

Abstract
Hydrops Fetalis (HF), accumulation of fluid in two or more fetal compartments, is life-threatening to the fetus. Genetic etiologies include many chromosomal and monogenic disorders. Despite this, the clinical workup typically evaluates limited genetic targets. To support broader molecular testing of pregnancies with HF, we cataloged the spectrum of monogenic disorders associated with nonimmune hydrops fetalis (NIHF).
We performed a systematic literature review under PROSPERO tag CRD42018099495 of cases reporting NIHF meeting strict phenotypic criteria and well-defined genetic diagnosis. We ranked the evidence per gene based on number of reported cases, phenotype and molecular/biochemical diagnosis.
We identified 131 genes with strong evidence for an association with NIHF and 46 genes with emerging evidence spanning the spectrum of multisystem syndromes, cardiac disorders, hematologic disorders, and metabolic disorders. Several genes previously implicated with NIHF did not have any reported cases in the literature with both fetal hydrops and molecular diagnosis. Many genes with strong evidence for association with NIHF would not be detected using current sequencing panels.
Nonimmune HF has many possible monogenic etiologies, several with treatment implications, but current diagnostic approaches are not exhaustive. Studies are needed to assess if broad sequencing approaches like whole exome sequencing are useful in clinical management of HF.
Keywords: Hydrops Fetalis, Nonimmune Hydrops Fetalis, prenatal
Introduction
Hydrops Fetalis (HF) is a life-threatening fetal condition defined as an abnormal accumulation of fluid in two or more fetal compartments [1]. This can be diagnosed by prenatal ultrasound and is characterized by the presence of ascites, pleural effusion, pericardial effusion, or generalized skin edema. In addition, HF may be associated with polyhydramnios and placental edema [2]. HF is a clinical description and not a diagnosis suggestive of a specific cause. The pathophysiology of HF can be classified according to immune and nonimmune etiology. The prevalence of immune etiologies has dramatically decreased with the development of effective Rh(D) immunization in mothers at risk. Consequently, nonimmune hydrops fetalis (NIHF) now accounts for almost 90% of hydrops cases [1]. Mortality of NIHF remains high ranging between 55–90 percent, depending on the cause [2]. Understanding the etiology of NIHF is important for effective management in pregnancies and in the neonatal period as both morbidity and mortality depend on the underlying cause.
The Society of Maternal-Fetal Medicine (SMFM) provided guidelines for the diagnostic workup of NIHF in 2015[2]. SMFM recommends first line workup of HF with karyotype and chromosomal microarray (CMA), fetal echocardiography, viral polymerase chain reaction (PCR) with an option to pursue single gene etiologies given appropriate history and clinical suspicion. Yet, currently the cause of up to half of the cases of NIHF remains unknown after evaluation by standard testing [1]. With additional genetic testing, idiopathic NIHF cases are often reclassified as inborn errors of metabolism, such as lysosomal storage disorders and congenital disorders of glycosylation, which are a class of disorders that can be diagnosed with specialized testing [3, 4]. Over the past 5 years since the SMFM guidelines were published, clinical genetic sequencing technologies have made rapid advancements but have not yet been incorporated into official recommendations.
The current genetic testing strategy focusing on karyotyping and CMA is limited by resolution. Single gene variants that cause NIHF are not within the diagnostic resolution of CMA or karyotype. It is a challenge for clinicians to investigate all possible etiologies given the vast number of single gene disorders that cause NIHF. According to the Genetic Testing Registry, several commercial laboratories in the United States offer gene panel testing for NIHF. However the yield for these panels has not been studied and proprietary nature of panel creation leads to variation of genes included on panels adding to the uncertainty of establishing a diagnostic yield for this approach. Therefore, more unbiased genetic testing with whole exome sequencing (WES) may be required for efficient identification of single gene causes of NIHF in a timely manner [5, 6]. The tradeoffs between exome and panel testing are related to the number of genes evaluated which also affects cost, turnaround time, and insurance coverage. Increasing the number of genes evaluated increases test sensitivity by allowing much rarer diagnoses to be found but increases the analytic burden of the test. While both tests are often built on the same laboratory platforms, panels offer lower risk of incidental findings or variants of uncertain significance in genes unrelated to the indication for testing. In contrast, exome sequencing typically will evaluate parental samples at the time of initial analysis, which decreases the risk of uncertain classification of de novo variants and unphased recessive variants. In practice, uncertain variants on gene panels frequently require subsequent parental testing to clarify their significance which adds time to the testing process or forces clinical decision making based on less certain results.
Diagnosis of the molecular cause of a hydropic pregnancy is important for several reasons. Several disorders which present as NIHF have specific treatments, such as enzyme replacement for lysosomal storage disorders, transplants, or transfusions for congenital anemias. Particularly for inborn errors of metabolism [3, 4], prenatal diagnosis could prevent delays in initiating treatment and avoid irreversible damage after birth. Additionally, a molecular diagnosis allows better targeted counseling of both likely pregnancy outcomes as well as long-term health and developmental outcomes for the child. This information can be crucial for families deciding whether or not to continue a pregnancy. Knowing a molecular diagnosis provides psychological benefit to a parent who can be reassured with the evidence that an adverse outcome was due to a random genetic event. Finally, knowing a molecular etiology can allow better prediction of recurrence risk for future pregnancies and in many cases allow earlier fetal genetic screening or preimplantation genetic diagnosis in future pregnancies
The focus of this review is to catalog identified single gene etiologies for NIHF. Previous reviews have made efforts to categorize the etiologies of NIHF and have used unifying disease pathologies or body systems. These suggested using a system-based approach in evaluating each NIHF case by considering phenotypic features for a more targeted genetic evaluation, diagnosis and management of NIHF [6]. This categorization remains important as additional phenotypic features become important in disambiguating uncertain variants identified in broader sequencing approaches. The yield of whole exome sequencing, as a phenotype driven analysis, is related to the whole list of genes known to be associated with a diagnosis [5]. As such, we sought to establish a baseline of evidence for the association of specific genes with NIHF to support future exome studies. In this study, we provide a systematic review of all single gene disorders known to be associated with NIHF, and we further unify and classify each monogenic disorder based on its level of evidence to serve as a reference for clinicians performing WES or building gene panels for NIHF. We then categorize each monogenic disorder based on the affected body system, to establish a more comprehensive catalog of single gene disorders associated with NIHF.
Methods
We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) system [7], including a checklist and flowchart. We registered the review using PROSPERO under the identification tag CRD42018099495 (https://www.crd.york.ac.uk/prospero/display_record.php?ID=CRD42018099495).
Articles were identified from PubMed, Scopus, OMIM, and GeneReviews from inception to December 15th, 2019. Search criteria included using one hydrops related term and one gene related term (see Appendix 1 for detailed search strategy). Language was restricted to English. Articles were included when NIHF was a diagnosis associated with a single gene etiology. Case studies, case reports, cohort studies, and review articles on hydrops were all included. For secondary literature, the citations were evaluated to identify the primary cases of hydrops referenced by the review. Articles that did not mention a specific gene contributing to an underlying etiology were excluded. Duplicate studies were excluded from total counts.
Articles discussing multigene deletions, duplications, and aneuploidy as etiologies of NIHF were also excluded as beyond the scope of this study. Articles on immune hydrops and on non-human subjects were also excluded. Two reviewers (A.Q., B.V.) independently screened all of the articles with disputes resolved by a third and fourth reviewer (H.A.K., S.I.B.) to create an initial list of cases reported with NIHF based on strict ultrasound criteria of fluid accumulation in two or more fetal compartments and their associated variant genes. The strict phenotypic criteria of two or more fetal compartment involvement including skin edema, ascites, pleural effusion, and pericardial effusion was applied in all reports where enough phenotypic descriptors were reported. For the sake of completeness, reports were also included where the only phenotypic description was fetal hydrops without further elaboration. As fetal hydrops can be the end result of fetal demise, it is not always clear in the literature if hydropic features in a fetus pre-existed fetal demise. Given this, we chose to include any case meeting the phenotypic criteria without regard to the status of pregnancy outcome. Genetic evidence of hydrops was included even if the reported case and genetic testing were published separately as in the case of PTH1R where the hydropic case was initially reported and the same case was sequenced in a future report [8].
For each gene from the initial literature search, additional targeted search in PubMed for at least two unrelated cases of confirmed hydrops if one or more than one distinct case reporting hydrops in association with the gene could be identified. Manual searches were completed from March to May 2020. Some original articles were replaced with newer articles.
Sibling pregnancies reported in a single paper were counted as a single family, while unrelated cases reported together in a case series were counted separately. Genetic diagnosis in papers was considered sufficient for the genetic association if a variant meeting ACMG variant classification criteria for pathogenic or likely pathogenic was identified [9]. For cases reported without a molecular genetic diagnosis, biochemical testing to implicate a specific gene, such as enzymology, was accepted for a second case when only a single molecularly confirmed case could be identified.
While we avoided including disorders due to chromosomal anomalies or contiguous gene deletion syndromes as outside the scope of the review, we sought to avoid confusion by including well known digenic disorders such as the alpha thalassemia genes HBA1 and HBA2 despite the hydropic phenotype requiring loss of both genes at the loci to have a hydropic phenotype [10].
We ranked the strength of a gene’s relationship with NIHF into 4 categories. We chose a strategy (Figure 2) to separate genes with clear evidence of multiple reports of hydrops from genes which may have been reported in a hydropic patient, but unrelated to the presentation. Dual genetic diagnosis is typically found in 4% of exomes done for a wide variety of indications [11]. As such, we chose the criteria of 2 molecularly or one molecular plus one biochemically defined unrelated cases in the literature with well described NIHF as the standard of evidence to assume a strong association. Given the overall rarity of many of these disorders, we accepted genes with a single case of hydrops as strong evidence if another gene associated with the same disorder had strong evidence. For example, generalized arterial calcification of infancy due to ENPP1 had enough cases for table 1 criteria, but ABCC6, the other gene for this disorder, had only a single case in the literature [12]. Therefore, we considered the ABCC6 association strong because the other gene which leads to the same pathogenesis has a strong association with hydrops.
Figure 2.
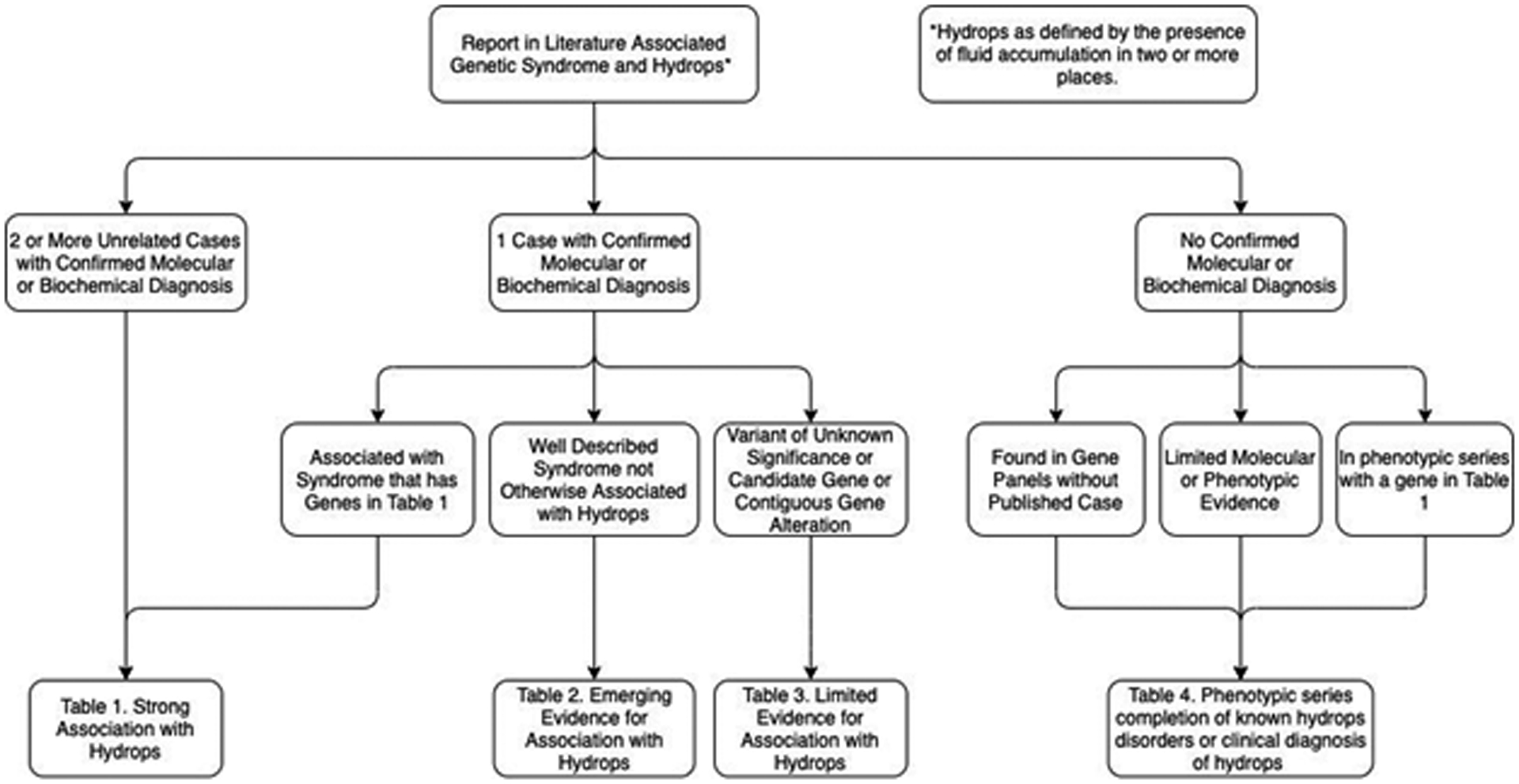
Algorithm for deciding which table of evidence a gene belonged to.
*Hydrops as defined by the presence of fluid accumulation in two or more compartments.
Table 1.
Syndromes and genes with more than one reported NIHF cases and (*) associated genes with only 1 reported case. Abbreviations: Mito (Mitochondrial); Sterol (Sterol Biosynthesis Disorder); CDG (Congenital Disorder of Glycosylation); LSD (Lysosomal Storage Disease)
| Type | Condition | Gene | |
|---|---|---|---|
| Syndromic | Aicardi–Goutieres syndrome | RNASEH2B | |
| Alkuraya-Kucinskas syndrome | KIAA1109 | ||
| Cornelia de Lange syndrome | NIPBL | ||
| RASopathies / Noonan syndrome | PTPN11 | ||
| RAF1 | |||
| RIT1 | |||
| SHOC2 | |||
| CBL | |||
| BRAF | |||
| MAP2K1 | |||
| SOS1 | |||
| HRAS | |||
| NRAS | |||
| LZTR1 | |||
| Kabuki Syndrome | KMT2D | ||
| Baraitser-Winter syndrome | ACTB | ||
| Takenouchi-Kosaki Syndrome | CDC42 | ||
| Fraser syndrome | FRAS1 | ||
| GRIP1* | |||
| Pierson syndrome | LAMB2 | ||
| Cardiovascular | Timothy syndrome | CACNA1C | |
| Left ventricular noncompaction | MYH7 | ||
| Generalized Arterial Calcification of Infancy | ENPP1 | ||
| ABCC6* | |||
| Congenital long-QT syndrome | KCNH2 | ||
| SCN5A | |||
| X-linked Cardiac valvular dysplasia | FLNA | ||
| NIHF with Congenital Cardiac and Hemangiomas | THSD1 | ||
| Left ventricular noncompaction | MYBPC3 | ||
| Familial Hypertrophic Cardiomyopathy | ALPK3 | ||
| Inborn Errors of Metabolism | LSD | Mucopolysaccharidosis I | IDUA |
| Mucolipidosis II | GNPTAB | ||
| Mucopolysaccharidosis IV | GALNS | ||
| Mucopolysaccharidosis VII | GUSB | ||
| Niemann Pick Type A | SMPD1 | ||
| Niemann Pick Type C | NPC1 | ||
| Farber lipogranulomatosis | ASAH1 | ||
| GM1 gangliosidosis | GLB1 | ||
| Galactosialidosis | CTSA | ||
| Sialidosis | NEU1 | ||
| Infantile Sialic acid storage diseases | SLC17A5 | ||
| Gaucher disease, perinatal lethal | GBA | ||
| Multiple sulfatase deficiency | SUMF1 | ||
| CDG | PMM2-CDG | PMM2 | |
| ALG1-CDG | ALG1 | ||
| ALG8-CDG | ALG8 | ||
| ALG9-CDG | ALG9 | ||
| MGAT2-CDG | MGAT2 | ||
| Sterol | Mevalonic aciduria | MVK | |
| Greenberg skeletal dysplasia | LBR | ||
| X-Linked Chondrodysplasia Punctata | EBP | ||
| Mito | Combined oxidative phosphorylation deficiency | MRPS22 | |
| AARS2 | |||
| Barth syndrome | TAZ | ||
| Complex I deficiency | NDUFB10 | ||
| Mitochondrial tRNA deficiency | MT-TE | ||
| MT-TL1 | |||
| NIHF, lactic acidosis, sideroblastic anemia | LARS2 | ||
| Other | LCHAD/Trifunctional protein deficiency | HADHA | |
| HADHB | |||
| Hypermethioninemia | AHCY | ||
| Glycogen storage disease type IV | GBE1 | ||
| Sphingosine-1-phosphate lyase deficiency | SGPL1 | ||
| Abnormal Growths | Tuberous Sclerosis Complex | TSC2 | |
| Capillary malformation-AV malformation | RASA1 | ||
| CLOVES Syndrome | PIK3CA | ||
| Plaque-Type Glomuvenous Malformations | GLMN | ||
| Hematologic | Anemias | Alpha-thalassemia | HBA1 |
| HBA2 | |||
| Hereditary spherocytosis | SLC4A1 | ||
| Hereditary spherocytosis/elliptocytosis | SPTA1 | ||
| SPTB | |||
| Congenital dyserythropoietic anemia | CDAN1 | ||
| SEC23B | |||
| KLF1 | |||
| GATA1-Related X-Linked Cytopenia | GATA1 | ||
| Familial Hemophagocytic Lymphohistiocytosis | PRF1 | ||
| UNC13D* | |||
| Diamond–Blackfan anemia | RPL11* | ||
| RPL35A* | |||
| RPL15 | |||
| RPS19 | |||
| Metabolic | Glucose-6-phosphate dehydrogenase deficiency | G6PD | |
| Transaldolase deficiency | TALDO1 | ||
| Congenital Erythropoietic Porphyria | UROS | ||
| Pyruvate kinase deficiency | PKLR | ||
| Glucose phosphate isomerase deficiency | GPI | ||
| Lymphatic | Hereditary lymphedema type IA | FLT4 | |
| Hennekam lymphangiectasia-lymphedema syndrome | CCBE1 | ||
| ADAMTS3* | |||
| Lymphatic Malformation / Dehydrated hereditary stomatocytosis | PIEZO1 | ||
| Lymphatic Malformation 7 | EPHB4 | ||
| Hypotrichosis-lymphedema-telangiectasia-renal defect syndrome | SOX18 | ||
| Lymphedema-distichiasis syndrome | FOXC2 | ||
| Skeletal | Chondrodysplasia, Blomstrand type | PTH1R | |
| Achondrogenesis | COL2A1 | ||
| SLC26A2 | |||
| Desbuquois dysplasia 1 | CANT1 | ||
| Cranioectodermal dysplasia-1, Sensenbrenner | IFT122 | ||
| Achondrogenesis type IA, Schneckenbecken | SLC35D1 | ||
| Opsismodysplasia | INPPL1 | ||
| Congenital chylothorax | ITGA9 | ||
| Short rib-polydactyly syndrome with or without polydactyly | DYNC2H1 | ||
| IFT80 | |||
| NEK1* | |||
| WDR35* | |||
| KIAA0586* | |||
| Neuromuscular | Multiple Contracture Syndrome, Finnish Type | GLE1 | |
| Lethal multiple pterygium syndrome | RYR1 | ||
| CHRNG | |||
| CHRNA1 | |||
| CHRND | |||
| Fetal akinesia deformation sequence | RAPSN | ||
| DOK7 | |||
| MUSK | |||
| Spinal Muscular Atrophy (SMA) | SMN1 | ||
| SMN2 | |||
| SMA, lower extremity-predominant | BICD2 | ||
| SMA with congenital bone fractures | ASCC1 | ||
| Nemaline myopathy | NEB | ||
| TPM2* | |||
| ACTA1 | |||
| LMOD3* | |||
| KLHL40* | |||
| Cerebro-oculofacioskeletal syndrome | ERCC5 | ||
| Myotonic dystrophy 1 | DMPK | ||
| Other | Congenital adrenal hyperplasia | CYP21A2 | |
| FZD6-associated hydrops | FZD6 | ||
| SERPINA11-prenatal lethal disorder | SERPINA11 | ||
| IPEX syndrome | FOXP3 | ||
| Incontinentia pigmenti | IKBKG | ||
Our ranking strategy is summarized in Figure 2 with the literature evidence for each gene’s ranking included in the supplemental tables. Group 1, genes with a strong association with NIHF, (Table 1; Table S1) includes genes with pathogenic changes identified in multiple reported cases of NIHF. Group 2, genes with emerging evidence for association with NIHF, (Table 2; Table S2) includes genes with a pathogenic variant identified in a single reported case of NIHF. Genes in Group 2 were moved to Group 1 if they were associated with a syndrome already included in Group 1 due to locus heterogeneity. Group 3, genes with limited evidence for association with NIHF, (Table S3) includes genes with uncertain changes or genes of uncertain significance reported in cases of NIHF. Group 4, candidate genes for association with NIHF, (Table S4) includes genes which are discussed in the literature as associated with NIHF but for which no reported molecularly confirmed cases were identified. This group also includes genes where the original case report did not meet the strict hydrops definition of two fetal compartments with fluid collections but were referred to as hydrops in subsequent literature and genes associated with a suspected clinical diagnosis without definitive molecular diagnosis. Genes included on three representative commercially available hydrops panels from Invitae Genetics, Prevention Genetics, and Greenwood Genetics were included in the candidate group as well [13]. Many of these represented associations of genes within a phenotypic spectrum where other genes have been associated with hydrops. Therefore, we used the Online Inheritance in Man (OMIM) Database phenotypic series lists [14] to add to Group 4 additional candidate genes for hydrops found in phenotypic series strongly associated with hydrops from Group 1.
Table 2.
Genes and associated syndromes with emerging evidence for an association with NIHF based on a single reported case in the literature. Abbreviations: Mito (Mitochondrial); Sterol (Sterol biosynthesis disorder); CDG (Congenital Disorder of Glycosylation); LSD (Lysosomal Storage Disease); FAOD (Fatty Acid Oxidation Disorder); GSD (Glycogen Storage Disease)
| Type | Condition | Gene | |
|---|---|---|---|
| Syndromic | Arthrogryposis, cleft palate, craniosynostosis, and impaired intellectual development | PPP3CA | |
| X-linked Opitz G/BBB Syndrome | MID1 | ||
| Bartsocas–Papas syndrome | RIPK4 | ||
| Scalp-ear-nipple Syndrome | KCTD1 | ||
| Mental retardation and distinctive facial features with or without cardiac defects | MED13L | ||
| Denys-Drash syndrome | WT1 | ||
| X-Linked Ohdo Syndrome | MED12 | ||
| Cardiovascular | Severe cardiomyopathy with left ventricular noncompaction | PKP2 | |
| Hypertrophic Cardiomyopathy 6 | PRKAG2 | ||
| Hydrops fetalis, congenital heart defects and genital anomalies, Congenital heart defects, multiple types, 5 | GATA5 | ||
| Inborn Errors of Metabolism | LSD | Sialic aciduria | NPL |
| CDGs | STT3B-CDG | STT3B | |
| Muscular dystrophy-dystroglycanopathy | FKTN | ||
| COG6-CDG | COG6 | ||
| FAOD | Primary carnitine deficiency | SLC22A5 | |
| Sterol | Smith-Lemli-Opitz syndrome | DHCR7 | |
| Peroxisomal | Zellweger syndrome | PEX3 | |
| GSD | Glycogen Storage Disease II Pompe disease | GAA | |
| Mito | Pontocerebellar Hypoplasia Type 6 | RARS2 | |
| Combined oxidative phosphorylation deficiency | SLC25A26 | ||
| MRPS16 | |||
| Leukodystrophy, hypomyelinating, 4 | HSPD1 | ||
| Other | Hexokinase deficiency | HK1 | |
| Neu-Laxova Syndrome | PHGDH | ||
| Cobalamin C disease | MMACHC | ||
| GLYT1 Encephalopathy | SLC6A9 | ||
| Hematologic | Anemia | Sideroblastic anemia with B-cell immunodeficiency, periodic fevers, and developmental delay | TRNT1 |
| Immunodeficiency, common variable, 13 | IKZF1 | ||
| X-linked hemolytic anemia | ATP11C | ||
| Bone marrow failure syndrome 4 | MYSM1 | ||
| Lymph | Emberger syndrome | GATA2 | |
| Lymphatic Malformation 8 | CALCRL | ||
| Skeletal | Multiple congenital anomalies-hypotonia-seizures syndrome 1 | PIGN | |
| Caffey disease | COL1A1 | ||
| Complex Lethal Osteochondrodysplasia | TAPT1 | ||
| Neuromuscular | Cerebral cavernous malformations-1 | KRIT1 | |
| Lethal Congenital Contracture Syndrome 10 | NEK9 | ||
| Epileptic encephalopathy, early infantile | GRIN2B | ||
| Cerebellar ataxia, mental retardation, and dysequilibrium syndrome 2 | WDR81 | ||
| Spinocerebellar ataxia 29, congenital nonprogressive | ITPR1 | ||
| Arthrogryposis multiplex congenital | TTN | ||
| Abnormal Growths | Basal Cell Nevus Syndrome | PTCH1 | |
| Other | X-linked Chronic granulomatous disease | CYBB | |
| Proliferative vasculopathy and hydrocephaly syndrome | FLVCR2 | ||
| Adrenal insufficiency | CYP11A1 | ||
| Nephronophthisis 2 | INVS | ||
Results were then organized into the following categories based on primary organ system thought to be responsible for the hydrops: cardiovascular, inborn errors of metabolism, hematologic, lymphatic, skeletal, neuromuscular, abnormal growth, syndromic for multisystem disorders and other for disorders not clearly in any of these categories in a systems based manner [6]. Gene and protein names were standardized using the HUGO Gene Nomenclature Committee approved symbol and name respectively. Etiology names were standardized using OMIM phenotype names [14], the name described in IEMbase for inborn errors of metabolism [15], or the GeneReviews name for simplicity if multiple overlapping OMIM names exist. If a disorder had multiple distinct disorder associations in OMIM, the name most closely related to the phenotype in the paper was chosen. If no syndrome association existed in OMIM, then the phenotype described in the paper was used. If multiple studies reference the same genetic etiology and protein, then the most recent article was cited.
Results
After duplicates were removed, 18,950 articles resulted from the searches. The flowchart in Figure 1 shows that 272 articles met the inclusion criteria. Individual literature searches identified 33 additional articles for a total of 305 articles utilized for this review. A total of 131 genes met criteria for Group 1 (strong molecular evidence for an association with NIHF), an additional 46 met Group 2 criteria (emerging molecular evidence for an association with NIHF), another 39 genes met Group 3 criteria (limited molecular evidence for an association with NIHF), and finally 301 genes met criteria for Group 4 (candidate genes for an association with NIHF) for a total 517 genes. The main categories of genetic causes of NIHF, based on strong and emerging evidence genes, are inborn errors of metabolism (27.6%), syndromic (15.3%), neuromuscular (14.1%), hematologic (13.6%), skeletal (9.0 %), cardiovascular (7.3%), lymphatic (5.1%) and abnormal growths (2.8%). The syndromic category contains multisystem syndromes where the association with hydrops could be due to one or more of the categories, but lacks specific evidence to group into a specific type.
Figure 1.
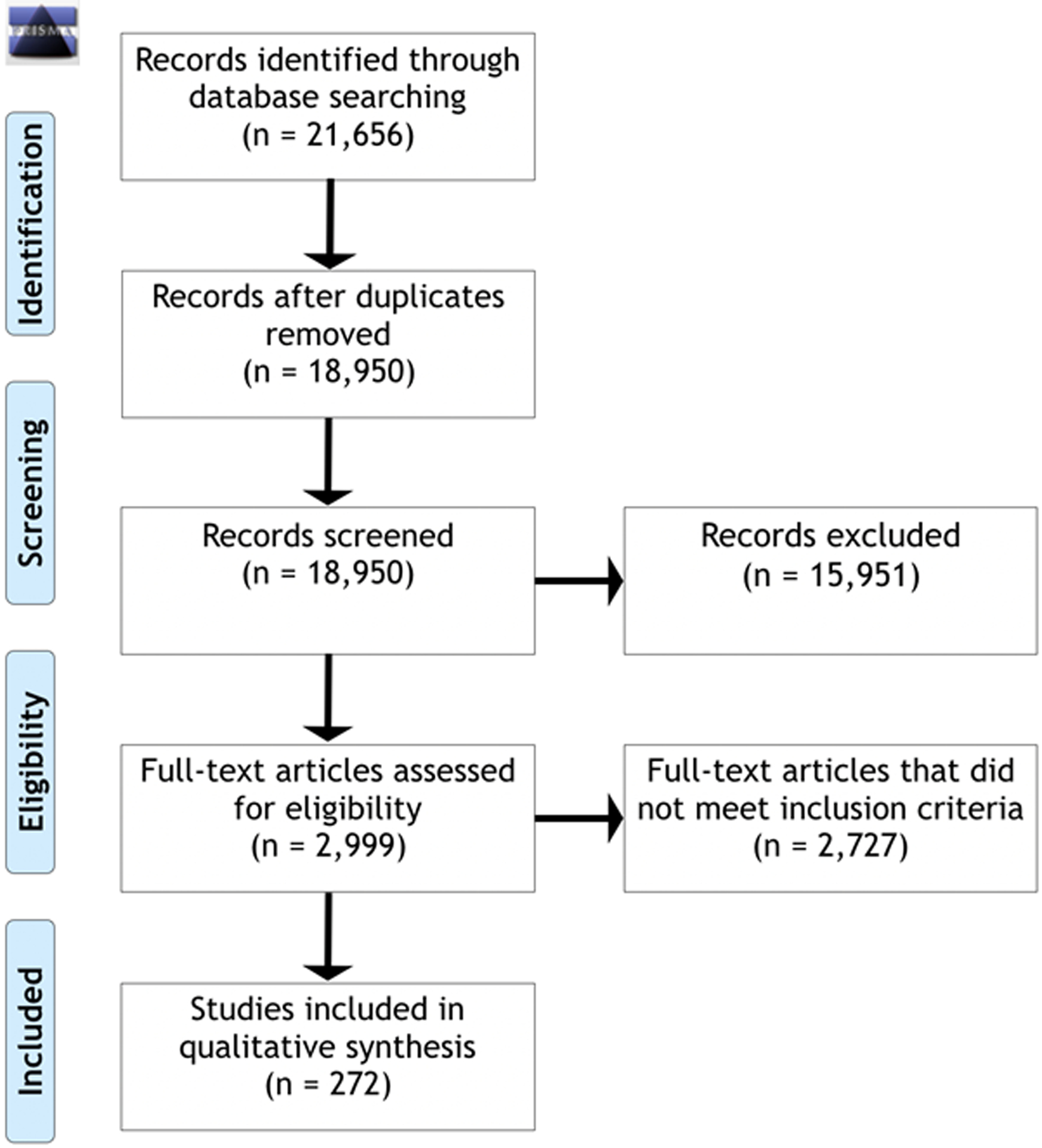
Prisma 2009 Flow Diagram of included and excluded articles.
NIHF genes with strong and emerging evidence (Table 1 and Table 2) were associated with the full range of inheritance patterns including autosomal dominant (26.0%), autosomal recessive (61.8 %), X-linked (5.3%), either autosomal dominant or recessive (3.8%), mitochondrial (2.3%), or somatic mutation (0.8%). Dominant disorders associated with NIHF have been both reported with sporadic de novo mutations as well as parentally inherited. One gene, DMPK, is associated with genetic anticipation due to trinucleotide repeat expansion [16]. Discerning each of the possibilities allows for better estimation of recurrence risks in future pregnancies.
Several genes discussed in the literature or found on panels have no reports in the literature associated with hydrops (Table 4). The largest and most inclusive commercial panel consisted of 130 genes [13] where 60.1% (79 genes) have strong evidence and 6.2% (8 genes) have emerging evidence for an association with NIHF, 0.8% (1 gene) with limited evidence, and 32.3% (42 genes) with no reports in the literature. Nevertheless, this panel does not cover 90 of the 177 (50.8%) of strong and emerging evidence genes reported in NIHF cases.
Discussion
There are many monogenic etiologies of nonimmune hydrops fetalis. For the classification in our tables, we sought to be inclusive of all genes currently discussed in the literature for a possible association with hydrops and to be systematic about their categorization. For many single gene disorders, however, the evidence associating many genetic etiologies with NIHF in the literature is limited. There are several reasons for this. First, all the monogenetic etiologies with severe fetal and infantile presentations are inherently rare diseases due to the significant detriment they present to reproductive fitness. We did not attempt to calculate the frequency of NIHF for each specific genetic etiology from our literature review due to the intrinsic reporting bias of the literature. A rare complication of a rare disorder may have similar representation in the literature as a common complication of a very rare disorder. Additionally, due to the prenatal presentation of hydrops fetalis, often ending with fetal demise, detailed genetic studies are not always performed, and a molecular genetic diagnosis is often not reached. Thus, the literature, especially the older literature, is frequently limited to cases where fetuses are born and a diagnosis was made postnatally. In many cases, diagnoses were made based on clinical exam, such as specific malformations or dysmorphic features suggestive of a specific diagnosis. While several of these have been listed in other reviews as associated with hydrops, in these cases, we did not feel this was sufficient evidence to associate a gene with a hydrops fetalis phenotype due to the likely occurrence of phenocopies, often with more severe presentations, due to different genes. For biochemical diagnoses where the biochemical phenotype is highly specific, we did count these cases particularly when there was a separate case with a molecular diagnosis. There are many cases where a gene was associated with hydrops fetalis in only a single case reported in the literature. While this could be due to the rarity of the disorder combined with the rarity of the disorder causing hydrops, this could also be due to a dual diagnosis [11], where the reported disorder was present in a case where the hydrops was in fact due to a separate cause. For this reason, we used the 2 hydrops cases as criteria for Group 1 genes. While most Group 1 genes met this criterion, a few genes with only 1 case of hydrops in the literature but association with Table 1 disorders were also included in Table 1. For instance, genes for Diamond Blackfan Anemia with only a single hydropic report in the literature, RPL11 and RPL35A, were upgraded to Table 1 since other Diamond Blackfan Anemia genes, RPL15 and RPS19, had multiple reports of hydrops [17]. Conversely, while there are many reported cases of hydrops fetalis associated with Tuberous Sclerosis, after review of the molecular etiologies, all the cases with molecular diagnoses had TSC2 variants [18]. Therefore, TSC1 remains on the candidate gene list of Table 4, as there was no evidence in the literature to suggest an association between TSC1 and hydrops. We suspect this could be the typically more severe phenotype associated with TSC2 compared to TSC1 which could contribute to a difference in risk of hydrops fetalis. A similar pattern was seen with some of the RASopathies. One of the most commonly reported monogenic causes of fetal hydrops is Noonan syndrome and other RASopathies [19]. As such, it was not surprising that we identified multiple reported cases of hydrops for almost all the known RASopathy genes. However, in this group we did not find any reported cases of HF associated with KRAS, SOS2, or MAP2K2. As such, these 3 genes are currently candidate genes for HF and are included in Group 4.
We were surprised by several findings in our analysis. First, we encountered several disorders where literature reviews described a known association with hydrops fetalis and several of these genes show up on panels for hydrops fetalis as well. Based on thorough review of the literature surrounding these genes, we found that this “known association” was often based on a single case report where no molecular diagnosis was available. For example, Simpson-Golabi-Behmel was reported in deceased hydropic fetuses and given a clinical diagnosis based on dysmorphic features and malformation pattern, but this was reported prior to known molecular loci for the gene and may be a distinct genetic loci than the known Simpson-Golabi-Behmel genes which we include on Table 4 due to the lack of molecular evidence at this time [20]. Similarly, the only reports for Meckel Gruber syndrome either lacked clearly defined molecular diagnosis [21] or had isolated pericardial effusion without other fluid compartment involvement. As such, despite previous discussions in the literature of the gene’s association with hydrops [6], the genes for this syndrome were placed in our candidate gene Table 4 due to lack of a definitive molecular diagnosis or hydropic case meeting our criteria.
For most disorders, hydrops represented a more severe end of the phenotypic spectrum known for the disorder. This includes multiple disorders that present with anemia. Mild forms of these disorders cause anemias which are diagnosed in children or even adults. However, these may present as hydrops if they present with a severe fetal anemia [10]. On the other hand, hydrops may be an unexpected prenatal presentation with an unclear relationship to the known disorder. For example, FZD6 is known to be associated with a congenital nail disorder [5]. Despite this relatively specific phenotype without a clear relationship to HF, pathogenic changes in this gene have been identified in two unrelated cases of hydrops fetalis and thus HF is considered an expansion of the phenotype associated with changes in this gene. The precise mechanism of the hydrops in this disorder is unclear.
For most disorders with reported cases of NIHF, the mechanism of the hydrops could be due to neuromuscular dysfunction, lymphatic malformation, cardiovascular defects, fetal anemia, or skeletal fetal restriction. A few disorders were identified that are thought to have primarily renal disease such as nephronophthisis, endocrine disease, and congenital adrenal hyperplasia. It is unclear if these represent a full phenotypic expansion of these disorders to other systems or if the renal defect or endocrinopathy itself leads to the hydrops [22, 23].
On the other hand, for some genes, hydrops represented an expansion of the known molecular disease mechanism for the gene. For example, PKP2 haploinsufficiency causes an autosomal dominant arrhythmogenic right ventricular dysplasia, where a single variant causes disease. The severe hydropic case reported with a PKP2 deletion had both alleles affected suggesting an expansion of inheritance pattern to a codominant pattern, where having both alleles affected leads to a more severe phenotype than having a single allele affected [24].
The mutational spectrum leading to the hydropic phenotype is also important to consider. In the case of TTN, the variants identified in a hydropic fetus with severe congenital contractures fell within an exon incorporated into a protein isoform expressed only during fetal development and not expressed in adult tissue [25]. This provides a plausible explanation for the unusual severe fetal presentation seen in that case, which is distinct from adult onset TTN associated disease, but also should caution interpretation of any molecular results in hydrops associated genes to ensure that an identified variant fits the mutational spectrum that causes hydrops.
Another gene with a distinct inheritance pattern than discussed for other phenotypes is ATP1A2. While dominant missense variants in ATP1A2 are associated with alternating hemiplegia of childhood and familial hemiplegic migraines, there have been 2 reports identified of fetuses affected with diffuse skin edema and biallelic loss of function variants, suggesting a recessive model for this phenotype and a distinct mutational spectrum from the dominant disease [26]. However, the described cases only reported diffuse skin edema, and lacking involvement of a second compartment, this gene remained on Table 4 for lack of phenotypic criteria.
Another important observation in this list is the inclusion of disorders that can be missed on standard sequencing approaches. While most of the genes on these lists would have variants that are detectable on standard exome analyses, DMPK, associated with myotonic dystrophy, causes disease due to a trinucleotide repeat expansion [16]. This type of variant is not routinely evaluated on exome or next generation sequencing panel based testing. As such, if one was doing a thorough molecular evaluation of fetal hydrops, DMPK repeat expansion would need to be evaluated separately.
In addition to well-defined disorders that can be missed on standardized panels, many genes not previously associated with documented disorders have been linked to hydrops. Since NIHF represents a severe phenotype that is often not compatible with live birth, genes that can cause hydrops are often not associated with human diseases that have traditionally been studied in living people. As such, many recent studies specifically evaluating molecular genetics of hydrops during pregnancies have identified single cases with strong candidate gene variants. These genes have mostly been included as genes of uncertain significance and uncertain variants on Table 3. However, after reviewing the literature, in some cases these prenatal lethal findings have been found in more than one case, such as in SERPINA11, which demonstrates how whole exome sequencing, rather than targeted panels, can increase the evidence of a gene’s association with hydrops [22].
Given the large number of genes associated with NIHF, and the rapid rate of new gene associations being published, predefined panel-based testing is limited for the diagnosis of monogenic hydrops fetalis. Large commercial panels currently available only are designed to detect half of the known monogenic causes of NIHF at best [13]. It would be possible to update panel offerings to include the genes we categorized as having strong evidence, emerging, limited, and candidate evidence for an association with NIHF in a tiered testing approach depending on clinical findings and family wishes. However, constant curation of such panels would be required as new cases provide evidence to reclassify current candidate genes and new genetic causes of NIHF are reported. Unbiased approaches such as exome sequencing will be able to detect the vast majority of monogenic NIHF cases. Careful genetic counseling is required for this testing due to the complicated nature of the results including uncertain variants and secondary findings.
Supplementary Material
References:
- 1.Bellini C, Donarini G, Paladini D, Calevo MG, Bellini T, Ramenghi LA, Hennekam RC: Etiology of non-immune hydrops fetalis: An update. Am J Med Genet A 2015, 167A(5):1082–1088. [DOI] [PubMed] [Google Scholar]
- 2.Norton ME, Chauhan SP, Dashe JS, (SMFM) SfM-FM: Society for maternal-fetal medicine (SMFM) clinical guideline #7: nonimmune hydrops fetalis. Am J Obstet Gynecol 2015, 212(2):127–139. [DOI] [PubMed] [Google Scholar]
- 3.Al-Kouatly HB, Felder L, Makhamreh MM, Kass SL, Vora NL, Berghella V, Berger S, Wenger DA, Luzi P: Lysosomal storage disease spectrum in nonimmune hydrops fetalis: a retrospective case control study. Prenat Diagn 2020, 40(6):738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Makhamreh MM, Cottingham N, Ferreira CR, Berger S, Al-Kouatly HB: Nonimmune hydrops fetalis and congenital disorders of glycosylation: A systematic literature review. J Inherit Metab Dis 2020, 43(2):223–233. [DOI] [PubMed] [Google Scholar]
- 5.Yates CL, Monaghan KG, Copenheaver D, Retterer K, Scuffins J, Kucera CR, Friedman B, Richard G, Juusola J: Whole-exome sequencing on deceased fetuses with ultrasound anomalies: expanding our knowledge of genetic disease during fetal development. Genet Med 2017, 19(10):1171–1178. [DOI] [PubMed] [Google Scholar]
- 6.Mardy AH, Chetty SP, Norton ME, Sparks TN: A system-based approach to the genetic etiologies of non-immune hydrops fetalis. Prenat Diagn 2019, 39(9):732–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moher D, Liberati A, Tetzlaff J, Altman DG, Group P: Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med 2009, 6(7):e1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoogendam J, Farih-Sips H, Wÿnaendts LC, Löwik CW, Wit JM, Karperien M: Novel mutations in the parathyroid hormone (PTH)/PTH-related peptide receptor type 1 causing Blomstrand osteochondrodysplasia types I and II. J Clin Endocrinol Metab 2007, 92(3):1088–1095. [DOI] [PubMed] [Google Scholar]
- 9.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E et al. : Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015, 17(5):405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Farashi S, Harteveld CL: Molecular basis of α-thalassemia. Blood Cells Mol Dis 2018, 70:43–53. [DOI] [PubMed] [Google Scholar]
- 11.Balci TB, Hartley T, Xi Y, Dyment DA, Beaulieu CL, Bernier FP, Dupuis L, Horvath GA, Mendoza-Londono R, Prasad C et al. : Debunking Occam’s razor: Diagnosing multiple genetic diseases in families by whole-exome sequencing. Clin Genet 2017, 92(3):281–289. [DOI] [PubMed] [Google Scholar]
- 12.Nitschke Y, Baujat G, Botschen U, Wittkampf T, du Moulin M, Stella J, Le Merrer M, Guest G, Lambot K, Tazarourte-Pinturier MF et al. : Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am J Hum Genet 2012, 90(1):25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Test | Non-Immune Hydrops Fetalis Panel - PreventionGenetics [https://www.preventiongenetics.com/testInfo?sel=test&val=Non-Immune+Hydrops+Fetalis+Panel]
- 14.Amberger JS, Bocchini CA, Scott AF, Hamosh A: OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Res 2019, 47(D1):D1038–D1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JJY, Wasserman WW, Hoffmann GF, van Karnebeek CDM, Blau N: Knowledge base and mini-expert platform for the diagnosis of inborn errors of metabolism. Genet Med 2018, 20(1):151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Son SB, Chun JM, Kim KA, Ko SY, Lee YK, Shin SM: A case report on 30-week premature twin babies with congenital myotonic dystrophy conceived by in vitro fertilization. J Korean Med Sci 2012, 27(10):1269–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuramitsu M, Sato-Otsubo A, Morio T, Takagi M, Toki T, Terui K, Wang R, Kanno H, Ohga S, Ohara A et al. : Extensive gene deletions in Japanese patients with Diamond-Blackfan anemia. Blood 2012, 119(10):2376–2384. [DOI] [PubMed] [Google Scholar]
- 18.Gu X, Han L, Chen J, Wang J, Hao X, Zhang Y, Zhang J, Ge S, He Y: Antenatal screening and diagnosis of tuberous sclerosis complex by fetal echocardiography and targeted genomic sequencing. Medicine (Baltimore) 2018, 97(15):e0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carrasco Salas P, Gómez-Molina G, Carreto-Alba P, Granell-Escobar R, Vázquez-Rico I, León-Justel A: Noonan syndrome: Severe phenotype and PTPN11 mutations. Med Clin (Barc) 2019, 152(2):62–64. [DOI] [PubMed] [Google Scholar]
- 20.Tenorio J, Arias P, Martínez-Glez V, Santos F, García-Miñaur S, Nevado J, Lapunzina P: Simpson-Golabi-Behmel syndrome types I and II. Orphanet J Rare Dis 2014, 9:138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moreno CA, Kanazawa T, Barini R, Nomura ML, Andrade KC, Gomes CP, Heinrich JK, Giugliani R, Burin M, Cavalcanti DP: Non-immune hydrops fetalis: A prospective study of 53 cases. Am J Med Genet A 2013, 161A(12):3078–3086. [DOI] [PubMed] [Google Scholar]
- 22.Shamseldin HE, Kurdi W, Almusafri F, Alnemer M, Alkaff A, Babay Z, Alhashem A, Tulbah M, Alsahan N, Khan R et al. : Molecular autopsy in maternal-fetal medicine. Genet Med 2018, 20(4):420–427. [DOI] [PubMed] [Google Scholar]
- 23.Esser T, Chaoui R: Enlarged adrenal glands as a prenatal marker of congenital adrenal hyperplasia: a report of two cases. Ultrasound Obstet Gynecol 2004, 23(3):293–297. [DOI] [PubMed] [Google Scholar]
- 24.Ramond F, Janin A, Di Filippo S, Chanavat V, Chalabreysse L, Roux-Buisson N, Sanlaville D, Touraine R, Millat G: Homozygous PKP2 deletion associated with neonatal left ventricle noncompaction. Clin Genet 2017, 91(1):126–130. [DOI] [PubMed] [Google Scholar]
- 25.Chervinsky E, Khayat M, Soltsman S, Habiballa H, Elpeleg O, Shalev S: A homozygous TTN gene variant associated with lethal congenital contracture syndrome. Am J Med Genet A 2018, 176(4):1001–1005. [DOI] [PubMed] [Google Scholar]
- 26.Monteiro FP, Curry CJ, Hevner R, Elliott S, Fisher JH, Turocy J, Dobyns WB, Costa LA, Freitas E, Kitajima JP et al. : Biallelic loss of function variants in ATP1A2 cause hydrops fetalis, microcephaly, arthrogryposis and extensive cortical malformations. Eur J Med Genet 2020, 63(1):103624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.