

Abstract
Purpose
Traumatic optic neuropathy (TON) is often caused by blunt head trauma and has no currently effective treatment. Common animal models of TON induced by surgical crush injury are plagued by variability and do not mimic typical mechanisms of TON injury. Traumatic head impact models have recently shown evidence of TON, but the degree of head impact necessary to consistently induce TON is not well characterized, and it is examined here.
Methods
Traumatic skull impacts to C57BL/6J mice were induced using an electromagnetic controlled impact device. One impact performed at two depths (mild and severe), as well as three and five repetitive impacts with an interconcussion interval of 48 hours, were tested. Optokinetic responses (OKRs) and retinal ganglion cell (RGC) loss were measured.
Results
Five repetitive mild impacts significantly decreased OKR scores and RGC numbers compared with control mice 10 weeks after initial impact, with maximal pathology observed by 6 weeks and partial but significant loss present by 3 weeks. One severe impact induced similar TON. Three mild impacts also induced early OKR and RGC loss, but one mild impact did not. Equivalent degrees of TON were induced bilaterally, and a significant correlation was observed between OKR scores and RGC numbers.
Conclusions
Repetitive, mild closed head trauma in mice induces progressive RGC and vision loss that worsens with increasing impacts.
Translational Relevance
Results detail a reproducible model of TON that provides a reliable platform for studying potential treatments over a 3- to 6-week time course.
Keywords: traumatic optic neuropathy, retinal ganglion cell, traumatic head impact
Introduction
Traumatic optic neuropathy (TON) is a condition in which acute injury to the optic nerve caused by ocular or head trauma results in vision loss.1 TON can present with severe visual loss in the form of visual field abnormalities, decreased visual acuity, loss of color vision, and afferent pupillary defect.2–4 In the International Optic Nerve Trauma Study (IONTS), 85% of patients with TON were males with an average age of 34 years.5 Common etiologies include motor vehicle accidents, falls, war injuries, sports injuries, and assaults.6,7 Direct trauma to the optic nerve is not common; rather, most TON presents with a multifactorial pathophysiology following blunt force head trauma, with primary damage being due to disruption or shearing of the optic nerve which can lead to secondary damage in the form of inflammation, vascular dysfunction, or optic nerve swelling.4 Trauma leads to a series of pathologic events that include increase in oxidative stress, release of inflammatory mediators,8 blood–brain barrier disruption,9 and intracellular calcium influx,10 all of which can contribute to retinal ganglion cell (RGC) death, causing irreversible blindness.5 Multiple signaling pathways can act in parallel in the same retina or RGCs, resulting in cell death.11
There is no evidence available from randomized clinical trials or other scientific studies that help clinicians provide treatment recommendations for TON. Currently, the main treatments considered for TON include systemic corticosteroids and/or surgical decompression of the optic nerve.1 However, studies show no convincing evidence that high-dose corticosteroids are any more beneficial than conservative management in the treatment of TON,12 and several studies show detrimental effects of high doses of steroids.13,14 The IONTS also supports a firmer conclusion that neither corticosteroids nor optic canal surgery should be considered as standard treatment for TON.5 Therefore, new therapies are needed to improve neurological outcomes in TON, but development has been limited by the ability of available animal models to mimic major features of TON injuries that occur in patients.
Animal models are pivotal to bridge preclinical translational research to the clinic. Limited availability of animal models that replicate precise injury mechanisms and pathophysiology of RGC damage is one major limitation in preclinical studies of TON. Optic nerve crush surgery, an acute direct mechanical injury to the optic nerve, is often used to study the pathophysiology of axonal trauma-induced RGC degeneration and vision loss.15,16 Difficulty quantifying the mechanical force applied to induce the crush injury and difficulty in entering the mouse orbit without disrupting the blood plexus surrounding the globe and thus causing severe ischemia are major drawbacks of this model,17 and the mechanism of injury does not represent the typical shearing injury most patients experience with blunt head trauma.4 Recent studies have focused on creating models that simulate indirect trauma to the optic nerve caused by ocular or head injury, including ocular blasts of compressed air directed against the eye,18 sonication-induced TON by delivery of ultrasonic pulses,19 exposure of the optic nerve to a graded controlled impact of known velocity,20 and mild repetitive controlled blunt trauma to the head.21 Each model has advantages and disadvantages related to reproducibility, mortality rate, undesirable ocular injuries, and mirroring the exact pathological progression in TON.19,20
Studies of TON in the general population have reported that a significant number of patients with TON have had blunt head injuries, and the most common etiology is vehicle accidents.7 Characterization of animal models of blunt head trauma are needed to better understand pathophysiological events that result in vision loss and to evaluate possible therapeutic interventions. Tzekov and colleagues21 demonstrated that mild repetitive head trauma induces optic nerve damage 10 to 13 weeks after initial trauma, but the time course of RGC loss and degree of trauma necessary to induce TON remains unclear. This is assessed in the present study by investigation of the morphological and functional consequences on the visual system after mild, controlled, repetitive impacts to the mouse head.
Methods
Mice
Eight-week-old C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed at the University of Pennsylvania. Mice were raised on a 12-hour light/dark cycle with an ambient temperature of 22°C; they were fed laboratory-grade rodent chow and given water ad libitum. All procedures were approved and carried out in accordance with the University of Pennsylvania's Institutional Animal Care and Use Committee, the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and the Association for Assessment and Accreditation of Laboratory Animal Care International.
Traumatic Head Impact/Optic Nerve Injury
Mice were randomly assigned to one of five injury groups: control (received all procedures except impact), 1×-S (single, severe impact), 1×-M (single, mild impact), 3×-M (three consecutive mild impacts, 48 hours apart), or 5×-M (five consecutive mild impacts, 48 hours apart) (Fig. 1C). Mice were anesthetized with isoflurane, the fur on their head was shaved, and they were placed on stereotaxic equipment. One impact was delivered to the center of the skull of mice in the 1×-S and 1×-M groups. Three to five consecutive impacts were delivered to mice in the 3×-M and 5×-M groups, respectively, with an interconcussion interval of 48 hours between each impact, similar to previously established methods.21 Impacts were induced using an electromagnetic controlled impact device with a 5-mm blunt metal impactor tip (Impact One Stereotaxic Impactor; Leica Biosystems, Buffalo Grove, IL). The strike velocity was 5 mm/s with a strike depth of 2.0 mm (severe impact) for the 1×-S group, and it was 1.0 mm (mild impact) for the 1×-M, 3×-M, and 5×-M groups, with a dwell time of 300 ms. The location of the impact on the skull was central, equally distant from both eyes (Figs. 1A, 1B). Control mice received anesthesia of equal intervals and duration as the injured mice; they were placed on the same stereotaxic equipment, and the impactor tip was triggered with the same velocity but at a reduced depth so that the impactor tip did not touch the skull.
Figure 1.

Diagrammatic representation of the mouse model of THI. (A) Device used to induce closed head impact. (B) The site of head impact. (C) Experimental design.
RGC Function (Optokinetic Response)
The optokinetic response (OKR) was used to evaluate visual function prior to and throughout the duration of the study. The OKR was assessed using OptoMotry software and equipment (Cerebral Mechanics, Inc., Medicine Hat, AB, Canada) as described in previous studies.22,23 Mice were placed on an elevated platform in the center of an arena consisting of four monitors with a camera above to image mouse behavior. The monitors display sine-wave gratings that rotate in a circular motion and vary in spatial frequencies, and the OKR is measured by determining the highest frequency at which mice can track a 100% contrast grid with head movements in the direction of the gratings. Data are reported in units of cycles/degree. Average OKR scores between the two eyes of each animal were used for all comparisons, except where noted for experiments comparing effects between right and left eyes.
Quantification of RGC Numbers
RGC quantification was performed as previously described.24,25 Eyes were harvested, fixed in 4% paraformaldehyde at room temperature for 1 hour, and then washed in phosphate buffered saline (PBS) followed by isolation and dissection of the retina. Retinas were prepared as flattened whole mounts, washed three times in PBS, and permeabilized in 0.5% Triton X-100 in PBS by freezing at –70°C for 15 minutes. Retinas were then incubated overnight at 4°C in a humidified chamber with rabbit anti-Brn3a antibody (RGC marker; Synaptic Systems, Goettingen, Germany) diluted 1:2000 in a blocking buffer containing 2% bovine serum albumin and 2% Triton X-100. Retinas were washed three times in PBS and incubated for 1 hour at room temperature in Alexa Fluor 488 anti-rabbit secondary antibody (Thermo Fisher Scientific, Waltham, MA) at a dilution of 1:500 in blocking buffer. After the incubation, tissues were washed five times in PBS and then mounted vitreous side up onto glass slides with Fluoromount-G mounting medium (Southern Biotech, Birmingham, AL). RGCs were photographed using a fluorescent microscope (Nikon, Melville, NY) at 40× magnification in 12 standard fields: 1/6, 3/6, and 5/6 of the retinal radius from the center of the retina in each quadrant. RGCs were counted by a masked investigator using image analysis software (Image-Pro Plus 5.0; Media Cybernetics, Rockville, MD). Average RGC numbers between the two eyes of each animal were used for all comparisons, except where noted for experiments comparing effects between right and left eyes.
Quantification of RGC Axons
The density of intact RGC axons was quantified in individual experiments using one of two standard methods as indicated in the results, each according to previously published methods.24–26 Neurofilament staining was performed and quantified in longitudinal paraffin embedded sections. Briefly, optic nerves were isolated, processed, and embedded in paraffin, after which 5-µm longitudinal paraffin sections were deparaffinized and rehydrated, and nonspecific binding was reduced using Blocking Reagent (Vector Laboratories, Burlingame, CA). Specimens were then incubated in rabbit anti-neurofilament antibody 1: 500 (Abcam, Cambridge, UK) at 4°C overnight. Sections were washed three times with PBS and then incubated with anti-rabbit secondary antibody (Vectastain Elite ABC rabbit kit; Vector Laboratories) for 30 minutes at 37°C. Avidin/biotin complex detection was performed by incubating with Vectastain Elite ABC reagent at 37°C for 30 minutes and diaminobenzidine (Vector Laboratories) substrate for 3 minutes at room temperature followed by washing in running water for 5 minutes. Dehydrated slides were mounted using Refrax Mounting Medium (Vector Laboratories). Photographs of three fields per nerve (one each at the distal, central, and proximal regions of the longitudinal optic nerve section) at 20× magnification were taken by a masked investigator. Neurofilament staining optical density was quantified by using ImageJ software (National Institutes of Health, Bethesda, MD).
Alternatively, transverse sections of the optic nerve were stained and axons were counted using toluidine blue staining as previously described.26 Briefly, optic nerves were isolated and post-fixed in 2% PFA and 2% glutaraldehyde for 24 hours. The nerves were dehydrated and incubated with infiltration solution (Polysciences, Inc., Warrington, PA), for 24 hours at 4°C. The nerves were then embedded in resin (Polysciences, Inc.), and 1-µM thin transverse sections were made and stained with 1% toluidine blue–1% borate solution (Sigma-Aldrich, St. Louis, MO) for 5 minutes. The sections were washed in running water for 5 minutes and mounted using refrax mounting medium. Assessment of morphologic evidence of axonal degeneration was done by counting the number of axons in five fields per optic nerve, each with an area of 100 µM2 (one at the center and four regions; left, right, up, and down 3/6th radial distance from the center point) at 40× magnification by a masked investigator. The number of axons per square micromolar was calculated by dividing the total number of axons counted by the sum of the area of five fields.
Statistics
Optokinetic responses over time were compared using repeated-measures analysis of variance (ANOVA) followed by Tukey post hoc comparisons for each group. RGC counts, axon numbers and staining density, and OKR final scores were compared using one-way ANOVA with Tukey post hoc comparisons between each injury group. To examine the relationship between RGC numbers and cumulative OKR scores, correlation and linear regression analyses were performed by calculating a Pearson's correlation coefficient and a line of best fit between the two variables in all datasets. All statistical analyses were calculated using Prism (GraphPad Software, San Diego, CA). All data are expressed as means ± standard error of mean (SEM). P values less than 0.05 were considered statistically significant.
Results
Repetitive Mild, or Single Severe, Traumatic Head Impacts Induce Significant Vision Loss with Loss of RGCs and Their Axons
Significant RGC loss in the eyes of mice 10 weeks after repetitive mild traumatic head impacts (THIs) was previously reported.21 To evaluate the reproducibility of this TON and observe whether a single, more severe impact can induce a similar effect, C57BL/6J mice were randomly grouped into control, 1×-S, and 5×-M THI groups. The 1×-S mice received one single severe impact with the impactor with a strike depth of 2.0 mm and velocity 5 m/s at day 0. The 5×-M group of mice received five consecutive mild impacts by the impactor with a strike depth of 1.0 mm and velocity 5 m/s, with an inter-concussion interval of 48 hours between each impact, starting on day 0 and finishing on day 8 (Fig. 1C). No skull fractures occurred in the 5×-M group, consistent with prior reports,21 whereas some mice in the 1×-S group were observed to have skull fractures after impact. The OKR was measured at baseline (prior to first impact) at day 0, and measurement was repeated on day 1 and again one time per week until sacrifice on day 70. Repeated-measures ANOVA showed a significant decrease in OKR scores across 10 weeks in both groups of mice that received five mild impacts (P < 0.001) and one severe impact (P < 0.01) when compared with control mice (Fig. 2A). Of note, the one severe impact induced a significantly larger decrease in the OKR on day 1, and optokinetic responses partially recovered at later time points. Comparison of OKR scores in TON mice versus controls at individual time points showed that five mild repeated impacts induced more consistent and sustained vision loss with significant OKR decreases compared with controls at more time points than the mice that received one severe impact (Fig. 2A). To evaluate whether the visual effects of one severe or five mild head impacts were due to RGC loss, mice were sacrificed on day 70 and RGCs were quantified. Both the 1×-S and 5×-M head trauma groups showed a significant (P < 0.001) decrease in RGC numbers (Fig. 2B) compared with controls. In order to determine whether blunt head trauma induces shearing injury differentially across distinct regions of the optic nerve and retina, numbers of surviving RGCs in the peripheral, mid-peripheral (middle), and central retina were counted and compared. Each region in 1×-S and 5×-M mice showed a significant (P < 0.001) decrease in RGC numbers compared with the corresponding region in control mice (Figs. 2C, 2D). To evaluate whether axonal loss in the optic nerve accompanies the vision loss and RGC loss in the retina of mice that received one severe or five mild head impacts, longitudinal optic nerve sections from mice sacrificed on day 70 were stained using neurofilament antibodies, and the density of axon staining was quantified. Both 1×-S and 5×-M head trauma groups showed a significant (P < 0.01) decrease in neurofilament staining (Figs. 2E, 2F) compared with controls. Because the 1×-S mice developed skull fractures, severe OKR decreases at day 1 suggestive of possible effects on visual processing, and less consistent OKR decreases across each time point than the 5×-M mice, only mild force head impacts were used for subsequent studies.
Figure 2.

Repetitive mild and single severe THIs both induced loss of RGCs and visual function. (A) OKR scores compared by repeated-measures ANOVA show a significant decrease in vision over a 10 week time course in 5×-M mice (n = 4) (xxxP < 0.001) and 1×-S mice (n = 4) (##P < 0.01) compared with control mice (n = 4). Decreased OKR scores at individual time points, compared with control mice, were determined by ANOVA (*P < 0.05; **P < 0.01; ***P < 0.001). (B) Brn3-stained, flat-mounted retinal sections from mice sacrificed on day 70 demonstrated a significantly (***P < 0.001) reduced number of RGCs in both the 5×-M (n = 4) and 1×-S (n = 4) head trauma groups when compared with control mice (n = 4). (C) RGCs in the peripheral, middle (mid-peripheral), and central (posterior pole) retinal regions were counted. Each region in 1×-S and 5×-M mice showed a significant (***P < 0.001) decrease in RGC numbers compared with the corresponding region in control mice. (D) Representative images show RGCs in three regions (peripheral, middle, and central) from one retina of each treatment group. Original magnification, 20×. Scale bars: 50 µM. (E) Neurofilament stained optic nerve sections from mice sacrificed on day 70 demonstrated a significantly (**P < 0.01) reduced staining intensity in both the 5×-M (n = 4) and 1×-S (n = 4) head trauma groups when compared with control (n = 4) mice. (F) A collage of images (three fields per nerve, one each at the distal, central, and proximal regions of the longitudinal optic nerve section; original magnification, 20×) of optic nerves from each group show variable neurofilament staining reflective of focal areas of axon loss. N represents the number of mice; each average of left and right eye is plotted as a single data point. Data represent mean ± SEM.
Repetitive Mild Traumatic Head Impacts Induce Significant Loss of Vision, RGCs, and Their Axons by Week 6
The time course of OKR decreases observed over 10 weeks following five mild repetitive head impacts (Fig. 2A) suggests that RGC loss likely occurs earlier than 10 weeks, with OKR scores stabilized by week 6. To assess this, five mild impacts were performed on mice with interconcussion intervals of 48 hours, and the OKR was measured before impact (baseline) and for 6 weeks after initial impact. Repeated-measures ANOVA showed a significant decrease in the OKR scores across 6 weeks in mice that received five mild impacts (P < 0.001) compared with control mice (Fig. 3A). RGC loss was evaluated following sacrifice on day 42. The five mild impacts induced a significant (P < 0.001) decrease in RGC numbers compared with control mice (Fig. 3B). The severity of RGC loss observed at 6 weeks was similar to that observed at 10 weeks (5×-M 6 weeks vs. 5×-M 10 weeks; P = 0.845, not significant) (Fig. 3B vs. Fig. 2B). To evaluate axonal loss, optic nerves were isolated on day 42, and transverse sections were stained with toluidine blue to assess morphologic evidence of axonal degeneration by counting the number of axons. The 5×-M mice showed a significant (P < 0.01) decrease in axonal numbers counted in five standardized fields compared with control mice (Figs. 3C, 3D).
Figure 3.

Repetitive, mild THIs induced loss of RGCs and vision by week 6. (A) The 5×-M mice (n = 4) showed a significant (*P < 0.05) decrease in OKR scores measured at various time points, and repeated-measures ANOVA showed a significant (xxxP < 0.001) decrease in the OKR scores in the 5×-M mice over time compared with the control mice (n = 4). (B) Brn3a-stained, flat-mounted retinal sections from mice sacrificed on day 42 demonstrated a significant (***P < 0.001) decrease in number of RGCs in 5×-M mice compared with control mice. (C) Toluidine blue-stained transverse sections of the optic nerve from mice sacrificed on day 42 demonstrated significantly (**P < 0.01) reduced axonal counts in 5×-M mice compared with control mice. (D) Representative high magnification images of toluidine blue-stained optic nerve transverse section from a control mouse and a 5×-M mouse. Original magnification, 40×. Scale bars: 10 µM. N represents the number of mice; each average of left and right eye is plotted as a single data point. Data represent mean ± SEM.
RGC and Vision Loss Occurs by 3 Weeks After Initial Head Impact and Can Be Induced by Fewer Repetitive Impacts
To evaluate the onset of RGC and visual loss and the number of mild impacts necessary to induce these effects, mice were randomly assigned to control, 1×-M, 3×-M, or 5×-M groups. The 1×-M mice received a single mild impact on day 0, whereas the 3×-M mice received repetitive impacts from day 0 to 4, and the 5×-M mice received repetitive impacts from day 0 to 8 with an interconcussion interval of 48 hours. The OKR was measured at various time points until sacrifice of all mice on day 21. The 5×-M and 3×-M mice showed a progressive decrease in OKR scores (Fig. 4A). Repeated-measures ANOVA showed a significant attenuation in the OKR scores of the 5×-M mice (P < 0.001) and 3×-M mice (P < 0.05) compared with control mice, whereas the 1×-M mice showed no significant decrease (Fig. 4A). RGC survival was assessed following sacrifice on day 21. The 1×-M mice showed no RGC loss compared with control mice. Significant RGC loss was observed in the 3×-M mice (P < 0.01) and 5×-M mice (P < 0.001) when compared with control mice (Fig. 4B). The degree of RGC loss observed at 6 weeks was more severe than that observed at 3 weeks (5×-M 6 weeks vs. 5×-M 3 weeks, P < 0.05) (Fig. 3B vs. Fig. 4B).
Figure 4.
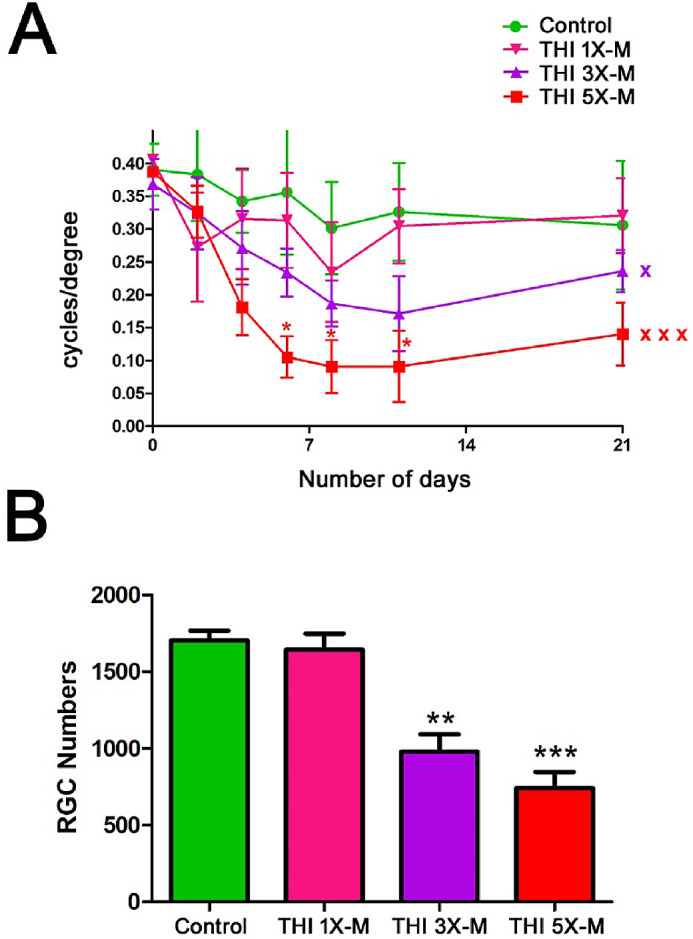
Significant RGC and visual function loss can be induced by 3 weeks after initial impact with as few as three repetitive impacts. (A) OKR scores compared by repeated-measures ANOVA over a 21-day time course were significantly decreased in 5×-M mice (n = 5; xxxP < 0.001) and 3×-M mice (n = 4; xP < 0.05) compared with control mice (n = 3). OKR scores were not significantly decreased in 1×-M mice (n = 3). Only 5×-M mice showed decreased OKR scores at any individual time points compared with control mice and as determined by ANOVA (*P < 0.05). (B) Brn3-stained, flat-mounted retinal sections from mice sacrificed on day 21 demonstrated significant RGC loss in 3×-M mice (n = 4; **P < 0.01) and 5×-M mice (n = 5; ***P < 0.001) compared with control mice, but 1×-M mice (n = 3) did not show significant RGC loss. N represents the number of mice; each average of left and right eye is plotted as a single data point. Data represent mean ± SEM.
Blunt Head Trauma Induces Bilateral RGC and Vision Loss
Because the head injuries in the current studies were made by impacts on the center of the skull, equidistant from both eyes, damage was expected to be similar in both eyes. OKR and RGC statistical comparisons were performed using the average of both eyes of each animal as a single data point (Figs. 23–4), To confirm whether the effect of the impacts is indeed bilateral, data from mice followed for 10 weeks (Fig. 2) and 6 weeks (Fig. 3) following five mild impacts were reanalyzed with OKR scores and RGC numbers of right and left eyes plotted separately. Repeated-measures ANOVA showed a similar significant (P < 0.001) decrease in OKR scores for both the right and left eyes of 5×-M mice over 70 days compared with control mice left and right eyes, respectively, with no significant difference between the left and right eyes (Fig. 5A). A significant decrease (P < 0.001) in RGC number was also seen in the right and left eyes of 5×-M mice sacrificed on day 70 compared with control mice left and right eyes, respectively, with no significant difference between the left and right eyes (Fig. 5B). Similar results were seen 6 weeks after initial impact. Both OKR and RGC scores for right and left eyes over 42 days decreased significantly (P < 0.001) compared with controls, whereas right and left eyes did not differ from each other (Figs. 5C, 5D).
Figure 5.

RGC and vision loss is bilateral. (A) OKR scores compared by repeated-measures ANOVA over 70 days were significantly decreased in right eyes (OD) (n = 4; xxxP < 0.001) and left eyes (OS) (n = 4; xxxP < 0.001) of 5×-M mice compared with right and left eyes of control mice (n = 4), respectively. Decreased OKR scores at individual time points, compared with the corresponding eyes of control mice, were determined by ANOVA (*P < 0.05; **P < 0.01; ***P < 0.001). No significant difference was found between right eyes and left eyes within the 5×-M mice over time nor at any individual time points. (B) Brn3a-stained, flat-mounted retinal sections from mice sacrificed on day 70 showed a significant decrease in number of RGCs in both right eyes (n = 4; ***P < 0.001) and left eyes (n = 4; ***P < 0.001) of 5×-M mice compared with corresponding eyes of control mice (n = 4). (C) OKR scores compared by repeated-measures ANOVA over 42 days were significantly decreased in right eyes (n = 4; xxxP < 0.001) and left eyes (n = 4; xxxP < 0.001) of 5×-M mice compared with right and left eyes of control mice (n = 4), respectively. Decreased OKR scores at individual time points, compared with the corresponding eyes of control mice, were determined by ANOVA (*P < 0.05; **P < 0.01; ***P < 0.001). No significant difference was found between right eyes and left eyes within the 5×-M mice over time nor at any individual time points. (D) Brn3a-stained, flat-mounted retinal sections from mice sacrificed on day 42 showed a significant decrease in RGC numbers in right eyes (***P < 0.001) and left eyes (***P < 0.001) of 5×-M mice compared with corresponding eyes of control mice. N represents the number of left or right eyes, each plotted as a single data point. Data represent mean ± SEM.
OKR Scores Correlate with RGC Loss Following Head Trauma
Reduced visual function following head trauma may be multifactorial, as the trauma has the potential to affect visual pathways and visual processing in the brain, in addition to inducing TON as marked by RGC loss (Figs. 234–5). To determine whether OKR scores decrease due to RGC loss in this model of TON, a correlation analysis was performed between RGC numbers and the cumulative OKR score of each eye of control and TON mice. The analysis showed a statistically significant (P < 0.0001) positive correlation between RGC number and cumulative OKR score at 3 weeks (Fig. 6A), 6 weeks (Fig. 6B), and 10 weeks (Fig. 6C) after initial head impact. Although almost all experimental eyes lost RGCs compared with control eyes, only eyes with more significant RGC loss also showed a decrease in OKR score.
Figure 6.
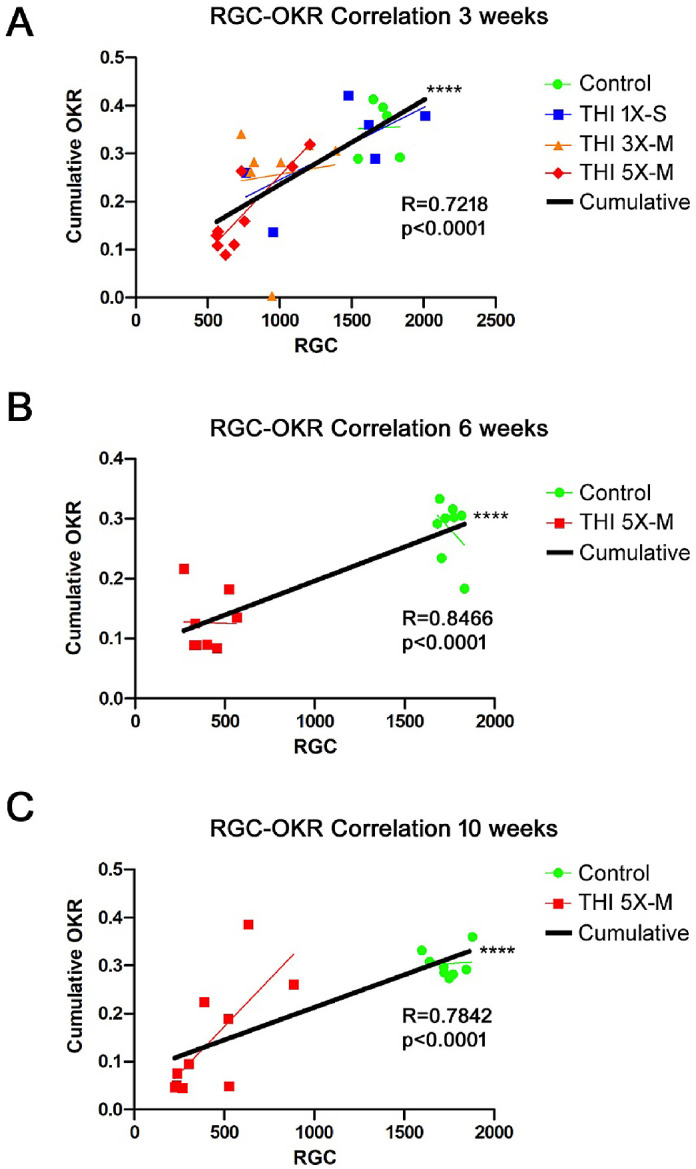
OKR scores correlate with RGC loss. (A) Cumulative OKR scores (total score added from each measured time point) from each mouse in the control, 5×-M, 3×-M, and 1×-M groups over 3 weeks were plotted against the number of surviving RGCs present on day 21. Results show a significant (****P < 0.0001) positive correlation between RGC number and OKR score. (B) Cumulative OKR scores for control and 5×-M mice over 6 weeks plotted against RGC numbers on day 42 showed a significant (****P < 0.0001) positive correlation between RGC number and OKR score. (C) Cumulative OKR scores for control and 5×-M impacted mice over 10 weeks plotted against RGC numbers on day 70 showed a significant (****P < 0.0001) positive correlation between RGC number and OKR score.
Discussion
Current results demonstrate that traumatic closed head trauma with repetitive mild impacts induces significant loss of vision and RGCs in mice 10 weeks after initial head impact, similar to a prior report,21 and they show that these changes develop by 3 weeks after initial head impact and reach peak damage by week 6. That is, significant RGC and vision loss occurred in mice that received five mild impacts by 6 weeks after initial impact, and, importantly, the severity of RGC loss was similar to that found at 10 weeks, suggesting that this structural change is complete by at least 6 weeks. Interestingly, 3 weeks after the initial impact, the 5×-M mice also showed a significant loss of RGCs and vision, although less severe than at 6 weeks. These findings are important for future design of treatment studies of potential drugs to evaluate best outcomes, as shorter duration treatment may be useful in this model. Our findings indicate that three mild impacts also induced significant RGC and vision loss in mice 3 weeks after initial impact, whereas a single mild impact resulted in no change in the RGCs or OKR scores. Our results further demonstrate that RGC and vision loss occurred in both left and right eyes equally and simultaneously, unlike some other models of TON, such as sonication-induced TON, where neuropathic progression in the collateral eye requires time.19 Therefore, in the current study, the average of the left and right eye was considered as a single data point. Although TON can have unilateral or bilateral involvement, an animal model with bilateral TON provides an experimental advantage whereby contralateral eyes can be used for different outcome measures and can be compared with each other.
Loss of visual acuity is a key manifestation seen in indirect TON patients.12 TON is frequently associated with degeneration of RGCs, and retinal morphological studies have shown that RGC soma and dendritic loss can precede axonal loss and optic nerve atrophy in animal models27 of TON and in humans.28 Therefore, we focused on two major outcomes, RGC loss and vision loss (OKR), as potentially clinically relevant measures that recapitulate human TON. Our results show that one severe impact induces similar RGC loss compared with five repetitive mild impacts, although the significant reduction in OKR scores induced by the one severe impact was not as consistently maintained over the course of 10 weeks. This, along with the presence of skull fractures in some mice, suggests that mild repetitive head impacts may provide a more consistent and more humane model for studying TON. Surprisingly, in the 10 weeks following one severe impact or five mild impacts, OKR scores trended toward increased responses at later time points after larger decreases early on, with the differences from control mice gradually attenuating. This was observed despite significant RGC loss. Although the OKR may be used as a potential surrogate marker of RGC function, discrepancies may occur between RGC numbers and OKR scores in some scenarios, and the mechanism behind these differences is not yet fully understood. Studies show that susceptibility of RGCs to various pathological insults or injuries are subtype specific.29 Therefore, it is possible that selective subsets of RGCs in the retina that are responsible for optokinetic responses such as direction-selective ganglion cells, may adapt or may become active after an injury shock period. Alternatively, surviving RGCs may increase in visual responses by sprouting new synaptic connections. It is also important to consider the fact that the OKR or any other visual behavioral evaluation in this model may be influenced by the traumatic brain injury, which itself can evoke visual pathway changes. Chronic neuropathological and neurobehavioral changes and learning and memory deficits accompanied by several persistent histological changes have been shown in repetitive mild traumatic brain injury models.30–36 The current studies do not include brain histopathological readouts, and it is important to note that the terms “mild” and “severe” impacts are used as an indication of the relative difference in the impact parameters, not necessarily the degree of brain injury. Although beyond the scope of the current studies, future investigations of brain pathology and its potential mechanisms resulting in or influencing traumatic optic neuropathy are warranted.
Characterization and utilization of this animal model may serve as a critical approach to study pathophysiological and molecular mechanisms in TON. Despite prior studies, the pathophysiology of TON is not well understood, in part due to a lack of animal models that approximate key clinical features of TON. Indirect traumatic optic nerve injury (ITON) is the most common form of TON and refers to optic nerve injuries that result from impact remote to the optic nerve, whereas direct optic nerve injury (DTON) typically occurs from injuries of the optic nerve by penetrating trauma.37 An important mechanism of damage in ITON during closed head trauma is suggested to be due to compression forces that are transmitted to the orbital apex and optic canal, causing contusion of the nerve and resulting in swelling, ischemia, necrosis, and intraneural edema.38–40 Because blunt closed head trauma is a major etiology of TON, clinically relevant animal models focused on ITON are essential to overcome limitations of the DTON injury models that are more widely used, such as optic nerve crush. The current model reproduces the clinical scenario of blunt head trauma without direct ocular/orbital trauma leading to optic neuropathy, and our results reconfirm those found in a prior study of mild repetitive THIs.21 Importantly, results further demonstrate that functional and structural changes of TON occur short term, within 3 weeks, after repetitive injury. Findings that RGC loss is not region specific but instead is spread across the entire retina, with equivalent damage in each eye, offer potential advantages over other models of TON where region-specific RGC loss discrepancies have been observed.11,19 Further investigations and comparisons of this model to other current traumatic optic neuropathy models and their mechanisms,41–47 not limited to pathophysiology of axonal degeneration, cytokine/chemokine profiles, alterations in astrocytes/microglia, molecular pathways initiated by the injury events including oxidative stress, and potential therapeutic interventions to prevent the progression of TON, are warranted. We intend to explore these mechanisms using the current model of TON in future studies.
Overall, our results suggest that mild repetitive head impacts may serve as an important model of TON by simulating mechanisms of damage and the pathological progression of clinical TON. Ease of implementation and reproducibility, along with a relatively short-term experimental duration after repetitive impacts to induce significant neuropathological outcomes, support this model as an innovative platform to further examine mechanisms of RGC damage and to evaluate therapeutic targets for neuroprotection following TON.
Acknowledgments
Supported provided by grants from the National Institutes of Health (EY019014 and EY030163), by Research to Prevent Blindness and the F. M. Kirby Foundation, and by a Robert Wood Johnson Harold Amos Faculty Development Award.
Disclosure: R.S. Khan, None; A.G. Ross, None; P. Aravand, None; K. Dine, None; E.B. Selzer, None; K.S. Shindler, None
References
- 1. Kumaran AM, Sundar G, Chye LT. Traumatic optic neuropathy: a review. Craniomaxillofac Trauma Reconstr. 2015; 8(1): 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Carta A, Ferrigno L, Salvo M, et al.. Visual prognosis after indirect traumatic optic neuropathy. J Neurol Neurosurg Psychiatry . 2003; 74(2): 246–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee KF, Muhd Nor NI, Yaakub A, Wan Hitam WH. Traumatic optic neuropathy: a review of 24 patients. Int J Ophthalmol . 2010; 3(2): 175–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Atkins EJ, Newman NJ, Biousse V. Post-traumatic visual loss. Rev Neurol Dis. 2008; 5(2): 73–81. [PMC free article] [PubMed] [Google Scholar]
- 5. Levin LA, Beck RW, Joseph MP, et al.. The treatment of traumatic optic neuropathy: the International Optic Nerve Trauma Study. Ophthalmology . 1999; 106(7): 1268–1277. [DOI] [PubMed] [Google Scholar]
- 6. Pirouzmand F. Epidemiological trends of traumatic optic nerve injuries in the largest Canadian adult trauma center. J Craniofac Surg. 2012; 23(2): 516–520. [DOI] [PubMed] [Google Scholar]
- 7. Lee V, Ford RL, Xing W, Bunce C, Foot B. Surveillance of traumatic optic neuropathy in the UK. Eye (Lond). 2010; 24(2): 240–250. [DOI] [PubMed] [Google Scholar]
- 8. Bricker-Anthony C, Hines-Beard J, Rex TS. Molecular changes and vision loss in a mouse model of closed-globe blast trauma. Invest Ophthalmol Vis Sci . 2014; 55(8): 4853–4862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wang J, Hamm RJ, Povlishock JT. Traumatic axonal injury in the optic nerve: evidence for axonal swelling, disconnection, dieback, and reorganization. J Neurotrauma . 2011; 28(7): 1185–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maxwell WL, McCreath BJ, Graham DI, Gennarelli TA. Cytochemical evidence for redistribution of membrane pump calcium-ATPase and ecto-Ca-ATPase activity, and calcium influx in myelinated nerve fibres of the optic nerve after stretch injury. J Neurocytol. 1995; 24(12): 925–942. [DOI] [PubMed] [Google Scholar]
- 11. Thomas CN, Thompson AM, Ahmed Z, Blanch RJ. Retinal ganglion cells die by necroptotic mechanisms in a site-specific manner in a rat blunt ocular injury model. Cells . 2019; 8(12): E1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu-Wai-Man P, Griffiths PG. Steroids for traumatic optic neuropathy. Cochrane Database Syst Rev . 2011; 19(6): CD006032. [DOI] [PubMed] [Google Scholar]
- 13. Edwards P, Arango M, Balica L, et al.. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet . 2005; 365(9475): 1957–1959. [DOI] [PubMed] [Google Scholar]
- 14. Kenneth S. Treatment of traumatic optic neuropathy with high-dose corticosteroid. J Neuro-Ophthalmol . 2006; 26(1): 65–67. [DOI] [PubMed] [Google Scholar]
- 15. Templeton JP, Geisert EE. A practical approach to optic nerve crush in the mouse. Mol Vis . 2012; 18: 2147–2152. [PMC free article] [PubMed] [Google Scholar]
- 16. Zuo L, Khan RS, Lee V, Dine K, Wu W, Shindler KS. SIRT1 promotes RGC survival and delays loss of function following optic nerve crush. Invest Ophthalmol Vis Sci. 2013; 54(7): 5097–5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Levkovitch-Verbin H. Animal models of optic nerve diseases. Eye (Lond). 2004; 18(11): 1066–1074. [DOI] [PubMed] [Google Scholar]
- 18. Hines-Beard J, Marchetta J, Gordon S, Chaum E, Geisert EE, Rex TS. A mouse model of ocular blast injury that induces closed globe anterior and posterior pole damage. Exp Eye Res. 2012; 99: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tao W, Dvoriantchikova G, Tse BC, et al.. A novel mouse model of traumatic optic neuropathy using external ultrasound energy to achieve focal. Indirect Optic Nerve Injury Sci Rep. 2017; 7(1): 11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ibrahim AS, Elmasry K, Wan M, et al.. A controlled impact of optic nerve as a new model of traumatic optic neuropathy in mouse. Invest Ophthalmol Vis Sci . 2018; 59(13): 5548–5557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tzekov R, Quezada A, Gautier M, et al.. Repetitive mild traumatic brain injury causes optic nerve and retinal damage in a mouse model. J Neuropathol Exp Neurol . 2014; 73(4): 345–361. [DOI] [PubMed] [Google Scholar]
- 22. Prusky GT, Alam NM, Beekman S, Douglas RM. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest Ophthalmol Vis Sci . 2004; 45(12): 4611–4616. [DOI] [PubMed] [Google Scholar]
- 23. Quinn T, Dutt M, Shindler KS. Optic neuritis and retinal ganglion cell loss in a chronic murine model of multiple sclerosis. Front Neurol. 2011; 2: 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Khan RS, Dine K, Bauman B, et al.. Intranasal delivery of a novel amnion cell secretome prevents neuronal damage and preserves function in a mouse multiple sclerosis model. Sci Rep. 2017; 7: 41768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Khan RS, Baumann B, Dine K, et al.. Dexras1 deletion and iron chelation promote neuroprotection in experimental optic neuritis. Sci Rep. 2019; 9(1): 11664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sappington RM, Pearce DA, Calkins DJ. Optic nerve degeneration in a murine model of juvenile ceroid lipofuscinosis. Invest Ophthalmol Vis Sci. 2003; 44(9): 3725–3731. [DOI] [PubMed] [Google Scholar]
- 27. Liu Y, McDowell CM, Zhang Z, Tebow HE, Wordinger RJ, Clark AF. Monitoring retinal morphologic and functional changes in mice following optic nerve crush. Invest Ophthalmol Vis Sci. 2014; 55(6): 3766–3774. [DOI] [PubMed] [Google Scholar]
- 28. Kanamori A, Nakamura M, Yamada Y, Negi A. Longitudinal study of retinal nerve fiber layer thickness and ganglion cell complex in traumatic optic neuropathy. Arch Ophthalmol. 2012; 130(8): 1067–1069. [DOI] [PubMed] [Google Scholar]
- 29. Yang N, Young BK, Wang P, Tian N. The susceptibility of retinal ganglion cells to optic nerve injury is type specific. Cells. 2020; 9(3): E677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mouzon BC, Bachmeier C, Ferro A, et al.. Chronic neuropathological and neurobehavioral changes in a repetitive mild traumatic brain injury model. Ann Neurol . 2014; 75(2): 241–254. [DOI] [PubMed] [Google Scholar]
- 31. Ojo JO, Mouzon B, Greenberg MB, Bachmeier C, Mullan M, Crawford F. Repetitive mild traumatic brain injury augments tau pathology and glial activation in aged hTau mice. J Neuropathol Exp Neurol . 2013; 72(2): 137–51. [DOI] [PubMed] [Google Scholar]
- 32. Mouzon B, Chaytow H, Crynen G, et al.. Repetitive mild traumatic brain injury in a mouse model produces learning and memory deficits accompanied by histological changes. J Neurotrauma. 2012; 29(18): 2761–2773. [DOI] [PubMed] [Google Scholar]
- 33. McAteer KM, Corrigan F, Thornton E, et al.. Short and long term behavioral and pathological changes in a novel rodent model of repetitive mild traumatic brain injury. PLoS One. 2016; 11(8): e0160220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bolton Hall AN, Joseph B, Brelsfoard JM, Saatman KE. Repeated closed head injury in mice results in sustained motor and memory deficits and chronic cellular changes. PLoS One. 2016; 11(7): e0159442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Luo J, Nguyen A, Villeda S, et al.. Long-term cognitive impairments and pathological alterations in a mouse model of repetitive mild traumatic brain injury. Front Neurol. 2014; 5: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mannix R, Meehan WP, Mandeville J, et al.. Clinical correlates in an experimental model of repetitive mild brain injury. Ann Neurol. 2013; 74(1): 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Levin LA. Neuro-ophthalmologic diagnosis and therapy of central nervous system trauma. Ophthalmol Clin North Am . 2004; 17(3): 455–464. [DOI] [PubMed] [Google Scholar]
- 38. Anderson RL, Panje WR, Gross CE. Optic nerve blindness following blunt forehead trauma. Ophthalmology. 1982; 89(5): 445–455. [DOI] [PubMed] [Google Scholar]
- 39. Walsh FB, Hoyt WF. Clinical Neuro-Ophthalmology. 3rd ed. Baltimore, MD: Williams & Wilkins; 1969;2380. [Google Scholar]
- 40. Crompton MR. Visual lesions in closed head injury. Brain. 1970; 93(4): 785–792. [DOI] [PubMed] [Google Scholar]
- 41. Bernardo-Colon A, Vest V, Cooper ML, et al.. Progression and pathology of traumatic optic neuropathy from repeated primary blast exposure. Front Neurosci. 2019; 13: 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Harper MM, Woll AW, Evans LP, et al.. Blast preconditioning protects retinal ganglion cells and reveals targets for prevention of neurodegeneration following blast-mediated traumatic brain injury. Invest Ophthalmol Vis Sci. 2019; 60(13): 4159–4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vest V, Bernardo-Colón A, Watkins D, Kim B, Rex TS. Rapid repeat exposure to sub-threshold trauma causes synergistic axonal damage and functional deficits in the visual pathway in a mouse model. J. Neurotrauma. 2019; 36(10): 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bernardo-Colón A, Vest V, Clark A, et al.. Antioxidants prevent inflammation and preserve the optic projection and visual function in experimental neurotrauma. Cell Death Dis. 2018; 9(11): 1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cansler SM, Evanson NK. Connecting endoplasmic reticulum and oxidative stress to retinal degeneration, TBI and traumatic optic neuropathy. J Neurosci Res. 2020; 98(3): 571–574. [DOI] [PubMed] [Google Scholar]
- 46. Jha KA, Pentecost M, Lenin R, et al.. TSG-6 in conditioned media from adipose mesenchymal stem cells protects against visual deficits in mild traumatic brain injury model through neurovascular modulation. Stem Cell Res Ther. 2019; 10(1): 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guley NM, Del Mar NA, Ragsdale T, et al.. Amelioration of visual deficits and visual system pathology after mild TBI with the cannabinoid type-2 receptor inverse agonist SMM-189. Exp Eye Res. 2019; 182: 109–124. [DOI] [PMC free article] [PubMed] [Google Scholar]