

Abstract
Triple-negative breast cancer (TNBC) is the deadliest form of breast cancer. Unlike other types of breast cancer that can be effectively treated by targeted therapies, no such targeted therapy exists for all TNBC patients. The ADAR1 enzyme carries out A-to-I editing of RNA to prevent sensing of endogenous double-stranded RNAs. ADAR1 is highly expressed in breast cancer including TNBC. Here, we demonstrate that expression of ADAR1, specifically its p150 isoform, is required for the survival of TNBC cell lines. In TNBC cells, knockdown of ADAR1 attenuates proliferation and tumorigenesis. Moreover, ADAR1-knockdown leads to robust translational repression. ADAR1-dependent TNBC cell lines also exhibit elevated IFN stimulated gene expression. IFNAR1 reduction significantly rescued the proliferative defects of ADAR1 loss. These findings establish ADAR1 as a novel therapeutic target for TNBC tumors.
Introduction
Generally defined by the lack of estrogen receptor (ER), progesterone receptor (PR) and HER2 expression, triple-negative breast cancer (TNBC) accounts for 15 to 20 percent of all breast cancer diagnoses in the United States each year (1). Unlike ER-positive (tamoxifen, fulvestrant, and other ER modulators) and HER2-positive (Herceptin and other HER2 inhibitors) breast cancers, there are no targeted therapies for all TNBC patients (2). The lack of targeted therapies for TNBC leaves chemotherapy as the main treatment option, which carries a generally worse prognosis (3). Efforts to develop effective targeted therapies against TNBC have focused on further sub-categorizing TNBC based on gene expression signatures, as well as looking to exploit common genetic vulnerabilities (4, 5).
A potential therapeutic target for TNBC is Adenosine Deaminase Acting on RNA (ADAR1, encoded by ADAR). ADAR1 caries out the enzymatic reaction of deaminating adenosine to inosine within cellular dsRNA, in a process known as A-to-I editing. Induction of ADAR1 expression is prevalent in breast cancer (6–10) and ADAR1-mediated A-to-I editing has been found to influence the levels of its targets in breast cancer (11–14). Recent studies have indicated expression of ADAR1 is elevated in TNBC and may be correlated with poor prognosis when RNA editing is increased (15, 16).
ADAR1 acts in a negative feedback loop to inhibit activation of the type I interferon (IFN) pathway triggered by endogenous dsRNAs or dsRNAs introduced upon viral infection (17, 18). ADAR1 has been shown to suppress type I IFN pathway through multiple mechanisms, including destabilization of the dsRNA structure, reduced expression, and activation of the dsRNA sensors MDA5 and RIG-I, and inhibition of IFN expression (17–22). ADAR1-mediated A-to-I RNA editing by the IFN-inducible p150 isoform (not the constitutive p110 isoform) is essential for its ability to modulate dsRNA-induced IFN signaling (18–20). ADAR1’s ability to regulate this response was recently linked to the development of ADAR1-dependency in some cancer cell lines; two groups showed that by removing ADAR1 from cancer cells with elevated IFN signaling, cells became susceptible to inflammation-induced cell death (23, 24). This is consistent with previous findings that ADAR1 prevents immune and translational catastrophes by blocking dsRNA-activated pathways (17, 25).
Here we demonstrate that TNBC cell lines are dependent on ADAR1 expression; loss of ADAR1 in these cell lines inhibits cellular growth and tumorigenesis, highlighting the therapeutic potential of ADAR1 inhibitors for the treatment of TNBC.
Results
ADAR1 is highly expressed in all breast cancer subtypes
Using publicly available data from TCGA (The Cancer Genome Atlas) (6, 7), we found that high expression of ADAR1 correlated with poor prognosis of breast cancers (Fig. 1a). Recent studies indicated that ADAR1 promotes tumorigenesis of metaplastic breast cancers, and that high expression of ADAR1 correlates with poor prognosis in basal-like breast cancers (13, 16). Since both basal-like and metaplastic breast cancers share similar characteristics with TNBC, we sought to determine the importance of ADAR1 in the tumorigenesis of TNBC. By evaluating the TCGA database, we found that while mRNA expression of ADAR1 was higher in TNBC compared to normal, it was not significantly different between TNBC and non-TNBC tumors (Fig. 1b). Additionally, ADAR1 expression was not significantly higher in any one subtype of breast cancer based on PAM50 classification (26) (Fig. 1c). This observation is consistent with data from the Cancer Cell Line Encyclopedia (CCLE), which uses both RNA-seq and Reverse Phase Protein Array (RPPA) to determine RNA and protein expression levels in numerous cancer cell lines (Fig. 1d and Supplementary Fig. S1a). Data from both the TCGA and CCLE datasets also revealed that both p150 and p110 isoforms of ADAR1 were expressed at similar levels between TNBC and non-TNBC specimen (Fig. 1e and Supplementary Fig. S1b–e), with p110 expression being consistently higher than p150 in all samples. The lack of distinctive expression patterns for p150 and p110 between TNBC and non-TNBC was also confirmed by performing quantitative RT-PCR in a panel of breast cancer cell lines (Supplementary Fig. S1f, g). To independently investigate ADAR1 expression in BRCA tumor samples, we assessed p150 isoform expression by immunohistochemistry in TNBC and non-TNBC patient tumors using an antibody that specifically recognizes only the p150 isoform (Fig. 1f and Supplementary Fig. S1h). Consistent with RNA-seq data from TCGA, p150 was upregulated in the majority of TNBC and non-TNBC tumors compared to normal tissues. Next we sought to determine the protein expression level of the ADAR1-p150 isoform in a panel of established breast cancer cell lines representing TNBC and non-TNBC. Immunoblot analysis showed that ADAR1 (p150 isoform) is overexpressed, compared to normal human mammary epithelial cells (HMECs), in over half of all TNBC (6/8) and non-TNBC (5/8) cell lines assayed (Fig. 1g). These results indicate that ADAR1-p150 is overexpressed in many breast cancer cell lines regardless of subtype.
Figure 1: ADAR1 is highly expressed in all breast cancer subtypes.
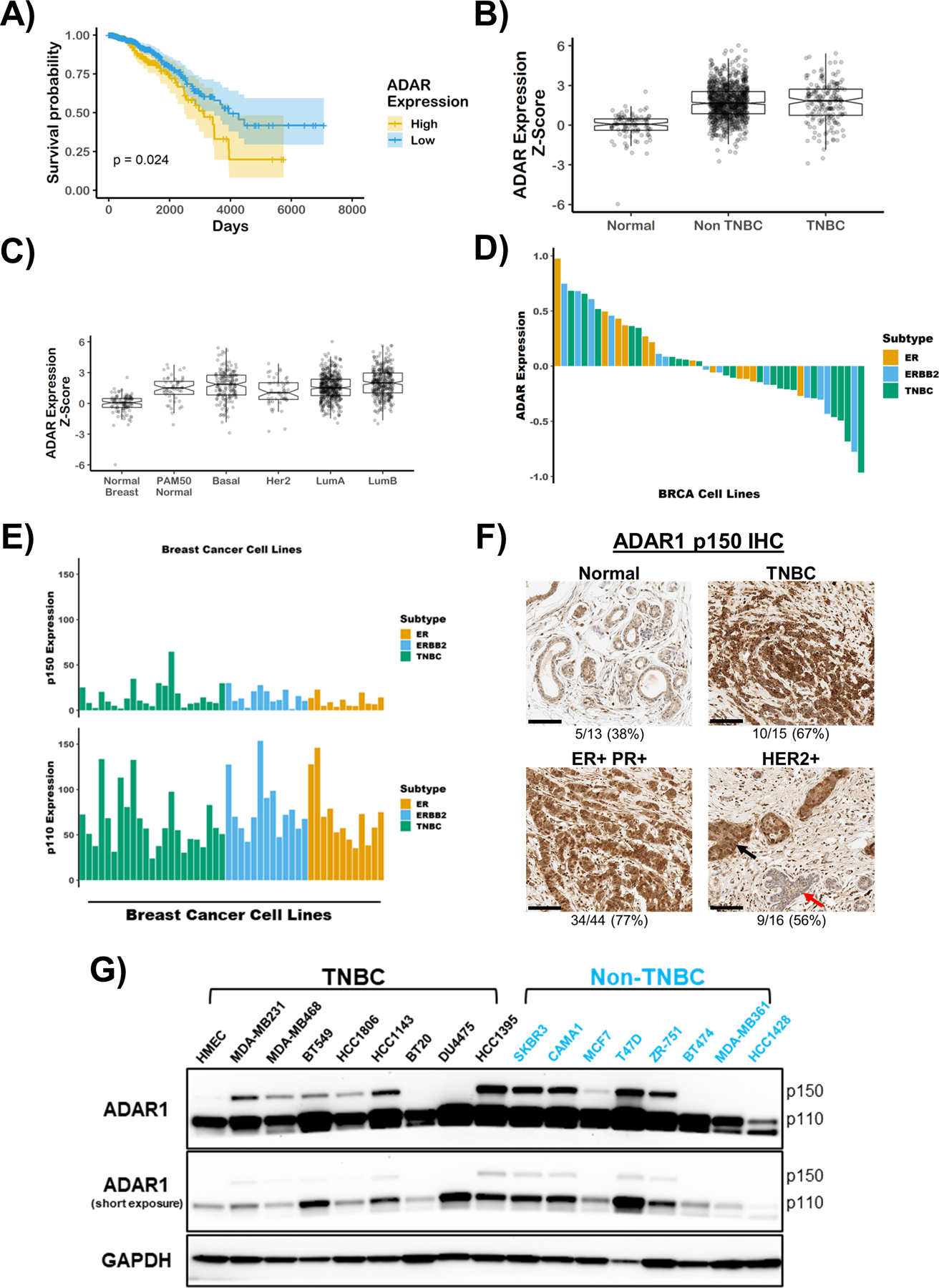
A) Kaplan-Meier survival curves of breast cancer patients. Patients were stratified by ADAR1 expression, above or below z-score = 2.34. B) and C), Z-score modified mRNA expression of ADAR1 in normal, TNBC and Non-TNBC breast cancer, or by PAM50 classification. LumA = luminal A and LumB = luminal B. Data were extracted from TCGA database. Tumor classification described previously(26). D) Protein expression of ADAR1 in breast cancer cell lines. ERBB2 = HER2. Data were extracted from CCLE database. E) mRNA expression of ADAR1 p150 and p110 isoforms in breast cancer cell lines. Data were extracted from CCLE database. F) Representative images of IHC staining of ADAR1 (p150 isoform) in normal, TNBC and Non-TNBC (ER+PR+ and HER2+) breast cancer tissues (scale bar: 100 μM). Numbers below the image indicate the ratio of samples identified as high p150-ADAR1 based on IHC scoring. Black and red arrows in the HER2+ image point to adjacent p150-high cancerous and p150-low non-cancerous ductal epithelial cells, respectively. G) Immunoblots showing protein levels of ADAR1 and GAPDH (loading control) in breast cancer cell lines. Images are representative of three replicates.
ADAR1 is required for TNBC proliferation
Several recent studies have suggested that some established cancer cell lines display strong dependencies on ADAR1 expression (23, 24, 27). Given the high expression of ADAR1-p150 in most breast cancer cell lines, we sought to determine whether these breast cancer cell lines exhibit ADAR1-dependency. We analyzed publicly available RNAi and CRISPR-Cas9 datasets to determine if ADAR1 was required for the survival of breast cancer cell lines representing various subtypes (28, 29). TNBC and basal-like cell lines made up the majority of breast cancer cells exhibiting high ADAR1 sensitivity scores (DEMETER2 Score < −0.5) (Fig. 2a and Supplementary Fig. S2a–c). Importantly, we did not observe a correlation between ADAR1 expression and ADAR1-dependency across these breast cancer cell lines (Supplementary Fig. S2d). This lack of correlation was noted for both p110 and p150 isoforms of ADAR1 (Supplementary Fig. S2e, f). To independently validate ADAR1-dependency among breast cancer cell lines, we knocked-down ADAR1 expression in eight cell lines (Four TNBC: MDA-MB231, MDA-MB468, BT549, HCC1806; Four non-TNBC: SKBR3, CAMA1, MCF7, T47D); all of these cell lines showed noticeable ADAR1-p150 isoform overexpression over HMEC controls in our immunoblot analysis (Fig. 1g). Long-term (7–28 days, foci formation) and short-term (4 days, growth curve) cell proliferation was evaluated for each cell line following ADAR1 knockdown. Notably, similar levels of ADAR1 knockdown were achieved for each cell line (Fig. 2b). All four TNBC cell lines displayed significant attenuation in both long- and short-term proliferation following ADAR1 knockdown (Fig. 2c, d and Supplementary Fig. S2g). Conversely, ADAR1 expression proved dispensable for proliferation in all four non-TNBC cell lines.
Figure 2: ADAR1 is required for TNBC survival and proliferation.
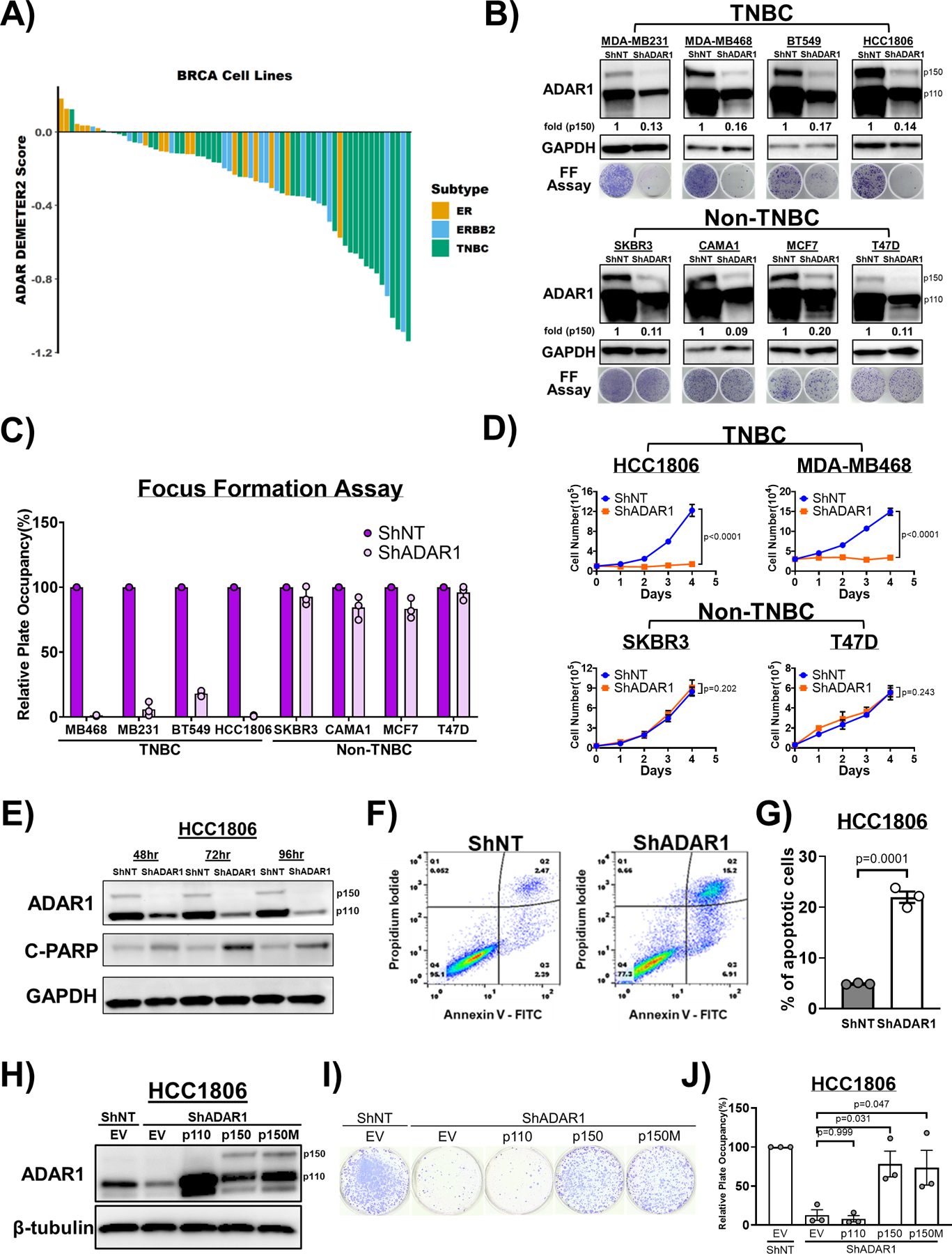
A) ADAR1-dependency scores in breast cancer cell lines. Lower DEMETER2 scores indicate stronger ADAR1-dependency. ERBB2 = HER2. B) Immunoblots showing protein levels of ADAR1 and GAPDH (loading control) with or without ADAR1-knockdown in breast cancer cell lines. Fold change of ADAR1 p150 isoform (ShADAR1/ShNT) is indicated, normalized to GAPDH. Focus formation (FF) assay showed that ADAR1-knockdown reduced proliferation of TNBC but not Non-TNBC cells. Images are representative, N=3. C) Quantification of FF in B).Relative plate occupancy was determined using ImageJ software and normalized to ShNT samples for each cell line. Data are represented as mean ± SD, N=3. D) Cell proliferation assay showing that ADAR1-knockdown reduced proliferation of TNBC but not Non-TNBC cells. Data are represented as mean ± SD, N=2. E) Immunoblots showing protein levels of ADAR1, GAPDH (loading control) and cleaved PARP (C-PARP; apoptosis marker) in ShADAR1-treated HCC1806 cells. F) Flow cytometry analysis with propidium iodide (PI) and Annexin V-FITC (AV) staining to detect portions of apoptotic cells in ShNT- or ShADAR1-treated HCC1806 cells. PIlowAVlow: Live cells. PIlowAVhigh: Early apoptotic cells. PIhighAVhigh: Late apoptotic cells. G) Quantification of relative level of apoptosis (early + late) in ShNT- or ShADAR1-treated HCC1806 cells in F). Data are represented as mean ± SD, N=3. H) Immunoblots showing protein levels of ADAR1 and β-tubulin (loading control) with overexpression of p150, p110 or editing-defective p150E912A (p150M) in ShADAR1-treated HCC1806 cells. Images are representative, N=3. EV, empty virus. I) FF assay showing that p150 and p150M, but not p110, partially rescued proliferation of ShADAR1-treated HCC1806 cells. Images are representative, N=3. J) Quantification of FF in I). Relative plate occupancy was determined using ImageJ software and normalized to ShNT-EV. Data are represented as mean ± SD, N>3.
Previous work has shown that loss of ADAR1 leads to cell death via apoptosis (30, 31). Knockdown of ADAR1 in two TNBC cell lines (HCC1806 and MB231) caused apoptosis (Fig. 2e–g and Supplementary Fig. S3a–d). Apoptosis induction was detected by co-staining of Annexin V and propidium iodide (PI), as well as immunoblot analysis showing increased levels of cleaved PARP. Taken together these data show that ADAR1 is essential for the survival and growth of many TNBC cells.
As expected, knockdown of ADAR1 reduced RNA editing level of its targets, such as antizyme inhibitor 1 (AZIN1) (Supplementary Fig. S3e, f). Edited AZIN1 (S367G) has been shown to block antizyme-mediated degradation of ornithine decarboxylase (ODC) and cyclin D1, leading to increased cell proliferation, tumor initiation and metastasis (32, 33). To determine if AZIN1 editing contributes to TNBC-associated ADAR1-dependency, we performed RNA Editing Site-Specific quantitative PCR (RESS-qPCR) to assess the editing level of AZIN1 with or without ADAR1 knockdown (34). The editing level of AZIN1 is generally lower in TNBC compared to non-TNBC cell lines, and ADAR1-knockdown did not result in more significant reduction of AZIN1 editing in TNBC cell lines (Supplementary Fig. S3f, g). These data suggest that it is unlikely that ADAR1-mediated AZIN1 editing contributes to the TNBC-associated ADAR1-dependency.
ADAR1-p150 is required for TNBC proliferation
While both isoforms of ADAR1 are expressed in TNBC, our knockdown experiment does not distinguish between p150 or p110 dependence. To address this, we set up a knockdown-rescue system. We overexpressed either the p110 or p150 isoform following ADAR1 knockdown in HCC1806 and MDA-MB231 cells and evaluated their ability to rescue cell proliferation. Overexpression of p110 and p150 caused increased editing of MDM2 mRNA as determined by Sanger sequencing of cDNA (Supplementary Fig. S3h, i). Overexpression of ADAR1-p150, but not p110, resulted in significant rescue of cell proliferation in HCC1806 and MDA-MB231 TNBC cells (Fig. 2h–j and Supplementary Fig. S3j–l). Having established a rescue system for ADAR1-dependent proliferation, we next aimed to determine whether the editing activity of ADAR1-p150 was required for this rescue. Overexpression of the editing-defective mutant (E912A) p150 had no effect on editing of MDM2 mRNA (Supplementary Fig. S3h, i). Overexpression of p150-E912A was capable of rescuing the ADAR1 knockdown phenotype in HCC1806 to the same extent as WT p150 (Fig. 2h–j). For the MDA-MB231 cell line, p150-E912A rescued to a lesser extent than WT p150 (Supplementary Fig. S3j–l). These data indicate that dependency on A-to-I editing by ADAR1 p150 isoform varies across TNBC cell lines.
ADAR1 is required for TNBC transformation and tumorigenesis
To assess the functional relevance of our findings, we investigated the requirement of ADAR1 for the transformation of breast cancer cell lines. We utilized anchorage-independent growth in soft agar as a measure of cellular transformation. Knockdown of ADAR1 dramatically reduced soft agar colonies of MDA-MB231 and HCC1806 TNBC cells while not significantly affecting the numbers of colonies formed by SKBR3 and T47D non-TNBC cells (Fig. 3a–d). The effect of ADAR1 knockdown on anchorage-independent growth could be rescued by WT and editing defective ADAR1-p150 in HCC1806 cells (Fig. 3e, f), implying that ADAR1 does possess critical non-editing functions.
Figure 3: ADAR1 is required for TNBC transformation and tumorigenesis.
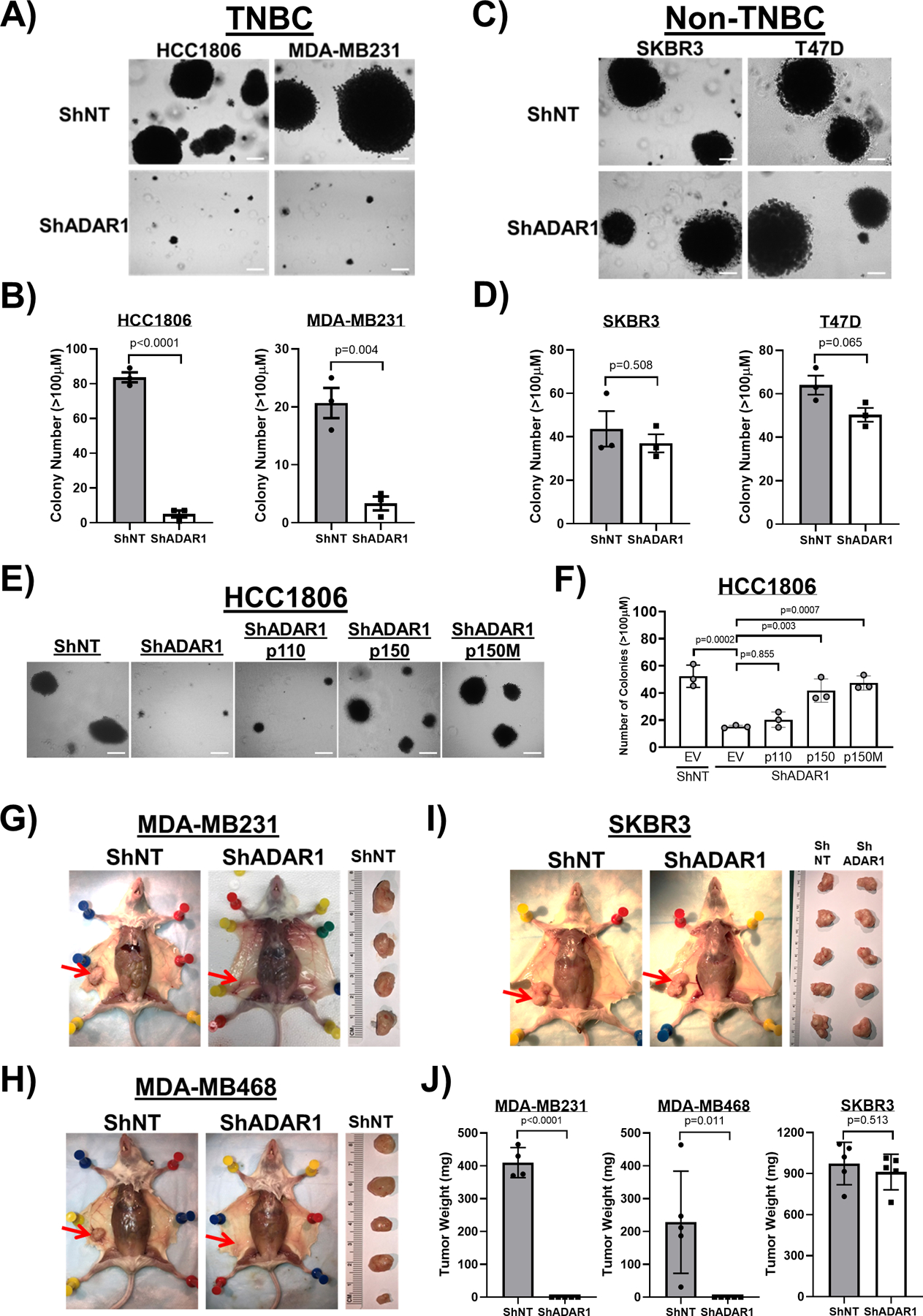
A) Soft agar assay (SAA) showing that ADAR1-knockdown reduced anchorage-independent growth of TNBC cells (HCC1806 and MDA-MB231). Images are representative, N=3. Scale-bar, 100μM. B) Quantification of SAA in A). Colonies bigger than 100μM in diameter were counted. Data are represented as mean ± SD, N=3. C) SAA showing that ADAR1-knockdown did not affect anchorage-independent growth of Non-TNBC cells (SKBR3 and T47D). Images are representative, N=3. Scale-bar, 100μM. D) Quantification of SAA in C). Colonies bigger than 100μM in diameter were counted. Data are represented as mean ± SD, N=3. E) SAA showing that overexpression of p150 and p150M, but not p110, partially rescued reduced anchorage-independent growth of HCC1806 cells due to ADAR1-knockdown. Images are representative, N=3. Scale-bar, 100μM. F) Quantification of SAA in E). Colonies bigger than 100μM in diameter were counted. Data are represented as mean ± SD, N=3. G) Orthotopic implantation of MDA-MB231 cells into abdominal mammary fat pad. Tumors were removed from the mice ~4 weeks post injection and weighed (ShNT, N=4; ShADAR1, N=5). Red arrows indicate the location of mammary fat pad. H) Orthotopic implantation of MDA-MB468 cells into abdominal mammary fat pad. Tumors were removed from the mice ~12 weeks post injection and weighed (N=5). Red arrows indicate the location of mammary fat pad. I) Orthotopic implantation of SKBR3 cells into abdominal mammary fat pad. Tumors were removed from the mice ~4 weeks post injection and weighed (N=5). Red arrows indicate the location of mammary fat pad. J) Quantification of the result shown in G)–I). Data are represented as mean ± SD.
To extend these in vitro findings, we next determined whether ADAR1 was required for TNBC cell lines to form tumors in vivo. We performed mammary gland orthotopic transplantations using TNBC and non-TNBC cells following ADAR1 knockdown. MDA-MB231, MDA-MB468, and SKBR3 cells were all able to form visible tumors in the mammary glands of independently transplanted female immune compromised mice (Fig. 3g–j). Knockdown of ADAR1 in MDA-MB231 and MDA-MB468 TNBC cells completely abrogated their ability to form tumors in transplanted mice. In contrast, ADAR1 knockdown in SKBR3 cells did not significantly affect tumor formation in transplanted mammary glands. Collectively, these results demonstrate that ADAR1 expression is required for in vitro transformation and in vivo tumor formation of TNBC cells but is completely dispensable for these properties in non-TNBC cells.
PKR is overexpressed in TNBC and activated upon ADAR1 loss
Previous reports have shown that ADAR1 dependency in human cancer cells could be mediated through several downstream pathways, including translational inhibition triggered by activated PKR or ribonuclease L (RNASEL), as well as type I IFN signaling (23, 24, 35). To investigate if these pathways contribute to the ADAR1 dependency observed in TNBC cells, we first analyzed the TCGA and CCLE datasets to determine if these pathways are intrinsically elevated in TNBC. Across TCGA breast cancer samples, RNA expression of PKR is significantly higher in TNBC samples compared to non-TNBC (Fig. 4a). This is consistent with RNA-seq data for breast cancer cell lines within the CCLE (Fig. 4b). Moreover, elevated PKR expression positively correlates with the ADAR1 sensitivity scores, suggesting a strong relationship between PKR and TNBC-associated ADAR1 dependency (Fig. 4c and Supplementary Fig. S4a–c). We further confirmed this observation by immunoblot analysis among our panel of sixteen breast cancer cell lines which showed a general elevation of PKR expression across all TNBC cell lines (Fig. 4d). We also detected heightened levels of PKR phosphorylation as well as its substrate alpha subunit of eukaryotic translation initiation factor 2 (eIF2α) in TNBC cells compared to non-TNBC cells. Upon ADAR1 knockdown, phosphorylation of PKR and eIF2α was markedly induced in all TNBC cell lines but remained unchanged in the non-TNBC cell lines (Fig. 4e). These observations suggest that TNBC-associated ADAR1 dependency might be facilitated by PKR activation. We observed another potential connection between PKR-eIF2α signaling and ADAR1-dependency by comparing levels of PKR- and eIF2α-phosphorylation in aforementioned rescue experiments using different isoforms of ADAR1 (Fig. 2h–j and Supplementary Fig. S3j–l). Overexpression of WT p150 and p150-E912A, both capable of rescuing ADAR1-dependent phenotypes in TNBC cells, resulted in decreased phosphorylation of PKR and eIF2α (Fig. 4f).
Figure 4: PKR is overexpressed in TNBC and activated upon ADAR loss.
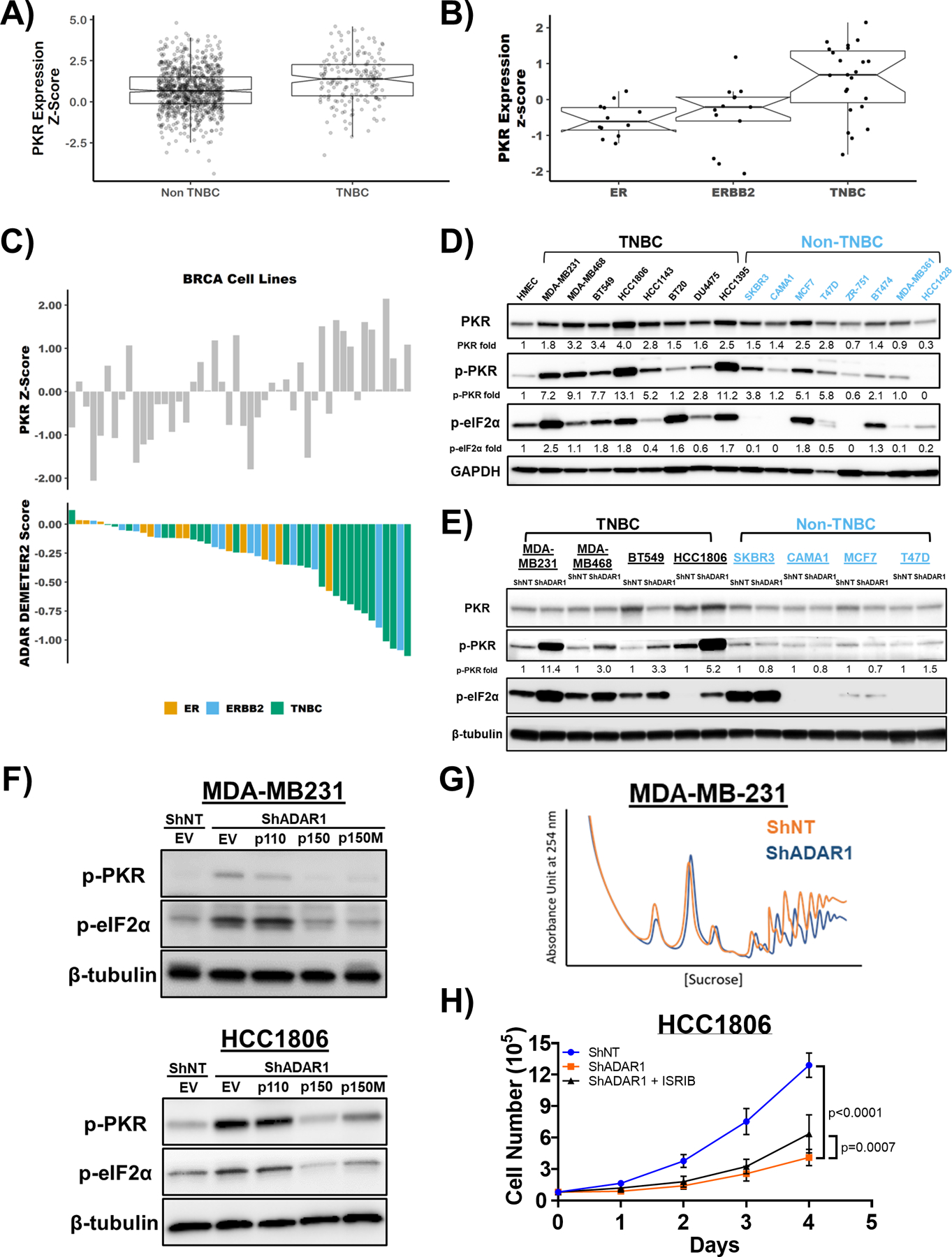
A) mRNA expression of PKR in TNBC and Non-TNBC. Data were extracted from TCGA database. B) mRNA expression of PKR in ER-positive, ERBB2(HER2)-positive and TNBC cell lines. Data were extracted from CCLE database. C) ADAR1-dependency score positively correlates with PKR expression. Upper panel: PKR expression z-score in breast cancer cell lines. Lower panel: ADAR1-dependency scores. Lower DEMETER2 scores indicate stronger ADAR1-dependency. D) Immunoblots showing protein levels of PKR, p-PKR (T446), p-eIF2α (S51) and GAPDH (loading control) in breast cancer cell lines. Densitometry quantification of gel images was normalized to GAPDH and set relative to HMEC signal. Data shown are representative, N=3. E) Immunoblots showing protein levels of PKR, p-PKR (T446), p-eIF2α (S51) and β-tubulin (loading control) in TNBC and non-TNBC breast cancer cell lines with or without ADAR1-knockdown. Densitometry quantification of gel images was normalized to GAPDH and compared to HMEC signal set as 1-fold. Data shown are representative, N=3. F) Immunoblots showing protein levels of p-PKR (T446), p-eIF2α (S51) and β-tubulin (loading control) with overexpression of p150, p110 or editing-defective p150E912A (p150M) in ShADAR1-treated MDA-MB231 and HCC1806 cells. Data shown are representative, N=3. G) Polysome profiling of MDA-MB231 cells with or without ADAR1-knockdown. Data shown are representative of three replicates. H) Cell proliferation assay showing that treatment of ISRIB (5nM) resulted in a modest rescue of ADAR1-knockdown phenotype in HCC1806 cells. Data are represented as mean ± SD. N=3.
Activation of PKR causes global translational repression through phosphorylation of eIF2α (36). To investigate if translational repression occurs following ADAR1 knockdown, we performed polysome profiling. ADAR1 knockdown in MDA-MB231 and HCC1806 TNBC cells led to inhibition of translation, demonstrated by the substantial reduction of polysome peaks (Fig. 4g and Supplementary Fig. S4d). These results suggest that translational repression may contribute to TNBC-associated ADAR1 dependency, however it is not clear if the observed translational repression is caused by PKR activation. To address this, we attempted a rescue experiment by knocking down PKR in conjunction with ADAR1 knockdown. While attempting to rescue the ADAR-knockdown phenotype in MDA-MB231 and HCC1806 cells by knockdown of PKR, we observed that knockdown of PKR alone greatly reduced foci formation (Supplementary Fig. S4e, f). Treatment of MDA-MB231 with a PKR inhibitor also caused reduced foci formation (Supplementary Fig. S4g). These data suggest that basal PKR expression and activity is required for the proliferation of these cell lines, thus precluding us from directly determining if expression of PKR is required for the ADAR-knockdown phenotype, or if increased PKR activity drives translational repression following ADAR-knockdown.
Due to the essentiality of PKR in the cell lines used in this study, we used a pharmacological approach to blunt the effects of eIF2α phosphorylation by PKR. We used the small molecule ISRIB which inhibits the translational repressive function of p-eIF2α (37). ISRIB was only capable of modestly rescuing the proliferation defect of ADAR1 knockdown in HCC1806 (Fig. 4h and Supplementary Fig. S4h) but not MDA-MB231 cells (data not shown). These data suggest that phosphorylation of eIF2α by PKR, following ADAR knockdown, contributes only modestly to reduced proliferation and is cell line dependent.
An important downstream effector of PKR activation is the pro-survival gene ATF4. Unlike most cellular mRNAs, the ATF4 mRNA is translationally upregulated following activation of PKR and phosphorylation of eIF2α (41). Because of the pro-survival role of ATF4, it is possible that it may play a role in protecting cells from cellular death following ADAR1-knockdown and PKR activation. Assessment of ATF4 expression across breast cancer cell lines revealed no correlation between ATF4 expression and ADAR1-dependency (Supplementary Fig. S5a, b). There was no clear pattern in ATF4 protein expression between TNBC and non-TNBC following ADAR1-knockdown, suggesting that ATF4 expression alone cannot be used to determine ADAR1-dependency in TNBC cells (Supplementary Fig. S5c). Together these data suggest that ATF4 expression does not contribute to ADAR-dependency or growth inhibition following ADAR1 knockdown.
RNASEL is not activated following loss of ADAR1 in TNBC
Activation of RNASEL and subsequent translational inhibition has also been shown to result in cell lethality upon ADAR1 loss (35). The CCLE dataset indicated that RNASEL activators OAS1, OAS2 and OAS3 were highly expressed in ADAR1 dependent cell lines, while the expression of RNASEL showed modest correlation with ADAR1 dependency (Supplementary Fig. S5d, e). A hallmark of RNASEL activation is degradation of rRNA (42). However, we did not observe rRNA degradation in ADAR1-dependent TNBC cells after ADAR1 knockdown (Supplementary Fig. 5f), further suggesting that the RNASEL pathway does not significantly contribute to TNBC-associated ADAR1 dependency and the induction of OAS genes likely reflects the fact that OAS genes are known ISGs (see below).
ADAR1-dependent TNBCs exhibit elevated ISG expression
Another factor contributing to ADAR1 dependency in cancer cell lines is the type I IFN pathway (24). It has been shown previously that this connection is mediated through either altering the expression of type I IFN regulators or activating the feed-forward loop of IFN signaling (23, 24). RNA expression data from the TCGA and CCLE datasets showed that TNBC have higher ISG expression (Core ISG Score (24)) compared to non-TNBC (Fig. 5a, b). This is consistent with the elevated expression of PKR and ISG15 in our immunoblot analysis among breast cancer cell lines (Fig. 4d, Supplementary Fig. S6a). Like PKR expression, the Core ISG Score positively correlated with ADAR1 sensitivity among TNBC cell lines (Fig. 5c and Supplementary Fig. S6b, c).
Figure 5: ADAR1-dependent TNBCs exhibit elevated ISG expression and INFAR1 loss rescues ADAR1 knockdown phenotype.
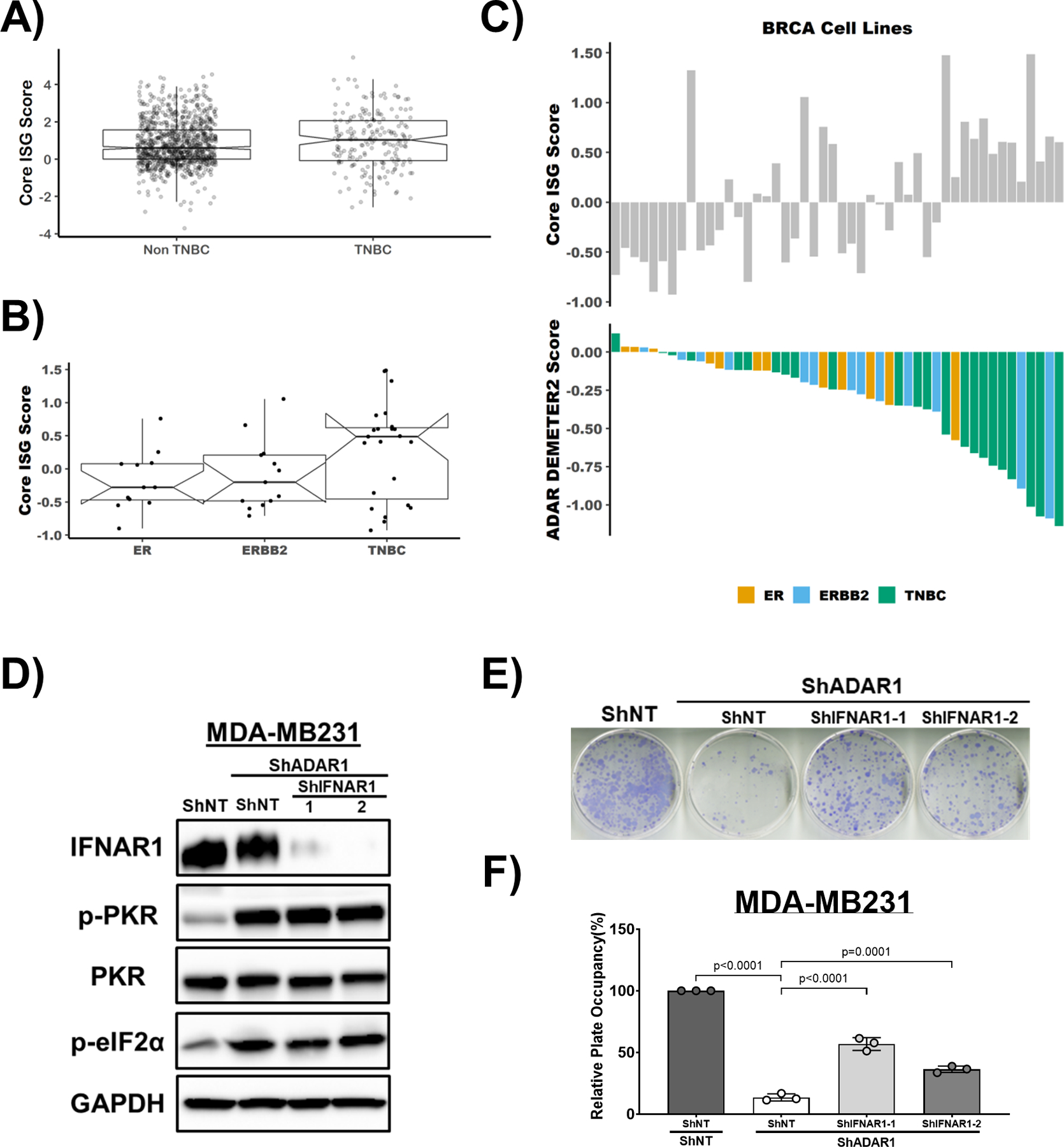
A) Core ISG Score in TNBC and Non-TNBC breast cancer samples. Data were extracted from TCGA database. B) Core ISG Score in ER-positive, ERBB2(HER2)-positive and TNBC cell lines. Data were extracted from CCLE database. C) ADAR1-dependency score positively correlates with Core ISG Score in breast cancer cell lines. Upper panel: Core ISG Score in breast cancer cell lines. Lower panel: ADAR1-dependency scores. D) Immunoblots showing protein levels of IFNAR1, PKR, p-PKR (T446), p-eIF2α (S51) and GAPDH (loading control) in MDA-MB231 cells. IFNAR1 was knocked down in ShADAR1-treated MDA-MB231 cells to determine if IFNAR1 loss reverses ADAR1-knockdown phenotype. Images are representative, N=3. F) FF assay showing that IFNAR1 loss partially rescued ADAR1-knockdown phenotype in MDA-MB231 cells. Images are representative, N=3. G) Quantification of FF in F). Relative plate occupancy was determined using ImageJ software and normalized to ShNT-ShNT. Data are represented as mean ± SD. N=3.
INFAR1 loss rescues ADAR1 knockdown phenotype
To establish whether the type I IFN pathway accounts for the significant differences of ADAR1-dependency between TNBC and non-TNBC cell lines, non-TNBC cell lines (SKBR3 and MCF7) were treated with IFNβ in ADAR1-intact and ADAR1-deficient cells (Supplementary Fig. S6d–g). Expression of ADAR1 and ISG15 were induced upon IFNβ treatment, as well as phosphorylation of STAT1. However, while the treatment of IFNβ generally reduced cell proliferation, it did not sensitize non-TNBC cells to ADAR1 deficiency (Supplementary Fig. S6e, g), implying that IFNβ alone is not capable of switching ADAR1-resistant cells to ADAR1-dependent cells.
To determine if the type I IFN pathway functionally contributes to ADAR1-dependency in TNBC, we knocked-down ADAR1 and the IFN alpha-receptor subunit 1 (IFNAR1) simultaneously in both MDA-MB231 and MDA-MB468 cells (Fig. 5d and Supplementary Fig. S6h). The knockdown of IFNAR1 partially rescued the proliferation of both cell lines, suggesting that TNBC-associated ADAR1 dependency can be partially attributed to type I IFN pathway activation (Fig. 5e, f and Supplementary Fig. S6i). However, knockdown of IFNAR1 in TNBC cells did not alter the levels of phosphorylated PKR (Fig. 5d and Supplementary Fig. S6h), suggesting that in these TNBC cells, type I IFN and PKR pathways might independently contribute to ADAR1 dependency.
Discussion
Recent studies have highlighted the dependence of some cancer cell lines on ADAR1 expression (23, 24). Here we characterized the requirement for ADAR1 in a panel of established breast cancer cell lines. ADAR1-dependent cell lines shared an elevated ISG-expression signature. Loss of ADAR1 in these cell lines led to activation of the translational repressors PKR and eIF2α, as well as translational repression. The ADAR1-dependence phenotype could be partially abrogated by knockdown of IFNAR1.
It is not currently understood what makes select cancer cell lines ADAR1-dependent, or conversely why others are refractory to ADAR1-loss. It has been proposed that the higher ISG expression might potentiate these cells towards ADAR1-dependency – loss of ADAR1 would further elevate ISG expression leading to the growth inhibition phenotype (23, 24). However, we have demonstrated that for cell lines refractory to ADAR1 loss, treatment with IFN-β did not render them sensitive to ADAR1 knockdown. Furthermore, we observed no activation of PKR in the ADAR1 refractory cell lines following ADAR1 loss. These findings suggest that the link between ADAR1 loss and the IFN pathway or PKR activation in ADAR1-refractory cell lines is missing. Loss of ADAR1 is thought to activate the IFN pathway and PKR by causing an increase in dsRNA – stemming from a reduction in A-to-I editing (17, 18). It is possible that ADAR1-refractory cell lines either do not accumulate dsRNA following ADAR1 loss, fail to accumulate a specific subset of dsRNA responsible for PKR or type I IFN pathway activation, or there exists a system that prevents dsRNAs from activating the IFN pathway or PKR. Understanding the molecular basis of this process would help to predict which cell lines – or more importantly which tumors – should be sensitive to ADAR1 loss or inhibition.
Translational repression mediated through PKR activation and phosphorylation of eIF2α has been shown to contribute to ADAR1-dependency in other cancers and remains a possible explanation for TNBC-associated ADAR1-dependency (23). We observed elevated PKR expression in TNBC tumors and cell lines, induced phosphorylation of PKR and eIF2α in TNBC cell lines upon ADAR1 loss and decreased overall translation in ADAR1-deficient TNBC cells. The lack of significant reversal of ADAR1-dependency by mitigating the effects of eIF2α phosphorylation, however, suggests that this phenotype is 1) only modestly influenced by PKR-eIF2α-translation pathway; 2) attributed to a specific subset of targets downstream of PKR other than eIF2α (43). We investigated the functional relevance of one such target, ATF4, in the context of ADAR1-dependency. The lack of association between ATF4 expression and ADAR1-dependency in breast cancer cells suggests that ATF4 does not play a significant role in this TNBC-specific phenotype.
It is not well understood how the RNA editing activity of ADAR1 contributes to its essentiality in certain cancer cells. Consistent with previous reports, we found that both catalytic and non-enzymatic functions of ADAR1 contribute to the ADAR1-dependent phenotype in a cell line specific manner. ADAR1-mediated editing of individual targets, including AZIN1, has been shown to regulate different aspects of tumorigenesis, including initiation, metastasis and drug response (44). However, we have shown that editing of AZIN1 does not contribute to TNBC proliferation, opening up future investigations into other ADAR1-edited targets in TNBC. Moreover, rescue with either wild-type ADAR1 or editing-defective p150 in ADAR1 knockdown TNBC cells resulted in reduced phosphorylation of PKR and eIF2α. Thus, the potential connection between ADAR1 and PKR/eIF2α in TNBC-associated ADAR1-dependency likely involves ADAR1 editing-independent functions and warrants further investigation.
Important clinical implications can be drawn from these observations. Our data suggest that ADAR1 is a legitimate candidate for targeted therapies in TNBC. First, we found that TNBC cell lines and patient samples exhibit elevated ISG and PKR expression, which is consistent with ADAR1-dependent cell lines. With increased understanding of ADAR1 functions, novel therapeutic strategies against ADAR1 could benefit ADAR1-dependent cancers, including TNBC (44). Secondly, the relationship between ADAR1-dependency and type I IFN pathway activity could point to new directions for TNBC interventions. Recent studies revealed that the increased IFNβ target gene signature correlates with improved recurrence-free survival in TNBC, and IFNβ treatment inhibits tumor progression in TNBC by reducing cancer stem cell (CSC) plasticity (45, 46). In addition to cell-intrinsic effects of ADAR1-loss in cancer cells, removal of ADAR1 has been shown to sensitize tumors to immunotherapy by overcoming resistance to checkpoint blockade (27).
It was recently demonstrated that chemotherapies elicit a state of immunological dormancy in ER-negative breast cancers, marked by sustained type I IFN signaling, reduced cell growth, and longer progression-free survival (47). This indicates a possible shared mechanism between chemotherapy-induced immunological dormancy and ADAR1-dependency in TNBC. It is important to note that careful considerations need to be given when applying the concepts of ADAR1 inhibition and type I IFN application in the treatment of TNBC. It is recognized that type I IFN can elicit paradoxical effects on cancer development (48). For example, it has been suggested that type I IFN pathway, potentially through ISG15-mediated ISGylation, can promote the aggressiveness of TNBC (49, 50). Therefore, further understanding of the relationship between ADAR1 functions and TNBC tumorigenesis should better inform the context in which this strategy can provide the maximum benefit.
Materials and Methods
Cell lines and reagents
Human mammary epithelial cells (HMEC) and breast cancer cells lines were obtained from American Tissue Cells Consortium (ATCC, Manassas, VA USA). HMECs were cultured in MammaryLife Basal Medium (Lifeline Cell Technology, Oceanside, CA USA) and passaged by using 0.05% trypsin-EDTA (Gibco, Waltham, MA USA) and Defined Trypsin Inhibitor (DTI, Gibco). All breast cancer cell lines were maintained in Dulbecco’s Modification of Eagle’s Medium (DMEM, GE Life Sciences, Pittsburgh, PA USA) supplemented with 10% fetal bovine serum (Gibco, 10091–148), Sodium Pyruvate (Cellgro, Waltham, MA USA, 30–002-CI), Non-Essential Amino Acids (NEA, Cellgro, 25–030-CI), and L-glutamine (Cellgro, 25–005-CI). Lipofectamine 2000 (Invitrogen, Waltham, MA USA) was used for transfection to generate lentivirus. Fugene 6 transfection reagent (Promega, Madison, WI USA) was used for all other transfection experiments. PKR inhibitor (MilliporeSigma, CAS-608512–97) and eIF2α inhibitor ISRIB (MilliporeSigma, SML0843) were used for phenotypic rescue experiments.
Immunoblot analysis
Cell lysates were extracted from cells at ~90% confluence. Cell were washed with phosphate-buffered saline (PBS, GE Life Sciences), scrape harvested, centrifuged at 1000 × g for 5 min, and lysed with RIPA buffer (20mM Tris-HCl pH7.5, 150mM NaCl, 1mM EDTA, 1% NP-40, 0.1% SDS, 0.1% Deoxycholate) supplemented with 10mM PMSF and HALT protease inhibitor cocktail (Thermo Fisher Scientific, Waltham, MA USA). Lysates were clarified by centrifugation and the protein concentration was determined using DC protein assay system (Bio-Rad, Hercules, CA USA). Equal amount of protein was resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using Criterion TGX Stain-Free Precast Gels (Bio-Rad) and transferred onto Immobilon-P membranes (MilliporeSigma). Primary antibodies used in this study include ADAR1 (Santa Cruz, Dallas, TX USA, sc-73408; Bethyl Laboratories, Montgomery, TX USA, A303–883A; Abcam, Cambridge, MA USA, ab126745), cleaved PARP (Cell Signaling, #9541), PKR (Cell Signaling, #3072), PKR Thr-446-P (Abcam, ab32036), IFNAR1 (Bethyl, A304–290A), ISG15 (Santa Cruz, sc-166755), GAPDH (Bethyl, A300–641A), β-Tubulin (Abcam, ab6046), EIF2S1/eIF2α Ser-51-P (Abcam, 32157), EIF2S1 (Abcam, ab5369), ATF4 (Cell Signaling, #11815). Secondary antibodies conjugated to Horseradish peroxidase were used at a dilution of 1:5–10,000 (Jackson Immunochemicals, West Grove, PA USA). Clarity Western ECL Substrate (Bio-Rad) was then applied to blots and protein levels were detected using autoradiography with ChemiDoc XRS+ Imager (Bio-Rad). Densitometry quantification of protein signals was performed using ImageJ software (NIH, Bethesda, MD USA).
Quantitative reverse-transcription polymerase chain reaction
Total RNA was isolated from cells using RNeasy Mini Plus kit (Qiagen, Hilden, Germany) including on-column DNase digestion following the manufacturer’s protocol. High Capacity cDNA Reverse Transcription Kit (Life technologies) was used to transcribe RNA to cDNA. Quantitative PCR (qPCR) was performed using iTaq Universal SYBR Green Supermix (Bio-Rad, #1725121) on the C1000 Thermal Cycler (CFX96 Real-Time System, Bio-Rad), and data analysis was performed using the 2(-ΔΔCT) method. Messenger RNA expression levels were normalized to GAPDH. Primers used in this study are listed in Supplementary Table S1.
Lentiviral production and transduction
To generate lentivirus, transformed human embryonic kidney HEK293T cells were transfected using Lipofectamine 2000 (Invitrogen) with pCMV-VSV-G, pCMV-ΔR8.2, and expression constructs (with pLKO.1-puromycin or pLKO.1-hygromycin backbone for short-hairpin RNAs and with pLVX-hygromycin backbone for overexpression constructs). Growth medium was replaced with fresh medium 24hr after transfection, and supernatants containing lentivirus were harvested 24hr later. For transduction, one million cells were infected with lentivirus for 24hr in the presence of 10 μg/ml protamine sulfate to facilitate viral entry. Sequences of shRNAs are listed in Supplementary Table 1. ADAR1 overexpression constructs (p150; p110; p150-E912A mutant) were subcloned from pBac-ADAR1 constructs (generous gifts from Kazuko Nishikura at The Wistar Institute, Philadelphia) into pLVX-hygromycin vectors (51).
Flow cytometric analysis of apoptosis
MDA-MB231 or HCC1806 cells were transduced with either ShNT or ShADAR (as described above). After 96 hours the percentage of apoptotic cells was determined using the Annexin V-FITC Apoptosis Staining / Detection Kit (Abcam) per the manufacturer’s instructions. Flow cytometry was performed on a Facscalibur Flow Cytometer (BD Biosciences). Data analysis was performed using FlowJo.
Cell proliferation and focus formation assays
For cell proliferation assays, 2–5 × 104 cells were plated in triplicate in six-well plates. Cells were trypsinized, harvested and counted using a hemocytometer or the Cello Cell counter every 24 hours for 4 days post-plating. For the focus formation assay, 3–5 × 103 cells were plated in triplicate in 10-cm cell culture dishes 7–28 days (depending on the cell line: MDA-MB231 and BT549, ~7 days; HCC1806, ~14 days; SKBR3, ~14–21 days; T47D, ~21 days; MDA-MB468, CAMA1 and MCF7, ~28 days) post-plating until foci became visible. Cells were washed with PBS twice, fixed with 100% methanol, dried, and stained with Giemsa staining reagent (Sigma Aldrich). Stained plates were scanned, and surface areas occupied by cell foci were measured using ImageJ software (NIH, Bethesda, MD).
Soft agar transformation assay
Equal volumes of 2X concentrated DMEM culture media and 1% noble-agar solution (made with sterile cell-culture-grade water) were mixed to make 0.5% agar-media solution and plated in the bottom of six-well plates. Equal volumes of 2X concentrated DMEM culture media and 0.6% noble-agar solution were mixed to make 0.3% agar-media solution for cell suspension. 2–5 × 104 cells were suspended in 0.3% agar-media solution and layered, in triplicate, onto the bottom layer. Cells were fed with fresh media every 7 days and incubated in 37°C for 21–30 days, before being stained with 0.005% crystal violet and examined under a microscope. Colonies bigger than 100 μM in diameter were manually counted.
Mammary gland orthopedic implantation
The abilities of human breast cancer cell lines to form tumors in vivo were evaluated by performing mammary gland orthopedic implantation as described previously (52). Immuno-deficient NOD scid gamma (NSG; NOD.Cg-Prkdcscid/J) female mice at 6–8 weeks old were purchased from Jackson Laboratory (Bar Harbor, ME) and used for this experiment. Five mice were used in each experiment group to achieve type I error rate at 0.05 with 90% power based on results from in vitro cell proliferation and transformation assays. The same batch of purchased mice were used for the experiment of each cell line (ShNT vs ShADAR1) without randomization or blinding selections.1–3 × 106 cells were harvested and resuspended in PBS, mixed with standard base-membrane Matrigel Matrix (Corning, Waltham, MA USA) at 1:1 volume ratio, and kept at 4°C until implantation. In total 100μl of cells-Matrigel solutions were injected into the right inguinal mammary glands of NSG mice, which were monitored closely to observe tumor formation. Mice were euthanized before tumors in control groups reached 2 cubic cm in size, and palpable tumors were dissected from the mice for weight measurement. All animal-related experimental procedures were performed in compliance with the guidelines given by the American Association for Accreditation for Laboratory Animal Care and the U.S. Public Health Service Policy on Human Care and Use of Laboratory Animals. All animal studies were approved by the Washington University Institutional Animal Care and Use Committee (IACUC) in accordance with the Animal Welfare Act and NIH guidelines (Protocol 20160916)
Statistical analysis
All experiments were performed with biological replicates, with the exact sample size stated in figure legends. All in vitro experiments were performed in triplicate, unless otherwise stated. For the mammary gland orthopedic implantation experiment, we included 5 mice in each experiment group to achieve type I error rate at 0.05 with 90% power based on results from in vitro cell proliferation and transformation assays. One mouse from MDA-MB231-ShNT experiment group was excluded due to premature death (Fig. 3g). Nonlinear regression test for Malthusian growth was used for statistical analysis for cell proliferation assay. One-way ANOVA with Tukey’s multiple comparisons was used for statistical analysis in ADAR1-overexpressing rescue experiments. The two-tailed unpaired Student t test was performed for statistical analysis for other experiments. All in vitro and in vivo data are reported as the mean ± SD unless stated otherwise, Statistical analyses were performed using GraphPad Prism.
Polysome profiling
Either MDA-MB231 or HCC1806 cells were treated with 100 μg/mL cycloheximide in growth media for 5 minutes at 37 °C. The cells were washed with ice-cold 1x PBS containing 100 μg/mL cycloheximide prior to harvesting by scraping. The cells were lysed in polysome lysis buffer (20 mM Tris pH 7.26, 130 mM KCl, 10 mM MgCl2, 0.5% NP-40, 0.2 mg/mL heparin, 200 U/mL RNasin, 2.5 mM DTT, 1x HALT, 100 μg/mL cycloheximide, 0.5% sodium deoxycholate) for 20 minutes on ice prior to clarification at 8000 g for 10 minutes at 4 °C. The absorbance at 260 nm was determined for each lysate. An equal number of A260 units for each lysate was overlaid on a 10–50% sucrose gradient (10 mM Tris pH 7.26, 60 mM KCl, 10 mM MgCl2, 2.5 mM DTT, 0.2 mg/mL heparin, 10 μg/mL cycloheximide). The gradients were subjected to centrifugation at 30,000 RPM for 3 hours at 4 °C. The absorbance at 254 nm was measured along the gradient using a fractionation system (Teledyne ISCO, Lincoln, NE USA).
Analysis of CCLE RNAseq Data and ADAR1 Dependency
Raw CCLE RNaseq count data from breast cancer cell lines were normalized by the ‘cpm’ function of ‘edgeR’ (53). From the cpm values z-scores were determined for each gene across all cell lines. To determine ‘Core ISG Score’ we calculated the median z-score of previously identified ‘Core ISGs’ (24). Molecular subtypes of breast cancer cell lines were defined previously (54).
Analysis of TCGA RNAseq Data
Unnormalized RSEM values were normalized by the ‘cpm’ function of edgeR (53). From the cpm values modified z-scores were determined using the following formula.
We calculated ‘Core ISG Score’ as described above. Molecular subtypes of TCGA samples were defined previously (26).
Data and Code Availability
CCLE RNAseq count data (CCLE_RNAseq_genes_counts_20180929.gct.gz, CCLE_RNAseq_rsem_transcripts_tpm_20180929.txt.gz) were obtained from the Broad Institute Cancer Cell Line Encyclopedia and is available online at https://portals.broadinstitute.org/ccle/data. Dependency data (D2_combined_gene_dep_scores.csv, Achilles_gene_effect.csv) were obtained from Broad Institute DepMap Portal and is available on at https://depmap.org/portal/download/. TCGA breast cancer RNAseq (illuminahiseq_rnaseqv2-RSEM_genes, illuminahiseq_rnaseqv2-RSEM_isoforms_normalized) and clinical data (Merge_Clinical) were obtained from the Broad Institute FireBrowse and is available online at http://firebrowse.org/.
All custom R scripts used in this study are available on GitHub (https://github.com/cottrellka/ADAR_TNBC). All other data are available in the main text and figures or Supplementary Information.
Immunohistochemistry
Human breast formalin fixed paraffin embedded tissue array sections (5μm) on positively charged slides were obtained from US Biomax Inc. (Derwood, MD USA, BC081116d). For immunohistochemistry, sections were stained using a Bond RXm autostainer (Leica, Buffalo Grove, IL USA). Briefly, slides were baked at 65°C for 15min and automated software performed dewaxing, rehydration, antigen retrieval, blocking, primary antibody incubation, post primary antibody incubation, detection (DAB), and counterstaining using Bond reagents (Leica). Samples were then removed from the machine, dehydrated through ethanols and xylenes, mounted and cover-slipped. An antibody specifically recognizing ADAR1-p150 (Abcam ab126745) was diluted 1:100 in Antibody Diluent (Leica). Intensity of p150 was scored on a scale of 0 to 3 (range established by samples with the strongest and weakest staining) by increments of 0.5. Any sample scored >1.0 was considered p150-high.
Supplementary Material
Acknowledgements
This work was supported by R01CA190986 (JDW), F32GM131514 (KAC) and TL1TR002344 (C-PK) from the National Institute of Health, and W81XWH-18-1-0025 from the Department of Defense (JDW). This work was supported by the Longer Life Foundation: A RGA/Washington University partnership. We thank Kazuko Nishikura (The Wistar Institute) for providing ADAR1 expressing constructs. The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Footnotes
Competing Interests
The authors declare no competing interests.
References
- 1.Ademuyiwa FO, Tao Y, Luo J, Weilbaecher K, Ma CX. Differences in the mutational landscape of triple-negative breast cancer in African Americans and Caucasians. Breast Cancer Res Treat. 2017;161(3):491–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waks AG, Winer EP. Breast Cancer Treatment: A Review. JAMA. 2019;321(3):288–300. [DOI] [PubMed] [Google Scholar]
- 3.Garrido-Castro AC, Lin NU, Polyak K. Insights into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019;9(2):176–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perou CM. Molecular stratification of triple-negative breast cancers. Oncologist. 2011;16 Suppl 1:61–70. [DOI] [PubMed] [Google Scholar]
- 5.Anders CK, Abramson V, Tan T, Dent R. The Evolution of Triple-Negative Breast Cancer: From Biology to Novel Therapeutics. Am Soc Clin Oncol Educ Book. 2016;35:34–42. [DOI] [PubMed] [Google Scholar]
- 6.Fumagalli D, Gacquer D, Rothe F, Lefort A, Libert F, Brown D, et al. Principles Governing A-to-I RNA Editing in the Breast Cancer Transcriptome. Cell reports. 2015;13(2):277–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han L, Diao L, Yu S, Xu X, Li J, Zhang R, et al. The Genomic Landscape and Clinical Relevance of A-to-I RNA Editing in Human Cancers. Cancer cell. 2015;28(4):515–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paz-Yaacov N, Bazak L, Buchumenski I, Porath HT, Danan-Gotthold M, Knisbacher BA, et al. Elevated RNA Editing Activity Is a Major Contributor to Transcriptomic Diversity in Tumors. Cell reports. 2015;13(2):267–76. [DOI] [PubMed] [Google Scholar]
- 9.Peng X, Xu X, Wang Y, Hawke DH, Yu S, Han L, et al. A-to-I RNA Editing Contributes to Proteomic Diversity in Cancer. Cancer cell. 2018;33(5):817–28 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anantharaman A, Gholamalamdari O, Khan A, Yoon JH, Jantsch MF, Hartner JC, et al. RNA-editing enzymes ADAR1 and ADAR2 coordinately regulate the editing and expression of Ctn RNA. FEBS Lett. 2017;591(18):2890–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gumireddy K, Li A, Kossenkov AV, Sakurai M, Yan J, Li Y, et al. The mRNA-edited form of GABRA3 suppresses GABRA3-mediated Akt activation and breast cancer metastasis. Nature communications. 2016;7:10715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Binothman N, Hachim IY, Lebrun JJ, Ali S. CPSF6 is a Clinically Relevant Breast Cancer Vulnerability Target: Role of CPSF6 in Breast Cancer. EBioMedicine. 2017;21:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dave B, Gonzalez DD, Liu ZB, Li X, Wong H, Granados S, et al. Role of RPL39 in Metaplastic Breast Cancer. J Natl Cancer Inst. 2017;109(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakano M, Fukami T, Gotoh S, Nakajima M. A-to-I RNA Editing Up-regulates Human Dihydrofolate Reductase in Breast Cancer. The Journal of biological chemistry. 2017;292(12):4873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Song IH, Kim YA, Heo SH, Park IA, Lee M, Bang WS, et al. ADAR1 expression is associated with tumour-infiltrating lymphocytes in triple-negative breast cancer. Tumour Biol. 2017;39(10):1010428317734816. [DOI] [PubMed] [Google Scholar]
- 16.Sagredo EA, Blanco A, Sagredo AI, Perez P, Sepulveda-Hermosilla G, Morales F, et al. ADAR1-mediated RNA-editing of 3’UTRs in breast cancer. Biol Res. 2018;51(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, et al. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell reports. 2014;9(4):1482–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, et al. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015;349(6252):1115–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. Isoforms of RNA-Editing Enzyme ADAR1 Independently Control Nucleic Acid Sensor MDA5-Driven Autoimmunity and Multi-organ Development. Immunity. 2015;43(5):933–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.George CX, Ramaswami G, Li JB, Samuel CE. Editing of Cellular Self-RNAs by Adenosine Deaminase ADAR1 Suppresses Innate Immune Stress Responses. The Journal of biological chemistry. 2016;291(12):6158–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Z, Okonski KM, Samuel CE. Adenosine deaminase acting on RNA 1 (ADAR1) suppresses the induction of interferon by measles virus. Journal of virology. 2012;86(7):3787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pujantell M, Riveira-Munoz E, Badia R, Castellvi M, Garcia-Vidal E, Sirera G, et al. RNA editing by ADAR1 regulates innate and antiviral immune functions in primary macrophages. Scientific reports. 2017;7(1):13339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, et al. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nature communications. 2018;9(1):5450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nature medicine. 2019;25(1):95–102. [DOI] [PubMed] [Google Scholar]
- 25.Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell. 2018;172(4):811–24 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann BD, Jovanović B, Chen X, Estrada MV, Johnson KN, Shyr Y, et al. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS One. 2016;11(6):e0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, et al. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature. 2019;565(7737):43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McFarland JM, Ho ZV, Kugener G, Dempster JM, Montgomery PG, Bryan JG, et al. Improved estimation of cancer dependencies from large-scale RNAi screens using model-based normalization and data integration. Nat Commun. 2018;9(1):4610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49(12):1779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, et al. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. The Journal of biological chemistry. 2004;279(6):4952–61. [DOI] [PubMed] [Google Scholar]
- 31.Sakurai M, Shiromoto Y, Ota H, Song C, Kossenkov AV, Wickramasinghe J, et al. ADAR1 controls apoptosis of stressed cells by inhibiting Staufen1-mediated mRNA decay. Nat Struct Mol Biol. 2017;24(6):534–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen L, Li Y, Lin CH, Chan TH, Chow RK, Song Y, et al. Recoding RNA editing of AZIN1 predisposes to hepatocellular carcinoma. Nature medicine. 2013;19(2):209–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shigeyasu K, Okugawa Y, Toden S, Miyoshi J, Toiyama Y, Nagasaka T, et al. AZIN1 RNA editing confers cancer stemness and enhances oncogenic potential in colorectal cancer. JCI Insight. 2018;3(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crews LA, Jiang Q, Zipeto MA, Lazzari E, Court AC, Ali S, et al. An RNA editing fingerprint of cancer stem cell reprogramming. J Transl Med. 2015;13:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S, et al. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dar AC, Dever TE, Sicheri F. Higher-order substrate recognition of eIF2alpha by the RNA-dependent protein kinase PKR. Cell. 2005;122(6):887–900. [DOI] [PubMed] [Google Scholar]
- 37.Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perkins DJ, Barber GN. Defects in translational regulation mediated by the alpha subunit of eukaryotic initiation factor 2 inhibit antiviral activity and facilitate the malignant transformation of human fibroblasts. Molecular and cellular biology. 2004;24(5):2025–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gil J, Alcami J, Esteban M. Induction of apoptosis by double-stranded-RNA-dependent protein kinase (PKR) involves the alpha subunit of eukaryotic translation initiation factor 2 and NF-kappaB. Molecular and cellular biology. 1999;19(7):4653–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Donze O, Jagus R, Koromilas AE, Hershey JW, Sonenberg N. Abrogation of translation initiation factor eIF-2 phosphorylation causes malignant transformation of NIH 3T3 cells. EMBO J. 1995;14(15):3828–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nature reviews Molecular cell biology. 2005;6(4):318–27. [DOI] [PubMed] [Google Scholar]
- 42.Silverman RH, Skehel JJ, James TC, Wreschner DH, Kerr IM. rRNA cleavage as an index of ppp(A2’p)nA activity in interferon-treated encephalomyocarditis virus-infected cells. J Virol. 1983;46(3):1051–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holcik M Could the eIF2alpha-Independent Translation Be the Achilles Heel of Cancer? Front Oncol. 2015;5:264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kung CP, Maggi LB Jr., Weber JD. The Role of RNA Editing in Cancer Development and Metabolic Disorders. Front Endocrinol (Lausanne). 2018;9:762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Doherty MR, Cheon H, Junk DJ, Vinayak S, Varadan V, Telli ML, et al. Interferon-beta represses cancer stem cell properties in triple-negative breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2017;114(52):13792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Doherty MR, Parvani JG, Tamagno I, Junk DJ, Bryson BL, Cheon HJ, et al. The opposing effects of interferon-beta and oncostatin-M as regulators of cancer stem cell plasticity in triple-negative breast cancer. Breast Cancer Res. 2019;21(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lan Q, Peyvandi S, Duffey N, Huang YT, Barras D, Held W, et al. Type I interferon/IRF7 axis instigates chemotherapy-induced immunological dormancy in breast cancer. Oncogene. 2019;38(15):2814–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Snell LM, McGaha TL, Brooks DG. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017;38(8):542–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Forys JT, Kuzmicki CE, Saporita AJ, Winkeler CL, Maggi LB Jr.,Weber JD. ARF and p53 coordinate tumor suppression of an oncogenic IFN-beta-STAT1-ISG15 signaling axis. Cell reports. 2014;7(2):514–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lo PK, Yao Y, Lee JS, Zhang Y, Huang W, Kane MA, et al. LIPG signaling promotes tumor initiation and metastasis of human basal-like triple-negative breast cancer. Elife. 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cho DS, Yang W, Lee JT, Shiekhattar R, Murray JM, Nishikura K. Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA. The Journal of biological chemistry. 2003;278(19):17093–102. [DOI] [PubMed] [Google Scholar]
- 52.Brenot A, Knolhoff BL, DeNardo DG, Longmore GD. SNAIL1 action in tumor cells influences macrophage polarization and metastasis in breast cancer through altered GM-CSF secretion. Oncogenesis. 2018;7(3):32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell. 2016;164(1–2):293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CCLE RNAseq count data (CCLE_RNAseq_genes_counts_20180929.gct.gz, CCLE_RNAseq_rsem_transcripts_tpm_20180929.txt.gz) were obtained from the Broad Institute Cancer Cell Line Encyclopedia and is available online at https://portals.broadinstitute.org/ccle/data. Dependency data (D2_combined_gene_dep_scores.csv, Achilles_gene_effect.csv) were obtained from Broad Institute DepMap Portal and is available on at https://depmap.org/portal/download/. TCGA breast cancer RNAseq (illuminahiseq_rnaseqv2-RSEM_genes, illuminahiseq_rnaseqv2-RSEM_isoforms_normalized) and clinical data (Merge_Clinical) were obtained from the Broad Institute FireBrowse and is available online at http://firebrowse.org/.
All custom R scripts used in this study are available on GitHub (https://github.com/cottrellka/ADAR_TNBC). All other data are available in the main text and figures or Supplementary Information.