

Summary:
Cell fate conversion generally requires reprogramming effectors to both introduce fate programs of the target cell type and erase the identity of starting cell population. Here, we reveal insights into the activity of microRNAs, miR-9/9* and miR-124 (miR-9/9*-124) as reprogramming agents that orchestrate direct conversion of human fibroblasts into motor neurons, by first eradicating fibroblast identity and promoting uniform transition to a neuronal state in sequence. We identify KLF-family transcription factors as direct target genes for miR-9/9*-124 and show their repression is critical for erasing fibroblast fate. Subsequent gain of neuronal identity requires upregulation of a small nuclear RNA, RN7SK, which induces accessibilities of chromatin regions and neuronal gene activation to push cells to a neuronal state. Our study defines deterministic components in the microRNA-mediated reprogramming cascade.
Keywords: neuronal reprogramming, epigenetics, microRNA, single-cell RNA-sequencing, cell fate, direct conversion, non-coding RNA, chromatin regulation
Graphical Abstract
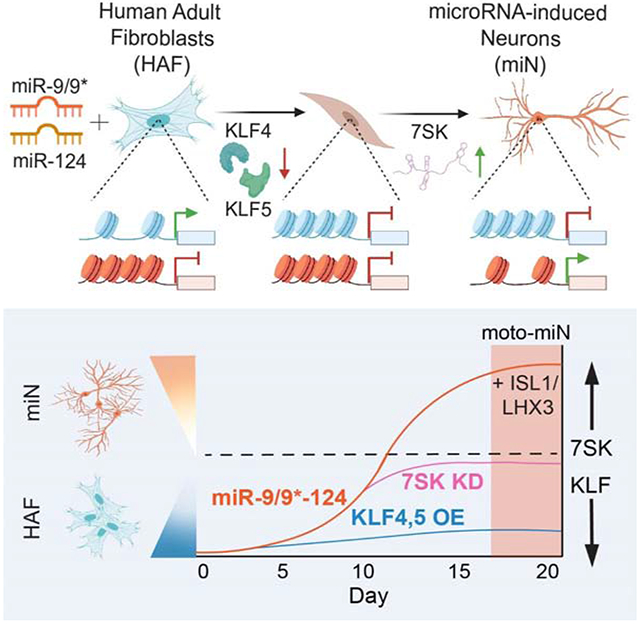
Introduction:
Direct neuronal reprogramming bypasses pluripotent and multipotent stem cell stages to drive somatic cells into a post-mitotic state and directly evoke neuronal identity (Mertens et al., 2015). Unlike reprogramming through induced pluripotency, which reverts cellular age to a fetal stage and therefore erases age- or pathology-dependent epigenetic information (Horvath, 2013; Lapasset et al., 2011; Miller et al., 2013; Patterson et al., 2012), direct conversion propagates age-associated signatures in reprogrammed neurons (Huh et al., 2016; Mertens et al., 2015) offering an advantage for modeling cellular processes that occur with aging (Liu et al., 2016; Victor et al., 2018). Understanding the processes underlying direct lineage conversion of human somatic cells can provide insights into genetic and epigenetic interactions that drive pathology of late-onset neurodegenerative disorders. As such, delineating cellular dynamics of neuronal conversion provides critical information about disease-related phenotypes that occur as a result of cells adopting the neuronal identity. Single-cell transcriptome analysis of mouse embryonic fibroblasts undergoing transcription factor (TF)-mediated direct reprogramming demonstrated the activity of TFs in activating neuronal target genes with competing fate programs during the early stages of reprogramming (Biddy et al., 2018; Treutlein et al., 2016). Yet, such information is not available for neuronal conversion of human somatic cells. Brain-enriched microRNAs (miRNAs), miR-9/9* and miR-124 (miR-9/9*-124), have been shown to be potent reprogramming effectors that induce a neuronal state permissive to subtype-defining TFs to generate subtypes of neurons from human fibroblasts (Abernathy et al., 2017; Yoo et al., 2011). In the current study, we sought to understand cellular dynamics underpinning neuronal reprogramming of human adult fibroblasts and identify the process by which microRNAs resolve conflicting fates between fibroblasts and neurons.
Results:
Neuronal conversion of human adult fibroblasts occurs through distinct sequential states
To assess cellular trajectory as miRNAs instruct the fate switch, we employed the reprogramming paradigm based on miR-9/9*-124 with ISL1 and LHX3 (ISL1/LHX3) that guide the miRNA-mediated neuronal conversion (miN) to motor neurons (moto-miNs) (Abernathy et al., 2017) and performed single-cell RNA sequencing (scRNA-seq) to dissect transcriptomic changes in individual cells at various time points of conversion (day 0, 5, 10, 15, and 20) (Figure 1A). Of the 15,238 cells analyzed, we found reprogramming cells to be segregated by three defined clusters: cluster 1 (D0, starting human adult fibroblasts (HAFs) at day 0), cluster 2 (D5&10), and cluster 3 (D15&20) as depicted by uniform manifold approximation and projection (UMAP) analysis (Becht et al., 2018) (Figure 1B). Using quadratic programming to measure lineage conversion (Treutlein et al., 2016), comparing scRNA-seq datasets from all time points to bulk transcriptome data from fully reprogrammed human motor neurons (Abernathy et al., 2017) showed the highest neuronal identity in cluster 3 (Figure 1C). Notably, cells in cluster 2 as an intermediate stage between clusters 1 and 3 were characterized by downregulation of fibroblast-enriched genes (e.g. S100A4, ITGB1, and TNFRSF11B) whereas the expression of neuronal genes (e.g. MAP2, SNAP25, and BEX2) became apparent in cluster 3 (Figure 1D). Similarly, GSEA analysis showed that the top GO terms in cluster one compared to cluster 2 were enriched for fibroblast processes (Figure S1A, Table S2), whereas the terms in cluster 3 as compared to cluster 2 are enriched for neuronal function (Figure S1B, Table S2). Together, these results suggest sequential steps of reprogramming characterized by fibroblast fate erasure followed by the appearance of neuronal identity. To globally and quantitatively determine cell lineage dynamics, we applied RNA velocity analysis, which uses the amounts of unspliced and spliced RNA to measure the rate and directionality of transcriptomic changes during biological processes (La Manno et al., 2018). Interestingly, with projection of the velocity estimates, we did not detect continuous changes between three cluster groups, but instead found quantile clustering (Figure 1E). Pseudotime ordering by principle curve analysis (La Manno et al., 2018) also showed clear segregation between clusters, corroborating the distinct states at defined time points during reprogramming (Figure 1F). Notably, we did not find evidence of an alternative fate as cells reached cluster 3, but rather cells were uniformly enriched for neuronal gene expression (Figure 1D). Interrogation of cluster 3 identified upregulation of the top 5% longest genes in the genome (Figure 1G) which continued to be enriched by day 20. Long gene expression (LGE) has been shown to distinguish neuronal from non-neuronal cells (Gabel et al., 2015; McCoy and Fire, 2020; McCoy et al., 2018). Accordingly, LGE coincided with increasing enrichment of neuronal markers (e.g. MAP2 and SNAP25) (Figure 1G) demonstrating that cells in cluster 3 exhibit transcriptomic signatures of neurons. Also, the activity of ISL1/LHX3 used with miRNAs to generate motor neurons only became apparent in cluster 3 (marked, for instance, by LMO1, RSPO2, PACRG, PPP2R2B, and SLIT2, all of which are known targets of ISL1/LHX3) (Abernathy et al., 2017; Mazzoni et al., 2013) and continued to be enriched with LGE (Figure 1H). Altogether, our scRNA-seq analyses revealed that neuronal conversion by miR-9/9*-124 and ISL1/LHX3 occurs via an initial step of fate erasure followed by the acquisition of neuronal identity.
Figure 1. Distinct reprogramming states of human fibroblasts to neurons by miR-9/9*-124 and ISL1/LHX3.

(A) Experimental scheme for studying cellular dynamics of neuronal reprogramming by miR-9/9*-124 and ISL1/LHX3. Human adult fibroblasts (HAFs) undergoing neuronal reprogramming were immunostained with TUBB3 to show morphological changes from day 0, 5, 10, 15, and 20. Scale bars, 20 μm. Right panel, top: Moto-miNs immunostained with motor neuron marker MNX1 and morphology marker TUBB3 at day 22. Scale bar, 23 μm. Right panel, bottom: Moto-miNs immunostained with neuronal marker MAP2 and TUBB3 at day 30. Scale bar, 45 μm. (B) UMAP projection of cells colored by time points (PID = post-induction day for microRNAs). (C) UMAP projection of cells colored by quadratic programing (QP) score. (D) UMAP projection of cells colored by expression of fibroblast-enriched genes (S100A4, ITGB1, TNFRSF11B) and neuronal genes (MAP2, BEX2, SNAP25). (E) RNA velocity analysis applied to the UMAP projection of cells, colored by sub-clusters. (F) Biplot showing the correlation between cell orders based on principle curve analysis (pseudotime) and fraction neuronal identity (QP). Cells are colored by time points. Pearson correlation coefficient = 0.94. (G) UMAP projection of cells colored by the median expression for the top 5% longest genes (LGE = long gene expression), and LGE violin plot of cells grouped by time point during reprogramming. (H) Heatmap showing expression (scaled 0-1 for each gene) of mature motor neuron markers and LGE scores (scaled 0-1) between PID 15 and 20. Cells are ordered from left to right by increasing QP scores. Aligned below are two plots also organized left to right by increasing QP scores: a Loess regression of neuronal gene expression, including total LGE, and a scaled density plot of QP scores grouped by time points.
Transition from cluster 2 to cluster 3 represents gradual acquisition of neuronal fate
Given that cluster 3 is characterized by the activation of neuronal program, we sought to increase the temporal resolution of our analyses as cells progress from cluster 2 to 3. We performed additional scRNA-seq analyses at days 10, 12.5, and 15 with miR-9/9*- 124+ISL1/LHX3. A total of 16,628 cells formed clusters corresponding to each time point. We found that day 12.5 cells segregated between days 10 and 15 (Figure S1C), suggesting an intermediate stage between two time points and a continuous trajectory to neuronal fate. RNA velocity analysis showed the directionality of cell velocities from each cluster towards the next clusters (Figure S1D). To validate if day 12.5 cluster is in the midst of the neuronal transition, we projected the day 10, 12.5, and 15 scRNA-seq datasets from Figure S1 onto the moto-miN scRNA-seq datasets from Figure 1 (day 0, 5, 10, 15, and 20) on UMAP and performed integrated cell state analysis, which uses anchors as pairwise correspondences between individual cells within the same biological state in each dataset to enable multiple data to be jointly analyzed (Stuart et al., 2019). The cell state analysis also showed day 12.5 cells localized between days 10 and 15 within the array of cell states from day 0 to 20 (Figure S1E). We examined a list of panneuronal marker genes (e.g. SNAP25, STMN2, NEFL, CALY, SERPINF1, EEF1A2, and KCNIP2) including long genes (e.g. DCLK2 and KIF1A) and motor neuron marker genes (e.g. SLIT2, SLIT3, and LMO1). As demonstrated by violin plots (Figure S1F), cells corresponding to each time point showed the pattern of increasing gene expression over time. Together, our results reveal that the initiation stage of neuronal conversion by miR-9/9*-124-ISL1/LHX3 is characterized by the downregulation of fibroblast identity genes followed by the gradual and synchronous transition towards neuronal fate revealed by day 12.5 cluster as a bridge of cellular transition.
miR-9/9*-124 drive fate erasure and sequential neuronal program
Despite the function of ISL1/LHX3 in defining the motor neuron identity (Figure 1) (Abernathy et al., 2017), ISL1/LHX3 without miR-9/9*-124 (but using the non-specific control miRNA, miR-NS) were insufficient to drive neuronal conversion in HAFs (Figure S2A). We then reasoned that miR-9/9*-124 are the primary reprogramming effector orchestrating the fate erasure and emergence of the neuronal state. To further stratify the effect of miR-9/9*-124 on the dynamics of reprogramming cells, we carried out time-course scRNA-seq analyses when HAFs were converted with miR-9/9*-124 only at days 0, 5, 10, 15, and 20. From 20,838 cells analyzed, we found that miR-9/9*-124 alone still led to temporal segregation into three distinct clusters: cluster 1 (D0, starting HAFs at day 0), cluster 2 (D5), and cluster 3 (D10, 15, and 20) as shown in the UMAP plot (Figure 2A, left). Interestingly, in the reprogramming using miR-9/9*-124 alone, activation of neuronal fate was slightly accelerated where D10 belonged to cluster 3 instead of cluster 2 as in Figure 1. We suspect that this shift was due to a higher titer of miR-9/9*-124-expressing virus in the absence of the additional ISL1/LHX3-expressing viruses. In RNA velocity visualization, each cluster showed independent velocity directionality that remained within each cluster while cluster 3 showed directionality of continuous transcriptomic changes from day 10 to 20 (Figure 2A, middle). To gain insights into overall changes in cell states between reprogramming conditions with miR-9/9*-124+ISL1/LHX3 (from Figure 1) and miR-9/9*-124 only, we performed integrated cell state analysis between the two scRNA-seq datasets. Based on the batch-corrected expression analysis, cells in each cluster shared the overlapping states in the integrated dataset (Figure 2A, right) demonstrating miR-9/9*-124 as a driver of the conversion trajectory. miRNA-induced cluster 2 and 3 were characterized by the downregulation of fibroblast genes (e.g. COL4A1, SERINC2, RAB34, PLP2, ITGB1, and CTGF) and activation of neuronal genes (e.g. BEX2, MAP2, NEFL, SNAP25, SERPINI1, and KIF1A), respectively (Figure 2B). It is noteworthy that while moto-miN and miN conditions share the overall clustering patterns, miNs do not express markers of motor neurons supporting the subtype-defining role of ISL1/LHX3 in the neuronal state of miN (Figure S2B; Abernathy et al., 2017). Also, as alternative fates have been previously identified in neuronal conversion methods based on ASCL1 (Herdy et al., 2019; Treutlein et al., 2016), we assayed miNs and moto-miNs for myocyte and stem cell markers. Interestingly, we did not detect myocyte (MYH3) or stem cell (SOX2) markers in moto-miNs (Fig. S2C-D). Similarly, we detected no induction of myocyte cell clusters and markers, including ACTA1, MYOG, MYOD1, and MYO18B in miNs by scRNA-seq, or the upstream ASCL1, likely accounting for the absence of the alternative myogenic induction (Figure S2E).
Figure 2. miR-9/9*-124 alone are sufficient to induce homogenous fibroblast identity erasure and neuronal fate activation.

(A) Examples of conversion of HAFs cells with miR-9/9*-124-only. Left: A representative picture showing the morphologies of converted cells immunostained for TUBB3 (in gray). Scale bar, 20 μm. Top: Zoomed in pictures of converted cells stained for TUBB3 (left), MAP2 (middle), and merged (right). Scale bar, 50 μm. Bottom, left: Visualization of scRNA-seq data. UMAP projection of cells colored by time points. Bottom, center: RNA velocity analysis applied to the UMAP projection colored by sub-clusters. Bottom, right: Integrated cell state analysis plots of full time-course miN cell populations onto the full time-course moto-miN UMAP. (B) UMAP projection of cells colored by expression of fibroblast-enriched genes (COL4A1, SERINC2, RAB34, PLP2, ITGB1, CTGF) and neuronal genes (BEX2, MAP2, NEFL, SNAP25, SERPINI1, KIF1A). (C) Scatter plots showing the correlation between cell orders based on Monocle (pseudotime) and relative gene expression analyses. Cells are colored by time points. Top: Expression of fibroblast-enriched gene examples (ITGB1, PLP2, RAB34, CTGF) over pseudotime. Bottom: Expression of neuronal gene examples (SNAP25, BEX2, SERPINI1, KIF1A). D) Top Gene Ontology (GO) terms for biological processes of DEGs enriched in Day 0 compared to Day 20 (top, salmon) or Day 20 compared to Day 0 (bottom, purple).
To gain global insights into cell clustering with miR-9/9*-124 only, we carried out pseudotime analysis by Monocle (Qiu et al., 2017) in all time points. Our results demonstrate that miR-9/9*-124 start fate conversion by fibroblast fate erasure in cluster 2 cells as exemplified by the downregulated pseudotime projection of fibroblast genes (Figure 2C, top). This initial step was sequentially coordinated with upregulated neuronal genes as shown by the trajectory of neuronal genes in cluster 3 cells (Figure 2C, bottom). Gene-set enrichment analysis (GSEA) with differentially expressed genes (DEGs) enriched at day 0 corresponded to non-neuronal terms including collagen fibril organization and skin development (Figure 2D, top panel; Table S2). In contrast, genes enriched in day 20 corresponded to neuronal terms including synaptic signaling, nervous system development, and neuron differentiation (Figure 2D, bottom panel; Table S2). Further, the process of erasing the fibroblast identity was reflected by cells adopting the post-mitotic state by reprogramming day 5 as demonstrated by EdU assay, FUCCI-cell cycle reporter, and real time tracking of cell numbers (Figure S3 and Movie S1). In sum, our results demonstrate that miR-9/9*-124 were sufficient to drive the conversion process through distinct cell clusters, separating the intermediate cluster from fibroblast population when cells start losing fibroblast identity in a post-mitotic state, then proceeding onto a cluster in which neuronal genes are activated.
miR-9/9*-124 induce chromatin closure for fibroblast-enriched genes distinctively from ISL/LHX3 or MYT1L
To further stratify the contribution of miR-9/9*-124 and ISL1/LHX3 to the erasure of fibroblast state when separately expressed in HAFs, we compared the abilities of ISL1/LHX3 and miR-9/9*-124 to downregulate the expression of fibroblast marker genes. We found that ISL1/LHX3 alone were insufficient to induce neuronal morphologies (Figure 3A), and eliminate S100A4 (a fibroblast marker also known as FSP1) and induce a neuronal marker MAP2 by day 22 in contrast to cells expressing miR-9/9*-124 only (Figure 3B-C). Transcriptionally as measured by qPCR, miR-9/9*-124 alone repressed fibroblast markers S100A4 and VIM gene expression within 10 days of expression (Figure 3D), supporting the role of miR-9/9*-124 in deactivating fibroblast identity.
Figure 3. miR-9/9*-124 alone induce early fibroblast gene program erasure.

(A) Phase contrast images of HAFs expressing either miR-9/9*-124 or ISL1/LHX3 alone at PID 7. Inset shows HAF controls. Scale bars, 130 μm. (B) Representative images of HAFs expressing ISL1/LHX3 or miR-9/9*-124 alone at PID 22. Top: Cells immunostained for fibroblast identity marker FSP1 (green) and morphology marker TUBB3 (red) over DAPI (blue). Bottom: Cells immunostained for neuronal marker MAP2 (green) and TUBB3 (red) over DAPI (blue). Both: Insets shows control HAFs. Scale bars, 45 μm. (C) Quantification of FSP1- and MAP2-positive cells over the total number of cells (DAPI) (*p < 0.05, ** p <0.01, *** p < 0.001). D) qPCR results for fibroblast gene expression (S100A4 and VIM) for HAF + miR-9/9*-124-only and control HAFs at PID 13 (* p < 0.05, ** p <0.01). Error bars represent s.e.m. (E) Heatmaps of ATAC-seq signal intensity for each sample mapped to differentially accessible regions (DAR) closed between HAFs and miR-9/9*-124 conditions (n = 20,869). Signals mapped within 2 kb of DAR peak center. (F) Heatmaps for gene expression levels for downregulated DEGs that positively-correlate with closed DAR signal intensity in promoter (± 2 kb transcriptional start site) or gene body regions. (G) Top GO terms for biological processes enriched in downregulated DEGs associated with closed DARs.
To probe the differential activity of miR-9/9*-124 and ISL1/LHX3 in fibroblast identity erasure at the chromatin level, we performed omni-ATAC-seq (Corces et al., 2017) and RNA-seq on HAFs expressing miR-9/9*-124 or ISL1/LHX3 (I1L3) at day 7 into conversion. Here, we also assayed for the pro-neuronal MYT1L that has been shown to safeguard neuronal identity through repression of various somatic lineages (Mall et al., 2017) and used in cortical and striatal neuron reprogramming with miRNAs (Victor et al., 2014, Richner et al., 2015, Victor et al., 2018). ATAC- and RNA-seq read coverage correlation analyses showed that miR-9/9*-124-only cells shared the least signal similarity with HAFs compared to I1L3- and MYT1L-only cells (Figure S4A-B). miR-9/9*-124 expression instructed the closure of approximately 21,000 differentially accessible regions (DAR) compared to HAFs (Figure 3E). Examining promoter and intragenic chromatin accessibility correlated to gene expression, we identified about 200 genes that miR-9/9*-124 differentially closed and downregulated (Log2FC ≤ −1, adj. p-value < 0.01) including canonical fibroblast fate markers, S100A4, FBLN1, MFAP5, and COL1A1 (Figure 3F; Figure S4C). GO enrichment analysis showed that these genes are enriched for biological processes associated with fibroblast function, such as extracellular matrix organization and collagen fibril organization (Figure 3G, Table S3). Our results show that miR-9/9*-124 orchestrate fibroblast fate erasure through chromatin closure and gene suppression during the early stages of HAF conversion differently from ISL1/LHX3 or MYT1L.
Identification of direct targets of miR-9/9*-124 by AGO-HITS-CLIP during fate erasure
Next, we investigated how miR-9/9*-124 initiate the loss of fibroblast identity. To reliably identify the cell context-dependent miRNA targets that would be difficult to predict computationally (Pinzón et al., 2017), we performed high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation for Argonaute (AGO-HITS-CLIP) (Moore et al.,2014) to map miR-9/9*-124-target landscape during reprogramming (Figure 4A). Sequencing of transcripts and miRNAs loaded into AGO complexes isolated from cells at day 7 (Figure S4D) showed miR-9 (miR-9-5p), miR-9* (miR-9-3p), miR-124 (miR-124-3p), and miR-124* (miR-124-5p) as mature miRNAs (Figure 4B). Sequences identified by AGO-HITS-CLIP primarily targeted the 3’UTRs and exons of 1,120 gene transcripts containing 2,042 total peaks enriched over the control miRNA (miR-NS) expression (Figure S4E, Table S4). By comparing the identified target genes to downregulated DEG based on RNA-seq analysis at day 7 (HITS-CLIP targets: log2FC < −1, adj. p-value < 0.001; DEG: log2FC < −0.5, adj. p-value < 0.05), we identified 311 target genes that were repressed at the transcript level (Figure 4C), enriched with non-neuronal terms such as actin filament function and skeletal system development (Figure 4D, Table S5). We found that 61% of peaks aligned to 6-7 nucleotide seed regions of miR-9/9*-124 (Figure 4E, Fig. S4F for examples) whereas 21% of these sites contained a non-canonical pairing to nucleotides 3-8 of miRNAs (Figure 4E). Motif analysis also identified miR-124 sites containing a G-bulge between position 5 and 6 nucleotides (Figure 4E) consistent with a previous finding (Chi et al., 2012). These results demonstrate that miR-9/9*-124 obey the targeting activity through seed-match sequences as the primary targeting mode during reprogramming.
Figure 4. Targets of miR-9/9*-124 are enriched in non-neuronal gene networks.

(A) Overview of AGO-HITS-CLIP to identify targets of miR-9/9*-124 during neuronal conversion. (B) Aggregate analysis of miR-9/9*-124 and miR-NS conditions shows that miR-9, miR-9*, miR-124, and miR-124* (red) were enriched in RISC complex immunoprecipitated with AGO antibody relative to total input in contrast to the control miR-NS (green). (C) Heatmap of genes downregulated in miR-9/9*-124 condition over miR-NS at PID 7. Signal intensity is based on normalized CPM values, and data is shown as a z-score normalized log2CPM. (D) Top GO terms corresponding to downregulated miR-9/9*-124-target genes at PID 7. (E) RNAhybrid analysis of miR-9/9*-124 targets detected by AGO-HITS-CLIP (left). Various modes of seed positions on miRNAs were detected to pair with enriched peaks. Canonical seed = seed 1-6 or 2-7 of miRNAs. The miRNA binding motif analysis for miR-9, miR-124 seed-matches, and miR-124 bulge detected by MEME motif analysis. (F) Comparative gene expression analysis (scaled 0-1) of the top fibroblast-enriched network between days 0 and 10 of reprogramming. The fibroblast network is significantly enriched for targets of miR-9, miR-9*, and miR-124 as identified by AGO-HITS-CLIP (red).
Dissolution of fibroblast-enriched gene network in response to miR-9/9*-124
To gain a global view of target genes in relation to fibroblast erasure, we analyzed gene co-expression networks in our single-cell transcriptome data from HAFs and evaluated changes in the gene networks (Song and Zhang, 2015) during reprogramming. We identified a major fibroblast gene network with the majority of genes being downregulated within 10 days into reprogramming by miR-9/9*-124 (Figure 4F, Table S4). The fibroblast gene network was significantly enriched for miR-9/9*-124 targets identified by AGO-HITS-CLIP (Figure 4F, Table S4). Several 3’ UTR targets identified within the fibroblast subnetworks were further validated as direct targets of miR-9/9*-124 by luciferase assays (Figure S4G).
KLF4 and KLF5 are key targets of miR-9/9*-124 for fibroblast fate erasure
From the target genes identified, we sought to define miR-9/9*-124 targets critical for erasing fibroblast fate. Because miR-9/9*-124 expression reduced chromatin accessibility at enhancers of fibroblast genes (Figure 3) (Abernathy et al., 2017), we hypothesized that there would be an enrichment of sequences within chromatin loci closed in response to miR-9/9*-124. Out of 38,882 total closed peaks, 41% (15,953 peaks) contained binding sites of KLF-family transcription factors, KLF4 and KLF5, which corresponded to 726 genes downregulated by day 20 of reprogramming (Abernathy et al., 2017) (Figure 5A, Table S5). When TF-binding motifs were compared among other TFs targeted by miR-9/9*-124, such as TCF and ELK family TFs, KLF4 and KLF5 motifs were most robustly enriched in closed regions by day 10 (Figure 5A, Figure S5A) and overlapped with fibroblast enhancer loci (Figure 5B, Figure S5B). Importantly, we identified KLF4 and KLF5 to be direct targets of miR-9/9*-124 within the fibroblast gene network (Figure 5B-D) among other KLF-family TFs repressed during reprogramming (Figure S5C). Prolonging KLF4 or KLF5 using cDNAs lacking respective UTRs (thus miRNA-insensitive) inhibited fibroblast fate repression as key fibroblast genes failed to be repressed (Figure 5E) unlike other TFs targeted by miR-9/9*-124 (Figure S5D). Also, the failure to suppress fibroblast genes was not due to cell cycle re-entry (Figure S5E-F).
Figure 5. KLF4 and KLF5 are key miR-9/9*-124 targets in fibroblast.

(A) Heatmaps of KLF4 and KLF5 binding site density within chromatin loci that close between Days 0 and 10 of miR-9/9*-124 expression. Legend depicts representative motifs for KLF4 and KLF5 binding sites within closed loci. (B) The top fibroblast network targeted by miR-9, miR-9*, and miR-124 as identified by AGO-HITS-CLIP (red) contains KLF4 and KLF5 as hubs in the network. (C) Representative IGV snapshots showing peaks enriched with miR-9/9*-124 (red) over miR-NS (green) expression within the KLF4 and KLF5 3’ UTRs. (D) Luciferase assay validation of KLF4 and KLF5 3’UTRs as repressive targets of miR-9/9*-124 in comparison to PGK 3’UTR as a negative control. (E) qPCR quantification of fibroblast genes (HSPB7, MFAP5, PAMR1, S100A3, S100A4) with LacZ (control), microRNA-resistant-KLF4 or -KLF5 overexpression with miR-9/9*-124.
To test the effect of prolonging KLF expression in response to miR-9/9*-124, we established HAFs that stably overexpressed KLF4, KLF5, KLF4 and KLF5, or luciferase (LUC) as a control (Figure 6A, Figure S6A). Forcing KLF expression in the presence of miR-9/9*-124 led to stunted conversion by day 10 in which cells exhibited large rounded morphology in contrast to the narrowed process outgrowth in control cells (Figure S6B). We performed omni-ATAC-seq at day 10 of reprogramming by miR-9/9*-124 with KLF overexpression. Extending KLF4 and KLF5 (in particular, KLF4) expression severely impaired the ability of miR-9/9*-124 to induce the closure of fibroblast-associated chromatin loci (Figure 6B; Figure S6C; log2FC ≤ −1.5, adj. p-value < 0.01) containing fibroblast enhancers (Figure 6C; log2FC ≤ −1.5, adj. p-value < 0.01). The loci that remained accessible by KLF4 were enriched for KLF4 and KLF5 binding sites (Figure S6D; see fibroblast marker examples in Figure 6D) and were associated with non-neuronal functions such as skeletal system development and positive regulation of cell migration (Figure S6E, Table S5). GO enrichment for DEGs (log2FC ≤ −0.5, adj. p-value < 0.01) that normally require KLF repression for promoter closure (Figure S6F; log2FC ≥ 1, adj. p-value < 0.01) showed fibroblast functions such as actin cytoskeleton and extracellular structure organization (Figure 6E, Table S5). As a validation by qPCR, extending KLF4 expression led to the failure of the fibroblast marker S100A4 repression at day 10 of reprogramming, which was also confirmed by immunostaining analysis for S100A4 (FSP1) (Figure 6F-G). Interestingly, we found that MAP2 expression was still upregulated (Figure 6F) and the opening of chromatin loci for neuronal genes in response to miR-9/9*-124 persisted even when KLF-expression was prolonged (Figure S6G). Thus, our results imply that KLF4 and KLF5 control non-neuronal chromatin accessibility while neuronal chromatin activation likely occurs independently.
Figure 6. miR-9/9*-124 repression of KLF4 and KLF5 is required for complete erasure of fibroblast identity.

(A) Experimental conditions for studying the role of KLF4 and KLF5 in miR-9/9*-124 reprogramming. (B) Heatmaps of ATAC-seq signal intensity mapped to DAR closed between HAF and miR-9/9*-124 conditions by PID 10 of reprogramming (n = 9,854). (C) Heatmaps of ATAC-seq signal intensity mapped to DAR closed between HAF and miR-9/9*-124 conditions by day 10 of reprogramming that overlap with fibroblast enhancer loci (n = 469). D) IGV snapshot showing ATAC-seq read coverage (blue) and KLF4/KLF5 transcription factor binding sites (grey) for the fibroblast-associated genes ECM1 and MFAP5. The LUC compared to KLF4 DAR peaks are highlighted in pink. (E) Top GO biological processes associated with downregulated DEGs in response to miR-9/9*-124, containing DARs that fail to close with prolonged expression of KLF4 compared to LUC control expression in day 10 of miRNA expression. (F) qPCR results for fibroblast (S100A4) and neuronal (MAP2) gene expression for LUC or KLF overexpression with miR-9/9*-124 and HAF controls at PID 10 (**p <0.01, *** p < 0.001). Error bars are s.e.m. (G) Representative images of HAFs expressing miR-9/9*-124 with LUC, KLF4, KLF5, or KLF4 and KLF5 at Day 10 immunostained for FSP1 (red) and DAPI (blue). Scale bars, 60 μm.
RN7SK is critical for neuronal fate activation
For the subsequent transition to the neuronal fate, we identified RN7SK (7SK) as the most highly enriched nuclear effector in cluster 3 (Figure 7A, Figure S7A, Table S6), whose upregulation was primarily due to miR-9/9*-124 expression (Figure 7B). Reducing 7SK expression by shRNA (sh7SK, Figure S7B-C) in miNs suppressed both morphological neuronal conversion (Figure 7C; Figure S7D) and transcriptional reprogramming (Figure S7E). 7SK is a brain-enriched small nuclear RNA (Bazi et al., 2018; Briese et al., 2018; Carithers et al., 2015) shown to play a role in DNA-targeting of BAF chromatin remodeling complex (Flynn et al., 2016). As we previously found BAF to be critical for reshaping chromatin accessibilities during neuronal reprogramming (Abernathy et al., 2017) and the molecular identity that determines the specificity of BAF function in neurons is unknown, we reasoned that 7SK may facilitate the opening of chromatin regions encompassing neuronal genes. We carried out omni-ATAC-seq in parallel with RNA-seq to see whether chromatin regions encompassing neuronal genes that become accessible in response to miR-9/9*-124 would be affected upon 7SK knockdown by day 15. RNA-seq and ATAC-seq read coverage correlation analyses indicated that sh7SK-treated cells showed far more similarity in read coverage to microRNA-induced neurons (miNs + shCTRL) than HAFs, suggesting that sh7SK-treated cells at day 15 still represented cells being reprogrammed via fibroblast identity erasure (Figure S7F-G).
Figure 7. 7SK is a critical determinant of neuronal identity during reprogramming.

(A) UMAP projection of cells for RN7SK in the miR-9/9*-124-only scRNA-seq. (B) qPCR analysis of 7SK in HAFs expressing ISL1/LHX3 or miR-9/9*-124 for 12 days normalized to GAPDH expression. Error bars represent s.e.m. (*p < 0.05). (C) Representative images of HAFs expressing miR-9/9*-124 with either shCTRL or sh7SK at Day 20 immunostained for TUBB3 (red) and DAPI (blue). Scale bars, 50 μm. (D) Heatmaps of ATAC-seq signal intensity mapped to DAR (log2FC ≤ −1.5, FDR-adj. p-value < 0.01) closed between shCTRL and sh7SK conditions (n = 2,693). (E) PID 20-enriched neuronal network over PID 20 (top) and overlay of genes differentially downregulated by 7SK identified by RNA-seq (red). Insets show critical neuronal genes as hubs in the network targeted by 7SK. (F) P-values for enrichment of sh7SK-downregulated genes in each subnetwork against the mean log2FC in expression of PID20/PID0 of that subnetwork (Bonferroni-adj. p-values < 0.1; red subnetworks FC > 0.5). (G) Top GO function annotations with differentially accessible sh7SK DEGs. (H) The genomic distribution of significant (log2FC ≥ 1.5, FDR-adj. p-value < 0.01) DAR open in HAFs vs shCTRL (n=48,723). (I) IGV snapshot for ATAC- and RNA-seq signal for the neuronal gene STMN4 (sh7SK TSS-proximal DAR highlighted in pink). (J) IGV snapshot for ATAC- and RNA-seq signal for the fibroblast gene MFAP5 (shCTRL and sh7SK TSS-proximal DAR highlighted in pink). (K) Heatmaps of ATAC-seq signal intensity for miR-9/9*-124 with shCTRL or sh7SK (left) compared to miR-9/9*-124 with shCTRL or shBRG1 (right) overlapping at DAR between shCTRL and sh7SK conditions (n = 2,693). (L) Top GO function annotations for DAR-associated genes that fail to open in shBRG1 and sh7SK conditions (both comparisons: log2FC ≤−1, FDR-adj. p-value < 0.01).
ATAC-seq analyses of cells expressing miR-9/9*-124 with shCTRL or sh7SK for 15 days revealed approximately 5,000 significant DAR, of which about 3,000 failed to open with sh7SK (Figure 7D). Strikingly, the major fraction of DAR (~85%) that failed to open with sh7SK corresponded to differentially downregulated genes that are normally activated in response to miR-9/9*-124 (Figure 7E, Figure S7H). The sh7SK-mediated differentially closed and downregulated genes are highly enriched in the gene networks active in cluster 3 (Figure 7F), and primarily function in neuronal fate activation, exemplified by neuronal terms and a neuronal gene network (Figure 7G, Figure S7I). The networks that require 7SK for chromatin accessibility and transcriptional activation included key neuronal genes such as STMN4, HOOK1, and CNTN1 (Figure 7I, Figure S7J, Table S6). Additionally, sh7SK also decreased LGE (Figure S7K) indicating the loss of neuronal identity. Conversely, sh7SK did not disrupt miR-9/9*-124-mediated chromatin closure of fibroblast genes, indicating that 7SK specifically regulates neuronal program activation, not fibroblast program erasure (Figure 7H). For instance, key fibroblast identity genes, such as MFAP5, VIM, and S100A4, retained chromatin closure and gene deactivation with or without 7SK (Figure 7J, Figure S7L). Similarly, sh7SK-treated cells maintained a post-mitotic state and did not revert to cell division (Figure S7M). Because BAF complex activity was also critical for opening miR-9/9*-124-activated neuronal loci (Abernathy et al., 2017), we compared DARs between BRG1- and 7SK knockdown conditions. Many regions affected by 7SK overlapped with BRG1-affected regions (Figure 7K-L, Table S6) consistent with 7SK function in chromatin remodeling associated with BAF complex. Overall, our results delineate the role of 7SK in promoting neuronal identity through chromatin reconfiguration downstream of miR-9/9*-124 overexpression.
Discussion:
Efficient somatic cell reprogramming requires a fate trajectory reflecting erasure of pre-existing fate and induction of new fate. The current study uncovers the fate erasure of HAFs by miR-9/9*-124 as a prerequisite for subsequent and uniform transition into a target neuronal fate. Previous direct reprogramming studies have focused on the mechanism by which pioneer TF elicits the reprogramming process in mouse fibroblasts, which proceeds through binding to genomic sites in closed chromatin and activating neuronal genes (Wapinski et al., 2013). Here, we demonstrate that miRNAs act through a different reprogramming process in human fibroblasts that relies on post-transcriptional control of fate erasure leading to chromatin reconfiguration, followed by neuronal fate activation. We show that miR-9/9*-124 alone are sufficient to guide HAFs through fate erasure and neuronal gene program activation in sequence, offering insights into miRNA-mediated fate control different from repressing anti-neurogenic genes (Xue et al., 2013) or addition of pro-neuronal TFs (Babos et al., 2019; Mertens et al., 2015; Treutlein et al., 2016). In addition, we found that miR-9/9*-124 did not induce alternative cell lineages, such as myocyte fate (Treutlein et al., 2016) likely due to the absence of ASCL1 in miNs. Our results also indicate that at least in HAFs, the subtype-defining ISL1/LHX3 (Abernathy et al., 2017) or safeguard TF MYT1L (Mall et al., 2017) by themselves did not induce fibroblast erasure through chromatin closure. Therefore, our results position miRNAs as reprogramming effectors that allow the synergism with neural TFs through fate erasure and neuronal induction in sequence.
Our AGO-HITS-CLIP analysis identified miR-9/9*-124 target genes in reprogramming HAFs comprised of a gene network enriched in fibroblasts. Surprisingly, the target TFs KLF4 and KLF5 revealed significant binding motif occupancy at chromatin regions that are closed in response to miR-9/9*-124. Also, prolonged expression of KLF4 and KLF5 stunted the ability of reprogramming cells to fully erase fibroblast fate through chromatin closure highlighting KLF4 and KLF5 as key miR-9/9*-124 targets that maintain non-neuronal chromatin accessibilities. Along this line of evidence, KLF4 and KLF5 repression could be further studied as a method to expedite human fibroblast fate erasure in neuronal reprogramming. Also, miR-124 directly targets and represses PTBP1 during development (Makeyev et al., 2007) and neuronal conversion of fibroblasts (this study). As knocking down PTBP1 has been shown to induce astrocyte-to-neuron conversion (Qian et al., 2020; Zhou et al., 2020), we suspect that miR-9/9*-124 may also act as reprogramming effectors for astrocytes through conserved mechanisms.
We uncovered RN7SK (7SK) as an upregulated nuclear effector necessary for opening and activation of neuronal chromatin and program as cells transition to neuronal state. Counter to the chromatin landscape of KLF-expressing cells which still showed chromatin accessibility of neuronal genes, reprogramming cells lacking 7SK maintained the ability to close and deactivate fibroblast loci, suggesting two separate pathways for fate erasure and neuronal induction by miRNAs. The cascade of miRNAs working through another non-coding RNA to promote neuronal transition was unexpected finding, yet highlights the activity of BAF complexes with 7SK to activate neuronal genes. Interestingly, the effect of sh7SK in reducing LGE and impacting BAF complex-dependent loci is similar to TOP1 inhibition, providing potential insights into a role for 7SK in coordinating the BAF complex, which has been shown to recruit TOP1 to chromatin (Husain et al., 2016) and promote neuronal maturity (Mabb et al., 2014). In this regard, 7SK potentially acts as an aid in the genomic targeting of neuronal BAF complex, possibly with neuron-specific components that are activated in parallel to 7SK, such as BAF45B and BAF45C (Ho and Crabtree, 2010; Yoo and Crabtree, 2009).
Overall, we propose that neuronal reprogramming should be reflected by successful fate erasure and induction of neuronal identity. Our study also indicates that future work implementing directly converted human neurons for disease-modeling may explore KLF repression and LGE as unbiased markers of fate erasure and neuronal activation, respectively. Further developing this alternative paradigm of cell fate reprogramming by miRNAs will be essential to identifying the principles underlying reprogramming, and to understanding how neuronal conversion uniquely propagates age- and pathology-associated signatures.
Limitations of the Study:
The experimental design of the current study mainly focused on the early stages of neuronal conversion whereas modeling of neurodegenerative disease pathology in reprogrammed neurons is typically assayed at later time points (Victor et al., 2018). Further directions stemming from the current study could include the investigation of cell trajectories throughout the disease modeling process. Regarding KLF4 and KLF5 as critical targets for fibroblast identity erasure, there may be other unidentified targets that contribute to fibroblast identity through pathways unrelated to KLF function. We tested other TF targets of miR-9/9*-124 including ELK and TCF factors, we suspect that there may be additional co-factors contributing to fibroblast fate. Future studies should further uncover the core network components that miR-9/9*-124 downregulate to eliminate non-neuronal fates. Finally, while we found that 7SK is critical for initiating neuronal program, what activates 7SK downstream of miR-9/9*-124 remains unknown, and future work should be directed to answer how miRNAs orchestrate this neurogenic cascade.
STAR Methods
Resource Availability
Lead Contact
Further information and requests for reagents may be directed to, and will be fulfilled by, the Lead Contact, Andrew S. Yoo (yooa@wustl.edu).
Materials Availability
All unique and stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The single-cell and bulk RNA-, ATAC-, and HITS-CLIP sequencing generated in this study are as follows: 8 single-cell RNA-seq libraries from starting HAFs and moto-miNs throughout reprogramming (D0, D5, D10x2, D12.5, D15x2, D20); 5 single-cell RNA-seq libraries from starting HAFs and miNs throughout the conversion process (D0, D5, D10, D15, D20); 8 paired-end omni-ATAC-seq libraries of HAFs with control (no transduction), miR-9/9*-124, ISL1/LHX3 only, or MYT1L only at day 7 after transduction, 2 replicates per condition; 8 single-end RNA-seq libraries of HAFs with control (no transduction), miR-9/9*-124, ISL1/LHX3 only, or MYT1L only at day 7 after transduction, 2 replicates per condition; 6 paired-end HITS-CLIP libraries of HAFs expressing miR-NS or miR-9/9*-124 at day 7 after transduction, 3 replicates per condition; 6 single-end RNA-seq libraries of HAFs expressing miR-NS or miR-9/9*-124 at day 7 after transduction, 3 replicates per condition; 8 paired-end omni-ATAC-seq libraries of miNs overexpressing luciferase, KLF4, KLF5, or KLF4 and KLF5 at day 10 of conversion, two replicates per condition; 6 paired-end omni-ATAC-seq libraries for HAFs and miNs overexpressing shCTRL or sh7SK at day 15 of reprogramming, 2 replicates per condition; 6 single-end RNA-seq libraries of HAFs and miNs overexpressing shCTRL or sh7SK after day 15 of reprogramming, 2 replicates per condition. All data has been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE154178 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154178).
Experimental Model and Subject Details
Primary cell culture
Primary fibroblasts utilized in this study were Human Dermal Fibroblasts-neonatal (Catalog #2310, ScienCell) and 22-year-old female (GM02171, NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research). Adult and neonatal human fibroblasts were cultured in fibroblast media comprised of Dulbecco’s Modified Eagle Medium (Invitrogen) supplemented with 15% fetal bovine serum (Life Technologies), 0.01% β-mercaptoethanol (Life Technologies), 1% non-essential amino acids, 1% sodium pyruvate, 1% GlutaMAX, 1% 1M HEPES buffer solution, and 1% penicillin/streptomycin solution (all from Invitrogen). Cells were only maintained up to 15 passages.
Plasmid construction and virus production
Complementary cDNA was generated for transcription factors from adult human fibroblast genomic DNA (Fig. S4G, S5D) and subcloned into N106 lentiviral vectors using standard cloning techniques. Lentivirus was produced in Lenti-X 293T cells (293LE, Clontech). 293LE cells were plated on 10cm dishes (at 7x10^6 cells/dish). About 16 hours after plating, 293LE cells were transfected using polyethyleneimine (48 μ L of 2 mg/mL, Polysciences), packaging vectors (1.5 μ g pMD2.G and 4.5 μ g psPAX2), and lentiviral backbone plasmid (6 μ g) (e.g. pT-BCL-9/9*-124, N174-ISL1). Media was replaced the following day. After another two days, media was collected, filtered through a 0.45 μ m polyethersulfone (PES) syringe filter and concentrated by centrifugation at 70,000xg for 2 hours at 4°C. Virus collected from a 10cm dish was resuspended and aliquoted in 1 mL of sterile PBS and stored at −80°C for up to a year. For transduction, virus aliquots were thawed on ice and spun at 5,000xg for 5 minutes at 4°C to remove cellular debris.
Lentiviral sh7SK was generated by annealing and ligating the sequences below into the pLKO.1 vector (Addgene 8453, blasticidin resistance in place of puromycin, Yoo lab): sh7SK Forward 5’ CCGG-GAACCTCCAAACAAGCTCTTT-CTCGAGAAAGAGCTTGTTTGGAGGTTC-TTTTTG 3’ sh7SK Reverse 5’ AATTCAAAAA-GAACCTCCAAACAAGCTCTTT-CTCGAGAAAGAGCTTGTTTGGAGGTTC 3’ Similarly, shCTRL was previously constructed using shRNA to copGFP in pLKO.1-blast. Lentiviral shCTRL and sh7SK titer was optimized to 192 μ L per 6-well plate.
Method Details
Direct Neuronal Conversion and Cell Collection
Direct neuronal reprogramming of human fibroblasts to neurons was performed as previously described (Abernathy et al., 2017). Primary 22-year-old female adult human fibroblasts (HAFs; NIGMS, GM02171) were seeded onto Costar 6-well cell culture vessels (Corning) at a density of 300,000 cells/well. The following day, each plate was transduced with the following reprogramming cocktail: 750 μL of concentrated lentivirus containing the reverse tetracycline-controlled transactivator (rtTA; Addgene, 66810), 500 μL of virus containing pT-BCL-9/9*-124 (Addgene, 60859), and 500 μL of each virus containing ISL1 and LHX3 under the control of the EF1α promoter (Abernathy et al., 2017) in the presence of polybrene (8 μg/mL ; Sigma-Aldrich) all diluted up to 18 mL with MEF media. 3 mLs of the virus/media solution was added to each well then spinfected at 37°C for 30 minutes at 1,000xG using a swinging bucket rotor.
One day post-transduction media was changed to fresh fibroblast media (2 mL per well) supplemented with doxycycline (Dox; Sigma Aldrich, Cat# D9891; 1 μg/mL). After 2 days, fresh fibroblast media was changed and supplemented with Dox and antibiotics for respective vectors (Puromycin, 3 μg/ml; Geneticin, 400 ug/mL; all from Invitrogen). Five days post-transduction cells were replated on to poly-ornithine/laminin/fibronectin (PLF) coated glass coverslips. Before PLF coating, glass coverslips were acid treated as previously described (Richner et al., 2015). To transfer cells, for each well of a 6 well plate, cells were first washed 2x with 1 mL sterile PBS. Then 320 μL of 0.25% Trypsin (Gibco) was added to each well then placed in an incubator. Cells were monitored every 2 minutes, as soon as cells began to detach (no more than 6 minutes) 1 ml of MEF media supplemented with 1 μg/mL Dox was added to each well. One by one, each well was gently triturated three times to remove remaining attached cells then transferred to a sterile 1.5 mL Eppendorf tube. Cells were then spun at 200xG for 5 minutes at 37°C. The supernatant was aspirated and cells were gently resuspended in 300 μL MEF media supplemented with Dox. Cells were then drop-plated onto 12 mm (60 μL per c.s.; placed in 24 well plate) coverslips. Cells were left to settle for 15 minutes in an incubator then each well was flooded with fibroblast media supplemented with 1 μg/mL Dox. The following day media was then changed to Neuronal Media (Sciencell) supplemented with Dox, valproic acid (1 mM; EMD Millipore) dibutyryl cAMP (200 μM; Sigma-Aldrich), BDNF (10 ng/ml, Peprotech) , NT-3 (10 ng/ml, Peprotech), and Retinoic Acid (1 uM; Sigma-Aldrich) and antibiotics for each vector. For moto-miNs, CDNF and GDNF were also added (both 10 ng/ml, Peprotech). For miNs, RVC was added (100X, Thermo Fisher Scientific) after the replating step to enhance the survival of attached cells. Dox was replenished every two days and half the media was changed every 4 days. Drug selection was halted 14 days into conversion.
Single-cell RNA-seq sample preparation
For collecting reprogramming cells with the time-course single-cell RNA-seq (Figure 1), at 0, 5, 10, 15 and 20 days into reprogramming, media was aspirated out of 2-3 wells containing cells being converted, then these cells were washed with 500 μL of sterile PBS and then 200 μL of 0.25%. Trypsin was added to each well and cells were closely monitored. Upon detachment from the culture vessel. each well was flooded with complete neuronal media supplemented with 5% FBS. Cells were then counted using a hemocytometer followed centrifugation at 200xG for 5 minutes at 37°C. Cell pellets were resuspended in Cryostar CS10 cryopreservation medium (C2874-100ML; Sigma-Aldrich) at a concentration of 100 cells per μL and 100 μL aliquots were placed in a cryogenic freezing container (ThermoFisher Scientific, # 5100-0001) and stored at −80°C until all timepoints were collected (Figure 1). Alternatively, reprogramming times were stacked so that cells at different time points of neuronal conversion could be collected on the same day (Figure 2 and S1). Here, after trypsinization, detached cells were spun down at 300xg for 5 minutes and supernatant was removed. Cells were washed using 0.04% BSA in PBS by gentle pipetting. Collection and wash steps were repeated once, then cells were counted on a hemocytometer and volume was adjusted to achieve 1,000 cells/μL with 10% FBS in DMEM. Cells were placed on ice to be immediately proceeded with 10X Genomics Single Cell Processing through the Genome Technology Access Center at Washington University in St. Louis (https://gtac.wustl.edu/) on the same day (Figure 2 and S1).
10x procedure
For single-cell library preparation, we used the 10x Genomics platform: the Chromium Single Cell 3′ Library & Gel Bead Kit v2 (PN-120237), Chromium Single Cell 3′ Chip kit v2 (PN-120236) and Chromium i7 Multiplex Kit (PN-120262) were used according to the manufacturer’s instructions in the Chromium Single Cell 3′ Reagents Kits V2 User Guide. cDNA libraries were quantified on an Agilent BioAnalyzer. The moto-miN timecourse single cell preparation was sequenced on an Illumina HiSeq 2500. The neuronal activation and miR-9/9*-124-only time course, sequencing libraries were prepared using Chromium 3’ v3 system and sequencing was run on a NovaSeq S4.
Single-cell RNA-seq data processing and analysis
The raw 10x reads were processed with the Cell Ranger count pipeline using default parameters (Cell ranger v3.1.0, 10x Genomics). Reads were aligned to the hg38 reference index provided by 10x Genomics (refdata-cellranger-GRCh38 v3.0.0). Cell barcode and UMI (unique molecular identifier) were extracted and corrected from the feature library using the same methods as gene expression read processing. Feature-barcode matrices were generated by counting distinct UMIs of each gene within a given individual cell (Zheng et al., 2017).
The R package, Seurat (v3.1.4) was used for quality control, analysis, and exploration of scRNA-seq data (Satija et al., 2015). We first removed cells where features less than 200 are detected and mitochondrial counts are high, then filtered features detected in cells less than 10. Seurat was used to remove unwanted variation from the gene expression by regressing out proportion of mitochondrial UMIs and overall UMIs. Highly variable genes were identified and used as input for dimensionality reduction via Principal Component Analysis (PCA). The resulting PCs and the correlated genes were examined to determine the number of components to be included in downstream analysis. These principal components were then used as inputs to cluster individual cells, using a K-nearest neighbor graph and the Louvain algorithm. The resulting cell clusters were visualized and explored using UMAP as a non-linear dimensional reduction technique. We found cell type-specific marker genes that define clusters via differential gene expression. To identify the changes of these marker genes in order of progress through a biological process (such as fate conversion of HAFs to neurons) as a function of pseudotime, we used the R package, Monocle (v2.10.1) (Qiu et al., 2017). Gene Set Enrichment Analysis (GSEA) (v4.0.0) was employed to identify Gene Ontology (GO) using differentially expressed genes that characterize the separation of each cluster (Subramanian et al., 2007).
Integrated cell state analysis was carried to identify shared cell states that are present across different datasets (Stuart et al., 2019). This method first identifies anchors between pairs of datasets. These anchors are then used to harmonize the datasets and the returned object holds an integrated (or batch-corrected) expression matrix for all cells, enabling them to be jointly analyzed. For the analysis of RNA velocity, the python library, Velocyto (v0.17) was used to annotate spliced and unspliced reads, storing the quantifications in a standard loom file. The R package, velocyto.R (v0.6) was executed to estimate and visualize RNA velocity on that embedding (Risso et al., 2014, La Manno et al., 2018). Quadratic programming was employed using the R Package, QuadProg was used to score cell identity, as described by Treutlein et al. (Treutlein et al., 2016). This approach was modified using bulk expression data collected previously (Abernathy et al., 2017). Pseudotime ordering of moto-miN scRNA-seq data was generated by applying principle curve analysis to the first four principle components of expression data using the R Package, princurve (Hastie and Stuetzle, 1989). Gene interaction networks were generated using MEGENA (Multiscale Embedded Gene Co-Expression Network Analysis (Song and Zhang, 2015). Enrichment of microRNA-targets within subnetworks was determined via bootstrap analysis: the probability of observing the number of microRNA-targets within each subnetwork if targeting were random was estimated by generating 1000 distributions of random genes within the subnetworks, and Bonferroni-corrected p-values were reported as q-values. Long gene expression was estimated by measuring the median gene expression of the top 5% longest genes per cell.
Immunocytochemistry
Cells were fixed using 4% formaldehyde for 20 min, then blocked and permeabilized for one hour at room temperature in PBS containing 0.3% Triton X-100, 5% bovine serum albumin (Sigma-Aldrich), and 2% of goat serum (Sigma-Aldrich). Primary antibodies were incubated overnight at 4°C in blocking buffer. Cells were then washed 3x and incubated with secondary antibodies conjugated to either Alexa 488 or 568 for one hour at room temperature. DAPI (Sigma-Aldrich) was added at 1:10,000. Images were obtained on a Leica SP-2 Confocal Microscope (Leica Microsystems). Images were processed using LAS X (Leica Microsystems) and ImageJ (Schneider et al., 2012).
Cell Proliferation Assays
For each EdU experiment, the Click-iT Plus EdU Alexa Fluor 488 Imaging Kit (Invitrogen) was used and were carried out according to the Click-iT Plus EdU Imaging protocol.
For the EdU assay in HAFs and miNs (Fig. S3), EdU (10 μM) was added at day 3 after transduction and cells were collected for imaging at day 5 (48-hour pulse). For the EdU assay for HAFs and miNs with shCTRL or sh7SK, EdU (10 μM) was applied at day 14 of reprogramming and fixed for imaging at day 15 (24 hour pulse). Cells were fixed and permeabilized according to the Immunocytochemistry methods above. DNA was stained using Hoechst 33342 (1:2000, Invitrogen). Likewise, Ki67 were fixed and stained at day 15 as outlined above in Immunocytochemistry (1:500, Anti-Ki67 rabbit polyclonal, Abcam).
For monitoring FUCCI cell cycle reporter expression, HAFs were transduced with lentivirus to express both mVenus and mCherry that report S/G2/M and G1/G0 phases of cell cycle, respectively (Sakaue-Sawano et al., 2008). After the expression of miRNAs, reporter expression was monitored in live cells using the Incucyte S3 Live-Cell Analysis System (Essen Bioscience/ Santoris) for 4 days.
Cell Death Assay
SYTOX green nucleic acid staining (Thermo Fisher Scientific) was performed as recommended by the manufacturer. In brief, a final concentration of 0.1 μ M SYTOX green was added to the media of live HAFs or miNs at day 5 of reprogramming. In parallel, Hoechst 33342 nuclear counterstain (Thermo Fisher Scientific) was added to a final concentration of 1 μ g/mL in the live culture media. Cells were incubated with SYTOX and Hoechst for 20 minutes. Images were captured with a Leica DMI 400B inverted microscope with LAX Advanced Fluorescence. Four images were taken from each quadrant of the coverslip for at least two replicates per experiment. Quantification performed by counting the number of SYTOX-positive cells per Hoechst.
Immunoblot analysis
HAF control cells and HAFs expressing miR-9/9*-124 were lysed at days 1-5 after transduction in RIPA buffer (Thermo Fisher Scientific) supplemented with protease inhibitors (Roche). The concentrations of whole cell lysates were measured using the Pierce BCA protein assay it (Thermo Fisher Scientific). Equal amounts of whole cell lysates were resolved by SDS-PAGE and transferred to nitrocellulose membrane (GE Healthcare Life Sciences) using a transfer apparatus according to manufacturer’s protocols (Bio-rad). After incubation with 5% BSA in TBS containing 0.1% Tween-20 (TBST) for 30 min, the membrane was incubated with primary antibodies at 4°C overnight. Following incubation, membranes were incubated with a horseradish peroxidase-conjugated anti-mouse antibody for 1 hour. Blots were developed with the ECL system (Thermo Fisher Scientific) according to the manufacturer’s instructions.
HITS-CLIP identification of miR-9/9*-124 targets
Human Dermal Fibroblasts-neonatal (ScienCell, Carlsbad, CA) were transduced with miR-9/9*-124 or miR-NS as described previously (Richner et al., 2015). After expressing miRNAs for 5 days in DMEM, cells were plated to 100 mm Primaria dishes (Corning, Corning, NY) at the density of 10 million cells/plate. Cells were cultured in Neuronal Media (ScienCell, Carlsbad, CA) with growth factor supplements for another two days and then harvested for HITS-CLIP following the published protocol (Moore et al., 2014) with modifications. Briefly, cross-linked cells came from 3 dishes were lysed in lysis buffer, digested with RQ1 DNase (Promega, Madison, WI) and diluted (1:1000) RNAseA (Thermo, Waltham, MA) respectively, and then the RISC complex containing bound miRNAs and mRNAs (ternary complex) was immunoprecipitated overnight at 4 °C with mouse anti-Ago1/2/3 antibody 2A8 (Active Motif, Carlsbad, CA). The ternary complex was labelled 32P by ligating miRNA with 32P-labelled 3’-linker and then separated by NuPAGE™ gel (Thermo, Waltham, MA). The 130 kDa bands on the nitrocellulose membrane corresponding to ternary complex containing Ago:miRNA:mRNA were excised and used to extract RNA for sequencing library prep for deep sequencing with TruSeq Small RNA Library Preparation Kits (San Diego, CA).
Libraries were sequenced on the Illumina HiSeq 2500 platform to produce single-end reads of 100 bp. Reads were trimmed using Trimmomatic (v0.33) to remove primer and adapter sequences (Bolger et al., 2014). Trimmed reads were aligned to human genome hg38 using STAR (v2.4.2a) with default parameters (Dobin and Gingeras, 2015). Reads uniquely aligning to genomic features (phred score ≥ 10) were quantified using featureCounts (v1.4.6-p4) from the Subread package with gencode v23 annotations (Liao et al., 2014). Additionally, in-house software was used to divide the UTRs or CDs from the gencode annotations into continuous 100bp bins. For each feature, two sets of bins were formed such that the second set of bins was offset by 50bp from the first set of bins. Reads aligning to bins were quantified using featureCounts. Differentially associated features or bins were determined by using DESeq2 (Love et al., 2014) in the negative binomial distribution model with a cutoff of adj. p-value < 0.001 and log2FC ≥ 1. A UTR or CD was considered to have differential association if one or more bins from that UTR or CD was called as differentially associated by DESeq2. Reads Per Kilobase per Millions mapped reads, RPKM, of features or bins were determined by quantifying uniquely aligned reads. Wiggle and bigwig files were created from bam files using igvtools (v2.3.60) (https://software.broadinstitute.org/software/igv/igvtools) (Thorvaldsdóttir et al., 2013) and wigToBigWig (v4) (Kent et al., 2010).
To correlate identified peaks with overexpressed miR-9/9*-124, sequences corresponding to differential bin was extracted from UCSC genome browser (https://genome.ucsc.edu) with 25 extra nucleotides on each end and analyzed using RNAhybrid (energy threshold −15) (Rehmsmeier et al., 2004). The miRNA binding motifs on target genes were detected by MEME suite (Bailey et al., 2009).
Luciferase assay to validate miRNA targets
The 3’-UTR of miR-9/9*-124 targets was cloned downstream of the firefly luciferase gene using the following primers. These constructs were co-transfected with a control reporter expressing Renilla luciferase to 293T cells in the presence of either miR-9/9*-124 or miR-NS. The construct containing 3’-UTR of human PGK without any miR-9/9*-124 predicted binding site was used as negative control. After 48 hrs of transfection, luciferase assay was performed using Dual-Luciferase® Reporter Assay System (Promega, Madison, WI) according to the user instruction. The repressive activity of miR-9/9*-124 to target gene 3’UTRs was calculated by normalizing firefly luminescence to control renilla luminescence. To control for off-target effects associated with overexpressing any miRNA on luciferase activity and to identify the specific activities of miR-9/9*-124 this value was then normalized to transfections using identical 3’UTR constructs and a miR-NS (non-specific control miRNA) expression vector. Primer sequences are available upon request.
Quantitative PCR
Total RNA was extracted using TRIzol (Invitrogen, USA) according to manufacturer guidelines. Reverse-transcribed complementary DNA (cDNA) was synthesized from 250 – 500 ng of RNA with SuperScript III First-Strand Synthesis SuperMix (Invitrogen, USA). cDNA was analyzed using a StepOnePlus Real-Time PCR System (AB Applied Biosystems, Germany). Replicates for each condition represent independent experimental samples collected from separate plates within one reprogramming experiment.
RNA-sequencing and data analysis
Briefly, total RNA was extracted using the RNeasy plus micro kit (QIAGEN, USA) as instructed by the manufacturer. Each condition consisted of two replicates collected from independent experiments. RNA sample integrity was assessed on a 2100 Bioanalyzer (Agilent, USA) before proceeding to library preparation. mRNA was prepared for Illumina sequencing using poly-A library preparation for the TF versus miR RNA-seq (Girihlet Inc., USA) and SMARTer Ultra Low Input RNA Kit (Clontech, USA) for the miR-NS versus miR-9/9*-124 RNA-seq and 7SK knock-down RNA-seq. cDNA libraries were sequenced for single-end reads, with 75-bp reads on an Illumina NextSeq 500 for the TF versus miR RNA-seq and 50-bp reads on an Illumina Hi-Seq 2500 for the miR-NS versus miR-9/9*-124 RNA-seq and the 7SK knock-down RNAseq.
For the TF versus miR RNA-seq analysis, the sequencing reads were mapped to the human reference genome (GRCh38) using STAR (Dobin and Gingeras, 2015). Raw gene counts were derived by deepTools multiBamSummary with the option –outRawCounts (Ramírez et al., 2016). To account for technical variations, the raw gene counts were normalized using the RUVSeq (Remove Unwanted Variation (v1.20.0) (Risso et al., 2014)based on the factor analysis of replicate samples. Differential expression analysis was performed using DESeq2 (v1.26.0) (Love et al., 2014). Differentially expressed genes (DEGs) are considered with log2FC ≥ 1 and adj. p-value < 0.01 between each pairwise.
For the 7SK knock-down RNA-seq analysis, the raw reads were aligned to human reference genome (GRCh38) by STAR (Dobin and Gingeras, 2015). Gene counts were derived from the number of uniquely aligned reads by Subread:featureCounts (Liao et al., 2014). The counts were normalized by trimmed mean of M-values (TMM) using edgeR (Robinson et al., 2010). EdgeR and limma were used to identify differentially expressed genes. Genes with any count per million (CPM) < 1 in at least one of any two replicates per biological condition across the experiment were eliminated. The resulting filtered read counts were used to generate Pearson correlation coefficients using R package ggcorr for the 7SK knock-down RNA-seq (https://github.com/ggobi/ggally). Quantitative difference of counts between miNs with shCTRL and miNs with sh7SK were analyzed by limma and graphically displayed using Glimma (Ritchie et al., 2015; Su et al., 2017). Gene function annotation was evaluated using Metascape (Zhou et al., 2019).
Omni-ATAC-sequencing preparation
Omni-ATAC was performed as outlined in Corces et al. (Corces et al., 2017). In brief, each sample was treated with DNase for 30 minutes prior to collection. Approximately 50,000 cells were collected for library preparation. Transposition reaction was completed with Nextera Tn5 Transposase (Illumina Tagment DNA Enzyme and Buffer Kit, Illumina) for 30 minutes at 37°C and library fragments were amplified under optimal amplification conditions. Final libraries were purified by the DNA Clean & Concentrator 5 Kit (Zymo, USA). Libraries were sequenced with 50-bp paired-end reads on Illumina HiSeq 2500 for HAFs in the 7SK knock-down project (Genome Technology Access Center at Washington University in St. Louis) or with 75-bp
paired-end reads on Illumina NextSeq 500 for the TF versus miR project, the KLF overexpression project, and miNs with shRNA in the 7SK knock-down project (Girihlet Inc., USA).
Omni-ATAC-sequencing data analyses
At least 45 million omni-ATAC reads per sample were processed by Atac-seq Integrative Analysis Pipeline (AIAP, https://github.com/Zhang-lab/ATAC-seq_QC_analysis). Briefly, AIAP trims adapters using Cutadapt v2.4 (Martin, 2011), aligns fragments using BWA-MEM to the hg38 human reference genome (Li, 2013), removes low quality reads using methylQA (Li et al., 2015), normalizes count signal by 10 million input single end reads, and call peaks by macs2 with q-value less than 0.01 (Zhang et al., 2008).
The normalization based on housekeeping genes and differential accessible region (DAR) analysis between each pairwise were conducted using DESeq2 (v1.26.0) (Love et al., 2014). DARs were filtered for significance by a minimum of log2FC ≥ 1 with an adj. p-value < 0.01. UCSC hg38 genes nearest to DARs and genomic features of DARs were annotated with ChIPSeeker (Yu et al., 2015) for the TF versus miR project, KLF overexpression, and 7SK knock-down projects. To select only one DAR peak per gene, DAR peaks were sorted by most significant FDR-adjusted p-value. Functional annotation was performed by Metascape (Zhou et al., 2019), and Ingenuity Pathway Analysis was used to identify a top gene network downregulated the 7SK knock-down project (Krämer et al., 2014). The joint ATAC and RNA heatmap was made based on normalized read count of each sample mapped onto relevant regions. Metagene heatmaps were generated through mapping from sequencing depth-normalized signal intensity of each sample onto DARs of interest using deepTools2 (Ramírez et al., 2016). The metagene heatmap for closed fibroblast enhancer loci is based on H3K4me1- and H3K27me3-marked loci of human fibroblasts in the Roadmap Epigenome Project (Roadmap Epigenomics Consortium et al., 2015). For the TF versus miR and KLF overexpression projects, the Pearson correlation was created using deepTools2 (Ramírez et al., 2016). For the 7SK knock-down project, the Pearson correlation heatmap was constructed using ggcorr (https://github.com/ggobi/ggally) on low-count filtered and TMM-normalized read counts to match the RNA-seq processing. Read counts were calculated by plotting read counts within peak regions combined across all samples through deepTools2 (Ramírez et al., 2016).
Comparative analysis of BRG1 and 7SK knock-down ATAC-seq
Comparative analysis of shBRG1- and sh7SK-associated chromatin accessibility was completed from mapping signal intensity for ATAC-seq for shEGFP and shBRG1 at day 20 of reprogramming (Abernathy et al., 2017) onto the closed sh7SK DAR (log2FC ≤ −1, adj. p-value ≤ 0.01). shBRG1 closed DAR were defined as log2FC ≤ −1, adj. p-value ≤ 0.01 against shEGFP. shBRG1- and sh7SK-associated closed DAR were intersected using bedtools (v2.25.0) (Quinlan and Hall, 2010), and Metascape Gene Ontology analysis was used to define the biological processes associated with the intersected loci (Zhou et al., 2019). Heatmaps for both analyses were constructed using deepTools2 (Ramírez et al., 2016).
Homer Motif Analysis
For analysis of fibroblast identity network regulators, HOMER motif analysis (Heinz et al., 2010) was used to identify enriched transcription factor binding motifs in the total closed loci between days 0 and 10 of reprogramming from a previous time-course ATAC-seq dataset (Abernathy et al., 2017) and a subset of these closed loci that overlap with fibroblast enhancer loci (Roadmap Epigenomics Consortium et al., 2015). High-fidelity KLF4 and KLF5 binding sites in these closed loci were validated and filtered using JASPAR (score > 900) (Khan et al., 2018). Percentages of TF enrichment represent the number of TF motifs in target sequences normalized to the number of background sequences with the motif.
Quantification and Statistical Analysis
Unless otherwise noted, results are shown as mean ± standard error mean (SEM) of values obtained in two to three independent experiments. For qPCR analyses, n represents the number of RNA extraction from a condition per independent reprogramming event. qPCR data were analyzed using ΔΔCt method, and significance was tested with a two-tailed student’s t-test for samples with Holm-Sidak correction for multiple comparisons, paired t-test, or a two-way ANOVA (GraphPad Prism v8; GraphPad Software). All values < 0.05 were considered statistically significant.
For immunostaining analyses, n represents the number of cells as identified by nuclear staining. Differences between two groups were compared using a two-tailed student’s t-test for unpaired samples with Holm-Sidak correction for multiple comparisons, paired t-test, or ordinary one-way ANOVA (GraphPad Prism v8; GraphPad Software). Differences between multiple treatment groups were evaluated using a two-way ANOVA (GraphPad Prism v8; GraphPad Software). All values < 0.05 were considered statistically significant.
Cells were manually counted for each immunostaining quantification. For quantification of fibroblast and neuronal markers in miR-only or TF-only cells (Fig. 3): MAP2 quantification, only cells with processes at least three times the length of the soma were included. Data are represented as mean with s.e.m.: HAF control: FSP1, n= 273 and MAP2, n= 407; I1L3 only: FSP1, n= 289 and MAP2, n= 206; miR-9/9*-124 only: FSP1, n= 385 and MAP2, n= 357. For quantification of alternative fates in I1L3-only cells and motomiNs (Fig. S2): For MNX1 and MAP2 quantification, only cells with processes at least three times the length of the soma were included. Data are represented as mean with s.e.m.: I1L3 only: MNX1, n = 63; MAP2, n= 47; FSP1, n= 67; SOX2, n= 91; and MYH3, n= 54; miR-9/9*-124 + I1L3: MNX1, n = 134; MAP2, n= 94; FSP1, n= 103; SOX2, n= 100; and MYH3, n= 72. For quantification of EdU assay (Fig. S3): Data are represented as mean with s.e.m. HAF control: n= 840 and miN: n= 193. For quantification of Sytox-positive cells (Fig. S3): Data are represented as mean with s.e.m. HAF control: n= 1,560 and miN: n= 837. For quantification of Ki67-positive cells for KLF-expressing cells (Fig. S5): Data are represented as mean with s.e.m. HAFs: n = 248; miR-9/9*-124 + LUC, n = 234 ; miR-9/9*-124 + KLF4, n = 347; miR-9/9*-124 + KLF5, n= 344; miR-9/9*-124 + KLF4 + KLF5, n= 403.
Supplementary Material
Movie S1. Related to Figure 2. miR-mediated reprogramming movie
Table S3. Related to Figure 3. miR-instructed downregulated DEG with closed DAR
Table S4. Related to Figure 4 and S4. miR AGO-HITS-CLIP and fibroblast network data
Table S6. Related to Figure 7. 7SK-associated data
Key Resource Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-MAP2 | Cell Signaling Technology | Cat#4542S; RRID: AB_ 10693782 |
| Rabbit polyclonal anti-TUBB3 | BioLegend | Cat# 802001; RRID: AB_ 2564645 |
| Mouse monoclonal anti-TUBB3 | BioLegend | Cat# 801202; RRID: AB_ 10063408 |
| Mouse monoclonal anti-MNX1 | DSHB | Cat# 81.5C10, RRID: AB_2145209 |
| Rabbit polyclonal anti-KI-67 | Abcam | Cat# ab15580, RRID: AB_443209 |
| Rabbit monoclonal anti-S100A4 (FSP1) | Abcam | Cat# ab124805, RRID: AB_410978091 |
| Rabbit monoclonal anti-SOX2 | Cell Signaling Technology | Cat#23064S; RRID: AB_ 2714146 |
| Mouse monoclonal anti-MYH3 | Santa Cruz Biotechnology | Cat#sc53091; RRID: AB_ 670121 |
| Mouse monoclonal anti-AGO1/2/3 | Active Motif | Cat# 39937; RRID: AB_2793399 |
| Mouse monoclonal anti-p27 (KIP1) | BD Biosciences | Cat# 610241, RRID: AB_397636 |
| Mouse monoclonal anti-GAPDH | Santa Cruz Biotechnology | Cat #: sc-47724, RRID: AB_627678 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fetal bovine serum (FBS), qualified, US Department of Agriculture (USDA)-approved regions | Life Technologies | Cat# 10437028 |
| Polybrene | Sigma-Aldrich | Cat# H9268 |
| Neuronal Media | ScienCell Research Laboratories | Cat# 1521 |
| Doxycycline hyclate (Dox) | Sigma-Aldrich | Cat# D9891 |
| Poly-L-ornithine solution | Sigma-Aldrich | Cat # P4957 |
| Laminin | Sigma-Aldrich | Cat# L2020 |
| Fibronectin | Sigma-Aldrich | Cat# F4759 |
| Valproic acid (VPA), sodium salt | Sigma-Aldrich | Cat# 676380 |
| Dibutyryl-cAMP sodium salt | Sigma-Aldrich | Cat# D0627 |
| Retinoic acid (RA) | Sigma-Aldrich | Cat# R2625 |
| Neurotrophin-3 (NT-3) | PeproTech | Cat# 450-03 |
| Brain-derived neurotrophic factor (BDNF) | PeproTech | Cat# 450-02 |
| Ciliary Neurotrophic factor (CNTF) | PeproTech | Cat# 450-13 |
| Glial-derived Neurotrophic Factor (GDNF) | PeproTech | Cat# 450-10 |
| RevitaCell Supplement | Thermo Fisher Scientific | Cat# A2644501 |
| Critical Commercial Assays | ||
| RNeasy plus micro kit | QIAGEN | Cat# 74034 |
| SMARTer Ultra Low RNA Kit for Illumina sequencing | Clontech | Cat# 635029 |
| TruSeq Small RNA Library Preparation Kit | Illumina | Cat# RS-200-0012 |
| Illumina Tagment DNA Enzyme and Buffer Kit | Illumina | Cat# 20034197 |
| DNA Clean and Concentrator Kit | Zymo Research | Cat# D4014 |
| MinElute Gel Extraction Kit | QIAGEN | Cat# 28604 |
| SuperScript III Reverse Transcriptase | Thermo Fisher Scientific | Cat# 18080044 |
| Premo™ FUCCI Cell Cycle Sensor (BacMam 2.0) | Thermo Fisher Scientific | Cat# P36237 |
| Click-iT EdU Cell Proliferation Kit for Imaging | Thermo Fisher Scientific | Cat# C10337 |
| Deposited Data | ||
| Raw and processed data (scRNA-seq, omni-ATAC-seq, RNA-seq, HITS-CLIP) | This paper | GEO: GSE154178 |
| Experimental Models: Cell Lines | ||
| Primary 22 year old female human adult fibroblasts | NIGMS Human Genetic Cell Repository at the Coriell Institute for Medical Research | GM02171 |
| Lenti-X 293LE cell line | Clontech | Cat# 632180 |
| Human dermal fibroblasts-neonatal | ScienCell Research Laboratories | Cat# 2310 |
| Oligonucleotides | ||
| Primers used for qPCR, see Table S1 | This paper | N/A |
| Recombinant DNA | ||
| N174 | Addgene | 60859 |
| N106 | Addgene | 66808 |
| pMD2.G | Addgene | 12259 |
| psPAX2 | Addgene | 12260 |
| pT-BCL-9/9*-124 | Addgene | 60859 |
| pT-BCL-NS | Victor et al., 2014 | N/A |
| LHX3-N174 | Abernathy, Kim, and McCoy, et al., 2017 | N/A |
| ISL1-N174 | Abernathy, Kim, and McCoy, et al., 2017 | N/A |
| MYT1L-N174 | Richner et al., 2015 | Addgene Plasmid #66809 |
| LUC-N106 | This paper | N/A |
| ELK3-N106 | This paper | N/A |
| TCF3-N106 | This paper | N/A |
| TCF4-N106 | This paper | N/A |
| TCF12-N106 | This paper | N/A |
| KLF11-N106 | This paper | N/A |
| KLF4-N106 | This paper | N/A |
| KLF5-N106 | This paper | N/A |
| pLKO-sh7SK | This paper | N/A |
| pLKO-shCTRL | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| LAS X | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| Seurat v3.1.4 | Satija et al., 2015; Stuart and Butler, 2019 | https://satijalab.org/seurat/install.html |
| Monocle v2.10.1 | Qiu et al., 2017 | http://cole-trapnell-lab.github.io/monocle-release/docs/#installing-monocle |
| Quadprog | Turlach and Weingessel, 2007 | https://www.rdocumentation.org/packages/quadprog/versions/1.5-8/source |
| princurve | Hastie and Stuetzle, 1989 | https://github.com/cran/princurve |
| MEGENA | Song and Zhang, 2015 | https://github.com/songw01/MEGENA |
| Graphpad Prism 8 | GraphPad Software Inc | http://www.graphpad.com/ |
| edgeR 3.11 | Robinson et al., 2010; McCarthy, Chen, and Smyth, 2012 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| limma | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Metascape | Zhou et al., 2019 | http://metascape.org/gp/index.html#/main/step1 |
| Ingenuity Pathway Analysis | Kramer et al., 2014 | QIAGEN |
| ChIPseeker | Yu, Wang, and He, 2015 | http://www.bioconductor.org/packages/release/bioc/html/ChIPseeker.html |
| deepTools v3.1.3 | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/develop/content/installation.html |
| Atac-seq Integrative Analysis Pipeline (AIAP) v1.1 | https://github.com/Zhang-lab/ATAC-seq_QC_analysis | |
| HOMER | Heinz et al., 2010 | http://homer.salk.edu |
| Cutadapt v2.4 | Martin, 2011; National Bioinformatics Infrastructure Sweden | https://github.com/marcelm/cutadapt/ |
| BWA-MEM | Li, 2010 | https://github.com/lh3/bwa |
| methylQA | Li, 2015 | http://methylqa.sourceforge.net/ |
| macs2 v.2.1.0 | Zhang et al., 2008 | https://github.com/taoliu/MACS/ |
| DESeq2 | Love, Huber, and Anders, 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| RNA-seq analysis pipe v4 | https://github.com/Zhang-lab/RNA-seq_QC_analysis | |
| FastQC v0.11.4 | Simon Andrews, 2010 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| RSeQC | Wang, Wang, and Li, 2012; Wang et al., 2016 | http://rseqc.sourceforge.net/ |
| RSEM | Li and Dewey, 2011 | https://github.com/deweylab/RSEM |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Samtools v1.9 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| Picard v2.18 | Broad Institute | https://broadinstitute.github.io/picard/ |
| GSEA v4.0.0 | Subramanian et al., 2007 | https://www.gsea-msigdb.org/gsea/downloads.jsp |
| Cell ranger 3.1.0 | Zheng et al., 2017 | https://support.10xgenomics.com/single-cell-gene-expression/software/downloads/latest |
| bedGraphToBigWig | Kent et al., 2010 | http://hgdownload.soe.ucsc.edu/admin/exe/linux.x86_64/ |
| Bedtools v2.27.0 | Quinlan and Hall, 2010; Quinlan, 2014 | https://bedtools.readthedocs.io/en/latest/content/installation.html |
| IGV | Thorvaldsdóttir et al., 2013 | https://software.broadinstitute.org/software/igv/download |
| RUVSeq v1.20.0 | Risso et al., 2014 | https://bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| Velocyto | La Manno et al., 2018 | http://velocyto.org/ |
| Glimma | Ritchie et al., 2015; Su et al., 2017 | https://github.com/Shians/Glimma |
| STAR | Dobin et al., 2015 | https://github.com/alexdobin/STAR |
| Subread:featureCounts | Liao et al., 2014 | http://subread.sourceforge.net/ |
Acknowledgments:
We thank G. Dorn and V. Church for helpful suggestions on the manuscript. We thank the Genome Technology Access Center at Washington University School of Medicine for deep-sequencing experiments. We recognize BioRender.com for creation of the graphical abstract. This study was supported by the following programs, grants, and fellowships: Grass Fellowship in Neuroscience and the Interface of Psychology, Neuroscience and Genetics training fellowship (T32GM081739) (M.J.M.); Cellular and Molecular Biology Training Program (T32 GM007067) (K.C.); R01GM126112 (NIGMS), Paul G. Allen Frontiers Group, Vallee Foundation, and Alfred P. Sloan Foundation Awards (S.A.M.); NIH Director’s Innovator Award (DP2NS083372), Cure Alzheimer’s Fund (CAF), Presidential Early Career Award for Scientists and Engineers (PECASE; 4DP2NS083372), RF1AG056296 (NIA), R01NS107488 (NINDS), Farrell Foundation Fund, and Mallinckrodt Scholar Award (A.S.Y.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests:
We declare no competing interests.
References
- Abernathy DG, Kim WK, McCoy MJ, Lake AM, Ouwenga R, Lee SW, Xing X, Li D, Lee HJ, Heuckeroth RO, et al. (2017). MicroRNAs Induce a Permissive Chromatin Environment that Enables Neuronal Subtype-Specific Reprogramming of Adult Human Fibroblasts. Cell Stem Cell 21, 332–348.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babos KN, Galloway KE, Kisler K, Zitting M, Li Y, Shi Y, Quintino B, Chow RH, Zlokovic BV, and Ichida JK (2019). Mitigating Antagonism between Transcription and Proliferation Allows Near-Deterministic Cellular Reprogramming. Cell Stem Cell 25, 486–500.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 37, W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazi Z, Bertacchi M, Abasi M, Mohammadi-Yeganeh S, Soleimani M, Wagner N, and Ghanbarian H (2018). Rn7SK small nuclear RNA is involved in neuronal differentiation. J. Cell. Biochem 119, 3174–3182. [DOI] [PubMed] [Google Scholar]
- Becht E, McInnes L, Healy J, Dutertre C-A, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol [DOI] [PubMed] [Google Scholar]
- Biddy BA, Kong W, Kamimoto K, Guo C, Waye SE, Sun T, and Morris SA (2018). Single-cell mapping of lineage and identity in direct reprogramming. Nature 564, 219–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinforma. Oxf. Engl 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briese M, Saal-Bauernschubert L, Ji C, Moradi M, Ghanawi H, Uhl M, Appenzeller S, Backofen R, and Sendtner M (2018). hnRNP R and its main interactor, the noncoding RNA 7SK, coregulate the axonal transcriptome of motoneurons. Proc. Natl. Acad. Sci. U. S. A 115, E2859–E2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter-Demchok J, Gelfand ET, et al. (2015). A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation Biobanking 13, 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi SW, Hannon GJ, and Darnell RB (2012). An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol 19, 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, and Gingeras TR (2015). Mapping RNA-seq Reads with STAR. Curr. Protoc. Bioinforma 51, 11.14.1–11.14.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RA, Do BT, Rubin AJ, Calo E, Lee B, Kuchelmeister H, Rale M, Chu C, Kool ET, Wysocka J, et al. (2016). 7SK-BAF axis controls pervasive transcription at enhancers. Nat. Struct. Mol. Biol 23, 231–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, and Greenberg ME (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie T, and Stuetzle W (1989). Principal Curves. J. Am. Stat. Assoc 84, 502–516. [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herdy J, Schafer S, Kim Y, Ansari Z, Zangwill D, Ku M, Paquola A, Lee H, Mertens J, and Gage FH (2019). Chemical modulation of transcriptionally enriched signaling pathways to optimize the conversion of fibroblasts into neurons. ELife 8, e41356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L, and Crabtree GR (2010). Chromatin remodelling during development. Nature 463, 474–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S (2013). DNA methylation age of human tissues and cell types. Genome Biol. 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh CJ, Zhang B, Victor MB, Dahiya S, Batista LF, Horvath S, and Yoo AS (2016). Maintenance of age in human neurons generated by microRNA-based neuronal conversion of fibroblasts. ELife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husain A, Begum NA, Taniguchi T, Taniguchi H, Kobayashi M, and Honjo T (2016). Chromatin remodeller SMARCA4 recruits topoisomerase 1 and suppresses transcription-associated genomic instability. Nat. Commun. 7, 10549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinforma. Oxf. Engl. 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Fornes O, Stigliani A, Gheorghe M, Castro-Mondragon JA, van der Lee R, Bessy A, Chèneby J, Kulkarni SR, Tan G, et al. (2018). JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 46, D260–D266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krämer A, Green J, Pollard J, and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinforma. Oxf. Engl 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Manno G, Soldatov R, Zeisel A, Braun E, Hochgerner H, Petukhov V, Lidschreiber K, Kastriti ME, Lönnerberg P, Furlan A, et al. (2018). RNA velocity of single cells. Nature 560, 494–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapasset L, Milhavet O, Prieur A, Besnard E, Babled A, Aït-Hamou N, Leschik J, Pellestor F, Ramirez J-M, De Vos J, et al. (2011). Rejuvenating senescent and centenarian human cells by reprogramming through the pluripotent state. Genes Dev. 25, 2248–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv13033997 Q-Bio. [Google Scholar]
- Li D, Zhang B, Xing X, and Wang T (2015). Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods 72, 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinforma. Oxf. Engl 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Liu M-L, Zang T, and Zhang C-L (2016). Direct Lineage Reprogramming Reveals Disease-Specific Phenotypes of Motor Neurons from Human ALS Patients. Cell Rep. 14, 115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, Kullmann PHM, Twomey MA, Miriyala J, Philpot BD, and Zylka MJ (2014). Topoisomerase 1 inhibition reversibly impairs synaptic function. Proc. Natl. Acad. Sci. U. S. A 111, 17290–17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeyev EV, Zhang J, Carrasco MA, and Maniatis T (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27, 435–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mall M, Kareta MS, Chanda S, Ahlenius H, Perotti N, Zhou B, Grieder SD, Ge X, Drake S, Euong Ang C, et al. (2017). Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature 544, 245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal 17, 10–12. [Google Scholar]
- Mazzoni EO, Mahony S, Closser M, Morrison CA, Nedelec S, Williams DJ, An D, Gifford DK, and Wichterle H (2013). Synergistic binding of transcription factors to cell-specific enhancers programs motor neuron identity. Nat. Neurosci 16, 1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MJ, and Fire AZ (2020). Intron and gene size expansion during nervous system evolution. BMC Genomics 21, 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MJ, Paul AJ, Victor MB, Richner M, Gabel HW, Gong H, Yoo AS, and Ahn T-H (2018). LONGO: an R package for interactive gene length dependent analysis for neuronal identity. Bioinforma. Oxf. Engl 34, i422–i428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertens J, Paquola ACM, Ku M, Hatch E, Böhnke L, Ladjevardi S, McGrath S, Campbell B, Lee H, Herdy JR, et al. (2015). Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 17, 705–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JD, Ganat YM, Kishinevsky S, Bowman RL, Liu B, Tu EY, Mandal PK, Vera E, Shim J, Kriks S, et al. (2013). Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MJ, Zhang C, Gantman EC, Mele A, Darnell JC, and Darnell RB (2014). Mapping Argonaute and conventional RNA-binding protein interactions with RNA at single-nucleotide resolution using HITS-CLIP and CIMS analysis. Nat. Protoc 9, 263–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson M, Chan DN, Ha I, Case D, Cui Y, Van Handel B, Mikkola HK, and Lowry WE (2012). Defining the nature of human pluripotent stem cell progeny. Cell Res. 22, 178–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinzón N, Li B, Martinez L, Sergeeva A, Presumey J, Apparailly F, and Seitz H (2017). microRNA target prediction programs predict many false positives. Genome Res. 27, 234–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Kang X, Hu J, Zhang D, Liang Z, Meng F, Zhang X, Xue Y, Maimon R, Dowdy SF, et al. (2020). Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 582, 550–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat. Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinforma. Oxf. Engl 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmsmeier M, Steffen P, Hochsmann M, and Giegerich R (2004). Fast and effective prediction of microRNA/target duplexes. RNA N. Y. N 10, 1507–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, and Dudoit S (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. (2015). Integrative analysis of 111 reference human epigenomes. Nature 518, 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W-M, and Zhang B (2015). Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput. Biol 11, e1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S, Law CW, Ah-Cann C, Asselin-Labat M-L, Blewitt ME, and Ritchie ME (2017). Glimma: interactive graphics for gene expression analysis. Bioinforma. Oxf. Engl 33, 2050–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Kuehn H, Gould J, Tamayo P, and Mesirov JP (2007). GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinforma. Oxf. Engl. 23, 3251–3253. [DOI] [PubMed] [Google Scholar]
- Thorvaldsdóttir H, Robinson JT, and Mesirov JP (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform 14, 178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein B, Lee QY, Camp JG, Mall M, Koh W, Shariati SAM, Sim S, Neff NF, Skotheim JM, Wernig M, et al. (2016). Dissecting direct reprogramming from fibroblast to neuron using single-cell RNA-seq. Nature 534, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor MB, Richner M, Hermanstyne TO, Ransdell JL, Sobieski C, Deng P-Y, Klyachko VA, Nerbonne JM, and Yoo AS (2014). Generation of human striatal neurons by microRNA-dependent direct conversion of fibroblasts. Neuron 84, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victor MB, Richner M, Olsen HE, Lee SW, Monteys AM, Ma C, Huh CJ, Zhang B, Davidson BL, Yang XW, et al. (2018). Striatal neurons directly converted from Huntington’s disease patient fibroblasts recapitulate age-associated disease phenotypes. Nat. Neurosci 21, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski OL, Vierbuchen T, Qu K, Lee QY, Chanda S, Fuentes DR, Giresi PG, Ng YH, Marro S, Neff NF, et al. (2013). Hierarchical Mechanisms for Direct Reprogramming of Fibroblasts to Neurons. Cell 155, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ouyang K, Huang J, Zhou Y, Ouyang H, Li H, Wang G, Wu Q, Wei C, Bi Y, et al. (2013). Direct Conversion of Fibroblasts to Neurons by Reprogramming PTB-Regulated MicroRNA Circuits. Cell 152, 82–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, and Crabtree GR (2009). ATP-dependent chromatin remodeling in neural development. Curr. Opin. Neurobiol 19, 120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo AS, Sun AX, Li L, Shcheglovitov A, Portmann T, Li Y, Lee-Messer C, Dolmetsch RE, Tsien RW, and Crabtree GR (2011). MicroRNA-mediated conversion of human fibroblasts to neurons. Nature 476, 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, and He Q-Y (2015). ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinforma. Oxf. Engl 31, 2382–2383. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nat. Commun 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Su J, Hu X, Zhou C, Li H, Chen Z, Xiao Q, Wang B, Wu W, Sun Y, et al. (2020). Glia-to-Neuron Conversion by CRISPR-CasRx Alleviates Symptoms of Neurological Disease in Mice. Cell 181, 590–603.e16. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Zhou B, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, Benner C, and Chanda SK (2019). Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun 10, 1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. Related to Figure 2. miR-mediated reprogramming movie
Table S3. Related to Figure 3. miR-instructed downregulated DEG with closed DAR
Table S4. Related to Figure 4 and S4. miR AGO-HITS-CLIP and fibroblast network data
Table S6. Related to Figure 7. 7SK-associated data
Data Availability Statement
The single-cell and bulk RNA-, ATAC-, and HITS-CLIP sequencing generated in this study are as follows: 8 single-cell RNA-seq libraries from starting HAFs and moto-miNs throughout reprogramming (D0, D5, D10x2, D12.5, D15x2, D20); 5 single-cell RNA-seq libraries from starting HAFs and miNs throughout the conversion process (D0, D5, D10, D15, D20); 8 paired-end omni-ATAC-seq libraries of HAFs with control (no transduction), miR-9/9*-124, ISL1/LHX3 only, or MYT1L only at day 7 after transduction, 2 replicates per condition; 8 single-end RNA-seq libraries of HAFs with control (no transduction), miR-9/9*-124, ISL1/LHX3 only, or MYT1L only at day 7 after transduction, 2 replicates per condition; 6 paired-end HITS-CLIP libraries of HAFs expressing miR-NS or miR-9/9*-124 at day 7 after transduction, 3 replicates per condition; 6 single-end RNA-seq libraries of HAFs expressing miR-NS or miR-9/9*-124 at day 7 after transduction, 3 replicates per condition; 8 paired-end omni-ATAC-seq libraries of miNs overexpressing luciferase, KLF4, KLF5, or KLF4 and KLF5 at day 10 of conversion, two replicates per condition; 6 paired-end omni-ATAC-seq libraries for HAFs and miNs overexpressing shCTRL or sh7SK at day 15 of reprogramming, 2 replicates per condition; 6 single-end RNA-seq libraries of HAFs and miNs overexpressing shCTRL or sh7SK after day 15 of reprogramming, 2 replicates per condition. All data has been deposited in NCBI’s Gene Expression Omnibus (Edgar et al., 2002) and are accessible through GEO Series accession number GSE154178 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154178).