

Abstract
Kaposi’s sarcoma-associated herpesvirus (KSHV) causes several human cancers, including Kaposi’s sarcoma (KS) and primary effusion lymphoma (PEL), which are mostly seen in immunocompromised patients such as HIV+ individuals. Tuberculosis (TB), caused by the bacterial pathogen Mycobacterium tuberculosis (Mtb), remains one of the deadliest infectious diseases in the world. The risk of developing TB is dramatically higher in people living with HIV than among those without HIV infection. Case reports link cutaneous or pulmonary KS in HIV+ patients with mycobacterial co-infections, however, impacts of Mtb infection or its products on KSHV-infected cells are not known. We report here that ESAT-6, a secreted Mtb virulence factor, induces viral reactivation from KSHV-infected cells. KSHV-infected pulmonary endothelial cells were resistant to ESAT-6 induced inhibition of cell growth. Our data demonstrate that Mtb virulence factors influence the biology of KSHV-infected cells, highlighting the need to study the interactions between these two pathogens commonly found in people living with HIV.
Keywords: KSHV, Kaposi’s Sarcoma, Mycobacterium tuberculosis, ESAT-6, HIV
Introduction
Kaposi’s sarcoma–associated herpesvirus (KSHV, also known as human herpes virus-8, HHV8) is one of the most common etiologic agents for human cancers, including Kaposi’s sarcoma (KS), primary effusion lymphoma (PEL) and multicentric Castleman disease (MCD).1,2 Similar to other human herpesviruses, KSHV has two alternating life-cycle programs following infection of host cells, the latent and lytic phases, which are characterized by different patterns of viral gene expression.3 Among KSHV-related malignancies, KS lesions are characterized by endothelial-derived “spindle” tumor cells, abnormal and leaky vessels and extravasated red blood cells with haemosiderin deposits.4 Clinically, the lesions are described as patch, plaque, and nodule stages, and an individual patient can have different types of lesions simultaneously.5 Currently, there are four known types of KS disease: classic KS affecting elderly men of Mediterranean; endemic KS, existing in countries of Central and Eastern Africa; iatrogenic KS, usually developed in organ transplant recipients under immune suppression regimens; and epidemic or AIDS-KS with more aggressive features, which includes visceral lesions, such as in the gastrointestinal tract and lungs.3 Interestingly, almost 45% of HIV+ patients with cutaneous KS show pleural or pulmonary manifestations of KS. Conversely, 85–95% of patients with pulmonary KS also present with cutaneous involvement.6,7 Another KSHV-related malignancy, PEL, harbors KSHV episomes in transformed B cells, and typically presents as effusions in the pleural, pericardial, or abdominal cavities of patients, although solid/extracavitary lesions have also been reported.8,9
Tuberculosis (TB), caused by Mycobacterium tuberculosis (Mtb), is a major global health problem; one-third of the world’s population is infected with Mtb, and TB kills 1.6 million people around the world each year.10 Moreover, the risk of developing TB is estimated to be 16–27 times greater in people living with HIV than among those without HIV infection.10 Recent studies have established that secretion of early secreted antigenic target 6-kDa (ESAT-6) through the ESAT-6 secretion system (ESX-1) is essential for Mtb pathogenesis.11 ESAT-6 is encoded in a region of difference (RD) 1, which also includes the genes for ESX-1, and is present only in the virulent strains of mycobacteria.12 Our previous studies suggest that ESAT-6 has the potential to manipulate host immunity during Mtb infection to promote its pathogenesis.13–17 Interestingly, case reports describe cutaneous or pulmonary KS with mycobacterial comorbid infections (Mtb or Mycobacterium genavense) in the same patients.18–20 Moreover, KSHV infection is prevalent in patients with pulmonary TB.21 However, the potential impacts of Mtb (or its products) on KSHV-infected cells and/or this oncogenic virus itself are not known. In the current study, we have investigated whether Mtb secreted ESAT-6 influences KSHV gene expression, viral replication, and proliferation of latently infected cells.
Materials and Methods
Cell culture, reagents and infection protocols.
KSHV+ PEL cell line BCBL-1 and a KSHV negative Burkitt lymphoma cell line, BL-41, were kindly provided by Dr. Dean Kedes (University of Virginia) and cultured in RPMI 1640 media with supplemented with 10% fetal bovine serum (FBS), 10 mM HEPES, 100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine, 0.05 mM β-mercaptoethanol, and 0.02% (wt/vol) sodium bicarbonate. Primary human umbilical vein endothelial cells (HUVEC) and human pulmonary artery endothelial cells (HPAEC) were purchased from American Type Culture Collection (ATCC), and cultured as recommended by ATCC. Recombinant Mtb ESAT-6 and CFP10 proteins were expressed in Escherichia coli and prepared as described earlier.13 To obtain KSHV for the infection experiments, BCBL-1 cells were incubated with 0.6 mM valproic acid for 4–6 days, and KSHV was purified from the culture supernatant by ultracentrifugation at 20,000 × g for 3 h, 4°C. The viral pellets were resuspended in 1/100 the original volume with the appropriate culture media. The infectious titers were determined as described previously.22
qRT-PCR.
Total RNA was isolated using the RNeasy Mini kit (Qiagen), and cDNA was synthesized using a SuperScript III First-Strand Synthesis SuperMix Kit (Invitrogen). Primers used for amplification of target genes were listed in Table S1. Amplification was carried out using an iCycler IQ Real-Time PCR Detection System, and cycle threshold (Ct) values were tabulated in duplicate for each gene of interest in each experiment. “No template” (water) controls were used to ensure minimal background contamination. Using mean Ct values tabulated for each gene, and paired Ct values for β-actin as a loading control, fold changes for experimental groups relative to assigned controls were calculated using automated iQ5 2.0 software (Bio-rad).
Cell proliferation assays.
Cell proliferation was measured using the WST-1 Assay (Roche). Briefly, after the period of treatment of cells, 10 μL/well of cell proliferation reagent, WST-1 (4-[3-(4-Iodophenyl)-2-(4-nitro- phenyl)-2H-5-tetrazolio]-1,3-benzene disulfonate), was added into 96-well microplate and incubated for 3 h at 37°C in 5% CO2. The absorbance of samples was measured by using a microplate reader at 490 nm.
Immunofluorescence.
Cells were incubated in 1:1 methanol-acetone at −20°C for fixation and permeabilization, then with a blocking reagent (10% normal goat serum, 3% bovine serum albumin, and 1% glycine) for an additional 30 min. Cells were then incubated for 1 h at 25°C with 1:1000 dilution of a rat anti-LANA monoclonal antibody (ABI) followed by 1:200 dilution of a goat anti-rat secondary antibody conjugated to Texas Red (Invitrogen). For identification of nuclei, cells were subsequently counterstained with 0.5 mg/mL 4′,6-diamidino-2- phenylindole (DAPI; Sigma) in 180 mM Tris-HCl (pH 7.5). Slides were washed once in 180 mM Tris-HCl for 15 min and prepared for visualization using a Leica TCPS SP5 AOBS confocal microscope.
Statistical analysis.
Significance for differences between experimental and control groups was determined using the two-tailed Student’s t-test.
Results and Discussion:
To determine the impact of Mtb gene products on KSHV infection, we treated KSHV-infected primary endothelial cells (HUVEC) and/or PEL cells (BCBL-1) with purified recombinant ESAT-6 protein. The chemical inducer, valproic acid (VA) was used as a positive control. Viral gene expression was examined using qRT-PCR. Treatment with ESAT-6 significantly induced the expression of KSHV lytic genes, such as Rta, vGpcr, K8.1, from both cell lines when compared to the PBS control groups (Figure 1A–B). Consistent with enhanced lytic gene expression, ESAT-6 treatment promoted KSHV reactivation, as demonstrated by increased detection of LANA-positive cells (a marker for successful establishment of KSHV latent infection in host cells) following using purified virions released from control or ESAT-6 treated HUVEC or BCBL-1 cells to infect fresh HUVEC (Figure 1C–D). Culture filtrate protein of 10 kD (CFP10) is a molecular partner of ESAT-6 and form a dimer with ESAT-6 after their expression and secreted as a dimer through Mtb specific ESX-1 secretion system, and at low pH, these two proteins can be disassociated.13 Interestingly, we found that CFP10 treatment was not able to induce KSHV lytic gene expression from infected cells (Figure S1). Since Mtb and KSHV may co-exist in the same anatomic sites on a human body, such as skin, lung, or gastrointestinal tract, our data indicate that comorbid infection with Mtb may promote KSHV lytic reactivation, leading to increased virus dissemination in infected patients. Since ESAT-6 protein can be released into the extracellular space from cells infected with Mtb,23 the effect of ESAT-6 on KSHV-infected cells would not require these two pathogens to infect the same cells. However, co-infection of a single cell could be possible, given that macrophage, dendritic cells, epithelial cells and lymphatic endothelial cells (LEC) are potential targets of both pathogens.24–26
Figure 1. Mycobacterium tuberculosis ESAT-6 protein induces KSHV lytic reactivation from latently infected cells.
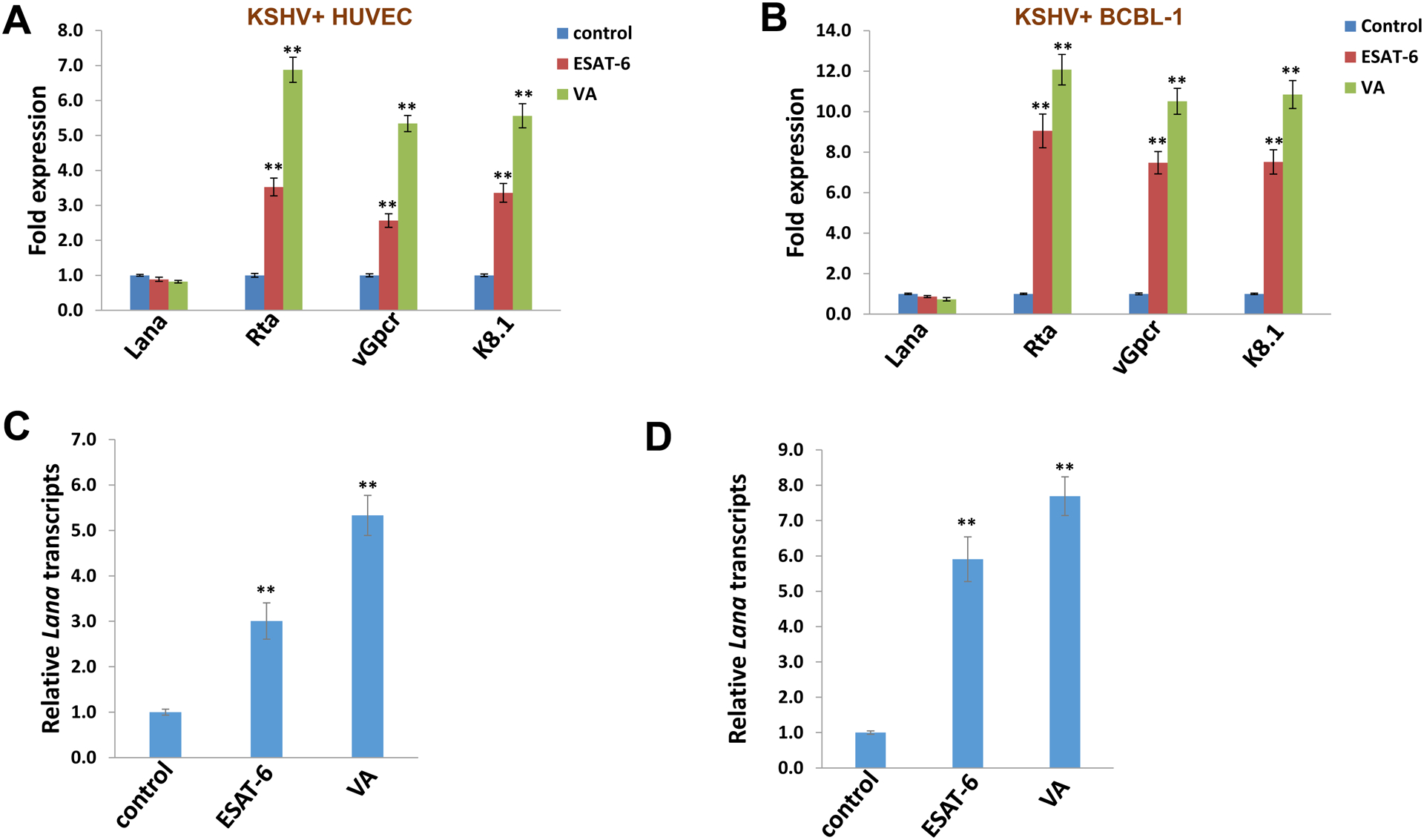
(A-B) KSHV-infected primary endothelial cells (HUVEC) or PEL cells (BCBL-1) were treated with purified ESAT-6 protein (2.5 μg/mL) or PBS control for 48 h. The valproic acid (VA) was used as a positive control. Quantitative RT-PCR was performed to determine the expression of representative viral latent (Lana) and lytic genes (Rta, vGpcr and K8.1). (C-D) HUVECs were infected with purified virions isolated from supernatants of HUVEC (C) or BCBL-1 (D) cells treated with ESAT-6, VA or PBS control for 72–96 h by ultracentrifugation. After 24 h post infection, Lana transcripts were quantified using qRT-PCR. Error bars represent the S.D. for 3 independent experiments. ** = p<0.01.
Since ESAT-6 causes lysis of alveolar epithelial cells and macrophages,11,12 we investigated how ESAT-6 treatment would impact the growth of KSHV-infected cells, especially cells from lung, since this is a common organ for pathologies caused by both Mtb and KSHV. As mentioned above, KS consists of endothelial-derived “spindle” tumor cells, so here we used human pulmonary artery endothelial cells (HPAEC) for our study as a potential physiologically relevant model for lung disease. We first demonstrated that HPAEC were susceptible to KSHV infection using immunofluorescence (IFA) to quantify the number of LANA-positive cells. We found that > 95% of HPAEC contained LANA in their nuclei by 48 h post infection (p.i.) with a MOI ~10, while no LANA dots were observed in mock-infected control cells (Figure 2A). Compared to uninfected cells, KSHV-infected HPAEC were resistant to the inhibition of cell growth caused by ESAT-6 (Figure 2B), even at a relatively high concentration of 10 μg/mL. In contrast, ESAT-6 treatment caused a dose-dependent reduction in growth of both mock-infected and KSHV-infected HUVEC, as well as BCBL-1 and BL-41 cells (Figure 2C–D). Again, we found that CFP10 treatment failed to affect the growth of cells with or without KSHV infection (Figure S2), which is in accordance with our previous findings that only ESAT-6 but not CFP10 inhibited human immune cell responses.13–15 The underlying mechanisms for the resistance of KSHV-infected HPAEC (but not HUVEC) to ESAT-6 will require further investigation. Interestingly, we found that ESAT-6 as well as VA treatments induced less extent of viral lytic gene expression from KSHV-infected HPAEC when compared to infected HUVEC (Figure S3), which may represent one of the potential mechanisms for its impact on cell growth differences. In addition, ESAT-6 is able to stimulate cells to produce a variety of inflammatory cytokines, such as IL-1β, IL-6, IL-8 and MCP-1,16,17 all of which are also induced during KSHV infection and highly expressed in patients with KSHV-related malignancies.27–29 Therefore, it will be interesting to understand whether co-infection with Mtb manipulates host immune responses and tissue-specific microenvironments in a manner that further drives KSHV-induced tumorigenesis, especially in immunocompromised patients. To our knowledge, this is the first study reporting that Mtb products have impacts on KSHV-infected cells, highlighting the need to further study the interactions of these two pathogens commonly found in people living with HIV.
Figure 2. KSHV-infected pulmonary endothelial cells are resistant to ESAT-6 mediated inhibition of cell growth.
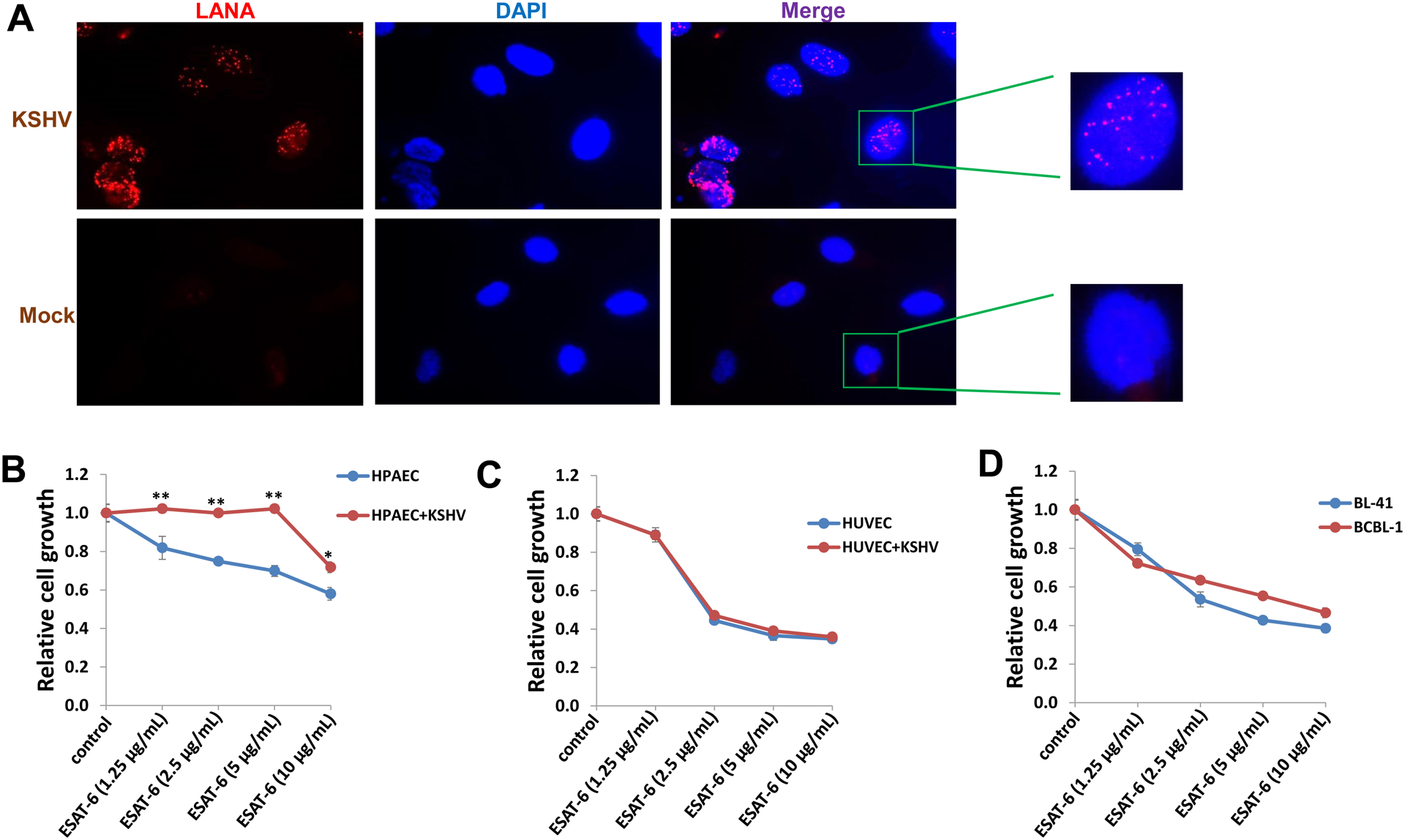
(A) Human pulmonary artery endothelial cells (HPAEC) were mock infected or infected with purified KSHV (MOI~10). Immunofluorescence was performed 48 h post-infection to quantify the number of cells expressing KSHV-encoded LANA, as indicated by the typical intranuclear, punctate staining pattern (red dots). Nuclei were identified using DAPI (blue). (B-D) Infected and uninfected HPAEC and HUVEC, BCBL-1 or BL-41 (a KSHV negative lymphoma cell line) were treated with the indicated concentrations of ESAT-6 protein for 48 h. Cellular proliferation relative to untreated cells was examined using the WST-1 cell proliferation assays (Roche). Error bars represent the S.D. for 3 independent experiments. * = p<0.05, ** = p<0.01.
Supplementary Material
Acknowledgement:
This work was supported by NIH/NCI R01CA228166. Additional support was provided in part by the Arkansas Bioscience Institute, the major research component of the Arkansas Tobacco Settlement Proceeds Act of 2000 and by the Presidential Research Council of the University of Texas Health Science Center at Tyler. Funding sources had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Conflict of interest
All the authors have declared no conflict of interest.
References
- 1.Dittmer DP, Damania B. Kaposi’s Sarcoma-Associated Herpesvirus (KSHV)-Associated Disease in the AIDS Patient: An Update. Cancer Treat Res. 2019;177:63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goncalves PH, Ziegelbauer J, Uldrick TS, Yarchoan R. Kaposi sarcoma herpesvirus-associated cancers and related diseases. Curr Opin HIV AIDS. 2017;12(1):47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mesri EA, Cesarman E, Boshoff C. Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010;10(10):707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Safai B, Johnson KG, Myskowski PL, et al. The natural history of Kaposi’s sarcoma in the acquired immunodeficiency syndrome. Ann Intern Med. 1985;103(5):744–750. [DOI] [PubMed] [Google Scholar]
- 5.Krown SE, Testa MA, Huang J. AIDS-related Kaposi’s sarcoma: prospective validation of the AIDS Clinical Trials Group staging classification. AIDS Clinical Trials Group Oncology Committee. J Clin Oncol. 1997;15(9):3085–3092. [DOI] [PubMed] [Google Scholar]
- 6.Restrepo CS, Martinez S, Lemos JA, et al. Imaging manifestations of Kaposi sarcoma. Radiographics. 2006;26(4):1169–1185. [DOI] [PubMed] [Google Scholar]
- 7.Palmieri C, Dhillon T, Thirlwell C, et al. Pulmonary Kaposi sarcoma in the era of highly active antiretroviral therapy. HIV Med. 2006;7(5):291–293. [DOI] [PubMed] [Google Scholar]
- 8.Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332(18):1186–1191. [DOI] [PubMed] [Google Scholar]
- 9.Oksenhendler E, Boutboul D, Galicier L. Kaposi sarcoma-associated herpesvirus/human herpesvirus 8-associated lymphoproliferative disorders. Blood. 2019;133(11):1186–1190. [DOI] [PubMed] [Google Scholar]
- 10.World Health Organization. 2018. Global tuberculosis report 2018. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 11.Hsu T, Hingley-Wilson SM, Chen B, et al. The primary mechanism of attenuation of bacillus Calmette-Guerin is a loss of secreted lytic function required for invasion of lung interstitial tissue. Proc Natl Acad Sci U S A. 2003;100(21):12420–12425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao LY, Guo S, McLaughlin B, Morisaki H, Engel JN, Brown EJ. A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT-6 secretion. Mol Microbiol. 2004;53(6):1677–1693. [DOI] [PubMed] [Google Scholar]
- 13.Wang X, Barnes PF, Dobos-Elder KM, et al. ESAT-6 inhibits production of IFN-gamma by Mycobacterium tuberculosis-responsive human T cells. J Immunol. 2009;182(6):3668–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Peng H, Wang X, Barnes PF, Tang H, Townsend JC, Samten B. The Mycobacterium tuberculosis early secreted antigenic target of 6 kDa inhibits T cell interferon-gamma production through the p38 mitogen-activated protein kinase pathway. J Biol Chem. 2011;286(27):24508–24518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X, Barnes PF, Huang F, et al. Early secreted antigenic target of 6-kDa protein of Mycobacterium tuberculosis primes dendritic cells to stimulate Th17 and inhibit Th1 immune responses. J Immunol. 2012;189(6):3092–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boggaram V, Gottipati KR, Wang X, Samten B. Early secreted antigenic target of 6 kDa (ESAT-6) protein of Mycobacterium tuberculosis induces interleukin-8 (IL-8) expression in lung epithelial cells via protein kinase signaling and reactive oxygen species. J Biol Chem. 2013;288(35):25500–25511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jung BG, Wang X, Yi N, Ma J, Turner J, Samten B. Early Secreted Antigenic Target of 6-kDa of Mycobacterium tuberculosis Stimulates IL-6 Production by Macrophages through Activation of STAT3. Sci Rep. 2017;7:40984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramdial PK, Sing Y, Subrayan S, et al. Granulomas in acquired immunodeficiency syndrome-associated cutaneous Kaposi sarcoma: evidence for a role for Mycobacterium tuberculosis. J Cutan Pathol. 2010;37(8):827–834. [DOI] [PubMed] [Google Scholar]
- 19.Ramdial PK, Miles E, Pillay B. The Emerging Histopathologic Diagnostic Challenges of Kaposi Sarcoma in the Acquired Immunodeficiency Syndrome Era. Austin J HIV/AIDS Res. 2016;3(1):1020. [Google Scholar]
- 20.Tribuna C, Angela C, Eira I, Carvalho A. Pulmonary Kaposi sarcoma and disseminated Mycobacterium genavense infection in an HIV-infected patient. BMJ Case Rep. 2015;2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Su CC, Lai CL, Tsao SM, et al. High prevalence of human herpesvirus type 8 infection in patients with pulmonary tuberculosis in Taiwan. Clin Microbiol Infect. 2015;21(3):266 e265–267. [DOI] [PubMed] [Google Scholar]
- 22.Chen J, Dai L, Goldstein A, et al. Identification of new antiviral agents against Kaposi’s sarcoma-associated herpesvirus (KSHV) by high-throughput drug screening reveals the role of histamine-related signaling in promoting viral lytic reactivation. PLoS Pathog. 2019;15(12):e1008156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Srivastava S, Ernst JD. Cell-to-cell transfer of M. tuberculosis antigens optimizes CD4 T cell priming. Cell Host Microbe. 2014;15(6):741–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bussi C, Gutierrez MG. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol Rev. 2019;43(4):341–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chakraborty S, Veettil MV, Chandran B. Kaposi’s Sarcoma Associated Herpesvirus Entry into Target Cells. Front Microbiol. 2012;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aguilar B, Choi I, Choi D, et al. Lymphatic reprogramming by Kaposi sarcoma herpes virus promotes the oncogenic activity of the virus-encoded G-protein-coupled receptor. Cancer Res. 2012;72(22):5833–5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bhutani M, Polizzotto MN, Uldrick TS, Yarchoan R. Kaposi sarcoma-associated herpesvirus-associated malignancies: epidemiology, pathogenesis, and advances in treatment. Semin Oncol. 2015;42(2):223–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee HR, Amatya R, Jung JU. Multi-step regulation of innate immune signaling by Kaposi’s sarcoma-associated herpesvirus. Virus Res. 2015;209:39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Broussard G, Damania B. KSHV: Immune Modulation and Immunotherapy. Front Immunol. 2019;10:3084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.