

SUMMARY
Bone marrow failure in Fanconi anemia (FA) patients results from dysfunctional hematopoietic stem and progenitor cells (HSPCs). To identify determinants of bone marrow failure, we performed single-cell transcriptome profiling of primary HSPCs from FA patients. In addition to overexpression of p53 and TGFβ pathway genes, we identified high levels of MYC expression. We correspondingly observed coexistence of distinct HSPC subpopulations expressing either high levels of TP53 or MYC in FA bone marrow. Inhibiting MYC expression with the BET bromodomain inhibitor (+)-JQ1 reduced clonogenic potential of FA patient HSPCs but rescued physiological and genotoxic stress in HSPCs from FA mice, showing MYC promotes proliferation while increasing DNA damage. MYC-high HSPCs showed significant downregulation of cell adhesion genes consistent with enhanced egress of FA HSPCs from bone marrow to peripheral blood. We speculate that MYC overexpression therefore impairs HSPC function in FA patients and contributes to exhaustion in FA bone marrow.
Keywords: Fanconi anemia, hematopoietic stem cells, bone marrow failure, single cell RNA sequencing, MYC, DNA damage, genotoxic stress, physiological stress, CXCR4
Graphical Abstract
eTOC:
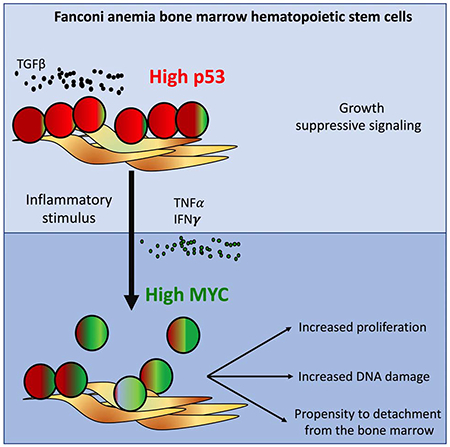
Rodriguez and colleagues demonstrate that MYC is upregulated in hematopoietic stem and progenitor cells (HSPCs) from Fanconi anemia (FA) patients. On one hand, MYC counteracts the p53 and TGFβ-mediated growth suppression in FA bone marrow. On the other hand, MYC expression worsens the replication and genotoxic stress of FA HSPCs.
INTRODUCTION
Fanconi anemia (FA), a DNA repair disorder, is the most frequently inherited bone marrow failure (BMF) syndrome. Patients with FA suffer from early childhood onset of bone marrow (BM) failure, developmental abnormalities, and heightened susceptibility to solid tumors. FA patients also have a strong predisposition to myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) (Alter, 2014, Risitano et al., 2016). Allogeneic hematopoietic stem and progenitor cell (HSPC) transplantation is the long-term curative treatment for the BMF in FA (Ebens et al., 2017, Shimamura and Alter, 2010, Guardiola et al., 2000). Alternative treatment approaches, such as gene therapy, are underway (Rio et al., 2019).
FA is caused by biallelic mutations in one of twenty-three FANC genes, the protein products of which cooperate in the FA/BRCA DNA repair pathway and regulate cellular resistance to DNA cross-linking agents (Rodríguez and D’Andrea, 2017). Due to their underlying DNA repair defect, FA cells exhibit chromosomal instability and hypersensitivity to genotoxic DNA crosslinking agents, such as Mitomycin C (MMC) (Oostra et al., 2012). FA BM HSPCs are also hypersensitive to oxidative stress (Li et al., 2017) and inflammatory cytokines (Briot et al., 2008).
FA patients develop BMF due to HSPCs exhaustion. Progressive age-related attrition is observed in CD34+ cell content in FA patients (Ceccaldi et al., 2012). Additionally, FA patients and FA mice exhibit HSPC functional defects (Ceccaldi et al., 2012, Parmar et al., 2010, Parmar et al., 2009, Haneline et al., 1999). BMF in FA results from accumulation of DNA damage in HSPCs caused by the endogenous crosslinking agents (Garaycoechea et al., 2018, Pontel et al., 2015) or from physiological stress (Walter et al., 2015). In response to genotoxic stress, FA HSPCs hyperactivate growth suppressive pathways, such as the p53 pathway (Ceccaldi et al., 2012) and the TGF-β pathway, further contributing to BMF (Zhang et al., 2016).
The molecular pathways in FA HSPCs leading to BMF and MDS/AML remain unknown. Although primary HSPCs from BM of FA patients provide a useful model system, studying these cells is challenging, due to their heterogeneity and low number. Subpopulations of HSPCs with heterogeneous transcriptional profiles may co-exist in the BM of FA patients. These subpopulations may include- 1) a stressed HSPCs sustaining hematopoiesis, 2) HSPCs committed to apoptosis resulting from the accumulation of unrepaired DNA damage, and 3) premalignant/malignant cells that eventually lead to clinically detectable MDS or AML.
Single cell RNA sequencing (scRNA-seq) allows high-resolution whole-transcriptome profiling of individual cells and direct analysis of cell subpopulations from heterogeneous samples (Zheng et al., 2017). ScRNAseq can allow the discovery of new cell populations, and the comparison of the relative abundance of cell populations from different groups of patients including the patients with BMF (Zheng et al., 2018, Watcham et al., 2019, Tikhonova et al., 2019, Zhao et al., 2017, Joyce et al., 2019).
Here, we used scRNAseq to transcriptionally profile HSPCs from 7 patients with FA and 5 healthy donors. Importantly, FA HSPCs overexpressed the MYC pathway seemingly as a counteracting force against the growth suppressive activities of the p53 and TGFβ pathways. The “High-MYC” expressing HSPCs from patients with FA also have a reduction in the expression of cell-adhesion genes, resulting in a propensity for egress from the BM.
RESULTS AND DISCUSSION
Early hematopoietic differentiation is preserved in FA HSPCs
Using scRNAseq technologies we analyzed HSPCs from 7 FA patients and 5 healthy donors (Supplementary Table 1) and resolved the heterogeneity of their gene expression profile. We combined the sequenced FA and healthy HSPCs in a single bioinformatic pipeline (Figure 1A). Using Seurat (Butler et al., 2018), we deconvoluted the different clustering subpopulations with CD34 expression. Using gene expression profiles of human HSPCs subpopulations from healthy donors (Velten et al., 2017, Zheng et al., 2018, Setty et al., 2019), we identified HSPC clusters (Supplementary Figure 1A). Thirteen different HSPCs clusters were identified, including a HSC-containing cluster (identified by green in Figure 1B) and two divergent progenitor populations (composed by several clusters). Clusters committed to the lympho-myeloid fate (shaded blue colors in Figure 1B) and the megakaryocyte-erythroid fate (shaded red colors in Figure 1B) were also identified.
Figure 1. Single cell RNA sequencing reveals that early hematopoiesis is not perturbed in FA.
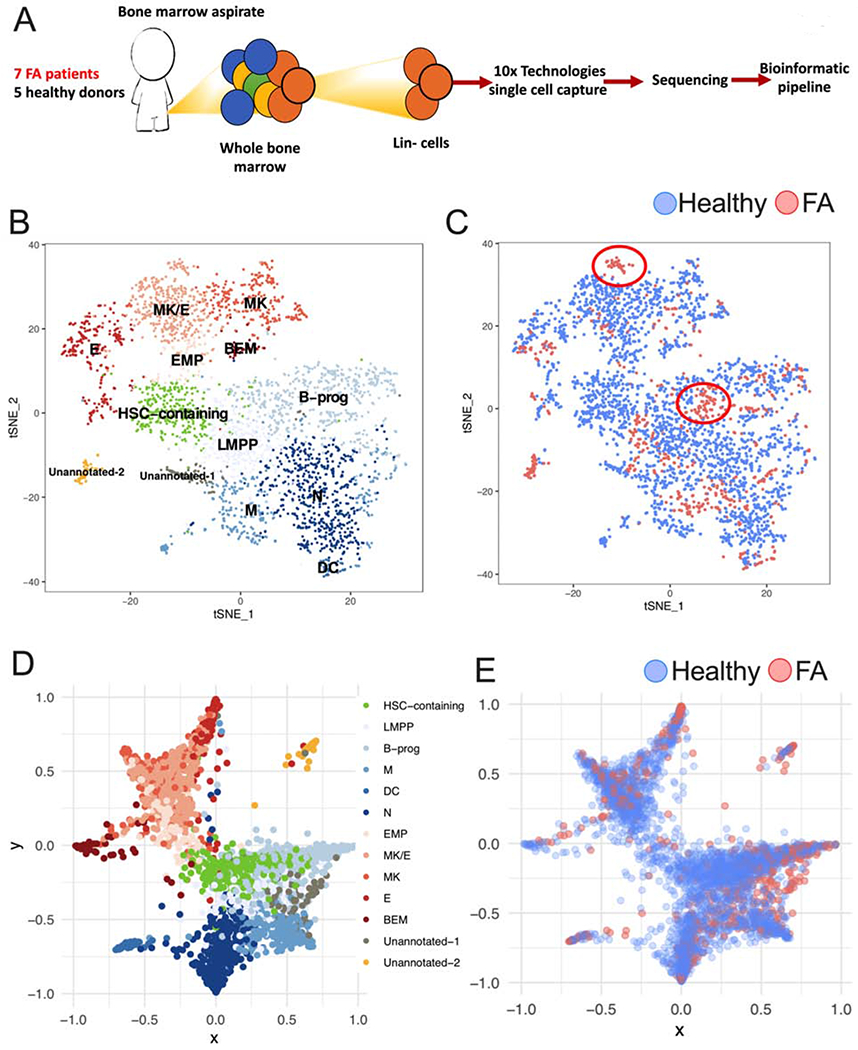
(A) Lin- cells from 7 patients with FA and 5 healthy donors (Supplementary Table 1) were individually captured, lysed and barcoded using the 10x Chromium™ controller platform. Barcoded bulk cDNA was sequenced in an Illumina platform. Unique Molecular Identifiers (UMIs) were used for sequencing deconvolution and clustering analysis, DEG analysis and lineage analysis.
(B) t-SNE plot showing the clustering analysis for CD34 expressing HSPCs. FA and healthy CD34 expressing HSPCs were combined for clustering based on their gene expression profiles. A total of 13 clusters were identified spanning the different HSPC subpopulations. Identified clusters include a HSC-containing cluster (HSC-containing); clusters with megakaryocytic-erythroid identity include: EMP (Erythroid-megakaryocyte progenitor), MK (Megakaryocyte progenitor), MK/E (common megakaryocyte/erythroid cluster), E (erythroid progenitor) and BEM (basophil, eosinophil and mast cell progenitor); clusters with lympho-myeloid identity include LMPP (lymphoid-primed multipotential progenitor), B-prog (B-cells progenitor), N (neutrophil progenitor), DC (dendritic cell progenitor) and M (monocyte progenitor). Two clusters with distinctive gene expression profiles, one of them enriched in FA cells, were assigned as “unannotated” since they required further research for identity definition.
(C) FA-derived CD34 expressing HSPCs (red dots) cluster together with healthy donors derived CD34 expressing HSPCs (blue dots). Two FA-specific sub-clusters are highlighted with circles.
(D) Lineage trajectory analysis showing that HSPC differentiation follows a star-like path. HSCs (green) located in the center region of this star follow at least two main differentiation pathways, one characterized by lympho-myeloid commitment (shades of blue), and the other characterized by megakaryocytic-erythroid commitment (shades of red). The lympho-myeloid commitment includes B, M, DC and N progenitors; the megakaryocytic-erythroid commitment spans MK, E and BEM progenitors.
(E) Projection of FA HSPCs in the lineage trajectory analysis showing that FA HSPCs follow the same differentiation star-like profile as healthy cells do. FA HSPCs (red dots), healthy HSPCs (blue dots).
See also Supplementary Figure 1.
Supplementary Table 1 describes the characteristics of heathy donors and FA patients whose bone marrow samples were used for the experiments described in panels A-E.
Interestingly, FA and healthy cells clustered together in every subpopulation, demonstrating that lineage commitment during early hematopoietic differentiation is preserved in FA (Figure 1C). An unusual cell cluster “Unannotated-2 cluster” (Identified by yellow in Figure 1B) with unique gene expression profile of unknown significance was enriched in FA-derived cells. Because FA patients have a strong predisposition to MDS and AML, we speculate this cluster may consist of pre-leukemic cells or leukemia stem cells. Besides this cluster, there were two additional FA-specific sub-clusters; one in the B-progenitors and one in the MK/E cluster (identified by circles in Figure 1C). Both of these clusters were part of a larger cell-type cluster. Analysis of cells in these FA-specific sub-clusters revealed specific functional differences from the corresponding big clusters, such as the elevation of expression of genes involved in TNF-α pathway, Toll-like receptor pathway, and apoptosis pathway, consistent with bone marrow failure in FA patients.
Using StemNet (Velten et al., 2017), and the gene expression profile of each cell, we performed a lineage trajectory analysis and tracked the differentiation pathways in FA and normal BM (Figure 1D). Expression of markers previously described (Velten et al., 2017, Zheng et al., 2018) were used for lineage trajectory analysis (Supplementary Figure 1B). Cells positive for AVP and negative for CDK6 expression in the HSC-containing cluster have the most primitive gene expression profile and were used as the root of the lineage trajectory analysis (Zheng et al., 2018). In concordance with the previous study (Velten et al., 2017), committed progenitors emerged early from primitive HSCs and not from intermediary progenitors. In addition, two very distinctive differentiation outcomes were identified. One pathway is composed of cells committed to the lympho-myeloid fate (shades of blue in Figure 1D), and the other pathway is composed of cells committed to the megakaryocyte-erythroid fate (shades of red in Figure 1D). Taken together, these results support the hypothesis that these cell populations are the major branches of differentiation emerging from the most primitive human HSCs.
FA cells (red dots) follow the same differentiation trajectories as normal cells (blue dots), indicating that FA HSPCs exhibit an appropriate hematopoietic differentiation program (Figure 1E). Expression of CD38, a progenitor-commitment marker, was gained by CD34-expressing cells as they differentiate into specific cell types (Supplementary Figure 1C). FA patients had a decreased number of cells in the HSC-containing cluster and an increased frequency of cells in cluster Unannotated-2, in comparison to healthy donors (Supplementary Figure 1D). Quantitation of CD34-expressing cells and HSCs confirmed the decreased number of these populations in FA patients compared to healthy donors, consistent with a previous report (Ceccaldi et al., 2012). Additionally, older FA patients had a reduced number of CD34-expressing cells compared to the younger patients (Supplementary Figure 1E and 1F). The reduced CD34-expressing cell content in FA patients did not correlate with their aplastic anemia status (Supplementary Figure 1G).
The MYC pathway is overexpressed in most HSPC clusters from FA Bone Marrow
Inter-cluster gene expression analysis identified MYC as one of the top over-expressed genes in the CD34-expressing cells from FA patients (Figure 2A). As a consequence, the subset of genes transcriptionally-regulated by MYC are also significantly over-expressed in FA across all of the HSPC clusters, as shown by single cell enrichment score of CD34-expressing cells (Figure 2B).
Figure 2. The MYC pathway is overexpressed in FA HSPCs.
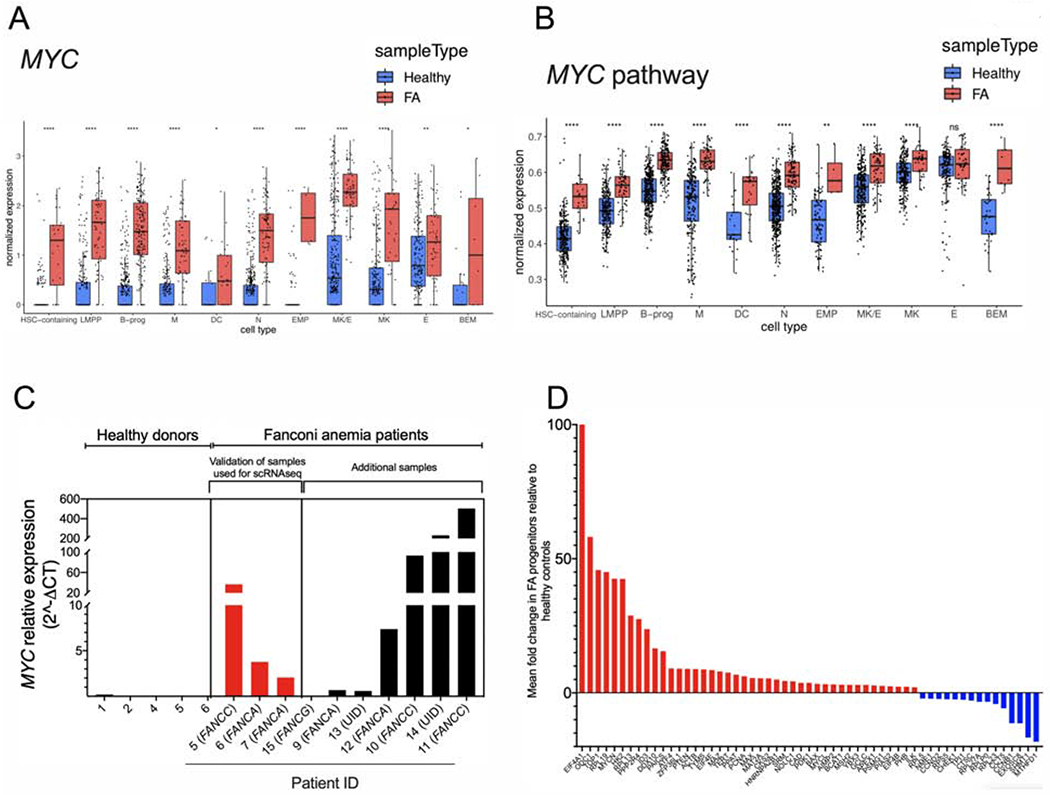
(A) Single cell gene expression analysis of the different HSPCs clusters.
(B) Single cell enrichment score analysis of the different HSPCs clusters.
(C) Quantitative real-time RT-PCR for MYC expression in primary HSPCs from FA patients and healthy donors. The Supplemental Table 1 and Supplemental Table 2 describe the characteristics of healthy donors and FA patients whose bone marrow samples were used in this study. Complementation group of each patient with FA is indicated between parenthesis. UID: Unidentified pathogenic variant.
(D) Real-time RT-PCR for expression of the MYC pathway genes in bulk population of FA HSPCs in comparison to HSPCs from healthy controls (see Supplementary Table 2 for the details about the samples).
Data in (A) and (B) are represented as box plots. p-values of <0.001 were considered extremely significant (***, ****). See also Supplementary Figure 2.
In addition to MYC, other genes over-expressed in the CD34 expressing cells from FA patients include the members of the immunoglobulin super-family, previously linked to stimulatory division signals in premalignant states. Expression of DYNLL1, a recently described gene regulating anti-resection and NHEJ, was also upregulated in FA cells (He et al., 2018), consistent with the well-known role of the hyperactive NHEJ in FA pathogenesis (Adamo et al., 2010, Pace et al., 2010, Eccles et al., 2018). HSPA8, was also upregulated, consistent with the observation that chaperone proteins are upregulated in FA (Karras et al., 2017); RPL9, a ribosome gene, exhibited increased expression, consistent with a nucleolar function of an FA protein in ribosome biogenesis (Sondalle et al., 2019). The top 100 differentially expressed genes (DEG) in FA HSPC versus HSPC from healthy donors, are shown in Supplementary Figure 2A.
MYC overexpression was validated through real-time PCR in progenitor-enriched fractions from BM of FA patients (Figure 2C). In addition to MYC overexpression, which varies greatly among FA patients, we detected, in some samples, upregulation of the MYC paralog, MYCN, and MAX, the dimerization partner of MYC (Mathsyaraja and Eisenman, 2016) (Supplementary Figure 2B). Inter-patient scRNAseq analysis demonstrated a clear overexpression of MYC in most of the samples derived from patients with FA, one healthy control exhibited increased levels of MYC expression demonstrating interindividual variability (Supplementary Figure 2C). No significant variation was observed in terms of MYC levels with respect to the age of the FA patients (Supplementary Figure 2C). However, the MYC phenotype was associated with the severity of the aplastic anemia status. FA patients with mild/moderate disease, but not the patients with severe disease, had high MYC levels in their HSPCs (Supplementary Figure 2D).
Since MYC is a transcription factor (Nasi et al., 2001), we validated changes in the expression of MYC targets in bulk HSPCs from eleven FA patients. EIF4A1 showed the highest upregulation level (Figure 2D), consistent with the known transcriptional regulatory pattern of MYC. Overexpression of EIF4A1, ODC1, and TOP1, in primary HSPCs may indicate that the patients are responding to inflammatory stimuli (Figure 2D).
Single cell enrichment score of scRNAseq dataset using AUCell verified enrichment of other pathways previously known to be over-expressed in FA, including the p53 pathway (Ceccaldi et al., 2012) (Supplementary Figure 2E) and the TGFβ pathway (Zhang et al., 2016) (Supplementary Figure 2F). Finally, we observed that FA patients who have mosaicism (Supplementary Tables 1 and 2) (Supplementary Figure 2G) or clonal hematopoiesis (Supplementary Figure 2H) still exhibit the characteristic increase in MYC expression in their HSPCs, comparable to the level observed in other FA patients.
HSPCs in two opposing functional states co-exist in the BM of patients with FA.
Overexpression of TP53, previously demonstrated in FA and linked to BM failure (Ceccaldi et al., 2012), was validated in the scRNAseq data set (Supplementary Figure 2E). However, real time-PCR analysis of bulk population of primary HSPCs did not show significant overexpression of TP53 in all the FA patients (Supplementary Figure 3A). Since MYC and TP53 are transcription factors with opposing activities (Aguda et al., 2011), cells with their differential outcomes may co-exist in the BM of FA patients. By dividing the scRNAseq LogRatio of TP53 expression by the LogRatio of MYC expression, we identified a gradient of High TP53 expressing FA HSPC and High MYC expressing FA HSPC (Figure 3A). The TP53-MYC gradient was not exclusive of FA BM cells; however, both the “high TP53” and “high MYC” cell states were exacerbated in FA. Bone marrow from healthy donors exhibited more “balanced” state whereas bone marrow samples from FA patients exhibited “unbalanced” expression of both the genes, with a clear indication of high MYC expression in FA. This gradient behavior was observed in every HSPC cluster from FA patients (Supplementary Figure 3B) and along the entire trajectory of HSPC differentiation (Supplementary Figure 3C).
Figure 3. HSPCs expressing two opposing functional cell states exist in the BM of patients with FA.

(A) “High-TP53” expressing HSPCs and “High-MYC” expressing HSPCs co-exist in the BM of patients with FA. The LogRatio of TP53 expression was divided by the LogRatio of MYC expression per cell, resulting in a gradient spanning from “High-TP53” expressing HSPCs to “High-MYC” expressing HSPCs. The expression of MYC and TP53 per cell is represented with a red or blue bar.
(B) Flow cytometric analysis confirming the co-existence of MYC and p53 expressing CD34+ cells from FA patients. FACS plot for gating CD34+ cells is shown in left panel. FACS plot for MYC and p53 expression in CD34+ cells is shown in right panel. Two seemingly exclusive populations of FA CD34+ cells were detected, one with high MYC levels (shaded green box) and the other with high p53 levels (shaded pink box) in comparison to healthy cells. A third population (shaded in orange box) in samples from FA patients co-expresses medium-high levels of MYC and p53 in comparison to healthy CD34+ cells. Data are from pooled BM samples of two healthy controls and eight FA patients.
(C) FACS plot (left panel) and quantitation (right panel) of DNA content showing high MYC expression in FA CD34+ cells during S and G2/M phases of the cell cycle. The data are from pooled healthy BM controls (n=2) and patients with FA (n=8).
Data in (C) are represented as box plots. p-values of <0.001 were considered extremely significant (***, ****). See also Supplementary Figure 3.
In every patient with FA, this dichotomy of “High TP53” and “High MYC” cellular states is present (Supplementary Figure 3D). Of note, bone marrow from one of the healthy individuals also showed a moderate dichotomy of the cellular states. The presence of “High-MYC” and “High-p53” HSPC populations in FA was also confirmed by flow cytometric analysis of p53 and MYC protein levels (Figure 3B). Increased p53 expression was more evident at the protein levels than at the RNA levels. CD34+ cells in S phase and G2 phase showed the highest levels of MYC, suggesting that MYC is required for progression into the cell cycle of FA CD34+ cells (Figure 3C). The dichotomic cellular state, orchestrated by TP53 versus MYC, suggests that FA patients have HSPC populations controlled by different cellular states. Collectively. these results also support a model of mutual exclusivity and suggest that some FA HSPCs are prone to apoptosis or quiescence (High-p53) and some are prone to enhanced proliferation and survival (High-MYC).
MYC is required for proliferation of FA hematopoietic cells at the expense of DNA damage
Previous studies have failed to detect MYC gene amplifications or translocations in the BM or leukemias derived from FA patients (Quentin et al., 2011). Accordingly, we hypothesized that the increase in MYC expression in FA BM cells may result from transcriptional upregulation, perhaps via a super enhancer mechanism, and activation of the BET proteins (Bahr et al., 2018). We tested this hypothesis using a BRD4 inhibitor with super enhancer inhibitory capacity, (+)-JQ1, known to suppress MYC transcription (Qi, 2014).
scRNAseq analysis of linage negative (Lin−) cells from bone marrow of FA mice (Fancd2−/− mice) showed a gradient of High Trp53 expressing and High Myc expressing cells (Supplementary Figure 4A). In comparison to the bone marrow from FA patients, however, the co-existence of Trp53-high and Myc-high HSPCs was less prominent in bone marrow of FA mice. This is consistent with the previous observations that most FA mice do not exhibit spontaneous bone marrow failure unless they are exposed to physiological stress-inducing agents (Parmar et al., 2009, Walter et al., 2015).
We next injected (+)- JQ1 or its inactive enantiomer (−)-JQ1 daily over a 1 month period to WT or Fancd2−/− mice (Figure 4A) and analyzed the effect on the long-term-HSC (LT-HSC) compartment (Figure 4B). Higher basal levels of Myc protein were observed in the LT-HSC from Fancd2−/− mice in comparison to WT mice, and (+)-JQ1 reduced in vivo the levels of Myc (Figure 4C). This effect was accompanied by a reduction in the percentage of LT-HSCs (Figure 4D) and a reduction in the percentage of cycling Ki67 positive cells in the same compartment (Supplementary Figure 4B), suggesting that Myc pathway activity is required for proliferation of the hematopoietic stem cell pool in FA mice. As MYC is required for the proliferation of HSPCs and normal hematopoiesis (Wilson et al., 2004), the reduction in the percentage in LT-HSC and in cycling Ki67 cells was also observed in WT mice (Figure 4D and Supplementary Figure 4B). Reduction of LT-HSC by (+)-JQ1 was accompanied by reduction in the production of cells from the lympho-myeloid blood compartment (Supplementary Figure 4C). (+)-JQ1 exposure in FA mice induced a CD48+CD150+ LSK cell population- namely, the HPC-2 population with megakaryocyte potential (Figure 4B) (Oguro et al., 2013).
Figure 4. (+)-JQ1 inhibits the growth of FA-deficient hematopoietic stem cells.
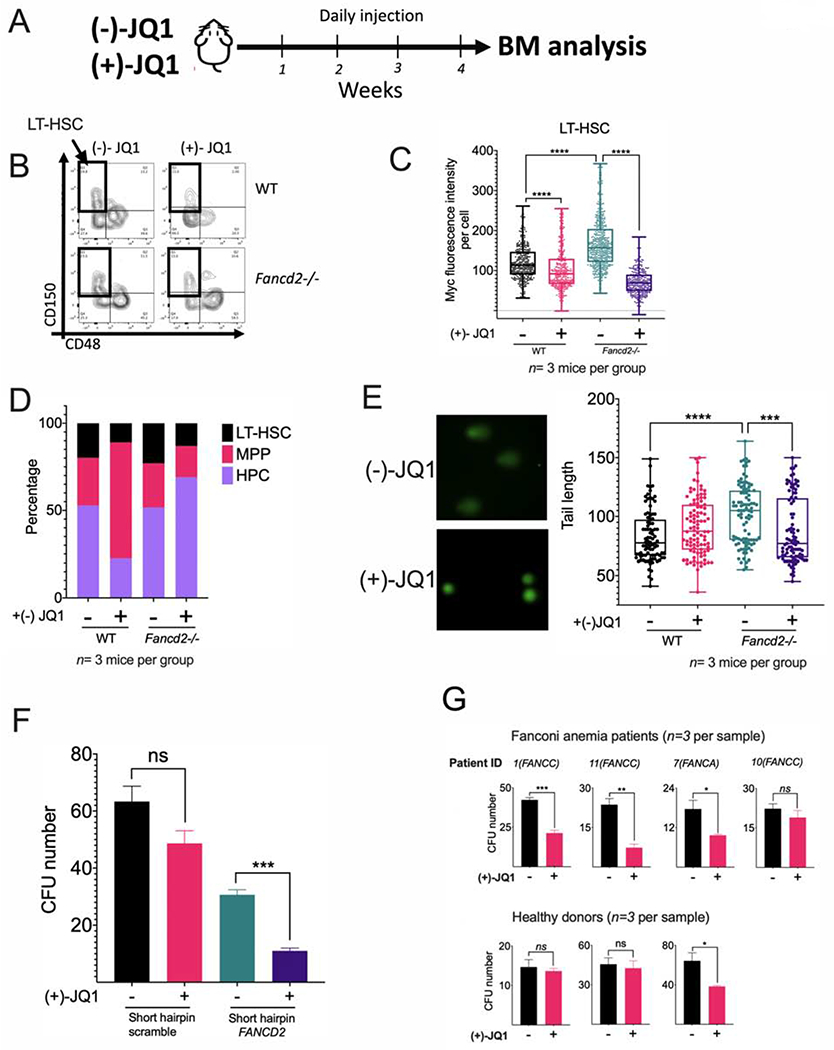
(A) WT and Fancd2−/− mice were injected daily with (+)-JQ1 (50 mg/kg) for 1 month or the inactive enantiomer (−)-JQ1 (50 mg/kg) as a negative control (n=3 mice per group) and BM was analyzed.
(B) Flow cytometry plots for identification of LT-HSCs in wild-type (WT) and Fancd2−/− mice exposed to (−)-JQ1 or (+)-JQ1. LSK (Lin-Sca-1+c-Kit+) population was analyzed for CD150 and CD48 expression and LT-HSCs (LSK CD150+CD48−) were identified.
(C) Quantitation of Myc expression by FACS analysis in LT-HSCs from wild-type (WT) and Fancd2−/− mice exposed to (−)-JQ1 or (+)-JQ1. (n=3 mice per group).
(D) LT-HSC (Long-term HSC), MPP (multipotent progenitors), HPC (restricted hematopoietic progenitors) content in wild-type (WT) and Fancd2−/− mice exposed to (−)-JQ1 or (+)-JQ1 (n=3 mice per group).
(E) DNA damage in LT-HSCs from wild-type (WT) and Fancd2−/− mice exposed to (−)-JQ1 or (+)-JQ1. DNA damage was measured by tail length in a comet assay (n=3 mice per group). Representative images of the alkaline comets (left panel) and quantitation of comet tail length (right panel) are shown.
(F) CFU assay of FA-like CD34+ HSPCs, generated through infection with a shRNA lentivirus against FANCD2. FA-like HSPCs were cultured in triplicates in complete methylcellulose medium with or without (+)-JQ1 (50 nM) for 14 days and clonogenic growth was assessed.
(G) CFU assay of HSPCs from bone marrow of healthy donors or FA patients. Healthy and FA BM derived Lin- cells (3000 cells for healthy BM and 5000 cells for FA BM) were plated in complete methycellulose with (+)-JQ1 (50 nM) or DMSO and CFU numbers were quantified after 14 days of culture. Note that the cells were plated at different time depending upon the availability of the samples.
Data in (C) and (E) are represented as box plots. Data in (F) and (G) are represented as mean ± SEM. p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***, ****). See also Supplementary Figure 4.
Cell proliferation required for blood cell production needs several rounds of DNA replication. This process appears to be promoted by the Myc pathway and contributes to the production of physiologically-induced DNA damage in FA pathway deficient cells. Accordingly, the in vivo inhibition of the Myc pathway with (+)-JQ1 reduced the amount of DNA damage in Fancd2−/− LT-HSC (Figure 4E). In line with the former, in vitro proliferation of LSK cells from Fancd2−/− mice was reduced by exposure to (+)-JQ1 in comparison to WT LSK cells (Supplementary Figure 4D).
To determine whether human FA pathway-deficient HSPCs depend on MYC for cell division, we generated FA-like HSPCs, using a lentivirus expressing a short-hairpin RNA against FANCD2 (Zhang et al., 2016). FA-like HSPCs exhibited reduced clonogenic potential when exposed to (+)-JQ1, confirming that MYC enhances cell division (Figure 4F). In addition, FANCG-knockout (FANCG-KO) cord blood CD34+ cells, generated by CRISPR/Cas9 genome editing, also exhibited high MYC and high p53 expression profile similar to what was observed in primary HSPCs from FA patients (Supplementary 4F and Figure 3B). Knocking out MYC in the FANCG-KO cord blood CD34+ cells reduced their clonogenic potential and confirmed that MYC is required for survival of primary FA HSPCs (Supplementary Figure 4G). Similarly, the clonogenic potential of primary HSPCs from FA patients was also reduced after (+)-JQ1 exposure (Figure 4G and Supplementary Figure 4H). Taken together, our results suggest that MYC upregulation has some benefits for FA HSPCs, resulting in enhancement of cell proliferation and survival.
Overexpression of MYC was validated in the FA lymphoblast cell line EUFA316+EV (FANCG-deficient), derived from a patient with FA and reversed by functional complementation of the cells with the FANCG cDNA (EUFA316+G) (Figure 5A). As expected, several MYC pathway genes were over expressed in the EUFA316+EV cell line (Supplementary Figure 5A). Overexpression of MYC was efficiently blocked by the BET bromodomain inhibitor (+)-JQ1 (Figure 5B, Supplementary Figure 5B).
Figure 5. MYC inhibition prevents entry into S phase and reduces replication associated stress in FA lymphoblastoid cells.
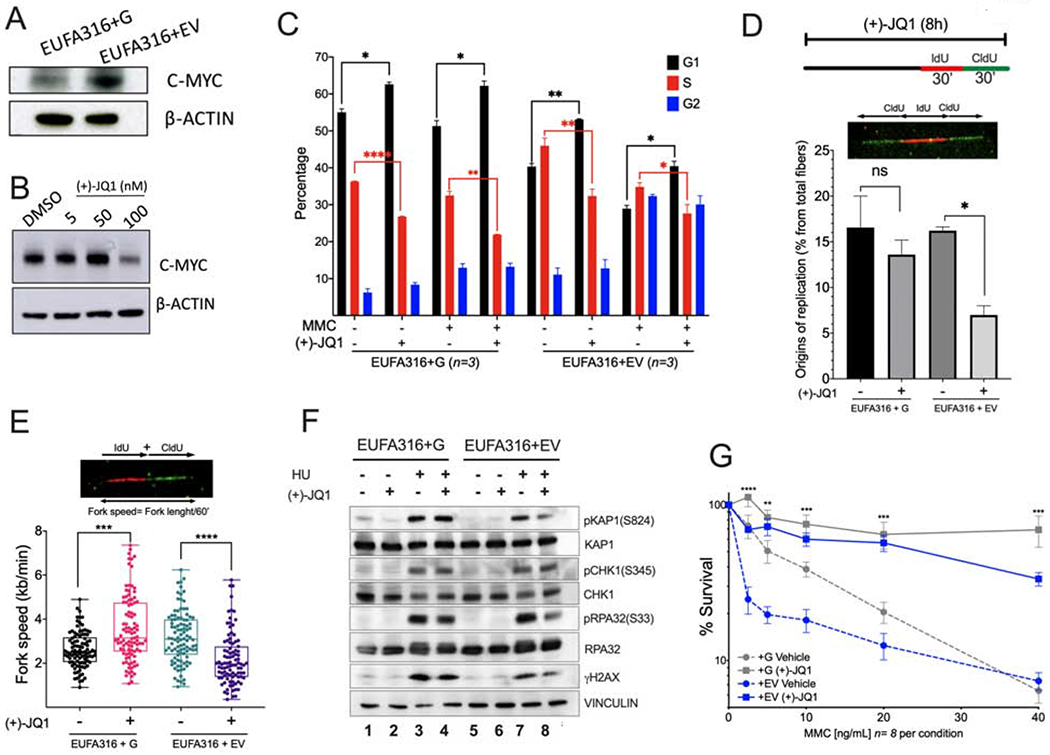
(A) Western blots of the lysates of FANCG-deficient human lymphoblastoid cells (EUFA316+EV) and FANCG-complemented lymphoblastoid cells (EUFA316+G).
(B) Western blots of the lysates of EUFA316+EV cells exposed to the BET bromodomain inhibitor (+)-JQ1 for 48 h.
(C) EUFA316+G and EUFA316+EV cells were treated for 24h with Mitomycin C (MMC) (20 ng/ml) and (+)-JQ1 (50 nM) and cell cycle distribution was analyzed.
(D) (+)-JQ1 reduces the amount of new replication origins in the FANCG-deficient lymphoblastoid cells. EUFA316+G and EUFA316+EV cells were exposed to (+)-JQ1 for 8 h and then incubated with the nucleotide analogs IdU for 30 min followed by CldU for 30 min in presence of (+)-JQ1. DNA fiber assay was carried out and the percentage of fibers with replication origins were quantified per condition.
(E) (+)-JQ1 slows-down replication fork speed in the FANCG-deficient lymphoblastoid cells. EUFA316+G and EUFA316+EV cells were treated as described in panel D. DNA fiber assays were performed and the fiber length composed by IdU tracts plus CldU tracts was measured and divided by the incubation time in the presence of the analogs. At least 100 fibers were analyzed per sample.
(F) Western blots of the lysates of EUFA316+EV and EUFA316+G cells after treatment with hydroxyurea (HU) and (+)-JQ1 for 24 h.
(G) Survival of the FANCG-deficient EUFA316+EV and EUFA316+G cells exposed to increased concentrations of MMC and (+)-JQ1 (50 nM) for 5 days. The experiment was performed three times and the data from a representative experiment are shown.
Data in (C), (D) and (G) are represented as mean ± SEM. Data in (E) are represented as boxplots. p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***, ****). See also Supplementary Figure 5.
Reduced proliferation capacity of FA pathway deficient cells exposed to (+)-JQ1 appeared to have protective effects against DNA damaging agents. We therefore analyzed cell cycle status of the FANCG-deficient cell line, EUFA316+EV, when exposed to (+)-JQ1 and genotoxic agent Mitomycin C (MMC). As expected, FANCG+EV cells showed an increase in the number of cells in G2 after exposure with MMC (Figure 5C). Exposure of EUFA316+EV cells to (+)-JQ1 and MMC resulted in an increase in the number of cells in G1, with a concomitant reduction in the number of cells in S-phase (Figure 5C). DNA fiber assays also showed that exposure to (+)-JQ1 has a direct effect in cell proliferation by reducing the opening of new origins of replication (Figure 5D) and reducing replication fork speed of EUFA316+EV cell line (Figure 5E), confirming that FA cells require MYC for progression through the cell cycle. Since EUFA316+EV cells expressed high levels of MYC, the major effect of (+)-JQ1 appears to be the reduction in MYC expression, leading to decreased fork speed and to an increase in the G1 fraction of the cell cycle. Interestingly, the wild-type cells (FANCG-corrected cells) exhibited an increase in fork speed following short-term (+)-JQ1 exposure, consistent with a previous study (Bowry et al., 2018).
We next determined whether MYC inhibition contributed to reduce replication stress. Replication stress from MYC upregulation is known to activate the ATR-CHK1 pathway, leading to the generation of single strand DNA (Maya-Mendoza et al., 2015, Herold et al., 2009, Puccetti et al., 2019). Interestingly, (+)-JQ1 reduced the replication stress and DNA damage induced by HU or MMC as indicated by decreased phosphorylation of pRPA and γH2AX in FANCG-deficient lymphoblast cells (Figure 5F, Supplementary Figure 5C). In addition, (+)-JQ1 improved the survival to MMC of FA cells in 5 days survival assays (Figure 5G). Of note, induction of DNA damage by both HU and MMC is dependent on active S-phase. As (+)-JQ1 treatment increases the number of cells in G1 and decreases cells in S phase (Figure 5C), the improved survival of FA cells under genotoxic stress may be attributed to the inhibition of toxic effects due to reduced replication fork speed and reduced replication origin firing. Collectively, inhibition of MYC improves survival and avoids DNA damage in FA cells by inhibiting the accumulation of toxic effects, which are S-phase dependent.
Physiological stress by the immunostimulant molecule pI:pC activates the MYC pathway in a mouse model of FA
Polyinosinic:polycytidylic acid (pI:pC) is a double-stranded RNA mimetic and immunostimulant that promotes the activation of inflammatory responses. pI:pC is a potent inducer of BM failure in FA mice since it prompts HSCs to enter into the cell cycle and accumulate DNA damage (Walter et al., 2015). We tested the potential of pI:pC to induce MYC activation in FA mice (Figure 6A). One month after pI:pC injection, BM cells from Fancd2−/− mice exhibited a pathological expansion of the LSK compartment accompanied by a reduction in the number of LT-HSCs, consistent with a previous report (Walter et al., 2015) (Supplementary Figure 6A). Also, a striking increase in Myc expression was observed in the LT-HSCs of Fancd2−/− mice (Figure 6B), suggesting that Myc pathway plays a role in expansion and subsequent exhaustion of HSCs in the FA bone marrow.
Figure 6. Physiological/inflammatory stress activates the MYC pathway in Fancd2−/− mice and causes BM failure.
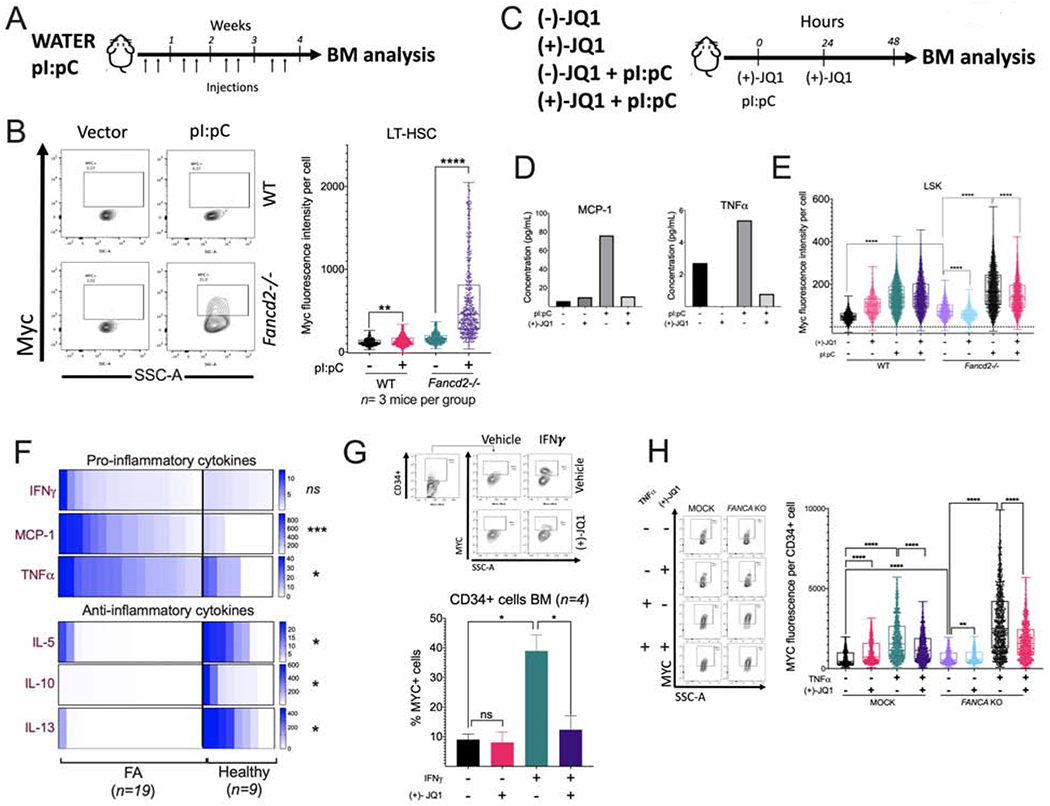
(A) Wild-type (WT) and Fancd2−/− mice were injected with pI:pC (5 mg/kg) twice per week for 1 month and bone marrow was analyzed.
(B) Flow cytometric plots (left panel) and the quantitation (right panel) for Myc expression in LT-HSCs of the bone marrow from wild-type or Fancd2−/− mice (n=3 mice per group) after pI:pC treatment as shown in panel A.
(C) Wild-type (WT) and Fancd2−/− mice were treated with pI:pC (5 mg/kg) and (+)-JQ1 and 48 h after the treatment BM was analyzed. (−)-JQ1 was used as a negative control. (n=3 mice per group)
(D) Wild-type or Fancd2−/− mice were treated with pI:pC and (+)-JQ1 as shown in panel C and peripheral blood was analyzed for cytokines.
(E) Wild-type or Fancd2−/− mice were treated with pI:pC and (+)-JQ1 as shown in panel C and Myc expression in LSK cells from BM was analyzed using flow cytometry.
(F) Cytokine levels in the BM plasma of FA patients and or healthy donors. FA n=19, Healthy n=9.
(G) Representative flow cytometry plots for MYC staining (upper panels) and quantitation of MYC expression (lower panel) in BM CD34+ cells from healthy donors after in vitro culture for 24 h with IFNγ (20 ng/ml) and/or JQ1 (50 nM). (n= 4)
(H) Representative flow cytometry plots for MYC staining (left panels) and quantitation of MYC expression (right panel) in cord blood CD34+ with CRISPR-mediated knockout of FANCA (FANCA-KO) after in vitro culture for 24 h with TNF-α and/or JQ1.
Data in (B), (E) and (H) are represented as boxplots. Data in (G) are represented as mean ± SEM. p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***, ****). See also Supplementary Figure 6.
We next co-injected pI:pC and (+)-JQ1 into WT and Fancd2−/− mice (Figure 6C), and observed that 48 h after treatment initiation, (+)-JQ1 was able to reduce the activation of a subset of pro-inflammatory cytokines in Fancd2−/− mice, including MCP1 and TNF-α (Figure 6D). This short-term exposure of mice to (+)-JQ1 reduced the pI:pC-induced Myc expression in the LSK cells but not in LT-HSCs in Fancd2−/− mice (Figure 6E, Supplementary Figure 6B). (+)-JQ1 also reduced in vivo the pI:pC induced pathologic expansion of HSPCs (LSK cells) in Fancd2−/− mice (Supplementary Figure 6C) and ameliorated the pI:pC-induced reduction of LT-HSC (Supplementary Figure 6D). Finally, (+)-JQ1 exposure improved the peripheral blood WBC counts in pI:pC-injected Fancd2−/− mice (Supplementary Figure 6E). In agreement with reduced expansion of the HSPC compartment after exposure to (+)-JQ1 upon pI:pC treatment, we observed a reduction in DNA damage (Supplementary Figure 6F).
As the acute inflammatory response induced by pI:pC drove Myc pathway activation in FA mouse model, we returned to samples from FA patients and explored inflammatory cues that might be responsible for MYC pathway activation in the human setting. Inflammatory cytokines are increased in the BM of FA patients (Dufour et al., 2003, Briot et al., 2008, Garbati et al., 2016). We confirmed the increased concentration of pro-inflammatory cytokines in the BM plasma of patients with FA with respect to healthy subjects, including IFNγ, MCP1 and TNF-α (Figure 6F upper panel). Interestingly, anti-inflammatory cytokines IL-5, IL-10, and IL-13, which have the capacity to mitigate the activation of immune cells (Banchereau et al., 2012, Couper et al., 2008), had significantly decreased concentrations in the BM plasma from FA patients, perhaps contributing to a sustained proinflammatory BM environment (Figure 6F lower panel). In addition, FA patients showed increased C-reactive protein (CRP) protein levels (Supplementary Figure 6G). CRP is known to be elevated during inflammatory conditions (Sproston and Ashworth, 2018). To confirm a role for inflammatory cytokines in the transcriptional activation of MYC, we exposed CD34+ cells in vitro to these pro-inflammatory cytokines. IFNγ increased the percentage of CD34+ cells expressing MYC and (+)-JQ1 counteracted this effect (Figure 6G). Since TNF-α is implicated in the pathogenesis of FA (Rosselli et al., 1994, Dufour et al., 2003, Li et al., 2007), we next evaluated its effect in cord blood CD34+ cells with CRISPR knockout of FANCA. Interestingly, FANCA-KO HSPCs exhibited higher expression of MYC compared to the mock controls and this was even further increased after in vitro exposure to TNF-α (Figure 6H). As expected, the addition of (+)-JQ1 counteracted this effect (Figure 6H). Treatment with MCP1 did not induce any changes (data not shown).
MYC overexpression causes detachment of FA HSPCs from their bone marrow niche
Gene expression profiles of “High-TP53” expressing HSPCs and “High-MYC” expressing HSPCs from bone marrow of FA patients revealed that these cellular states have opposite gene expression profiles (Figure 7A). High-TP53 expressing HSPCs exhibited high expression of CXCR4, a pro-quiescence gene (Nie et al., 2008), whereas High-MYC expressing HSPCs had a strong downregulation of CXCR4 and other cell adhesion genes, such as VIM. CXCR4 is a critical regulator of adhesion of stem cells to their BM niche (Karpova and Bonig, 2015). MYC overexpression is known to negatively impact the adhesion of HSPCs to their BM niche (Wilson et al., 2004). Taken together, these results suggest that, in addition to enhanced proliferation, “High-MYC” expressing HSPCs have weak adhesion to their BM niche and a predisposition to detachment.
Figure 7. Physiological stress-induced MYC overexpression predisposes FA HSPCs to egress from the BM.
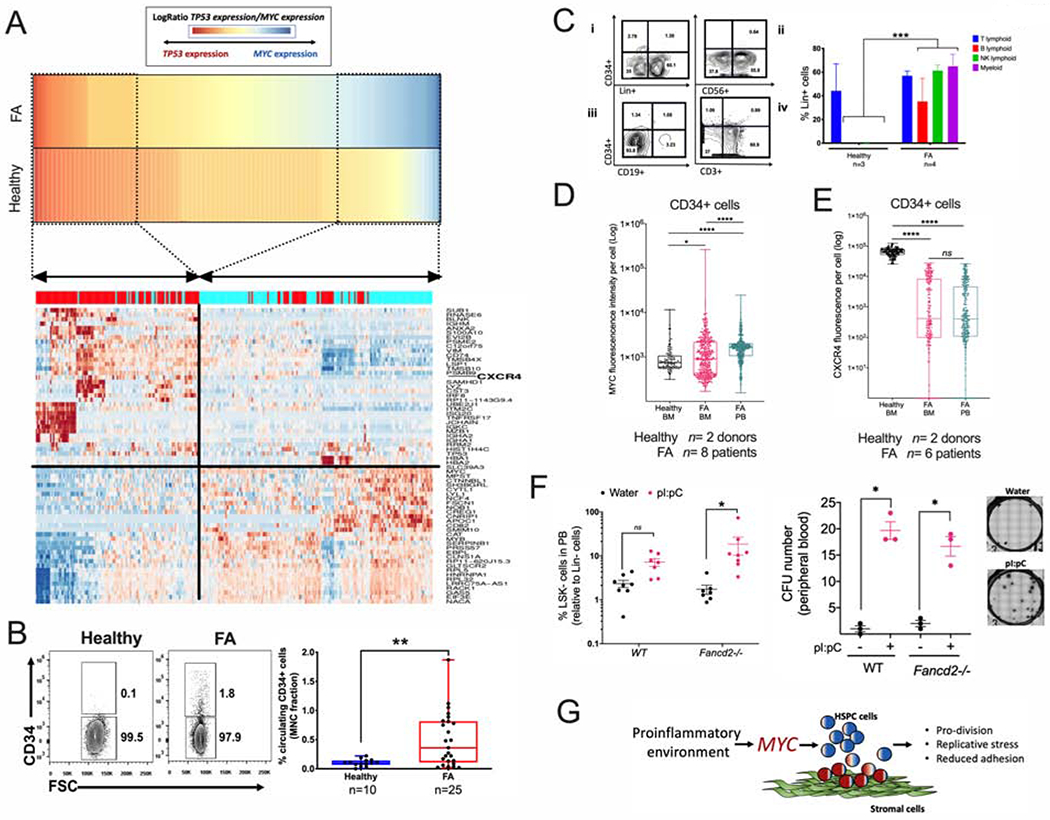
(A) High MYC expression significantly correlates with reduced expression of the HSPC adhesion molecule CXCR4. Gene expression profiles of “High-TP53” and “High-MYC” expressing HSPCs were compared and DEG were obtained. Heat map for the gene expression in High-TP53 and High-MYC expressing HSPCs is shown in the lower panel. Note that both cellular states have totally opposite gene expression profiles. “High-MYC” expressing cells downregulate several cell adhesion genes including CXCR4 and VIM.
(B) Representative FACS plots (left panel) and quantitation (right panel) of the percentage of CD34+ cells in peripheral blood of healthy donors and FA patients.
(C) Representative FACS plots (left panel) and quantitation (right panel) of Lin+ cells from the CD34+ cells of peripheral blood MNC, (i) Lin+ cell production in liquid myeloid promoting culture conditions, (ii) NK-cell (CD56+) production in MS-5 supporting stroma. (iii) B-cell (CD19+) production in MS-5 supporting stroma, (iv) T-cell (CD3+) production in OP9-DL4 supporting stroma. Note that the circulating CD34+ cells from FA patients show multilineage potential while the healthy counterpart shows preferential T-cell potential.
(D) MYC expression in CD34+ cells from peripheral blood or bone marrow as analyzed by flow cytometry. n= 8 FA samples and 2 healthy samples.
(E) CXCR4 expression in CD34+ cells from peripheral blood or bone marrow as analyzed by flow cytometry. n= 6 FA samples and 2 healthy samples.
(F) Quantitation of LSK- cells (left panel) and CFU content (right panel) in peripheral blood (PB) of WT or Fancd2−/− mice after two weeks treatment with pI:pC. Representative images of the hematopoietic colonies are also shown.
(G) Working model: MYC has a pro-survival role for the HPSC pool of patients with FA, driving the cells through S phase at the expense of DNA damage. Inflammatory episodes might hyperactivate the MYC pathway and precipitate bone marrow failure by increasing replicative stress and reducing adhesion of the HSPCs to their niche. In the FA context, MYC must be a counteracting force against the previously described growth suppressive activities of p53 and TGFβ pathways.
The Supplemental Table 3 describes the characteristics of the FA patients whose bone marrow or peripheral blood samples were used in the experiments described in panels B-E.Data in (B), (D), (E) and (F) are represented as boxplots.
Data in (C) are represented as mean ± SEM. p-values of 0.01 to 0.05 were considered significant (*), p-values of 0.001 to 0.01 were considered very significant (**) and p-values of <0.001 were considered extremely significant (***, ****). See also Supplementary Figure 7.
To explore the predisposition to detachment hypothesis, we explored whether there was an increase in the CD34+ cells trafficking in the peripheral blood of FA patients with respect to healthy controls. Indeed, an increased percentage of circulating CD34+ cells, in comparison to healthy individuals, was observed (Figure 7B). Interestingly, although a few patients exhibited cytogenetic evidence of clonal hematopoiesis, none of them displayed pathologic evidence of AML transformation and there was no correlation between the high numbers of CD34+ cells and the presence of clonal hematopoiesis (Supplementary Table 3, Figure 7B). Clonogenic capacity of these FA circulating CD34+ cells was assessed using several assays. CFU assays resulted in erythroid colonies, granulocyte colonies, and monocyte colonies. Colony numbers were reduced when the samples were obtained from patients with severe BM failure (Supplementary Figure 7A). Differentiation assays indicated that circulating CD34+ cells from peripheral blood of FA patients exhibit lymphoid and myeloid differentiation potential (Figure 7C). Additional LTC-IC assays demonstrated that primitive HSC are more frequently found in the circulating CD34+ population from patients with FA than from healthy donors (Supplementary Figure 7B). Immunophenotype profiling of the FA circulating CD34+ cells revealed the presence of pro-B cells, early lymphoid progenitors, and myeloid progenitors (Supplementary Figure 7C).
As expected, FA patients had a decreased percentage of CD34+ cells residing in the BM in comparison to healthy individuals, which worsens with the progression of the disease (Supplementary Figure 7D, left panel). The opposite occurred in peripheral blood, where FA patients have a higher percentage of circulating CD34+ cells in comparison to healthy controls. Importantly, the number of circulating CD34+ cells in FA patients tends to decrease as the severity of BM failure worsened, correlating with the cellularity of the BM (Supplementary Figure 7D, right panel).
Interestingly, circulating CD34+ cells from FA patients had a significantly higher MYC expression in comparison to CD34+ cells residing in the BM from the same patients, and also in comparison to healthy BM CD34+ cells (Figure 7D). In addition, CD34+ cells from FA patients, both from BM and peripheral blood, had reduced levels of CXCR4 expression compared to BM CD34+ cells from healthy controls (Figure 7E).
Reduced levels of CXCR4 might be responsible for the reduced adhesion capacity of FA CD34+ cells to the BM niche and egression into the peripheral blood. CXCR4 is required for maintenance of HSPCs in their BM niche and is critical for maintenance of HSPC in quiescence (Nie et al., 2008). When CXCR4 expression is lost, cells enter into the cell cycle. MYC promotes entrance into the cell cycle of HSPC, therefore, MYC upregulation and CXCR4 downregulation appear to act in a concerted manner in FA HSPCs.
We have shown above that pI:pC induces Myc expression in vivo, and others have previously shown that pI:pC provokes the exit from dormancy of LT-HSC (Walter et al., 2015, Sato et al., 2009), which consequently should reduce adhesion of progenitor cells to their BM niche. We therefore tested if pI:pC exposure was able to increase the trafficking of mouse progenitor cells into the peripheral blood of WT and Fancd2−/− mice. Indeed, we found that upon pI:pC injection two main hematopoietic progenitor populations circulate in peripheral blood of WT and Fancd2−/− mice: LSK and LSK- cells, the LSK population is enriched in progenitor and LT-HSC cells, whereas the LSK-population has been reported to be an LSK derived population (Peng et al., 2012) (Figure 7F left panel, Supplementary Figure 7E). Moreover, the MNCs from the peripheral of blood of the pI:pC-treated mice had CFU capacity (Figure 7F right panel). Our results suggest that inflammatory conditions or physiological stress, such as pI:pC exposure, change the FA HSPC compartment from a quiescent state to a hyperproliferative state orchestrated by Myc. This transition results in increased replication stress and propensity to detachment of progenitor cells (Fig. 7G).
In the current study, we demonstrate that MYC expression is increased across multiple HSPC lineages in FA. The upregulation of MYC in the FA BM has important consequences. On the one hand, MYC upregulation plays a beneficial role in HSPCs, providing a critical survival function and stimulating cell cycle progression (Bretones et al., 2015, Laurenti et al., 2008). On the other hand, MYC upregulation, resulting in chronic replication stress and increased DNA damage (Puccetti et al., 2019), might have a detrimental role in the BM of patients with FA. Previous studies have also demonstrated that FA cells have increased replication stress and replication fork instability (Balcerek et al., 2018). In our study, inhibition of MYC expression with (+)-JQ1 reduced the percentage of cells in S phase, reduced replication stress, reduced DNA damage, and improved the survival of FA cells upon genotoxic stress in preclinical models of FA. Multiple FA proteins function in mitophagy (Sumpter et al., 2016) and the absence of mitophagy may affect the survival of FA cells during genotoxic stress. BRD4 is a negative regulator of autophagy gene expression. The BET inhibitor (+)-JQ1 induces an upregulation of autophagy gene expression, leading to an increase in autophagy and lysosomal function (Sakamaki et al., 2017). Therefore, MYC inhibition by (+)-JQ1 may enhance survival of FA HSPCs by upregulation of autophagy.
Another potentially detrimental consequence of MYC upregulation in FA HSPCs is the transcriptional downregulation of the cell surface adhesion protein, CXCR4 (Karpova and Bonig, 2015). We demonstrate that CD34+ cells from FA patients have reduced CXCR4 levels with an increase in the number of HSPCs with clonogenic capacity in the peripheral blood. Moreover, FA mice also exhibited increased HSPCs in the peripheral blood following pI:pC exposure and elevated Myc expression. Therefore, chronic release of HSPCs from the BM, exacerbated by acute MYC induction, may contribute to the chronic state of BM failure in FA patients.
Increase in MYC expression in FA BM cells may result from transcriptional upregulation, perhaps via a super enhancer mechanism and BET activation (Wroblewski et al., 2018, Roe et al., 2015, Qi, 2014, Delmore et al., 2011, Bahr et al., 2018). Consistently, in our study, the BET inhibitor (+)-JQ1 strongly downregulated MYC expression and acutely reduced many of the functional consequences of elevated MYC activity. The increased MYC mRNA expression in FA BM cells may also result from the local increased levels of inflammatory cytokines, such as IFNγ and TNFα (Lin et al., 2014, Ramana et al., 2000, Briot et al., 2008). Accordingly, the inhibition of these cytokine signaling pathways can reduce MYC expression and offer potential therapeutic opportunities for FA patients.
In acute episodes of stress, resulting from a viral infection and inflammation (Matatall et al., 2016, Walter et al., 2015, Sato et al., 2009), FA patients may experience a strong MYC induction, leading to rapid cycling and HSPC loss. Indeed, FA patients often exhibit a rapid decline in BM HSPCs numbers after an acute viral infection, such as a varicella infection. In this setting, HSPCs are likely to be released into the peripheral blood, due to the reduction in expression of the cell surface adhesion protein CXCR4 that accompanies MYC overexpression. Release of HSPCs to the peripheral blood may create a chronic state of BM failure. A BET inhibitor may reduce HSPC cycling during the acute viral episode, limit DNA damage, and preserve the BM HSPC pool in FA patients. In contrast, the chronic use of BET inhibitors may have negative consequences for patients with FA. Chronic MYC loss or inhibition would be expected to reduce the overall survival of FA BM cells. Paradoxically, FA cells seem to require MYC to overcome the cell cycle blockade resulting from high p53 expression (Ceccaldi et al., 2012) and increased TGFβ signaling (Zhang et al., 2016).
Limitations of the study
Our study has limitations. During our initial scRNAseq screening, we were limited to the specific FA patients coming into the clinics, as the scRNAseq experiments needed to be run on fresh samples. Working with a bank of frozen clinical bone marrow samples was not possible due to RNA degradation. We were therefore unable to perform the scRNAseq analysis of the bone marrow from FA patients with severe aplastic anemia, myelodysplastic syndrome, or acute myeloid leukemia. Also, patients with severe aplastic anemia were not used for the single cell RNAseq due to the limitation of the cell numbers. The inter-individual variability was observed among healthy individuals and that HSCs were low in some individuals and one healthy donor had high MYC expression. We have mainly studied patients with pathogenic variants in FANCA and FANCC genes, as patients with mutations in genes downstream in the FA pathway are rare. Despite these limitations, this study uncovers a unique mechanism of the pathophysiology of Fanconi anemia, using rare bone marrow samples derived from patients. For our studies, we have used (+)-JQ1, which is one of the best known MYC inhibitors. However, for potential therapeutic purpose, more specific inhibitors are needed for FA patients. Inhibition of inflammatory cytokines might be more desirable and have less off-target effects in FA patients.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Alan D’Andrea (alan_dandrea@dfci.harvard.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The datasets generated during this study have been deposited in Gene Expression Omnibus (GEO) under the accession number: GSE157591.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
2-6 month old C57BL/6 mice were used for this study. Fancd2−/− mice in C57BL/6 background were previously generated (Parmar et al., 2010). n=3 mice per group were used for in vivo experiments. All mice were housed, treated, and handled in accordance with the guidelines set by the Animal Care and Use Committee of the Dana Farber Cancer Institute. Mice were euthanized by CO2 asphyxiation or by isoflurane overdose followed by cervical dislocation at the experimental timepoint or when loss of body weight, diarrhea, progressive dermatitis and any condition interfering with eating or drinking appeared. All experimental procedures were approved by the Animal Care and Use Committee of the Dana Farber Cancer Institute.
Cell lines
Lymphoblast cell line EUFA316+EV (FANCG-deficient), derived from a patient with FA and reversed by functional complementation of the cells with the FANCG cDNA (EUFA316+G) were used.
Subject details
Remnant bone marrow or peripheral blood samples from patients with FA in clinical follow up were used in this study (age range 2.7 years old −17.8 years old). Written informed consent was received from participants or their parents prior to inclusion in this study. Use of patients’ samples was approved by the ethics and research committees from Dana Farber Cancer Institute, Boston Children’s Hospital and Instituto Nacional de Pediatría, Mexico. Healthy bone marrow and peripheral blood was purchased from Lonza.
Clinical information from patients with FA was obtained from the clinical records of the Boston Children’s Hospital and Instituto Nacional de Pediatría, Mexico. Classification of aplastic anemia in patients was obtained using absolute neutrophil count (ANC), platelet count (PC), and hemoglobin (Hb) levels as follows: mild aplastic anemia if ANC <1,500/mm3, PC 150,000-50,000/mm3 and Hb≥8 g/dL; moderate aplastic anemia if ANC <1,000/mm3, PC <50,000/mm3 and Hb<8 g/dL; and severe aplastic anemia if ANC <500/mm3, PC <30,000/mm3 and Hb<8 g/dL. The nearest available values of C-reactive protein (CRP) in blood were used as a marker of inflammation, normal ranges were considered from 0.07-0.8 mg/dL.
METHOD DETAILS
Enrichment of human HSPCs
Whole BM and peripheral blood samples were obtained from patients with FA after informed consent of sample use for research. Fresh healthy BM samples were purchased from Lonza (1M-105, Lonza). Supplementary Tables 1, 2 and 3 describe characteristics of all the FA patients and normal donors whose bone marrow or blood samples were used for the analysis. Based on the DEB test or MMC sensitivity test, all the patient samples used in our study met clinical criteria for FA.
Red blood cell lysis was performed by incubating the samples with Ammonium Chloride (07800, StemCell Technologies) for 10 min on ice followed by washing with PBS. After red blood cell lysis, Lin- cells were enriched for mononuclear cells (MNCs) by negative selection using the EasySep kit (19056, StemCell Technologies), according to manufacturer’s instructions. For isolation of CD34+ cells, MNCs were incubated 30 min on ice with anti-CD34+ antibody coupled to magnetic beads and FcR blocking reagent (Miltenyi). MNCs were washed with PBS and sieved with a MACS MultiStand isolation system (Miltenyi).
Capture of HSPCs and scRNAseq library preparation.
Chromium™ Single Cell 3’ Reagent Kits v2 were used following manufacturer’s instructions. GEM generation and barcoding were performed using Chromium™ Single Cell A Chip Kit (PN-120236). Enriched HSPCs awere resuspended in 1X PBS with 0.04% bovine serum albumin (BSA) and 3000-5000 cells per individual were loaded into the single Cell 3’ Chip, individually captured and barcoded using the 10x Chromium™ Controller (10x Technologies). Post-GEM-RT clean-up and cDNA amplification reaction were performed using the Chromium™ Single Cell 3’ Library & Gel Bead Kit v2 (PN-120237). Library construction was performed using the Chromium™ i7 Multiplex Kit (PN-120262). Sequencing was performed in a NextSeq High Output 75 Cycle Flow Cell in an Illumina platform.
Single cell RNA-seq data pre-processing and quality control
The raw base call (BCL) files were processed (demultiplexation, alignment, barcode assignment and UMI quantification) with Cell Ranger (version 2.1.1) pipelines. The reference index was built upon the GRCh38.d1.vd1 reference genome with GENCODE v25 annotation. To filter lower quality cells, we removed any cells expressing fewer than 2000 genes or with fewer than 80,000 UMIs or with greater than 4% expression originating from mitochondrial genes.
Clustering
Raw UMI counts for each gene were normalized to the total UMI counts of each cell, multiplied by a scale factor (10^4) and log-transformed. Cells with normalized expression of CD34 higher than 0.125 were identified as CD34+ cells. The number of UMI per cell and the clinics batch of the samples were regressed out using a negative binomial model, and the shared nearest neighbor (SNN) modularity optimization based clustering from Seurat v2.3.4 (Butler et al., 2018) were used for clustering. Expression levels of genes required for hematopoietic development were used for cluster identification and cell types. These genes included markers of HSC (AVP), differentiating progenitors (CDK6), early Megakaryocyte/Erythroid progenitors (GATA2), early Lymphoid progenitors (SELL), and downstream lineage progenitors. Specifically, we used BLVRB and GATA1 levels for identifying Erythroid progenitors, LMO4 for EBM progenitors, PLEK for megakaryocyte progenitors, MPO for neutrophil progenitors, IRF8 for monocyte progenitors, and LTB for B-cell progenitors
For further downstream analysis, the highly variable genes were selected using FindVariableGenes (Seurat R package). The graph-based method from Seurat was used to cluster cells. PCA was selected as dimensional reduction technique to use in construction of Shared Nearest-Neighbor (SNN) graph. Differentially expressed genes were identified using FindMarkers function.
Single cell gene set enrichment score
Single cell enrichment score was run using AUCell (version 1.6.0) in R (Aibar et al., 2017) on normalized expression values and Molecular Signatures Database C2 (version 5) signatures and custom signatures.
StemNet
STEMNET (Velten et al., 2017) was used to reconstruct the differentiation trajectory, in particular, a gradient commitment towards the B, MD, ME/EBM and Neutrophil lineages.
Clonogenic assays with human BM samples
Clonogenic potential of human HSPCs was assessed in colony-forming unit (CFU) assays, 3000 (for healthy samples) or 5000 (for FA samples) HSPCs were plated per triplicate in human methylcellulose MethoCult H4434 Classic medium (04434, StemCell Technologies). The BET bromodomain inhibitor (+)-JQ1 was added to a final concentration of 50 nM and hematopoietic colonies were scored after 14 days of culture at 37°C and 5% CO2. Pictures were taken with the STEMvision System (StemCell Technologies).
Flow cytometric analysis of human bone marrow and blood samples
BM and peripheral blood MNCs were blocked with fetal bovine serum (FBS) for 15 min and incubated with anti-CD34-FITC antibody (Biolegend) and anti-CXCR4-PE-Cy7 antibody (Biolegend) for 30 min at room temperature. After antibody incubation cells were thoroughly resuspended and fixed with CytoFix/CytoPerm (BD) buffer during 30 min on ice, washed with PermWash buffer (BD) and permeabilized for 30 on ice with Transcription Factor Fix/Perm buffer (BD). After permeabilization cells were incubated at room temperature for 40 min with primary antibodies goat-anti-human-p53 (R&D) and rabbit-anti-human-MYC (Cell Signaling), washed with Transcription Factor Perm/Wash buffer (BD) and incubated for additional 40 min with secondary antibodies Donkey anti-goat-PE (Thermo-Fisher) and Donkey anti-rabbit-Brilliant Violet 421 (Biolegend). DNA was stained with DRAQ5 (Thermo-Fisher). Samples were acquired in a LSR Fortessa Analyzer (BD) and data analysis was performed using FlowJo software version 10.5.3.
Culture of primary human cells
MYC induction in vitro.
60,000 BM CD34+ cells were seeded in alfa-MEM medium (Gibco) without FBS and exposed for 24 h to 50 nM (+)-JQ1 in combination with 20 ng/mL of human-TNF-α, IFNγ or MCP-1 (Peprotech). After the incubation time, cells were stained with anti-CD34-FITC, fixed and permeabilized, and MYC was detected using a rabbit anti-MYC primary antibody and a Donkey anti-rabbit-Brilliant Violet or Donkey anti-rabbit-PE secondary antibody.
Differentiation assays.
60,000 cells derived from the CD34+ enriched fraction were seeded in 200 μl of alfa-MEM medium with 10% FBS and specific cytokines. For myeloid differentiation, cells were seeded in 96-well round bottom plates supplemented with SCF, GM-CSF, IL-6, IL-3 and FLT3-l (Peprotech). For B/NK lymphoid differentiation, cells were seeded in 96-well flat bottom plates in co-culture with the mouse derived MS-5 stromal cell line and the medium was supplemented with FLT-3, SCF, IL-7 and IL-15 (Peprotech). For T lymphoid differentiation, cells were seeded in 96-well flat bottom plates in co-culture with the mouse derived OP9-DL4 stromal cell line expressing the Delta-like 4 Notch ligand and supplemented with cytokines. Cells were maintained at 37°C and 5% CO2 until harvest. Myeloid cultures were harvested after 7 and 14 days of culture and cell differentiation status was analyzed with the following antibody mixture: CD34-APC, CD19-PE, CD3-PE, CD11b-PE, CD14-PE, CD56-PE and GlyA-PE. NK/B lymphoid cultures were harvested after 14 and 28 days of culture and cell differentiation status was analyzed with the following antibody mixture: CD34-APC, CD19-FITC and CD56-PE. T lymphoid cultures were harvested after 28 days of culture and cell differentiation status was analyzed with the following antibody mixture: CD34-APC and CD3-FITC.
Lentiviral transduction of CD34+ cells
Isolated human CD34+ cells were cultured in StemSpan SFEMII (09655, Stem Cell Technologies) with 100ng/ml of recombinant human cytokines SCF (300-07, Peprotech), TPO (300-18, Peprotech), Flt3 (300-19, Peprotech) and IL-6 (200-06, Peprotech) at a density of 1-2 million cells/ml in non-tissue culture (non-TC) treated plates for 36 hours. After initial culture, cells were plated in non-TC treated 96 well plates with a density of 1-2x10^5 cells in 100-150 μl of new media with 8 μg/ml polybrene (TR-1003-G, Sigma) and shRNA producing lentivirus targeting human FANCD2. A MOI of 50 was used for the scrambled shRNA and a MOI of 100 was used for the FANCD2 shRNA. Plates were spun down at 2300 rpm for 30 minutes at RT after addition of the viral prep. New media was added after 12-16 hours of incubation. Selection media with 1μg/ml puromycin (MIR 5940, MirusBio) was added to cultures 12-24 hours after viral infection. Puromycin selection was applied for 72 hours. Puromycin resistant cells were used for assays with FA-like CD34+ cells.
Gene targeting in CD34+ umbilical cord blood (UCB) cells using CRISPR/Cas9
Single donor CD34+ umbilical cord blood cells (UBC) were purchased from commercial suppliers (Stemcell Technologies and Cincinnati Children’s Hospital Translation Core Laboratories). Unique CD34+ UCB samples were used for each experimental replicate. Unless otherwise indicated, cells were cultured in 5% CO2 and 5% O2 at 37°C at a cell density of ≤ 3x105 cells/ml in StemSpan SFEM II (Stemcell Technologies) supplemented with 200mM L-glutamine, 1x103 U/ml penicillin, 10mg/ml streptomycin, 25μg/ml amphotericin B, 10 μg/ml human LDL (Stemcell technologies), 35nM UM171 (Selleckchem) and 100ng/ml each human recombinant FLT3L, SCF, IL-6, TPO (Peprotech and Stemcell Technologies). For inflammatory cytokine cultures, targeted cells were plated in StemSpan SFEMII supplemented with 20 ng/ml human recombinant TNF-α, IFNγ or MCP1 (Peprotech) and 50nM (+)-JQ1 (ApexBio). Cells were plated at a density of ~1x106 cells/ml into round bottom 96 well plates (200μl/well), cultured for 24 hours at 37°C in 5% CO2, washed twice in PBS, then fixed and permeabilized prior to staining. For colony forming assays, cells were plated in duplicate or triplicate into methocult H4434 classic (Stemcell Technologies).
The introduction of indels into CD34+ UCB cells using Crispr/Cas9 was performed as described by (Bak et al., 2018). Specifically, chemically modified short guide RNAs (sgRNA) were purchased from Synthego (Supplementary Table 4) and complexed with Alt-R S. pyogenes Cas9 nuclease V3 (IDT, catalog 1081059) at a molar ratio of 1:2.5. Ribonucleoprotein complexes were introduced into cells using an Amaxa 4D nucleofector (program DZ100) and P3 primary cell solution (Lonza). Mock controls were nucleofected without RNPs. To evaluate targeting efficiency, genomic DNA was harvested from mock and targeted cells and the targeted region was amplified with Platinum SuperFi or Platinum SuperFi II DNA polymerase (Invitrogen) using the primers listed in Supplementary Table 5. Reactions were column purified (PureLink PCR purification kit, Thermofisher) or treated with Exo-SAP-IT express (Applied Biosystems) and then sequenced using the primers in Supplementary Table 5. Indel analysis was performed on sequenced DNA using the Synthego ICE Analysis Tool (https://ice.synthego.com/#/). Targeting efficiency data are presented in Supplementary Table 6.
Isolation of mouse HSPCs
BM cells were harvested from tibia and femurs of mice by gentle flushing with HBSS++ buffer [Hanks balanced salt solution (10-547F, Lonza) + HEPES (BP299-100, Fisher Scientific) + Fetal bovine serum (F2442, Sigma) + penicillin-streptomycin (15140-122, GIBCO)]. Samples were filtered through a 70 μM filter and Lin- enrichment was performed using the lineage cell depletion kit (130-090-858, Miltenyi). LT-HSCs were recognized by being LSK (Lin-Sca-1+c-Kit+) and CD150+CD48- using PE-Cy7-Sca (Clone D7, 558162, BD Biosciences), APC-c-kit (Clone ACK2, 135108, BD Biosciences), Pacific Blue-CD150 (Clone TC15-12F12.2, 115924, Biolegend) and APC-Cy7-CD48 (Clone HM48-1, 47-0481-82, e-Bioscience).
Clonogenic assays with mouse bone marrow and peripheral blood cells
For clonogenic assays, Lin- cells were plated in Methocult GF M3434 methylcellulose (03444, Stem Cell Technologies) and cultured. The BET bromodomain inhibitor (+)-JQ1 was added to a final concentration of 50 nM and hematopoietic colonies were scored after 7 days of culture at 37°C and 5% CO2.
For clonogenic assays using peripheral blood derived cells, mononuclear cells were isolated by lysing the whole blood with ACK lysis buffer (10-548E, Lonza) and incubating during 5 min at 37 °C, cells were washed with PBS and the process repeated if the cell pellet looks red. 300, 000 total mononuclear cells were plated in Methocult GF M3434 methylcellulose (03444, Stem Cell Technologies) during 14 days at 37 °C and 5% CO2. Pictures were taken with the STEMvision System (StemCell Technologies).
Murine in vivo experiments
For 30 days experiments, pI:pC injections were performed intraperitoneally at a concentration of 5 mg/kg twice per week for one month. (+)-JQ1 or the inactive enantiomer (−)-JQ1 were injected intraperitoneally at a 50 mg/kg concentration daily for one month. For 2 days experiments, a single pI:pC injections was performed intraperitoneally at a concentration of 5 mg/kg, or a single injection with (+)-JQ1 or (−)-JQ1 were injected intraperitoneally at a 50 mg/kg.
Cytokines quantification
Plasma was collected from human whole BM blood or from mouse peripheral blood and subjected to a bead-based Multiplex Immunoassay for detection of human and mouse pro-inflammatory cytokines using the discovery assay of Eve Technologies (Eve Technologies Corporation, Canada). The bead analyzer Bio-Plex 200 (BIORAD) was used for detection and results were quantified according to a standard curve.
Alkaline comet assay
Sorted LT-HSCs were mixed with low-melting-temperature agarose, plated on slides and incubated at 4°C overnight in lysis solution using the CometAssay kit (4250-050-K, Trevigen). Next day cells were subjected to a current voltage of 12 V washed and stained with SYBR-Green dye (S7567, Invitrogen). Pictures were taken with a Zeiss Imager Z1 fluorescence microscope and analysis was performed using the OpenComet plugin in the software Image J.
Survival assays
For survival assays, EUFA316+EV and EUFA316+G cells were plated at a density of 1x103 cells per well in 96-well plates and exposed to increasing doses of mitomycin C (MMC) and (+)-JQ1 (50 nM). Cell viability was assessed after 5 days of culture using the Cell Titer-Glo® Luminescent Cell Viability Assay (G7573, Promega).
Quantitative real-time PCR
RNA was extracted from human Lin- cells using the micro-RNA extraction kit (74034, QIAGEN) and from lymphoblast cell lines using the Mini-RNA extraction kit (74134, QIAGEN) following the manufacturer’s instructions. cDNA was synthesized using the RT2 PreAMP cDNA synthesis kit (33045, QIAGEN), target sequences were amplified using the RT2 PreAMP Pathway primer Mix (PBM-029Z, 30241, QIAGEN) and detected using the qPCR Master Mix RT2 SYBR® Green (330533, QIAGEN). Multiplex real-time PCR was performed with the MYC targets PCR array (QIAGEN) following manufacturer’s instructions. PCR was performed in a QuantStudio 7 Flex real Time machine (Life Technologies).
Cell cycle analysis
Cell cycle analysis in lymphoblast cell lines were thoroughly resuspended and fixed with ice-cold 70% ethanol and stored at −20 during 30 min. After fixation, cells were washed with PBS, stained with FxCycle PI/RNase staining solution (Invitrogen, F10797) and analyzed in a CytoFlex 5 Flow Cytometer (Beckman Coulter). Cell cycle analysis in mouse LT-HSC was performed using the cell surface markers PE-Cy7-Sca (Clone D7, 558162, BD Biosciences), APC-c-kit (Clone ACK2, 135108, BD Biosciences), FITC-CD150 (Clone A12 (7D4), 306306, Biolegend) and APC-Cy7-CD48 (Clone HM48-1, 470481-82, e-Bioscience), along with intracellular staining using PE-Cy7-Ki67 (350526, Biolegend) and DAPI (422801, Biolegend). Data analysis was performed using FlowJo software version 10.5.3.
CFSE labeling
A lyophilized vial of CFSE dye was reconstituted in 36 μL of DMSO to make a 5 mM solution. A 5 μM CFSE working solution was prepared in StemSpan SFEMII medium (09655, Stem Cell Technologies). Sorted LSK were resuspended in the later solution and incubated during 20 min at 37°C protected from light. The staining was quenched by adding 5 times the original staining volume of StemSpan SFEMII medium containing 10% FBS. Cells were incubated for additional 10 min, and for experiments were distributed in StemSpan SFEMII medium containing TPO (50 μl/mL), SCF (50 μl/mL), 1% L-glutamine (Gibco, 25030-081) and 2% Penicillin streptomycin (Gibco, 15140-122). Cells were maintained in culture at 5% CO2 and 37°C for 7 days and collected. CFSE intensity was assessed in a CytoFlex 5 Flow Cytometer (Beckman Coulter). Data analysis was performed using FlowJo software version 10.5.3.
Western blotting
Whole cell lysates were prepared using RIPA cell lysis buffer (9803, Cell Signaling) and 1 mM PMSF (8553s, Cell Signaling) and western blots were performed using the following antibodies MYC (Cell Signaling), pKAP1 (S824), KAP1, pCHK1 (S345), CHK1, pRPA32 (S33), RPA32, γH2AX and Vinculin (sc-25336, Santa Cruz).
DNA fiber assay
DNA fiber assay was performed using the FiberComb machine (Genomic Vision). Cells were cultured in presence of IdU (Sigma I7125), CldU (Sigma C6891) and (+)-JQ1 and fiber assay was performed as described previously (Lim et al., 2018, Kais et al., 2016). Briefly, after treatments, cells were embedded in low melting point agarose plugs and incubated with proteinase K (Fisher E0491) overnight. Next day plugs were washed and digested with Agarase (BioLab M0392L). Agarase-treated samples were combed onto silanized coverslips in a FiberComb well. Slides were incubated with rat anti-BrdU antibody (clone BU1/75 (ICR1) specific to CldU, Abcam ab6326) and mouse anti-BrdU Antibody (specific to IdU, BD Biosciences 347580). Pictures were taken in a fluorescence microscope with at least 100 fibers per condition. DNA fibers were measured with ImageJ.
QUANTIFICATION AND STATISTICAL ANALYSIS
For single cell RNAseq data.
Two tailed Wilcoxon rank-sum test was used for detection of differences between groups. Differential expression analysis was performed using the likelihood ratio test assuming an underlying negative binomial distribution, genes with Bonferroni adjusted p-value < 0.01 and log fold-change > 0.25 or < −0.25 were considered as differentially expressed genes. Statistical analyses were done in R (> v3.6). Gene-specific differential expression and gene set enrichment scores among clusters were calculated by Wilcoxon rank-sum test and is indicated by asterisks (****, p < 0.0001; ***, p < 0.001; **, p < 0.01; *, p < 0.05 or ns, p > 0.05).
For other experiments.
Normality was assessed using the D’Agostino-Pearson, Shapiro-Wilk and Kolmogorov Smirnoff normality tests. Outliers were identified with the ROUT method. 2-way ANOVA and Dunn’s multiple comparisons test were used for detection of differences between experimental groups. Two-tailed P values for statistical analysis were obtained using Student’s t-test. Graphpad Prism 8 was used for statistical analysis.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| FITC anti-human CD34 | BioLegend | Cat# 343604; RRID:AB_1732005 |
| PE/Cy7 anti-human CXCR4 | BioLegend | Cat# 306514; RRID:AB_2089651 |
| Goat anti-human/mouse/rat p53 | R&D Systems | Cat# AF1355-SP; RRID:AB_354749 |
| Rabbit anti-human c-Myc | Cell Signaling | Cat# 5605S; RRID:AB_1903938 |
| Donkey anti-goat IgG PE | Thermo Fisher Scientific | Cat# PA1-29953; RRID:AB_10983834 |
| Brilliant Violet 421 donkey anti-rabbit IgG | BioLegend | Cat# 406410; RRID:AB_10897810 |
| PE-Cy7 rat anti-mouse Ly-6A/E | BD Biosciences | Cat# 558162; RRID:AB_647253 |
| APC anti-mouse CD117 (c-kit) (Clone ACK2) | BD Biosciences | Cat# 135108; RRID:AB_2028407 |
| Pacific Blue anti-mouse CD150 (Clone TC15-12F12.2) | BioLegend | Cat# 115924; RRID:AB_2270307 |
| APC-Cy7 anti-mouse CD48 (Clone HM48-1) | e-Bioscience | Cat# 47-0481-82; RRID:AB_2573962 |
| Rabbit anti-human phospho-KAP1 (S824) | Abcam | Cat# ab70369; RRID:AB_1209417 |
| Rabbit anti-human/mouse KAP1 | Abcam | Cat# ab10484; RRID:AB_297223 |
| Rabbit anti-human/mouse/rat/monkey phospho-Chk1 (Ser345) (133D3) | Cell Signaling | Cat# 2348S; RRID:AB_331212 |
| Anti-human/mouse/rat CHK1 (G-4) | Santa Cruz | Cat# sc-8408; RRID:AB_627257) |
| Rabbit anti-human phospho RPA32 (S33) | Bethyl Laboratories | Cat# A300-246A-M; RRID:AB_2779098 |
| Rabbit anti-human/mouse RPA32 | Bethyl Laboratories | Cat# A300-244A-M; RRID:AB_2779096 |
| Rabbit anti-human/mouse phospho-Histone H2A.X (Ser139) (20E3) | Cell Signaling | Cat# 9718S; RRID:AB_2118009 |
| Mouse anti-human vinculin (H-10) | Santa Cruz | Cat# sc-25336; RRID:AB_628438 |
| PE Donkey anti-rabbit IgG | BioLegend | Cat# 406421; RRID:AB_2563484 |
| Rat anti-BrdU (clone BU1/75 (ICR1) | Abcam | Cat# ab6326; RRID:AB_305426 |
| Mouse anti-BrdU | BD Biosciences | Cat# 347580; RRID:AB_400326 |
| Anti-mouse IgG, HRP-linked | Cell Signaling | Cat# 7076S; RRID:AB_330924 |
| Anti-rabbit IgG, HRP-linked | Cell Signaling | Cat# 7074S; RRID:AB_2099233 |
| APC anti-human CD34 (Clone 561) | BioLegend | Cat# 343608; RRID:AB_2228972 |
| PE anti-human CD19 (Clone 4G7) | BioLegend | Cat# 392506; RRID:AB_2750097 |
| PE anti-human CD11b (Clone ICRF44) | BioLegend | Cat# 301306; RRID:AB_314158 |
| PE anti-human CD14 (Clone 63D3) | BioLegend | Cat# 367103; RRID:AB_2565887 |
| PE anti-human CD56 (NCAM) (Clone 5.1H11) | BioLegend | Cat# 362508; RRID:AB_2563925 |
| PE anti-human CD235a (Glycophorin A) (Clone HI264) | BioLegend | Cat# 349106 RRID:AB_10640739 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Human whole bone marrow and blood from patients with FA | Boston Children’s Hospital (USA) and Instituto Nacional de Pediatria (Mexico) | N/A |
| Human whole bone marrow and blood from healthy donors | Lonza | Cat# 1M-105 |
| Human CD34+ UCB (single donor) | Cincinnati Children’s Translational Core Cell Processing Core | https://www.cincinnatichildrens.org/research/cores/translational-core-laboratory/cell-processing-core |
| Human CD34+ UCB (single donor) | StemCell Technologies | Cat# 70008.5 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| UM171 | Selleck Chemicals | Cat# S7608; CAS:1448724-09-1 |
| FLT3L | Peprotech; StemCell Technologies | Cat# 300-19; Cat# 78009 |
| SCF | Peprotech; StemCell Technologies | Cat# 300-07; Cat# 78062 |
| TPO | Peprotech; StemCell Technologies | Cat# 300-18; Cat# 78210 |
| IL-6 | Peprotech; StemCell Technologies | Cat# 200-06; Cat# 78050 |
| TNF-α | Peprotech | Cat# 300-01A |
| IFN-γ | Peprotech | Cat# 300-02 |
| (+)-JQ1 | Apex Bio; Selleck Chemicals | Cat# A1910; Cat# S7110. CAS:1268524-70-4 |
| IdU | Sigma-Aldrich | Cat# I7125; CAS:54-42-2 |
| CldU | Sigma-Aldrich | Cat# C6891; CAS:50-90-8 |
| Crystal violet | Sigma-Aldrich | Cat# C0775; CAS:548-62-9 |
| Mitomycin C | Sigma-Aldrich | Cat# M0503; CAS:50-07-7 |
| PMSF | Cell Signaling | Cat# 8553S; CAS:329-98-6 |
| Alt-R S. pyogenes Cas9 nuclease V3 | Integrated DNA Technologies, Inc. | Cat# 1081059 |
| Platinum SuperFi DNA polymerase | Thermo Fisher Scientific | Cat# 12351010 |
| Platinum SuperFi II DNA polymerase | Thermo Fisher Scientific | Cat# 12361050 |
| ExoSAP-IT Express | Thermo Fisher Scientific | Cat# 75001 |
| Critical Commercial Assays | ||
| Human Cytokine Array Proinflammatory Focused 13-plex (HDF13) | Eve Technologies Corporation | https://www.evetechnologies.com/multiplex-assay/ |
| Mouse Cytokine Array Proinflammatory Focused 10-plex (MDF10) | Eve Technologies Corporation | https://www.evetechnologies.com/multiplex-assay/ |
| Deposited Data | ||
| Single-cell RNA-seq of primary human hematopoietic stem and progenitor cells from patients with Fanconi anemia and healthy donors | This paper | GEO: GSE157591 |
| Experimental Models: Cell Lines | ||
| EUFA316 +EV cell line | Zhang et al., 2016 | N/A |
| EUFA316 +G cell line | Zhang et al., 2016 | N/A |
| Mouse derived MS-5 stromal cell line | Dr. Paul Kincade, Oklahoma Medical Research Foundation | N/A |
| Mouse derived OP9-DL4 stromal cell line | Dr. Paul Kincade, Oklahoma Medical Research Foundation | N/A |
| Experimental Models: Organisms/Strains | ||
| Fancd2−/− mice C57BL/6 background | Parmar et al., 2010 | N/A |
| Oligonucleotides | ||
| Guidestrands for FANCA, FANCG and MYC targeting, see Supplementary Table S4 | This paper | N/A |
| Primers for PCR amplification and sequencing of targeted FANCA, FANCG and MYC, see Supplementary Table S5 | This paper | N/A |
| Recombinant DNA | ||
| pMD2.G plasmid | Addgene | Cat# 12259 |
| psPAX2 plasmid | Addgene | Cat# 12260 |
| pLKO.1 cloning vector | Addgene | Cat# 10878 |
| Software and Algorithms | ||
| Synthego ICE | Synthego | https://ice.synthego.com |
| Cell Ranger (version 2.1.1) | https://support.10xgenomics.com/singlecell-geneexpression/software/overview/welcome | |
| R package Seurat (version 2.3.4) | Butler et al., 2018 | https://satijalab.org/seurat/ |
| R version 3.6 | https://www.r-project.org/ | |
| R package AUCell (version 1.6.0) | Aibar et al., 2017 | |
| Molecular Signatures Database (version 5) | https://www.gsea-msigdb.org/gsea/msigdb/index.jsp | |
| R package STEMNET (version 0.1) | Velten et al., 2017 | |
| FlowJo, version 10.5.3 | https://www.flowio.com/ | |
| OpenComet plugin | http://www.cometbio.org/ | |
| Image J | http://fiji.sc | |
| Graphpad Prism 8 | Graphpad | https://www.graphpad.com/scientificsoftware/prism/ |
| Other | ||
| P3 Primary Cell 4D Nucleofector Kit | Lonza | Cat# V4XP-3024 & V4XP-3032 |
| PureLink PCR purification kit | Thermo Fisher Scientific | Cat# K310001 |
| EasySep Kit | StemCell Technologies | Cat# 19356 |
| EasySep buffer | StemCell Technologies | Cat# 20144 |
| EasySep magnet | StemCell Technologies | Cat# 18000 |
| MACS MultiStand | Miltenyi | Cat# 130-042-303 |
| CD34 MicroBead Kit, human | Miltenyi | Cat# 130-046-702 |
| FcR Blocking Reagent, human | Miltenyi | Cat# 130-059-901 |
| Human LDL | StemCell Technologies | Cat# 02698 |
| Stemspan SFEM II | StemCell Technologies | Cat# 09655 |
| MethoCult H4434 Classic | StemCell Technologies | Cat# 04434 |
| Methocult GF M3434 methylcellulose | StemCell Technologies | Cat# 03444 |
| Ammonium chloride solution | StemCell Technologies | Cat# 07800 |
| CytoFix/CytoPerm | BD Biosciences | Cat# 554714 |
| PermWash | BD Biosciences | Cat# 554723 |
| Transcription Factor Fix/Perm buffer | BD Biosciences | Cat# 562574 |
| DRAQ5 | Thermo Fisher Scientific | Cat# 62251 |
| Polybrene Infection/Transfection Reagent | Sigma-Aldrich | Cat# TR-1003-G |
| Puromycin Dihydrochloride Antibiotic Solution | Mirus Bio | Cat# MIR 5940 |
| RIPA cell lysis buffer | Cell Signaling | Cat# 9803 |
| SYBR-Green dye | Invitrogen | Cat# S7567 |
| Proteinase K | Fisher Scientific | Cat# E0491 |
| Agarase | BioLab | Cat# M0392L |
| FBS | Sigma-Aldrich | Cat# F2442 |
| Hanks balanced salt solution | Lonza | Cat# 10-547F |
| HEPES | Fisher Scientific | Cat# BP299-100 |
| Penicillin-streptomycin | Gibco | Cat# 15140-122 |
| L-Glutamine | Gibco | Cat# 25030081 |
| 70 μm cell strainer | Fisher Scientific | Cat# 087712 |
| 500 mL Bottle Top Vacuum Filter, 0.45 μm | Corning | Cat# 430514 |
| Lineage cell depletion kit | Miltenyi | Cat# 130-090-858 |
| RNeasy Micro kit | Qiagen | Cat# 74034 |
| RNeasy Mini kit | Qiagen | Cat# 74134 |
| RT2 PreAMP cDNA synthesis kit | Qiagen | Cat# 330451 |
| RT2 PreAMP Pathway primer Mix | Qiagen | Cat# 330241 PBH-177Z (human) PBM-177Z (mouse) |
| qPCR Master Mix RT2 SYBR Green | Qiagen | Cat# 330533 |
| RT2 Profiler MYC Targets PCR Array | Qiagen | Cat# 330231 PAHS-177ZC (human) PAMM-177ZC (mouse) |
| Cell Titer-Glo Luminescent Cell Viability Assay | Promega | Cat# G7573 |
| MEM α | Gibco | Cat# 12571048 |
| RPMI 1640 medium | Gibco | Cat# 11875119 |
| IMDM | Gibco | Cat# 12440053 |
| CometAssay kit | Trevigen | Cat# 4250-050-K |
| 10x Chromium Controller | 10X Technologies | https://www.evetechnologies.com/multiplex-assay/ |
| Chromium Single Cell 3’ Library & Gel Bead Kit v2 | 10X Technologies | Cat# PN-120237 |
| Chromium Single Cell A Chip Kit | 10X Technologies | Cat# PN-120236 |
| Chromium i7 Multiplex Kit | 10X Technologies | Cat# PN-120262 |
| NextSeq 500/550 High Output 75 Cycle kit v2.5 | Illumina | Cat# 20024906 |
Highlights:
MYC is overexpressed in bone marrow HSPCs from Fanconi anemia patients
MYC promotes proliferation of Fanconi anemia cells at the expense of DNA damage
Inflammation increases MYC expression in bone marrow HSPCs in Fanconi anemia mice
MYC overexpression causes detachment of Fanconi anemia CD34+ cells from bone Marrow
ACKNOWLEDGMENTS
We thank the FA Patients and their families and also the physicians, Angélica Monsiváis and Gerardo López, who referred the patients. This research was supported by grants from the U.S. National Institutes of Health (R01HL052725, P01HL048546), the Leukemia and Lymphoma Society (6237-13), and the Fanconi Anemia Research Fund (to ADD); the European Union’s Horizon 2020 research and innovation program (667403 for HERCULES) and the Academy of Finland; SEP-CONACYT (243102), PAPIIT (IA202615) and FOSISS-CONACyT (233721). We thank Dr. Cailin E. Joyce for providing guidance on processing primary human bone marrow for single cell RNAseq and Dr. Jun Qi for kindly providing (+)-JQ1 and (−)-JQ1 enantiomers.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- ADAMO A, COLLIS SJ, ADELMAN CA, SILVA N, HOREJSI Z, WARD JD, MARTINEZ-PEREZ E, BOULTON SJ & LA VOLPE A 2010. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell, 39, 25–35. [DOI] [PubMed] [Google Scholar]
- AGUDA BD, KIM Y, KIM HS, FRIEDMAN A & FINE HA 2011. Qualitative network modeling of the Myc-p53 control system of cell proliferation and differentiation. Biophys J, 101, 2082–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AIBAR S, GONZALEZ-BLAS CB, MOERMAN T, HUYNH-THU VA, IMRICHOVA H, HULSELMANS G, RAMBOW F, MARINE JC, GEURTS P, AERTS J, VAN DEN OORD J, ATAK ZK, WOUTERS J & AERTS S 2017. SCENIC: single-cell regulatory network inference and clustering. Nat Methods, 14, 1083–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALTER BP 2014. Fanconi anemia and the development of leukemia. Best Pract Res Clin Haematol, 27, 214–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAHR C, VON PALESKE L, USLU VV, REMESEIRO S, TAKAYAMA N, NG SW, MURISON A, LANGENFELD K, PETRETICH M, SCOGNAMIGLIO R, ZEISBERGER P, BENK AS, AMIT I, ZANDSTRA PW, LUPIEN M, DICK JE, TRUMPP A & SPITZ F 2018. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature, 553, 515–520. [DOI] [PubMed] [Google Scholar]
- BAK RO, DEVER DP & PORTEUS MH 2018. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat Protoc, 13, 358–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALCEREK J, JIANG J, LI Y, JIANG Q, HOLDREITH N, SINGH B, CHANDRA V, LV K, REN JG, ROZENOVA K, LI W, GREENBERG RA & TONG W 2018. Lnk/Sh2b3 deficiency restores hematopoietic stem cell function and genome integrity in Fancd2 deficient Fanconi anemia. Nat Commun, 9, 3915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BANCHEREAU J, PASCUAL V & O’GARRA A 2012. From IL-2 to IL-37: the expanding spectrum of anti-inflammatory cytokines. Nat Immunol, 13, 925–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOWRY A, PIBERGER AL, ROJAS P, SAPONARO M & PETERMANN E 2018. BET Inhibition Induces HEXIM1- and RAD51-Dependent Conflicts between Transcription and Replication. Cell Rep, 25, 2061–2069 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRETONES G, DELGADO MD & LEON J 2015. Myc and cell cycle control. Biochim Biophys Acta, 1849, 506–16. [DOI] [PubMed] [Google Scholar]
- BRIOT D, MACE-AIME G, SUBRA F & ROSSELLI F 2008. Aberrant activation of stress-response pathways leads to TNF-alpha oversecretion in Fanconi anemia. Blood, 111, 1913–23. [DOI] [PubMed] [Google Scholar]
- BUTLER A, HOFFMAN P, SMIBERT P, PAPALEXI E & SATIJA R 2018. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol, 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CECCALDI R, PARMAR K, MOULY E, DELORD M, KIM JM, REGAIRAZ M, PLA M, VASQUEZ N, ZHANG QS, PONDARRE C, PEFFAULT DE LATOUR R, GLUCKMAN E, CAVAZZANA-CALVO M, LEBLANC T, LARGHERO J, GROMPE M, SOCIE G, D'ANDREA AD & SOULIER J 2012. Bone marrow failure in Fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell, 11, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COUPER KN, BLOUNT DG & RILEY EM 2008. IL-10: the master regulator of immunity to infection. J Immunol, 180, 5771–7. [DOI] [PubMed] [Google Scholar]
- DELMORE JE, ISSA GC, LEMIEUX ME, RAHL PB, SHI J, JACOBS HM, KASTRITIS E, GILPATRICK T, PARANAL RM, QI J, CHESI M, SCHINZEL AC, MCKEOWN MR, HEFFERNAN TP, VAKOC CR, BERGSAGEL PL, GHOBRIAL IM, RICHARDSON PG, YOUNG RA, HAHN WC, ANDERSON KC, KUNG AL, BRADNER JE & MITSIADES CS 2011. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell, 146, 904–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUFOUR C, CORCIONE A, SVAHN J, HAUPT R, POGGI V, BEKA'SSY AN, SCIME R, PISTORIO A & PISTOIA V 2003. TNF-alpha and IFN-gamma are overexpressed in the bone marrow of Fanconi anemia patients and TNF-alpha suppresses erythropoiesis in vitro. Blood, 102, 2053–9. [DOI] [PubMed] [Google Scholar]
- EBENS CL, MACMILLAN ML & WAGNER JE 2017. Hematopoietic cell transplantation in Fanconi anemia: current evidence, challenges and recommendations. Expert Rev Hematol, 10, 81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ECCLES LJ, BELL AC & POWELL SN 2018. Inhibition of non-homologous end joining in Fanconi Anemia cells results in rescue of survival after interstrand crosslinks but sensitization to replication associated double-strand breaks. DNA Repair (Amst), 64, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARAYCOECHEA JI, CROSSAN GP, LANGEVIN F, MULDERRIG L, LOUZADA S, YANG F, GUILBAUD G, PARK N, ROERINK S, NIK-ZAINAL S, STRATTON MR & PATEL KJ 2018. Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature, 553, 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARBATI MR, HAYS LE, RATHBUN RK, JILLETTE N, CHIN K, AL-DHALIMY M, AGARWAL A, NEWELL AE, OLSON SB & BAGBY GC JR. 2016. Cytokine overproduction and crosslinker hypersensitivity are unlinked in Fanconi anemia macrophages. J Leukoc Biol, 99, 455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUARDIOLA P, PASQUINI R, DOKAL I, ORTEGA JJ, VAN WEEL-SIPMAN M, MARSH JC, BALL SE, LOCATELLI F, VERMYLEN C, SKINNER R, LJUNGMAN P, MINIERO R, SHAW PJ, SOUILLET G, MICHALLET M, BEKASSY AN, KRIVAN G, DI BARTOLOMEO P, HEILMANN C, ZANESCO L, CAHN JY, ARCESE W, BACIGALUPO A & GLUCKMAN E 2000. Outcome of 69 allogeneic stem cell transplantations for Fanconi anemia using HLA-matched unrelated donors: a study on behalf of the European Group for Blood and Marrow Transplantation. Blood, 95, 422–9. [PubMed] [Google Scholar]
- HANELINE LS, GOBBETT TA, RAMANI R, CARREAU M, BUCHWALD M, YODER MC & CLAPP DW 1999. Loss of FancC function results in decreased hematopoietic stem cell repopulating ability. Blood, 94, 1–8. [PubMed] [Google Scholar]
- HE YJ, MEGHANI K, CARON MC, YANG C, RONATO DA, BIAN J, SHARMA A, MOORE J, NIRAJ J, DETAPPE A, DOENCH JG, LEGUBE G, ROOT DE, D'ANDREA AD, DRANE P, DE S, KONSTANTINOPOULOS PA, MASSON JY & CHOWDHURY D 2018. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature, 563, 522–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEROLD S, HERKERT B & EILERS M 2009. Facilitating replication under stress: an oncogenic function of MYC? Nat Rev Cancer, 9, 441–4. [DOI] [PubMed] [Google Scholar]
- JOYCE CE, SAADATPOUR A, RUIZ-GUTIERREZ M, BOLUKBASI OV, JIANG L, THOMAS DD, YOUNG S, HOFMANN I, SIEFF CA, MYERS KC, WHANGBO J, LIBERMANN TA, NUSBAUM C, YUAN GC, SHIMAMURA A & NOVINA CD 2019. TGFbeta signaling underlies hematopoietic dysfunction and bone marrow failure in Shwachman-Diamond Syndrome. J Clin Invest, 130, 3821–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAIS Z, RONDINELLI B, HOLMES A, OĽEARY C, KOZONO D, D'ANDREA AD& CECCALDI R 2016. FANCD2 Maintains Fork Stability in BRCA1/2-Deficient Tumors and Promotes Alternative End-Joining DNA Repair. Cell Rep, 15, 2488–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KARPOVA D & BONIG H 2015. Concise Review: CXCR4/CXCL12 Signaling in Immature Hematopoiesis--Lessons From Pharmacological and Genetic Models. Stem Cells, 33, 2391–9. [DOI] [PubMed] [Google Scholar]
- KARRAS GI, YI S, SAHNI N, FISCHER M, XIE J, VIDAL M, D'ANDREA AD, WHITESELL L & LINDQUIST S 2017. HSP90 Shapes the Consequences of Human Genetic Variation. Cell, 168, 856–866 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAURENTI E, VARNUM-FINNEY B, WILSON A, FERRERO I, BLANCO-BOSE WE, EHNINGER A, KNOEPFLER PS, CHENG PF, MACDONALD HR, EISENMAN RN, BERNSTEIN ID & TRUMPP A 2008. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell, 3, 611–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI J, SEJAS DP, ZHANG X, QIU Y, NATTAMAI KJ, RANI R, RATHBUN KR, GEIGER H, WILLIAMS DA, BAGBY GC & PANG Q 2007. TNF-alpha induces leukemic clonal evolution ex vivo in Fanconi anemia group C murine stem cells. J Clin Invest, 117, 3283–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI Y, AMARACHINTHA S, WILSON AF, LI X & DU W 2017. Persistent response of Fanconi anemia haematopoietic stem and progenitor cells to oxidative stress. Cell Cycle, 16, 1201–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIM KS, LI H, ROBERTS EA, GAUDIANO EF, CLAIRMONT C, SAMBEL LA, PONNIENSELVAN K, LIU JC, YANG C, KOZONO D, PARMAR K, YUSUFZAI T, ZHENG N & D'ANDREA AD 2018. USP1 Is Required for Replication Fork Protection in BRCA1- Deficient Tumors. Mol Cell, 72, 925–941 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN FC, KARWAN M, SALEH B, HODGE DL, CHAN T, BOELTE KC, KELLER JR & YOUNG HA 2014. IFN-gamma causes aplastic anemia by altering hematopoietic stem/progenitor cell composition and disrupting lineage differentiation. Blood, 124, 3699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATATALL KA, JEONG M, CHEN S, SUN D, CHEN F, MO Q, KIMMEL M & KING KY 2016. Chronic Infection Depletes Hematopoietic Stem Cells through Stress-Induced Terminal Differentiation. Cell Rep, 17, 2584–2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATHSYARAJA H & EISENMAN RN 2016. Parsing Myc Paralogs in Oncogenesis. Cancer Cell, 29, 1–2. [DOI] [PubMed] [Google Scholar]
- MAYA-MENDOZA A, OSTRAKOVA J, KOSAR M, HALL A, DUSKOVA P, MISTRIK M, MERCHUT-MAYA JM, HODNY Z, BARTKOVA J, CHRISTENSEN C & BARTEK J 2015. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol Oncol, 9, 601–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NASI S, CIARAPICA R, JUCKER R, ROSATI J & SOUCEK L 2001. Making decisions through Myc. FEBS Lett, 490, 153–62. [DOI] [PubMed] [Google Scholar]
- NIE Y, HAN YC & ZOU YR 2008. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med, 205, 777–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OGURO H, DING L & MORRISON SJ 2013. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell, 13, 102–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OOSTRA AB, NIEUWINT AW, JOENJE H & DE WINTER JP 2012. Diagnosis of fanconi anemia: chromosomal breakage analysis. Anemia, 2012, 238731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PACE P, MOSEDALE G, HODSKINSON MR, ROSADO IV, SIVASUBRAMANIAM M & PATEL KJ 2010. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science, 329, 219–23. [DOI] [PubMed] [Google Scholar]
- PARMAR K, D'ANDREA A & NIEDERNHOFER LJ 2009. Mouse models of Fanconi anemia. Mutat Res, 668, 133–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARMAR K, KIM J, SYKES SM, SHIMAMURA A, STUCKERT P, ZHU K, HAMILTON A, DELOACH MK, KUTOK JL, AKASHI K, GILLILAND DG & D'ANDREA A 2010. Hematopoietic stem cell defects in mice with deficiency of Fancd2 or Usp1. Stem Cells, 28, 1186–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PENG C, CHEN Y, SHAN Y, ZHANG H, GUO Z, LI D & LI S 2012. LSK derived LSK-cells have a high apoptotic rate related to survival regulation of hematopoietic and leukemic stem cells. PLoS One, 7, e38614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PONTEL LB, ROSADO IV, BURGOS-BARRAGAN G, GARAYCOECHEA JI, YU R, ARENDS MJ, CHANDRASEKARAN G, BROECKER V, WEI W, LIU L, SWENBERG JA, CROSSAN GP & PATEL KJ 2015. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol Cell, 60, 177–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUCCETTI MV, ADAMS CM, KUSHINSKY S & EISCHEN CM 2019. Smarcal1 and Zranb3 Protect Replication Forks from Myc-Induced DNA Replication Stress. Cancer Res, 79, 1612–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QI J 2014. Bromodomain and extraterminal domain inhibitors (BETi) for cancer therapy: chemical modulation of chromatin structure. Cold Spring Harb Perspect Biol, 6, a018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUENTIN S, CUCCUINI W, CECCALDI R, NIBOUREL O, PONDARRE C, PAGES MP, VASQUEZ N, DUBOIS D'ENGHIEN C, LARGHERO J, PEFFAULT DE LATOUR R, ROCHA V, DALLE JH, SCHNEIDER P, MICHALLET M, MICHEL G, BARUCHEL A, SIGAUX F, GLUCKMAN E, LEBLANC T, STOPPA-LYONNET D, PREUDHOMME C, SOCIE G & SOULIER J 2011. Myelodysplasia and leukemia of Fanconi anemia are associated with a specific pattern of genomic abnormalities that includes cryptic RUNX1/AML1 lesions. Blood, 117, e161–70. [DOI] [PubMed] [Google Scholar]
- RAMANA CV, GRAMMATIKAKIS N, CHERNOV M, NGUYEN H, GOH KC, WILLIAMS BR & STARK GR 2000. Regulation of c-myc expression by IFN-gamma through Statl-dependent and -independent pathways. EMBO J, 19, 263–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIO P, NAVARRO S, WANG W, SANCHEZ-DOMINGUEZ R, PUJOL RM, SEGOVIA JC, BOGLIOLO M, MERINO E, WU N, SALGADO R, LAMANA ML, YANEZ RM, CASADO JA, GIMENEZ Y, ROMAN-RODRIGUEZ FJ, ALVAREZ L, ALBERQUILLA O, RAIMBAULT A, GUENECHEA G, LOZANO ML, CERRATO L, HERNANDO M, GALVEZ E, HLADUN R, GIRALT I, BARQUINERO J, GALY A, GARCIA DE ANDOIN N, LOPEZ R, CATALA A, SCHWARTZ JD, SURRALLES J, SOULIER J, SCHMIDT M, DIAZ DE HEREDIA C, SEVILLA J & BUEREN JA 2019. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat Med, 25, 1396–1401. [DOI] [PubMed] [Google Scholar]
- RISITANO AM, MAROTTA S, CALZONE R, GRIMALDI F, ZATTERALE A & CONTRIBUTORS R 2016. Twenty years of the Italian Fanconi Anemia Registry: where we stand and what remains to be learned. Haematologica, 101, 319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODRIGUEZ A & D’ANDREA AD 2017. Quick guide. Fanconi anemia pathway. Current Biology, 27, R979–R1001. [DOI] [PubMed] [Google Scholar]
- ROE JS, MERCAN F, RIVERA K, PAPPIN DJ & VAKOC CR 2015. BET Bromodomain Inhibition Suppresses the Function of Hematopoietic Transcription Factors in Acute Myeloid Leukemia. Mol Cell, 58, 1028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSSELLI F, SANCEAU J, GLUCKMAN E, WIETZERBIN J & MOUSTACCHI E 1994. Abnormal lymphokine production: a novel feature of the genetic disease Fanconi anemia. II. In vitro and in vivo spontaneous overproduction of tumor necrosis factor alpha. Blood, 83, 1216–25. [PubMed] [Google Scholar]
- SAKAMAKI JI, WILKINSON S, HAHN M, TASDEMIR N, O'PREY J, CLARK W, HEDLEY A, NIXON C, LONG JS, NEW M, VAN ACKER T, TOOZE SA, LOWE SW, DIKIC I & RYAN KM 2017. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol Cell, 66, 517–532 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATO T, ONAI N, YOSHIHARA H, ARAI F, SUDA T & OHTEKI T 2009. Interferon regulatory factor-2 protects quiescent hematopoietic stem cells from type I interferon-dependent exhaustion. Nat Med, 15, 696–700. [DOI] [PubMed] [Google Scholar]
- SETTY M, KISELIOVAS V, LEVINE J, GAYOSO A, MAZUTIS L & PE'ER D 2019. Characterization of cell fate probabilities in single-cell data with Palantir. Nat Biotechnol, 37, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHIMAMURA A & ALTER BP 2010. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev, 24, 101–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONDALLE SB, LONGERICH S, OGAWA LM, SUNG P & BASERGA SJ 2019. Fanconi anemia protein FANCI functions in ribosome biogenesis. Proc Natl Acad Sci U S A, 116, 2561–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPROSTON NR & ASHWORTH JJ 2018. Role of C-reactive protein at sites of inflammation and infection. Frontiers in Immunology, 9, 754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUMPTER R JR., SIRASANAGANDLA S, FERNANDEZ AF, WEI Y, DONG X, FRANCO L, ZOU Z, MARCHAL C, LEE MY, CLAPP DW, HANENBERG H & LEVINE B 2016. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell, 165, 867–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIKHONOVA AN, DOLGALEV I, HU H, SIVARAJ KK, HOXHA E, CUESTA-DOMINGUEZ A, PINHO S, AKHMETZYANOVA I, GAO J, WITKOWSKI M, GUILLAMOT M, GUTKIN MC, ZHANG Y, MARIER C, DIEFENBACH C, KOUSTENI S, HEGUY A, ZHONG H, FOOKSMAN DR, BUTLER JM, ECONOMIDES A, FRENETTE PS, ADAMS RH, SATIJA R, TSIRIGOS A & AIFANTIS I 2019. The bone marrow microenvironment at single-cell resolution. Nature, 569, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VELTEN L, HAAS SF, RAFFEL S, BLASZKIEWICZ S, ISLAM S, HENNIG BP, HIRCHE C, LUTZ C, BUSS EC, NOWAK D, BOCH T, HOFMANN WK, HO AD, HUBER W, TRUMPP A, ESSERS MA & STEINMETZ LM 2017. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol, 19, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALTER D, LIER A, GEISELHART A, THALHEIMER FB, HUNTSCHA S, SOBOTTA MC, MOEHRLE B, BROCKS D, BAYINDIR I, KASCHUTNIG P, MUEDDER K, KLEIN C, JAUCH A, SCHROEDER T, GEIGER H, DICK TP, HOLLAND-LETZ T, SCHMEZER P, LANE SW, RIEGER MA, ESSERS MA, WILLIAMS DA, TRUMPP A & MILSOM MD 2015. Exit from dormancy provokes DNA-damage-induced attrition in haematopoietic stem cells. Nature, 520, 549–52. [DOI] [PubMed] [Google Scholar]
- WATCHAM S, KUCINSKI I & GOTTGENS B 2019. New insights into hematopoietic differentiation landscapes from single-cell RNA sequencing. Blood, 133, 1415–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WILSON A, MURPHY MJ, OSKARSSON T, KALOULIS K, BETTESS MD, OSER GM, PASCHE AC, KNABENHANS C, MACDONALD HR & TRUMPP A 2004. c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev, 18, 2747–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WROBLEWSKI M, SCHELLER-WENDORFF M, UDONTA F, BAUER R, SCHLICHTING J, ZHAO L, BEN BATALLA I, GENSCH V, PASLER S, WU L, WANIOR M, TAIPALEENMAKI H, BOLAMPERTI S, NAJAFOVA Z, PANTEL K, BOKEMEYER C, QI J, HESSE E, KNAPP S, JOHNSEN S & LOGES S 2018. BET-inhibition by JQ1 promotes proliferation and self-renewal capacity of hematopoietic stem cells. Haematologica, 103, 939–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG H, KOZONO DE, O’CONNOR KW, VIDAL-CARDENAS S, ROUSSEAU A, HAMILTON A, MOREAU L, GAUDIANO EF, GREENBERGER J, BAGBY G, SOULIER J, GROMPE M, PARMAR K & D’ANDREA AD 2016. TGF-b Inhibition Rescues Hematopoietic Stem Cell Defects and Bone Marrow Failure in Fanconi Anemia. Cell Stem Cell, 18, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHAO X, GAO S, WU Z, KAJIGAYA S, FENG X, LIU Q, TOWNSLEY DM, COOPER J, CHEN J, KEYVANFAR K, FERNANDEZ IBANEZ MDP, WANG X & YOUNG NS 2017. Single-cell RNA-seq reveals a distinct transcriptome signature of aneuploid hematopoietic cells. Blood, 130, 2762–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG GX, TERRY JM, BELGRADER P, RYVKIN P, BENT ZW, WILSON R, ZIRALDO SB, WHEELER TD, MCDERMOTT GP, ZHU J, GREGORY MT, SHUGA J, MONTESCLAROS L, UNDERWOOD JG, MASQUELIER DA, NISHIMURA SY, SCHNALL-LEVIN M, WYATT PW, HINDSON CM, BHARADWAJ R, WONG A, NESS KD, BEPPU LW, DEEG HJ, MCFARLAND C, LOEB KR, VALENTE WJ, ERICSON NG, STEVENS EA, RADICH JP, MIKKELSEN TS, HINDSON BJ & BIELAS JH 2017. Massively parallel digital transcriptional profiling of single cells. Nat Commun, 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG S, PAPALEXI E, BUTLER A, STEPHENSON W & SATIJA R 2018. Molecular transitions in early progenitors during human cord blood hematopoiesis. Mol Syst Biol, 14, e8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during this study have been deposited in Gene Expression Omnibus (GEO) under the accession number: GSE157591.