

Abstract
RAF family protein kinases signal through the MAPK pathway to orchestrate cellular proliferation, survival, and transformation. Identifying BRAF alterations in pediatric cancers is critically important as therapeutic agents targeting BRAF or MEK may be incorporated into the clinical management of these patients. In this study, we performed comprehensive genomic profiling on 3,633 pediatric cancer samples and identified a cohort of 221 (6.1%) cases with known or novel alterations in BRAF or RAF1 detected in extracranial solid tumors, brain tumors, or hematological malignancies. Eighty percent (176/221) of these tumors had a known‐activating short variant (98, 55.7%), fusion (72, 40.9%), or insertion/deletion (6, 3.4%). Among BRAF altered cancers, the most common tumor types were brain tumors (74.4%), solid tumors (10.8%), hematological malignancies (9.1%), sarcomas (3.4%), and extracranial embryonal tumors (2.3%). RAF1 fusions containing intact RAF1 kinase domain (encoded by exons 10–17) were identified in seven tumors, including two novel fusions TMF1‐RAF1 and SOX6‐RAF1. Additionally, we highlight a subset of patients with brain tumor with positive clinical response to BRAF inhibitors, demonstrating the rationale for incorporating precision medicine into pediatric oncology.
Implications for Practice
Precision medicine has not yet gained a strong foothold in pediatric cancers. This study describes the landscape of BRAF and RAF1 genomic alterations across a diverse spectrum of pediatric cancers, primarily brain tumors, but also encompassing melanoma, sarcoma, several types of hematologic malignancy, and others. Given the availability of multiple U.S. Food and Drug Administration‐approved BRAF inhibitors, identification of these alterations may assist with treatment decision making, as described here in three cases of pediatric cancer.
Keywords: Brain neoplasms, Leukemia, Precision medicine, Pediatrics, Proto‐oncogene, Proteins B‐raf, Biomarkers, Tumor
Short abstract
Despite gains in clinical success of biomarker‐informed targeted therapy in children with cancer, access to relevant targeted therapy is limited. This article describes the landscape of genomic alterations across a range of pediatric cancers, highlighting a subset of patients with brain tumors with positive clinical response to BRAF inhibitors.
Introduction
Pediatric cancers are a leading cause of death in the U.S. among children aged 1 to 14 years [1]. Despite significant improvements in 5‐year overall survival for this population, outcomes vary considerably depending on cancer type, with cure rates not exceeding 20% in patients with recurrent disease [2, 3].
Precision medicine, defined as biomarker‐informed treatment, accounts for significant advances in management of patients with cancer during the past two decades, including trastuzumab for HER2‐positive breast cancer [4], imatinib for BCR‐ABL–driven chronic myeloid leukemia [5], crizotinib targeting ALK‐rearranged non‐small cell lung cancer [6], and BRAF V600E–targeting agents in melanoma [7, 8].
With certain exceptions, such as the use of tyrosine kinase inhibitors in Philadelphia chromosome–positive acute lymphoblastic leukemia, the targeted therapy paradigm has not been fully realized for pediatric patients with cancer. Improvements in cytotoxic chemotherapy and radiation therapy techniques have dramatically improved survival rates in many pediatric cancers over the past 50 years; however, certain tumor types continue to be resistant to standard therapeutic approaches [9], and when therapy is effective, long‐term toxicities in survivors remain problematic [10, 11, 12].
Comprehensive genomic profiling (CGP) with next‐generation sequencing is an effective tool for identifying clinically relevant genomic alterations (GAs) across diverse types of pediatric cancers, including low grade glioma (LGG) and high grade glioma (HGG) [13, 14, 15], osteosarcoma [16], neuroblastoma [17], medulloblastoma [18], thyroid carcinoma [19], acute myeloid leukemia (AML) [20], T‐lineage acute lymphoblastic leukemia [21], gonadal tumors [22], and histiocytic neoplasms [23], with implications for more precise diagnoses, prognoses, and personalized therapeutic decision making.
BRAF encodes a member of the RAF family of protein kinases, which includes ARAF, BRAF, and CRAF (RAF1). These kinases function downstream of RAS as part of the MAPK (RAF‐MEK‐ERK) signaling cascade that facilitates cell proliferation, survival, and transformation [24, 25]. BRAF mutations have been reported in up to 20% of all cancers, with a majority occurring at the V600 position [26, 27]. BRAF fusions, which activate the MAPK pathway, have been reported in multiple tumor types [28] and are the most common genomic alteration in juvenile pilocytic astrocytoma (PA), a type of LGG [29]. RAF1 fusions, which are functionally similar to BRAF fusions, are recurrent in adult solid tumors [30, 31, 32] and juvenile PA [15, 33, 34, 35]. Among LGGs, BRAF V600E may predict poorer long‐term outcome after chemotherapy and radiation therapies compared with non–BRAF V600E tumors and those harboring BRAF fusions (KIAA1549‐BRAF), although further study is needed [36, 37, 38]. Furthermore, BRAF V600E has been observed concurrent with CDKN2A loss in patients with ganglioglioma, although no significant difference in prognosis was identified compared with patients with BRAF V600E and intact CDKN2A [39].
Therapeutic strategies targeting BRAF‐driven tumors rely mostly on U.S. Food and Drug Administration (FDA)‐approved small molecule tyrosine kinase inhibitors (e.g., dabrafenib), approved in metastatic melanoma and non‐small cell lung cancer, and vemurafenib, approved in metastatic melanoma and Erdheim‐Chester disease. These and additional investigational BRAF V600E–targeting agents [40, 41] are under clinical evaluation for pediatric indications in multiple early phase trials. BRAF‐altered pediatric malignancies have derived clinical benefit from BRAF V600E–targeting agents in central nervous system disease [2, 42, 43, 44, 45, 46, 47, 48, 49] and histiocytic neoplasms [50, 51]. Targeting BRAF fusions remains a challenge in pediatric brain tumors, although reports demonstrating clinical benefit with MEK inhibitors are increasing [52, 53, 54, 55, 56]. Therapeutic modalities targeting RAF1 fusion–positive tumors are even rarer, with no clinical studies (pediatric or adult) available. However, three reports demonstrated clinical benefit from trametinib in two adult patients with melanoma [30, 57] and a pediatric patient with LGG, respectively [58].
Despite gains in clinical success of biomarker‐informed targeted therapy in children with cancer, access to relevant targeted therapy is limited [2, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 58, 59, 60, 61]. In this study, we sequenced tumors from 3,633 patients with pediatric cancer and identified a cohort of 221 cases with known and novel BRAF or RAF1 alterations in extracranial solid tumors, brain tumors, or hematological malignancies. Furthermore, we highlight a subset of patients with brain tumors with positive clinical response to BRAF inhibitors, demonstrating the rationale for incorporating precision medicine into pediatric oncology.
Materials and Methods
CGP was performed in a Clinical Laboratory Improvement Amendments–certified, College of American Pathologists–accredited laboratory (Foundation Medicine, Inc., Cambridge, MA). Approval for this study, including a waiver of informed consent and a Health Insurance Portability and Accountability Act waiver of authorization, was obtained from the Western Institutional Review Board (protocol no. 20152817). The pathologic diagnosis of each case was confirmed on routine hematoxylin and eosin–stained slides, and all samples forwarded for DNA extraction contained a minimum of 20% tumor nuclear area as a proportion of all nucleated cells. In brief, ≥50 ng DNA was extracted from 40 microns of specimen from formalin‐fixed, paraffin‐embedded tissue blocks or unstained slides. The samples were assayed by CGP using adaptor ligation, and hybrid capture was performed for all coding exons from 287 (version 1) to 315 (version 2) cancer‐related genes plus select introns from 19 (version 1) to 28 (version 2) genes frequently rearranged in cancer. Sequencing of captured libraries was performed using the Illumina HiSeq technology (Illumina, San Diego, CA) to a median exon coverage depth of at least 500× and analyzed for GAs, including short variant alterations (base substitutions, insertions, and deletions), copy number alterations (focal amplifications and homozygous deletions), and select gene fusions or rearrangements, as previously described [62]. Benign germline variants documented in publicly accessible population databases or recurrent variants of unknown significance that were predicted by an internally developed algorithm to be germline were removed, with the exception of known driver germline events (e.g., documented hereditary and deleterious BRCA1/2 or TP53 mutations) [63]. Somatic alterations present in the Catalog of Somatic Mutations in Cancer were highlighted as biologically significant [64]. Tumor mutational burden (TMB) was determined on 1.1 megabases of sequenced DNA for each case and categorized as low (0–5 mutations per megabase [mut/Mb]), intermediate (6–19 mut/Mb), or high (≥20 mut/Mb) as previously described [65]. Clinical histories, disease stage, and primary versus recurrent disease status of samples tested were not available.
Results
Characteristics of the Pediatric Cohort
CGP was performed on 3,633 pediatric (median 10.5 years, range < 1–21 years) cancer samples and revealed 221 (6.1%) unique samples that harbored alterations in BRAF. Alterations were classified as “known‐activating” or “functionally impairing” if supported by publicly available, peer‐reviewed biochemical data. An alteration was classified as “uncharacterized” if biochemical data supporting a specific functional status were not available at the time of this study's publication.
Of the BRAF mutation–positive cohort, 176 (79.6%) samples harbored a known‐activating short variants (SVs), insertions/deletions (indels), or fusion; 34 (15.4%) harbored an uncharacterized SV, indel, or nonfusion rearrangement; 8 (3.6%) harbored a SV known to result in decreased protein function (i.e., functionally impairing); and 3 (1.4%) contained multiple uncharacterized or functionally impairing SVs (supplemental online Fig. 1A). Of the 176 samples bearing a BRAF known‐activating alteration, 98 (55.7%) encompassed SVs, 72 (40.9%) fusions, and 6 (3.4%) indels (supplemental online Fig. 1B).
Figure 1.
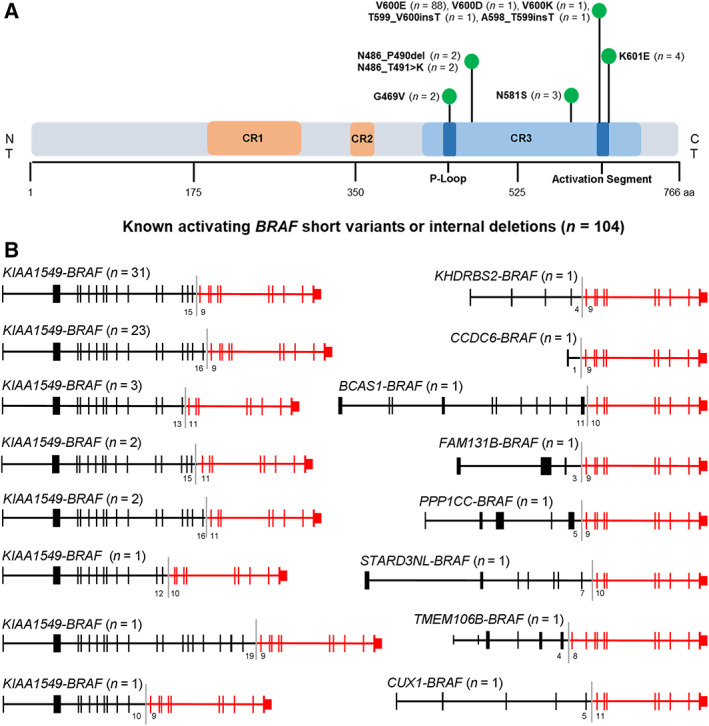
Landscape of known‐activating BRAF alterations. (A): Schematic diagram of domains and alterations of BRAF. (B): Schematic of known‐activating BRAF fusions, including those identified in this study (STARD3NL‐BRAF and KHDRBS2‐BRAF). Exon numbers at the fusion boundary are depicted below each fusion diagram.Abbreviation: CR, conserved region.
Known‐activating BRAF alterations were identified in samples encompassing six primary histological categories: brain tumors (74.4%; 18 subtypes), other solid tumors (10.8%; 6 subtypes), hematological malignancies (9.1%; 5 subtypes), sarcomas (3.4%; 3 subtypes), and extracranial embryonal tumors (2.3% 2 subtypes) (supplemental online Fig. 2A). Brain tumors (n = 131) included pilocytic astrocytoma (PA), grade I (45; 34.4%); low grade glioma (LGG) not otherwise specified (NOS) (19; 14.5%); glioblastoma (GBM) (13; 9.9%); pilomyxoid astrocytoma, grade 2 (13; 9.9%); ganglioglioma, grade 1 (10; 7.6%); and 13 additional subtypes of varying frequency (supplemental online Fig. 2B).
Figure 2.
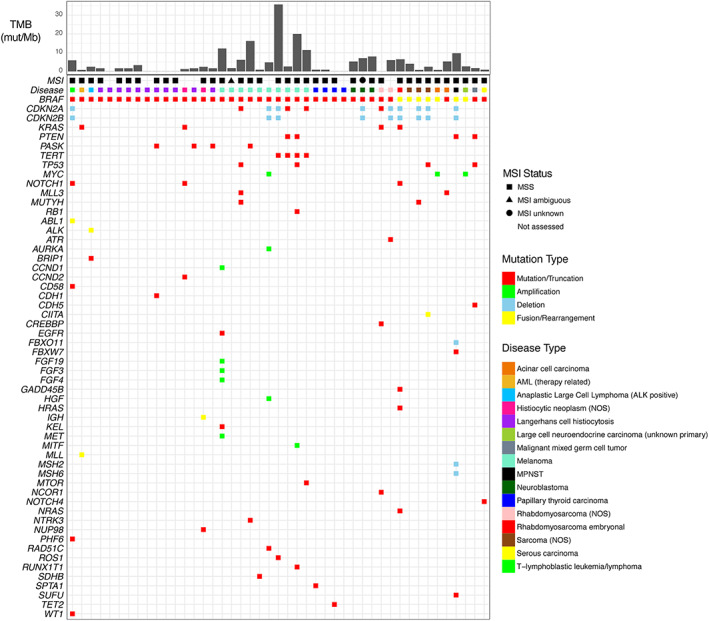
Genomic landscape of hematological malignancies and extracranial solid tumors with a known‐activating BRAF alteration. Specimens are arranged from young to old (left to right) within each cancer type.Abbreviations: AML, acute myeloid leukemia; MPNST, malignant peripheral nerve sheath tumor; MSI, microsatellite instability; MSS, microsatellite stable; mut/Mb, mutations per megabase; NOS, not otherwise specified; TMB, tumor mutational burden.
Sarcomas (n = 6) included rhabdomyosarcoma (NOS) (2; 33.3%); sarcoma (NOS) (3; 50%); and rhabdomyosarcoma, embryonal (1; 16.7%) (supplemental online Fig. 2C). Embryonal tumors (n = 4) included neuroblastoma (3; 75%) and malignant mixed germ cell tumor (1; 25%) (supplemental online Fig. 2D). Hematological tumors (n = 16) included Langerhans cell histiocytosis (11; 68.8%), histiocytic cell neoplasm (NOS) (2; 12.5%), anaplastic large cell lymphoma (ALK‐positive) (1; 6.3%), AML (treatment‐related) (1; 6.3%), and T‐lymphoblastic leukemia/lymphoma (1; 6.3%) (supplemental online Fig. 2E). Other solid tumors (n = 19) included melanoma (10; 52.6%), papillary thyroid carcinoma (4; 21.1%), acinar cell carcinoma (2; 10.5%), and three additional subtypes (supplemental online Fig. 2F).
Landscape of BRAF Known‐Activating Variants
BRAF V600E accounted for 50% (88/176) of all identified known‐activating variants, followed by K601E (2.3%; n = 4) and N581S (1.7%; n = 3). Less common known‐activating SVs included G469V (1.1%; n = 2), V600D (0.6%; n = 1), and V600K (0.6%; n = 1). Known‐activating indels identified included N486_T491 > K and N486_P490del (each at 1.1%, n = 2) and A598_T599insT and T599_V600insT (each at 0.6%; n = 1) (Fig. 1A). A known‐activating BRAF fusion was identified in 72 cases (32.6% of the BRAF mutation–positive cohort), all of which contained an intact BRAF kinase domain (encoded by exons 11–18) and breakpoints in BRAF introns 7, 8, 9, or 10. Sixty‐four (88.9%) of these included the KIAA1549 fusion partner, with 8 distinct KIAA1549‐BRAF fusion variants identified. BRAF fusions with unique fusion partners were identified in eight cases, with two involving the novel fusion partners STARD3NL and KHDRBS2 (Fig. 1B).
Genomic Landscape of Hematologic Malignancies and Extracranial Solid Tumors with Known‐Activating BRAF Alteration
Among 45 patients with extracranial solid tumors or hematologic malignancies harboring a known‐ activating BRAF alteration, BRAF V600E was the most common SV (n = 25/45, 55.6%) and was observed in AML (therapy‐related) (n = 1); histiocytic neoplasms (n = 9); melanoma (n = 8); papillary thyroid carcinoma (n = 4); and rhabdomyosarcoma, acinar cell carcinoma, and serous carcinoma (n = 1 each). A known‐activating BRAF fusion was identified in seven samples: KIAA1549‐BRAF in embryonal rhabdomyosarcoma (n = 1), sarcoma (NOS) (n = 1), and malignant peripheral nerve sheath tumor (n = 1); CUX1‐BRAF in sarcoma (NOS) (n = 1); STARD3NL‐BRAF in sarcoma (NOS) (n = 1); PPP1CC‐BRAF in acinar cell carcinoma (n = 1); and KHDRBS2‐BRAF in large cell neuroendocrine carcinoma (unknown primary) (n = 1) (supplemental online Table 1).
Few co‐occurring genomic driver alterations or signatures were identified in this cohort with the notable exception of the hematological and melanoma sample subsets. Thirteen of 45 samples contained either CDKN2A/B deletion (n = 9) or a CDKN2A truncating alteration (n = 4). Co‐occurring known‐activating KRAS SVs were found in four samples. The majority of cases contained low or intermediate TMB with the exception of melanoma samples, of which 50% (5/10) displayed high TMB. All cases that could be assessed for microsatellite instability demonstrated a microsatellite stable status (Fig. 2).
Genomic Landscape of Primary Brain Tumors with Known‐Activating BRAF Alterations
Analysis of 131 primary brain tumors revealed 66 samples with known‐activating BRAF SV or indels and 65 with a known‐activating gene fusion. BRAF V600E was the most common (62/66 cases) SV or indel observed in 14 distinct histological subtypes. Less common, known‐activating BRAF variants were identified in one patient with high grade glioneuronal tumor (NOS) (N581S), one patient with GBM (N486_T491 > K), and two patients with LGG (NOS) (T599_V600insT, A598_T599insT, respectively) (supplemental online Table 2). All cases had low TMB, and those that could be assessed for microsatellite instability all demonstrated a microsatellite stable status (Fig. 3).
Figure 3.
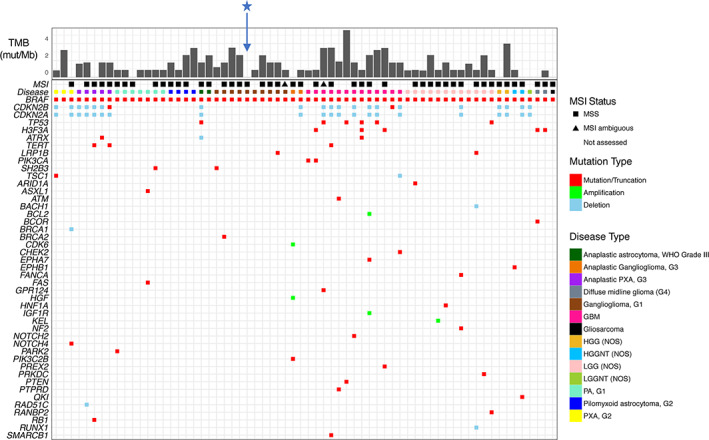
Genomic landscape of primary brain tumors bearing a known‐activating BRAF short variant or indel. Specimens are arranged from young to old (left to right) within each cancer type. Blue star indicates the genomic profile associated with Index Case 1.Abbreviations: G, grade; GBM, glioblastoma; HGG, high grade glioma; HGGNT, high grade glioneuronal tumor; LGG, low‐grade glioma; LGGNT, low grade glioneuronal tumor; MSI, microsatellite instability; MSS, microsatellite stable; mut/Mb, mutations per megabase; NOS, not otherwise specified; PA, pilocytic astrocytoma; PXA, pleomorphic xanthoastrocytoma; TMB, tumor mutational burden; WHO, World Health Organization.
KIAA1549‐BRAF was the most common fusion, identified in 61 of 65 fusion‐positive cases represented by 16 tumor subtypes, with highest frequency observed in PA, grade 1 (55% of fusion‐positive specimens). Noncanonical BRAF fusions were identified in four other tumors: PA, grade 1 (FAM131B‐BRAF); PA, grade 1 (BCAS1‐BRAF); anaplastic pleomorphic xanthoastrocytoma, grade 3 (CCDC6‐BRAF); and anaplastic ganglioglioma, grade 3 (TMEM106B‐BRAF) (supplemental online Table 3). All the BRAF fusion–positive cases except for one contained low or intermediate TMB, the exception being a single tumor from a 20‐year‐old patient diagnosed with anaplastic astrocytoma World Health Organization (WHO) grade III that contained a TMB of >40 mut/Mb. All cases that were able to be assessed for microsatellite instability demonstrated microsatellite stable status (Fig. 4).
Figure 4.
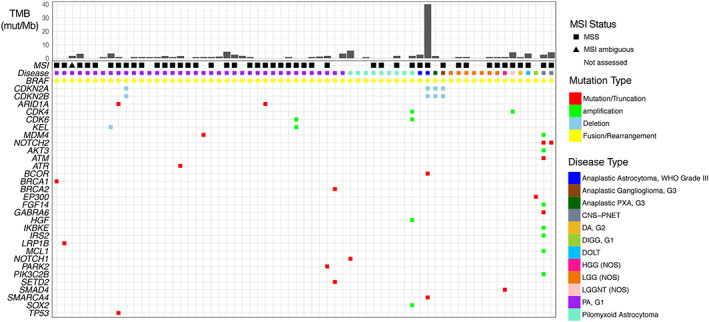
Genomic landscape of primary brain tumors bearing a known‐activating BRAF fusion. Specimens are arranged from young to old (left to right) within each cancer type.Abbreviations: CNS‐PNET, central nervous system–primitive neuroectodermal tumor; DA, diffuse astrocytoma; DIGG, desmoplastic infantile ganglioglioma; DOLT, disseminated oligodendroglial‐like leptomeningeal tumor; G, grade; HGG, high grade glioma; LGG, low‐grade glioma; LGGNT, low grade glioneuronal tumor; MSI, microsatellite instability; MSS, microsatellite stable; mut/Mb, mutations per megabase; NOS, not otherwise specified; PA, pilocytic astrocytoma; PXA, pleomorphic xanthoastrocytoma; TMB, tumor mutational burden; WHO, World Health Organization.
BRAF Nonfusion Rearrangements
Nonfusion BRAF rearrangements were identified in five patients, including one patient with PA, grade 1; one patient with LGG (NOS); two patients with neuroblastoma; and one patient with osteosarcoma (Fig. 5A). These noncanonical BRAF alterations manifested from one of three distinct chromosomal rearrangements, and all resulted in genomic loss or disruption of the BRAF N‐terminal autoinhibitory domain with breakpoints in intron 7, 8, or 9, which has been shown to result in constitutive kinase activation in a RAS‐independent manner [66] (Fig. 5B). All five specimens demonstrated low or intermediate TMB, and none demonstrated microsatellite instability (Fig. 5C).
Figure 5.
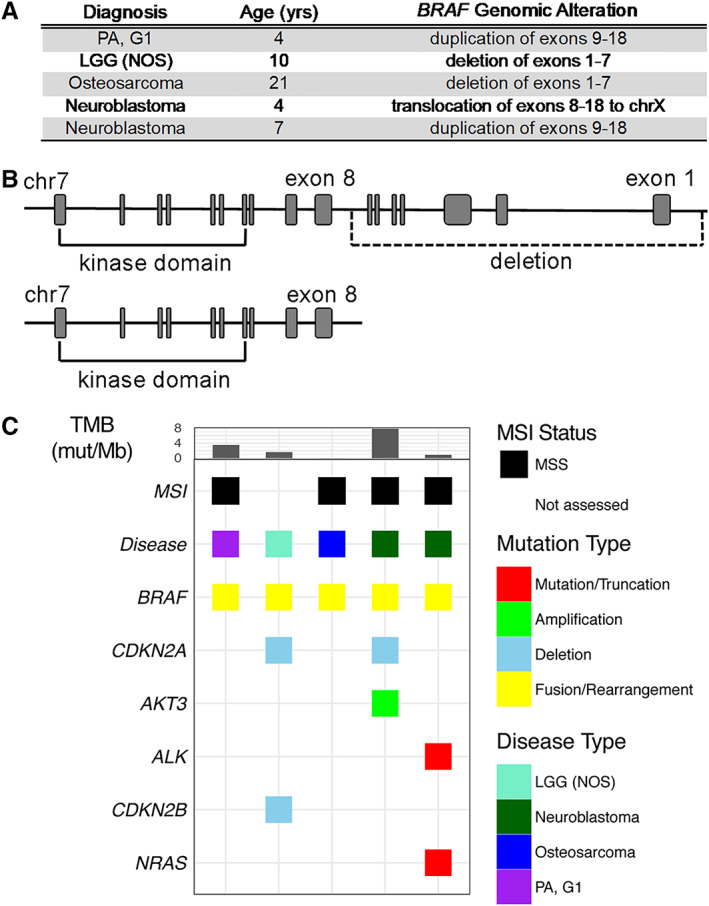
Landscape of BRAF nonfusion rearrangements. (A): Description of BRAF rearrangements in pediatric malignancy. (B): Schematic representing loss of BRAF exons 1–7. (C): Genomic landscape of pediatric cancers bearing a BRAF nonfusion rearrangement.Abbreviations: G, grade; LGG, low‐grade glioma; MSI, microsatellite instability; MSS, microsatellite stable; mut/Mb, mutations per megabase; NOS, not otherwise specified; PA, pilocytic astrocytoma; TMB, tumor mutational burden.
RAF1 Known‐Activating Fusions in Solid Tumors
Known‐activating RAF1 fusions (n = 7) were identified in five distinct brain tumor subtypes, one sarcoma, and one histiocytic neoplasm. All fusions contained an intact RAF1 kinase domain (encoded by exons 10–17) with unique fusion partners and breakpoints in RAF1 introns 7 or 9. Two involved the novel fusion partners TMF1 (sarcoma [NOS]) and SOX6 (HGG [NOS]) (Fig. 6A). All seven specimens demonstrated low TMB, and none demonstrated microsatellite instability (Fig. 6B).
Figure 6.
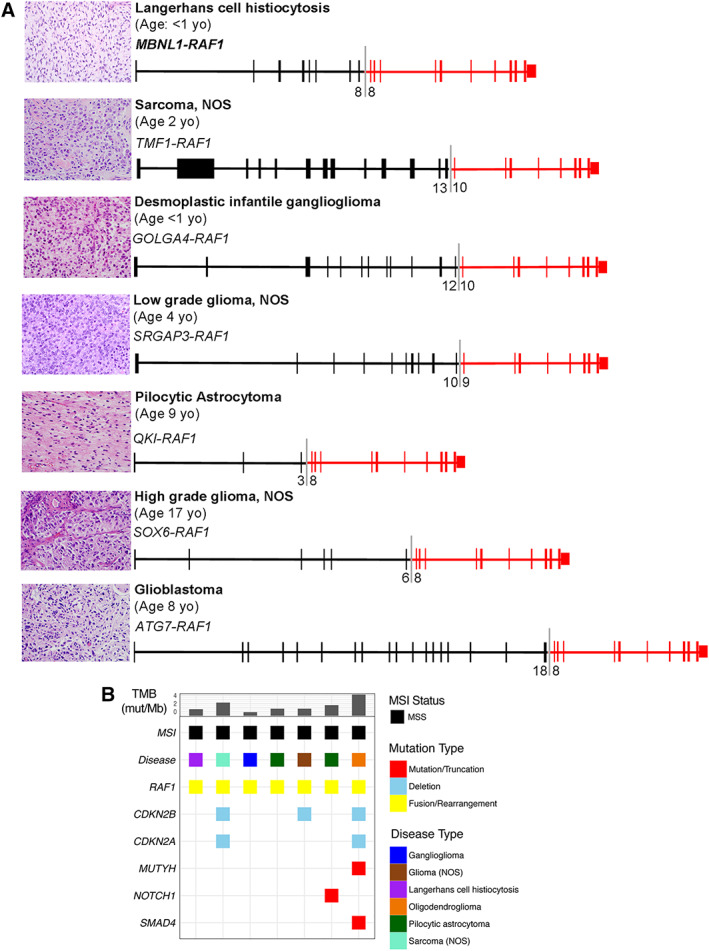
Landscape of RAF fusions. (A): Hematoxylin and eosin staining and corresponding schematics representing recurrent and novel RAF1 kinase fusions identified in pediatric cancer. Exon numbers at the fusion boundary are depicted below each fusion diagram. (B): Genomic landscape of pediatric cancers bearing a known‐activating RAF1 fusion. Specimens are arranged from young to old (left to right) within each cancer type.Abbreviations: MSI, microsatellite instability; MSS, microsatellite stable; mut/Mb, mutation; mutations per megabase; NOS, not otherwise specified; TMB, tumor mutational burden.
Index Cases
Three patients with BRAF V600E–mutated brain tumors, including a 10‐year‐old boy and 10‐year‐old girl, each with ganglioglioma (WHO grade I) (Index Cases 1 and 2), and a 7‐year‐old girl with a GBM (WHO grade IV) (Index Case 3), who each experienced progression after conventional treatment, were independently treated with the BRAF inhibitor vemurafenib on a compassionate basis. Index Cases 1 and 2 showed clinical and radiological response to the targeted therapy (960 mg b.i.d.) and remained on treatment 22 months and > 18 months, respectively, with ongoing response. After treatment with vemurafenib for 22 months, therapy was discontinued in Index Case 1 because of recurrent photosensitivity, and this patient has remained off treatment for >9 months with no radiologic or clinical evidence of tumor progression. The patient described in Index Case 3 was treated with 480 mg p.o. b.i.d. and showed stable magnetic resonance imaging findings >7 months with ongoing sustained response (Fig. 7).
Figure 7.
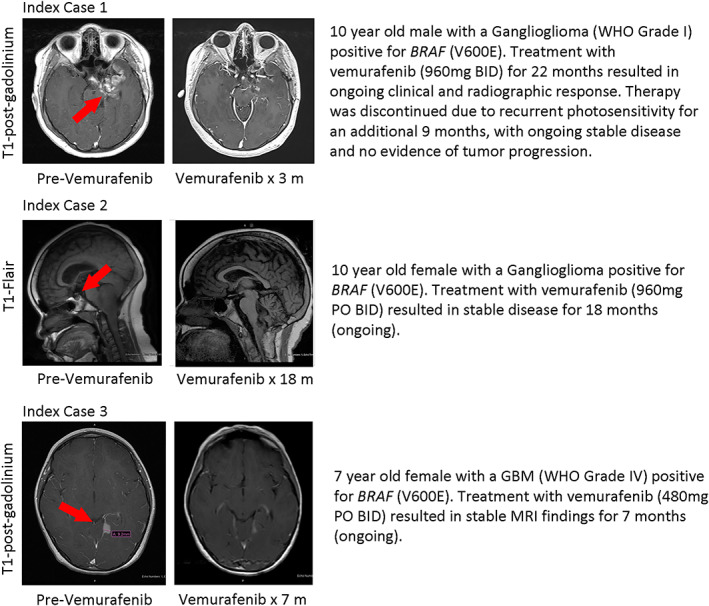
Postcontrast magnetic resonance imaging images showing decrease in tumor size in three separate BRAF V600E–positive pediatric patients after treatment with vemurafenib.Abbreviations: BID, bis in die (twice daily); GBM, glioblastoma; MRI, magnetic resonance imaging; PO, per os (by mouth); WHO, World Health Organization.
Prior Molecular Testing
To better understand the extent to which prior molecular testing was used in this data set of tumors that harbored BRAF or RAF1 known‐activating alterations, we assessed cases (n = 35) with available clinical histories. Of those with prior BRAF molecular testing results, 19 (54.3%) reported results from prior testing methodologies inconsistent with the respective BRAF alteration type later identified with CGP (supplemental online Fig. 3A). Specifically, of eight PA cases with either KIAA1549‐BRAF or QKI‐RAF1 fusion detected by CGP, six were previously tested for BRAF V600E by immunohistochemistry or polymerase chain reaction, and therefore the underlying BRAF fusion was not detected (supplemental online Fig. 3B).
Discussion
In this study we highlight the diverse landscape of pediatric cancer types that harbor genomic alterations in BRAF or RAF1 and describe three index cases with durable benefit with RAF inhibitors. In our data set, alterations in BRAF likely to represent driver events were identified in approximately 6% of all pediatric tumors screened with CGP during routine clinical care. Key among these findings is that 25% of the tumor samples represent extracranial solid or hematologic tumor types for which single gene or broad panel testing for druggable biomarkers are unlikely to be employed routinely in a clinical setting. For example, KIAA1549‐BRAF, CUX1‐BRAF, STARD3NL‐BRAF, or TMF1‐RAF1 fusions, which were identified in four separate patients with sarcoma in our study, would have likely gone unrecognized with standard of care molecular testing.
Multiple biomarker‐informed targeted therapies have been developed for adult patients with cancer, but there continues to be significant lag time for similar development for pediatric cancers. Notable exceptions are recent age‐agnostic therapy approvals, including larotrectinib and entrectinib for NTRK fusion–positive patients and the emergence of umbrella protocols, including the Children's Oncology Group–National Cancer Institute Pediatric Molecular Analysis for Therapeutic Choice (Pediatric MATCH) protocol [67]. To address this disparity, one potential strategy is the repurposing of off‐label FDA‐approved targeted therapies for pediatric patients with cancer with malignancies harboring relevant predictive biomarkers. Notably, a key challenge in implementing such a strategy is the ability to identify patients likely to benefit from a given targeted therapy. Per patient, single gene tests or other protein expression–based diagnostics suffer the limitations of requiring significant tissue and/or a limited range of biomarker detection. Sequential testing of individual biomarkers via multiple molecular diagnostic tests can result in significant loss of treatment time or in unnecessary toxicity because of use of conventional therapy. Our data are consistent with this; of cases with prior BRAF molecular testing results available, more than half of reported results were inconsistent with the respective BRAF alteration later identified by CGP. Moreover, even within one indication, diverse and druggable biomarkers are potentially discoverable. For example, at least 60% of PAs harbor KIAA1549‐BRAF fusion. However, tumors found to be BRAF fusion–negative by standard molecular testing (e.g., fluorescence in situ hybridization) may instead harbor alternative activating variants in diverse genes including BRAF, NTRK1–3, FGFR1, NF1, or KRAS [14, 15, 68], all of which are directly or indirectly druggable with currently approved targeted therapies [69]. Large gene panel–based molecular profiling is currently the most efficient means of identifying the breadth of potentially clinically relevant variants in pediatric cancers.
Conclusion
There remains wide disparity in survival depending on cancer type in pediatric cancers. Improved therapeutic strategies are therefore urgently needed. Broad panel‐based molecular profiling can efficiently identify multiple key genomic drivers and should therefore be considered a component of standard molecular testing in advanced or recurrent pediatric cancer types, regardless of disease indication.
Author Contributions
Conception/design: Andrew Rankin, Adrienne Johnson, Siraj Ali, Shakti Ramkissoon
Provision of study material or patients: Andrew Rankin, Adrienne Johnson, Geoffrey Kannan, Jeffrey Knipstein, Nicholas Britt, Dean Pavlick, Shakti Ramkissoon
Collection and/or assembly of data: Andrew Rankin, Adrienne Johnson, Geoffrey Kannan, Jeffrey Knipstein, Nicholas Britt, Dean Pavlick, Shakti Ramkissoon
Data analysis and interpretation: Andrew Rankin, Adrienne Johnson, Alison Roos, Geoffrey Kannan, Jeffrey Knipstein, Nicholas Britt, Mark Rosenzweig, James Haberberger, Dean Pavlick, Eric Severson, Jo‐Anne Vergilio, Rachel Squillace, Rachel Erlich, Pratheesh Sathyan, Stuart Cramer, David Kram, Jeffrey Ross, Vince Miller, Prasanth Reddy, Brian Alexander, Siraj Ali, Shakti Ramkissoon
Manuscript writing: Andrew Rankin, Alison Roos, James Haberberger, Siraj Ali, Shakti Ramkissoon
Final approval of manuscript: Andrew Rankin, Adrienne Johnson, Alison Roos, Geoffrey Kannan, Jeffrey Knipstein, Nicholas Britt, Mark Rosenzweig, James Haberberger, Dean Pavlick, Eric Severson, Jo‐Anne Vergilio, Rachel Squillace, Rachel Erlich, Pratheesh Sathyan, Stuart Cramer, David Kram, Jeffrey Ross, Vince Miller, Prasanth Reddy, Brian Alexander, Siraj Ali, Shakti Ramkissoon
Disclosures
Andrew Rankin: Foundation Medicine, Inc. (E); Jeffrey Knipstein: Atheneum Partners (C/A); Nicholas Britt: Foundation Medicine, Inc. (E, OI); Mark Rosenzweig: Foundation Medicine, Inc. (E, OI); James Haberberger: Foundation Medicine, Inc. (E, OI); Dean Pavlick: Foundation Medicine, Inc. (E, OI); Eric Severson: Foundation Medicine, Inc. (E, OI); Jo‐Anne Vergilio: Foundation Medicine, Inc. (E, OI); Rachel Squillace: Foundation Medicine, Inc. (E, OI); Rachel Erlich: Foundation Medicine, Inc. (E, OI); Jeffrey Ross: Foundation Medicine, Inc. (E, OI); Vince Miller: Foundation Medicine, Inc., Roche, Revolution Medicines (E, OI), Mirati Therapeutics (OI), Memorial Sloan Kettering Cancer Center, USPO 8501413 (other—patent royalties); Prasanth Reddy: Foundation Medicine, Inc. (E, OI); Brian Alexander: Foundation Medicine, Inc. (E, OI); Siraj Ali: Foundation Medicine, Inc. (E, OI); Shakti Ramkissoon: Foundation Medicine, Inc. (E, OI). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Figure S1. Distribution of BRAF alteration types in pediatric cancers according to known functional status (A), and by alteration type within the cohort bearing a known‐activating alteration (B). Abbreviations: alt, alteration
Figure S2. Distribution of cancer types with known‐activating BRAF alteration. (A) Samples were grouped into one of five major categories. Each major category was subsequently divided into multiple subcategories pertaining to diagnosis. (B) Brain tumors contained 18 subtypes, (C) Sarcomas contained 3 subtypes, (D) Embryonal tumors contained 2 subtypes, (E) Hematological malignancies contained 5 subtypes, and (F) other solid tumors included 6 subtypes. Abbreviations: CNS‐PNET, central nervous system‐primitive neuroectodermal tumor; DA, diffuse astrocytoma; DIGG, desmoplastic infantile ganglioglioma; DOLT, disseminated oligodendroglial‐like leptomeningeal tumor; GBM, glioblastoma; HGG, high grade glioma; HGGNT, high grade glioneuronal tumor; LGG, low‐grade glioma; LGGNT, low grade glioneuronal tumor; MPNST, malignant peripheral nerve sheath tumor; NOS, not otherwise specified; PA, pilocytic astrocytoma; PXA, pleomorphic xanthoastrocytoma
Figure S3. Comparison of BRAF molecular testing results (A) Concordance/discordance of prior BRAF molecular testing with CGP across pediatric malignancy. (B) Detailed analysis of prior BRAF molecular testing in PA and LGG (NOS). Abbreviations: CGP, comprehensive genomic profiling; DOLT, disseminated oligodendroglial‐like leptomeningeal tumor; NOS, not otherwise specified; PA, pilocytic astrocytoma; PXA, pleomorphic xanthoastrocytoma; WHO, world health organization.
Table S1. Hematologic malignancies and extracranial solid tumors with known‐activating BRAF alteration
Table S2. Primary brain tumors with known‐activating BRAF SV or Indels
Table S3. Primary brain tumors with known‐activating BRAF fusion
Acknowledgments
We thank Dr. Megan Stephan (Foundation Medicine) for helpful discussion of the manuscript.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin 2015;65:5–29. [DOI] [PubMed] [Google Scholar]
- 2. Worst BC, van Tilburg CM, Balasubramanian GP et al. Next‐generation personalised medicine for high‐risk paediatric cancer patients ‐ The INFORM pilot study. Eur J Cancer 2016;65:91–101. [DOI] [PubMed] [Google Scholar]
- 3. Miller KD, Siegel RL, Lin CC et al. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin 2016;66:271–289. [DOI] [PubMed] [Google Scholar]
- 4. Slamon DJ, Leyland‐Jones B, Shak S et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 2001;344:783–792. [DOI] [PubMed] [Google Scholar]
- 5. Druker BJ, Talpaz M, Resta DJ et al. Efficacy and safety of a specific inhibitor of the BCR‐ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med 2001;344:1031–1037. [DOI] [PubMed] [Google Scholar]
- 6. Solomon BJ, Mok T, Kim DW et al. First‐line crizotinib versus chemotherapy in ALK‐positive lung cancer. N Engl J Med 2014;371:2167–2177. [DOI] [PubMed] [Google Scholar]
- 7. Hauschild A, Grob JJ, Demidov LV et al. Dabrafenib in BRAF‐mutated metastatic melanoma: A multicentre, open‐label, phase 3 randomised controlled trial. Lancet 2012;380:358–365. [DOI] [PubMed] [Google Scholar]
- 8. McArthur GA, Chapman PB, Robert C et al. Safety and efficacy of vemurafenib in BRAF(V600E) and BRAF(V600K) mutation‐positive melanoma (BRIM‐3): Extended follow‐up of a phase 3, randomised, open‐label study. Lancet Oncol 2014;15:323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Oberlin O, Rey A, Sanchez de Toledo J et al. Randomized comparison of intensified six‐drug versus standard three‐drug chemotherapy for high‐risk nonmetastatic rhabdomyosarcoma and other chemotherapy‐sensitive childhood soft tissue sarcomas: Long‐term results from the International Society of Pediatric Oncology MMT95 study. J Clin Oncol 2012;30:2457–2465. [DOI] [PubMed] [Google Scholar]
- 10. Frobisher C, Gurung PM, Leiper A et al. Risk of bladder tumours after childhood cancer: The British Childhood Cancer Survivor Study. BJU Int 2010;106:1060–1069. [DOI] [PubMed] [Google Scholar]
- 11. Teepen JC, van Leeuwen FE, Tissing WJ et al. Long‐term risk of subsequent malignant neoplasms after treatment of childhood cancer in the DCOG LATER study cohort: Role of chemotherapy. J Clin Oncol 2017;35:2288–2298. [DOI] [PubMed] [Google Scholar]
- 12. Devine KA, Mertens AC, Whitton JA et al. Factors associated with physical activity among adolescent and young adult survivors of early childhood cancer: A report from the childhood cancer survivor study (CCSS). Psychooncology 2018;27:613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Paugh BS, Qu C, Jones C et al. Integrated molecular genetic profiling of pediatric high‐grade gliomas reveals key differences with the adult disease. J Clin Oncol 2010;28:3061–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang J, Wu G, Miller CP et al. Whole‐genome sequencing identifies genetic alterations in pediatric low‐grade gliomas. Nat Genet 2013;45:602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson A, Severson E, Gay L et al. Comprehensive genomic profiling of 282 pediatric low‐ and high‐grade gliomas reveals genomic drivers, tumor mutational burden, and hypermutation signatures. The Oncologist 2017;22:1478–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kager L, Tamamyan G, Bielack S. Novel insights and therapeutic interventions for pediatric osteosarcoma. Future Oncol 2017;13:357–368. [DOI] [PubMed] [Google Scholar]
- 17. Padovan‐Merhar OM, Raman P, Ostrovnaya I et al. Enrichment of targetable mutations in the relapsed neuroblastoma genome. PLoS Genet 2016;12:e1006501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kool M, Jones DT, Jager N et al. Genome sequencing of SHH medulloblastoma predicts genotype‐related response to smoothened inhibition. Cancer Cell 2014;25:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vanden Borre P, Schrock AB, Anderson PM et al. Pediatric, adolescent, and young adult thyroid carcinoma harbors frequent and diverse targetable genomic alterations, including kinase fusions. The Oncologist 2017;22:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Farrar JE, Schuback HL, Ries RE et al. Genomic profiling of pediatric acute myeloid leukemia reveals a changing mutational landscape from disease diagnosis to relapse. Cancer Res 2016;76:2197–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu Y, Easton J, Shao Y et al. The genomic landscape of pediatric and young adult T‐lineage acute lymphoblastic leukemia. Nat Genet 2017;49:1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chmielecki J, Bailey M, He J et al. Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res 2017;77:509–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Allen CE, Parsons DW. Biological and clinical significance of somatic mutations in Langerhans cell histiocytosis and related histiocytic neoplastic disorders. Hematology Am Soc Hematol Educ Program 2015;2015:559–564. [DOI] [PubMed] [Google Scholar]
- 24. Holderfield M, Deuker MM, McCormick F et al. Targeting RAF kinases for cancer therapy: BRAF‐mutated melanoma and beyond. Nat Rev Cancer 2014;14:455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burotto M, Chiou VL, Lee JM et al. The MAPK pathway across different malignancies: A new perspective. Cancer 2014;120:3446–3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davies H, Bignell GR, Cox C et al. Mutations of the BRAF gene in human cancer. Nature 2002;417:949–954. [DOI] [PubMed] [Google Scholar]
- 27. Kandoth C, McLellan MD, Vandin F et al. Mutational landscape and significance across 12 major cancer types. Nature 2013;502:333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ross JS, Wang K, Chmielecki J et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int J Cancer 2016;138:881–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Helgager J, Lidov HG, Mahadevan NR et al. A novel GIT2‐BRAF fusion in pilocytic astrocytoma. Diagn Pathol 2017;12:82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim KB, Semrad T, Schrock A et al. Significant clinical response to a MEK inhibitor therapy in a patient with metastatic melanoma harboring an RAF1 fusion. JCO Precis Oncol 2018. DOI: 10.1200/PO.17.00138. [DOI] [PubMed] [Google Scholar]
- 31. Chmielecki J, Hutchinson KE, Frampton GM et al. Comprehensive genomic profiling of pancreatic acinar cell carcinomas identifies recurrent RAF fusions and frequent inactivation of DNA repair genes. Cancer Discov 2014;4:1398–1405. [DOI] [PubMed] [Google Scholar]
- 32. Palanisamy N, Ateeq B, Kalyana‐Sundaram S et al. Rearrangements of the RAF kinase pathway in prostate cancer, gastric cancer and melanoma. Nat Med 2010;16:793–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jones DT, Kocialkowski S, Liu L et al. Oncogenic RAF1 rearrangement and a novel BRAF mutation as alternatives to KIAA1549:BRAF fusion in activating the MAPK pathway in pilocytic astrocytoma. Oncogene 2009;28:2119–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cin H, Meyer C, Herr R et al. Oncogenic FAM131B‐BRAF fusion resulting from 7q34 deletion comprises an alternative mechanism of MAPK pathway activation in pilocytic astrocytoma. Acta Neuropathol 2011;121:763–774. [DOI] [PubMed] [Google Scholar]
- 35. Forshew T, Tatevossian RG, Lawson AR et al. Activation of the ERK/MAPK pathway: A signature genetic defect in posterior fossa pilocytic astrocytomas. J Pathol 2009;218:172–181. [DOI] [PubMed] [Google Scholar]
- 36. Lassaletta A, Scheinemann K, Zelcer SM et al. Phase II weekly vinblastine for chemotherapy‐naive children with progressive low‐grade glioma: A Canadian Pediatric Brain Tumor Consortium Study. J Clin Oncol 2016;34:3537–3543. [DOI] [PubMed] [Google Scholar]
- 37. Lassaletta A, Zapotocky M, Mistry M et al. Therapeutic and prognostic implications of BRAF V600E in pediatric low‐grade gliomas. J Clin Oncol 2017;35:2934–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jones DTW, Kieran MW, Bouffet E et al. Pediatric low‐grade gliomas: Next biologically driven steps. Neuro Oncol 2018;20:160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pekmezci M, Villanueva‐Meyer JE, Goode B. The genetic landscape of ganglioglioma. Acta Neuropathol Commun 2018;6:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sun Y, Alberta JA, Pilarz C et al. A brain‐penetrant RAF dimer antagonist for the noncanonical BRAF oncoprotein of pediatric low‐grade astrocytomas. Neuro Oncol 2017;19:774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Koelblinger P, Thuerigen O, Dummer R. Development of encorafenib for BRAF‐mutated advanced melanoma. Curr Opin Oncol 2018;30:125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bautista F, Paci A, Minard‐Colin V et al. Vemurafenib in pediatric patients with BRAFV600E mutated high‐grade gliomas. Pediatr Blood Cancer 2014;61:1101–1103. [DOI] [PubMed] [Google Scholar]
- 43. Aguilera D, Janss A, Mazewski C et al. Successful retreatment of a child with a refractory brainstem ganglioglioma with vemurafenib. Pediatr Blood Cancer 2016;63:541–543. [DOI] [PubMed] [Google Scholar]
- 44. Meletath SK, Pavlick D, Brennan T et al. Personalized treatment for a patient with a BRAF V600E mutation using dabrafenib and a tumor treatment fields device in a high‐grade glioma arising from ganglioglioma. J Natl Compr Canc Netw 2016;14:1345–1350. [DOI] [PubMed] [Google Scholar]
- 45. Shih KC, Shastry M, Williams JT et al. Successful treatment with dabrafenib (GSK2118436) in a patient with ganglioglioma. J Clin Oncol 2014;32:e98–e100. [DOI] [PubMed] [Google Scholar]
- 46. Kieran MW, Cohen KJ, Doz F et al. Complete radiographic responses in pediatric patients with BRAFV600‐positive tumors including high‐grade gliomas: Preliminary results of an ongoing phase 1/2a safety and pharmacokinetics (PK) study of dabrafenib. J Clin Oncol 2014;32(suppl 15):10056a. [Google Scholar]
- 47. Lassaletta A, Guerreiro Stucklin A, Ramaswamy V et al. Profound clinical and radiological response to BRAF inhibition in a 2‐month‐old diencephalic child with hypothalamic/chiasmatic glioma. Pediatr Blood Cancer 2016;63:2038–2041. [DOI] [PubMed] [Google Scholar]
- 48. del Bufalo F, Carai A, Figa‐Talamanca L et al. Response of recurrent BRAFV600E mutated ganglioglioma to vemurafenib as single agent. J Transl Med 2014;12:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Robinson GW, Orr BA, Gajjar A. Complete clinical regression of a BRAF V600E‐mutant pediatric glioblastoma multiforme after BRAF inhibitor therapy. BMC Cancer 2014;14:258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Heritier S, Emile JF, Barkaoui MA et al. BRAF mutation correlates with high‐risk Langerhans cell histiocytosis and increased resistance to first‐line therapy. J Clin Oncol 2016;34:3023–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heritier S, Helias‐Rodzewicz Z, Lapillonne H et al. Circulating cell‐free BRAF(V600E) as a biomarker in children with Langerhans cell histiocytosis. Br J Haematol 2017;178:457–467. [DOI] [PubMed] [Google Scholar]
- 52. Banerjee A, Jakacki RI, Onar‐Thomas A et al. A phase I trial of the MEK inhibitor selumetinib (AZD6244) in pediatric patients with recurrent or refractory low‐grade glioma: A Pediatric Brain Tumor Consortium (PBTC) study. Neuro Oncol 2017;19:1135–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Miller C, Guillaume D, Dusenbery K et al. Report of effective trametinib therapy in 2 children with progressive hypothalamic optic pathway pilocytic astrocytoma: Documentation of volumetric response. J Neurosurg Pediatr 2017;19:319–324. [DOI] [PubMed] [Google Scholar]
- 54. Wagner LM, Myseros JS, Lukins DE et al. Targeted therapy for infants with diencephalic syndrome: A case report and review of management strategies. Pediatr Blood Cancer 2018;65:e26917. [DOI] [PubMed] [Google Scholar]
- 55. Kondyli M, Larouche V, Saint‐Martin C et al. Trametinib for progressive pediatric low‐grade gliomas. J Neurooncol 2018;140:435–444. [DOI] [PubMed] [Google Scholar]
- 56. Fangusaro J, Onar‐Thomas A, Poussaint TY. Selumetinib in paediatric patients with BRAF‐aberrant or neurofibromatosis type 1‐associated recurrent, refractory, or progressive low‐grade glioma: A multicentre, phase 2 trial. Lancet Oncol 2019;20:1011–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. McEvoy CR, Xu H, Smith K et al. Profound MEK inhibitor response in a cutaneous melanoma harboring a GOLGA4‐RAF1 fusion. J Clin Invest 2019;129:1940–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yde CW, Sehested A, Mateu‐Regue A et al. A new NFIA:RAF1 fusion activating the MAPK pathway in pilocytic astrocytoma. Cancer Genet 2016;209:440–444. [DOI] [PubMed] [Google Scholar]
- 59. Harris MH, DuBois SG, Glade Bender JL et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: The Individualized Cancer Therapy (iCat) study. JAMA Oncol 2016;2:608–615. [DOI] [PubMed] [Google Scholar]
- 60. Denburg A, Arora B, Arora RS et al. Access to essential medicines for children with cancer: A joint SIOP‐CCI position statement. Lancet Oncol 2017;18:20–22. [DOI] [PubMed] [Google Scholar]
- 61. Mody RJ, Prensner JR, Everett J et al. Precision medicine in pediatric oncology: Lessons learned and next steps. Pediatr Blood Cancer 2017;64:e26288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Sun J, He Y, Sanford E et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLoS Comput Biol 2018;14:e1005965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Forbes SA, Beare D, Gunasekaran P et al. COSMIC: Exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Res 2015;43:D805–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chalmers ZR, Connelly CF, Fabrizio D et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 2017;9:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tran N, Wu X, B‐Raf Frost J. and Raf‐1 are regulated by distinct autoregulatory mechanisms. J Biol Chem 2005;280:16244–16253. [DOI] [PubMed] [Google Scholar]
- 67. Allen CE, Laetsch TW, Mody R et al. Target and agent prioritization for the Children's Oncology Group‐National Cancer Institute Pediatric MATCH Trial. J Natl Cancer Inst 2017;109:djw274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Jones DT, Hutter B, Jager N et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 2013;45:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Raabe E, Kieran MW, Cohen KJ. New strategies in pediatric gliomas: Molecular advances in pediatric low‐grade gliomas as a model. Clin Cancer Res 2013;19:4553–4558. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Figure S1. Distribution of BRAF alteration types in pediatric cancers according to known functional status (A), and by alteration type within the cohort bearing a known‐activating alteration (B). Abbreviations: alt, alteration
Figure S2. Distribution of cancer types with known‐activating BRAF alteration. (A) Samples were grouped into one of five major categories. Each major category was subsequently divided into multiple subcategories pertaining to diagnosis. (B) Brain tumors contained 18 subtypes, (C) Sarcomas contained 3 subtypes, (D) Embryonal tumors contained 2 subtypes, (E) Hematological malignancies contained 5 subtypes, and (F) other solid tumors included 6 subtypes. Abbreviations: CNS‐PNET, central nervous system‐primitive neuroectodermal tumor; DA, diffuse astrocytoma; DIGG, desmoplastic infantile ganglioglioma; DOLT, disseminated oligodendroglial‐like leptomeningeal tumor; GBM, glioblastoma; HGG, high grade glioma; HGGNT, high grade glioneuronal tumor; LGG, low‐grade glioma; LGGNT, low grade glioneuronal tumor; MPNST, malignant peripheral nerve sheath tumor; NOS, not otherwise specified; PA, pilocytic astrocytoma; PXA, pleomorphic xanthoastrocytoma
Figure S3. Comparison of BRAF molecular testing results (A) Concordance/discordance of prior BRAF molecular testing with CGP across pediatric malignancy. (B) Detailed analysis of prior BRAF molecular testing in PA and LGG (NOS). Abbreviations: CGP, comprehensive genomic profiling; DOLT, disseminated oligodendroglial‐like leptomeningeal tumor; NOS, not otherwise specified; PA, pilocytic astrocytoma; PXA, pleomorphic xanthoastrocytoma; WHO, world health organization.
Table S1. Hematologic malignancies and extracranial solid tumors with known‐activating BRAF alteration
Table S2. Primary brain tumors with known‐activating BRAF SV or Indels
Table S3. Primary brain tumors with known‐activating BRAF fusion