

SUMMARY
Heart contraction depends on a complicated array of interactions between sarcomeric proteins required to convert chemical energy into mechanical force. Cyclic interactions between actin and myosin molecules, controlled by troponin and tropomyosin, generate the sliding force between the actin-based thin and myosin-based thick filaments. Alterations in this sophisticated system due to missense mutations can lead to cardiovascular diseases. Numerous structural studies proposed pathological mechanisms of missense mutations at the myosin-myosin, actin-tropomyosin and tropomyosin-troponin interfaces. However, despite the central role of actomyosin interactions a detailed structural description of the cardiac actomyosin interface remained unknown. Here we report a cryo-EM structure of a cardiac actomyosin complex at 3.8 Å resolution. The structure reveals the molecular basis of cardiac diseases caused by missense mutations in myosin and actin proteins.
Keywords: cardiac muscle, actin, myosin, cryo electron microscopy
Graphical Abstract
eTOC Blurb
Heart contraction depends on interactions between actin and myosin molecules. The cardiac actomyosin interface remained unknown. The structure of cardiac actomyosin complex at near-atomic resolution by Risi et al. provides molecular details of the actomyosin interface and reveals molecular basis of cardiac diseases caused by mutations in actin and myosin.
INTRODUCTION
The interactions between myosin motors and actin filaments govern fundamental biological processes within a wide variety of cell types. Only a small minority of eukaryotic taxons (i.e. red algae and certain flagellate protozoans) lack myosin protein-coding sequences within their genomes (Richards and Cavalier-Smith, 2005), reflecting the functional importance of myosins across the eukaryotic phylogeny. The overall architectures of the tail domains and the lever arm, the myosin head’s regulatory domain that binds light chains, vary considerably among these different myosin classes (Syamaladevi et al., 2012). By contrast, the ATP- and actin-binding regions that comprise the head’s motor domain are generally conserved features in all myosins (Foth et al., 2006). The motor domain also contains a C-terminal converter domain that transduces small (angstrom-scale) conformational changes within the motor domain into a large movement (nanometer-scale) of the lever arm that drives the powerstroke of the actomyosin cycle (Sun and Goldman, 2011; Uyeda et al., 1996). The combination of the motor and regulatory domains of the myosin head is referred to as subfragment 1 (S1), and isolated myosin S1 can serve as a minimal motor unit capable of driving ATP-dependent force production (Kishino and Yanagida, 1988) and actin filament motility (Toyoshima et al., 1987) in vitro.
The initial structural characterizations of the domains involved in the actomyosin rigor complex came from the work of Rayment and colleagues (Rayment et al., 1993). With the advent of cryo-EM and molecular dynamics techniques, Lorenz and Holmes performed flexible fitting of the crystal structure of skeletal muscle myosin into a 13 Å rigor actomyosin complex cryo-EM map (Lorenz and Holmes, 2010). The invention of novel cryo-EM microscopes and direct electron detectors yielded significant insights into the structure of the actomyosin complex. In 2012 a complex of rabbit skeletal muscle F-actin and Dictyostelium discoideum myosin-Ie was resolved at 8 Å resolution (Behrmann et al., 2012). The rigor complex was further refined to an atomic-level resolution of 3.9 Å using human-derived myosin IIc and cytoplasmic beta-actin (von der Ecken et al., 2016). It has been followed up by high-resolution reconstructions of skeletal (5.2 Å) (Fujii and Namba, 2017) and smooth (Banerjee et al., 2017) (6.0 Å) muscle actomyosin complexes as well as those belonging to the non-muscle myosins Ib (3.9 Å) (Mentes et al., 2018) and VI (4.6 Å) (Gurel et al., 2017).
Despite the recent influx of high-resolution cryo-EM structures for these functionally diverse actomyosin complexes, high-resolution reconstruction for the cardiac muscle actomyosin complex is still missing. Cardiac myosin possesses two isoforms: the α-isoform (encoded by MYH6 in humans) is expressed in the atria, while the β-isoform (encoded by MYH7) is mainly expressed in the heart ventricles and slowtwitch type I skeletal muscle fibers. Mutations in MYH7 gene have been linked to cardiomyopathies in humans (Colegrave and Peckham, 2014; Walsh et al., 2017). The missense mutations in the cardiac actin isoform encoded by ACTC1 gene have also been reported to cause cardiomyopathies in human patients (Augiere et al., 2015; Burns et al., 2017; Coppini et al., 2014; Kindel et al., 2012; Lakdawala et al., 2012; Lopes et al., 2015; Matsson et al., 2008; Wang et al., 2017; Wang et al., 2014; Zou et al., 2013). The molecular mechanisms of these diseases caused by pathogenic variants in both β-cardiac myosin and cardiac actin remain unresolved in part due to the absence of the cardiac actomyosin complex atomic model. The existence of such a model would allow for a systematic evaluation of pathological mutations involved in the actin-binding interface without having to extrapolate information from homologous myosins. Here we report an atomic model of cardiac actomyosin-S1 complex in rigor state derived from the high-resolution (3.8 Å) cryo-EM map.
RESULTS
Intermolecular interactions in the cardiac actomyosin rigor complex.
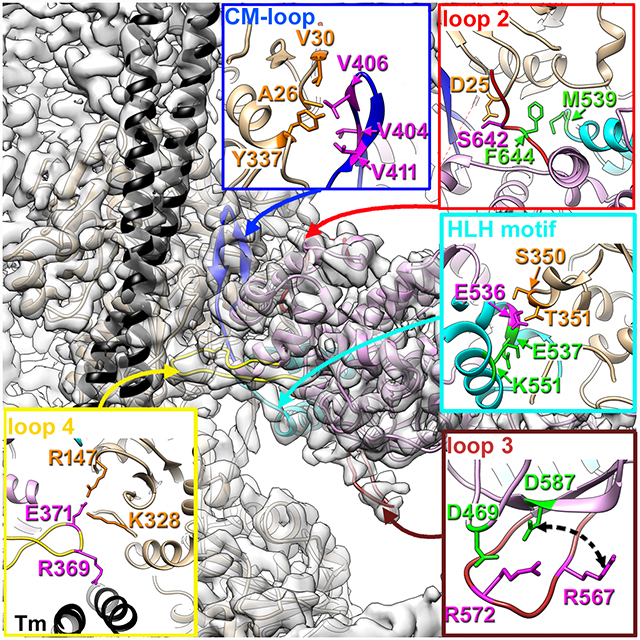
We used RELION software package (Scheres, 2012) to calculate high-resolution cryo-EM density map of the cardiac myosin S1 bound to the native cardiac TF in rigor state (Fig. S1A–C). Resolution through the map varied from 3.5–4.0 Å within the actin filament and actomyosin interface to ~7 Å at the essential light chain (ELC) region (Fig. S2A–C). Due the absence of high-resolution structures of porcine cardiac actin and myosin we used homology modeling to generate an initial model of the complex (Fig. S1E) which subsequently was docked into the segmented cryo-EM map (Fig. S1D) using flexible fitting approach (Schroder et al., 2007). Most regions of the structure had near-atomic resolution, which enabled us to build a model de novo. Only for the lower resolution periphery did we have to rely on the information from the flexibly fit homology model. The resolution at the actomyosin interface allowed us to determine the structural elements of cardiac myosin involved in interactions with actin and tropomyosin. These structural elements include multiple regions: cardiomyopathy (CM) loop and loop 4 located in the upper domain of myosin (Fig. 1A blue and yellow ribbons, respectively), loop 2 at the junction between the upper and lower domains (Fig. 1A, red ribbons), and finally the helix-loop-helix (HLH) motif in the myosin lower domain (Fig. 1A, cyan ribbons). Although the general layout of the interacting regions in the rigor state is shared between myosin isoforms (Fujii and Namba, 2017; Gurel et al., 2017; Mentes et al., 2018; von der Ecken et al., 2016), we found that cardiac myosin interactions with the thin filament have significant differences from the other published actomyosin structures explaining unique properties of cardiac myosin.
Fig. 1. Intermolecular interactions in the cardiac actomyosin rigor complex.
(A) Atomic model of the cardiac actomyosin complex shows structural elements of cardiac myosin involved in interactions with actin and tropomyosin: CM loop (blue ribbons), loop 2 (red ribbons), loop 4 (yellow ribbons), HLH motif (cyan ribbons), and loop 3 (brown ribbons). Actin atoms are tan, tropomyosin is black, while myosin motor domain is plum. Myosin residues involved in interactions with actin are in magenta while myosin residues involved in intra-myosin contacts are in green. (B-H) Detailed interfaces between myosin tropomyosin and actin are shown for CM loop (B and C), loop 2 (D), loop 4 (E), HLH motif (F and G), and loop 3(H).
The cardiac myosin CM loop (H401-N416) (Fig. 1A, blue ribbons) forms a major contact between the upper myosin domain and F-actin and, therefore, is an important element of the actomyosin complex. Its interactions with actin are comprised of a hydrophobic patch formed by residues located in the upper part of the loop (Fig. 1B) and electrostatic interactions at the base of the loop (Fig. 1C). Residues V404, V406 and V411 residing in the upper part of the CM loop (Fig. 1B, magenta atoms) form a hydrophobic cluster with A26, V30 and Y337 residues of actin (Fig. 1B, orange atoms). Similar hydrophobic interactions between the upper part of the CM loop and actin were found in myosin IIc (von der Ecken et al., 2016), myosin VI (Gurel et al., 2017), and myosin 1b (Mentes et al., 2018) complexes. Somewhat different interactions have been proposed for the skeletal complex (Fujii and Namba, 2017) where Y337 of actin makes hydrophobic contact with Y412 (cardiac Y410), while V408 (cardiac V406) with P27 of actin. In our model actin P27 maintains the folding of the actin molecule by making hydrophobic interaction with W340. Nevertheless, the skeletal model is consistent with the hydrophobic interactions of the upper part of the CM loop with actin revealed in the other complexes. Residue E409, located near the top of the CM loop, is highly conserved nearly in all myosins (Bement and Mooseker, 1995), and has been shown to interact with K336 and Y337 of actin in actomyosin 1b complex (E334 in myo1b) (Mentes et al., 2018). In contrast, cardiac myosin E409 is presumably not involved in actomyosin interactions because actin Y337 forms a hydrophobic cluster with V406 and V411 of myosin (Fig. 1B), while K336 in cardiac actin coordinates nucleotide (Fig. S3A). Equivalently positioned glutamic acid in myosin IIc does not interact with actin either (von der Ecken et al., 2016). Consistent with previous observations (von der Ecken et al., 2016), we show that the highly conserved and disease-related R403 does not interact with actin, but rather stabilizes the structure of the CM loop by cation-pi interactions with Y410 (Fig. S3B). Electrostatic interactions between cardiac CM loop and actin are formed by K413 (Fig. 1C, magenta atoms) residing at the base of the loop, which forms salt bridges with D25 and E334 of actin (Fig. 1C, orange atoms). Equivalent residue K429 in myosin IIc (von der Ecken et al., 2016) and K415 skeletal myosin (Fujii and Namba, 2017) forms the same electrostatic interactions with actin. In contrast, in actomyosin VI complex actin E334 makes a salt bridge with myosin R393 (cardiac R403) (Gurel et al., 2017), while myosin 1b does not possess electrostatic interactions at the base of CM loop (Mentes et al., 2018). Myosin IIc also contains a unique electrostatic interaction of its R424 (cardiac N408) with a negatively-charged region of actin formed by E55 and D92 (von der Ecken et al., 2016), which may explain why the CM loop of myosin IIc is pulled closer towards this E55/D92 region of actin relative its position in the cardiac complex. Overall, the interaction between cardiac CM loop and actin is similar to what has been observed by cryo-EM for other myosin isoforms.
Loop 2 (L620-T646, red ribbons in Fig. 1A), which varies in its length and amino acid composition between myosin isoforms, has been shown to play an important role in early stages of actomyosin interactions (Furch et al., 2000; Joel et al., 2001; Onishi et al., 2006). The length of loop 2 is correlated with its flexibility and, hence, its visibility in the cryo-EM density maps. In myosin 1b the shorter loop is well resolved (Mentes et al., 2018), while in actomyosin IIc (von der Ecken et al., 2016) and our cardiac complex (Fig. 1D) the ‘flexible’ tip of loop 2 is not visible. However, the base of the loop is very well preserved in our map due to hydrophobic interaction between F644 of loop 2 and M539 of HLH motif (Fig. 1D, green atoms). In addition, cardiac loop 2 and actin form a hydrogen bond between myosin S642 and actin D25 (Fig. 1D insert, magenta and orange atoms, respectively). In contrast to myosin IIc (von der Ecken et al., 2016) and skeletal (Fujii and Namba, 2017) complexes, the negatively charged N-terminus of actin is not resolved in our map suggesting that it is flexible and, therefore, does not make prominent interactions with the positively charged K639 and K640 residues in loop 2. Overall, cardiac myosin loop 2 possesses weaker interactions with actin compared to skeletal or cytoplasmic myosin II. Interestingly, loop 2 was not well resolved in the actomyosin VI complex, suggesting high flexibility of this segment (Gurel et al., 2017).
Loop 4 (N361-E379, yellow ribbons in Fig. 1A) is well resolved in our density map (Fig. 1E, yellow atoms). Electrostatic interaction between E371 of cardiac myosin located at the tip of loop 4 (Fig. 1E, magenta atoms) and R147 and K328 residues on actin (Fig. 1E, orange atoms) reinforces the binding of the myosin upper domain to actin. Both skeletal myosin (E373) and cytoplasmic myosin IIc (D387) exhibit a negatively charged residue analogous to porcine cardiac E371 that interacts with the same lysine residues on SD3 of actin (Fujii and Namba, 2017; von der Ecken et al., 2016). Similarly, to myosin IIc, which angles its R384 side chain towards the Tm strand, equivalent R369 of cardiac myosin (Fig. 1E, magenta atoms) makes an electrostatic interaction with Tm (Fig. 1E, black ribbons). While loop 4 interaction with actin is very conserved between cardiac, skeletal and cytoplasmic myosin isoforms, it retains its uniqueness in myosins VI and 1b. In myosin VI loop 4 (A355-C362) still forms an electrostatic interaction with K328 of actin, but it does it via E354 located in the base of loop 4 rather than at its tip (Gurel et al., 2017). Loop 4 of myosin 1b (I284-K301) is mainly hydrophobic so that its G294 and L295 are engulfed by a hydrophobic pocket on actin formed by I329, I330, A331, and P332 (Mentes et al., 2018).
The HLH motif (I530-H556, cyan ribbons in Fig. 1A) shows high degree of structural similarity among the myosins because several hydrophobic residues that comprise the loop between the two α-helixes actively participate in the interaction with actin. In our cardiac model the loop comprised of M539, F540, and P541 is fully resolved. The P541, a conserved residue in both skeletal and cytoplasmic myosins, is crucial for actomyosin interactions as its mutation results in the drastic reduction of myosin motility (Onishi et al., 2006). In the cardiac complex F540 interacts with Y143 and L349 of subdomain 1 of the upper actin subunit and G46 of the lower actin D-loop, while adjacent P541 makes hydrophobic interaction with T148 of subdomain 1 of actin (Fig. 1F, F540/P541 are in magenta; actin atoms are in orange). The third hydrophobic residue M539 does not bind to actin, but rather interacts with myosin F644, which stabilizes the HLH motif (Fig. 1D, green atoms). A highly conserved negatively charged patch in the first helix of the HLH motif (cardiac E535, E536, and E537), which is present in all myosin isoforms, has been shown to stabilize HLH interaction with actin (Fujii and Namba, 2017; Gurel et al., 2017; Mentes et al., 2018; von der Ecken et al., 2016). In our complex E536 makes hydrogen bonds with S350 and T351 of actin (Fig. 1G, magenta and orange atoms, respectively). The adjacent E537 stabilizes the HLH fold through the salt bridge with K551 (Fig. 1G, green residues) – another conserved residue over the myosin spectrum. We do not see any evidence for the interaction of the so-called “activation loop” (Varkuti et al., 2012) (I524-M528) with actin found in the myosin IIc complex (von der Ecken et al., 2016), because in cardiac isoform this loop is shorter than that in myosin IIc. Nevertheless, M528 of this loop stabilizes HLH motif interacting with its N555 (Fig. S3C). Our data suggests that HLH motif interactions with actin are conserved over myosin isoforms.
In most of the actomyosin complexes (Fujii and Namba, 2017; Gurel et al., 2017; von der Ecken et al., 2016) compared with our cardiac model, complementary electrostatic interactions between positively charged residues on loop 3 and negatively charged residues on actin SD1 form the “Milligan contact” (Milligan et al., 1990). In myosin 1b hydrophobic interactions are primarily responsible for forming the loop 3 actin interface (Mentes et al., 2018). Therefore, in all the models for the variety of actomyosin complexes loop 3 proximal enough to actin SD1 to form contacts with it. In our map loop 3 (K565-H576, brown ribbons in Fig. 1A) is not uniformly resolved. Residues located in its 565–571 region do not show full size side chains densities at intermediate contour levels, while the 571–576 part of the loop 3 is nicely defined in our map presumably due to the salt bridge between R572 and D469 (Fig. 1H, R572 magenta, D469 green). Because of this salt bridge the loop 3 is pulled back towards the SH3 domain of myosin away from the surface of actin. Interestingly, R572 is not unique for the cardiac isoform - myosin IIc has R589 at equivalent position, but it lacks an acidic equivalent of D467 that would otherwise hold loop 3 away from the actin surface. In contrast to myosin IIc, while myosin Ib does have a negatively charged D396 equivalent to cardiac D469, it lacks a positively charged residue equivalent to R572, and, hence cannot keep loop 3 attached to the body of myosin motor domain. The skeletal actomyosin complex, which contains both equivalents (K574 and D471, respectively) does not show extensive interaction of loop 3 with actin (Fujii and Namba, 2017) since position of loop 3 in this complex is very similar to that in the cardiac complex (Fig. S3D). The large loop 3 of myosin VI has its unique conformation different from the other myosins and makes unique extensive contacts with actin (Gurel et al., 2017). Comparison of the densities of the 565–571 region of loop 3 at either high or low contouring (Fig. S3E) suggests that it is not disordered, but rather may fluctuate between the two modes – in one mode (minor) it may form non-specific ionic interactions with subdomain 1 (SD1) of actin, while in the other mode (major) R567 may form a salt bridge with D587 (Fig. 1H, R567 is magenta, D587 is green). The overall position of loop 3 in the cardiac isoform is very different from what has been reported for the other actomyosin complexes (Fig. S3D).
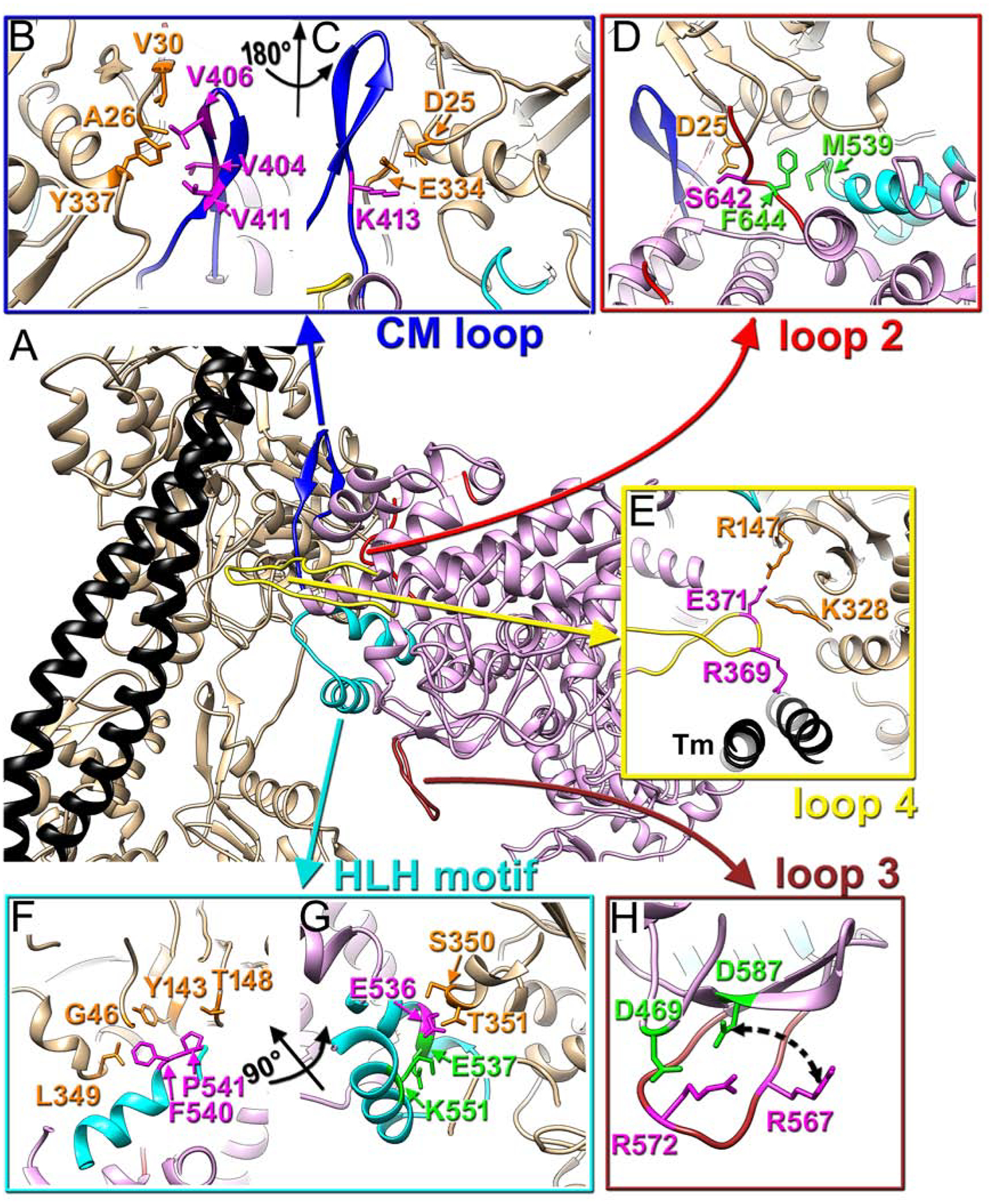
Due to intrinsic mobility of the essential (ELC) and regulatory (RLC) light chains in our complex those regions were partially disordered which reduced the effective resolution in the outer domains of myosin to 7 Å for the ELC region (Fig. S2B). Nevertheless, we compared the interface between the converter domain and the ELC in our complex with the interface obtained by the X-ray crystallography (Robert-Paganin et al., 2018) (Fig. 2). We docked a portion of the myosin motor domain and the attached ELC from (Robert-Paganin et al., 2018) without any perturbations into our density map (Fig. 2, red arrow). The crystal structure yielded a good fit into our map (Fig. 2, insert) suggesting that the interaction between the ELC and myosin motor domain observed in the crystal structure of post-rigor myosin is very similar to that in the rigor actomyosin complex.
Fig. 2. Interface between the converter domain of myosin and myosin ELC.
3D map of actomyosin complex filtered to 7 Å resolution shows the full size ELC (cyan) and traces of the RLC (green letters). A portion of bovine cardiac motor domain (residues 1–130, 647–806) along with the bound ELC (PDB: 6FSA) (Robert-Paganin et al., 2018) matches our density map (red arrow) to confirm that myosin R723 and R780 (blue atoms) of converter domain interact with E135, D136, E139 and R142 (orange atoms) of the myosin ELC (insert). Actin shown in tan, tropomyosin in black, myosin motor domain in plum, while ELC is cyan.
Overall, our data shows that while all the structural elements of myosin previously shown to interact with actin are involved in formation of the rigor cardiac actomyosin complex, there are two significant differences between cardiac and other myosin isoforms: (1) cardiac loop 2 shows weaker interactions with actin; (2) cardiac complex lacks the “Milligan contact”.
Myosin pathological variants at the cardiac actomyosin interface.
Porcine TF and myosin S1 used in our complex provide previously missing information about the mechanism(s) of pathological mutations in actin and myosin that lead to hypertrophic (HCM) and dilated (DCM) cardiomyopathies. Porcine and human cardiac actins (ACTC1) are 100% identical. The motor domains of the two myosin isoforms are 96.1 % identical (98.3 % similar) possessing almost 99% identity in the key elements of the actomyosin interface (Fig. S4).
CM loop retains its name because ~33 % of its residues are linked to HCMs. Human pathogenic mutations at V404L/M (Van Driest et al., 2004; Woo et al., 2003), V406M (Walsh et al., 2017), and V411I (Walsh et al., 2017) (Fig. 3B) affect cardiac contraction because they are directly involved in the interactions with actin (Fig. 3B), while pathogenic substitutions R403W/G/L/Q and Y410D (Walsh et al., 2017), alter the integrity of the β-strand of CM loop (Fig. S3B). Consistently, R403Q variant demonstrated a two-fold decrease in its affinity to actin in the rigor state (Nag et al., 2015).
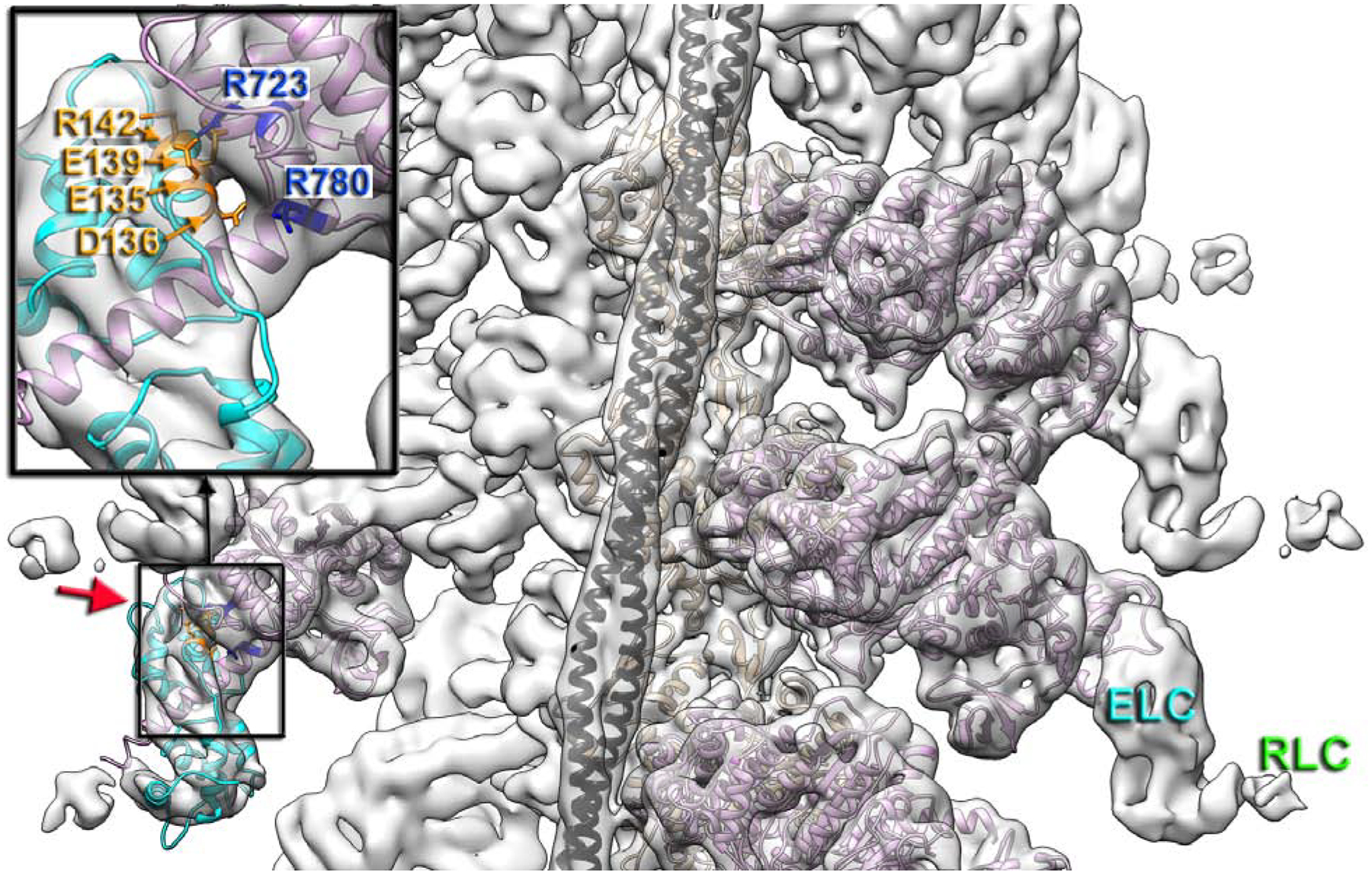
Fig. 3. Positioning of myosin pathological variants at the cardiac actomyosin interface.
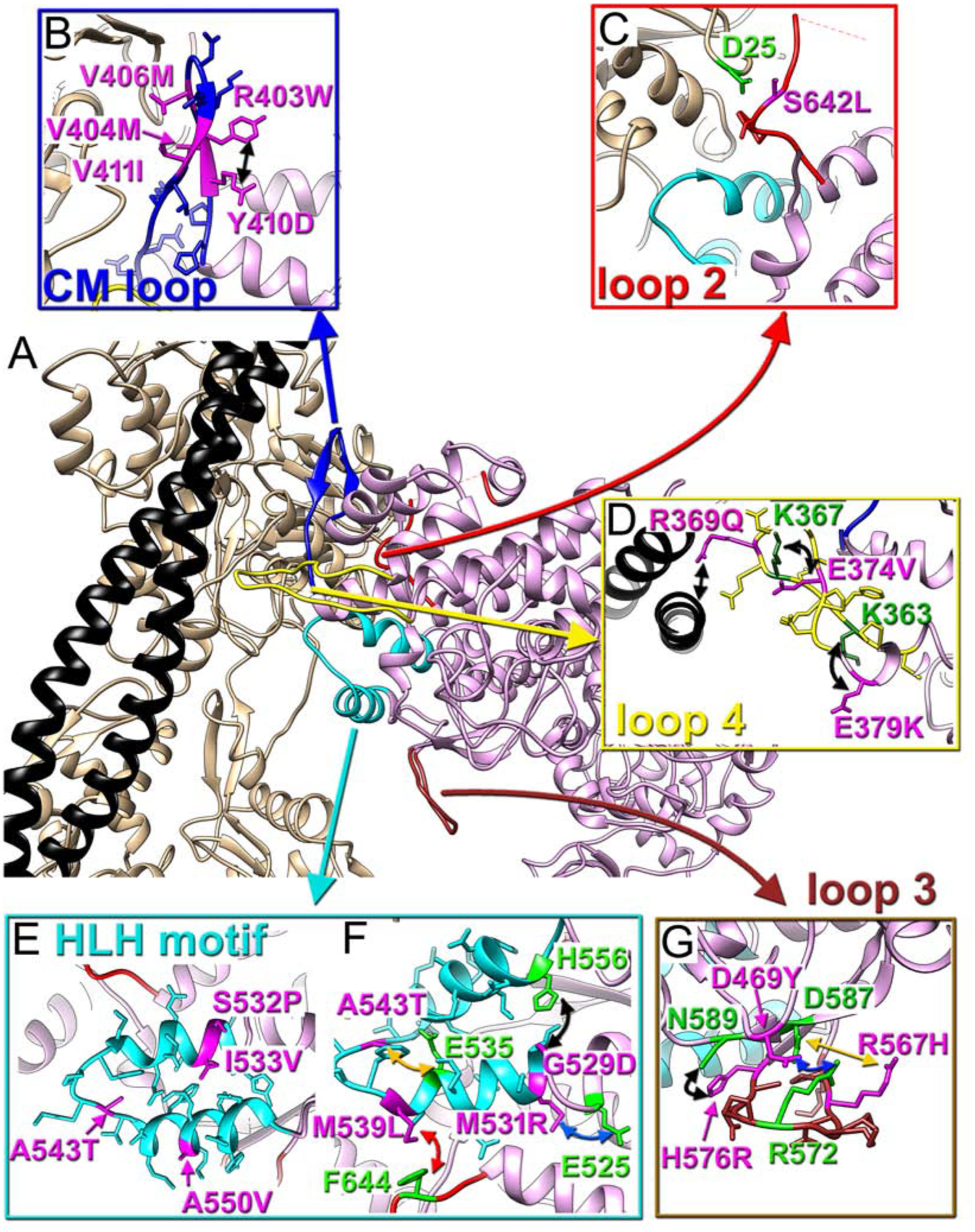
(A) Atomic model of the cardiac actomyosin complex shows structural elements of cardiac myosin involved in interactions with actin and tropomyosin: CM loop (blue ribbons), loop 2 (red ribbons), loop 4 (yellow ribbons), HLH motif (cyan ribbons), and loop 3 (brown ribbons). Actin atoms are tan, tropomyosin is black, while myosin motor domain is plum. (B-G) Myosin pathogenic variants are in magenta, while residues involved in interaction with those variants are in green. The proposed interactions are indicated with colored arrows. Pathogenic variants are shown for CM loop (B), loop 2 (C), loop 4 (D), HLH motif (E and F), and loop 3 (G).
Our model nicely explains a pathogenic variant S642L (Fig. 3C) which is linked to DCM (Daehmlow et al., 2002) – serine substitution to leucine would break the hydrogen bond between serine and aspartic acid (Fig. 3C).
Loop 4 harbors three pathogenic variants - two linked to HCM phenotype (E374V and E379K), and one (R369Q) found in patients with DCM (Walsh et al., 2017). R369 interacts with the Tm cable (Fig. 3D), and therefore, charge-altering mutation R369Q should affect loop 4 interaction with Tm (Fig. 3D, black straight arrow). The other two variants E374V and E379K (Fig. 3D, magenta atoms) are located against positively charged K367 and K363 (Fig. 3D, green atoms) residing at the opposite side of the loop 4. Therefore, charge-altering substitutions (E374V and E379K) should prevent the formation of the two salt bridges (Fig. 3D, black curved arrows) which preserve the geometry of loop 4 essential for its proper interactions with the TF.
As a most conservative element between myosin isoforms the HLH motif harbors several pathogenic substitutions linked to either HCM (M531R, M539L) (Iascone et al., 2007; Kaneda et al., 2008), or DCM (E525K, G529D, S532P, I533V, A543T, A550V) (Kamisago et al., 2006; Villard et al., 2005; Waldmuller et al., 2011; Walsh et al., 2017). Four residues linked to cardiomyopathy variants (S532P, I533V, A543T, and A550V shown in Fig. 3E) are involved in intra-stabilization of the HLH motif. The other four are likely to alter interaction of the HLH motif with surrounding residues of myosin (Fig. 3F). Three of those represent charge-gaining mutations capable of introducing artifactual salt bridges that may alter the position/mobility of the HLH motif: G529D mutation may result in a salt bridge between D529 and H556 (Fig. 3F, black arrow), M531R may form a bond with E525 (Fig. 3F, blue arrow), while A543T could introduce H-bond with E535 (Fig. 3F, orange arrow). M539 interaction with F644 stabilizes loop 2 (Fig. 3F, red arrow), therefore, we predict that M539L will loosen this contact.
Our data shows that cardiac myosin loop 3 does not form interactions with actin due to salt bridges between R572/D469 and presumably R567/D587 (Fig. 1H). DCM-related R567H and D469Y (Walsh et al., 2017) human pathogenic variants nicely map into the interface between loop 3 and the body of myosin motor domain (Fig. 3H). Charge altering D469Y mutation should disrupt aspartic acid salt bridge with R572 (Fig. 3G, blue arrow), while replacement of longer arginine with shorter histidine in R567H presumably would affect R567 interaction with D587 (Fig. 3G, yellow arrow). In our cardiac model H576R makes an H-bond with N589 (Fig. 3G, black arrow). In the HCM-related H576R variant the longer arginine may interfere with the salt bridges between R572/D469 and R567/D587, therefore, alter the positioning of loop 3.
Actin pathological variants.
At 3.5 – 3.8 Å resolution positions of most side chains of actin residues are fully resolved (Fig. S2B and C) and can be used to elucidate possible mechanism(s) of pathogenesis in cardiac actin variants. These mutations are not evenly spread over the actin molecule, but rather form several highly populated patches within the actin filament. These patches include the following parts of the actin molecule: short loop (residues 20–29) at the actomyosin interface (Fig. 4A); the DNAse-1 loop (D-loop) (Fig. 4B); two major helixes (78–92 and 112–126) in subdomain 1 (SD1) of actin (Fig. 4C); upper part of subdomain 4 (SD4) (Fig. 4D); lower portion of subdomain 3 of actin (Fig. 4E); Tm binding interface (Fig. 4F); and the hydrophobic plug (HP) (Fig. 4G).
Fig. 4. Actin pathological variants.
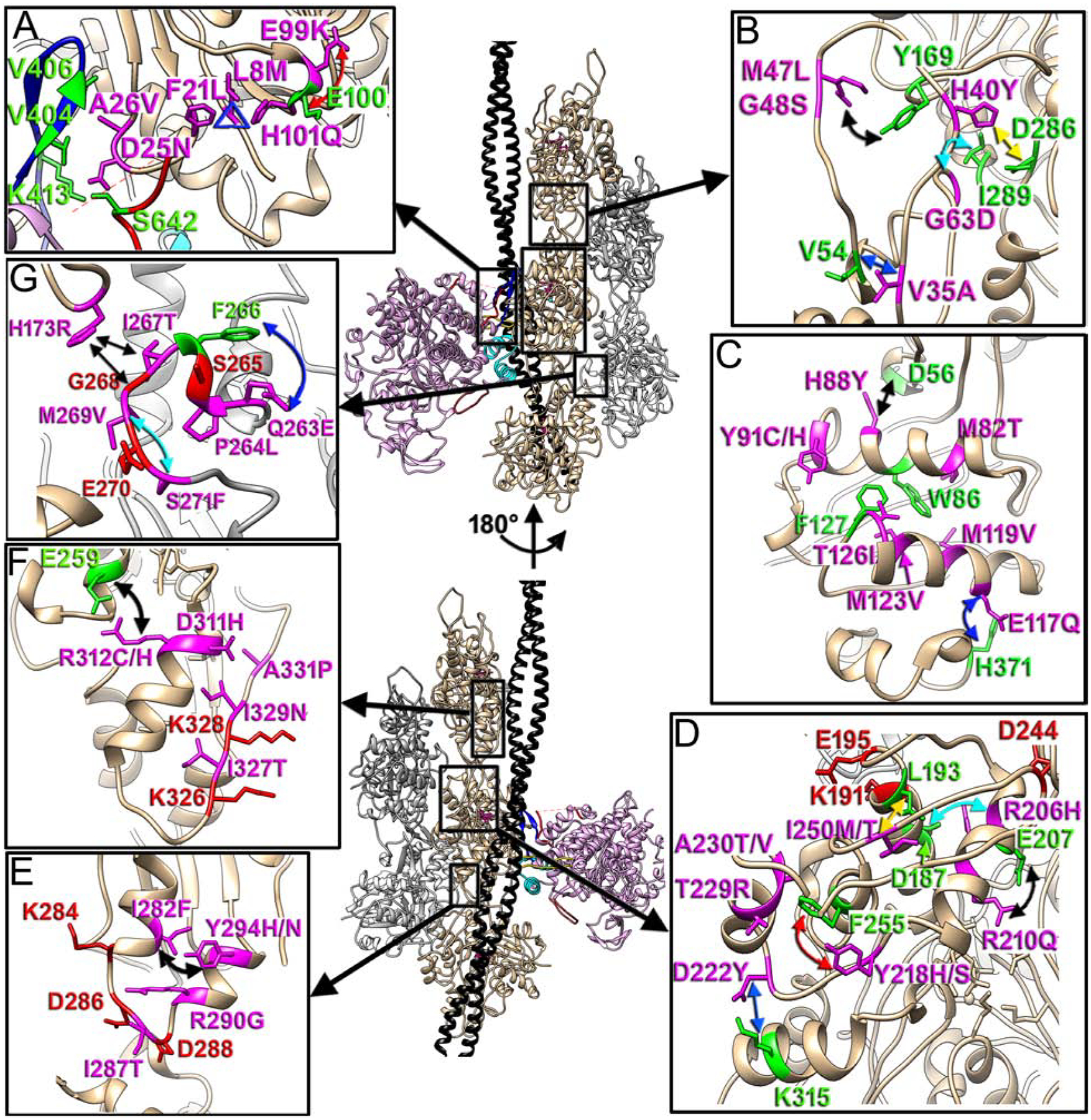
Positioning of the seven actin regions that harbor actin missense mutations linked to cardiomyopathies is shown with black boxes on our model and is linked to the inserts as follows: actomyosin interface with CM loop and loop 2 (A), D-loop in SD2 of actin (B), two major helixes (78–92 and 112–126) in SD1 of actin (C), upper part of SD4 of actin (D), lower portion of SD3 of actin (E), Tm binding interface (F), and hydrophobic plug (HP) (G). Actin molecules in the two strands are either tan or grey, tropomyosin is black, while myosin is plum. The two views of the model are related by 180° rotation around actin helical axis. Actin pathogenic variants are in magenta, while their interacting partners are in green. The proposed interactions are marked with colored arrows.
Loop 20–29 located at the actomyosin interface encompasses three residues that are responsible for HCM phenotype (Fig. 4A: F21L, D25N and A26V in magenta). F21 stabilizes the 20–29 loop by forming a hydrophobic triangle with H101 and L8 (Fig. 4A, blue triangle). The triad of pathogenic mutations associated with these three residues which includes L8M (Kindel et al., 2012), F21L (Coppini et al., 2014) and H101Q (Wang et al., 2014) would seemingly destabilize the 20–29 loop resulting in altered positioning of the D25 and A26 residues involved in the actomyosin interactions with CM-loop and loop 2. Not surprisingly, the D25N and A26V (Walsh et al., 2017) missense mutations themselves are pathogenic, since D25 forms a salt bridge with K413 (Fig. 1C) and H-bond with S642 (Fig. 1D), while A26 stabilizes the actin interaction with the CM loop of by forming a hydrophobic patch with V404 and V406 (Fig. 1B). One of the well-studied pathogenic mutations in actin is E99K (Olson et al., 2000). Our data suggests that E99 may stabilize the kink in the 98–101 loop via repulsive charge interaction with the E100 (Fig. 4A, red arrow). In this case the substitution of E99 with K99 in E99K actin variant may alter the geometry of 98–101 loop with a rippling effect on the hydrophobic triangle (Fig. 4A, blue triangle) and loop 20–29 harboring myosin binding D25 and A26. Alternatively, E99K may reduce the charged interaction between loop 3 and SD1 of actin (Bookwalter and Trybus, 2006; Milligan, 1996; Rayment et al., 1993; Volkmann et al., 2000) upon initial (weak) binding of myosin to actin.
A major hotspot for pathogenic variants in cardiac actin is located in the D-loop in subdomain 2 (SD2). Most of the residues linked to cardiomyopathies are involved in inter-subunit interactions: M47 interacts with Y169 of the upper actin’s SD1 (Fig. 4B, black arrow), while H40 bridges with D286 of SD3 of the above actin protomer (Fig. 4B, yellow arrow). The salt bridge between H40 and Y169 is reinforced by hydrophobic interaction between G63 and I289 (Fig. 4B, cyan arrow). Weaker inter-subunit interactions introduced by M47L (Zou et al., 2013), H40Y (Walsh et al., 2017), and G63D (Wang et al., 2014) presumably reduces force generation in cardiac muscle which is compensated by thickening of the cardiac wall and results in HCM phenotype. G48 and V35 residues structurally stabilize the D-loop – V35 reinforces the base of the D-loop via interaction with the V54 (Fig. 4B, blue arrow), while G47 contributes to the intrinsic flexibility of the D-loop (Merino et al., 2018).
Another patch of missense mutations linked to cardiomyopathies is located in the two major helices of SD1 of actin (78–92 and 112–126, respectively). All those actin residues are directly involved in preservation of geometry of the SD1/SD2 of actin. M82, Y91, M119, M123, and T126 (missense mutations M82T, Y91C/H, M119V, M123V, and T126I, respectively) (Augiere et al., 2015; Burns et al., 2017; Lakdawala et al., 2012; Lopes et al., 2015; Matsson et al., 2008; Wang et al., 2017) form a patch of hydrophobic interactions with W86 and P127 which holds the two major helices in SD1 of actin (Fig. 4C). H88 (H88Y) (Morita et al., 2008) stabilizes interface between SD1 and SD2 of actin via a salt bridge with D56 (Fig. 4C, black arrow), while E117 (E117Q) (Zimmerman et al., 2010) links the C-terminus of actin to the body of SD1 via salt bridge with H371 (Fig. 4C, blue arrow).
The top of SD4 harbors three residues that hold the integrity of F-actin (Fig. 4D, red atoms) (von der Ecken et al., 2015). A group of missense mutations in SD4 may destabilize the upper part of SD4 to alter the filament stability. R206 (R206H) (Dal Ferro et al., 2017) forms a salt bride with D187 (Fig. 4D, cyan arrow) which stabilizes helix(182–193)-loop(194–202)-helix(203–215) motif in SD4 of actin involved in the interstrand interactions, while R210 (R210Q) (Meng et al., 2017) directly stabilizes 203–215 helix via a bond with E207 (Fig. 4D, black arrow). Y218 (Y218H/S) (Takasaki et al., 2018; Vasilescu et al., 2018) and D222 (D222Y) (Richard et al., 2019) are involved in positioning of the long loop 216–223: Y218 forms a hydrophobic bond with F255 (Fig. 4D, red arrow), while D222 forms a salt bridge with K315 residing in SD3 of actin (Fig. 4D, blue arrow). Therefore, we hypothesize that Y218H/S and D222Y may alter the positioning of the following short 224–230 helix of actin and affect the inter-subdomain contacts within the actin molecule. T229 (T229R) (Yoshida et al., 2016) and A230 (A230T/V) (Aljeaid et al., 2019; Van Driest et al., 2003) structure the short 224–230 helix of actin. The following I250 (I250M/T) (Dal Ferro et al., 2017; Lakdawala et al., 2012) makes a hydrophobic interaction with L193 of the 182–193 helix (Fig. 4D, yellow arrow), so that it interconnects both elements of SD4 involved in direct interactions between actin protomers.
The bottom loop in SD3 of actin (283–290) harbors residues K284, D286, D288, and R290 (Fig. 4E, red atoms except for R290G) involved in forming inter and intrastrand interactions (von der Ecken et al., 2015). I282 (I282F) (Walsh et al., 2017) forms a hydrophobic interaction with Y294 (Y294H/N) (Norrish et al., 2019; Sheikh et al., 2018) (Fig. 4E, black arrow) which holds the two helices that flank loop 283–290. R290 (R290G) (Guo et al., 2007) is involved in the intrastrand contacts (Fig. 4E, shown in magenta). I287 (I287T) (Rodriguez-Serrano et al., 2014) (Fig. 4E, shown in magenta) is located between the two aspartic acids D286 and D288 (Fig. 4E, shown in red) separating those so that D286 contributes to interstrand interactions, while D288 to intrastrand ones. I287T mutation presumably allows H-bond formation between T287 and adjacent aspartic acids, which alters interfilament contacts.
A number of mutations are located at the actin-Tm interface (Fig. 4F, magenta atoms). D311 has been shown to interact with Tm (Risi et al., 2017). We predict that the charge swapping D311H mutation (Kaski et al., 2008) should severely alter actomyosin regulation by Tm by weakening its binding to this pivotal residue. Adjacent R312 (R312R/C) (Kaski et al., 2009; Olson et al., 1998) stabilizes the 308–321 helix which harbors R312 by making the salt bridge with E259 (Fig. 4F, black arrow). I327 (I327T) (Tian et al., 2015) and I329 (I329N) (Cecconi et al., 2016) (Fig. 4F, in magenta) are located next to K326 and K328 (Fig. 4F, red atoms) involved in interaction with Tm (Risi et al., 2017; von der Ecken et al., 2015). Both I327T and I329N pathogenic mutations introduce polar groups into the sidechains that are capable of forming H-bonds with K326 and K328, therefore, interfering with Tm interactions. A331P (Cecconi et al., 2016) (Fig. 4F, in magenta) presumably creates a kink in the straight long loop that holds K326 and K328, which alters their positioning.
The HP plug harbors S265, G268, and E270 actin residues directly involved into the interstrand contacts (von der Ecken et al., 2015) (Fig. 4G, red atoms), therefore, its proper orientation and folding is crucial for the health of the actin filament. Out of five missense mutations within the HP four are involved in maintaining the proper folding of the HP. Residues Q263 (Q263E) and P264 (P264L) (Walsh et al., 2017) maintain the integrity of the short α-helix: the hydrophobic part of the sidechain of Q263 forms a prominent bridge of density with F266 (Fig. 4G, blue arrow) while P264 makes a kink to form the helix. M269 (M269V) (Hoedemaekers et al., 2010) forms an H-bond with S271 (S271F) (Olivotto et al., 2008) (Fig. 4G, cyan arrow) which maintains the geometry of the HP tip. In contrast, I267 (I267T) (Pugh et al., 2014) is directly involved in interstrand interactions with H173 (Fig. 4G, black arrow), which missense mutation (H173R) (Vasilescu et al., 2018) has also being linked to DCM.
DISCUSSION
Sequence divergence between myosin isoforms is an evolutionary mechanism that tunes the physiological parameters of each isoform for its particular cellular duty. The two actin binding elements that possess the highest difference between the cardiac and the other myosin isoforms are loops 3 and 2 (Fig. 1A, brown and red ribbons, respectively). In contrast to the fast skeletal and cardiac myosins all the three non-muscle actomyosin complexes we compared with our cardiac model have loop 3 forming either electrostatic (Gurel et al., 2017; von der Ecken et al., 2016) or hydrophobic (Mentes et al., 2018) interactions with the actin SD1. This additional contact presumably reduces the sliding velocity of non-muscle myosins in comparison to skeletal or cardiac isoforms (Cuda et al., 1997; Yengo et al., 2012) while providing additional stability to the non-muscle actomyosin complexes required for .their cellular duties. Despite a difference in resolution between our cardiac and the fast skeletal (Fujii and Namba, 2017) actomyosin complex, the two skeletal isoforms showed similar interactions with actin with one exception - the cardiac myosin loop 2 which did not form salt bridges with the negatively charged N-terminus of actin (Fig. 1D). Indeed, the ionic interactions between the loop 2 and the N-terminus of actin may take place during the initial stages of actomyosin interaction (Furch et al., 1998). Nevertheless, the difference that we observe between the two isoforms in rigor state may in part explain the slower velocity of the cardiac isoform when compared to the fast skeletal myosin (Mijailovich et al., 2017). It has been suggested (Rao et al., 2011) that skeletal myosin acts as an allosteric catch bond due its ability to form force-dependent short- or long-lived bound rigor (Clobes and Guilford, 2014) states. Elimination of loop 2 in skeletal isoform leads to an allosteric change from a short- to a long-lived rigor bond state of myosin (Clobes and Guilford, 2014). For that reason, it is plausible that the weaker interaction of the cardiac loop 2 with actin in rigor complex may contribute to its slower velocity without affecting the almost identical low-duty ratio (~0.11) for the two isoforms (Mijailovich et al., 2017). Overall, the comparison of the cardiac complex to other actomyosin structures supports the idea (Robert-Paganin et al., 2020) that the key actin-binding elements of myosins find similar sites on F-actin, but interact differently in detail. This allows different myosins to use very similar track on the surface of the evolutionary conserved actin filament.
The healthy function of cardiac muscle depends on many physiologically relevant interactions of the sarcomeric proteins – interactions between actin molecules that form the thin filament, regulatory interactions between F-actin and tropomyosin/troponin complex, interactions between myosin molecules in the super relaxed state, and, finally, actomyosin interactions, which convert chemical energy into the contractile force. Any alteration in this sophisticated system due to missense mutations leads to cardiovascular diseases. A cohort of missense mutations in residues maintaining the sequestered state of the thick filament has been linked to cardiomyopathies in humans (Adhikari et al., 2019; Alamo et al., 2017; Nag et al., 2017; Robert-Paganin et al., 2018) including residues (e.g. R403) involved in both rigor binding (Nag et al., 2015) and stabilization of the sequestered state (Sarkar et al., 2020). The two structures of the cardiac myosin in the pre-power stroke and post-rigor states gave crucial information about the essential light chain/converter interface (Planelles-Herrero et al., 2017; Robert-Paganin et al., 2018). Nevertheless, the crucial structural information that describes the cardiac actomyosin interface and provides the structure of the cardiac actin filament has been missing. Here we report this missing part, which provides insights into molecular basis of cardiac diseases caused by missense mutations linked pathogenic variants in either myosin (Fig. 3) or actin (Fig. 4) pathogenic variants. Most of the pathogenic missense substitutions reside in catalytic or converter parts of the myosin motor domain or parts of myosin involved in stabilization of the sequestered state, while ~15% of variants arising from substitutions at the vicinity of the actomyosin interface (Alamo et al., 2017). Our data show that only 5 out of 18 analyzed variants are directly involved in the interactions of myosin with either actin (Fig. 3B and C: V404M, V406M, V411I, and S642L) or Tm (Fig. 3D: R369Q), while the rest 11 are involved in structural maintenance of the actin binding regions of myosin. Importantly, two pathogenic myosin variants (Fig. 3G: D469Y and R567H) are linked to substitutions in the residues involved in keeping loop 3 of myosin away from the actin surface (Fig. 1H) suggesting that the formation of the “Milligan contact” (Milligan et al., 1990) is energetically unfavorable for cardiac contraction.
Along the six actin genes expressed in humans ACTA1 (skeletal) has the highest number of reported mutations followed by ACTA2 (smooth muscle) and ACTC1 (cardiac). Analysis of the missense mutations across these actin isoforms shows that there are common mutational hotspots where the numbers of mutations tend to be higher than elsewhere (Parker and Peckham, 2020). Out of 41 structurally evaluated actin pathogenic variants only two reside at the actomyosin interface (Fig. 4A: D25N, and A26V) and only five are involved at direct actin-actin contacts (Fig. 4B, E and G: H40Y, M47L, H173R, I267T, and R290G). Importantly, only R290G is involved in the intrastrand interactions, while the rest are involved in the interstrand contacts. Therefore, over 80% of pathogenic variants arises from destabilization of the secondary and tertiary structure of the actin’s subdomains harboring regions involved in inter- and intrastrand interactions. Actin is one of the most highly conserved eukaryotic proteins presumably due to its ability to adopt multiple high order structures (Galkin et al., 2010; Galkin et al., 2002), since actin residues that diverged over the evolution are found mostly on the surface of actin (Egelman, 2001). For that reason, it is not surprising that missense substitutions in residues involved in the maintenance of the actin molecule tertiary structure lead to diseases in humans.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Vitold E. Galkin (galkinve@evms.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The atomic has been deposited at the Protein Data Bank (www.rcsb.org) with accession code 7JH7. The corresponding cryo-EM map was deposited in the Electron Microscopy Data Bank (www.ebi.ac.uk/pdbe/emdb) with accession code EMD-22335.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The samples were prepared from Sus Scofa heart left ventricle tissue.
METHODS DETAILS
Native porcine cardiac thin filaments purification
Whole frozen porcine hearts were obtain from Pel Freez and stored at −80°C until needed. Approximately 100–125 grams of ventricular tissue was removed and thawed on ice. The tissue was passed through a cold meat grinder and collected in a 1 liter beaker on ice. The tissue was suspended in 400 ml of Buffer F1 (10 mM Na2HPO4, 100 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 1 mM NaN3, 1 mM DTT, pH 7) containing 1% Triton-X 100 (v/v) and homogenized for 40 seconds using a Polytron foam-reducing homogenizer. The tissue was pelleted by spinning at 15,000xg for 8 minutes. The resulting supernatant was discarded and the homogenization step was repeated once more with Buffer F1 + Triton-X and an additional three times using Buffer F1 without detergent. The thin filaments were extracted from the washed myofibrils by resuspending and homogenizing twice in 180 ml of Buffer F1 + 5 mM ATP followed by spinning at 15,000xg for 8 minutes. The supernatants were combined and centrifuged at 186,000xg for 15 minutes. The supernatants were transferred to fresh tubes and centrifuged for 2.5 hours at 186,000xg. The pelleted crude thin filaments were gently resuspended using a tissue homogenizer to approximately 120 ml in Dialysis Buffer (20 mM TRIS (pH 7.0), 100 mM NaCl, 5 mM MgCl2, 1 mM EGTA, 1 mM NaN3, 1 mM DTT) and dialyzed overnight against the same buffer. The following morning, the solution was adjusted to 20 mM TRIS, 200 mM NaCl, 2 mM ATP, 2.5 mM MgCl2, 1 mM EGTA, 1 mM DTT, 1 mM NaN3 (pH 7.0) by the addition of an equal volume of ATP Release Adjustment Buffer (20 mM TRIS, 300 mM NaCl, 1 mM EGTA, 1 mM DTT, 1 mM NaN3, 4 mM ATP (pH 7.0)). The solution was stirred on ice for 10 minutes before spinning at 186,000xg for 15 minutes, discarding the pellets and spinning for another 2.5 hours at the same speed. The pellets were resuspended with Dialysis Buffer as described previously to approximately 90 ml and dialyzed overnight. The thin filaments were again diluted with an equal volume of ATP Release Adjustment Buffer, stirred on ice for 10 minutes and centrifuged as above. The final thin filament pellets were resuspended to approximately 10–15 ml to provide a concentration of 150–200 μM. The concentration was determined after accounting for light scattering (OD280 – (OD320 × 1.5)) using an extinction coefficient of 0.765 mg−1/ml−1 and a molecular weight of 63,000.
Native porcine cardiac myosin-S1 purification
All solutions were kept on ice. Approximately 500 grams of pig heart ventricles, with the atria, aorta and fatty tissue removed were passed through a cold meat grinder once. The ground tissue was suspended to a total volume of 3.5 liters in 0.1 M KCl, 7 mM KH2PO4 (pH 7.0), 8 mM Na2SO3, 7.5 mM EGTA, 1 mM DTT, 1 mM MgCl2, 1% Triton X-100 (v/v), 0.2 ug/ml Pepstatin A, and homogenized briefly using a Polytron homogenizer equipped with a foam-reducing generator to break up any large clumps of tissue. The suspension was spun at 1600g for 12 min immediately after homogenization. The resulting supernatant was discarded and the pellets were resuspended in 3.5 liters of the same buffer and centrifuged. The suspension was homogenized briefly before spinning at 1600g for 12 minutes and the pellets were resuspended in 3.5 liters in the same buffer without Triton X-100 and homogenized briefly. The resuspension and spin were repeated two more times in Triton-free buffer. After the final wash/spin, the pellets were resuspended by gentle homogenization in a 3x volume of 0.225 M KCl, 37.5 mM KH2PO4 (pH 6.8), 2.5 mM ATP, 0.25 mM MgCl2, 1 mM EGTA, 1 mM DTT, packed on ice and stirred for 30 minutes. The suspension was next spun at 14,000g for 45 minutes to remove any cellular debris and the resulting supernatant is dialyzed overnight against vs ~15 volumes of 0.1 mM DTT to precipitate the myosin. The precipitated myosin was collected by spinning at 26,000g for 20 minutes. The pellets were resuspended in a minimal volume of 0.5 M KCl, 0.1 M KH2PO4 (pH 7.0), 20 mM MgCl2, 1 mM DTT, 20 mM ATP followed by spinning at 140,000g for 4 hours at 4°C to remove any actin and HMM. The resulting supernatant was dialyzed overnight against 200 mM ammonium acetate, (pH 7.3), 2 mM MgCl2 with one change of dialysate. The myosin concentration was determined using an extinction coefficient of 0.53 mg−1/ml−1 at 280 nm after adjusting for light scattering ((OD280 – (OD320 × 1.5) / ε)). The myosin was diluted with 200 mM ammonium acetate, 2 mM MgCl2 to obtain a final concentration of 10 mg/ml for digestion. In order to avoid degradation of the heavy chains, a test digestion must be performed to determine the amount of time necessary to provide a 50% theoretical yield, maintain the presence of the two light chains in equal amounts and avoid degradation of the heavy chains. Approximately 100 μl of a 1 mg/ml solution of papain (Sigma #P3125) was incubated in papain activation buffer (50 mM TRIS (pH 8.0), 1 mM EDTA, 10 mM DTT) in a 25°C water bath for 15 minutes and 1 ml of the PV myosin solution was placed in a test tube and warmed to 25°C. 10 ul of the activated papain was added to 1 ml of myosin and 50 μl aliquots were collected every two minutes from 0–26 minutes, added to a tube containing 2 μl of 0.25 mM E-64, mixed well (not vortexed) and placed on ice. 450 μl of water was added to each tube which resulted in a protein concentration of 1 mg/ml. 20 μl of each fraction were mixed with 20 μl of 2x Sample Buffer and heated for 8 minutes (90°C) before loading 20 μl of each sample on a 1-D PAGE. A digestion time was selected to produce minimal degradation of the S1 heavy chain and equal amounts of both light chains. The required amount of 1 mg/ml papain in Papain Activation Buffer was prepared and activated as described previously at 25°C for 15 minutes. The myosin was warmed to 25°C and a 1/1000th volume of papain was added to obtain 10 μg/ml papain. The myosin solution was stirred throughout the digestion. After the appropriate digestion time, a 1/1000th volume of 10 mM E-64 was added to obtain a concentration of 10 μM. The solution was packed on ice and stirred for 30 minutes. The undigested myosin and rod were removed from the S1 by spinning at 215,000g for 2 hours. The crude PV-S1 was further purified by ion-exchange chromatography on Q-Sepharose. Approximately 500 mg of crude S1 is dialyzed overnight against column buffer (20 mM TRIS (pH 8.0), 0.5 mM MgCl2, 0.5 mM DTT) with one change. This was applied to a 2.6 × 40 cm Q-Sepharose column equilibrated with several volumes of column buffer. Once the S1 had entered the bed, the column was rinsed with one bed volume of column buffer and eluted with a 2 liter gradient of column buffer plus 0 – 0.4 M NaCl at 2 ml/min. 8–10 ml fractions were collected throughout the loading, rinsing and elution steps. SDS-PAGE gels were run on any fractions showing the presence of protein. Based on the results of the gels, fractions that had intact S1 bands and showed the two light chains present in equal amounts were pooled. The S1 was concentrated by the addition of saturated (NH4)2SO4 to 60%. The precipitated S1 was collected and dialyzed exhaustively against 50 mM KCl, 5 mM MOPS (pH 7.0), 2 mM MgCl2, 0.5 mM DTT. A final spin at 230,000g was used to clarify the pool before drip-freezing in liquid nitrogen.
Cryo EM Sample preparation
A-buffer was used for cryo EM experiments: 50 mM KAc, 10 mM MOPS, 3 mM MgCl2, pH 7.0. 3μL of [2 μM] TFs in A-buffer were applied to glow-discharged lacey carbon grid for 1 min, gently blotted with Whatman #1 filter paper, and incubated with 2 μL of myosin-S1 [1.2 μM] for 2 min on grid followed by final blotting with Whatman #1 filter paper for 5–6 sec and vitrification in a Vitrobot Mark IV (FEI,Inc.).
Image analysis
The detailed imaging parameters are shown in Table S1. Micrographs were collected with a 300-kV Titan Krios electron microscope equipped with a Falcon III direct electron detector. Automatic data collection was performed with EPU software. 2,373 micrographs were recorded with 40 subframes with a dose rate of ~1.2 e-/Å2 per frame over a defocus range of 0.6–3.5 μm with a pixel size of 1.056 Å. Images (Fig. S1A) were imported into RELION (Version 3.0.8) software suite (Scheres, 2012), where motion corrected and dose weighting was done using internal RELION implementation. Contrast transfer function parameters were calculated by using CTFFIND3 (Mindell and Grigorieff, 2003). Particles were manually picked in EMAN2 e2helixboxer (Tang et al., 2007) and EMAN2 coordinates were used in RELION package to extract 162,100 particles with the box size of 422 Å and an overlap of 27.5 Å. After 2D classification step, 115,746 segments were used in 3D refinement (Fig. S1C) to yield 5.15.Å resolution map. Post-processing and particle polishing procedures were used as described in RELION manual to properly correct the map for the CTF and improve beam-induced motion correction which yield the map with global resolution of 3.8 Å (Fig. S2A) with a helical symmetry of −166.7°/27.3Å. Local resolution was determined using RELION subroutines and shown in Fig. S2B.
Actomyosin model building
The cryo-EM density map served as a template for building an atomic model. At the time of the model building there was no available structure of porcine β-cardiac myosin or α-cardiac actin. SWISS-MODEL web service (Waterhouse et al., 2018) was used for homology modeling of the actomyosin complex. Homology model of porcine α-cardiac actin was build based on α-skeletal muscle actin (PDB: 3MFP) (Fujii et al., 2010) as a template using the porcine amino acid sequence (UniProt: B6VNT8). For the β-cardiac myosin S1 heavy chain the model was created using porcine amino acid sequence (UniProt: P79293) and the structure of rigor actomyosin complex (PDB: 5H53, chain A) (Fujii and Namba, 2017) as a template. The homology models of cardiac actin and myosin were structurally aligned to their counterparts in the skeletal actomyosin complex (PDB: 5H53) in UCSF Chimera (Pettersen et al., 2004) and helical symmetry of −166.7°/27.3Å was imposed to build up a helical filament. The structure of the cytoplasmic actomyosin complex (PDB: 5JLH) was docked into our density map filtered to 7Å resolution and tropomyosin model atom coordinates were extracted to use in our model using the USCF Chimera. The model containing cardiac actin, myosin and tropomyosin model was used for initial flexible fitting in USCF iMODFIT plugin (Lopez-Blanco and Chacon, 2013). Next, UCSF Chimera “segment” map tool was used to extract five actin subunits, three S1 motor domains, and one tropomyosin strand from the high-resolution map (Fig. S1D). Corresponding atom coordinates from the helical homology model were placed into the segmented map for final refinement using DireX (Schroder et al., 2007) and PHENIX (Liebschner et al., 2019) software. In detail, DireX was used to fit the model into the map while simultaneously applying deformable elastic network (DEN-) restraints to preserve the topology of the starting model and prevent overfitting. The strength of the DEN-restraints was set to 0.4, and the map strength was set to 0.03. Remaining issues with geometry of the structure, Ramachandran outliers were manually eliminated using Coot. Furthermore, alignments of individual sidechains to the density map were improved in Coot (Emsley et al., 2010). The following residues were removed due to a lack of reliable densities in our cryo-EM reconstruction: myosin loop 1 residues 201–215, myosin loop 2 residues 622–637, and L50D residues 507–510. A final refinement was performed with PHENIX (Liebschner et al., 2019). The parameters for the resultant model are shown in Table S1.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was only relevant for the atomic model, and those results are presented in Table S1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals | ||
| Magnesium sulfate hexahydrate | Baker | Cat. #1-2500 |
| Sodium azide | Sigma | Cat. #S2002 |
| Potassium chloride | Sigma | Cat. #P4504 |
| Tris(hydroxymethyl)aminomethane | Sigma | Cat. #T1503 |
| Sodium chloride | Sigma | Cat. #S9625 |
| Pepstatin A | Santa Cruz Biotech | Cat. #SC-45036A |
| Dimethylsulfoxide (DMSO) | Sigma | Cat. #D2650 |
| Phenylmethylsulfonyl fluoride (PMSF) | Sigma | Cat. #P7626 |
| E-64 protease inhibitor | Fisher | Cat. #501010687 |
| DTT | Sigma | Cat. #D9779 |
| Triton X-100 | Fisher | Cat. #BP151 |
| Sodium phosphate dibasic | Fisher | Cat. #S374 |
| Potassium phosphate monobasic | Fisher | Cat. #P285 |
| Sodium sulfite | Sigma | Cat. #S0505 |
| Ethylene glycol-bis(2-amino-ethylether)-N,N,N’,N’-tetra acetic acid (EGTA) | Sigma | Cat. #E4378 |
| Magnesium chloride hexahydrate | Sigma | Cat. #M2393 |
| Adenosine 5’-triphosphate disodium salt hydrate (ATP) | Sigma | Cat. #A2383 |
| Ammonium acetate | Sigma | Cat. #A1542 |
| Papain from papaya latex | Sigma | Cat. #P3125 |
| Q-Sepharose fast flow | GE Healthcare | Cat. #17-0510-01 |
| Ammonium sulfate | Sigma | Cat. #A4418 |
| 3-(N-Morpholino)propanesulfonic acid (MOPS) | Sigma | Cat #M1254 |
| Biological Samples | ||
| Sus Scofa heart left ventricle tissue | Pel-Freez Biologicals | Cat. #59416-1 |
| Deposited Data | ||
| cardiac actomyosin complex pseudo-atomic model | This paper | PDB 7JH7 |
| cardiac actomyosin complex density map | This paper | EMD-22335 |
| α-skeletal muscle actin | (Fujii et al., 2010) | PDB: 3MFP |
| rabbit skeletal muscle actomyosin rigor complex | (Fujii and Namba, 2017) | PDB: 5H53 |
| human cytoplasmic actomyosin complex | (Von der Ecken et al., 2016) | PDB: 5JLH |
| Software and Algorithms | ||
| RELION | (Scheres., 2012) | https://github.com/3dem/relion |
| CTFFIND3 | (Mindell et al., 2003) | http://grigoriefflab.janelia.org |
| EMAN2 | (Tang et al., 2007) | https://blake.bcm.edu/emanwiki/EMAN2 |
| SWISS-MODEL web service | (Waterhouse et al., 2018) | https://swissmodel.expasy.org/ |
| iMODFIT | (Lopez-Blanco and Chacon, 2013) | https://chaconlab.org/hybrid4EM/imodfit |
| DireX | (Schroder et al., 2007) | https://simtk.org/home/direx |
| UCSF Chimera | (Pettersen et al., 2004) | http://www.cgl.ucsf.edu/chimera |
| PHENIX | (Liebschner et al., 2019) | https://www.phenix-online.org/ |
| Coot | (Emsley et al., 2010) | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
Highlights.
The structure of cardiac actomyosin complex is resolved at near-atomic 3.8 Å resolution
Resultant pseudo-atomic model provides molecular details of actomyosin interface
The structure reveals how mutations in actin and myosin may lead to cardiac Diseases
Acknowledgments:
This work was supported by R01 HL140925 (VEG), U24 GM116790 (VEG), and U24 GM116788 (VEG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no competing interests.
REFERECES
- Adhikari AS, Trivedi DV, Sarkar SS, Song D, Kooiker KB, Bernstein D, Spudich JA, and Ruppel KM (2019). beta-Cardiac myosin hypertrophic cardiomyopathy mutations release sequestered heads and increase enzymatic activity. Nat Commun 10, 2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamo L, Ware JS, Pinto A, Gillilan RE, Seidman JG, Seidman CE, and Padron R (2017). Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aljeaid D, Sanchez AI, Wakefield E, Chadwell SE, Moore N, Prada CE, and Zhang W (2019). Prevalence of pathogenic and likely pathogenic variants in the RASopathy genes in patients who have had panel testing for cardiomyopathy. Am J Med Genet A 179, 608–614. [DOI] [PubMed] [Google Scholar]
- Augiere C, Megy S, El Malti R, Boland A, El Zein L, Verrier B, Megarbane A, Deleuze JF, and Bouvagnet P (2015). A Novel Alpha Cardiac Actin (ACTC1) Mutation Mapping to a Domain in Close Contact with Myosin Heavy Chain Leads to a Variety of Congenital Heart Defects, Arrhythmia and Possibly Midline Defects. PLoS One 10, e0127903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee C, Hu Z, Huang Z, Warrington JA, Taylor DW, Trybus KM, Lowey S, and Taylor KA (2017). The structure of the actin-smooth muscle myosin motor domain complex in the rigor state. J Struct Biol 200, 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrmann E, Muller M, Penczek PA, Mannherz HG, Manstein DJ, and Raunser S (2012). Structure of the rigor actin-tropomyosin-myosin complex. Cell 150, 327–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bement WM, and Mooseker MS (1995). TEDS rule: a molecular rationale for differential regulation of myosins by phosphorylation of the heavy chain head. Cell Motil Cytoskeleton 31, 87–92. [DOI] [PubMed] [Google Scholar]
- Bookwalter CS, and Trybus KM (2006). Functional consequences of a mutation in an expressed human alpha-cardiac actin at a site implicated in familial hypertrophic cardiomyopathy. J Biol Chem 281, 16777–16784. [DOI] [PubMed] [Google Scholar]
- Burns C, Bagnall RD, Lam L, Semsarian C, and Ingles J (2017). Multiple Gene Variants in Hypertrophic Cardiomyopathy in the Era of Next-Generation Sequencing. Circ Cardiovasc Genet 10. [DOI] [PubMed] [Google Scholar]
- Cecconi M, Parodi MI, Formisano F, Spirito P, Autore C, Musumeci MB, Favale S, Forleo C, Rapezzi C, Biagini E, et al. (2016). Targeted next-generation sequencing helps to decipher the genetic and phenotypic heterogeneity of hypertrophic cardiomyopathy. Int J Mol Med 38, 1111–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clobes AM, and Guilford WH (2014). Loop 2 of myosin is a force-dependent inhibitor of the rigor bond. J Muscle Res Cell Motil 35, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrave M, and Peckham M (2014). Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat Rec (Hoboken) 297, 1670–1680. [DOI] [PubMed] [Google Scholar]
- Coppini R, Ho CY, Ashley E, Day S, Ferrantini C, Girolami F, Tomberli B, Bardi S, Torricelli F, Cecchi F, et al. (2014). Clinical phenotype and outcome of hypertrophic cardiomyopathy associated with thin-filament gene mutations. J Am Coll Cardiol 64, 2589–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuda G, Pate E, Cooke R, and Sellers JR (1997). In vitro actin filament sliding velocities produced by mixtures of different types of myosin. Biophys J 72, 1767–1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daehmlow S, Erdmann J, Knueppel T, Gille C, Froemmel C, Hummel M, Hetzer R, and Regitz-Zagrosek V (2002). Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun 298, 116–120. [DOI] [PubMed] [Google Scholar]
- Dal Ferro M, Stolfo D, Altinier A, Gigli M, Perrieri M, Ramani F, Barbati G, Pivetta A, Brun F, Monserrat L, et al. (2017). Association between mutation status and left ventricular reverse remodelling in dilated cardiomyopathy. Heart 103, 1704–1710. [DOI] [PubMed] [Google Scholar]
- Egelman EH (2001). Actin allostery again? Nat Struct Biol 8, 735–736. [DOI] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foth BJ, Goedecke MC, and Soldati D (2006). New insights into myosin evolution and classification. Proc Natl Acad Sci U S A 103, 3681–3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Iwane AH, Yanagida T, and Namba K (2010). Direct visualization of secondary structures of F-actin by electron cryomicroscopy. Nature 467, 724–728. [DOI] [PubMed] [Google Scholar]
- Fujii T, and Namba K (2017). Structure of actomyosin rigour complex at 5.2A resolution and insights into the ATPase cycle mechanism. Nat Commun 8, 13969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furch M, Geeves MA, and Manstein DJ (1998). Modulation of actin affinity and actomyosin adenosine triphosphatase by charge changes in the myosin motor domain. Biochemistry 37, 6317–6326. [DOI] [PubMed] [Google Scholar]
- Furch M, Remmel B, Geeves MA, and Manstein DJ (2000). Stabilization of the actomyosin complex by negative charges on myosin. Biochemistry 39, 11602–11608. [DOI] [PubMed] [Google Scholar]
- Galkin VE, Orlova A, Schroder GF, and Egelman EH (2010). Structural polymorphism in F-actin. Nat Struct Mol Biol 17, 1318–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galkin VE, VanLoock MS, Orlova A, and Egelman EH (2002). A new internal mode in F-actin helps explain the remarkable evolutionary conservation of actin’s sequence and structure. Curr Biol 12, 570–575. [DOI] [PubMed] [Google Scholar]
- Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, et al. (2007). Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet 39, 1488–1493. [DOI] [PubMed] [Google Scholar]
- Gurel PS, Kim LY, Ruijgrok PV, Omabegho T, Bryant Z, and Alushin GM (2017). Cryo-EM structures reveal specialization at the myosin VI-actin interface and a mechanism of force sensitivity. eLife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoedemaekers YM, Caliskan K, Michels M, Frohn-Mulder I, van der Smagt JJ, Phefferkorn JE, Wessels MW, ten Cate FJ, Sijbrands EJ, Dooijes D, et al. (2010). The importance of genetic counseling, DNA diagnostics, and cardiologic family screening in left ventricular noncompaction cardiomyopathy. Circ Cardiovasc Genet 3, 232–239. [DOI] [PubMed] [Google Scholar]
- Iascone MR, Marchetti D, and Ferrazzi P (2007). Gene symbol: MYH7. Hum Genet 120, 916. [PubMed] [Google Scholar]
- Joel PB, Trybus KM, and Sweeney HL (2001). Two conserved lysines at the 50/20-kDa junction of myosin are necessary for triggering actin activation. J Biol Chem 276, 2998–3003. [DOI] [PubMed] [Google Scholar]
- Kamisago M, Schmitt JP, McNamara D, Seidman C, and Seidman JG (2006). Sarcomere protein gene mutations and inherited heart disease: a beta-cardiac myosin heavy chain mutation causing endocardial fibroelastosis and heart failure. Novartis Found Symp 274, 176–189; discussion 189–195, 272–176. [PubMed] [Google Scholar]
- Kaneda T, Naruse C, Kawashima A, Fujino N, Oshima T, Namura M, Nunoda S, Mori S, Konno T, Ino H, et al. (2008). A novel beta-myosin heavy chain gene mutation, p.Met531Arg, identified in isolated left ventricular non-compaction in humans, results in left ventricular hypertrophy that progresses to dilation in a mouse model. Clin Sci (Lond) 114, 431–440. [DOI] [PubMed] [Google Scholar]
- Kaski JP, Syrris P, Burch M, Tome-Esteban MT, Fenton M, Christiansen M, Andersen PS, Sebire N, Ashworth M, Deanfield JE, et al. (2008). Idiopathic restrictive cardiomyopathy in children is caused by mutations in cardiac sarcomere protein genes. Heart 94, 1478–1484. [DOI] [PubMed] [Google Scholar]
- Kaski JP, Syrris P, Esteban MT, Jenkins S, Pantazis A, Deanfield JE, McKenna WJ, and Elliott PM (2009). Prevalence of sarcomere protein gene mutations in preadolescent children with hypertrophic cardiomyopathy. Circ Cardiovasc Genet 2, 436–441. [DOI] [PubMed] [Google Scholar]
- Kindel SJ, Miller EM, Gupta R, Cripe LH, Hinton RB, Spicer RL, Towbin JA, and Ware SM (2012). Pediatric cardiomyopathy: importance of genetic and metabolic evaluation. J Card Fail 18, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishino A, and Yanagida T (1988). Force measurements by micromanipulation of a single actin filament by glass needles. Nature 334, 74–76. [DOI] [PubMed] [Google Scholar]
- Lakdawala NK, Funke BH, Baxter S, Cirino AL, Roberts AE, Judge DP, Johnson N, Mendelsohn NJ, Morel C, Care M, et al. (2012). Genetic testing for dilated cardiomyopathy in clinical practice. J Card Fail 18, 296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkoczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr D Struct Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopes LR, Syrris P, Guttmann OP, O’Mahony C, Tang HC, Dalageorgou C, Jenkins S, Hubank M, Monserrat L, McKenna WJ, et al. (2015). Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 101, 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Blanco JR, and Chacon P (2013). iMODFIT: efficient and robust flexible fitting based on vibrational analysis in internal coordinates. J Struct Biol 184, 261–270. [DOI] [PubMed] [Google Scholar]
- Lorenz M, and Holmes KC (2010). The actin-myosin interface. Proc Natl Acad Sci U S A 107, 12529–12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsson H, Eason J, Bookwalter CS, Klar J, Gustavsson P, Sunnegardh J, Enell H, Jonzon A, Vikkula M, Gutierrez I, et al. (2008). Alpha-cardiac actin mutations produce atrial septal defects. Hum Mol Genet 17, 256–265. [DOI] [PubMed] [Google Scholar]
- Meng L, Pammi M, Saronwala A, Magoulas P, Ghazi AR, Vetrini F, Zhang J, He W, Dharmadhikari AV, Qu C, et al. (2017). Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management. JAMA Pediatr 171, e173438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentes A, Huehn A, Liu X, Zwolak A, Dominguez R, Shuman H, Ostap EM, and Sindelar CV (2018). High-resolution cryo-EM structures of actin-bound myosin states reveal the mechanism of myosin force sensing. Proceedings of the National Academy of Sciences of the United States of America 115, 1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merino F, Pospich S, Funk J, Wagner T, Kullmer F, Arndt HD, Bieling P, and Raunser S (2018). Structural transitions of F-actin upon ATP hydrolysis at near-atomic resolution revealed by cryo-EM. Nat Struct Mol Biol 25, 528–537. [DOI] [PubMed] [Google Scholar]
- Mijailovich SM, Nedic D, Svicevic M, Stojanovic B, Walklate J, Ujfalusi Z, and Geeves MA (2017). Modeling the Actin.myosin ATPase Cross-Bridge Cycle for Skeletal and Cardiac Muscle Myosin Isoforms. Biophys J 112, 984–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan RA (1996). Protein-protein interactions in the rigor actomyosin complex. Proc Natl Acad Sci U S A 93, 21–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan RA, Whittaker M, and Safer D (1990). Molecular structure of F-acin and location of surface binding sites. Nature 348, 217–221. [DOI] [PubMed] [Google Scholar]
- Mindell JA, and Grigorieff N (2003). Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142, 334–347. [DOI] [PubMed] [Google Scholar]
- Morita H, Rehm HL, Menesses A, McDonough B, Roberts AE, Kucherlapati R, Towbin JA, Seidman JG, and Seidman CE (2008). Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 358, 1899–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S, Sommese RF, Ujfalusi Z, Combs A, Langer S, Sutton S, Leinwand LA, Geeves MA, Ruppel KM, and Spudich JA (2015). Contractility parameters of human beta-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci Adv 1, e1500511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nag S, Trivedi DV, Sarkar SS, Adhikari AS, Sunitha MS, Sutton S, Ruppel KM, and Spudich JA (2017). The myosin mesa and the basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Nat Struct Mol Biol 24, 525–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrish G, Jager J, Field E, Quinn E, Fell H, Lord E, Cicerchia MN, Ochoa JP, Cervi E, Elliott PM, et al. (2019). Yield of Clinical Screening for Hypertrophic Cardiomyopathy in Child First-Degree Relatives. Circulation 140, 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, Ommen SR, Theis JL, Vaubel RA, Re F, et al. (2008). Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc 83, 630–638. [DOI] [PubMed] [Google Scholar]
- Olson TM, Doan TP, Kishimoto NY, Whitby FG, Ackerman MJ, and Fananapazir L (2000). Inherited and de novo mutations in the cardiac actin gene cause hypertrophic cardiomyopathy. J Mol Cell Cardiol 32, 1687–1694. [DOI] [PubMed] [Google Scholar]
- Olson TM, Michels VV, Thibodeau SN, Tai YS, and Keating MT (1998). Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 280, 750–752. [DOI] [PubMed] [Google Scholar]
- Onishi H, Mikhailenko SV, and Morales MF (2006). Toward understanding actin activation of myosin ATPase: the role of myosin surface loops. Proceedings of the National Academy of Sciences of the United States of America 103, 6136–6141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker F, and Peckham M (2020). Disease mutations in striated muscle myosins. Biophys Rev 12, 887–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Planelles-Herrero VJ, Hartman JJ, Robert-Paganin J, Malik FI, and Houdusse A (2017). Mechanistic and structural basis for activation of cardiac myosin force production by omecamtiv mecarbil. Nat Commun 8, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh TJ, Kelly MA, Gowrisankar S, Hynes E, Seidman MA, Baxter SM, Bowser M, Harrison B, Aaron D, Mahanta LM, et al. (2014). The landscape of genetic variation in dilated cardiomyopathy as surveyed by clinical DNA sequencing. Genet Med 16, 601–608. [DOI] [PubMed] [Google Scholar]
- Rao VS, Clobes AM, and Guilford WH (2011). Force spectroscopy reveals multiple “closed states” of the muscle thin filament. J Biol Chem 286, 24135–24141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, and Milligan RA (1993). Structure of the Actin-Myosin Complex and Its Implications for Muscle Contraction. Science 261, 58–65. [DOI] [PubMed] [Google Scholar]
- Richard P, Ader F, Roux M, Donal E, Eicher JC, Aoutil N, Huttin O, Selton-Suty C, Coisne D, Jondeau G, et al. (2019). Targeted panel sequencing in adult patients with left ventricular non-compaction reveals a large genetic heterogeneity. Clin Genet 95, 356–367. [DOI] [PubMed] [Google Scholar]
- Richards TA, and Cavalier-Smith T (2005). Myosin domain evolution and the primary divergence of eukaryotes. Nature 436, 1113–1118. [DOI] [PubMed] [Google Scholar]
- Risi C, Eisner J, Belknap B, Heeley DH, White HD, Schroder GF, and Galkin VE (2017). Ca2+-induced movement of tropomyosin on native cardiac thin filaments revealed by cryoelectron microscopy. Proc Natl Acad Sci U S A 114, 6782–6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert-Paganin J, Auguin D, and Houdusse A (2018). Hypertrophic cardiomyopathy disease results from disparate impairments of cardiac myosin function and auto-inhibition. Nat Commun 9, 4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert-Paganin J, Pylypenko O, Kikuti C, Sweeney HL, and Houdusse A (2020). Force Generation by Myosin Motors: A Structural Perspective. Chem Rev 120, 5–35. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Serrano M, Domingo D, Igual B, Cano A, Medina P, and Zorio E (2014). Familial left ventricular noncompaction associated with a novel mutation in the alpha-cardiac actin gene. Rev Esp Cardiol (Engl Ed) 67, 857–859. [DOI] [PubMed] [Google Scholar]
- Sarkar SS, Trivedi DV, Morck MM, Adhikari AS, Pasha SN, Ruppel KM, and Spudich JA (2020). The hypertrophic cardiomyopathy mutations R403Q and R663H increase the number of myosin heads available to interact with actin. Sci Adv 6, eaax0069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder GF, Brunger AT, and Levitt M (2007). Combining efficient conformational sampling with a deformable elastic network model facilitates structure refinement at low resolution. Structure 15, 1630–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikh N, Papadakis M, Wilson M, Malhotra A, Adamuz C, Homfray T, Monserrat L, Behr ER, and Sharma S (2018). Diagnostic Yield of Genetic Testing in Young Athletes With T-Wave Inversion. Circulation 138, 1184–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, and Goldman YE (2011). Lever-arm mechanics of processive myosins. Biophys J 101, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syamaladevi DP, Spudich JA, and Sowdhamini R (2012). Structural and functional insights on the Myosin superfamily. Bioinform Biol Insights 6, 11–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasaki A, Hirono K, Hata Y, Wang C, Takeda M, Yamashita JK, Chang B, Nakaoka H, Okabe M, Miyao N, et al. (2018). Sarcomere gene variants act as a genetic trigger underlying the development of left ventricular noncompaction. Pediatr Res 84, 733–742. [DOI] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46. [DOI] [PubMed] [Google Scholar]
- Tian T, Wang J, Wang H, Sun K, Wang Y, Jia L, Zou Y, Hui R, Zhou X, and Song L (2015). A low prevalence of sarcomeric gene variants in a Chinese cohort with left ventricular non-compaction. Heart Vessels 30, 258–264. [DOI] [PubMed] [Google Scholar]
- Toyoshima YY, Kron SJ, McNally EM, Niebling KR, Toyoshima C, and Spudich JA (1987). Myosin subfragment-1 is sufficient to move actin filaments in vitro. Nature 328, 536–539. [DOI] [PubMed] [Google Scholar]
- Uyeda TQ, Abramson PD, and Spudich JA (1996). The neck region of the myosin motor domain acts as a lever arm to generate movement. Proc Natl Acad Sci U S A 93, 4459–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Driest SL, Ellsworth EG, Ommen SR, Tajik AJ, Gersh BJ, and Ackerman MJ (2003). Prevalence and spectrum of thin filament mutations in an outpatient referral population with hypertrophic cardiomyopathy. Circulation 108, 445–451. [DOI] [PubMed] [Google Scholar]
- Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, and Ackerman MJ (2004). Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol 44, 602–610. [DOI] [PubMed] [Google Scholar]
- Varkuti BH, Yang Z, Kintses B, Erdelyi P, Bardos-Nagy I, Kovacs AL, Hari P, Kellermayer M, Vellai T, and Malnasi-Csizmadia A (2012). A novel actin binding site of myosin required for effective muscle contraction. Nat Struct Mol Biol 19, 299–306. [DOI] [PubMed] [Google Scholar]
- Vasilescu C, Ojala TH, Brilhante V, Ojanen S, Hinterding HM, Palin E, Alastalo TP, Koskenvuo J, Hiippala A, Jokinen E, et al. (2018). Genetic Basis of Severe Childhood-Onset Cardiomyopathies. J Am Coll Cardiol 72, 2324–2338. [DOI] [PubMed] [Google Scholar]
- Villard E, Duboscq-Bidot L, Charron P, Benaiche A, Conraads V, Sylvius N, and Komajda M (2005). Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur Heart J 26, 794–803. [DOI] [PubMed] [Google Scholar]
- Volkmann N, Hanein D, Ouyang G, Trybus KM, DeRosier DJ, and Lowey S (2000). Evidence for cleft closure in actomyosin upon ADP release. Nat Struct Biol 7, 1147–1155. [DOI] [PubMed] [Google Scholar]
- von der Ecken J, Heissler SM, Pathan-Chhatbar S, Manstein DJ, and Raunser S (2016). Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature 534, 724–728. [DOI] [PubMed] [Google Scholar]
- von der Ecken J, Muller M, Lehman W, Manstein DJ, Penczek PA, and Raunser S (2015). Structure of the F-actin-tropomyosin complex. Nature 519, 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmuller S, Erdmann J, Binner P, Gelbrich G, Pankuweit S, Geier C, Timmermann B, Haremza J, Perrot A, Scheer S, et al. (2011). Novel correlations between the genotype and the phenotype of hypertrophic and dilated cardiomyopathy: results from the German Competence Network Heart Failure. Eur J Heart Fail 13, 1185–1192. [DOI] [PubMed] [Google Scholar]
- Walsh R, Thomson KL, Ware JS, Funke BH, Woodley J, McGuire KJ, Mazzarotto F, Blair E, Seller A, Taylor JC, et al. (2017). Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet Med 19, 192–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Hata Y, Hirono K, Takasaki A, Ozawa SW, Nakaoka H, Saito K, Miyao N, Okabe M, Ibuki K, et al. (2017). A Wide and Specific Spectrum of Genetic Variants and Genotype-Phenotype Correlations Revealed by Next-Generation Sequencing in Patients with Left Ventricular Noncompaction. J Am Heart Assoc 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang Y, Zou Y, Sun K, Wang Z, Ding H, Yuan J, Wei W, Hou Q, Wang H, et al. (2014). Malignant effects of multiple rare variants in sarcomere genes on the prognosis of patients with hypertrophic cardiomyopathy. Eur J Heart Fail 16, 950–957. [DOI] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. (2018). SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46, W296–W303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo A, Rakowski H, Liew JC, Zhao MS, Liew CC, Parker TG, Zeller M, Wigle ED, and Sole MJ (2003). Mutations of the beta myosin heavy chain gene in hypertrophic cardiomyopathy: critical functional sites determine prognosis. Heart 89, 1179–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yengo CM, Takagi Y, and Sellers JR (2012). Temperature dependent measurements reveal similarities between muscle and non-muscle myosin motility. J Muscle Res Cell Motil 33, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Hirono K, Nakamura K, Suzuki T, Hata Y, and Nishida N (2016). A novel ACTC1 mutation in a young boy with left ventricular noncompaction and arrhythmias. HeartRhythm Case Rep 2, 92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman RS, Cox S, Lakdawala NK, Cirino A, Mancini-DiNardo D, Clark E, Leon A, Duffy E, White E, Baxter S, et al. (2010). A novel custom resequencing array for dilated cardiomyopathy. Genet Med 12, 268–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou Y, Wang J, Liu X, Wang Y, Chen Y, Sun K, Gao S, Zhang C, Wang Z, Zhang Y, et al. (2013). Multiple gene mutations, not the type of mutation, are the modifier of left ventricle hypertrophy in patients with hypertrophic cardiomyopathy. Mol Biol Rep 40, 3969–3976. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic has been deposited at the Protein Data Bank (www.rcsb.org) with accession code 7JH7. The corresponding cryo-EM map was deposited in the Electron Microscopy Data Bank (www.ebi.ac.uk/pdbe/emdb) with accession code EMD-22335.