | Active | I/II | Rucaparib + copanlisib | Patients with mCRPC | Phase I: maximum tolerated dose; phase II: overall response | Phase I: none; phase II: BRCA1, BRCA2, ATM, BARD1, BRIP1, CHEK1, FANCL, FANCA, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, and RAD54L |
|
(LODESTAR) |
Active | II | Rucaparib only | Patients with various solid tumors and with deleterious mutations in HRR genes | Best overall response rate | BRCA1, BRCA2, PALB2, RAD51C, RAD51D, BARD1, BRIP1, FANCA, NBN, RAD51, RAD51B |
|
(TRITON2) |
Active, not recruiting | II | Rucaparib only | Patients with metastatic castration‐resistant prostate cancer, and evidence of a homologous recombination gene deficiency | Objective response rate, prostate specific antigen response | ATM, BRCA1, BRCA2, BARD1, BRIP1, CDK12, CHEK2, FANCA, NBN, PALB2, RAD51, RAD51B, RAD51C, RAD51D, RAD54L, or other |
|
(TRITON3) |
Active | III | Rucaparib vs. abiraterone or enzalutamide | Patients with mCRPC and evidence of a homologous recombination gene deficiency | Radiographic progression‐free survival | ATM, BRCA1, BRCA2, or other HR gene mutation |
| Talazoparib | ||||||
| NCT03330405 | Active, not recruiting | II | Avelumab + talazoparib | Patients with mCRPC | Dose limiting toxicity, Overall response | ATM, BRCA1, BRCA2 |
| NCT03148795 (TALAPRO) | Active, not recruiting | II | Talazoparib only | Patients with mCRPC and DNA repair defects who have previously received taxane‐based chemotherapy and have progressed on at least 1 hormonal agent | Objective response rate | General DNA repair deficiency |
| NCT03395197 (TALAPRO2) | Active | III | Talazoparib + enzalutamide vs. placebo + enzalutamide | Patients with mCRPC | Dose confirmation, radiographic progression‐free survival | None |
| NCT04332744 | Active, not yet recruiting | II | Talazoparib + enzalutamide | Patients with metastatic, castration‐sensitive PCa | PSA response rate | None |
| Niraparib | ||||||
| NCT04030559 | Active | II | Niraparib: neoadjuvant | Patients with localized PCa and alterations in DNA repair pathways | Pathologic complete response | ATM, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCA, FANCD2, FANCL, GEN1, NBN, RAD51, RAD51C, and other DDR genes |
| NCT04288687 | Active, Not yet recruiting | II | Niraparib only | Patients who have recently received platinum‐based therapy | Radiographic progression‐free survival | BRCA1/2, ATM, FANCA, PALB2, CHEK2, HDAC2, or BRIP1 |
| NCT02854436 (GALAHAD) | Active | II | Niraparib only | Patients with mCRPC and DNA repair anomalies | Objective response rate | BRCA1, BRCA2, and other DDR genes |
| NCT04037254 | Active | II | Niraparib + ADT | Patients with PCa with a high chance of recurrence | Preferred dose, PSA response | None |
| NCT03748641 | Active | III | Niraparib + abiraterone vs. placebo + abiraterone | Patients with mCRPC | Progression‐free survival | None |
| Other | ||||||
| NCT04182516 | Active | I | NMS‐03305293 | Patients with selected advanced or metastatic, relapsed, or refractory solid tumors who have exhausted standard treatment options or for whom standard therapy is considered unsuitable | First‐cycle dose limiting toxicity | BRCA1, BRCA2 |
Abbreviations: DDR, DNA damage repair; HR, homologous recombination; HRR, homologous recombination repair, mCRPC, metastatic castration‐resistant prostate cancer; PCa, prostate cancer; PSA, prostate‐specific antigen.
In 2014, a phase II clinical trial, TOPARP‐A (NCT01682772), investigated treatment of mCRPC with olaparib in 50 patients, irrespective of DNA damage repair mutations. Of these patients, 16 had tumor aberrations of DNA‐repair genes (BRCA2, 7; ATM, 5; BRCA1 or CHEK2, 3; and HDAC2, 1). Of those 16 patients, 14 had a response to olaparib, as measured by a composite methodology that included declines in circulating tumor cells (CTCs) [52]. These data resulted in olaparib receiving breakthrough therapy designation in prostate cancer from the FDA. To further examine the antitumor effects of olaparib in men harboring DNA damage repair mutations with mCRPC, TOPARP‐A was followed by TOPARP‐B (NCT01682772), a randomized phase II trial for men with prostate cancer that had progressed to mCRPC. Patients were screened via targeted next‐generation sequencing (NGS) of either primary or metastatic cancer biopsies. Those who exhibited a pathogenic mutation or homozygous deletion in a DNA damage repair gene that had previously been associated with PARP inhibition sensitivity were enrolled and separated into two cohorts receiving either 300 mg or 400 mg olaparib twice daily. Of the DDR mutation subgroups, the BRCA1/2 subgroup saw the most confirmed responses and the longest median radiographic progression‐free survival, with 25 of 30 BRCA1/2 patients achieving a composite overall response rate (ORR) of 83.3%. However, many of these responses focused on CTC declines rather than prostate‐specific antigen (PSA) declines or objective responses. Patients with alterations in ATM, CDK12, and PALB2 achieved radiographic objective responses in 1 of 12, 0 of 18, and 2 of 6 patients, respectively. PSA declines of at least 50% were detected in 1 of 19, 0 of 20, and 4 of 6 patients, respectively. These data suggest that PALB2 mutants may be susceptible to PARP inhibition. Both of the TOPARP trials demonstrated the antitumor effects of olaparib when used to treat men with mCRPC possessing certain DDR genetic aberrations, with evidence that certain patients with non‐BRCA mutations may benefit from PARP inhibition as well [53]. TOPARP‐B, however, demonstrated some of the limitations of obtaining accurate and timely somatic genetic testing, as only 13.7% (98/711) of the screened patients were placed on study.
The PROfound trial (NCT02987543) was a prospective, randomized phase III trial that examined the efficacy of olaparib in men with mCRPC and DDR mutations in 15 genes associated with HR: BRCA1, BRCA2, ATM, BRIP1, BARD1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, and RAD54L. The primary endpoint examined was imaging‐based progression‐free survival (PFS). Patients with alterations in BRCA1/2 or ATM were assigned to cohort A, and patients with alterations in any of the other 12 genes were assigned to cohort B. Within each cohort, patients received either 300 mg olaparib twice a day or a standard of care treatment (enzalutamide or abiraterone) at a 2:1 ratio. In cohort A, PFS was significantly longer in the olaparib group compared with the control (7.4 months vs. 3.6 months; hazard ratio for progression or death, 0.34; 95% confidence interval [CI], 0.25–0.47; p < .001), with BRCA1 and BRCA2 patients achieving hazard ratios of 0.41 and 0.21, respectively. Of the overall population (cohorts A and B), PFS was significantly longer in the olaparib patients than control patients (5.8 months vs. 3.5 months; hazard ratio, 0.49; 95% CI, 0.38–0.63; p < .001). Patients with mutations in RAD54L had a 0.33 hazard ratio, suggesting potential benefit outside the realm of BRCA1/2 [44]. As noted, 4,425 patients were screened, but only 387 of those 4,425 (8.7%) patients were sequenced successfully. Failure of DNA sequencing occurred in approximately 31% of the tumor samples received, highlighting one of the limitations of the study.
Findings from the PROfound trial led to the FDA's recent approval of olaparib for patients with mCRPC with progression after treatment with enzalutamide and/or abiraterone that have deleterious germline alterations in BRCA1/BRCA2 or somatic deleterious alterations in BRCA/BRCA2, ATM, BARD, BRIP, CDK12, CHEK1, CHEK2, FANCL, PALB2, RAD51B, RAD51C, RAD51D, and RAD54L.
The TRITON series of clinical trials is currently evaluating the use of rucaparib to treat men with mCRPC that have germline or somatic mutations in DDR genes. The eligibility criteria of TRITON2 (NCT02952534), a phase II trial, included mutations in any HR gene, whereas TRITON3 (NCT02975934), an ongoing phase III trial comparing rucaparib with standard of care treatments, is enrolling only men with mCRPC and mutations in BRCA1/2 and ATM. Rucaparib was granted breakthrough therapy designation by the FDA based on initial efficacy and safety results from the TRITON2 study. Preliminary published data from this study shows promising results, with 43.9% of BRCA patients achieving a confirmed radiographic response, with the majority of responses lasting longer than 24 weeks [54]. Analysis of patients with non‐BRCA DDR genes was conducted, and of 19 evaluable patients with ATM mutations, 10.5% had confirmed partial radiographic responses that were ongoing at the time of visit cutoff. No objective responses were observed in those harboring CDK12 mutations. Of nine evaluable patients with CHEK2 aberrations, one patient with a co‐occurring ATM alteration had a confirmed partial response and a confirmed PSA response, and one other patient also achieved a confirmed PSA response. Of 13 patients comprising a group that included mutations in FANCA, PALB2, BRIP1, or RAD51B, 38.5% had an ORR, with one complete response seen in a patient with a FANCA mutation and four partial responses seen in patients with PALB2 (n = 2), BRIP1 (n = 1), or RAD51B (n = 1) [55]. The TRITON2 trial contributed to the FDA's recent accelerated approval of rucaparib to treat patients with mCRPC with germline or somatic BRCA1/2 mutations.
The TOPARP, PROfound, and TRITON series trials, along with others listed in Table 3, have paved the way for the recent addition of PARP inhibitors to the PCa treatment regimen, which marks the beginning of a new molecular‐based approach to treating the disease. However, much remains to be considered, including challenges facing sequencing availability, tissue acquisition, lack of protein ascertainment, and suggested guidelines, as well as the appropriateness of using PARP inhibitors to treat patients with non‐BRCA mutations in DDR genes.
Targeting Noncanonical DDR Genes
Although the beneficial effects of treating patients with BRCA1/2 mutations are evident, a wider patient population with mutations in other DDR genes may benefit from treatment with PARP inhibitors. As with BRCA, the ideology behind treating patients harboring mutations in noncanonical DDR genes remains rooted in the concept of synthetic lethality: using loss‐of‐function mutations present in tumors in combination with inhibition of the cell's ability to detect and respond to DNA damage in order to induce a lethal event. Several studies investigating the effects of using PARP inhibitors to treat PCa, including the trials discussed above, have reported responses in patients with mutations in non‐BRCA DDR genes; however, less attention has been given to these “other” genes. A recent study using unsupervised clustering of whole‐genome sequencing data found that 7 of 22 patients clustered in a HR‐deficient category did not have a biallelic BRCA inactivation [56]. Although individual mutation frequencies may be low, understanding which genes and mutational subtypes most benefit, as well as the mechanisms by which these noncanonical DDR genes respond to PARP inhibition, would considerably increase the patient population considered for PARP inhibitor treatment across other types of cancer.
ATM
ATM, together with ATR and DNA‐PKcs, is recruited to DSB sites and works to repair the damage via both NHEJ and HR [57, 58]. According to whole‐exome sequencing data available on the cBio Cancer Genomics portal (Fig. 2), alterations in ATM were observed in 7% of primary prostate samples [59] and in 8% of tumor samples from patients with metastatic PCa [60, 61, 62]. Although patients with ATM mutations have not reliably responded in several landmark PARP inhibitor clinical trials in PCa, emerging evidence suggests treating ATM‐altered patients with both a PARP inhibitor and an ATR inhibitor may have a more efficacious result compared with PARP inhibition alone [63]. Cells proficient in ATM that were treated with olaparib and ATR inhibitors, both alone and in combination, did not experience significant cell death [64, 65]. Several clinical trials are examining the combinatory effects of treating patients with PARP inhibitors and ATR inhibitors in different forms of cancer. The TRAP trial (NCT03787680) is an ongoing phase II trial comparing the responses of patients with mCRPC with DDR mutations to those of mCRPC patients without DDR mutations. Patients in both groups receive olaparib and the ATR inhibitor AZD6738 [63]. Use of PARP inhibitors as a monotherapy has been disappointing to date in terms of either radiographic responses or PSA responses when used to treat patients with ATM alterations.
Figure 2.

OncoPrints of primary and metastatic prostate cancer samples. (A): cBioPortal OncoPrint of queried genes using whole‐exome sequencing of 333 primary prostate adenocarcinoma tumor samples analyzed by The Cancer Genome Atlas [59, 61, 62]. (B): cBioPortal OncoPrint of queried genes using whole‐exome sequencing of 444 metastatic prostate adenocarcinoma samples analyzed by Abida et al. [60, 61, 62].
CHEK2
CHEK2 plays an active role in many cellular processes, including cell cycle regulation, apoptosis, and DNA repair. CHEK2 is activated by ATM and ATR in response to DSBs, and its activated monomers activate TP53, serine 988 of BRCA1, and other cell cycle checkpoints responsible for DSB reparations [66, 67, 68]. CHEK2 variants are well characterized and often associated with poor prognoses in several forms of cancer, including PCa, in which alterations were found in around 3% in both primary tumors and metastatic sites [59, 60, 61, 62, 69, 70]. Because of the key role of CHEK2 in DDR, PARP inhibition is a possible treatment approach for patients with CHEK2 aberrations. However, representation of men with PCa and alterations in CHEK2 was low in the TOPARP, PROfound, and TRITON2 trials [44, 54]. In TOPARP‐B, one patient with a CHEK2 alteration achieved a PSA decrease of 50%, and preliminary TRITON2 data report one patient achieving a radiological response and a PSA reduction, with another also achieving a PSA reduction [55]. Without higher participation of these patients, it will be difficult to draw conclusions [53]. More studies in patients with CHEK2 mutated tumors are warranted.
NBN
NBN encodes the protein Nibrin (NBS1), a participant of the MRN complex that functions in DSB end processing and HR [71]. Amplifications in NBN have been associated with olaparib and veliparib resistance in ovarian cancer and occur in over 40% of patients across 16 cancer types [72, 73, 74]. Alterations in NBN were found in 6% of primary tumors and 22% of metastatic samples, almost all of which were amplifications in both data sets [59, 60, 61, 62]. A small phase II study of olaparib and durvalumab in mCRPC reported one responder (with ongoing responses of more than 12 months) having a deleterious mutation in NBN [75]. More data are needed to assess the potential benefit of PARP inhibitor treatments in those with NBN mutations.
FANCA, PALB2, and RAD51
FANCA, PALB2, and RAD51 occupy roles in the Fanconi anemia (FA)/BRCA HR pathway for repair of DSBs. Data concerning PARP inhibitor treatment of patients harboing each of these mutations are provocative but limited. FA is an autosomal recessive disease that is characterized by congenital abnormalities and hypersensitivity to DNA cross‐linking agents [76]. This observation led investigators to identify the FA class of genes as important mediators in DSB repair that ultimately activate and aid BRCA1 and BRCA2 in repair [77].
FANCA is a component of the FA core complex responsible for the activation of interstrand crosslink repair [78, 79]. FANCA and the seven other FANC essential proteins in the FA core complex activate the FA/BRCA repair pathway through monoubiquitination of FANCD2 and FANCI. FANCA is the most commonly altered FA gene [80], and its significant role in DDR implies patients with mutations in FANCA could benefit from PARP inhibition treatment plans. FANCA aberrations were found in 8% of primary tumors analyzed by The Cancer Genome Atlas (TCGA), the majority of which were deep deletions [59, 61, 62]. In the preliminary TRITON2 data, one patient with a monoallelic truncating mutation (of 4 patients with FANCA alterations) had complete radiographic and PSA responses [54, 55]. One patient with a nonsense FANCA mutation achieved a PSA response in the TOPARP‐B trial [53].
PALB2 (partner and localizer of BRCA2) is recruited by BRCA1 to the site of DNA damage, where PALB2 then recruits BRCA2. PALB2 and BRCA2 facilitate the formation of the RAD51 nucleoprotein filament that is responsible for homology search of the intact sister chromatid. The homologous template found is used for accurate DNA synthesis and repair of the DSB [81, 82]. Mutations in PALB2 were found in approximately 1.5% of primary tumors analyzed by TCGA and in approximately 6% of metastatic sites analyzed by Abida et al. [59, 60, 61, 62]. Patients with PALB2 mutations achieved composite overall responses in four of seven cases, and four of six achieved PSA responses in the TOPARP‐B trial, indicating potential benefit from PARP inhibition [53]. In TRITON2, two of two patients with PALB2 alterations experienced PSA responses. One patient also achieved a partial radiographic response. The other patient had a 47% reduction in tumor volume, but as of the preliminary data release, a follow‐up had not occurred, and response has not been confirmed [55].
RAD51 works together with BRCA2 to maintain replication fork stability and independently to promote fork reversal in the process of repairing DSBs. Because of its key role in DDR, deficiencies in RAD51 are detrimental for genome maintenance, although overexpression of RAD51 also contributes to an unstable genome, as high levels of RAD51 can lead to aberrant replication fork reversal [16, 83]. Both deletions and amplifications of RAD51 were found in 2.3% of metastatic samples and 2.1% of primary tumors [59, 60, 61, 62]. Participation of patients with RAD51 mutations in completed PARP inhibitor clinical trials in PCa has been low, but breast cancer studies have determined that lack of RAD51 nuclear foci implies PARP sensitivity [84]. Although potentially promising, more data is needed to determine the effects of PARP inhibitors when used to treat RAD51‐altered men with PCa.
RNASEH2B
RNASEH2B is one of three genes composing the ribonuclease H2 complex responsible for cleavage of single ribonucleases that have mistakenly been incorporated into DNA [85]. RNASEH2B‐deficient cells lead to increased incidence of genome‐embedded ribonucleotides, the repair of which relies on the topoisomerase 1 excision repair pathway and recruitment of PARP1, indicating a potentially synthetically lethal relationship involving PARP inhibitors separate from HR and MMR [86]. Zimmermann et al. found that loss of the ribonuclease H2 complex induced PARP sensitivity both in vitro and in vivo, with talazoparib having the greatest effect [87]. RNASEH2B loss occurred in 35% of mCRPC tumors analyzed in one study [88], suggesting potential clinical benefit for these patients not previously considered for treatments involving PARP inhibitors.
Mutational Burden and MMR Deficiency
PARP‐based therapies inhibit single‐strand DNA repair, leading to not only DNA damage but also increased tumor mutational burden (TMB), which enhances the efficacy of immune checkpoint inhibition. Defective HR and MMR mechanisms can also result in higher TMB. Recently, the FDA approved pembrolizumab for the treatment of adult and pediatric patients with TMB‐high (≥10 mutations per megabase) solid tumors. This follows prior FDA approvals for MSI‐H and MMR‐deficient patients. Several clinical trials are investigating PARP inhibitor and immune check point inhibitor combinations, as mutational burden can increase following PARP inhibition treatments [89, 90].
CDK12
CDK12, a kinase frequently mutated in a number of cancers, is most often associated with its role in elongating RNA polymerase II, thereby mediating transcription and translation of several protein‐coding genes and contributing to genome stability. Through this mechanism, the transcription of several DDR repair genes, including BRCA1 and ATR, is CDK12 dependent [91, 92]. CDK12 was altered in 2.4% of primary prostate tumors examined by the Cancer Genome Atlas, with 5 of 8 mutations determined to be either truncating or deep deletions [59]. In contrast, 10% of mCRPC metastatic sites (lymph node, bone, or liver) examined by Abida et al. showed abnormalities in CDK12, with 23 of 41 mutations evaluated as either truncating or deep deletions [60, 61, 62]. Bi‐allelic inactivation of CDK12 in PCa results in accumulation of focal tandem duplications, gene fusions, and elevated neoantigen burden [93, 94]. Because of CDK12's central role in DDR gene transcription and translation, loss‐of‐function mutations of CDK12 were determined to imply PARP inhibitor sensitivity in a genome‐wide screen [95]. In the TOPARP‐B trial, however, no patients (0/20) had a PSA or radiographic objective response. In the PROfound trial, a hazard ratio of 0.74 for rPFS with wide confidence limits was noted for patients with CDK12 alterations treated with olaparib [44, 53]. Studies of other PARP inhibitor treatments in patients with CDK12 mutations are similarly disappointing, and alternative agents are needed in this subset of patients [44, 53].
Some studies have shown CDK12‐deficient tumors to be phenotypically and genetically distinct from other PCa HR‐deficient tumors, with high levels of inflammatory gene activation, presence of chemokines, and abnormal levels of T cell infiltration in CDK12 mutant tumors. This suggests that patients harboring CDK12 mutations may benefit from checkpoint inhibitor immunotherapy [93]. In a retrospective study of men with alterations in CDK12 and PCa at various stages, Antonarakis et al. found that three of nine patients who received a programmed cell death protein 1 (PD‐1) inhibitor had a PSA response, with median progression‐free survival of 5.4 months [96]. Patients with PCa with MMR deficiencies have been reported to respond to checkpoint inhibitor immunotherapy in the past [97], and Wu et al. described responses seen in patients with PCa with CDK12 inactivating mutations when treated with anti‐PD‐1 monotherapy [93]. This is relevant to PARP inhibition, as upregulations in programmed death ligand 1 are often seen in patients with both DDR mutations and MMR mutations, particularly following treatment with PARP inhibitors [89, 90]. Combinations of PD‐1 inhibitors and PARP inhibitors are being tested in ongoing clinical trials in several forms of cancer known for presence of DDR mutations. An ongoing phase III clinical trial (NCT03834519) in men with mCRPC is evaluating the combination of pembrolizumab and olaparib compared with abiraterone or enzalutamide. Data will help elucidate the possibility of synergistic properties of this combination, particularly in those with CDK12 inactivation mutations and MMR deficiencies. Recent data suggest platinum‐based regimens may also be effective in these patients [98].
MSH2 and MSH6
MSH2 and MSH6 are two key genes involved in MMR, an excision‐based mechanism for DNA repair of mismatched nucleotides that escape polymerase detection. These errors can occur during replication and recombination [15]. Disruption of the function of MMR genes can lead to aberrant point mutations throughout the genome, the effects of which can vary widely. Germline mutations in any of the mismatch genes is most often associated with hereditary colorectal cancer and accounts for 2%–5% of all colorectal cancer cases [99]. In PCa, alterations in MSH2 and MSH6 were found in approximately 4% of primary PCa tumors and around 6% of metastatic samples [59, 60, 61, 62], although around 12% of metastatic prostate tumors have elevated rates of single nucleotide mutations, likely arising from deficient MMR genes [100]. Like CDK12 deficiencies, MMR‐deficient patients are known to respond favorably to checkpoint inhibitors. Combinations of PARP inhibitors and check point inhibitors are being explored, but PARP inhibitors are not indicated in MMR deficiencies.
Other Synthetically Lethal Opportunities
PARP inhibition exists as an attractive complement to current prostate cancer therapies, regardless of genetic DNA damage repair mutation presence, because of PARP1's cooperation with the AR and AR signaling. PARP1 has been shown to be recruited to sites of AR action within the cell and to facilitate AR chromatin occupancy in both castration sensitive and resistant cancer cell types. PARP1 has also been shown to promote ligand‐independent AR activation, suggesting a role in treatment resistance and disease progression to mCRPC [101].
Previous prostate cancer studies have shown the benefits of treating locally advanced disease with radiotherapy in combination with androgen deprivation therapy (ADT) [102]. The positive effects seen in these trials are likely due to the synergistic consequence of radiological‐induced DNA damage along with ADT impairment of HR and DNA damage repair mechanisms [103]. Resistance to this combination therapy has been observed and could be explained by reports showing an increase in PARP activity following ADT, thus allowing DNA damage repair to persist [104]. These findings, along with the benefits of PARP inhibition in prostate cancer that have already been discussed, have influenced the use of olaparib and other PARP inhibitors in combination with current standard of care therapies in a number of clinical trials.
A recent study investigating mechanisms of cancer treatment resistance supports a potentially synthetically lethal relationship between PARP inhibition and the mTOR signaling pathway, which orchestrates stress‐induced mutagenesis in response to stress that increases cell‐to‐cell variability, enabling adaptation in response to selective pressures, such as cancer therapies [105]. Although some treatments, such as those involving radiation, are pointedly genotoxic, others are not, and yet an accumulation of DSBs is still observed. Cipponi et al. found that DSBs were a common early response to nongenotoxic treatments in eventually resistant colonies. As MTOR inhibition has previously been shown to impair DDR response, it has been hypothesized that the repression of MTOR, or of genes regulated by MTOR (PTEN, AKT1, and PIK3s), results in disrupted DDR. This allows for mutagenesis and the fostering of resistant clones in the presence of selective pressures [105, 106, 107]. Because of MTOR's affiliations with DDR, specifically HR, Cipponi et al. proposed a synthetically lethal relationship that could be targeted through a combination of targeted cancer therapy and drugs that target DDR (PARP inhibitors). This was tested with rucaparib and palbociclib (a CDK4/CDK6 inhibitor) in a pancreatic cancer cell line, and antitumor effects were more drastic in the combination model compared with either agent alone (p < .0001) [105]. This proposed mechanism may explain the clinical benefit observed for PARP inhibitors in the mCRPC setting (in the PROfound and TRITON2 trials) that could potentially extend to DDR genes beyond BRCA1/2.
Challenges of PARP Inhibition
Although PARP inhibition is at the forefront of a new precision‐based era of the PCa treatment regimen, there are challenges associated with necessary DDR mutation detection methods, interpreting sequencing data itself, and resistance.
Costs and time required to perform NGS have reduced, but limitations still exist. NGS requires high quality, undamaged samples to be successful. Such rigorous thresholds are oftentimes not met, which limits the power and reliability of studies relying on NGS. In the TOPARP‐B trial, 119 of 711 patients that consented to the trial were unable to be screened via NGS because of lack of sample or insufficient tissue, or because they had samples that failed quality assessments [53]. In the PROfound trial, only 69% of acquired tumor samples were successfully sequenced [44]. There are other methods of mutation detection, however, and some studies are using investigational NGS methods that use DNA extracted from circulating tumor cells found in blood samples. As liquid NGS technology improves to become more sensitive and precise, preserved tissue NGS will likely become the inferior option, and the concerns associated with tissue preservation and quality will no longer be relevant. Questions still remain, however, concerning the frequency that patients should be sequenced to capture and address somatic modifications or clonal variations that occur in response to treatment or disease progression.
Even as sequencing methods continue to improve and become more available in the clinical setting, questions exist surrounding the significance of many detected mutations and effects on protein expression and function. NGS findings do not reflect epigenetic changes that could result in disruption of protein function. Although NGS might relay a normal genetic sequence, there could be methylation patterns present that result in the silencing of a certain protein. This could result in missed actionable opportunities. Protein‐based assessment methods may help to assess the limitations of NGS.
Furthermore, guidelines for genetic screening are not necessarily in agreement. The National Comprehensive Cancer Network's updated 2020 guidelines state that patients with strong family history of cancer or family history of known germline variants, including BRCA1, BRCA2, ATM, PALB2, CHEK2, MLH1, MSH2, MSH6, and PMS2, should consider germline testing. Somatic testing for BRCA1, BRCA2, ATM, PALB2, FANCA, RAD51D, CHEK2, and CDK12 is recommended for men with mCRPC or regional PCa. The Philadelphia Prostate Cancer Consensus Conference in 2019 encouraged all patients with familial and metastatic PCa to be screened for mutations BRCA1/2 and MMR gene deficiencies. Wider screens are encouraged for clinical trial participation [108]. Although these guidelines are in place, a study in 2018 found that NGS testing is only performed in 1.4% of PCa cases [109]. Similarly, the clinical benefits of germline profiling across several forms of cancer from 2015 to 2019 were assessed, and Stadler et al. found that 50.9% of patients with advanced cancer with BRCA1/2 germline mutations received targeted therapy. As drug development and identification of actionable mutations advance, the percentage of patients receiving the option of targeted treatment is expected to grow [110]. Understanding the significance of germline and somatic mutations of non‐BRCA genes, as well as optimal treatments and combinations for each, will allow for streamlined guidelines and increased sequencing practices for the treatment of a wider patient population [111].
Finally, as with most treatment options, resistance to PARP inhibition is inevitable in many patients. Of initially HR‐deficient ovarian cancers, 50% acquire HR proficiency as a result of PARP inhibition resistance, and similar figures may be seen in PCa [112]. Mechanisms of resistance in prostate cancer can be driven by HR pathway restoration that occurs through reversion mutations [113]. Resistance can also occur with upregulations of replication fork stability genes (most commonly those in the ATR/CHK1 pathway). ATR inhibitors are currently being investigated as a potential therapy for PARP inhibitor resensitization [114]. Persistent DDR transcriptional activity of CDK12 can also contribute to resistance mechanisms [115]. Mechanisms of PARP inhibitor resistance within the realm of PCa is not yet well understood, but understanding the effects of different combinations of PARP inhibitors, checkpoint inhibitors, and classic AR‐targeting therapies will aid in clinical decisions anticipating delayed resistance.
Conclusion
The FDA's recent approval of olaparib and rucaparib for biomarker‐positive mCRPC marks a new era of PCa treatment, which thus far has lacked targeted treatment options and the reliable biomarkers necessary for molecular‐stratified approaches. The effects of germline and somatic mutations of BRCA1 and BRCA2 and potential for PARP inhibitor treatments are well characterized across several forms of cancer, but treatment options for alterations in other DDR genes have yet to be realized. Further inquiries into potential synthetically lethal interactions between mutations in these “other” DDR genes, PARP inhibitors, and PARP inhibitor combinations could significantly increase the percentage of patients that might benefit from these treatments. Efforts must also be made to further understand sequencing results, as well as to streamline national and institutional screening guidelines and recommendations.
Author Contributions
Conception/design: Emily N. Risdon, Cindy H. Chau, Douglas K. Price, William D. Figg
Provision of study material: Emily N. Risdon, Cindy H. Chau, Douglas K. Price, Oliver Sartor, William D. Figg
Assembly of Data: Emily N. Risdon, Cindy H. Chau, Douglas K. Price, Oliver Sartor
Data Analysis and Interpretation: Emily N. Risdon, Cindy H. Chau
Manuscript Writing: Emily N. Risdon, Cindy H. Chau, Douglas K. Price, Oliver Sartor, William D. Figg
Final approval of manuscript: Emily N. Risdon, Cindy H. Chau, Douglas K. Price, Oliver Sartor, William D. Figg
Disclosures
Oliver Sartor: Advanced Accelerator Applications, Astellas, AstraZeneca, Bayer, Blue Earth Diagnostics, Inc., Bavarian Nordic, Bristol Myers Squibb, Clarity Pharmaceuticals, Clovis, Constellation, Dendreon, EMD Serono, Fusion Pharmaceuticals, Janssen, Myovant, Myriad, Noria Therapeutics, Inc., Novartis, Noxopharm, Progenics, POINT Biopharma, Pfizer, Sanofi, Tenebio, Telix Pharmaceuticals, and Theragnostics (C/A). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplemental online Table 1
Acknowledgments
This work was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health (ZIA BC 010547).
The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the U.S. Government.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Flaig TW, Potluri RC, Ng Y et al. Treatment evolution for metastatic castration‐resistant prostate cancer with recent introduction of novel agents: Retrospective analysis of real‐world data. Cancer Med 2016;5:182–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nuhn P, De Bono JS, Fizazi K et al. Update on systemic prostate cancer therapies: Management of metastatic castration‐resistant prostate cancer in the era of precision oncology. Eur Urol 2019;75:88–99. [DOI] [PubMed] [Google Scholar]
- 3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin 2020;70:7–30. [DOI] [PubMed] [Google Scholar]
- 4. Zimmer AS, Gillard M, Lipkowitz S et al. Update on PARP inhibitors in breast cancer. Curr Treat Options Oncol 2018;19:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kurnit KC, Coleman RL, Westin SN. Using PARP inhibitors in the treatment of patients with ovarian cancer. Curr Treat Options Oncol 2018;19:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dantzer F, de La Rubia G, Ménissier‐De Murcia J et al. Base excision repair is impaired in mammalian cells lacking poly(ADP‐ribose) polymerase‐1. Biochemistry 2000;39:7559–7569. [DOI] [PubMed] [Google Scholar]
- 7. Satoh MS, Lindahl T. Role of poly(ADP‐ribose) formation in DNA repair. Nature 1992;356:356–358. [DOI] [PubMed] [Google Scholar]
- 8. Rouleau M, Patel A, Hendzel MJ et al. PARP inhibition: PARP1 and beyond. Nat Rev Cancer 2010;10:293–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ciccia A, Elledge SJ. The DNA damage response: Making it safe to play with knives. Mol Cell 2010;40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang G, Liu C, Chen SH et al. Super‐resolution imaging identifies PARP1 and the Ku complex acting as DNA double‐strand break sensors. Nucleic Acids Res 2018;46:3446–3457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wu L, Luo K, Lou Z et al. MDC1 regulates intra‐S‐phase checkpoint by targeting NBS1 to DNA double‐strand breaks. Proc Natl Acad Sci USA 2008;105:11200–11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chatterjee N, Walker GC. Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen 2017;58:235–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chiruvella KK, Liang Z, Wilson TE. Repair of double‐strand breaks by end joining. Cold Spring Harb Perspect Biol 2013;5:a012757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res 2008;18:99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tiwari V, Wilson DM, 3rd . DNA damage and associated DNA repair defects in disease and premature aging. Am J Hum Genet 2019;105:237–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Richardson C, Stark JM, Ommundsen M et al. RAD51 overexpression promotes alternative double‐strand break repair pathways and genome instability. Oncogene 2004;23:546‐553. [DOI] [PubMed] [Google Scholar]
- 17. Moynahan ME, Pierce AJ, Jasin M. BRCA2 is required for homology‐directed repair of chromosomal breaks. Mol Cell 2001;7:263–272. [DOI] [PubMed] [Google Scholar]
- 18. Moynahan ME, Chiu JW, Koller BH et al. Brca1 controls homology‐directed DNA repair. Mol Cell 1999;4:511–518. [DOI] [PubMed] [Google Scholar]
- 19. Choi M, Kipps T, Kurzrock R. ATM mutations in cancer: Therapeutic implications. Mol Cancer Ther 2016;15:1781–1791. [DOI] [PubMed] [Google Scholar]
- 20. Maréchal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 2013;5:a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Harfe BD, Jinks‐Robertson S. DNA mismatch repair and genetic instability. Ann Rev Genet 2000;34:359–399. [DOI] [PubMed] [Google Scholar]
- 22. Drummond JT, Genschel J, Wolf E et al. DHFR/MSH3 amplification in methotrexate‐resistant cells alters the hMutSα/hMutSβ ratio and reduces the efficiency of base–base mismatch repair. Proc Natl Acad Sci USA 1997;94:10144–10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Plotz G, Raedle J, Brieger A et al. hMutSalpha forms an ATP‐dependent complex with hmutlalpha and hmutlbeta on DNA. Nucleic Acids Res 2002;30:711–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nielsen FC, Jäger AC, Lützen A et al. Characterization of human exonuclease 1 in complex with mismatch repair proteins, subcellular localization and association with PCNA. Oncogene 2004;23:1457–1468. [DOI] [PubMed] [Google Scholar]
- 25. Terada M, Fujiki H, Marks PA et al. Induction of erythroid differentiation of murine erythroleukemia cells by nicotinamide and related compounds. Proc Natl Acad Sci USA 1979;76:6411–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pommier Y, O'Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med 2016;8:362ps317. [DOI] [PubMed] [Google Scholar]
- 27. Murai J, Huang SN, Das BB et al. Trapping of PARP1 and PARP2 by clinical parp inhibitors. Cancer Res 2012;72:5588–5599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Purnell MR, Whish WJ. Novel inhibitors of poly(ADP‐ribose) synthetase. Biochem J 1980;185:775–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Shall S. Proceedings: Experimental manipulation of the specific activity of poly(ADP‐ribose) polymerase. J Biochem 1975;77:2p. [PubMed] [Google Scholar]
- 30. Fong PC, Boss DS, Yap TA et al. Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med 2009;361:123–134. [DOI] [PubMed] [Google Scholar]
- 31. Pilié PG, Gay CM, Byers LA et al. PARP inhibitors: Extending benefit beyond BRCA‐mutant cancers. Clin Cancer Res 2019;25:3759–3771. [DOI] [PubMed] [Google Scholar]
- 32. Leo E, Johannes J, Illuzzi G et al. A head‐to‐head comparison of the properties of five clinical PARP inhibitors identifies new insights that can explain both the observed clinical efficacy and safety profiles. Cancer Res 2018;78:LB‐273a. [Google Scholar]
- 33. Farmer H, McCabe N, Lord CJ et al. Targeting the DNA repair defect in brca mutant cells as a therapeutic strategy. Nature 2005;434:917–921. [DOI] [PubMed] [Google Scholar]
- 34. Bryant HE, Schultz N, Thomas HD et al. Specific killing of BRCA2‐deficient tumours with inhibitors of poly(ADP‐ribose) polymerase. Nature 2005;434:913–917. [DOI] [PubMed] [Google Scholar]
- 35. Wooster R, Weber BL. Breast and ovarian cancer. N Engl J Med 2003;348:2339–2347. [DOI] [PubMed] [Google Scholar]
- 36. Stratton JF, Gayther SA, Russell P et al. Contribution of BRCA1 mutations to ovarian cancer. N Engl J Med 1997;336:1125–1130. [DOI] [PubMed] [Google Scholar]
- 37. Edwards SM, Kote‐Jarai Z, Meitz J et al. Two percent of men with early‐onset prostate cancer harbor germline mutations in the BRCA2 gene. Am J Hum Genet 2003;72:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lord CJ and Ashworth A. Parp inhibitors: Synthetic lethality in the clinic. Science 2017;355:1152‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim G, Ison G, McKee AE et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA‐mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res 2015;21:4257–4261. [DOI] [PubMed] [Google Scholar]
- 40. Petrovics G, Price DK, Lou H et al. Increased frequency of germline BRCA2 mutations associates with prostate cancer metastasis in a racially diverse patient population. Prostate Cancer Prostatic Dis 2019;22:406–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robinson D, Van Allen EM, Wu YM et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pritchard CC, Mateo J, Walsh MF et al. Inherited DNA‐repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016;375:443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Abida W, Armenia J, Gopalan A et al. Prospective genomic profiling of prostate cancer across disease states reveals germline and somatic alterations that may affect clinical decision making. JCO Precis Oncol 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. de Bono J, Mateo J, Fizazi K et al. Olaparib for metastatic castration‐resistant prostate cancer. N Engl J Med 2020;382:2091–2102. [DOI] [PubMed] [Google Scholar]
- 45. Chung JH, Dewal N, Sokol E et al. Prospective comprehensive genomic profiling of primary and metastatic prostate tumors. JCO Precis Oncol 2019;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Dawson NA, Zibelman M, Lindsay T et al. An emerging landscape for canonical and actionable molecular alterations in primary and metastatic prostate cancer. Mol Cancer Ther 2020;19:1373–1382. [DOI] [PubMed] [Google Scholar]
- 47. Mateo J, Seed G, Bertan C et al. Genomics of lethal prostate cancer at diagnosis and castration resistance. J Clin Invest 2020;130:1743–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bono JSD, Mehra N, Higano CS et al. TALAPRO‐1: Phase II study of talazoparib (TALA) in patients (pts) with DNA damage repair alterations (DDRm) and metastatic castration‐resistant prostate cancer (mCRPC) – updated interim analysis (IA). J Clin Oncol 2020;38:5566a. [Google Scholar]
- 49. Smith MR, Sandhu SK, Kelly WK et al. Phase II study of niraparib in patients with metastatic castration‐resistant prostate cancer (mCRPC) and biallelic DNA‐repair gene defects (DRD): Preliminary results of GALAHAD. J Clin Oncol 2019;37:202a.30523719 [Google Scholar]
- 50. Bryce AH, Sartor O, de Bono J. DNA repair and prostate cancer: A field ripe for harvest. Eur Urol;2020. [DOI] [PubMed] [Google Scholar]
- 51. Wilkes DC, Sailer V, Xue H et al. A germline FANCA alteration that is associated with increased sensitivity to DNA damaging agents. Cold Spring Harb Mol Case Stud 2017;3:a001487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mateo J, Carreira S, Sandhu S et al. DNA‐repair defects and olaparib in metastatic prostate cancer. N Engl J Med 2015;373:1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mateo J, Porta N, Bianchini D et al. Olaparib in patients with metastatic castration‐resistant prostate cancer with DNA repair gene aberrations (TOPARP‐B): A multicentre, open‐label, randomised, phase 2 trial. Lancet Oncol 2020;21:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Abida W, Campbell C, Patnaik A et al. 846PD: Preliminary results from the TRITON2 study of rucaparib in patients (pts) with DNA damage repair (DDR)‐deficient metastatic castration‐resistant prostate cancer (mCRPC): Updated analyses. Ann Oncol 2019;30:v327–v328. [Google Scholar]
- 55. Abida W, Campbell D, Patnaik A et al. Non‐BRCA DNA damage repair gene alterations and response to the PARP inhibitor rucaparib in metastatic castration‐resistant prostate cancer: Analysis from the phase II TRITON2 study. Clin Cancer Res 2020;26:2487–2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. van Dessel LF, van Riet J, Smits M et al. The genomic landscape of metastatic castration‐resistant prostate cancers reveals multiple distinct genotypes with potential clinical impact. Nat Commun 2019;10:5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Beucher A, Birraux J, Tchouandong L et al. ATM and Artemis promote homologous recombination of radiation‐induced DNA double‐strand breaks in G2. EMBO J 2009;28:3413–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Karanam K, Kafri R, Loewer A et al. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell 2012;47:320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Cancer Genome Atlas Research Network . The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Abida W, Cyrta J, Heller G et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc Natl Acad Sci USA 2019;116:11428–11436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gao J, Aksoy BA, Dogrusoz U et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 2013;6:pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cerami E, Gao J, Dogrusoz U et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012;2:401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jette NR, Kumar M, Radhamani S et al. ATM‐deficient cancers provide new opportunities for precision oncology. Cancers (Basel) 2020;12:687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jette NR, Radhamani S, Arthur G et al. Combined poly‐ADP ribose polymerase and ataxia‐telangiectasia mutated/Rad3‐related inhibition targets ataxia‐telangiectasia mutated‐deficient lung cancer cells. Br J Cancer 2019;121:600–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mei L, Zhang J, He K et al. Ataxia telangiectasia and Rad3‐related inhibitors and cancer therapy: Where we stand. J Hematol Oncol 2019;12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pommier Y, Sordet O, Rao VA et al. Targeting chk2 kinase: Molecular interaction maps and therapeutic rationale. Curr Pharm Des 2005;11:2855–2872. [DOI] [PubMed] [Google Scholar]
- 67. Mohelnikova‐Duchonova B, Havranek O, Hlavata I et al. CHEK2 gene alterations in the forkhead‐associated domain, 1100delc and del5395 do not modify the risk of sporadic pancreatic cancer. Cancer Epidemiol 2010;34:656–658. [DOI] [PubMed] [Google Scholar]
- 68. Ansari N, Shahrabi S, Khosravi A et al. Prognostic significance of CHEK2 mutation in progression of breast cancer. Lab Med 2019;50:e36–e41. [DOI] [PubMed] [Google Scholar]
- 69. Dong X, Wang L, Taniguchi K et al. Mutations in CHEK2 associated with prostate cancer risk. Am J Hum Genet 2003;72:270–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Seppälä EH, Ikonen T, Mononen N et al. CHEK2 variants associate with hereditary prostate cancer. Br J Cancer 2003;89:1966–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Carney JP, Maser RS, Olivares H et al. The hMre11/hRad50 protein complex and Nijmegen breakage syndrome: Linkage of double‐strand break repair to the cellular DNA damage response. Cell 1998;93:477–486. [DOI] [PubMed] [Google Scholar]
- 72. Etemadmoghadam D, deFazio A, Beroukhim R et al. Integrated genome‐wide DNA copy number and expression analysis identifies distinct mechanisms of primary chemoresistance in ovarian carcinomas. Clin Cancer Res 2009;15:1417–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Tothill RW, Tinker AV, George J et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res 2008;14:5198–5208. [DOI] [PubMed] [Google Scholar]
- 74. Wu Z, Li S, Tang X et al. Copy number amplification of DNA damage repair pathways potentiates therapeutic resistance in cancer. Theranostics 2020;10:3939–3951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Karzai F, VanderWeele D, Madan RA et al. Activity of durvalumab plus olaparib in metastatic castration‐resistant prostate cancer in men with and without DNA damage repair mutations. J Immunother Cancer 2018;6:141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Donahue SL, Lundberg R, Saplis R et al. Deficient regulation of DNA double‐strand break repair in Fanconi anemia fibroblasts. J Biol Chem 2003;278:29487–29495. [DOI] [PubMed] [Google Scholar]
- 77. D'Andrea AD, Grompe M. The Fanconi anaemia/BRCA pathway. Nat Rev Cancer 2003;3:23–34. [DOI] [PubMed] [Google Scholar]
- 78. Kee Y, D'Andrea AD. Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest 2012;122:3799–3806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Moldovan GL, D'Andrea AD. How the Fanconi anemia pathway guards the genome. Ann Rev Genet 2009;43:223–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wang AT, Smogorzewska A. Snapshot: Fanconi anemia and associated proteins. Cell 2015;160:354–354.e351. [DOI] [PubMed] [Google Scholar]
- 81. Buisson R, Dion‐Côté AM, Coulombe Y et al. Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination. Nat Struct Mol Biol 2010;17:1247–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Dray E, Etchin J, Wiese C et al. Enhancement of RAD51 recombinase activity by the tumor suppressor PALB2. Nat Struct Mol Biol 2010;17:1255–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Bhat KP, Cortez D. RPA and RAD51: Fork reversal, fork protection, and genome stability. Nat Struct Mol Biol 2018;25:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cruz C, Castroviejo‐Bermejo M, Gutiérrez‐Enríquez S et al. RAD51 foci as a functional biomarker of homologous recombination repair and parp inhibitor resistance in germline BRCA‐mutated breast cancer. Ann Oncol 2018;29:1203–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Reijns MA, Jackson AP. Ribonuclease H2 in health and disease. Biochem Soc Trans 2014;42:717–725. [DOI] [PubMed] [Google Scholar]
- 86. Sparks JL, Burgers PM. Error‐free and mutagenic processing of topoisomerase 1‐provoked damage at genomic ribonucleotides. EMBO J 2015;34:1259–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zimmermann M, Murina O, Reijns MAM et al. CRISPR screens identify genomic ribonucleotides as a source of PARP‐trapping lesions. Nature 2018;559:285–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Armenia J, Wankowicz SAM, Liu D et al. The long tail of oncogenic drivers in prostate cancer. Nat Genet 2018;50:645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jiao S, Xia W, Yamaguchi H et al. PARP inhibitor upregulates Pd‐L1 expression and enhances cancer‐associated immunosuppression. Clin Cancer Res 2017;23:3711–3720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Stewart RA, Pilié PG, Yap TA. Development of parp and immune‐checkpoint inhibitor combinations. Cancer Res 2018;78:6717–6725. [DOI] [PubMed] [Google Scholar]
- 91. Blazek D, Kohoutek J, Bartholomeeusen K et al. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev 2011;25:2158–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Chirackal Manavalan AP, Pilarova K, Kluge M et al. CDK12 controls G1/S progression by regulating RNAPII processivity at core DNA replication genes. EMBO Rep 2019;20:e47592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wu YM, Cieślik M, Lonigro RJ et al. Inactivation of CDK12 delineates a distinct immunogenic class of advanced prostate cancer. Cell 2018;173:1770–1782.e1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pilarova K, Herudek J, Blazek D. CDK12: Cellular functions and therapeutic potential of versatile player in cancer. NAR Cancer 2020;2:zcaa003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bajrami I, Frankum JR, Konde A et al. Genome‐wide profiling of genetic synthetic lethality identifies CDK12 as a novel determinant of PARP1/2 inhibitor sensitivity. Cancer Res 2014;74:287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Antonarakis ES, Isaacsson Velho P, Fu W et al. CDK12‐altered prostate cancer: Clinical features and therapeutic outcomes to standard systemic therapies, poly (ADP‐ribose) polymerase inhibitors, and PD‐1 inhibitors. JCO Precis Oncol 2020;4:370–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Le DT, Uram JN, Wang H et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med 2015;372:2509–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Barata P, Ledet E, Manogue C et al. Long term disease control using taxane/platinum‐based chemotherapy in CDK12‐mutated advanced prostate cancer. The Oncologist 2020. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Aaltonen LA, Salovaara R, Kristo P et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med 1998;338:1481–1487. [DOI] [PubMed] [Google Scholar]
- 100. Pritchard CC, Morrissey C, Kumar A et al. Complex MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat Commun 2014;5:4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Schiewer MJ, Goodwin JF, Han S et al. Dual roles of PARP‐1 promote cancer growth and progression. Cancer Discov 2012;2:1134–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Mason MD, Parulekar WR, Sydes MR et al. Final report of the intergroup randomized study of combined androgen‐deprivation therapy plus radiotherapy versus androgen‐deprivation therapy alone in locally advanced prostate cancer. J Clin Oncol 2015;33:2143–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Polkinghorn WR, Parker JS, Lee MX et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov 2013;3:1245–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Asim M, Tarish F, Zecchini HI et al. Synthetic lethality between androgen receptor signalling and the PARP pathway in prostate cancer. Nat Commun 2017;8:374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Cipponi A, Goode DL, Bedo J et al. MTOR signaling orchestrates stress‐induced mutagenesis, facilitating adaptive evolution in cancer. Science 2020;368:1127–1131. [DOI] [PubMed] [Google Scholar]
- 106. Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer 2016;16:20–33. [DOI] [PubMed] [Google Scholar]
- 107. Guo F. MTOR‐Fanconi anemia DNA damage repair pathway in cancer. J Oncobiomarkers 2014;2:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Giri VN, Knudsen KE, Kelly WK et al. Implementation of germline testing for prostate cancer: Philadelphia Prostate Cancer Consensus Conference 2019. J Clin Oncol 2‐2‐:JCO2000046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Ikeda S, Elkin SK, Tomson BN et al. Next‐generation sequencing of prostate cancer: Genomic and pathway alterations, potential actionability patterns, and relative rate of use of clinical‐grade testing. Cancer Bio Ther 2019;20:219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Stadler ZK, Maio A, Kemel Y et al. Targeted therapy based on germline analysis of tumor‐normal sequencing (MSK‐IMPACT) in a pan‐cancer population. J Clin Oncol 2020;38:1500a. [Google Scholar]
- 111. Lang SH, Swift SL, White H et al. A systematic review of the prevalence of DNA damage response gene mutations in prostate cancer. Int J Oncol 2019;55:597–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Johnson N, Li YC, Walton ZE et al. Compromised CDK1 activity sensitizes BRCA‐proficient cancers to PARP inhibition. Nat Med 2011;17:875–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Vidula N, Rich TA, Sartor O et al. Routine plasma‐based genotyping to comprehensively detect germline, somatic, and reversion BRCA mutations among patients with advanced solid tumors. Clin Cancer Res 2020;26:2546–2555. [DOI] [PubMed] [Google Scholar]
- 114. Yazinski SA, Comaills V, Buisson R et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor‐resistant BRCA‐deficient cancer cells. Genes Dev 2017;31:318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. D'Andrea AD. Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) 2018;71:172–176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplemental online Table 1