

Abstract
Almond [Prunus dulcis Miller (D.A. Webb)] is the main tree nut species worldwide. Here, genotyping-by-sequencing (GBS) was applied to 149 almond cultivars from the ex situ collections of the Italian Council for Agricultural Research (CREA) and the Spanish National Research Council (CSIC), leading to the detection of 93,119 single-nucleotide polymorphisms (SNPs). The study of population structure outlined four distinct genetic groups and highlighted diversification between the Mediterranean and Californian gene pools. Data on SNP diversity and runs of homozygosity (ROHs) allowed the definition of kinship, inbreeding, and linkage disequilibrium (LD) decay in almond cultivated germplasm. Four-year phenotypic observations, gathered on 98 cultivars of the CREA collection, were used to perform a genome-wide association study (GWAS) and, for the first time in a crop species, homozygosity mapping (HM), resulting in the identification of genomic associations with nut, shell, and seed weight. Both GWAS and HM suggested that loci controlling nut and seed weight are mostly independent. Overall, this study provides insights on the almond cultivation history and delivers information of major interest for almond genetics and breeding. In a broader perspective, our results encourage the use of ROHs in crop science to estimate inbreeding, choose parental combinations minimizing the risk of inbreeding depression, and identify genomic footprints of selection for specific traits.
Subject terms: Plant breeding, Natural variation in plants, Agricultural genetics
Introduction
Almond (Prunus dulcis Miller (D.A. Webb), syn. Prunus amygdalus L., 2n = 2x = 16) is one of the oldest domesticated tree species, presumably originating in the first half of Holocene1. Starting from the Fertile Crescent, almond cultivation rapidly spread westwards and eastwards through human migration and commercial routes. At present, almond is the main tree nut species worldwide, with an estimated production of 2.2 million tonnes (FAOSTAT data 2017). Approximately 80% of the global almond cultivated area is concentrated in California and the Mediterranean region (FAOSTAT data 2017). Clonal cultivars are widely grown in developed countries, whereas seedling populations mainly occur in developing countries.
Information on the genetic structure of crop species is pivotal for the correct management of ex situ germplasm collections and the efficient implementation of breeding programs, which should be based on the hybridization of genetically diverse individuals2. In addition, geographic patterns in the genetic structure may reveal key events, including routes of dissemination, associated with crop cultivation histories3,4. So far, studies aiming at the characterization of the almond genetic structure used a few simple sequence repeat (SSR) markers5–9. Nowadays, high-throughput genotyping methods, such as genotyping-by-sequencing (GBS), are routinely applied in agrigenomics research for the fine-scale characterization of genetic structure with thousands of single-nucleotide polymorphism (SNP) markers10–12.
Inbreeding depression, i.e. the reduced fitness of offspring of related individuals13, has been widely documented in human, animal, and plant populations. In almond, inbreeding depression leads to declined vegetative vigor and dramatic reduction of flower and fruit set14–16. Therefore, information on kin relationships among cultivars, which can be reconstructed from pedigrees or inferred a posteriori from genotypic data17,18, is of great value for almond breeders to minimize the risk of inbreeding depression. In addition, it would be interesting to investigate whether some of the almond cultivars currently grown on a large scale are inbred, and thus may display deleterious phenotypes due to inbreeding depression.
The level of inbreeding of an individual (F) depends on the extent of homologous chromosome segments displaying identity by descent (IBD), i.e., deriving from the same recent common ancestor19. Therefore, F can be estimated by the pedigree inbreeding coefficient (FP), which expresses IBD probabilities based on pedigree data. However, the use of FP has two major limitations, i.e., the strong assumption that the pedigree founders are unrelated, and the difficulty to retrieve pedigree information20. Another estimator of F (here named FPLINK, as it can be calculated by the widely used PLINK bioinformatics toolset) is based on the positive correlation occurring between IBD and the ratio between homozygosity observed and expected at Hardy–Weinberg equilibrium17.
To date, high-throughput genotyping enables the assessment of inbreeding through the direct detection of IBD segments. These appear as long chromosomal stretches of homozygous marker loci, referred to as runs of homozygosity (ROHs). ROHs were proven to be more accurate than FP when genotyping with thousands of SNP markers19 and are therefore commonly used to estimate inbreeding in human and cattle population studies21,22. In contrast, only a few works used ROHs to estimate inbreeding in plants23.
Seed and nut weight are economically important traits associated with the almond cultivation and processing industry. Specifically, seed weight is a major determinant for the almond final market utilization, whereas nut weight, which is given by the sum of the seed and shell weight, is important to set up appropriate harvesting, dehulling, transportation, and storing strategies24. Previous studies indicated that seed and nut weight are highly heritable and weakly correlated with the production density25–27. In addition, QTLs significantly associated with almond seed and nut weight were mapped on chromosomes 1, 2, and 724.
The recent publication of the almond genome sequence28,29 provides the opportunity to carry out genome-wide association studies (GWASs), identifying associations between phenotypes and markers with the known chromosomal locations. Information on the average linkage disequilibrium (LD) decay in the organism on which a GWAS is performed is of main importance, as this parameter influences, for a given number of markers, the chance to reveal significant associations12,30. In addition, rapid LD decay increases the possibility that GWAS experiments lead to the identification of marker loci residing within, or in the proximity of, genes causally related to the phenotype.
Besides GWAS, homozygosity mapping (HM) is another approach enabling the disclosure of genotype–phenotype relationships, which consists in testing the association between traits of interest and ROHs31. Compared to GWAS, HM has the major advantage of using a lower number of covariates, thus reducing type II error associated with multiple correction tests. Practically, this means that HM may reveal genomic associations that escape GWAS detection. In addition, IBD identified by HM can highlight genomic regions associated with inbreeding depression or selective pressure32. Indeed, in medical genomics, HM is performed to map homozygosity derived from consanguinity and associated with recessive diseases33, whereas, in animal science, it is carried out to highlight genomic regions resulting from human selection for specific traits32,34. To the best of our knowledge, HM has been never applied in plant research.
Here, we aimed to study genetic structure, kinship, inbreeding, and average LD decay in almond cultivated germplasm, through GBS-based identification of SNPs and ROHs. In addition, we addressed the detection of genomic regions associated with nut, shell, and seed weight, based on HM and GWAS.
Results
GBS results and quality control
Sequencing of a GBS library based on 149 almond cultivars (Supplementary Table S1) generated about 2.5 million reads/sample. Mean read depth in individual cultivars is reported in Supplementary Fig. S1. On average, SNP calling was supported by 47.08 reads per locus.
The SNP filtering procedure generated 93,119 markers, resulting in an average density of one SNP/2.18 Kb. The almond chromosomes 1–8 contained 18,357, 12,244, 10,733, 10,559, 8818, 12,680, 9679, and 10,049 SNPs, respectively. The cultivars Del Cid, Peraleja, and Lauranne were excluded from downstream analyses, as they displayed low genotypic call rates.
Based on pairwise identity by state (IBS) distance among biological replicates of the same cultivar, 11 clonal groups (CG1–11) were identified (Supplementary Table S2). For each clonal group, the cultivar associated with the highest SNP call rate was selected to represent the group, leading to a panel of 131 genetically distinct cultivars used for further analyses.
Population structure
Analysis with the parametric clustering method implemented by the software ADMIXTURE35 indicated that a model with four ancestral populations (C1–C4) was the most suitable to describe genetic structure (Supplementary Fig. S2). Based on their membership coefficient (qi), 105 cultivars were assigned to one of the ancestral populations, whereas 26 cultivars were assigned to the admixed group. C1 and C2, composed by 19 and 27 cultivars, respectively, mainly include Italian germplasm; C3 encompasses 38 cultivars originating from several Mediterranean countries; finally, C4 is formed by 21 cultivars, all from U.S and Ukrainian origin, except for the French cultivar Sultana and the Greek cultivar Symmetrike (Fig. 1a). Support to the ADMIXTURE results was provided by a nonparametric study of the genetic structure by principal component analysis (PCA), as the first three principal components clearly differentiated cultivars assigned to different ancestral populations (Fig. 1b).
Fig. 1. Genetic structure of almond cultivated germplasm.

a Results of ADMIXTURE analysis for K = 4. Each cultivar is represented by a vertical bar. The length and the color of segments in each bar represent the proportion of the genome (qi) contributed by each of four ancestral populations (C1–C4). Cultivars assigned to one of the populations have a membership coefficient (qi) for that population >0.6. The remaining cultivars are classified as admixed. b Scatter plot for genetic variation explained by the top three principal components. Cultivars assigned to C1–C4 are colored in accordance with panel a. Admixed individuals are colored in gray. c Stacked bar plots indicating the ancestry of the Italian, French, Spanish, and U. S. almond populations, based on qi coefficients resulting from ADMIXTURE analysis. d Treemix output for the Italian, French, Spanish, and U. S. almond populations. Branch lengths represent the evolutionary change as resulting from the drift parameter. The migration weight describes the percentage of ancestry derived from the source population. The scale bar represents the average standard error (se) of the values of the covariance matrix
The French population displayed significant contributions from all the four ancestral populations C1–C4. Conversely, the Italian and Spanish populations displayed minimal contribution (<1%) from C4 and C1, respectively, and the U.S. population could be mostly referable to C4 (Fig. 1c). The modeling approach implemented in TreeMix36 highlighted the largest genetic distance between Italian and U.S. germplasm. Strong support was found for a model with one migration event between French and U. S. germplasm (Fig. 1d), which, compared with a model with no migration, increased the percentage of explained allele frequency covariance among populations from 0.97 to 1.
Kin relationships among cultivars
Kin relationships were predicted for known parent/offspring pairs present in the cultivar collection (“Cristomorto” (CG9)/”Ferragnès”, “Aï’/‘Ferragnès”, “Ferragnès”/”Antoñeta”, “Chino” (CG1)/“Antoñeta”, “Texas”/”Merced”, “Nonpareil”/”Merced”, “Nonpareil”/”Davey“, and “Nonpareil”/”Kapareil”) (Supplementary Table S1), with PI_HAT values ranging from 0.26 to 0.45. Several family clusters were identified, of which the largest included the U.S. cultivars Davey, Dhen, Drake, Kapareil, Merced, Ne Plus Ultra, Nonpareil, Peerlees (CG11), Ridenhome (CG5), Titan, and Vesta (Fig. 2). The Italian cultivar Rachelina displayed the highest number of kin relationships (22), with PI_HAT values ranging from 0.06 to 0.41 (Fig. 2 and Supplementary Table S3). The French cultivars Sultana and R1000, the Greek cultivar Symmetrike, and the Italian cultivar Chino (CG1) were the only Mediterranean cultivars displaying kinship with U.S. germplasm.
Fig. 2. Heatmap produced on estimated pairwise IBD values among almond cultivars.

Cladograms were produced with the unweighted pair group method with arithmetic mean (UPGMA) hierarchical clustering algorithm
Estimation of individual inbreeding
In total, 21,019 ROHs were detected in the panel of cultivars, leading to an average of 160.45 ROHs/cultivar. In Supplementary Fig. S3, the distribution of the percentage of missing data per ROH is reported. Weak correlation (adjusted R2 = 0.16) was found between mean read depth per cultivar and ROH count per cultivar (Supplementary Fig. S4). Ranking according to ROH count indicated a high level of inbreeding in several U.S. cultivars, with Mono displaying the highest ROH count (374) (Fig. 3a). The lowest ROH count (20) was observed in the Italian cultivar Piscalze. A similar ranking was obtained when sorting cultivars according to the cumulative ROH length, with the U.S. cultivar Wawona and the Italian cultivar Piscalze showing the highest (82.9 Mb) and the lowest (2.9 Mb) values, respectively (Supplementary Fig. S5). A strong correlation (adjusted R2 = 0.84) was found between ROH count and FPLINK (Fig. 3b).
Fig. 3. Study of inbreeding in almond cultivated germplasm.

a Ranking of cultivars based on the count of runs of homozygosity (ROHs). b Linear regression model between ROH count and the PLINK inbreeding coefficient (FPLINK)
Almond average LD decay
Correlation of the allelic state at pairs of different loci, expressed through the pairwise squared correlation coefficient R2, was equal to 0.141 on average. This value assumed as the lower threshold to declare LD between two loci was reached on average after 130 bp (Fig. 4). Mean pairwise R2 values were quite similar for marker loci within individual chromosomes, ranging from 0.132 (chromosome 6) to 0.147 (chromosome 2) (Supplementary Table S4).
Fig. 4. Pattern of LD decay.
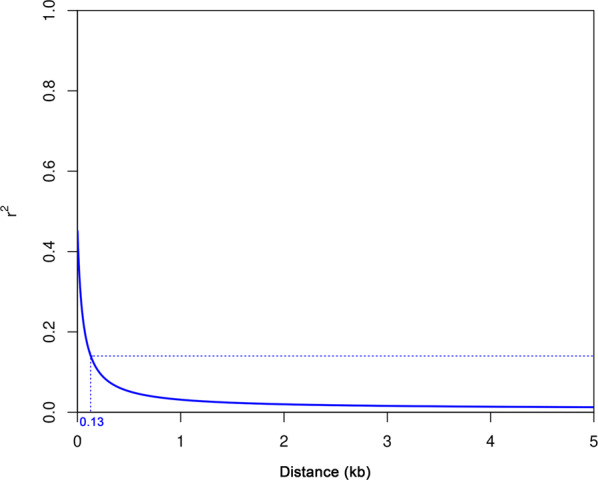
On average, R2 = 0.141, assumed as the lower threshold to declare LD between two loci, is reached after 130 bp
Homozygosity mapping and genome-wide association study
The distribution of best linear unbiased predictors (BLUPs) calculated for nut, shell, and seed weight is reported in Supplementary Fig. S6. The null hypothesis of normal distribution could be accepted for all the traits (Kolmogorov–Smirnov test P value > 0.05). A high correlation was found between nut and shell weight (adjusted R2 = 0.96), whereas a weak correlation was found between nut and seed weight (adjusted R2 = 0.1). No significant correlation was found between shell and seed weight (Supplementary Fig. S7).
HM suggested an association between nut weight and three clusters of ROHs located on the almond chromosomes 1, 2, and 7 (Fig. 5 and Supplementary Table S5), with the cluster ROH_2_16414730 displaying the highest indication of significance (−log10 (P value) = 5.17; false discovery rate (FDR) P value = 6 × 10−3). In accordance with the correlation pattern found for phenotypic data, the same clusters of ROHs associated with nut weight were also associated with shell weight (Fig. 5 and Supplementary Table S5), but not with seed weight. Indication of association with seed weight was found for two ROHs on chromosomes 1 and 6 (Fig. 5 and Supplementary Table S5). Notably, cultivars contributing to the clusters of ROHs identified by HM displayed significantly higher nut or seed weight (Table 1), indicating that such homozygous regions might represent genomic footprints of selection for larger nuts and seeds.
Fig. 5. Homozygosity mapping (HM) for nut, shell, and seed weight.

a Genomic distribution of clusters of runs of homozygosity (ROHs), whose loci occur in ROHs identified in at least ten cultivars. Red dots indicate association for FDR-corrected P value < 0.1. b Q–Q plots relative to the regression models
Table 1.
Comparison of nut and seed weight BLUP means calculated for cultivars contributing (+) or not contributing (−) to clusters of ROHs identified by HM analysis
| Mean ± se ROH ( + ) | Mean ± se ROH (-) | P value | ||
|---|---|---|---|---|
| Nut weight | ROH_S1_11260515 | 1.36 ± 1.12 | −0.12 ± 0.10 | 6.37E-04 |
| ROH_S2_16414730 | 1.43 ± 0.46 | −0.22 ± 0.10 | 1.29E-06 | |
| ROH_S7_11701812 | 1.62 ± 0.96 | −0.11 ± 0.11 | 4.86E-04 | |
| Seed weight | ROH_S1_26367663 | 0.14 ± 0.06 | −0.03 ± 0.02 | 8.80E-04 |
| ROH_S6_20767156 | 0.36 ± 0.13 | −0.02 ± 0.02 | 2.85E-05 |
For each comparison, P values associated with two-tailed t test are reported.
In total, 57 GWAS signals were identified for nut weight (Fig. 6 and Supplementary Table S6). By far, the highest indication of association (-log10 (P value) = 11.05; FDR P value = 8.22 × 10−7) was found for the marker S1_30936643, residing in the putative promoter sequence (218 bp upstream the start codon) of the aspartyl protease gene Prudu_003450_v1.0. In accordance with the results of HM, SNP loci associated with nut weight were in most cases also associated with shell weight (Fig. 6 and Supplementary Table S6). A single GWAS signal (−log10 (P value) = 6.19; FDR P value = 0.06) was detected for seed weight, for a marker (S1_2496687) located within the gene Prudu_000307_v1.0, encoding a putative pathogenesis-related thaumatin superfamily protein (Fig. 6 and Supplementary Table S6).
Fig. 6. Genome-wide association study (GWAS) for nut, shell, and seed weight.

a Manhattan plots. Red dots indicate association for FDR-corrected P value < 0.1. b Q–Q plots relative to the regression models
Discussion
In this study, we report the characterization of genome-wide diversity and homozygosity in almond, which were used to provide information of major interest for fundamental research and breeding purposes.
Calculation of pairwise IBS distances resulted in the identification of 11 clonal groups (Supplementary Table S2), thus providing valuable data for the rationalization of germplasm collections. The composition of CG1 is in accordance with previous scientific literature, indicating synonymy between the cultivars Tuono and Troito37. We cannot exclude that some of the cultivars included in the same CG may differ for a few clonal mutations. Concerning CG1, it was previously reported that the cultivar Supernova was derived from “Tuono” by experimental mutagenesis38.
Analysis of almond genetic structure assigned cultivars to four ancestral populations (Fig. 1a), although caution should be taken in such interpretation of ADMIXTURE clustering results39. Cultivars classified as admixed, corresponding to ~20% of the total, might reflect hybridization between different ancestral populations. It is generally thought that almond was introduced to Italy by ancient Greeks and Phoenicians37, and from there spread to France and Spain, possibly through Ancient Romans expanding in the Mediterranean area. However, the Italian almond population almost completely lacked the C4 ancestry, which was significant for the Spanish and French gene pools. This evidence can be explained by further historical introductions of almond germplasm to Spain and France, possibly from North Africa in relation to the Arabic domination of the Iberic Peninsula and the colonial period37.
In accordance with previous investigations based on SSR markers6–8, the study of population structure also indicated genetic differentiation between Mediterranean and U.S. cultivars, with these last mostly referable to the ancestral cluster C4 (Fig. 1). This result most likely reflects the founder effect associated with the recent introduction of almond to the New World. Importantly, migration modeling using the TreeMix algorithm (Fig. 1d) indicated that French germplasm played an important role as a parental source for Californian almond breeding, in accordance with previous literature40–42.
Estimation of pairwise IBD through the PI_HAT parameter correctly indicated kinship for known parent/offspring cultivar pairs occurring in the almond collection genotyped in this study, as these were associated with values ranging from 0.26 to 0.45. The discrepancy with the theoretical PI_HAT value of 0.5 (i.e., 50% of the alleles originating from the same ancestral chromosomes) might be due to the violation of the assumption of random mating used for PI_HAT estimation17 and under-calling of heterozygous loci associated with the GBS method43. Besides confirming known kinship, IBD analysis unveiled several familial relationships that were not reported in the literature (Fig. 2 and Supplementary Table S3). This result, besides contributing to uncover the pedigree of almond cultivated germplasm, provides useful information to avoid hybridization of related individuals in breeding programs, thus minimizing the risk of inbreeding depression.
A large family group was composed by several U.S. cultivars, including “Nonpareil”. This is consistent with the recurrent use of “Nonpareil”, considered as standard for superior tree and nut characteristics, in U.S. breeding programs14,44. Several cultivars displayed kinship with CG1 and CG9, containing the Italian cultivars Tuono and Cristomorto, in accordance with the extensive use of these two cultivars in breeding as a source of self-compatibility. Surprisingly, the Italian cultivar Rachelina, which is not reported in main pedigree records, displayed the highest number of kin relationships (22), not only with Italian germplasm, but also with the French cultivars Rabasse and Tournefort, and the Ukrainian cultivar Picantili. The identification of kinship between “Sultana” and “Texas” further indicates the role of French introductions as founders of U.S. breeding programs. Remarkably, “Sultana” was previously indicated as one of the few commercial cultivars introduced to California from the Languedoc area of Southern France from 1850 to 1900, representing the basis of the U.S. almond industry37,41,45. Finally, the kinship between Ukrainian cultivars (“Crimsky”, “Nikitsky”, “Nessebre”, “Picantili”) and cultivars from Italy and U.S. is consistent with the use of foreign germplasm in breeding programs held at the Ukrainian Nikita Botanical Garden46,47.
Similarly to the work by Wu et al.23, which focused on cultivated Citrus species, we searched for ROHs to estimate the level of inbreeding in individual cultivars (Fig. 3a and Supplementary Fig. S5). A high correlation was found between ROH count per individual and FPLINK inbreeding coefficient (Fig. 3b). However, we stress that, differently from ROHs, FPLINK is an indirect estimator of F, based on increased homozygosity associated with IBD. Several U. S. cultivars were characterized by high ROH count and length, indicating a high level of inbreeding. This is in accordance with our finding that a high level of IBD occurs within U.S. germplasm. In contrast, Lansari et al.14, based on the FP coefficient, concluded that most U.S. cultivars are non-inbred, possibly due to incomplete pedigree information.
It is known that one of the major technical drawbacks of GBS is uneven read depth among samples43. To evaluate whether this generated a severe bias in heterozygous loci, and thus ROH, calling, we performed a regression analysis between mean read depth per cultivar and ROH count per cultivar. We found a weak correlation between the two variables, although two cultivars, “Mono” and “Ramillete”, associated with extremely low mean read depth, also displayed the highest ROH count (Supplementary Fig. S4). This indicates that: (1) with a few exceptions, our GBS approach was successful in quantifying the level of inbreeding through ROH identification; (2) when available, SNP array platforms, allowing accurate heterozygosity call, should be preferred over GBS to identify ROHs. Missing data did not have a major impact on ROH call, as most ROHs contained a low percentage of missingness (Supplementary Fig. S3).
Homozygosity mapping31, a strategy successfully used in animal science to associate ROHs with traits under anthropic selection34,48, was herein applied for the first time to a crop species. Our results defined IBD segments which could have arisen from selection for larger nuts and seeds. In addition, our data suggest that selection for larger nuts, while increasing the weight of the fruit endocarp (the almond shell), did not have a substantial effect on the almond edible part, i.e., the seed (Fig. 5). ROH_ 2_16414730, displaying the highest evidence of association with nut and shell weight, includes two members of the PLAC8 protein family, previously associated with fruit size in tomato, maize, and rice49 (Supplementary Table S5). Concerning seed weight, an interesting candidate for future functional studies is a Cyclin D3 gene located within ROH_S6_20767156, as it was shown that D-type cyclins play a major role in seed development50.
In accordance with the results of HM, GWAS suggested that loci controlling nut weight and seed weight are mostly independent and that there is parallel control of nut and shell weight by several genomic loci (Fig. 6). Many of the GWAS peaks identified for these two traits were located within genes encoding transcription factors or response factors to the phytohormones abscisic acid, auxin and ethylene (Supplementary Table S6). These protein categories are renowned to be major players in fruit growth and development51, therefore they are obvious candidates to have a similar physiological role in almond. The highest significance level for nut and shell weight was found for a SNP variant located upstream of a putative aspartyl protease gene (Supplementary Table S6). Remarkably, the recent proteomic study by Rodriguez et al.52 indicated that the development of the peach endocarp (corresponding to the almond shell), is accompanied by an outstanding variation of protein degradation enzymes, including aspartyl proteases. It is thought that amino acids derived from the degradation of proteins stored in the early immature fruit act as substrates for the phenylpropanoid and lignin pathways activated during endocarp hardening53.
Concerning seed weight, the association was found with a SNP residing in a gene putatively encoding a member of the thaumatin-like protein (TLP) superfamily (Supplementary Table S6). Although some of the TLP proteins have been related to biotic stresses, the role of most members of the TLP superfamily remains unknown54, thus it cannot be excluded they might also have a role in determining seed growth. With this respect, we highlight that some TLP proteins, referred to as permeatins, accumulate in high concentration in seeds of cereals54,55.
No overlap was found between genomic regions identified by GWAS and HM. It should be pointed out that GWAS and HM search for different kind of genomic associations, in the first case with a specific marker allele, and in the second with one or more combinations of alleles at the homozygous state. In addition, different results from the two approaches may arise from the different number of covariates used for association tests. We could not assess whether signals on the same chromosome identified by our study and the one of Fernandez i Marti et al.24 are overlapping in the same genomic region, as the latter refers to a QTL linkage map obtained by a bi-parental population, rather than the almond genome sequence.
We found that almond displays one of the fastest LD decay ever characterized in a crop species, with R2 dropping to the threshold value after 130 bp on average (Fig. 4). This might reflect self-incompatibility displayed by most almond cultivars, which favors haplotype block-breaking through recombination. From a genetic perspective, rapid LD decline in almond reinforces the possibility that SNPs identified by this or future GWAS experiments are located within or in close association with genes determining phenotypic variation.
Materials and methods
Plant material
Plant material selected for this study includes 149 cultivars, of which 132 from the ex situ collection of CREA-AA (Italian Council for Agricultural Research and Analysis of Agricultural Economics—Section Agriculture and Environment), Bari, Italy, and 17 from the ex situ collection of CEBAS-CSIC (Spanish National Research Council—Center for Edaphology and Applied Biology of the Segura River), Murcia, Spain (Supplementary Table S1). Pedigree information, available for the cultivars Antoñeta, Davey, Ferragnés, Kapareil, and Merced, is reported in Supplementary Table S1.
GBS assay and quality control
Leaf tissue samples were collected from three biological replicates of the cultivar R1000, two biological replicates of the cultivars Ardechoise, Ferragnès, Filippo Ceo, Marcona, and Desmayo Largueta, and one individual of the remaining cultivars. DNA was extracted using the DNeasy Plant Mini Kit (Qiagen) and assayed for quality and concentration using agarose gel (0.8%) electrophoresis and the Qubit 3.0 fluorometer (Life Technologies). A GBS library was prepared as reported by Elshire et al.10, using the restriction enzyme ApeKI (The Elshire Group Ltd.). Paired-end sequencing was performed using the HiSeq2500 device (Illumina), including an empty negative control well. The TASSEL-GBS pipeline43 and the almond reference genome28 were used for SNP calling and mapping.
Quality control was carried out using TASSEL v.556. Specifically, marker quality control was performed by filtering for biallelic SNP loci with minor allele frequency >0.05 and call rate >0.7. As for genotype quality control, cultivars associated with an overall SNP call rate >0.6 were selected. In addition, mean and sd of the pairwise IBS distance17 between biological replicates were used to set up an IBS minimal threshold (mean−3 × sd) to declare clonal groups and select, within each group, the cultivar with the highest SNP call rate.
Analysis of population structure
Genetic structure was studied using SNPs in approximate linkage equilibrium, which were obtained using the LD pruning algorithm in PLINK v.1.90p17. This calculates pairwise R2 for all marker pairs in sliding windows with a size of 50 markers and an increment of 5 markers and removes the first marker of pairs, in which R2 < 0.5.
Analysis with the ADMIXTURE parametric model35 was performed with a number of ancestral populations (K) ranging from 1 to 15. One thousand bootstrap replicates were run to estimate parameter standard errors. The most suitable number of K was selected in correspondence with the lowest cross‐validation (CV) error. Cultivars were assigned to one specific ancestral population when the membership coefficient qi for that cluster was >0.6. If not, they were considered admixed.
PCA on SNP data was performed using SVS v.8.8.3 (Golden Helix Inc.), and a three-dimensional plot was obtained using the top three components identified with default parameters of the additive model.
TreeMix (v1.12)36 was used to infer splits and mixtures among Italian, French, Spanish, and U.S. germplasm, testing a model with no migration, and models with all the three possible migration events among the four populations. The “get_f()” R function was used to obtain the variance explained by each model.
Inference of kinship
In order to infer kinship among cultivars, pairwise genotype probabilities (P) of sharing 0, 1, or 2 IBD alleles were calculated at each locus, given IBS distances and allele frequencies, using the method-of-moments algorithm implemented in PLINK v.1.90p17. The matrix obtained with the PI_HAT parameter, given by P(IBD = 2) + 0.5 × P (IBD = 1) and providing an estimate of the proportion of IBD alleles, was used to draw a clustered heatmap, using the pheatmap v.1.0.12. R package57.
ROH detection and quantification of individual inbreeding
ROH detection analysis was carried out with a subset of SNPs, selected for having MAF > 0.15. The algorithm implemented in SVS v.8.8.3 (Golden Helix Inc.) was used to identify completely homozygous genomic stretches on chromosomes 1–8 with at least 15 SNP loci and with a minimal length of 100 Kb. As a measure of inbreeding, the ROH count and the ROH total length were computed for each individual. The ggplot2 R package58 was used to visualize the distribution of the percentage of missing data per ROH and to perform a regression analysis between mean read depth per cultivar and ROH count per cultivar.
Individual inbreeding was also estimated using the FPLINK inbreeding coefficient, which was computed using the LD-pruned marker dataset as input. Regression analysis between ROH count, or ROH total length, and FPLINK coefficient, was performed using the ggplot2 R package58.
Estimation of LD decay
PLINK v.1.90p17 was used to calculate pairwise linkage disequilibrium (LD) between SNPs59, expressed as the squared correlation coefficient R2. The R2 values were plotted against the marker physical distance, and the Hill and Weir formula was used to describe the decay of r260. The mean pairwise R2 for markers within and between chromosomes was used to define a lower threshold value for LD. Mean pairwise R2 was also calculated for markers on individual chromosomes.
Phenotypic data collection and analysis
Phenotyping was carried out in 2006, 2007, 2009, and 2012, on 98 cultivars of the CREA-AA collection, grown at the experimental farm “La Piantata” (Bari, Italy) (41°02’29.9”N; 16°46’01.4”E, 126 m a.s.l.) (Supplementary Table S1). The orchard was established in 1968 according to a completely randomized design with three clonal replicates for each cultivar. Plants were grown according to traditional practices in Southern Italy, without irrigation, and trained as classic vase. Nut and seed weight were determined for each cultivar as the average of its three clonal replicates. In turn, data from each clone were determined as the average of thirty fruits. Data on the shell weight were obtained by the difference between nut and seed weight.
BLUPs of phenotypic traits collected over 4 years were calculated using the Lme4 R package61 and the following model: y = lmer (Trait ~ (1 | Genotype) + (1 | Year)). Normal distribution of BLUP data was verified using the Kolmogorov–Smirnov test implemented in the stats R package. Linear models to study correlation between BLUPs for nut, shell, and seed weight were generated using the ggplot2 R package58.
Homozygosity mapping
HM was carried out using options available in SVS v.8.8.3 (Golden Helix Inc.). Clusters of ROHs, defined as genomic regions of at least 100 Kb whose loci occur in ROHs of at least ten cultivars characterized at the phenotypic level, were identified. Repeated binary spectral clustering62 was used to trim boundaries of clusters of ROHs, in order to define homozygous regions highly overlapping among cultivars. Finally, a linear regression model was fit between clusters of ROHs and BLUPs, using the top five principal components as covariates to correct for population structure. The FDR correction was used to account for multiple testing and suggest an association for P < 0.1. BLUP means of cultivars either contributing or not contributing to clusters of ROHs associated with phenotypic traits were computed and compared using a two-tail Student’s t test. Genes included in clusters of ROHs identified by HM were retrieved by the Prunus dulcis (cv. Lauranne) v1.0 genome annotation available at the genomic database of Rosaceae28,63.
Genome-wide association study
The EMMAX linear mixed model64 was used for GWAS, using BLUPs as phenotypic values and the IBS matrix as a covariance matrix of random effects. The FDR correction was used to suggest the association for P < 0.1. Genes containing or flanking SNPs associated with phenotypic traits were retrieved by the Prunus dulcis (cv. Lauranne) v1.0 genome annotation available at the genomic database of Rosaceae28,63.
Supplementary information
Acknowledgements
We are grateful to Dr. Federico Dicenta for providing access to the CEBAS-CSIC almond germplasm collection.
Author contributions
S.P. planned and designed the experiment. R.M., C.L., and R.S.P. carried out laboratory work. P.L. and L.G. conducted field trials. S.P., C.D., N.D.A., and F.T. performed elaboration of the data. S.P. wrote the paper. N.D.A., F.T., L.R., and C.L. critically revised the paper. All authors read and approved the paper.
Data availability
The unfiltered variant call format (VCF) file relative to the GBS experiment is publicly available at the Figshare repository (doi: 10.6084/m9.figshare.12205652).
Conflict of interest
The authors declare that they have no conflict of interest.
Contributor Information
Stefano Pavan, Email: stefano.pavan@uniba.it.
Concetta Lotti, Email: t.lotti@unifg.it.
Supplementary information
Supplementary Information accompanies this paper at (10.1038/s41438-020-00447-1).
References
- 1.Delplancke M, et al. Evolutionary history of almond tree domestication in the Mediterranean basin. Mol. Ecol. 2013;22:1092–1104. doi: 10.1111/mec.12129. [DOI] [PubMed] [Google Scholar]
- 2.Acquaah, G. (ed.) Principles of Plant Genetics and Breeding (Wiley-Blackwell, 2012).
- 3.De Lorenzis G, et al. SNP genotyping elucidates the genetic diversity of Magna Graecia grapevine germplasm and its historical origin and dissemination. BMC Plant Biol. 2019;19:7. doi: 10.1186/s12870-018-1576-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pavan S, et al. Genotyping by sequencing of cultivated lentil (Lens culinaris Medik.) highlights population structure in the Mediterranean gene pool associated with geographic patterns and phenotypic variables. Front. Genet. 2019;10:872. doi: 10.3389/fgene.2019.00872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hamadeh B, Chalak L, Coppens d’Eeckenbrugge G, Benoit L, Joly HI. Evolution of almond genetic diversity and farmer practices in Lebanon: impacts of the diffusion of a graft-propagated cultivar in a traditional system based on seed-propagation. BMC Plant Biol. 2018;18:155. doi: 10.1186/s12870-018-1372-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rigoldi MP, Rapposelli E, De Giorgio D, Resta P, Porceddu A. Genetic diversity in two Italian almond collections. Electron. J. Biotechn. 2015;18:40–45. doi: 10.1016/j.ejbt.2014.11.006. [DOI] [Google Scholar]
- 7.Fernández i Martí A, et al. Molecular analyses of evolution and population structure in a worldwide almond [Prunus dulcis (Mill.) D.A. Webb syn. P. amygdalus Batsch] pool assessed by microsatellite markers. Genet. Resour. Crop. Ev. 2015;62:205–219. doi: 10.1007/s10722-014-0146-x. [DOI] [Google Scholar]
- 8.Font i Forcada C, et al. Identification of genetic loci associated with quality traits in almond via association mapping. PLoS ONE. 2015;10:e0127656. doi: 10.1371/journal.pone.0127656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halász J, et al. Genetic variability is preserved among strongly differentiated and geographically diverse almond germplasm: an assessment by simple sequence repeat markers. Tree Genet. Genomes. 2019;15:12. doi: 10.1007/s11295-019-1319-8. [DOI] [Google Scholar]
- 10.Elshire J, et al. A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE. 2011;6:e19379. doi: 10.1371/journal.pone.0019379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taranto F, Nicolia A, Pavan S, De Vita P, D’Agostino N. Biotechnological and digital revolution for climate-smart plant breeding. Agron. 2018;8:277. doi: 10.3390/agronomy8120277. [DOI] [Google Scholar]
- 12.Pavan, S. et al. Recommendations for choosing the genotyping method and best practices for quality control in crop genome-wide association studies. Front. Genet. 10.3389/fgene.2020.00447 (2020). [DOI] [PMC free article] [PubMed]
- 13.Charlesworth D, Willis JH. The genetics of inbreeding depression. Nat. Rev. Genet. 2009;10:783–796. doi: 10.1038/nrg2664. [DOI] [PubMed] [Google Scholar]
- 14.Lansari A, Iezzoni AF, Kester DE. Morphological variation within collections of Moroccan almond clones and Mediterranean and North American cultivars. Euphytica. 1994;78:27–41. [Google Scholar]
- 15.Alonso Segura JM, Socias i Company R. Negative inbreeding effects in tree fruit breeding: self-compatibility transmission in almond. Euphytica. 2007;115:151–158. doi: 10.1007/s00122-007-0532-2. [DOI] [PubMed] [Google Scholar]
- 16.Martínez-García PJ, Dicenta F, Ortega E. Anomalous embryo sac development and fruit abortion caused by inbreeding depression in almond (Prunus dulcis) Sci. Hortic. 2012;133:23–30. doi: 10.1016/j.scienta.2011.10.001. [DOI] [Google Scholar]
- 17.Purcell S, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conomos MP, Reiner AP, Weir BS, Thornton TA. Model-free estimation of recent genetic relatedness. Am. J. Hum. Genet. 2016;98:127–148. doi: 10.1016/j.ajhg.2015.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kardos M, Luikart G, Allendorf FW. Measuring individual inbreeding in the age of genomics: marker-based measures are better than pedigrees. Heredity. 2015;115:63–72. doi: 10.1038/hdy.2015.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kardos M, Taylor HR, Ellegren H, Luikart G, Allendorf FW. Genomics advances the study of inbreeding depression in the wild. Evol. Appl. 2016;9:1205–1218. doi: 10.1111/eva.12414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goszczynski D, et al. Runs of homozygosity in a selected cattle population with extremely inbred bulls: descriptive and functional analyses revealed highly variable patterns. PLoS ONE. 2018;13:e0200069. doi: 10.1371/journal.pone.0200069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yengo L, Wray NR, Visscher PM. Extreme inbreeding in a European ancestry sample from the contemporary UK population. Nat. Commun. 2019;10:3719. doi: 10.1038/s41467-019-11724-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu GA, et al. Genomics of the origin and evolution of Citrus. Nat. 2018;554:311–316. doi: 10.1038/nature25447. [DOI] [PubMed] [Google Scholar]
- 24.Fernández i Martí A, Font i Forcada C, Socias i Company R. Genetic analysis for physical nut traits in almond. Tree Genet. Genomes. 2013;9:455–465. doi: 10.1007/s11295-012-0566-8. [DOI] [Google Scholar]
- 25.Sorkheh K, et al. Correlations between quantitative tree and fruit almond traits and their implications for breeding. Sci. Hortic. 2010;125:323–331. doi: 10.1016/j.scienta.2010.04.014. [DOI] [Google Scholar]
- 26.Spiegel-Roy P, Kochba J. Inheritance of nut and kernel traits in almond (Prunus amygdalus Batsch) Euphytica. 1981;30:167–174. doi: 10.1007/BF00033674. [DOI] [Google Scholar]
- 27.Dicenta F, Garcia JE, Carbonell EA. Heritability of fruit characters in almond. J. Hort. Sci. 1993;68:121–126. doi: 10.1080/00221589.1993.11516335. [DOI] [Google Scholar]
- 28.Sánchez-Pérez R, et al. Mutation of a bHLH transcription factor allowed almond domestication. Sci. 2019;364:1095–1098. doi: 10.1126/science.aav8197. [DOI] [PubMed] [Google Scholar]
- 29.Alioto T, et al. Transposons played a major role in the diversification between the closely related almond and peach genomes: results from the almond genome sequence. Plant J. 2020;101:455–472. doi: 10.1111/tpj.14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bazakos C, Hanemian M, Trontin C, Jiménez-Gómez JM, Loudet O. New strategies and tools in quantitative genetics: how to go from the phenotype to the genotype. Annu. Rev. Plant Biol. 2017;68:435–455. doi: 10.1146/annurev-arplant-042916-040820. [DOI] [PubMed] [Google Scholar]
- 31.Lander ES, Botstein D. Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science. 1987;236:1567–1570. doi: 10.1126/science.2884728. [DOI] [PubMed] [Google Scholar]
- 32.Purfield DC, McParland S, Wall E, Berry DP. The distribution of runs of homozygosity and selection signatures in six commercial meat sheep breeds. PLoS ONE. 2017;12:e0176780. doi: 10.1371/journal.pone.0176780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Keller MC, et al. Runs of homozygosity implicate autozygosity as a schizophrenia risk factor. PLoS Genet. 2012;8:e1002656. doi: 10.1371/journal.pgen.1002656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim E-S, et al. Effect of artificial selection on runs of homozygosity in U.S. Holstein cattle. PLoS ONE. 2013;8:e80813. doi: 10.1371/journal.pone.0080813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pickrell JK, Pritchard JK. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012;8:e1002967. doi: 10.1371/journal.pgen.1002967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Socias i Company, R. & Gradziel, T. M. (eds) Almonds: Botany, Production and Uses (CABI Publishing, 2017).
- 38.Marchese A, Bošković RI, Martínez‐García PJ, Tobutt KR. The origin of the self‐compatible almond ‘Supernova’. Plant Breed. 2008;127:105–107. doi: 10.1111/j.1439-0523.2008.01421.x. [DOI] [Google Scholar]
- 39.Lawson DJ, van Dorp L, Falush D. A tutorial on how not to over-interpret STRUCTURE and ADMIXTURE bar plots. Nat. Commun. 2018;9:3258. doi: 10.1038/s41467-018-05257-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wickson, E. J. (ed.) The California Fruits and How to Grow Them (Dewey & Co., 1889).
- 41.Wood, M. N. (ed.) Almond Varieties in the United States (U.S. Department of Agriculture, 1925).
- 42.Kester DE. Almond cultivar and breeding programs in California. Acta Hortic. 1994;373:13–28. doi: 10.17660/ActaHortic.1994.373.1. [DOI] [Google Scholar]
- 43.Glaubitz JC, et al. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS ONE. 2014;9:e90346. doi: 10.1371/journal.pone.0090346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kester DE, Gradziel TM, Grasselly C. ALMOND (PRUNUS) Acta Hortic. 1991;290:699–758. [Google Scholar]
- 45.Jules, J. & Moore, J. N. (eds) Fruit Breeding, Volume III: Nuts (John Wiley & Sons, Inc., 1996).
- 46.Richter, A. A. Almond. Trudy gosudarstvennogo Nikitskogo botanicheskogo sada [Proc. of the State Nikita Botanical Garden] 47 [in Russian] (1972).
- 47.Denisov VP. Almond genetic resources in the USSR and their use in production and breeding. Acta Hortic. 1988;224:299–306. doi: 10.17660/ActaHortic.1988.224.37. [DOI] [Google Scholar]
- 48.Xu L, et al. Genomic patterns of homozygosity in Chinese local cattle. Sci. Rep. 2019;9:16977. doi: 10.1038/s41598-019-53274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Song W, et al. Rice PCR1 influences grain weight and Zn accumulation in grains. Plant Cell Environ. 2015;38:2327–2339. doi: 10.1111/pce.12553. [DOI] [PubMed] [Google Scholar]
- 50.Collins C, Dewitte W, Murray JAH. D-type cyclins control cell division and developmental rate during Arabidopsis seed development. J. Exp. Bot. 2012;63:3571–3586. doi: 10.1093/jxb/ers015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu S, et al. Tomato AUXIN RESPONSE FACTOR 5 regulates fruit set and development via the mediation of auxin and gibberellin signaling. Sci. Rep. 2018;8:2971. doi: 10.1038/s41598-018-21315-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodriguez CE, et al. Peach fruit development: a comparative proteomic study between endocarp and mesocarp at very early stages underpins the main differential biochemical processes between these tissues. Front. Plant Sci. 2019;10:715. doi: 10.3389/fpls.2019.00715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lombardo VA, et al. Metabolic profiling during peach fruit development and ripening reveals the metabolic networks that underpin each developmental stage. Plant Physiol. 2011;157:1696–1710. doi: 10.1104/pp.111.186064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu JJ, Sturrock R, Ekramoddoullah AK. The superfamily of thaumatin-like proteins: its origin, evolution, and expression towards biological function. Plant Cell Rep. 2010;29:419–436. doi: 10.1007/s00299-010-0826-8. [DOI] [PubMed] [Google Scholar]
- 55.Anžlovar S, Dermastia M. The comparative analysis of osmotins and osmotin-like PR-5 proteins. Plant Biol. 2003;5:116–124. doi: 10.1055/s-2003-40723. [DOI] [Google Scholar]
- 56.Bradbury PJ, et al. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–2635. doi: 10.1093/bioinformatics/btm308. [DOI] [PubMed] [Google Scholar]
- 57.Kolde, R. Pheatmap: pretty heatmaps. R package version 1.0.12. http://CRAN.R-project.org/package=pheatmap (2019).
- 58.Wickham, H. ggplot2: Elegant Graphics for Data Analysis. https://ggplot2.tidyverse.org (2016).
- 59.Chang CC, et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience. 2015;4:1–16. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hill WG, Weir BS. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 1988;33:54–78. doi: 10.1016/0040-5809(88)90004-4. [DOI] [PubMed] [Google Scholar]
- 61.Bates D, Maechler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. J. Stat. Softw. 2015;67:1–48. doi: 10.18637/jss.v067.i01. [DOI] [Google Scholar]
- 62.Zhang L, et al. cgaTOH: extended approach for identifying tracts of homozygosity. PLoS ONE. 2013;8:e57772. doi: 10.1371/journal.pone.0057772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jung S, et al. GDR (Genome Database for Rosaceae): integrated web-database for Rosaceae genomics and genetics data. Nucleic Acids Res. 2008;36:D1034–D1040. doi: 10.1093/nar/gkm803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kang HM, et al. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010;42:348–354. doi: 10.1038/ng.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The unfiltered variant call format (VCF) file relative to the GBS experiment is publicly available at the Figshare repository (doi: 10.6084/m9.figshare.12205652).