

SUMMARY
The repeating structural unit of metazoan chromatin is the chromatosome, a nucleosome bound to a linker histone, H1. There are 11 human H1 isoforms with diverse cellular functions, but how they interact with the nucleosome remains elusive. Here, we determined the cryoelectron microscopy (cryo-EM) structures of chromatosomes containing 197 bp DNA and three different human H1 isoforms, respectively. The globular domains of all three H1 isoforms bound to the nucleosome dyad. However, the flanking/linker DNAs displayed substantial distinct dynamic conformations. Nuclear magnetic resonance (NMR) and H1 tail-swapping cryo-EM experiments revealed that the C-terminal tails of the H1 isoforms mainly controlled the flanking DNA orientations. We also observed partial ordering of the core histone H2A C- and H3 N-tails in the chromatosomes. Our results provide insights into the structures and dynamics of the chromatosomes and have implications for the structure and function of chromatin.
Graphical Abstract
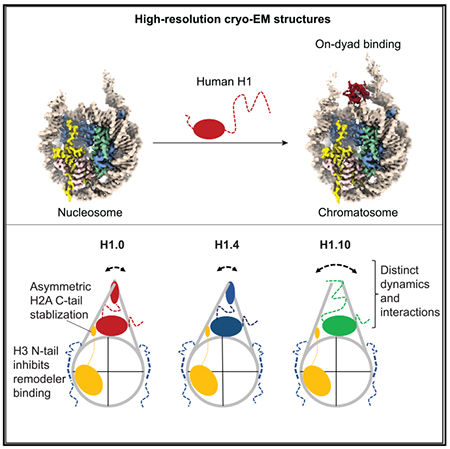
In Brief
Linker histones bind to the nucleosome to form chromatosomes. Zhou et al. determined the cryo-EM structures of the chromatosomes containing human H1.0, H1.4, and H1.10 isoforms, respectively, and investigated their dynamics using nuclear magnetic resonance and molecular dynamics simulation. The study reveals distinct structural and dynamic features of the chromatosomes.
INTRODUCTION
Eukaryotic genomic DNA is packaged into chromatin through association with core and linker histones to form nucleosomes and chromatosomes (Kornberg, 1974; Kornberg and Thomas, 1974; Olins and Olins, 1974). The canonical nucleosome core particle comprises an octamer of core histones with two copies of H2A, H2B, H3, and H4 (Arents et al., 1991), around which ~145–147 bp DNA winds in a left-handed manner (Luger et al., 1997). The chromatosome core particle consists of the nucleosome with ~168 bp DNA bound to a linker histone, H1 (Simpson, 1978). The chromatosome is the predominant repeating structural unit of metazoan chromatin because of the existence of an abundance of linker histones in the nucleus (Woodcock et al., 2006). As chromatin factors, linker histones play important roles in many cellular functions (Fyodorov et al., 2018; Hergeth and Schneider, 2015), including gene expression (Fan et al., 2005; Shen and Gorovsky, 1996), mitotic chromosome architecture and segregation (Maresca et al., 2005), muscle differentiation (Lee et al., 2004), embryonic stem cell differentiation (Zhang et al., 2012), the genetic activity of heterochromatin (Lu et al., 2013), cell pluripotency (Christophorou et al., 2014), and cancer cell heterogeneity (Torres et al., 2016).
Similar to core histones, linker histones have many isoforms (subtypes or variants). In humans, there are 11 isoforms of linker histones; 7 of them are expressed in somatic cells, and 4 are expressed in germ cells (Happel and Doenecke, 2009). The somatic H1s have a conserved tripartite structure consisting of a short flexible N-terminal tail, a central globular domain, and a long intrinsically disordered highly basic C-terminal tail. In contrast, the linker histones in the germ cells do not have the corresponding globular domain region. In general, the short N-terminal tail of linker histones contributes little to nucleosome binding. The middle globular domain preferentially binds to the nucleosome core and linker DNAs (Allan et al., 1980). The long C-terminal tail is important for higher-affinity binding of linker histones to the nucleosome (Allan et al., 1980; Singer and Singer, 1976), folding of nucleosome arrays (Allan et al., 1986), and association of linker histones with chromatin in vivo (Caterino and Hayes, 2011; Fang et al., 2012; Lu and Hansen, 2004). In contrast to the many high-resolution structures of the nucleosome core particles containing either canonical or variant core histones, only the crystal structure of the chromatosome core particle consisting of 167 bp of DNA and the globular domain of chicken H5 (cH5 or cH1.0) has been determined at near-atomic resolution (3.5 Å). Attempts to determine the structures of the chromatosomes consisting of longer linker DNA and full-length linker histones have only reached to 5.5 Å resolution in the case of the Xenopus H1.0 chromatosome by X-ray crystallography and single-particle cryoelectron microscopy (cryo-EM) methods (Bednar et al., 2017). Recent discoveries that expression levels of human H1.0 are associated with tumor cell heterogeneity (Torres et al., 2016) and mutations in human H1.4 can lead to autism and premature aging (Duffney et al., 2018; Flex et al., 2019) underscore the need for high-resolution structures of chromatosomes for a better understanding of how linker histones interact with the nucleosome.
It is difficult to obtain high-resolution structures of chromatosomes by cryo-EM, because linker histones tend to dissociate from the nucleosome in the thin vitreous ice. We have previously shown that a single-chain antibody fragment (scFv) that binds to the core histones can prevent the nucleosome from dissociating during the blotting-freezing process of sample preparation for single-particle cryo-EM imaging, allowing determination of the nucleosome structures with native DNA sequence to atomic resolution (Zhou et al., 2019). Here, we applied this antibody approach to study the structures of chromatosomes prepared with the full-length human linker histone isoforms H1.0 (H1°), H1.4 (H1e), and H1.10 (H1x). The rationale for choosing these three linker histones is that the sequences of the globular domains of these three isoforms are less conserved, whereas the sequences of H1.1–H1.5 are highly conserved. In addition, H1.0 has been shown to bind to the nucleosome on the dyad (Zhou et al., 2016), but H1.4 is reported to bind to the nucleosome off the dyad in reconstituted nucleosome arrays (Song et al., 2014).
We found that the scFv can indeed help prevent the dissociation of linker histones from the nucleosome in the thin vitreous ice, which allows us to solve the structures of the chromatosomes at 2.7–3.1 Å resolutions. We further used solution nuclear magnetic resonance (NMR) to investigate the dynamic roles of linker histone tails in the chromatosomes. Our results show that the globular domains of all three H1 isoforms bind to the nucleosome dyad region, while the C-terminal tails of H1 isoforms interact with the linker DNA dynamically and play a major role in controlling the linker DNA conformations. Also, we found that the binding of the H1 isoforms to the nucleosome altered the conformations of the C-terminal tails of core histone H2A and the N-terminal tails of H3, the latter lead to inhibition of chromatin remodeler binding.
RESULTS
Overall Structures of the Chromatosomes and the Free Nucleosome
To determine the structures of chromatosomes containing linker histone isoforms H1.0, H1.4, or H1.10, we reconstituted the chromatosomes using the linker histones, 197 bp DNA centered with Widom “601” DNA (W601), and human core histones (see STAR Methods). We performed single-particle cryo-EM experiments on these chromatosomes bound to two scFvs. We obtained the three-dimensional (3D) reconstruction density maps of the chromatosomes at resolutions from ~2.8 Å to 3.1 Å (Figures 1 and S1; Video S1; Table 1). These density maps allow us to build structural models with the globular domains of the linker histones and nucleosomes. Overall, the chromatosome structures showed that the globular domains of all three linker histones bound to the nucleosome on the dyad. One linker/flanking DNA, linker-α3, has closer contacts with the α3 helix of the globular domain, while the other linker/flanking DNA, linker-L1, has loose contacts with the L1 loop of the globular domain. The linker DNAs in the H1.10 chromatosome are more open than those in the H1.0 and H1.4 chromatosomes. To understand how linker histones control linker DNA conformations, we also determined the cryo-EM structure of the free nucleosome at 2.8 Å resolution (Figure 1), which showed more open conformation of the flanking DNAs. The numbers of base pairs of the two flanking DNAs that can ft into the density after low-pass filtered to 6.0 Å resolution are (11 and 20), (21 and 24), (13 and 17), and (13 and 17) for the chromatosomes containing H1.0, H1.4, H1.10, and the free nucleosomes, respectively.
Figure 1. Overall Density Maps and Structural Models.

(A) Top views of the cryo-EM reconstructions of the H1.0, H1.4, and H1.10 chromatosomes and the free nucleosome.
(B) Top views of the low-pass-filtered (6 Å) density maps (transparent surfaces) as in (A) and corresponding atomic structural models.
(C) Side views as in (A).
(D) Side views as in (B).
See also Figures S1 and S2.
Table 1.
Cryo-EM Data Collection, Refinement, and Validation Statistics
| H1.0 Chromatosome EMDB: EMD-22683, PDB: 7K5X | H1.4 Chromatosome, EMDB: EMD-22684, PDB: 7K5Y | H1.10 Chromatosome, EMDB: EMD-22685, PDB: 7K60 | Free Nucleosome, EMDB: EMD-22686, PDB: 7K61 | gH1.10-ncH1.4 Chromatosome, EMDB: EMD-22687, PDB: 7K63 | H1.4 Di-chromatosome, EMDB: EMD-22688 | |
|---|---|---|---|---|---|---|
| Data Collection and Processing | ||||||
| Magnification | 29,000 | 29,000 | 18,000 | 18,000 | 18,000 | 18,000 |
| Voltage (kV) | 300 | 300 | 300 | 300 | 300 | 300 |
| Electron exposure (e−/Å2) | 40 | 40 | 43 | 43 | 43 | 43 |
| Defocus range (μm) | −1 to −2.5 | −1 to −2.5 | −1.3 to −2.0 | −1.3 to −2.0 | −1.3 to −2.0 | −1.3 to −2.0 |
| Pixel size (Å) | 0.429 | 0.858 | 0.679 | 0.679 | 0.679 | 0.679 |
| Symmetry imposed | C1 | C1 | C1 | C1 | C1 | C1 |
| Initial particle images (no.) | 275,297 | 237,175 | 618,685 | 351,308 | 510,136 | 25,551 |
| Final particle images (no.) | 133,758 | 112,385 | 199,685 | 194,456 | 150,168 | 10,876 |
| Map resolution (Å) FSC threshold | 2.93 (0.143) | 2.76 (0.143) | 3.12 (0.143) | 2.85 (0.143) | 3.03 (0.143) | 9.7 (0.143) |
| Refinement | ||||||
| Initial model used (PDB code) | 4QLC, 6DZT, 5NL0 5Y0C | 4QLC, 6DZT, 5NL05Y0C | 2LSO, 6DZT, 5NL0 5Y0C | 6DZT, 5NL0, 5Y0C | 2LSO, 6DZT, 5NL0 5Y0C | |
| Model resolution (Å) FSC threshold | 3.04, (0.5) | 2.86, (0.5) | 3.21, (0.5) | 2.88, (0.5) | 3.16, (0.5) | |
| Model resolution range (Å) | 2.7–30 | 2.1–30 | 2.7–30 | 2.2–30 | 2.7–30 | |
| Map sharpening B factor (Å2) | −54 | −25 | −40 | −12 | −68 | |
| Model Composition | ||||||
| Non-hydrogen atoms | 18295 | 18263 | 18334 | 17727 | 18334 | |
| Protein residues | 1296 | 1297 | 1298 | 1222 | 1298 | |
| Nucleotides | 394 | 394 | 394 | 394 | 394 | |
| Ligands | NA | NA | NA | NA | NA | |
| B Factors (Å2) | ||||||
| Protein | 50.70 | 48.01 | 37.86 | 54.73 | 40.98 | |
| Nucleotide | 128.98 | 112.36 | 103 | 190.54 | 108.49 | |
| Ligands | NA | NA | NA | NA | NA | |
| RMSDs | ||||||
| Bond lengths (Å2) | 0.004 | 0.004 | 0.004 | 0.007 | 0.004 | |
| Bond angles (°) | 0.595 | 0.638 | 0.634 | 0.659 | 0.612 | |
| Validation | ||||||
| MolProbity score | 1.19 | 1.15 | 1.33 | 1.21 | 1.22 | |
| Clashscore | 3.55 | 3.58 | 5.86 | 4.31 | 4.47 | |
| Poor rotamers (%) | 0.09 | 0.00 | 0.09 | 0 | 0.09 | |
| Ramachandran Plot | ||||||
| Favored (%) | 97.80 | 98.58 | 97.96 | 98.50 | 98.03 | |
| Allowed (%) | 2.20 | 1.42 | 2.04 | 1.50 | 1.97 | |
| Disallowed (%) | 0 | 0 | 0 | 0 | 0 |
FSC, Fourier shell correlation; RMSD, root-mean-square deviation.
The reconstruction maps of the chromatosome containing H1.0 and H1.4 are of higher quality, allowing assignment of the side chains of some residues in the globular domains (local resolutions in 3.0–4.5 Å range) at their interfaces with the dyad and the linker-α3 DNA (Figures 2A–2C; Videos S2 and S3). The relatively lower-quality map of the chromatosome containing H1.10 is primarily caused by the preferred orientations of the chromatosome particles (Figure S2). Nevertheless, we also obtained a high-quality reconstruction map of the chromatosome containing a hybrid linker histone, including the tails of H1.4 and the globular domain of H1.10. In this structure, some of the side chains of the H1.10 globular domain residues could also be determined (see later). Alignment of the core histones structures in the chromatosomes showed that the globular domain of H1.0 displayed a noticeable shift in comparison with those of the H1.4 and H1.10, even though the structures of the H1 globular domains were similar (Figure 2D). The larger side chain of Gln47 in H1.0 that interacted with the dyad DNA likely led to the observed difference, as the corresponding residues in H1.4 and H1.10 were the smaller Ala residues (Figure 2A). The 2.76 Å resolution density map of the H1.4 chromatosome also allowed us to define the DNA sequence in the nucleosome core region (Figure 2E) and, therefore, the DNA sequences of the two linker DNAs. In contrast, the cryo-EM densities of the W601 DNA in the nucleosome represent the two positions’ average from opposite DNA orientations (Figure 2E). We found that in the H1.4 chromatosome, the linker-α3 contains more AT pairs than the linker-L1 and the Arg78 residue in the α3 helix insert into the minor groove consisting of more AT pairs (Figure 2F).
Figure 2. Interactions between the Globular Domains and DNA.
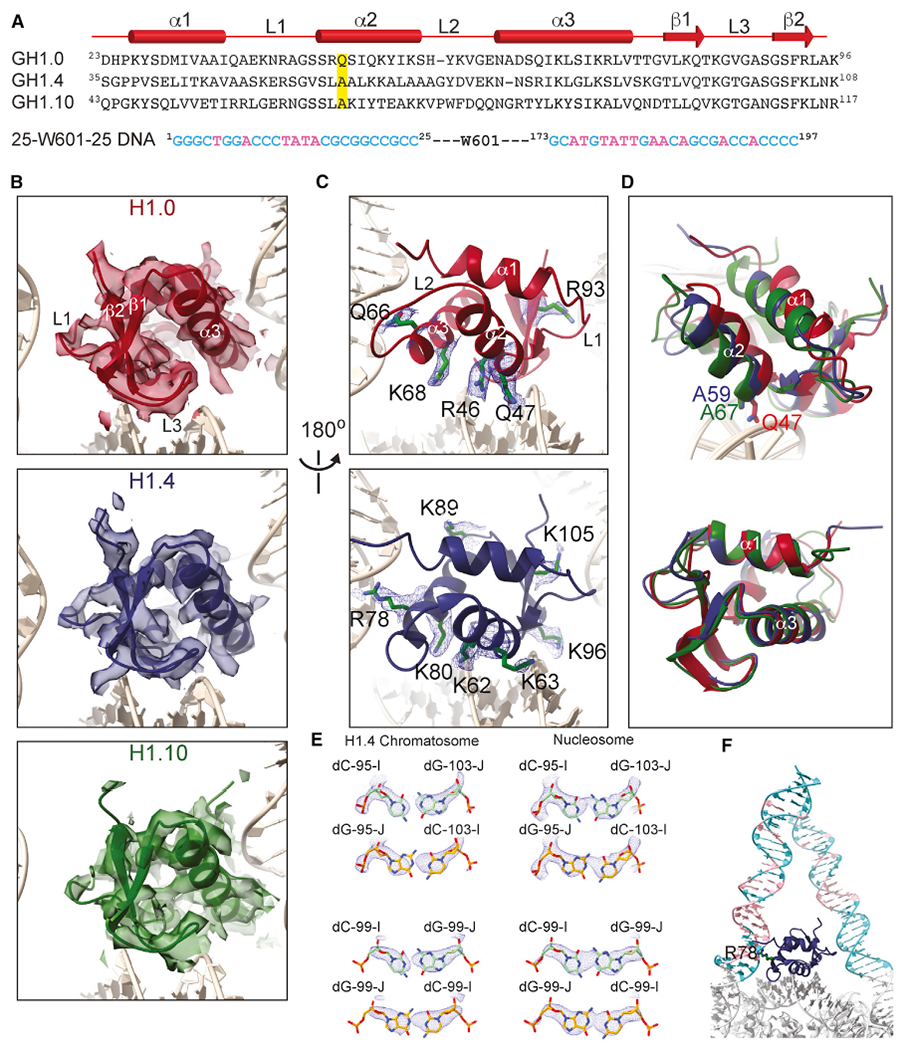
(A) Sequences of the globular domains of H1.0, H1.4, H1.10, and linker DNA. The diagram on the top shows the secondary structures. The residue Gln47 in H1.0 and corresponding Ala residues in H1.4 and H1.10, which are highlighted, interact with dyad DNA.
(B) Density maps (transparent surfaces) and cartoon structure models of the globular domains.
(C) Densities of the amino acid side chains of the globular domains that interact with the nucleosomal and linker DNAs.
(D) Illustration of the difference of the orientation of the globular domain of H1.0 relative to those of H1.4 and H1.10 in the chromatosomes and the residues that are likely responsible for the difference. Structures of the chromatosomes were aligned on core histones in the top panel. The bottom panel showed the alignment of the globular domain structures alone.
(E) Illustration of DNA orientation determination in the H1.4 chromatosome by the fitting of cryo-EM densities with DC-95:DG-103 and DC-99:DG-99 pairs in one direction (carbon colored in green), which do not fit when the orientation of the DNA is reversed (carbon colored in gold) (left). In contrast, in the case of free nucleosome, the corresponding cryo-EM densities represent the average of the two positions from opposite DNA orientations (right).
(F) The AT-rich base pair region (pink color) in the linker/flanking DNA is bound by the α3 helix through residues Arg78 of H1.4.
See also Figures S1 and S2.
MD Simulations of Local Interactions
To complement the cryo-EM structures and gain insights into the local dynamic interactions between the linker histone globular domains and DNA, we conducted several 0.5-μs all-atom molecular dynamics (MD) simulations in explicit solvent for the three chromatosomes and the free nucleosome without core and linker histone tails. For all chromatosomes, the DNA linker that has close contacts with the α3 helix of the globular domain consists of the sequence with the higher AT-pair content compared to the linker that faces the loop 1 of H1. To measure the degree of flexibility and openness of the linker- and the nucleosomal DNA, we used RMSF (root mean square fluctuation) and the distance between the ends of two DNA linkers. We found that the linker DNAs had substantially larger RMSFs than the DNA in the nucleosome core region, even for the DNA region that had close contacts with the globular domain (Figures 3A and 3B). The linker DNA with high AT-pair content exhibited more pronounced flexibility compared to the lower AT-pair content strand in the free nucleosome. The overall distribution of the distances between the two DNA linker ends shifted toward smaller values in the order of free nucleosome, H1.10, H1.4, and H1.0 chromatosomes with broader distribution in the free nucleosome and the H1.10 chromatosome (Figure 3C). To evaluate the orientation of DNA linkers, we used two angles, α and β, with respect to the dyad principal axis to quantify the in- and out-of-nucleosomal-plane deviations, respectively (Figures 3D and S3) (Bednar et al., 2017). The distributions of the difference of the α and β values for each linker DNA showed that the linker-L1s of the free nucleosome and the H1.10 chromatosome are significantly more open than those of the H1.4 and H1.0 chromatosomes (Figure 3E), whereas the linker-α3 of the H1.4 chromatosome is significantly more closed than those in the others (Figure 3F). Notably, for the free nucleosome and the H1.10 chromatosome, the distributions showed multimodal shapes (Figures 3C, 3E, and 3F).
Figure 3. MD of the Chromatosomes.
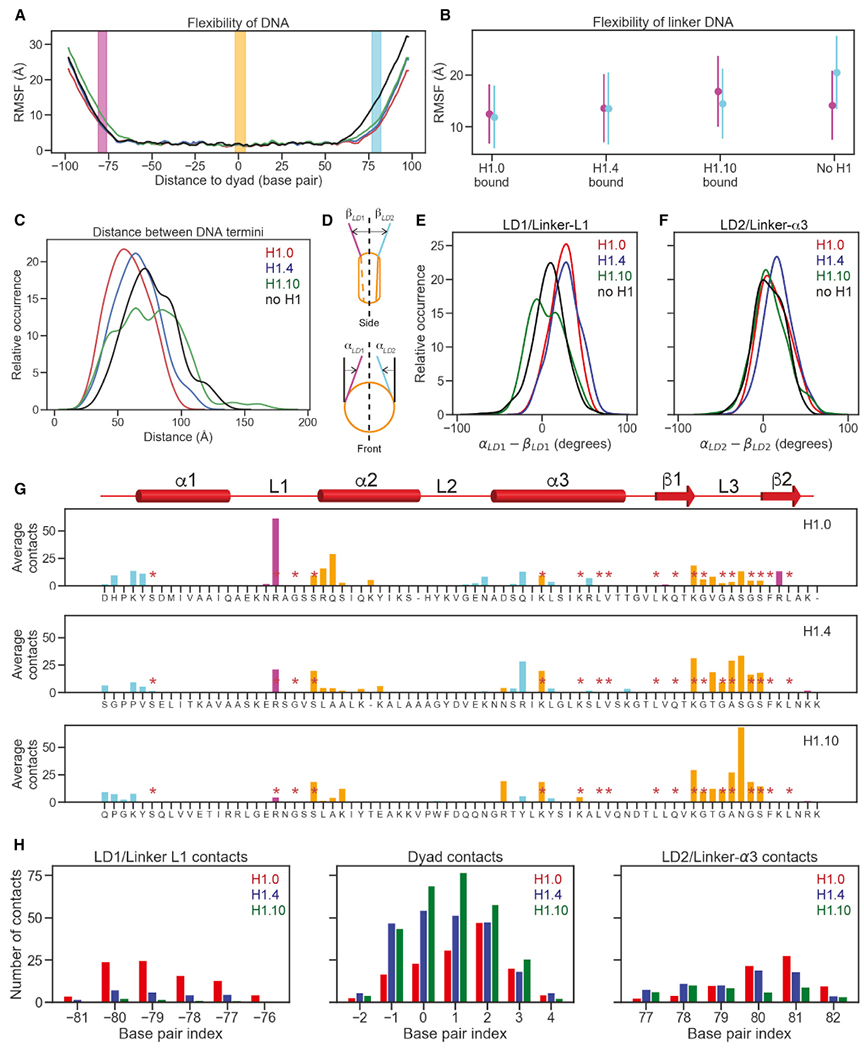
(A) Flexibility of DNA in the H1.0 (red), H1.4 (blue), H1.10 (green) chromatosomes, and free nucleosome (black) were illustrated via phosphate atom RMSFs over full molecular dynamics (MD) trajectories. Base pairs that contact globular domains are shown by DNA region: linker DNA 1 (LD1)/linker-L1 (magenta), dyad (orange), and linker DNA 2 (LD2)/linker-α3 (cyan). All panels follow this color convention. The dyad is numbered as 0.
(B) Mean and standard deviations of DNA RMSF averaged over base pairs.
(C) Distribution of distances between the two terminal base pairs of DNA.
(D) Cartoon illustrating the definition of linker strand opening angles, αLD1 and αLD2 for in-nucleosomal-plane motion (top), and βLD1 and βLD2 for out-of-nucleosomal-plane motion.
(E) Distributions of differences of linker strand angles, α and β for LD1/linker-L1. Higher values point to more compact structures.
(F) Distributions of differences of linker strand angles, α and β for LD2/ linker-α3. Higher values point to more compact structures
(G) The number of heavy-atom contacts between the globular domain and DNA averaged over MD trajectories. Residues conserved across all three H1 variants are annotated with red asterisks. Residues in contacts with LD1/linker-L1, dyad region, and LD2/linker-α3 are shown in purple, orange, and cyan, respectively.
(H) Average numbers of heavy-atom contacts for three DNA regions.
See also Figure S3.
Next, we calculated the average number of contacts of each globular domain residue of the linker histones with the DNA using the criterion that contact occurs if the distance between a heavy atom in the globular domain is within 5 Å of a heavy atom from DNA. We found that the majority of contacts occurred between the residues from the N-terminal region of the α2 helix and the C-terminal region of the L3 loop in the H1 globular domains and the DNA near the dyad (Figures 3G and 3H). This explains why the globular domain interface interacting with the dyad is highly conserved among most H1 isoforms from different organisms, as evident from the HistoneDB 2.0 database (Draizen et al., 2016). Linker-α3 forms more contacts with the residues in the α3 helix of the globular domains than those formed between linker-L1 with the L1 loop of the globular domains. However, there are significant variations in residue types and the number of contacts with the DNA among the three linker histone isoforms. For example, the conserved Arg in the L1 loop of H1.0 (Arg42) has a much larger average number of contacts than the corresponding residues in H1.4 (Arg53) and H1.10 (Arg62). In contrast, the C-terminal region of the globular domain of H1.0 has fewer contacts in comparison with those of H1.4 and H1.10. Thus, variant globular domains redistribute interactions with the nucleosomal and linker DNA differently while maintaining approximately the same overall number of interactions. Namely, H1.10 interacts mostly with the nucleosomal DNA near the dyad region, whereas H1.0 and H1.4 globular domains have more pronounced interactions with the linker strands. Also, upon nucleosome binding, H1.4 undergoes the most pronounced internal rearrangements, followed by H1.10 and then H1.0 (Figure S3).
Dynamic Conformation of Linker DNA and H1 Tails
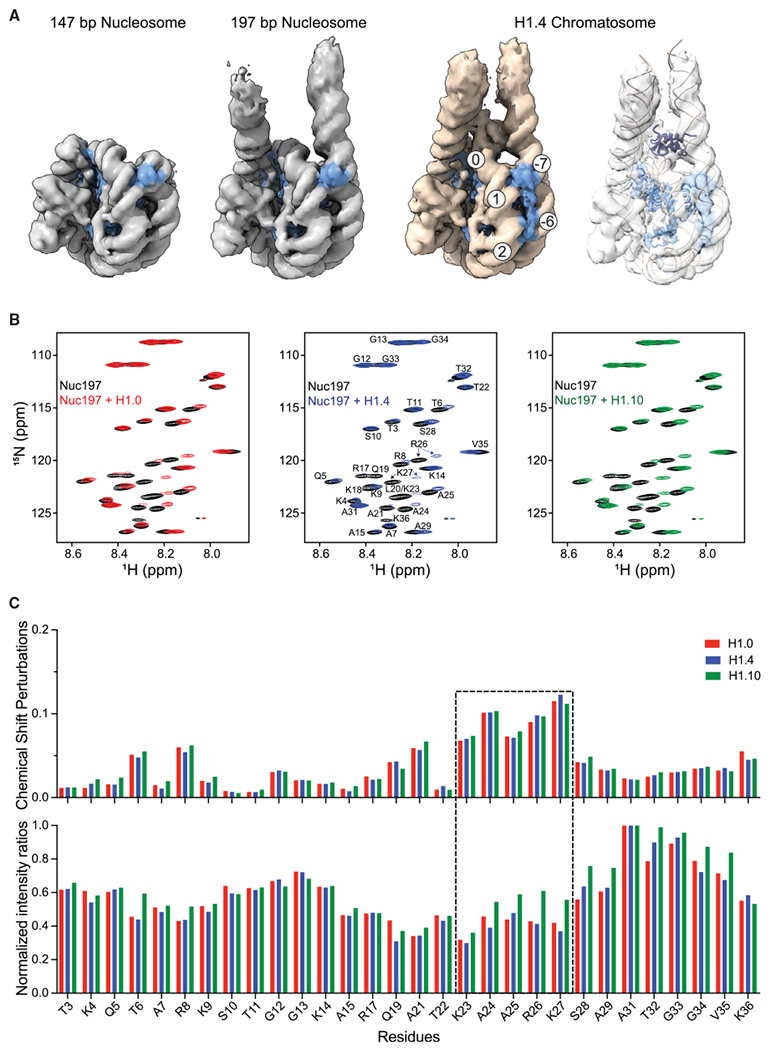
To explore the dynamic conformation of linker DNAs, we performed further 3D classification analysis for the free nucleosome and the H1.0, H1.4, and H1.0 chromatosomes using cryoSPARC (Figures 4 and S4). We identified two classes for the H1.0 and H1.4 chromatosomes and three classes for the free nucleosome. We found only one major class for the H1.10 chromatosome, which is likely due to the orientation preference of the particles (Figure S2). The differences in the structural models for the two classes of the H1.0 and H1.4 chromatosomes mainly occurred in the end regions of the linker DNA and were more pronounced for the linker-L1 DNA. We also observed densities between two DNA linkers in one class of the H1.0 chromatosome and in both classes of the H1.4 chromatosomes (Figures 4A and 4B). In the case of the free nucleosome and the H1.10 chromatosome, no such densities were observable (Figures 4C and 4D; Video S4).
Figure 4. Multiple Conformations and Interactions of Linker DNA.
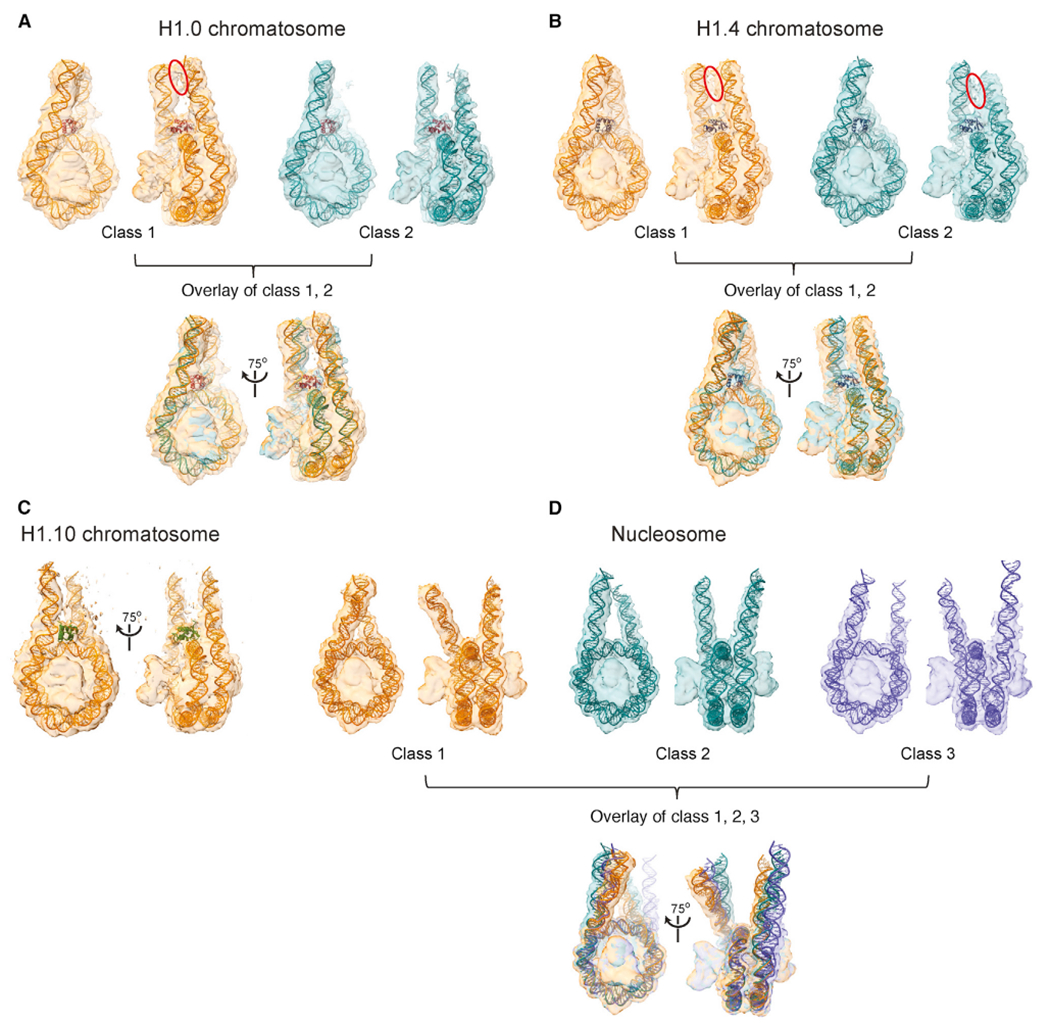
(A) 3D classification of the H1.0 chromatosome. The blue circle highlights the observation of densities between the two linker DNA.
(B) 3D classification of the H1.4 chromatosome. The blue circles highlight the observation of densities between the two linker DNA.
(C) The major class of the H1.10 chromatosome.
(D) 3D classification of the free nucleosome.
See also Figure S4.
To characterize the conformation of the H1 tails (Figure 5A), we performed NMR experiments on the chromatosomes using uniformly 15N- and 13C-labeled H1 isoforms. In comparison with the free H1s, all of the residues in the globular domains disappeared in the spectra as expected because of the large sizes of the chromatosomes and the folded structures of the globular domains (Figures 5B and S5A–S5C). In addition, some of the residues in the linker histone tails also disappeared or showed very weak intensities. We assigned chemical shifts of most of the observable peaks and calculated the deviations of Cα chemical shifts from random coil values, which are within the range from −1 to +1 (Figure 5C). These results indicate that the observable tail regions by NMR displayed a conformation close to random coils and form transient interactions with linker DNA. The observable tail regions include the N-terminal tails of all three linker histones and nearly all of the C-terminal tail residues of H1.10 and the regions immediately after the globular domains of H1.0 and H1.4. In contrast, the unobservable residues were mainly in the very C-terminal regions of the tails, including ~50 residues (Figure 5C), which are likely associated with the linker DNA most of the time, but dynamically.
Figure 5. Linker Histone Tails Control Linker DNA Orientation.
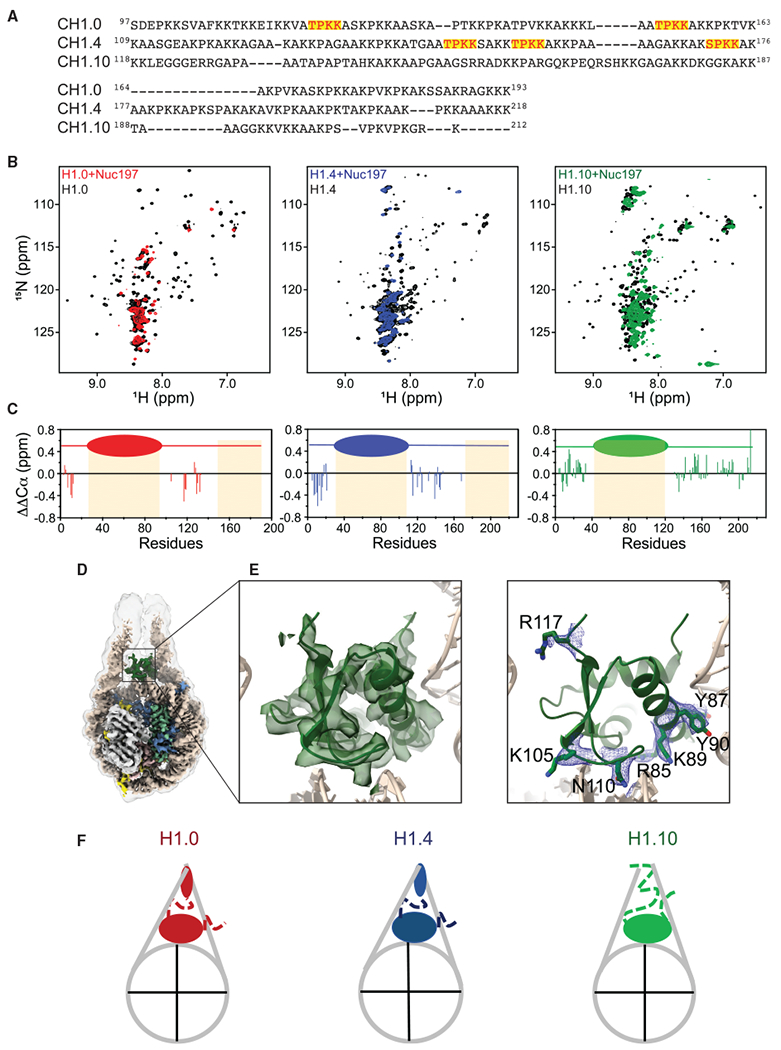
(A) Amino acid sequence alignment of the C-terminal tails of H1.0, H1.4, and H1.10. The S/TPKK motifs are highlighted in red with a yellow background.
(B) 1H-15N HSQC spectra of the H1 isoforms in the chromatosomes and the free form.
(C) Deviation of Cα chemical shifts from random coil values (ΔΔCα).
(D) Cryo-EM reconstruction of the gH1.10-ncH1.4 chromatosome. Low-pass-filtered (6 Å) cryo-EM density is shown with a transparent gray surface.
(E) Cryo-EM density fitted with the globular domain structural model. Density of the amino acid side chains of the globular domain that interact with the DNA.
(F) Cartoons of chromatosomes showing differences in linker DNA and H1 tails.
See also Figure S5.
To further test whether the linker histone tails play a predominant role in controlling the openness of DNA linkers, we made a chimeric linker histone that combines the globular domain of H1.10 with the tails of H1.4, termed gH1.10-ncH1.4. We determined the cryo-EM structure of the chromatosome containing the chimeric linker histone at an overall resolution of 3.0 Å (Figures 5D and S5E–S5H). Indeed, the two DNA linkers in this structure have more closed conformation. In this case, we also observed detailed interactions of the globular domain of H1.10 with the DNA (Figure 5E). In the low-pass-filtered maps, we also found the N-terminal loop of the H1.10 globular domain inserts between two linker DNA, which is opposite to that of H1.4 (Figures S5I–S5K). Overall, the NMR results are consistent with those of cryo-EM that the two DNA linkers in the H1.10 chromatosome have more open conformations than those in the H1.0 and H1.4 chromatosomes, and there are observable densities between the two DNA linkers in the H1.0 and H1.4 chromatosomes (Figures 4 and 5F; Video S4).
Conformations of H2A C- and H3 N-Tails in Chromatosomes
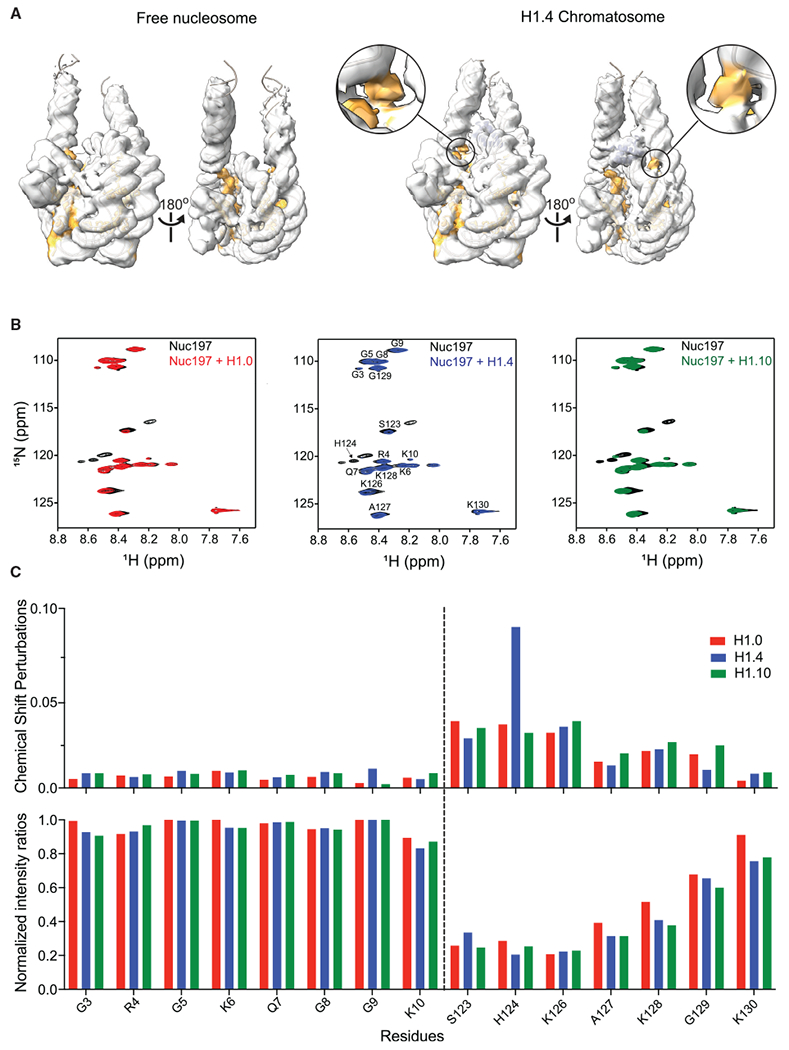
In the cryo-EM maps of the chromatosomes, we also observed additional densities between the DNA gyres of the super-helical location (SHL) 1 to 2 and −6 to −7 regions in comparison with the density map of the free nucleosome (Figure 6A; Video S5). Since the density is connected to the αN helix of H3, we speculated that it represented a region from the H3 tail. We could fit ~7 residues to the density after the original density map was low-pass filtered to 6 Å resolution. We performed NMR experiments using 15N- and 13C-labeled H3 in the nucleosomes and chromatosomes and assigned chemical shifts of the residues in the H3 tail. We found that a cluster of the residues (Lys23, Ala24, Ala25, Arg26, and Lys27) in the H3 N-tail showed large chemical shift changes and a decrease in peak intensities (Figures 6B and 6C), suggesting that these residues contributed to the densities observed between the two DNA gyres. Other residues (Thr6, Arg8, and Ala21) that also showed significant chemical shift changes and decreases in peak intensities may also make contributions to the observed density. Since SHL1-2 location is a part of the binding site of the ATPase domain of chromatin remodeler ISWI (Chittori et al., 2019; Yan et al., 2019), we reasoned that occupation of H3 histone tails at this location in the chromatosomes might inhibit remodeler binding. Indeed, electrophoresis mobility shift assay experiments revealed that the ATPase domain of the ISWI remodeler bound to the nucleosome specifically but to the chromatosomes nonspecifically (Figures S6A and S6B).
Figure 6. Effects of Linker Histone Binding on the Conformation of H3 Tails.
(A) Comparison of cryo-EM densities of the free nucleosome and the H1.4 chromatosome. The 147-bp nucleosome density map is from EMD-8938. All maps were low-pass filtered to 6 Å. The density maps were plotted at same intensity levels for core histones. H3 model and densities are zone colored in light blue. Extra densities between the two DNA gyres are colored in light blue, which is connected the H3 N-helix density. Numbers in the circles show the super-helical locations.
(B) 1H-15N spectra of H3 tails in the chromatosomes and free nucleosome.
(C) Chemical shift perturbations (upper panel) and NMR peak intensity changes (lower panel) for the residues in the H3 N-tails upon addition of H1.
See also Figure S6.
We also observed additional densities near linker-L1 and nucleosomal DNA when comparing the density maps of the free nucleosome and the H1.4 chromatosome (Figures 7A, S6C, and S6D; Video S5). Since the location of the density map is close to the structured C-terminal tail region of H2A, we speculated that the density represented a region in the C-terminal tail of H2A that is disordered in the free nucleosome. We could fit ~6 residues to the densities after the original density map was low-pass filtered to 6 Å resolution. Again, we performed NMR experiments on the nucleosome and chromatosomes using 15N- and 13C-labeled H2A and assigned the chemical shifts of the flexible residues in H2A (Figure 7B). We observed substantial changes in chemical shifts and peak intensities of the H2A C-terminal tail region between the nucleosome and the chromatosomes, whereas few changes occurred to the H2A N-terminal tail (Figure 7C).
Figure 7. Effects of Linker Histone Binding on the Conformation of H2A Tails.
(A) Comparison of the cryo-EM densities of the free nucleosome and the H1.4 chromatosome. All maps are low-pass filtered to 6 Å. The density maps were plotted at the same intensity levels for core histones. H2A model and density are zone colored in orange. Extra density is also colored in orange, which is closed to one of H2A C-terminal structured region.
(B) 1H-15N spectra of H2A tails in the chromatosomes and free nucleosome. The dashed lines separate the N-terminal (left) and C-terminal (right) tail regions.
(C) Chemical shift perturbations (upper panel) and NMR peak intensity changes (lower panel) for the residues in the H2A tails upon addition of H1.
See also Figure S6.
DISCUSSION
On-Dyad Binding by H1 Isoforms
In the cryo-EM structures of the chromatosomes consisting of human H1.0, H1.4, and H1.10, the globular domains of all three linker histones bind the nucleosome dyad. The globular domains mainly use the residues in the N-terminal region and the α3 helix to form close interactions with linker-α3 DNA and the residues in the L1 loop to make loose interactions with linker-L1 DNA. There are small differences in the orientations of the globular domains in the chromatosomes, which are caused by the detailed interactions between the globular domains and DNA. Our MD simulation studies showed that the globular domains of the linker histones and the associated linker DNA regions are highly dynamic in comparison with the nucleosome core region. The highly dynamic behavior of the globular domains in chromatosomes has also been shown in other computer simulation studies (Öztürk et al., 2016, 2018; Woods and Wereszczynski, 2020). These results are consistent with the observations from the fluorescence recovery after photobleaching (FRAP) experiments, which showed that linker histones have much shorter residence time than the core histones (Misteli et al., 2000), and the local resolution of the H1 globular domain is lower than that of the nucleosome core region (Figure S2).
In comparison with our H1.0 chromatosome cryo-EM structure, the previous crystal structures of the chromatosome core particle consisting of the globular domain of chicken H5 (cH1.0) (Zhou et al., 2015) and the chromatosome containing the full-length Xenopus H1.0 (xH1.0) (Bednar et al., 2017) showed slightly more closed linker DNA conformations (Figure S3C; Video S3), possibly influenced by molecular packing in the crystal that forces the linker DNA ends to form interactions with those of neighboring chromatosomes. Since the binding modes of linker histones are determined by the globular domains alone (Zhou et al., 2016) and human H1.1 to H1.5 isoforms have highly conserved sequences in the globular domain regions, our results strongly suggest that all human somatic linker histone isoforms bind to the nucleosome on the dyad region in mono-chromatosomes and open chromatin. Note that we previously found that Drosophila H1 binds to the nucleosome off the dyad (Zhou et al., 2013), and the globular domains of chicken H5/H1.0 and Drosophila H1 have different residues at five key positions that interact with DNA in the chromatosomes. When the five residues in the globular domain of H5/H1.0 (conserved between chicken and human) are substituted with the corresponding residues in Drosophila H1, the penta mutant (GH5pmut) binds to the nucleosome off the dyad (Zhou et al., 2016). Three of the five residues in human H1.0, H1.4, and H1.10 are largely conserved. It remains to be tested whether the corresponding five residues in H1.4and H1.10 play determinant roles in controlling their binding mode.
H1 C-Terminal Tails Control Openness of the Two DNA Linkers
Among the three chromatosome structures, the linker DNAs show different degrees of openness, which is roughly correlated with the numbers of linker histone C-terminal tail residues that are not observable by NMR. Our tail-swap cryo-EM experiment firmly demonstrates that it is the linker histone tails that play a predominant role in controlling the linker DNA closeness. Together with the observed densities between the two linker DNAs in the H1.0 and H1.4 chromatosomes, these results suggest that the very C-terminal tails form interactions with both linker DNAs simultaneously and bring them closer. Notably, the recent cryo-EM study has suggested that a C-terminal tail region of linker histone xH1.0 bind to the outer surface of linker-L1 DNA when using one structure to fit the density map (Bednar et al., 2017). In our study, however, we find that the density map of the human H1.0 chromatosome contains two different 3D classes; one class shows densities between the two linker DNAs, suggesting the H1.0 C-terminal tail forms interactions with both linker DNAs. It is still possible that the linker histone tails could also make transient and weaker interactions with the outer surface of the linker DNA. Notably, transient stabilizing interactions between the C-terminal tail of linker histone H1.0 and a 25-bp oligo DNA (Turner et al., 2018) and between the human linker histone H1.0 and its intrinsically disordered chaperone, ProTα (Borgia et al., 2018; Feng et al., 2018), have been observed.
The closeness of the two DNA linkers in the chromatosome is apparently correlated with the number of net positive charges in the C-terminal tails of the H1 isoforms, which are 40 for H1.0, 44 for H1.4, and 27 for H1.10, respectively. However, the recent cryo-EM study showed that the chromatosome containing H1.5ΔC50 (deletion of 50 residues at the C-terminal end), which reduces the number of net positive charges to 25, also displays a closed linker DNA conformation (Bednar et al., 2017), suggesting that the numbers of net positive charges are not the sole determinant of linker DNA closeness. Earlier hydroxyl radical footprinting studies have suggested that the T/SPKK motifs in the linker histone tails preferentially interact with DNA (Churchill and Suzuki, 1989). We found that the C-terminal tail regions of both H1.0 and H1.4 consist of two and three T/SPKK motifs (Figure 5A), respectively. Also, mutations of T/S to E in the three T/SPKK motifs in the C-terminal of H1.4 to mimic phosphorylation of T/S residue led to more observable NMR peaks (Figure S5D). Moreover, phosphorylation of linker histones is known to decrease their binding to DNA (Turner et al., 2018) and increase the activity of ATP-dependent chromatin remodeling enzymes on nucleosome arrays (Horn et al., 2002). In contrast, there are no such motifs in the C-terminal tail of H1.10. Interestingly, H1.5ΔC50 with more closed DNA ends in the chromatosome also consists of three T/SPKK motifs in the C-terminal tail. Moreover, our previous studies showed that most of the residues in the Drosophila H1 C-terminal tail, which does not include any T/SPKK motifs, are observable by NMR in the chromatosome (Zhou et al., 2013). It is likely that the number of T/SPKK motifs in the C-terminal tails of linker histones played an important role in controlling the linker DNA openness. Notably, frame-shift mutations in the C-terminal tail of H1.4, which lead to loss of the T/SPKK motifs and positively charged residues, have been linked to autism and premature aging (Duffney et al., 2018; Flex et al., 2019).
Linker DNA Sequence-Dependent Interactions
The two DNA linkers used in our studies have different DNA sequences. One linker has more AT pairs than the other. The density maps of the chromatosomes containing H1.4 and gH1.10-ncH1.4 are of sufficiently good quality to allow us to define the specific base pairs in the nucleosome core region and, therefore, the sequences in the two DNA linkers. In both cases, the globular domains use residues in the α3 helix residues to bind to the linker DNA with more AT pairs. For the H1.4 chromatosome, a possible interpretation of the binding preference is that the linker histone uses an Arg residue to interact with the minor groove of the linker DNA with more AT base pair (Figures 2C, 2E, and 2F), similar to the interactions between core histones and the DNA in the nucleosome core particles (Rohs et al., 2009). In the case of gH1.10-ncH1.4, the aromatic side chains of Tyr residues in the α3 helix interact with the linker DNA. The physical-chemical reason for this preferred interaction remains to be investigated in the future.
Implications of H1 Isoforms in Chromatin Structure, Dynamics, and Function
How linker histones condense chromatin to form higher-order structures remains a debated issue. Studies of chemically cross-linked nucleosome arrays or nucleosome arrays in crystals suggest the formation of 30-nm fibers with well-defined structures (Dorigo et al., 2004; Garcia-Saez et al., 2018; Song et al., 2014). However, it has been argued that nucleosome arrays condensed by linker histones in the absence of cross-linking are best described with an ensemble of dynamic structures (Zhou and Bai, 2019; Zhou et al., 2018), which is consistent with the observation that nucleosomes can pack in different ways in crystals or in oligomeric states without preference toward one specific way of packing (Bilokapic et al., 2018; Korzhnev et al., 2006). Our observation that chromatosomes can have multiple conformations in linker DNA further supports the ensemble view.
Also, the variation of linker DNA conformations in the chromatosomes containing various linker histone isoforms suggests that there are likely distinct structural regions in chromatin, which may be involved in different functions. The more closed linker DNA conformations in the H1.0 and H1.4 chromatosomes suggest that the two linker histones associate with the nucleosome more tightly than H1.10. Consistent with these results, FRAP studies have shown that linker histone H1.0 and H1.4 have much longer residence times (>20 s) on chromatin than H1.10 (~5 s) (Brown et al., 2006; Hergeth et al., 2011; Misteli et al., 2000; Takata et al., 2007). The stronger interactions of linker histones H1.0 and H1.4 with the linker DNA appears to be consistent with the known roles of H1.0 and H1.4 in gene transcription regulation. H1.0 inhibits the transcription of oncogenic effectors and self-renewal genes (Torres et al., 2016), and H1.4 is associated with the expression of genes that are involved in mental development (Duffney et al., 2018; Flex et al., 2019). In contrast, H1.10 is involved in mitotic progression (Takata et al., 2007).
In addition, our results that the disordered H3 N-terminal tails in the free nucleosome form interactions with their neighboring two DNA gyres in the chromatosome explain the earlier experimental results that binding of human linker histone H1.4 to the nucleosome leads to the decrease of NMR peak intensities of the residues in the H3 N-terminal tails and inhibition of their post-translational modifications (Stutzer et al., 2016). These results suggest that the N-terminal tails H3 have favorable interactions with flanking DNA in the absence of H1. In the presence of H1, the H1 competes with the N-terminal tails of H3 for binding of the flanking DNA, forcing them to interact with the nearby DNA in the nucleosome core region. The observation that the H3 N-terminal tail in the chromatosome inhibits the binding of the ATPase domain of chromatin remodeler is also consistent with the results that linker histones inhibit the activity of chromatin remodeling by the ATP-dependent chromatin remodeling enzymes (Horn et al., 2002). Moreover, our finding that one of the C-terminal tails of H2A binds to the DNA near the dyad in the chromatosome suggests that binding of linker histones to the nucleosome could break the symmetric conformation of H2A C-terminal tails in the same nucleosome, which has implications for their asymmetric post-translational modifications and gene transcription. The partial ordering of the H2Atail is likely due to the stabilization of the flanking DNA by H1.
Finally, ourcryo-EM structures of the chromatosomes provide the structural basis for understanding how post-translational modifications of linker histones may alter the structure and function of chromatin (Figure S7) Harshman et al., 2013; Hergeth and Schneider, 2015; Leidecker et al., 2016; Starkova et al., 2017). For example, the Arg53 residue in the globular domain of H1.2, conserved among H1.0-H1.5, interacts with the linker/flanking DNA in the chromatosome structure. Citrullination of Arg53 by the peptidyl arginine deiminase (PADI4) leads to the de-condensation of differentiated cell chromatin in the mouse myoblast nuclei, and an Arg53Ala mutant of H1.2 impairs its binding to the nucleosome (Christophorou et al., 2014).
Limitations of Study
Our studies of chromatosomes have several limitations. The structural models of the mono-chromatosomes may not fully represent the chromatosome in chromatin. The DNA used for reconstituting the chromatosome is non-native, and specific DNA sequences may affect the details of how linker histones bind to the nucleosome. Our MD simulation of chromatosomes does not consider histone tail effects, and simulation time is limited to sub-microseconds.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yawen Bai (baiyaw@mail.nih.gov).
Materials Availability
Unique and stable reagents generated in this study are available upon request.
Data and Code Availability
The accession numbers for the structures and the corresponding cryo-EM maps is PDB: 7K5Xand EMDB: EMD-22683 for the H1.0 chromatosome; PDB: 7K5Y and EMDB: EMD-22684 for the H1.4 chromatosome; PDB: 7K60 and EMDB: EMD-22685 for H1.10 chromatosome; PDB: 7K61 and EMDB: EMD-22686 for the nucleosome; PDB: 7K63 and EMDB: EMD-22687 for the gH1.10-ncH1.4 chromatosome. EMDB: EMD-22688forthe H1.4 Di-chromatosome. The raw 2D micrographs of all datasets will be uploaded to the EMPIAR database.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human core histone genes were gift from Dr. Hitoshi Kurumizaka, linker histone genes were purchased or synthesized. All genes were subcloned into expression plasmids and expressed in Escherichia coli.
METHOD DETAILS
Protein and DNA Preparation
Purification of linker histones
The gene for human H1.0 was obtained from Origene. The genes for human H1.4, and H1.10 were commercially synthesized (Bio Basic). These genes were subcloned into pET42b vector in frame with C-terminal hex-histidine tags. A chimeric linker histone gene containing H1.4(1–35)-H1.10(44–119)-H1.4(110–213), which replaces the globular domain gH1.4 in H1.4 with the globular domain gH1.10 in H1.10, was made using PCR, the HIFI DNA Assembly kit (NEB) and synthesized primers (IDT DNA). H1 mutants were made using PCR and Q5 mutagenesis kit (NEB). All genes were sequenced to ensure accuracy (Genewiz). Escherichia coli BL21-CodonPlus (DE3)-RIPL competent cells were transformed with individual linker histone plasmids and grown in 2XYT media at 37°C. When cell densities reached an OD600 of 0.8- 1.0, 0.5 mM IPTG was added to induce protein expression and the cells were grown further for another 3 hours. Cells were harvested by centrifugation and, when necessary, stored at −80°C until the next step. The cells from 1 l of growth medium were resuspended in 50 mL lysis buffer (50 mM NaH2PO4/Na2HPO4, pH 8.0, 750 mM NaCl, 10 mM imidazole, 4M guanidine hydrochloride) and lysed by sonication. The clarified lysate was added to 4 mL of Ni-NTA beads (QIAGEN) that had been equilibrated with wash buffer (50 mM NaH2PO4/Na2HPO4, pH 8.0, 750 mM NaCl, 10 mM imidazole, 6 M Urea). The beads were incubated with the lysate at 20°C for 45 min then loaded to a gravity flow column and washed with a 100 mL wash buffer. Proteins were eluted 6 times (8 mL each) with elution buffer (50 mM NaH2PO4/Na2HPO4, pH 8.0, 750 mM NaCl, 250 mM imidazole, 6 M Urea). After elution, 10 μL of each fraction was run on an SDS-PAGE gel. Fractions with linker histones were combined and dialyzed overnight at 4°C against the dialysis buffer (50 mM NaH2PO4/Na2HPO4, pH 8.0, 150 mM NaCl, 7 M Urea). The precipitate was removed by filtration through a 0.22 μm Millex syringe filter (Millipore). The sample was loaded onto a 5 mL Hitrap SP cation exchange column (GE Healthcare) equilibrated in dialysis buffer and eluted with a 150 mM - 1000 mM linear NaCl gradient of 20 column volumes. Peak fractions corresponding to linker histones were pooled and dialyzed overnight at 4°C against the storage buffer (20 mM Tris pH 8.0, 600 mM NaCl, 1 mM EDTA). Precipitates were removed by filtration through a 0.22 μm Millex syringe filter (Millipore) and protein were concentrated to ~1 mL using an Amicon Ultra spin concentrator (Millipore). Finally, the protein was purified using a Superdex 75 Increase 10/300 size exclusion chromatography column (GE Healthcare) that was pre-equilibrated with the storage buffer. Peak fractions were pooled and stored at −20°C until use.
To produce 15N/13C-labeled linker histones H1.0, H1.4 or H.10, Escherichia coli BL21-CodonPlus (DE3)-RIPL competent cells (Agilent) were transformed with corresponding plasmids and grown at 37°C in LB media to an OD600 of 1.0. The cells were pelleted by centrifugation, resuspended in fresh M9 media supplemented with 1g/L 15NH4Cl as the only nitrogen source and with 2g/L of 13C-glucose as the only carbon source. 1 mM IPTG was added to induce expression and the cells were grown at 37°C for 3-4 hours. Cells were harvested by centrifugation and isotope-labeled linker histones were purified using the same procedure as above.
Purification of core histone proteins
Human histone plasmids containing H2A, H2B, H3 and H4 were a generous gift from prof. Hitoshi Kurumizaka. To produce histones without extra tags or residues, H2A and H2B genes were amplified by PCR and cloned into pET42b vector using NdeI and BamHI restriction sites. H3 and H4 genes were amplified by PCR and cloned into pET21b vector using NdeI and BamHI restriction sites. Histone expression and purification were conducted following standard protocols (Dyer et al., 2004). We further purify all histones with an RP-protein column (YMC) using HPLC.
To produce 15N/13C labeled histone H2A or H3, Escherichia coli BL21-CodonPlus (DE3)-RIPL competent cells (Agilent) was transformed with the pET42b-H2A or pET21-H3 plasmid and grown at 37°C in LB media to an OD600 of 0.6, pelleted by centrifugation and then washed three times with Di-water. The cells were resuspended in fresh M9 media supplemented with 1g/L 15NH4Cl as the only nitrogen source and with 2g/L of 13C glucose as the only carbon source. 1 mM IPTG was added to induce expression and the cells were grown at 37°C for 3-4 hours. Cells were harvested by centrifugation and isotope-labeled histones were purified using the same procedure as nonlabelled histones.
Purification of 197bp and di-197 bp W601 DNA
197 bp Widom 601 DNA (W601) was amplified by PCR using one copy of 167bp W601 DNA as the template (the 12 × 167bp W601 plasmid was a gift from professor Timothy Richmond), with 147-bp W601 strong positioning DNA sequence (Dyer et al., 2004; Lowary and Widom, 1998) in the middle, and 25bp linker DNA at each end. To increase the yield of the 197bp W601 DNA, we generated a pUC19 plasmid harboring 16 copies of 197 bp W601 DNA sequence separated by SmaI (CCCGGG) cleavage sites (Wu et al., 2016). To generate di-197 bp W601 DNA, 197bp W601 DNA and pUC19 plasmid bearing one copy of 197bp W601 DNA were amplified separately by PCR. Both PCR products were purified and assembled into circular plasmid using NEB HIFI DNA Assembly kit (NEB), resulted in a pUC197 plasmid bearing two copy of 197 bp W601, separated by a ScaI (AGTACT) cleavage site (See nucleotide sequence below). Finally we produced a pUC19 plasmid harboring 8 copies of di-197 bp W601 DNA sequence separated by Smal (CCCGGG) cleavage sites (Wu et al., 2016). Plasmids bearing 16 copies of 197 bp or eight copies of di-197 bp W601 DNA were produced in Alpha-Select Chemically Competent cells (Bioline). Harvested cells were treated with lysis buffer (1% SDS + 0.2 M NaOH) and neutralization buffer (4 M potassium acetate, 2 M acetic acid). After centrifugation at 8000g for 30 min, the plasmids in the supernatant were precipitated, re-dissolved and further purified by phenol and chloroform extraction. 197 bp or di-197 bp W601 DNA was cleaved from the plasmid by SmaI (NEB) and separated from pUC plasmid backbone by PEG 6000 precipitation following the early protocol (Dyer et al., 2004). The nucleotide sequence of the 197bp W601 DNA is as follows (the W601 DNA is underlined): GGGCTGGACCCTATACGCGGCCGCCCTGGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCCTGTGCATGTATTGAACAGCGACCACCCC
The nucleotide sequence of the di-197bp W601 DNA is as the following (the W601 DNA is underlined): GGGCTGGACCCTATACGCGGCCGCCCTGGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCCTGTGCATGTATTGAACAGCGACCACAGTACTCTGGACCCTATACGCGGCCGCCCTGGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCCTGTGCATGTATTGAACAGCGACCACCCC
Purification of 237bp W601 DNA
237 bp W601 DNA was amplified by PCR using one copy of 197bp W601 DNA as the template and 2x LiTaq PCR Master Mix (LifeSct). DNA products from 96-well PCR plate were precipitated with 70% ethanol, redissolved in 1 mLTE buffer (pH 8.0), injected into a 4 mL POROS-HQ 50 μm column (Thermo Fisher Scientific). After washing with 5mM Tri-HCl, pH 8.0 buffer without salt, 237 W601 DNA was eluted by a linear 0–2 M NaCl gradient in 5 mM Tris-HCl, pH 8.0. The nucleotide sequence of the 237 bp W601 DNA is as the following (the W601 DNA is underlined): GCATCCCTTATGTGAGGTACGGGCTGGACCCTATACGCGGCCGCCCTGGAGAATCCCGGTGCCGAGGCCGCTCAATTGGTCGTAGACAGCTCTAGCACCGCTTAAACGCACGTACGCGCTGTCCCCCGCGTTTTAACCGCCAAGGGGATTACTCCCTAGTCTCCAGGCACGTGTCAGATATATACATCCTGTGCATGTATTGAACAGCGACCACCCCGCGTTTAAACCGGTGCCAGT
Purification of scFv of PL2-6 antibody
Using our previous scFv plasmid as the template (Zhou et al., 2019), the DNA sequence encoding the mouse mAb PL2-6 antibody light chain variable region (GenBank id: X60341), heavy chain variable region (GenBank id: X60334) and a linker with four repeats of GGGGS was amplified by PCR using NEB HIFI DNA Assembly kit (NEB). The DNA sequence was subcloned into pET15b plasmid. Purification of scFv using the same protocol described in our early study (Zhou et al., 2019).
Purification of ctISWI
To produce the full-length ctISWI and its fragment containing the ATPase domain (Chittori et al., 2019), Escherichia coli Rosetta(DE3) pLysS cells (Agilent) were transformed with pProEX-Htb-ctISWI and pProEX-Htb-ctISWI77-Δ-722 plasmid, respectively, grown in 1x LB Broth (IPM Scientific) with 100 μg/ml Ampicillin at 37°C while shaking. When the cell density reached an OD600 of ~1.0, the growth temperature was reduced to 14°C. 0.15-0.20 M NaCl was added into the cell culture and recombinant protein expression was induced by adding 0.3 mM IPTG for 18-20 h. Cells were harvested by centrifugation and the cell pellet was washed twice with 40 mL wash buffer containing 20 mM Tris–HCl pH 8.0, 500 mM NaCl and 2 mM β-ME. Cells were resuspended in 40 mL binding buffer containing 20 mM Tris–HCl pH 8.0, 1 M NaCl, 20 mM imidazole, 5 mM β-ME, 1 mM phenylmethylsulfonyl fluoride (PMSF), 1 U ml−1 Benzonase Nuclease (Sigma-Aldrich) and 10 μg ml−1 Ribonuclease A (Sigma-Aldrich), followed by sonication on ice for a total of 10 min with a pulse of 5 son, and 10 s off and 15% force. Soluble protein was separated from cell debris by centrifugation at a speed of 35,000 RPM for 2 h and at 4°C. The protein was first purified using Ni-NTA agarose (QIAGEN) following the protocol provided by the manufacturer and further purified by one round of size-exclusion chromatography (SEC) using S200 increase 10 300GL (GE Healthcare) column pre-equilibrated in a buffer containing 20 mM HEPES pH 7.5, 200 mM NaCl, and 5 mM β-ME. Peak fractions containing the full-length ctISWI or ctISWI77-Δ-722 were pooled and stored at 4°C.
Reconstitution of the Mono- and Di-nucleosome
The mono-nucleosome containing unlabeled human core histones, 197 bp or 237 bp W601 DNA was reconstituted as described previously (Dyer et al., 2004). Briefly, histone octamers were prepared from individual histone proteins by refolding and salt dialysis. Histone octamers were isolated from H2A/H2B dimers and H3/H4 tetramers by size exclusion chromatography, and concentrated using a 30 kDa cut-off Amicon Ultra spin concentrator (Millipore). Mono-nucleosomes were prepared by mixing histone octamers and purified 601 DNA under high salt, followed by salt gradient dialysis over 24 hours. The di-nucleosome were reconstituted following the same protocol except the salt gradient dialysis step took 48 hours. Fully formed mono-nucleosomes and di-nucleosomes were separated from misfolded nucleosomes using TSK DEAE-5PW HPLC column (TOSOH). Nucleosomes containing 15N/13C-labeled H2A or 15N/13C-labeled H3 were reconstituted the same way as the nucleosome containing unlabeled histones.
Reconstitution of Chromatosomes
The ratio of linker histone to nucleosome was determined by titration of individual linker histones with nucleosomes and checked on native PAGE gel (Figure S1A). To prevent sample from aggregation, the linker histone was mixed with chaperone ProTa in 1:1 ratio at a concentration of 10 μM or less. Then the linker histone was mixed with a nucleosome at 1:1 ratio in the buffer (10 mM Tris–HCl pH8.0, 1mM EDTA and 10mM NaCl). Chromatosomes were purified from extra ProTa by size-exclusion chromatography (SEC) using Superose 6 10 300 GL column (GE Healthcare) pre-equilibrated with the same buffer. Di-chromatosomes (di-nucleosome and linker histones) were reconstituted the same way as the mono-chromatosome.
AUC analysis
The solution conditions used for sedimentation experiments with H1.0/H1.4/H1.10 chromatosomes (Figure S1B) were 10 mM Tris–HCl pH8.0, 1mM EDTA, 10 mM NaCl and 0.5 mM TCEP. Sedimentation velocity data were collected at 20°C by centrifugation at 35,000 rpm in a Beckman Optima XL-A analytical ultracentrifuge following standard protocols (Zhao et al., 2013). The absorbance was monitored at 260 or 280 nm. Data were analyzed in SEDFIT(Schuck, 2000) in terms of a continuous c(s) distribution spanning a sedimentation coefficient range of 0 to 20 S with a resolution of 200 points and a confidence interval of 68%. The sedimentation coefficient reported was the value at the peak maximum. A partial specific volume of 0.65 cm3/g was used (Ausio et al., 1984). Solution densities and viscosities were calculated in SEDNTERP (Cole et al., 2008), and sedimentation coefficients were corrected to standard conditions s20,w.
Cryo-EM Structure Determination
Sample preparation
Each chromatosome was mixed with 3x scFv and concentrated using an Amicon Ultra spin concentrator (Millipore) to around 550 ng/μl (4.5 μM) based on the OD260 value. Quantifoil 1.2/1.3 holy carbon copper grids (Electron Microscopy Sciences) were pretreated using an easiGlow Glow Discharge Cleaning System (PELCO) at 25 mA for 15 s. 3 ul of each sample was vitrified on the glow discharged grids using a Vitrobot Mark IV (Thermo Fisher Scientific) operated at 4°C, 100% humidity with blotting force of 3 and 3 - 4 s blot time.
Cryo-EM Data Collections and Processing
All data were collected on a Titan Krios (Thermo Fisher Scientific) at 300 kV with a K2 Summit direct electron detector (Gatan). For the H1.0 chromatosome datasets, data were collected in super-resolution mode at a nominal magnification of 29,000x and a pixel size of 0.429 Å with a nominal dose of 40 e-/Å2 over 38 frames. For the H1.4 chromatosome dataset, data were collected in counting-mode at a nominal magnification of 29,000x and a pixel size of 0.858 Å with a nominal dose of 40 e-/Å2 over 76 frames. Datasets with 1,387 and 1229 movies were collected for the H1.0 and H1.4 chromatosomes, respectively. For free nucleosome, H1.10 chromatosomes, gH1.10-ncH1.4 chromatosomes, and H1.4 di-chromatosome, data were collected in super-resolution counting-mode at a nominal magnification of 18,000xand a pixel size of 0.679 Å with a nominal dose of 43 e-/Å2 over 40 frames. Datasets of 1985, 761, 1425 and 685 movies were collected for the free nucleosome, H1.10 chromatosome, gH1.4-ncH1.4 chromatosome, and H1.4 di-chromatosome respectively. The dataset for the free nucleosome was collected with 3x3 image shift using SerialEM v3.7 (Mastronarde, 2005).
For all datasets, movies were motion-corrected and dose-weighted using MotionCorr2 (Zheng et al., 2017) wrapped in Relion 3.0 (Zivanov et al., 2018). Contrast transfer function (CTF) correction was performed with Ctffind4 (Rohou and Grigorieff, 2015). Particle picking utilizing the positive-unlabeled convolutional neural networks of Topaz (Bepler et al., 2019). Particle coordinates labeled by Topaz were imported into Relion for image extraction. Extracted particles were imported into cryoSPARC (Punjani et al., 2017) and subjected to two rounds of 2D classification to remove junk particles. Cleaned particles were used for cryoSPARC Ab-initio reconstruction with 3 ~4 classes, followed by one to three rounds of heterogenous refinement to further clean junk particles and select the class with the best linker histone density. A homogeneous and non-uniform refinement was performed. The resolution was obtained at the Fourier shell correlation (FSC) value of 0.143 (Rosenthal and Henderson, 2003; van Heel and Schatz, 2005). The maps were sharpened using phenix.auto_sharpen with automatically calculated overall b-sharpen and b_iso factors or the cryoSPARC postprocessing tool with B-factor as list in Table 1.
After the cleanup step in cryoSPARC, the free nucleosome dataset was processed in Relion 3.1 (Scheres, 2020) utilizing the high-order aberration correction functions and per-particle defocus refinement, which improved the resolution by ~0.3 Å since this dataset was collected with 3x3 image shift. Angular distribution was calculated using csparc2star.py and star2bild.py in pyem (Asarnow and Cheng, 2019), and illustrated using ChimeraX (Goddard et al., 2018). The local resolution of all reconstructions was assessed with Bsoft BlocRes (Heymann, 2018) using the unfiltered half-maps as inputs (Figure S2).
Model Building and Refinement
For the H1.0 chromatosome, structures of the human core histones (PDB: 5Y0C) (Arimura et al., 2018), the globular domain of H5 in the chromatosome (PDB: 4QLC) and 601 DNA (PDB: 5NL0) were rigid-body fitted to the H1.0 chromatosome density map using UCSF Chimera (Pettersen et al., 2004). For H1.4 chromatosome, the globular domain of H1.4 was built de novo in Coot aided by a predicted structural model of H1.4 globular domain using PHENIX (Adams et al., 2010) (Liebschner et al., 2019). For H1.10 chromatosome, the NMR structure of the H1.10 globular domain (PDB: 2LSO) (Eletsky et al., 2012) was fitted to the density. Structural models were manually rebuilt using COOT (Emsley et al., 2010). Mutations of proteins and DNAs were made to generate the corresponding actual sequences. Extension of protein chains was made following the observed densities. All models were subjected to alternating local and global real-space refinement using PHENIX (Adams et al., 2010) (Liebschner et al., 2019) and COOT. PHENIX was used to examine the model to avoid overfitting. The final structures were assessed by Molprobity (Chen et al., 2010). DNA sequence in the H1.4 chromatosome and the gH1.10-ncH1.4 chromatosome were confirmed by the differences in purine and pyrimidine densities (in regions with adequate resolution). Linker DNA conformation adjustment was aided by ISOLDE (Croll, 2018).
Molecular Dynamics Simulations
Three chromatosome structures each containing one of linker histones H1.0, H1.4 and H1.10, as well as the free nucleosome structure containing the same 197 bp DNA sequence but with no H1 histone were solvated in cubic boxes in explicit water to ensure there is at least a 12 Å buffer zone between the molecules and system boundaries. Systems were charge-neutralized by randomly distributing Na+ and Cl− ions at a concentration of 0.15M, minimized via steepest descent and conjugate gradient methods and equilibrated at 100 Kfor 1 ns and 310 Kfor 1 ns using a timestep of 1 fs. Production trajectories were collected at the NPT ensemble at 310 K and 1 atm using atimestepof 2 fs for a duration of 0.5 μs for each starting configuration. AMBER14sb force field (Maier et al., 2015) and the OPC water model (Izadi et al., 2014) were used to describe the biological system and the solvent environment, respectively. Atomic coordinates were saved every 100 ps and analyzed using VMD and Python scripts. Molecular dynamics (MD) simulations were performed using Gromacs 2018/3 (Van Der Spoel et al., 2005). The same simulation was also conducted for the chromatosome containing the H1.4 R53A mutant for 0.22 μs.
DNA RMSFs were computed over phosphate atoms and contacts were defined using a 5 Å cutoff between heavy atoms. Distributions were plotted using their kernel density estimates with a bandwidth of 5. For quantifying linker strand dynamics in every frame of a trajectory, the nucleosomal DNA of each chromatosome was positioned such that molecular principal axes were in line with orthogonal cartesian axes. For this purpose, four nucleosome corners were defined using nucleosomal DNA base pair triplets or hexatuplets as points of reference: dyad (base pairs −1 through 1), base (base pairs −37 through −35 and 35 through 37), left (chain I base pairs −55 through −53 and 19 through 21, chain J base pairs −19 through −17 and −43 through −41), and right (chain I base pairs −19 through −17 and 55 through 57, chain J base pairs −55 through −53 and 19 through 21). The dyad-base axis (i.e., the nucleosome principal axis) was used as the x axis. The cross-product of the x axis and the axis connecting left and right corners was used as the y axis. To determine the linker strand deviation angles, α and β, an orientation vector was defined for each DNA strand by subtracting the position vector of the base pair closest to the nucleosome core particle (25th base pair) from the position vector of the most-terminal base pair (first base pair). A position vector connects the center of mass of the base pair and the origin at x = y = z = 0. To determine the angle α, the orientation vector is projected onto the nucleosomal plane (XZ plane). The angle between this projection and the nucleosomal principal axis is called α. To determine the angle β, the orientation vector is projected onto the XY plane. The angle between this projection and the nucleosomal principal axis is called β.
Nuclear Magnetic Resonance Experiments
The samples of the chromatosomes or nucleosomes containing the 15N/13C-labeld core or linker histones or 15N/13C-labeled free linker histones in the initial reconstituting buffer were exchanged into the NMR buffer (10 mM NaPi, pH 6.3,0.1 mM EDTA) by repeated concentration and dilution using an Amicon Ultra spin concentrator (Millipore). 10% D2O was added to the final samples. NMR data were collected at 310 K using Bruker 700 MHz NMR equipped with cryo-probe. The backbone chemical shift assignments were made by collecting the standard triple-resonance 3D experiments (assignments are available upon requests). The multi-dimensional NMR spectra were processed using NMRPipe (Delaglio et al., 1995). The peak intensities of the well-separated peaks in the 1H-15N HSQC spectra were measured using NMRViewJ (One Moon Scientific Inc.). Chemical shift perturbations (CSP) were calculated using the following equation:
Electrophoretic mobility shift assay
The ratio of linker histones and nucleosome was determined by titrating linker histones to the nucleosome in buffer containing 10 mM Tris, 1 mM EDTA, 10mM NaCl, 1% Ficoll 6000 at pH 7.4.100 nM nucleosome was titrated with 65, 130, or 195 nM of linker histones. 5 μL of each reaction was loaded on a 5% acrylamide gels in 0.2x TBE, and run at 100 V for 120 min at 4°C. After electrophoresis, PAGE gels were stained with Midori Green Advance (Bulldog) and the gel images were visualized using ImageJ (https://imagej.nih.gov/) (Schneider et al., 2012).
Binding of chromatin remodeler ctISWI77-Δ-722 with nucleosome or chromatosomes were carried out in buffer containing 20 mM Tris, 0.1 mM EDTA, 50mM NaCl, 1mM DTT and 1% Ficoll 6000 at pH 7.4. 100 nM nucleosome or chromatosomes were mixed with 200, 300, 400, or 500nM of ctISWI. After incubating on ice for 30 min, 5 μL of each solution was loaded to a 4.75% acrylamide gel in 0.2x TBE buffer and run at 100 V for 90 min at 4°C. PAGE gels were stained and visualized as described above.
Nucleosome sliding assay
Nucleosome sliding by the full-length chromatin remodeler ctISWI was carried out as described previously with minor modifications (Mueller-Planitz et al., 2013). 100 nM of 237 bp centrally positioned nucleosome in the presence or absence of linker histones (130 nM of H1.0, H1.4, or H1.0) and 100 nM of full-length ctISWI were used. Reactions were performed in buffer containing 20 mM HEPES, pH 7.5, 5 mM MgCl2, 50mM KCl, 2.5 mM DTT and 5% glycerol with 2 mM ATP. At 5,15, 30, and 60-minute time points, reactions were stopped by adding 400 ng of plasmid (6 kb length). The products were resolved with 4.8% native PAGE, 0.2 x TBE run at 4°C and 150 V for 55 minutes. PAGE gels were stained with Midori Green Advance (Bulldog). The band intensities were quantified using ImageJ (https://imagej.nih.gov/). The fraction of centered nucleosome was determined by the ratio of the centered nucleosome to the total loaded nucleosome intensity. Three independent nucleosome sliding experiments were performed.
QUANTIFICATION AND STATISTICAL ANALYSIS
The nucleosome sliding assays were repeated three times and represented as mean ± SD of the three independent experiments. The intensities of the gel bands were quantified using ImageJ and the data points were plotted using GraphPad Prism 8 (Figure S6C). Cryo-EM data collection, refinement, and validation statistics are listed in Table 1.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli BL21-CodonPlus (DE3)-RIPL Competent Cells | Agilent | Cat#230280 |
| Escherichia coli Rosetta(DE3) pLysS Competent Cells | Novagen | Cat#70956 |
| Chemicals, Peptides, and Recombinant Proteins Critical | ||
| HiTrap SP HP cation exchange chromatography column | GE Healthcare | Cat#17115101 |
| Ni-NTA Agarose | QIAGEN | Cat#30230 |
| Protein-RP, 5um, 250x20mm Column | YMC AMERICA | Cat#PR99S05-2520WT |
| TSKgel DEAE-5PW column | TOSOH | Cat#0007574 |
| Superdex 75 increase 10/300 GL column | GE Healthcare | Cat# 29148721 |
| Superdex 200 increase 10/300 GL column | GE Healthcare | Cat#28990944 |
| Superose 6 10/300 GL column | GE Healthcare | Cat#29091596 |
| ProTa | Feng et al., 2018 | https://pubs.acs.org/doi/10.1021/acs.biochem.8b01075 |
| Deposited Data | ||
| H1.0 Chromatosome | This Paper | EMDB: EMD-22683 PDB ID: 7K5X |
| H1.4 Chromatosome | This Paper | EMDB: EMD-22684 PDB ID: 7K5Y |
| H1.10 Chromatosome | This Paper | EMDB: EMD-22685 PDB ID: 7K60 |
| 197bp Nucleosome | This Paper | EMDB: EMD-22686 PDB ID: 7K61 |
| gH1.10-ncH1.4 Chromatosome | This Paper | EMDB: EMD-22687 PDB ID: 7K63 |
| H1.4 Di-chromatosome | This paper | EMBD: EMD-22688 |
| Crystal structure of chromatosome containing GH5 | Zhou et al., 2015 | PDB: 4QLC |
| Cryo-EM structure of nucleosome in complex with scFv | Zhou et al., 2019 | PDB: 6DZT |
| Solution NMR structure of GH1.10 | Eletsky et al., 2012 | PDB: 2LSO |
| Crystal structure of 197bp nucleosome in complex with H1 | Bednar et al., 2017 | PDB: 5NL0 |
| Crystal Structure of the human nucleosome | Arimura et al., 2018 | PDB: 5Y0C |
| Recombinant DNA | ||
| pET42b-H1.0-his6 | This Paper | N/A |
| pET42b-H1.4-his6 | This Paper | N/A |
| pET42b-H1.10-his6 | This Paper | N/A |
| pET42b-gH1.10-ncH1.4-his6 | This Paper | N/A |
| pET42b-H1.4T145E/T153E/S171E/S186E-his6 | This Paper | N/A |
| pET42b-H2B | This Paper | N/A |
| pET21b-H3 | This Paper | N/A |
| pET21b-H4 | This Paper | N/A |
| pUC19-16x197_W601 | This Paper | N/A |
| pUC19-di-197x8_W601 | This Paper | N/A |
| pET15b-scFv | Zhou et al., 2019 | http://www.nature.com/articles/s41467-019-10247-4 |
| pProEX-Htb-CtISWI77-Δ-722 | Chittori et al., 2019 | https://academic.oup.com/nar/article/47/17/9400/5545016 |
| Software and Algorithms | ||
| Relion 3.0 | Zivanov et al., 2018 | https://github.com/3dem/relion |
| CryoSparc V2 | Punjani et al., 2017 | https://cryospac.com/ |
| CTFind4 | Rohou and Grigorieff, 2015 | https://grigoriefflab.janelia.org/ctf |
| MotionCorr2 | Zheng et al., 2017 | https://msg.ucsf.edu/em/software/motioncor2.html |
| BlocRes | Heymann, 2018 | https://lsbr.niams.nih.gov/bsoft/ |
| Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Chimerax | Goddard et al., 2018 | https://www.cgl.ucsf.edu/chimerax |
| ISOLDE | Croll, 2018 | https://isolde.cimr.cam.ac.uk/ |
| Coot | Emsley et al., 2010 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Phenix | Liebschner et al., 2019 | https://www.phenix-online.org/ |
| Topaz | Bepler et al., 2019 | https://github.com/tbepler/topaz |
| pyem | Asarnow and Cheng, 2019 | https://github.com/asarnow/pyem |
| Prism 8 | GraphPad Software | https://www.graphpad.com/ |
| NMRPipe | Delaglio et al., 1995 | https://www.ibbr.umd.edu/nmrpipe/index.html |
| NMRViewJ | One Moon Scientific, Inc. | https://nmrfx.org/nmrfx/nmrviewj |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ |
Highlights.
2.8–3.1 Å resolution structures of chromatosomes containing human H1 isoforms
Human somatic H1 isoforms bind to the nucleosome on the dyad
C-terminal tails of the H1 isoforms control distinct flanking DNA orientations
Chromatosome H3 N-tails relocate and inhibit ISWI remodeler binding and activity
ACKNOWLEDGMENTS
We thank Dr. Weimin Wu for initial help with cryo-EM data processing and Dr. Shiv Grewal for helpful suggestions. The authors acknowledge the use of the cryo-EM resources at the Center for Microscopic Resources and the computational resources of the High-Performance Computing Biowulf cluster at the NIH (https://hpc.nih.gov/). This study was supported by the intramural research programs of the National Cancer Institute (Y.B.) and National Institute of Diabetes and Digestive and Kidney Diseases (R.G.), National Institutes of Health. A.R.P. and S.K. were in part supported by the intramural research program of the National Library of Medicine at the US National Institutes of Health. A.R.P. was supported by the Department of Pathology and Molecular Medicine, Queen’s University, Canada, a Senior Canada Research Chair in Computational Biology and Biophysics, and a Senior Investigator award from the Ontario Institute of Cancer Research, Canada.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2020.10.038.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan J, Hartman PG, Crane-Robinson C, and Aviles FX (1980). The structure of histone H1 and its location in chromatin. Nature 288, 675–679. [DOI] [PubMed] [Google Scholar]
- Allan J, Mitchell T, Harborne N, Bohm L, and Crane-Robinson C (1986). Roles of H1 domains in determining higher order chromatin structure and H1 location. J. Mol. Biol 187, 591–601. [DOI] [PubMed] [Google Scholar]
- Arents G, Burlingame RW, Wang BC, Love WE, and Moudrianakis EN (1991). The nucleosomal core histone octamer at 3.1 A resolution: a tripartite protein assembly and a left-handed superhelix. Proc. Natl. Acad. Sci. USA 88, 10148–10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura Y, Ikura M, Fujita R, Noda M, Kobayashi W, Horikoshi N, Sun J, Shi L, Kusakabe M, Harata M, et al. (2018). Cancer-associated mutations of histones H2B, H3.1 and H2A.Z.1 affect the structure and stability of the nucleosome. Nucleic Acids Res. 46, 10007–10018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asarnow DPE, and Cheng Y (2019). UCSF pyem v0.5 (Zenodo; ). [Google Scholar]
- Ausio J, Borochov N, Seger D, and Eisenberg H (1984). Interaction of chromatin with NaCl and MgCl2. Solubility and binding studies, transition to and characterization of the higher-order structure. J. Mol. Biol 111, 373–398. [DOI] [PubMed] [Google Scholar]
- Bednar J, Garcia-Saez I, Boopathi R, Cutter AR, Papai G, Reymer A, Syed SH, Lone IN, Tonchev O, Crucifix C, et al. (2017). Structure and dynamics of α197 bp nucleosome in complex with linker histone H1. Mol. Cell 66, 384–397.e388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bepler T, Morin A, Rapp M, Brasch J, Shapiro L, Noble AJ, and Berger B (2019). Positive-unlabeled convolutional neural networks for particle picking in cryo-electron micrographs. Nat. Methods 16, 1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilokapic S, Strauss M, and Halic M (2018). Cryo-EM of nucleosome core particle interactions in trans. Sci. Rep 8, 7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, et al. (2018). Extreme disorder in an ultrahigh-affinity protein complex. Nature 555,61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DT, Izard T, and Misteli T (2006). Mapping the interaction surface of linker histone H1(0) with the nucleosome of native chromatin in vivo. Nat. Struct. Mol. Biol 13, 250–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caterino TL, and Hayes JJ (2011). Structure of the H1 C-terminal domain and function in chromatin condensation. Biochem. Cell Biol 89, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittori S, Hong J, Bai Y, and Subramaniam S (2019). Structure of the primed state of the ATPase domain of chromatin remodeling factor ISWI bound to the nucleosome. Nucleic Acids Res. 4l, 9400–9409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christophorou MA, Castelo-Branco G, Halley-Stott RP, Oliveira CS, Loos R, Radzisheuskaya A, Mowen KA, Bertone P, Silva JC, Zernicka-Goetz M, et al. (2014). Citrullination regulates pluripotency and histone H1 binding to chromatin. Nature 501, 104–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill ME, and Suzuki M (1989). ‘SPKK’ motifs prefer to bind to DNA at A/T-rich sites. EMBO J. 8, 4189–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JL, Lary JW, P Moody T, and Laue TM (2008). Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium. Methods Cell Biol. 84, 143–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll TI (2018). ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr. D Struct. Biol l4, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, and Bax A (1995). NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, and Richmond TJ (2004). Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science 306, 1571–1573. [DOI] [PubMed] [Google Scholar]
- Draizen EJ, Shaytan AK, Mariño-Ramírez L, Talbert PB, Landsman D, and Panchenko AR (2016). HistoneDB 2.0: a histone database with variants—an integrated resource to explore histones and their variants. Database (Oxford) 2016, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffney LJ, Valdez P, Tremblay MW, Cao X, Montgomery S, McConkie-Rosell A, and Jiang YH (2018). Epigenetics and autism spectrum disorder: A report of an autism case with mutation in H1 linker histone HIST1H1E and literature review. Am. J. Med. Genet. B. Neuropsychiatr. Genet. Ill, 426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer PN, Edayathumangalam RS, White CL, Bao Y, Chakravarthy S, Muthurajan UM, and Luger K (2004). Reconstitution of nucleosome core particles from recombinant histones and DNA. Methods Enzymol. 315, 23–44. [DOI] [PubMed] [Google Scholar]
- Eletsky A, Acton TB, Xiao R, Everett JK, Montelione GT, and Szyperski T (2012). Solution NMR structures reveal a distinct architecture and provide first structures for protein domain family PF04536. J. Struct. Funct. Genomics 13, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Nikitina T, Zhao J, Fleury TJ, Bhattacharyya R, Bouhassira EE, Stein A, Woodcock CL, and Skoultchi AI (2005). Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 123, 1199–1212. [DOI] [PubMed] [Google Scholar]
- Fang H, Clark DJ, and Hayes JJ (2012). DNA and nucleosomes direct distinct folding of a linker histone H1 C-terminal domain. Nucleic Acids Res. 40, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng H, Zhou BR, and Bai Y (2018). Binding affinity and function of the extremely disordered protein complex containing human linker histone H1.0 and its chaperone ProTα. Biochemistry 51, 6645–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flex E, Martinelli S, Van Dijck A, Ciolfi A, Cecchetti S, Coluzzi E, Pannone L, Andreoli C, Radio FC, Pizzi S, et al. (2019). Aberrant function of the C-terminal tail of HIST1H1E accelerates cellular senescence and causes premature aging. Am. J. Hum. Genet 105, 493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyodorov DV, Zhou BR, Skoultchi AI, and Bai Y (2018). Emerging roles of linker histones in regulating chromatin structure and function. Nat. Rev. Mol. Cell Biol 19, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Saez I, Menoni H, Boopathi R, Shukla MS, Soueidan L, Noirclerc-Savoye M, Le Roy A, Skoufias DA, Bednar J, Hamiche A, et al. (2018). Structure of an H1-bound 6-nucleosome array reveals an untwisted two-start chromatin fiber conformation. Mol. Cell 72, 902–915.e907. [DOI] [PubMed] [Google Scholar]
- Goddard TD, Huang CC, Meng EC, Pettersen EF, Couch GS, Morris JH, and Ferrin TE (2018). UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Happel N, and Doenecke D (2009). Histone H1 and its isoforms: contribution to chromatin structure and function. Gene 431, 1–12. [DOI] [PubMed] [Google Scholar]
- Harshman SW, Young NL, Parthun MR, and Freitas MA (2013). H1 histones: current perspectives and challenges. Nucleic Acids Res. 41, 9593–9609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergeth SP, and Schneider R (2015). The H1 linker histones: multifunctional proteins beyond the nucleosomal core particle. EMBO Rep. 16, 1439–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hergeth SP, Dundr M, Tropberger P, Zee BM, Garcia BA, Daujat S, and Schneider R (2011). Isoform-specific phosphorylation of human linker histone H1.4 in mitosis by the kinase Aurora B. J. Cell Sci 124, 1623–1628. [DOI] [PubMed] [Google Scholar]
- Heymann JB (2018). Guidelines for using Bsoft for high resolution reconstruction and validation of biomolecular structures from electron micrographs. Protein Sci. 21, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn PJ, Carruthers LM, Logie C, Hill DA, Solomon MJ, Wade PA, Imbalzano AN, Hansen JC, and Peterson CL (2002). Phosphorylation of linker histones regulates ATP-dependent chromatin remodeling enzymes. Nat. Struct. Biol 9, 263–267. [DOI] [PubMed] [Google Scholar]
- Izadi S, Anandakrishnan R, and Onufriev AV (2014). Building water models: a different approach. J. Phys. Chem. Lett 5, 3863–3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornberg RD (1974). Chromatin structure: a repeating unit of histones and DNA. Science 184, 868–871. [DOI] [PubMed] [Google Scholar]
- Kornberg RD, and Thomas JO (1974). Chromatin structure; oligomers of the histones. Science 184, 865–868. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Bezsonova I, Evanics F, Taulier N, Zhou Z, Bai Y, Chalikian TV, Prosser RS, and Kay LE (2006). Probing the transition state ensemble of a protein folding reaction by pressure-dependent NMR relaxation dispersion. J. Am. Chem. Soc 128, 5262–5269. [DOI] [PubMed] [Google Scholar]
- Lee H, Habas R, and Abate-Shen C (2004). MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis. Science 304, 1675–1678. [DOI] [PubMed] [Google Scholar]
- Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Zaja R, Palazzo L, Stockum A, Ahel I, and Matic I (2016). Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat. Chem. Biol 12, 998–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebschner D, Afonine PV, Baker ML, Bunkóczi G, Chen VB, Croll TI, Hintze B, Hung LW, Jain S, McCoy AJ, et al. (2019). Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in Phenix. Acta Crystallogr. D Struct. Biol 75, 861–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowary PT, and Widom J (1998). New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. J. Mol. Biol 276, 19–42. [DOI] [PubMed] [Google Scholar]
- Lu X, and Hansen JC (2004). Identification of specific functional subdomains within the linker histone H10 C-terminal domain. J. Biol. Chem 279, 8701–8707. [DOI] [PubMed] [Google Scholar]
- Lu X, Wontakal SN, Kavi H, Kim BJ, Guzzardo PM, Emelyanov AV, Xu N, Hannon GJ, Zavadil J, Fyodorov DV, and Skoultchi AI (2013). Drosophila H1 regulates the genetic activity of heterochromatin by recruitment of Su(var)3–9. Science 340, 78–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mäder AW, Richmond RK, Sargent DF, and Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, and Simmerling C (2015). ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca TJ, Freedman BS, and Heald R (2005). Histone H1 is essential for mitotic chromosome architecture and segregation in Xenopus laevis egg extracts. J. Cell Biol 169, 859–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronarde DN (2005). Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol 152, 36–51. [DOI] [PubMed] [Google Scholar]
- Misteli T, Gunjan A, Hock R, Bustin M, and Brown DT (2000). Dynamic binding of histone H1 to chromatin in living cells. Nature 408, 877–881. [DOI] [PubMed] [Google Scholar]
- Mueller-Planitz F, Klinker H, Ludwigsen J, and Becker PB (2013). The ATPase domain of ISWI is an autonomous nucleosome remodeling machine. Nat. Struct. Mol. Biol 20, 82–89. [DOI] [PubMed] [Google Scholar]
- Olins AL, and Olins DE (1974). Spheroid chromatin units (v bodies). Science 183, 330–332. [DOI] [PubMed] [Google Scholar]
- Öztürk MA, Pachov GV, Wade RC, and Cojocaru V (2016). Conformational selection and dynamic adaptation upon linker histone binding to the nucleosome. Nucleic Acids Res. 44, 6599–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öztürk MA, Cojocaru V, and Wade RC (2018). Dependence of chromatosome structure on linker histone sequence and posttranslational modification. Biophys. J 114, 2363–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera: a visualization system for exploratory research and analysis. J. Comput. Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Punjani A, Rubinstein JL, Fleet DJ, and Brubaker MA (2017). cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296. [DOI] [PubMed] [Google Scholar]
- Rohou A, and Grigorieff N (2015). CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol 192, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohs R, West SM, Sosinsky A, Liu P, Mann RS, and Honig B (2009). The role of DNA shape in protein-DNA recognition. Nature 461, 1248–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenthal PB, and Henderson R (2003). Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J. Mol. Biol 333, 721–745. [DOI] [PubMed] [Google Scholar]
- Scheres SHW (2020). Amyloid structure determination in RELION-3.1. Acta Crystallogr. D Struct. Biol 76, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuck P (2000). Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J 78, 1606–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, and Gorovsky MA (1996). Linker histone H1 regulates specific gene expression but not global transcription in vivo. Cell 86, 475–483. [DOI] [PubMed] [Google Scholar]
- Simpson RT (1978). Structure of the chromatosome, a chromatin particle containing 160 base pairs of DNA and all the histones. Biochemistry 17, 5524–5531. [DOI] [PubMed] [Google Scholar]
- Singer DS, and Singer MF (1976). Studies on the interaction of H1 histone with superhelical DNA: characterization of the recognition and binding regions of H1 histones. Nucleic Acids Res. 3, 2531–2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Chen P, Sun D, Wang M, Dong L, Liang D, Xu RM, Zhu P, and Li G (2014). Cryo-EM study of the chromatin fiber reveals a double helix twisted by tetranucleosomal units. Science 344, 376–380. [DOI] [PubMed] [Google Scholar]
- Starkova TY, Polyanichko AM, Artamonova TO, Khodorkovskii MA, Kostyleva EI, Chikhirzhina EV, and Tomilin AN (2017). Post-translational modifications of linker histone H1 variants in mammals. Phys. Biol 14,016005. [DOI] [PubMed] [Google Scholar]
- Stützer A, Liokatis S, Kiesel A, Schwarzer D, Sprangers R, Söding J, Selenko P, and Fischle W (2016). Modulations of DNA contacts by linker histones and post-translational modifications determine the mobility and modifiability of nucleosomal H3 tails. Mol. Cell 61, 247–259. [DOI] [PubMed] [Google Scholar]
- Takata H, Matsunaga S, Morimoto A, Ono-Maniwa R, Uchiyama S, and Fukui K (2007). H1.X with different properties from other linker histones is required for mitotic progression. FEBS Lett. 581, 3783–3788. [DOI] [PubMed] [Google Scholar]
- Torres CM, Biran A, Burney MJ, Patel H, Henser-Brownhill T, Cohen AS, Li Y, Ben-Hamo R, Nye E, Spencer-Dene B, et al. (2016). The linker histone H1.0 generates epigenetic and functional intratumor heterogeneity. Science 353, 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner AL, Watson M, Wilkins OG, Cato L, Travers A, Thomas JO, and Stott K (2018). Highly disordered histone H1-DNA model complexes and their condensates. Proc. Natl. Acad. Sci. USA 115, 11964–11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, and Berendsen HJ (2005). GROMACS: fast, flexible, and free. J. Comput. Chem 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- van Heel M, and Schatz M (2005). Fourier shell correlation threshold criteria. J. Struct. Biol 151, 250–262. [DOI] [PubMed] [Google Scholar]
- Woodcock CL, Skoultchi AI, and Fan Y (2006). Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 14, 17–25. [DOI] [PubMed] [Google Scholar]
- Woods DC, and Wereszczynski J (2020). Elucidating the influence of linker histone variants on chromatosome dynamics and energetics. Nucleic Acids Res. 48, 3591–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Read C, McGeehan J, and Crane-Robinson C (2016). The construction of customized nucleosomal arrays. Anal. Biochem 496, 71–75. [DOI] [PubMed] [Google Scholar]
- Yan L, Wu H, Li X, Gao N, and Chen Z (2019). Structures of the ISWI-nucleosome complex reveal a conserved mechanism of chromatin remodeling. Nat. Struct. Mol. Biol 26, 258–266. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Cooke M, Panjwani S, Cao K, Krauth B, Ho PY, Medrzycki M, Berhe DT, Pan C, McDevitt TC, and Fan Y (2012). Histone h1 depletion impairs embryonic stem cell differentiation. PLoS Genet. 8, e1002691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Brautigam CA, Ghirlando R, and Schuck P (2013). Overview of current methods in sedimentation velocity and sedimentation equilibrium analytical ultracentrifugation. Curr. Protoc. Protein Sci Chapter 20, Unit20.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BR, and Bai Y (2019). Chromatin structures condensed by linker histones. Essays Biochem. 63, 75–87. [DOI] [PubMed] [Google Scholar]
- Zhou BR, Feng H, Kato H, Dai L, Yang Y, Zhou Y, and Bai Y (2013). Structural insights into the histone H1-nucleosome complex. Proc. Natl. Acad. Sci. USA 110, 19390–19395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BR, Jiang J, Feng H, Ghirlando R, Xiao TS, and Bai Y (2015). Structural mechanisms of nucleosome recognition by linker histones. Mol. Cell 59, 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BR, Feng H, Ghirlando R, Li S, Schwieters CD, and Bai Y (2016). A small number of residues can determine if linker histones are bound on or off dyad in the chromatosome. J. Mol. Biol 428, 3948–3959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BR, Jiang J, Ghirlando R, Norouzi D, Sathish Yadav KN, Feng H, Wang R, Zhang P, Zhurkin V, and Bai Y (2018). Revisit of reconstituted 30nm nucleosome arrays reveals an ensemble of dynamic structures. J. Mol. Biol 430 (18 Pt B), 3093–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou BR, Yadav KNS, Borgnia M, Hong J, Cao B, Olins AL, Olins DE, Bai Y, and Zhang P (2019). Atomic resolution cryo-EM structure of a native-like CENP-A nucleosome aided by an antibody fragment. Nat. Commun 10, 2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, and Scheres SH (2018). New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession numbers for the structures and the corresponding cryo-EM maps is PDB: 7K5Xand EMDB: EMD-22683 for the H1.0 chromatosome; PDB: 7K5Y and EMDB: EMD-22684 for the H1.4 chromatosome; PDB: 7K60 and EMDB: EMD-22685 for H1.10 chromatosome; PDB: 7K61 and EMDB: EMD-22686 for the nucleosome; PDB: 7K63 and EMDB: EMD-22687 for the gH1.10-ncH1.4 chromatosome. EMDB: EMD-22688forthe H1.4 Di-chromatosome. The raw 2D micrographs of all datasets will be uploaded to the EMPIAR database.