


Abstract
Genomic studies have indicated that certain bacterial lineages such as the Bacteroidetes lack Shine-Dalgarno (SD) sequences, and yet with few exceptions ribosomes of these organisms carry the canonical anti-SD (ASD) sequence. Here, we show that ribosomes purified from Flavobacterium johnsoniae, a representative of the Bacteroidetes, fail to recognize the SD sequence of mRNA in vitro. A cryo-electron microscopy structure of the complete 70S ribosome from F. johnsoniae at 2.8 Å resolution reveals that the ASD is sequestered by ribosomal proteins bS21, bS18 and bS6, explaining the basis of ASD inhibition. The structure also uncovers a novel ribosomal protein—bL38. Remarkably, in F. johnsoniae and many other Flavobacteriia, the gene encoding bS21 contains a strong SD, unlike virtually all other genes. A subset of Flavobacteriia have an alternative ASD, and in these organisms the fully complementary sequence lies upstream of the bS21 gene, indicative of natural covariation. In other Bacteroidetes classes, strong SDs are frequently found upstream of the genes for bS21 and/or bS18. We propose that these SDs are used as regulatory elements, enabling bS21 and bS18 to translationally control their own production.
Graphical Abstract
Graphical Abstract.
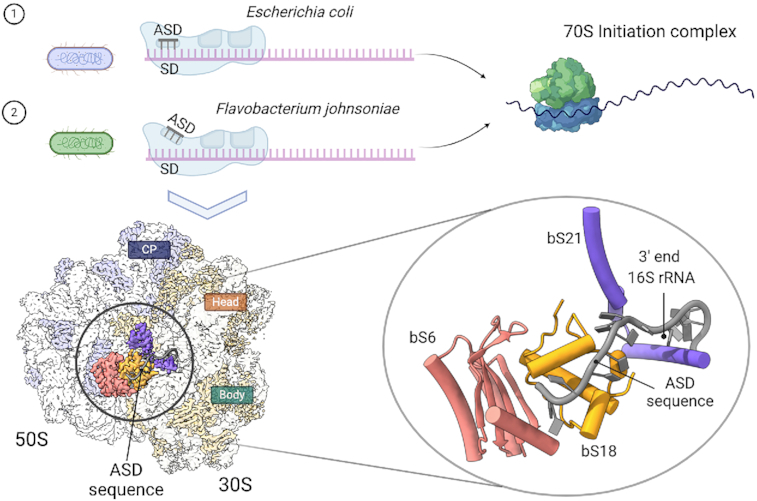
In most bacteria, start codon selection is guided by the Shine Dalgarno (SD) sequence. Bacteroidetes fail to recognize the SD sequence of mRNA. However, these bacteria retain the canonical anti-Shine-Dalgarno (ASD) sequence. The cryo-electron microscopy structure of the 70S ribosome from Flavobacterium johnsoniae, a representative of the Bacteroidetes reveals that the ASD is sequestered by ribosomal proteins bS21, bS18 and bS6, explaining the basis of ASD inhibition.
INTRODUCTION
The translation machinery must identify the correct start codon among all other AUG (and similar) trinucleotides to establish the reading frame for protein synthesis. In all cells, start codon selection is guided by intrinsic features of the mRNA. For prokaryotes, one such feature is the Shine-Dalgarno (SD) sequence, a purine-rich element (e.g. GGAGG) that lies 7–9 nucleotides (nt) upstream from the start codon (1–3). The SD base-pairs with the 30S subunit's anti-Shine-Dalgarno (ASD) sequence, a pyrimidine-rich element (CCUCC) near the 3′ end of 16S rRNA. SD-ASD interaction positions the start codon in the P site and stabilizes mRNA in the 30S initiation complex (IC) (4–8). The SD motif is commonly used to help delineate genes (9–13), and often the SD and start codon are collectively referred to as the ribosome binding site (RBS) (14–16). Numerous studies have shown that the SD, when present, plays an important role in initiation (17–19). However, faithful and efficient initiation can also occur on bacterial mRNAs that naturally have no SD (20–22).
In 2010, Nakagawa et al. analyzed the genomes of >200 representative bacteria and found that SD usage varies substantially (23). Remarkably, certain lineages including the Bacteroidetes and a subset of Cyanobacteria appear to lack SD sequences completely, a result confirmed in subsequent studies (24–26). With few exceptions (27,28), these bacteria retain the canonical anti-Shine-Dalgarno (ASD) sequence at the 3′ end of 16S rRNA. Yet, based on reporter gene studies in representative species, the corresponding ribosomes fail to recognize SD sequences in vivo (25,26). These observations imply that other mRNA determinants contribute to start codon selection in the Bacteroidetes and that some mechanism prevents SD-ASD pairing (or makes it inconsequential) during initiation.
The Bacteroidetes represent a large and understudied group of bacteria (29). They inhabit diverse environments, including the soil, ocean, and animal gut. Well known for their ability to import and degrade complex polysaccharides, members of the phylum account for a substantial portion of the intestinal microbiome of mammals (30–33). There, they play a crucial role in catabolic processes, impacting nutrient availability and uptake by the host intestinal epithelium. Human health problems such as irritable bowel syndrome, inflammatory bowel disease, obesity, type 2 diabetes and autism-spectrum disorders have been associated with under- or over-abundances of Bacteroidetes species in the gut microbiome (34,35). Gene expression in the Bacteroidetes differs substantially from that of well-studied Escherichia coli, with distinct mechanisms involved in not only translation initiation as discussed above but also transcription initiation (36–39). Consequently, genes moved from these organisms into E. coli yield no protein products and vice versa.
Baez et al. used ribosome profiling to identify mRNA determinants of translation initiation in Flavobacterium johnsoniae, a representative of the Bacteroidetes (40). They found that initiation is enhanced by reduced secondary structure in the translation initiation region (TIR) and by adenines at positions −3, −6, −12 and −13. Comparative analysis of E. coli showed that A-3 and A-6 also act as positive determinants of translation. Notably, A-3 is the key feature of the Kozak sequence (41–43), which in eukaryotes promotes start codon selection. Baez et al. also found that AUG trinucleotides other than the start codon are significantly underrepresented in the TIR, and the degree of underrepresentation follows the trend Bacteroidetes > Proteobacteria > Firmicutes. The opposite trend is seen for SD prevalence (23,24), suggesting that elimination of AUG trinucleotides from the TIR is one means by which Bacteroidetes compensate for the absence of SD-ASD pairing (40).
Genomic studies indicate that the ASD sequence is conserved in most Bacteroidetes and in tested cases the mature 16S rRNA includes the ASD. Yet, the Bacteroidetes ribosome seems blind to SD sequences in the cell, an apparent paradox that has defied explanation. Here, we show that F. johnsoniae ribosomes are intrinsically recalcitrant to SD recognition. A cryo-electron microscopy (cryo-EM) structure of the complete 70S ribosome from F. johnsoniae at 2.8 Å resolution shows that the ASD sequence is sequestered by ribosomal proteins (r proteins) bS21, bS18 and bS6. We also find that even though most genes lack SD sequences in the Bacteroidetes, rpsU and/or rpsR often contain strong SDs. These genes encode bS21 and bS18, respectively, the same proteins responsible for ASD occlusion, suggesting straightforward mechanisms of translational autoregulation.
MATERIALS AND METHODS
Purification of ribosomes and subunits
Ribosomes were purified from F. johnsoniae strain UW101 and E. coli strain MRE600. Cells were grown in rich media [CYE (44), F. johnsoniae; LB (45), E. coli] at optimum temperature (30°C, F. johnsoniae; 37°C, E. coli) to mid-logarithmic phase, cooled on ice, and pelleted. Ribosomes or individual subunits were then purified via conventional sedimentation methods as detailed previously (46,47).
Determination of the complete coding region of the IF3 gene in F. johnsoniae
In E. coli, translation of the IF3 gene (infC) begins at the noncanonical start codon AUU (48,49). This enables an autoregulatory circuit because IF3 reduces initiation from near-cognate start codons (50,51). Based on its sequence, the IF3 gene of F. johnsoniae appears to be similarly autoregulated. The infC open reading frame extends 57 codons upstream of the (mis)annotated start codon (genome position 32,505) (52) and is predicted to encode conserved amino acids of IF3 (Supplementary Figure S1A). To elucidate the correct N-terminus of F. johnsoniae IF3, we first cloned the infC gene with ample upstream DNA into the F. johnsoniae expression vector pSCH710 (40), such that the resulting protein would be C-terminally tagged with hexahistidine (His6). F. johnsoniae cells carrying this plasmid were induced to overexpress IF3-His6, and the protein was partially purified via Ni-NTA (Qiagen) affinity chromatography. IF3-His6 was resolved using SDS-PAGE, isolated, and subjected to LC/MS-MS analysis (Mass Spectrometry and Proteomics Facility, Campus Chemical Instrument Center, Ohio State University). One of the peptides identified was SNRGFQPRVEKK, narrowing down the start codon to two possible near-cognate codons: ATA (genome position 32,340) or ATA (genome position 32,346). To distinguish which of these codons corresponds to the natural start codon, we generated an infC-gfp fusion construct and targeted each candidate start codon by mutagenesis (Supplementary Figure S1B). F. johnsoniae cells carrying the WT infC-gfp construct produced strong fluorescence (350 000 RFU), measured as described previously (40). Changing the upstream ATA codon (32,340) to GCT nearly eliminated fluorescence (Mut_1; Supplementary Figure S1B), whereas changing the downstream ATA codon (32,346) to GCT failed to reduce fluorescence (Mut_2; Supplementary Figure S1B). Thus, we infer that ATA (genome position 32,340) corresponds to the natural infC start codon. This assignment is consistent with the annotated ATG start codon of infC of C. hutchinsonii (53), another member of the Bacteroidetes (Supplementary Figure S1A).
Overexpression and purification of initiation factors
Each initiation factor gene was amplified from the F. johnsoniae chromosome and cloned into pET28b (Novagen). Sequences of the primers used are listed in Supplementary Table S1. The resulting overexpression constructs—pBR6, pBR7, and pBR8—encode IF1 with no tag, IF2 with a C-terminal His6 tag, and IF3 with removable N-terminal His6 tag, respectively. The latter construct contains the complete coding region of infC (32 340–32 891), determined as described above, with codon 1 changed from ATA to ATG.
Plasmids pBR6, pBR7 and pBR8 were each transformed into E. coli strain BL21/DE3. Transformed cells were grown in LB at 37°C to mid-logarithmic phase, induced with IPTG (1 mM), and grown for an additional 5 h to allow ample protein overproduction. In each case, the overproduced protein was purified from the soluble lysate. IF1 was purified using a cation exchange column (Macro-Prep High S, Bio-RAD) and dialyzed against storage buffer SBA (20 mM Tris–HCl pH 7.5, 100 mM NH4Cl, 1 mM EDTA, 2 mM β-ME, 15% glycerol). IF2-His6 was purified in three sequential chromatography steps. The protein was partially purified on a Ni-NTA (Qiagen) affinity column, further purified on a heparin column (HiTrap, GE Healthcare), passed through a size exclusion column (Superdex 75, GE Healthcare), and finally dialyzed against storage buffer SBB (20 mM Tris–HCl pH 8.0, 100 mM NH4Cl, 1 mM EDTA, 2 mM β-ME, 5% glycerol). IF3 containing a N-terminal His6-tag was partially purified using Ni-NTA (Qiagen) and then subjected to thrombin cleavage. The cleavage reaction was performed in 20 mM Tris–HCl (pH 7.0), 300 mM NH4Cl, 1 mM EDTA and 2 mM β-ME at room temperature for 4 h, using 1U of thrombin (GE Healthcare) per 400 μg of protein. Following digestion, the reaction mixture was incubated with an excess of Ni-NTA resin at 4°C for 1 h, and the trimmed IF3 was recovered as unbound protein. IF3 was further purified using a cation exchange column (Macro-Prep High S, Bio-Rad) and dialyzed against storage buffer SBC (20 mM Tris–HCl pH 7.0, 300 mM NH4Cl, 1 mM EDTA, 2 mM β-ME, 15% glycerol). Each protein was >95% pure as judged by SDS-PAGE. Small aliquots were flash-frozen and stored at −80°C.
E. coli initiation factors were overexpressed and purified as described (54,55).
Preparations of fMet-tRNA
A DNA template containing a T7 promoter upstream of the F. johnsoniae tRNAfMet gene was generated using overlapping primers 201467 and 201468 (listed in Supplementary Table S1) in a PCR reaction. This DNA template (25 μg) was incubated with T7 RNA polymerase in a 1 ml reaction containing 5 mM ATP, 5 mM CTP, 5 mM GTP, 5 mM UTP, 60 mM Tris–HCl (pH 8.0), 25 mM MgCl2, 1 mM spermidine, 30 mM DTT and 0.01% Triton X-100 for 4 h at 37°C. The tRNAfMet transcript was purified, charged and formylated as described (56). Because T7 RNA polymerase strongly favors initiation with GTP, the 5′ nucleotide of the tRNA transcript was G2 (rather than C1). However, the absence of C1 caused no apparent problems for aminoacylation, formylation or initiation complex formation. Pure native E. coli tRNAfMet1 was purchased (Chemical Block Ltd) and charged and formylated as described previously (56).
Preparations of mRNA
Various model mRNAs from F. johnsoniae and E. coli were made by in vitro transcription, templated with BamHI-linearized plasmids, and gel purified (Supplementary Table S2). Model mRNAs from F. johnsoniae were named based on the gene (Fjoh) number, with an ‘m’ prefix. To make each plasmid template, a region of DNA from the predicted transcriptional start site (40) to a position downstream of codon 30 was amplified and cloned into the EcoRI and BamHI sites of pUC19. A T7 promoter sequence was included in the 5′ portion of the forward primer and, in some cases, a primer-binding site for primer 132 was included in the 5′ portion of the reverse primer (see sequences in Supplementary Table S1). Model E. coli mRNAs derive from gene 32 of bacteriophage T4 and have been described previously (57,58).
Toeprinting experiments
Initiation complexes were detected using the toeprinting technique (59). Typically, 5′[32P]-labeled primer 132 or 201445 (see sequences in Supplementary Table S1) was annealed to mRNA (0.1 μM) in 50 mM Tris–HCl (pH 7.5), 110 mM NH4Cl and 30 mM KCl by incubating at 37°C for 5 min and placing on ice. MgCl2 (7 mM), DTT (1 mM), GTP (0.1 mM), fMet-tRNA (2 μM), and ribosomes (1 μM) or heat-activated 30S subunits (1 μM) were then added, with or without IFs (2 μM each, as indicated) and further incubated for 1 h (without IFs) or 3 min (with IFs). To evaluate complexes formed, dNTPs (0.2 mM each) and AMV reverse transcriptase (2 U, Life Sciences Advanced Technologies, Inc.) were added, and after 3 min, cDNA products were resolved by 6% denaturing PAGE. Gel imaging and quantification were performed with a Typhoon FLA 9000 phosphorimager (GE Healthcare) and associated software (ImageQuant 5.2).
The reaction conditions described above were established empirically for 70S IC formation, by systematically varying NH4Cl concentration (50, 70, 110, 150 and 170 mM), MgCl2 concentration (7 and 11 mM), and temperature (30°C, optimum growth temperature of F. johnsoniae; 37°C, optimum growth temperature of E. coli). Polymix buffer (58) was also tested but did not enhance 70S IC formation. 30S IC formation was also compared at 30 and 37°C, and little-to-no difference was seen.
The overall equilibrium association constant for 30S IC formation was measured using toeprinting as described previously (60,61) (Supplementary Figure S2). In these experiments, the concentration of mRNA (0.02 μM), subunits (0.1 μM, Eco; 1 μM Fjo) and factors (0.5 μM, Eco 30S case; 1.5 μM, Fjo 30S case) were held constant, while the concentration of fMet-tRNA was varied (0.05 to 1 μM). Binding reactions at 37°C were allowed to equilibrate for 1 h (absence of IFs) or 3 min (presence of IFs). The fraction of bound complex (F) was calculated as F = toeprint/(toeprint + runoff), where toeprint and runoff represent signal intensities of the corresponding bands. Data were plotted as a function of fMet-tRNA concentration and fit to the equation F = Fmax [bc/(bc + 1/KA)], where b is the input tRNA concentration, c is the input subunit concentration, and KA is the equilibrium association constant. Fmax corresponds to the maximal level of detected complex and presumably reflects the probability that the complex resists disruption by reverse transcriptase. Experiments to measure the extent of 70S IC formation as a function of fMet-tRNA concentration were performed in an analogous way, except that 70S ribosomes were used instead of 30S subunits.
Puromycin reactions
70S ICs were formed with either F. johnsoniae or E. coli components by incubating mRNA (0.5 μM), formyl-[35S]-Met-tRNAfMet (0.1 μM), heat-activated 30S subunits (1 μM), native IFs (1.5 μM each), and 50S subunits (3 μM) in the presence of 50 mM Tris–HCl (pH 7.5), 110 mM NH4Cl, 30 mM KCl, 7 mM MgCl2, 1 mM GTP and 1 mM DTT for 10 min at 37°C. In each case, the complex formed was rapidly mixed with puromycin (1 mM) in a quench-flow machine (KinTek Laboratories, Inc.), and the reaction was quenched with 0.5 M KOH at various time points. The product, formyl-[35S]-Met-puromycin (fMet-Pmn), was extracted with ethyl acetate and quantified in a liquid scintillation counter (62).
Filter-binding experiments
Binding of RNA oligonucleotides to 30S subunits was measured using a filter-binding assay described previously (63,64). Briefly, radiolabeled RNA (0.5 nM; 5′[32P]-AGAAAGGAGGU-3′, SD underscored; or 5′[32P]-ACCUCCUUUCU-3′, negative control) and activated 30S subunits (from F. johnsoniae or E. coli; various concentrations) were separately preequilibrated in 20 mM K-HEPES (pH 7.6), 100 mM NH4Cl, 6 mM MgCl2, 1 mM GTP, 4 mM β-ME at 25°C. At time t = 0, 100 μl of labeled RNA was mixed with 100 μl of 30S subunits. Aliquots (20 μl) were removed at various time points, filtered through a bi-layer of nitrocellulose and positively charged nylon membranes (NitroBind and Hybond-N+, GE Healthcare), and immediately washed with 150 μl of the same buffer. Membranes were dried, and radioactive spots were quantified with a phosphorimager to determine the fraction of RNA bound as a function of time. Apparent rates were plotted versus 30S concentration, and the data were fit to a linear equation to deduce kon (slope) and koff (Y intercept).
Determination of the mature ends of 16S, 23S and 5S rRNA in F. johnsoniae
The mature ends of each rRNA molecule were identified using F. johnsoniae RNA-seq data collected previously (40). These RNA-seq libraries were made from 30–40 nucleotide RNA fragments, obtained after limited base hydrolysis of total F. johnsoniae RNA, without any rRNA-removal steps. Reads were mapped back to the genome, and 5′-read-end coverage and 3′-read-end-coverage were each plotted with respect to the corresponding genome-annotated gene ends. For each 5′ and 3′ coverage plot, we observed a large peak corresponding to the mature 5′ and 3′ ends, respectively. This enrichment is due to a higher probability that a small RNA fragment generated by limited hydrolysis comes from the terminus of the source molecule (65).
Mass spectrometry
Purified F. johnsoniae ribosomes and 50S subunits were subjected to LC–MS/MS analysis as described (66).
Cryo-electron microscopy
Cryo-EM grids (c-flat CF-2/2-2C-T) were prepared by washing them in chloroform for two hours and treating them with glow discharged in air at 5 mA for 15 seconds, right before the sample was applied. A total volume of 3.6 μl of purified F. johnsoniae 70S ribosomes at a concentration of 170 nM in dilution buffer (50 mM Tris–HCl pH 7.5, 10 mM MgCl2, 100 mM NH4Cl, 6 mM β-mercaptoethanol) was deposited on the grid. The β-mercaptoethanol was added to the buffer right before the dilution was performed. Sample vitrification was performed in liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific Inc.) using one blotting time for 3 s and with a blot force +1. The Vitrobot chamber was set to 25°C and 100% relative humidity. Data acquisition was performed using EPU software at FEMR-McGill using a Titan Krios microscope at 300 kV equipped with a Falcon II direct electron detector (Thermo Fisher Scientific Inc.). Movies were collected with seven frames acquired in 1 s exposure at a magnification of 75 000×, producing images with a calibrated pixel size of 1.073 Å. The nominal defocus range used during data collection was between –1.25 to –2.75 μm, and the total dose used per movie was 50 e−/Å2.
Image processing
The movies comprising the F. johnsoniae 70S dataset were corrected for beam-induced motion using RELION’s implementation of the MotionCor 2 algorithm (67,68). Estimation of the Contrast Transfer Function (CTF) was done using the Gctf program (69). From here, all processing was done with RELION 3.0. A description of the image processing workflow is shown in Supplementary Figure S3. Particle images were selected and extracted from the micrographs using auto-picking and subsequently subjected to one cycle of reference-free 2D classification to remove false positive and damaged particles. The clean dataset output by the 2D classification contained 1 138 048 particle images. These images were subjected to 3D classification and subsequent refinement. The initial 3D reference used for the classification and refinements was a 60 Å low pass filtered map of the mature 70S subunit created from 4V4Q.cif (70) using the Xmipp program (71) or the intermediate cryo-EM maps obtained during classification and refinement. To speed up the computer calculations, the 2D and 3D classifications were performed using particle images binned by 4 and a pixel size of 4.292 Å per pixel. However, we use the full-size images with a pixel size of 1.073 Å per pixel in the refinement steps. A soft-mask created with ‘relion_mask_create’ command was applied to all refinements. In this process, we extended the binary mask by two pixels and created a soft-edge with a width of ten pixels. Refinement was performed in three stages. In the first stage, we used the full-size particle images as they come out from 3D classification. In stage two of refinement, the particle images were first subjected to CTF refinement. In this process, we selected ‘Fit per-particle defocus’ as ‘Yes’ but selected ‘No’ for all the astigmatisms fits and phase-shift. We also did not correct for beam tilt estimation, as our data was not collected using this approach. In the last step particle images were also corrected using Bayesian polishing before subjecting them to final refinement. Bayesian polishing was performed using sigma values of 0.4515, 13545 and 0.21 for velocity, divergence and acceleration, respectively. Sharpening of the final cryo-EM maps and the local analysis was done with RELION (68). The average resolution for the structure of the F. johnsoniae 70S ribosome was estimated by gold-standard Fourier shell correlation. Resolution estimation is reported using a FSC threshold value of 0.143.
Map analysis and atomic model building
The starting point of the structural modeling for the E. coli 70S ribosome was the atomic model of the E. coli 70S ribosome (PDB ID 4V4Q) (70). The models for the 50S and 30S subunits were individually docked using rigid-body approaches into the obtained high-resolution cryo-EM map of the F. johnsoniae 70S ribosome using ‘dock-in-map tool’ in Phenix (72). The atomic model building and coordinate refinement for the F. johnsoniae 70S ribosome were performed with successive rounds of real space refinement in Phenix (72) and manual model building in Coot (73,74).
F. johnsoniae r proteins and rRNA sequences were obtained from UniProt (75) and gene-NCBI databases, respectively. F. johnsoniae sequences were aligned with E. coli sequences using Multiple Sequence Alignment program ClustalW (76). The conserved regions in the rRNA helices of the docked subunits were first located in the density map, and the rest of the segments were traced from these anchor points. The numbering in the model obtained for F. johnsoniae 16S, 5S and 23S rRNA follow the E. coli numbering. The rRNA expansion segments and segments that differed from the E. coli 70S ribosome structure were built manually into the density using Coot (73,74).
The r proteins were also built using a similar approach. Conserved sequences in the homologous proteins were located in the density map, and the positions of bulky sidechains were used both to determine and validate the correct assignment. The structure of the 70S ribosome from B. subtilis (PDB ID: 3J9W) (77) was used as a template to model the bL31 protein, as this protein is not present in the PDB 4V4Q, representing the E. coli 70S ribosome. Homologous r proteins were mutated to the correct sequences for F. johnsoniae, and specific regions were manually built into the density. Two unassigned regions of density corresponding to proteins were observed in the EM map. One of the areas was a well-resolved protein density, and it was possible to unambiguously build the F. johnsoniae protein with unknown function annotated with GenBank ID: ABQ04565.1. The resolution of the second area of density was lower. However, after identification of the Fjoh_4981 gene product by mass spectrometry as a new component of the F. johnsoniae 70S ribosome, it was possible to fit the sequence of the protein encoded by this gene into this second density in the map.
Finally, the atomic coordinates of the 50S and 30S models were combined to obtain the entire 70S. After fixing the regions of intersubunit contacts, the model was refined using Phenix real-space and B-factor refinement (78). The final atomic coordinates of the F. johnsoniae 70S ribosome were validated using Phenix cryo-EM Comprehensive validation tool (79,80) and the Molprobity server (80) (Supplementary Table S3).
Graphical representations of molecular models and cryo-EM density maps were generated using PyMOL (81), UCSF Chimera (82) and UCSF ChimeraX (83).
Secondary structure diagram of the rRNA
The secondary structure diagrams of the 16S, 5S and 23S rRNA were prepared by extracting base pairs information from the refined model using DSSR (84) and were drawn in VARNA (85).
Multiple sequence alignments
Peptide sequences for bS21, bS18 and bS6 were retrieved from the NCBI database (Supplementary Table S4). For each phylum, the organisms selected were diverse and well distributed. Sequences were analyzed in UGENE (86) using the MAFFT (87) multiple sequence alignment with default settings. Gaps were removed when present in 33% or more of the aligned sequences. Sequence logos were generated from edited MAFFT alignments using the WebLogo 3 application (88). Logos were aligned manually, based on alignments of phylum consensus sequences, generated in UGENE.
Comparisons of bS21 C-terminal domains
The isoelectric point (pI) of the C-terminal domain (CTD; defined as residues C-terminal of the conserved KPS/T motif) was determined using the Isoelectric Point Calculator (89). Because these are short peptide sequences (<40 residues), the IPC2 peptide scale was used.
Detection of SD sequences in the Bacteroidetes
A diverse and representative collection of almost 300 Bacteroidetes was chosen for analysis (Supplementary Table S5). Genomes and annotations were obtained from NCBI using the Assembly search feature (90) from BioProject numbers used in Hahnke et al. (29). To determine which genes contained SD sequences, sections of the TIR were analyzed by the free2bind suite of programs (91). Relevant sections of the TIR for SD detection were defined such that the spacing between the 5′ end of the ASD and the first nucleotide of the start codon was between 0 and 15 nucleotides, as described (92). Sequences were fed to the free_scan.pl program in the free2bind suite, which calculates the free energy of pairing between two RNA strands as a function of register / relative position. The ASD sequence 3′-UUCCUCCA-5′ was tested incrementally across each TIR, and if the ASD paired with a free energy of (for example) –7 kcal/mol or lower at any position, a SD sequence was assigned. As a control, the same analysis was performed on a window 25–40 nt upstream of the start codon, too distal for an authentic SD. The resulting ‘hits’ were termed mock SD (MSD) sequences, which served as a proxy for false positive SD assignments in the various organisms analyzed (Supplementary Table S5). An analogous approach was taken to screen for SD2 and MSD2 sequences, using the ASD2 sequence 3′-UUACUCUA-5′.
RESULTS
Reconstitution of Flavobacterium johnsoniae initiation in vitro
We purified F. johnsoniae and E. coli components of translation initiation (ribosomes, subunits, IF1, IF2, IF3, fMet-tRNAfMet, various mRNAs) to compare complex formation in the two systems. Using F. johnsoniae components and the toeprinting technique, we observed efficient 70S initiation complex (IC) formation at the cognate start codon of multiple tested mRNAs (Figure 1A). Virtually identical results were seen with either fully modified E. coli fMet-tRNAfMet1 or unmodified F. johnsoniae fMet-tRNAfMet (Supplementary Figure S4). Once formed, 70S ICs of F. johnsoniae and E. coli exhibit comparable puromycin reactivity (Supplementary Figure S5), indicating that fMet-tRNA is bound in the P/P site. In all tested cases, IC formation was strictly factor dependent. Surprisingly, 30S ICs were not readily detected with F. johnsoniae components in this set of experiments. However, replacement of F. johnsoniae factors with E. coli factors enabled 30S IC formation, an effect attributable to E. coli IF3 specifically (Figure 1B). Systematic mix-and-match experiments showed that swapping F. johnsoniae IF3 with E. coli IF3 is sufficient to stabilize the 30S IC (Supplementary Figure S6). The reason F. johnsoniae IF3 failed to stabilize the F. johnsoniae 30S IC remains unclear. This preparation of F. johnsoniae IF3 was active, based on its ability to promote F. johnsoniae 70S IC formation (Supplementary Figure S7).
Figure 1.

Biochemical evidence that E. coli and F. johnsoniae ribosomes differ in ASD function. (A) F. johnsoniae 30S and 70S complexes formed on various mRNAs in the presence of initiation factors (IFs) from F. johnsoniae (Fjo) or E. coli (Eco) as indicated. The toeprint band indicates the cognate complex (with start codon in P site), and the run-off band reflects unbound mRNA. Messages mFjoh_xxxx are named based on corresponding F. johnsoniae genes; m292 is a model E. coli mRNA (58). (B–D) Experiments to measure the overall equilibrium association constant (KA) for 30S IC formation. For each experiment, the amount of complex formed was quantified and plotted as a function of fMet-tRNA concentration. (B) F. johnsoniae 30S subunits (1 μM) were incubated with mFjoh_4413 and fMet-tRNA (various concentrations, as indicated), in the presence of all E. coli factors (+IFs, 1.5 μM each; open green triangles), IF1 and IF2 only (+IF1,2; open blue squares), or no factors (-IFs; open red circles). (C) E. coli 30S subunits (0.1 μM) were incubated with mRNA (m291, SD+ (58); or its derivative m295, SD-; as indicated) and fMet-tRNA (various concentrations, as indicated), in the absence or presence of E. coli factors (0.5 μM each), as indicated. Messages m291 and m295 are identical except that the latter has cytosines in place of guanines at positions –9 and –10. (D) F. johnsoniae 30S subunits (1 μM) were incubated with mRNA (m291, SD+; or m295, SD-; as indicated) and fMet-tRNA (various concentrations, as indicated), in presence of E. coli factors (1.5 μM each). (E) Summary of KA values (in units of μM−2) obtained from experiments like those of panels C-D. (F) 70S ribosomes (0.1 μM) from E. coli were incubated with mRNA (m291, SD+; or m295, SD-; as indicated) and fMet-tNA (various concentrations, as indicated) in presence of E. coli initiation factors (0.5 μM each). (G) 70S ribosomes (1 μM) from F. johnsoniae were incubated with mRNA (m291, SD+, filled green triangles; m295, SD-, filled gray diamonds; or mFjoh_4413, open black circles) and fMet-tRNA (various concentrations, as indicated) in the presence of F. johnsoniae initiation factors (1.5 μM each). (H) The rate of binding of a SD-containing RNA oligonucleotide to the 30S subunit was measured using a double-membrane filtration method (64). The apparent rate (kapp) was plotted versus 30S concentration (E. coli, open blue squares; F. johnsoniae, open red circles), to estimate kon (E. coli, 30 μM−1s−1; F. johnsoniae, 2.7 μM−1s−1) and koff (E. coli, 0.16 s−1; F. johnsoniae, 0.29 s−1).
Flavobacterium johnsoniae ribosomes fail to recognize the Shine-Dalgarno sequence of mRNA
The overall equilibrium association constant for 30S·mRNA·fMet-tRNA formation (KA) was measured as described previously (60,61), using message m291 (SD+) and its variant m295 (SD-) (57). For E. coli 30S, the SD increased KA by 30- to 40-fold in either the absence or presence of factors (Figure 1C and E). In contrast, for F. johnsoniae 30S, the SD had virtually no effect on KA (Figure 1D and E). Because the latter experiment involved (by necessity) E. coli factors, we also measured the extent of 70S IC formation as a function of fMet-tRNA concentration, employing native factors in each case. Again, we observed that the SD promotes complex formation in the E. coli case but not the F. johnsoniae case (Figure 1F and G). F. johnsoniae ribosomes generally exhibited lower efficiency in complex formation in these experiments than E. coli ribosomes. This is due at least in part the model mRNAs (derived from E. coli), because IC formation is considerably more efficient on mFjo_4413 (Figure 1G, open circles; KA = 130 μM−2, Fmax = 0.52). Collectively, these data suggest that F. johnsoniae ribosomes are inherently refractory to SD recognition.
To further compare ASD function in E. coli versus F. johnsoniae, we used a double-membrane filtration method (63,64) to measure binding between 30S subunits and an RNA oligo containing a ‘perfect’ SD (5′-AGAAAGGAGGU-3′; SD underscored). Subunits from either organism were able to bind this oligo, but the kinetics of binding differed (Figure 1H & Supplementary Figure S8). The rate of binding (kon) was 11-fold larger for E. coli 30S, and the dissociation rate (koff) was ∼2-fold smaller. These data suggest that the ASD in F. johnsoniae 30S is constrained in some way, making mRNA pairing slower and less favorable.
The structure of the Flavobacterium johnsoniae 70S ribosome
Ribosomes were purified from F. johnsoniae strain UW101 and their structure solved by cryo-EM and single particle techniques. Image classification revealed that all the ribosomes in the sample were empty and did not contain any tRNA molecule bound to their A, P or E site. The 30S subunit in all the ribosomal particles was in the same ratcheting position and exhibited the same rotation angle with respect to the 50S subunit, indicating that the sample represented a conformationally homogeneous population of 70S ribosomes (Supplementary Figure S3). The cryo-EM map (Figure 2A and B; Supplementary Video 1) refined to 2.8 Å resolution (Supplementary Figure S9). Local resolution analysis revealed that the core of the ribosomal particle was the most defined region reaching a resolution of 2.5 Å. Peripherical regions that are known to be more dynamic, including the beak and shoulder in the 30S subunit and the two stalks in the 50S subunit showed a resolution ranging between 3 and 3.8 Å.
Figure 2.
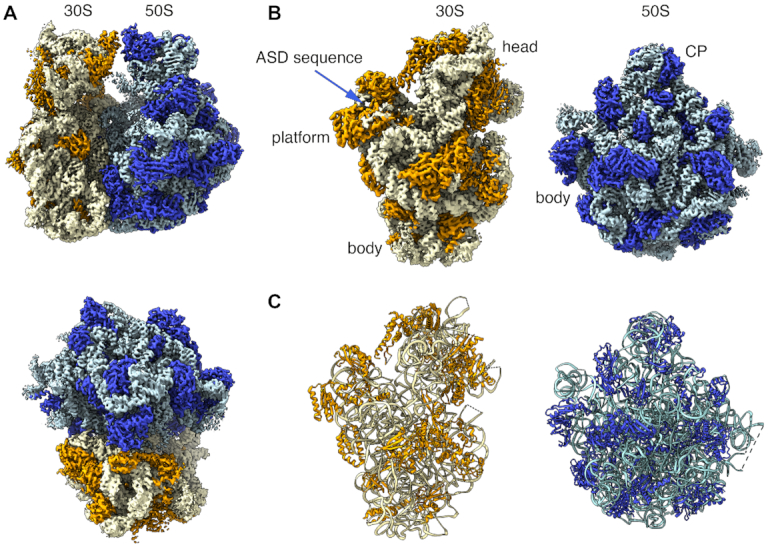
Structure of the Flavobacterium johnsoniae ribosome. (A) Side and top views of the cryo-EM map of the F. johnsoniae ribosome at 2.8 A resolution. The rRNA and r proteins in the 30S subunit are shown in yellow and orange, respectively. In the 50S subunit, the rRNA is colored in pale blue (rRNA) and the r proteins in dark blue. (B) Solvent view of the 30S and 50S subunit forming the F. johnsoniae ribosome. The main landmarks of the 30S and 50S subunits and the ASD sequence in the small subunit are indicated. CP indicates the central protuberance. (C) Solvent view of the molecular models derived for the 30S and 50S subunits derived from the cryo-EM map of the F. johnsoniae ribosome. The rRNA and r proteins and panel B and C are colored following the same scheme as in panel A.
The quality of the electron density map enabled us to build a molecular model of the 70S ribosome with confidence (Figure 2C and Supplementary Table S3). We were able to unambiguously distinguish most of the nucleobases in the rRNA helices and side chains in the r proteins, respectively (Supplementary Figure S10). A few regions of the rRNA and r proteins in both 50S and 30S subunits, in particular, the regions which are known to be flexible, had either weaker or no density and were therefore not built in the model. In line with common convention, helices of the small and large subunits were denoted with a lowercase ‘h’ and uppercase ‘H’, respectively.
To model the rRNA, we complemented the information from the density map with RNA-seq data that unambiguously identified the mature 5′ and 3′ ends of each rRNA molecule (Supplementary Figure S11). With one exception (5S, 3′), the termini differed from the annotated-genome predictions and showed the lengths of mature 16S, 23S, and 5S rRNA to be 1519, 2862 and 111 nucleotides, respectively. Notably, the mature 23S rRNA of F. johnsoniae lacks nucleotides 1–8 and nucleotides 2895–2902, which in other bacteria pair to form helix H1. Based on the genomic sequence, RNA strands corresponding to nucleotides 2–7 and 2895–2900 are complementary. Hence, a short (6 bp) helix 1 may form in the precursor rRNA during the 50S assembly and then be cleaved off during final maturation.
We were able to model two additional proteins. One of them is located in the 30S subunit and the other in the 50S subunit (see description below). The final three-dimensional structure of the F. johnsoniae 70S ribosome contains all three rRNAs (16S in the 30S subunit and 23S and 5S in 50S subunit), 20 r-proteins in the 30S subunit and 30 r-proteins in the 50S subunit (Supplementary Tables S6 and S7). No density was observed for bS1 and uS2 in the small subunit. Association of bS1 in most species is weak and reversible (93) and uS2 frequently dissociates upon exposure to the air-water interface during sample vitrification in cryo-EM (94). In the large subunit, density representing uL1, uL7/L12, uL10 and uL11 was also not present or highly fragmented due to the intrinsic flexibility of these ribosome components. Therefore, these proteins were excluded from the model.
To gain some insight on the basis of the missing small subunit proteins, we compared the protein composition of isolated subunits and ribosomes using tricine SDS-PAGE (Supplementary Figure S12). Protein bS1 is clearly underrepresented in the 30S subunits and is further underrepresented in the 70S ribosomes. Protein uS2 is present at stoichiometric levels in the 30S subunits but is somewhat underrepresented in the 70S ribosomes. One difference between the purification methods was a second high-salt wash in the 70S case, which likely explains the larger degree of protein depletion observed. We infer that loss of bS1 occurred mainly during the 70S purification, while loss of uS2 occurred during purification and sample vitrification.
Structural differences in the 30S subunit
Overall, the structure of F. johnsoniae 70S ribosome is very similar to other bacterial ribosomes (Figure 2 and Supplementary Video 1). However, several structural motifs in both the 30S and 50S subunits displayed differences compared to other known 70S ribosome structures.
Comparison of the 30S subunit from our 70S structure with the 30S subunit from 70S structures from E. coli (95), Thermus thermophilus (96), Mycobacterium smegmatis (97), Pseudomonas aeruginosa (98), Bacillus subtilis (77) and Staphylococcus aureus (99) (Supplementary Table S8) revealed structural differences in the 16S rRNA helices highlighted in Figure 3A. Significant differences were observed between helices h7–h10. Whereas helices h7-h8 were similar in length in all the structures, helices h9 and h10 were different, both in length and fold, compared to E. coli (Figure 3B) and other ribosomes (Supplementary Figure S13A). Helix h9 is much longer in F. johnsoniae, T. thermophilus, M. smegmatis, B. subtilis and S. aureus than in E. coli and P. aeruginosa; whereas helix h10 is shortest in F. johnsoniae and M. smegmatis compared to all others. Overall, helices h9 and h10 in F. johnsoniae resemble more closely to M. smegmatis. We also observed subtle differences in helix h17, which is comparatively shorter in F. johnsoniae than in E. coli (Figure 3C) and others, except in M. smegmatis, which has the shortest helix h17 (Supplementary Figure S13B). Apparent differences were also present in helices h26 and h44, where these helices are the shortest and less extended in F. johnsoniae compared to those in E. coli (Figure 3D and E) and other species (Supplementary Figures S13C and D). Whether any of these structural variations have functional consequences remains unclear.
Figure 3.

Comparison of structural differences in the 16S rRNA between F. johnsoniae and E. coli. (A) Tertiary structure of 16S rRNA from our refined model. The regions of structural variability (in terms of length, fold, and/or loop size) between F. johnsoniae and E. coli (PDB ID: 6O9K), are labelled and highlighted in different colors. (B–E) Superpositions of h7–10, h17, h26, and h44–45, respectively from F. johnsoniae and E. coli along with their secondary structure diagram. Color codes for F. johnsoniae 16S rRNA elements are same as in (A), and E. coli motifs are shown in grey. Comparisons with T. thermophilus, M. smegmatis, P. aeruginosa, B. subtilis and S. aureus are shown in Supplementary Figure S13.
Identification of a structural homolog of bS22
In the process of modelling the F. johnsoniae ribosome, we identified an unassigned area of density in the cryo-EM map in the 30S subunit. The clarity of density allowed us to build a de novo molecular model for this region (Figure 4A). The primary sequence best matched gene Fjoh_1533, annotated to encode a hypothetical protein. The protein was comprised of 30 amino acids and was highly basic with a pI of 12.7, owing to its high arginine and lysine content. The amino acid sequence of this protein is highly conserved among members of Bacteroidetes (Figure 4D).
Figure 4.
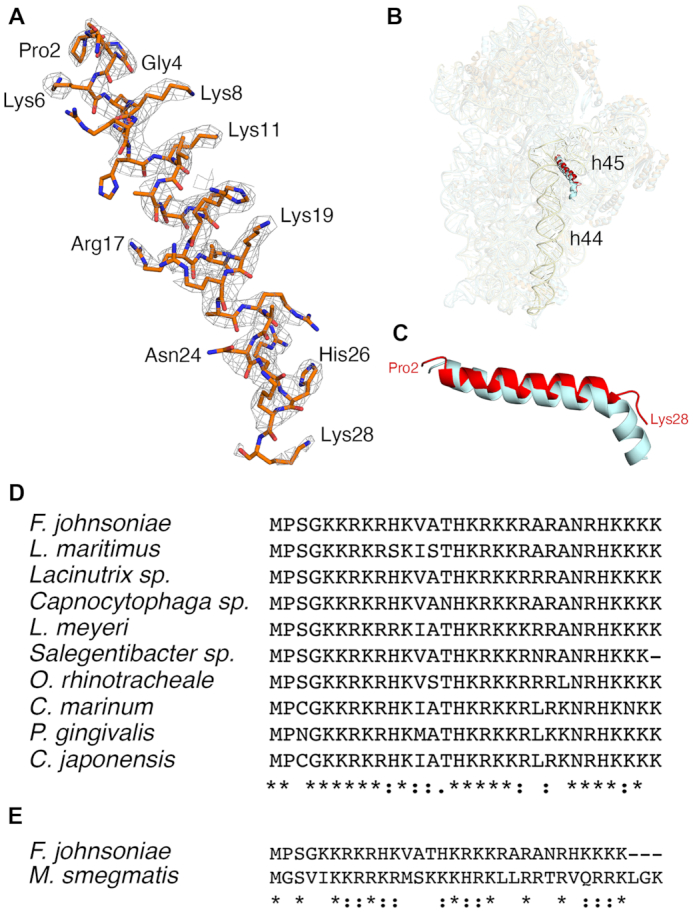
Homolog of bS22 in the 30S subunit of F. johnsoniae ribosome. (A) Electron density map and the derived molecular model of the Fjoh_1533 protein. (B) Structural comparison of the Fjoh_1533 protein in F. johnsoniae (highlighted in red) and bS22 in M. smegmatis (cyan) in the 30S subunit. Helices h44 and h45 in F. johnsoniae are highlighted in light yellow. (C) close-up view of C, showing the structural homology between the two proteins. (D) Amino acid sequence alignment of the uncharacterized protein in various Bacteroidetes, as indicated. (E) Amino acid sequence alignment of the uncharacterized protein of F. johnsoniae and bS22 of M. smegmatis.
Upon comparison with other ribosome structures, we found that protein Fjoh_1533 occupies the same position as bS22 in the M. smegmatis ribosome (97) (Figure 4B). The protein is similarly inserted between helices h44 and h45 of the 16S rRNA and in contact with helix H70 of the 50S subunit. Both proteins superimposed well and form an alpha helix (Figure 4C). Considering the structural homology between the two proteins (Figure 4E), we infer that this F. johnsoniae protein is the functional counterpart to bS22 in M. smegmatis (97) and hence name it bS22. Notably, SD sequences are quite prevalent in Mycobacteria (23), arguing against any role for bS22 in ASD occlusion.
Sequestration of the ASD sequence within the 30S subunit by ribosomal proteins bS21, bS18, and bS6
In all the structures previously obtained for the 30S subunit and 70S ribosome without bound mRNA, the 3′ end of the 16S rRNA, including the ASD region, remains flexible and is mostly unmodelled. In our structure, the ASD sequence presented an apparent density, and we were able to trace the entire 3′ sequence of the 16S rRNA with confidence (Figure 5A and B). Only the bases of C1535 and C1536 exhibited weak electron density, so the assigned orientation of these bases in the current model might be variable.
Figure 5.

Structural Basis for the Sequestration of the ASD sequence within the F. johnsoniae 30S subunit. (A) Solvent view of the cryo-EM structure of the F. johnsoniae 30S subunit showing the 3′ end segment of 16S rRNA (highlighted in yellow). The remaining 16S rRNA is shown in light grey. The r proteins bS6, bS18 and bS21 involved in the sequestration of the SD sequence are colored in cyan, pink and blue, respectively. The remaining r proteins are shown in red. Zoomed area shown in (B) is framed in the structure. (B) Interaction details between the 3′ end nucleotides (including the ASD sequence) of 16S rRNA (yellow sticks) and r proteins bS6 (cyan), bS18 (pink), and bS21 (dark blue). Zoomed areas shown in (C), (D) and (E) are indicated in panel (B).
We found that the 3′ end of the 16S rRNA, including the ASD sequences folds back away from the mRNA exit channel and binds along a groove formed by r proteins bS6 and bS18 (Figures 3E and 5A). The terminal 3′ nucleotides are stabilized through an extensive network of H-bonds and van der Waals contacts by the residues from bS6, bS18, and bS21 (Figure 5B–E). For example, A1534 and U1541 are stabilized through stacking interaction with the side chains of Tyr54 and Tyr32 from bS21 and bS18, respectively (Figure 5C and D). Similarly, the base and sugar of U1544 stack with the side chains of Trp100 from bS6 and Phe50 from bS18, respectively (Figure 5D). In addition, the side chains of Ser38, Arg34, Lys37, Asn56, Glu57 and Arg65 from bS18 and Arg103 from bS6 form H-bonds with U1537, C1538, U1541, U1542 and C1543. The van der Waals contacts from the side chains of Ile40, Leu62 and Leu66 from bS18 and Ile55 from bS21 contribute to further stabilize the terminal 3′ nucleotides (Figure 5C-E).
Multiple residues contributing to ASD occlusion are uniquely conserved in the Bacteroidetes
To further evaluate the ASD-binding site of the Bacteroidetes ribosome, we compared the conservation of amino acids in bS21, bS18 and bS6. Sequences from many representative species of the Bacteroidetes were aligned, as were those from the Firmicutes, γ-Proteobacteria, and Spirochaetae (Figure 6, Supplementary Figure S14, Supplementary Table S4). Organisms of these latter groups exhibit high SD prevalence, unlike the Bacteroidetes (23). Comparisons of the resulting logos/consensus sequences revealed features of all three proteins unique to the Bacteroidetes.
Figure 6.

Unique features of bS21 and bS18 of the Bacteroidetes. (A) Sequence logos for bS21 of the Bacteroidetes (Bact, n = 357), Firmicutes (Firm, n = 300), γ-Proteobacteria (γ-Pr, n = 247), and Spirochaetes (Spir, n = 112). Numbering based on F. johnsoniae. Arrowheads indicate residues that contact the 3′ end of 16S rRNA in the F. johnsoniae 70S structure. Stars indicate residues uniquely conserved in the Bacteroidetes, defined as any identical or similar (K/R; F/Y; S/T; D/E; N/Q) residue that occurs with a frequency of >0.9 in the Bacteroidetes and < 0.5 in each of the other groups analyzed. (B) Box plot comparing the isoelectric point (pI) of the C-terminal domain (CTD) of bS21 (residues C-terminal of S42). Based on two-tailed t tests, the Bacteroidetes data clearly differ from those of each other group (P < 10−70). α-Pr, α-Proteobacteria (n = 516); β-Pr, β-Proteobacteria (n = 172); γ-Pr, γ-Proteobacteria (n = 247); δ-Pr, δ-Proteobacteria (n = 164); Spir, Spirochaetes (n = 112); Bact, Bacteroidetes (n = 357); Chlo, Chorobi (n = 62); Firm, Firmicutes (n = 300); Cyan, Cyanobacteria (n = 312). (C) Sequence logos for bS18, annotated as described above. Bact, n = 317; Firm, n = 299; γ-Pr, n = 239; Spir, n = 111.
For bS21, the C-terminal portion of the protein is considerably less basic than in other organisms and contains a signature tyrosine (Tyr54, F. johnsoniae numbering), highly conserved in the phylum (Figure 6A and B). In the F. johnsoniae 70S structure, Tyr54 stacks onto A1534 of 16S rRNA (Figure 5C). This interaction occurs right where the 3′ tail begins to diverge from that seen in the E. coli and T. thermophilus ribosome and may be crucial for reorienting the ASD towards the platform in the Bacteroidetes ribosome. For bS18, residues uniquely conserved in the Bacteroidetes include Phe50, Asn56, Gln58 and Lys/Arg at position 37 (Figure 6C). In the F. johnsoniae ribosome structure, Asn56 contacts U1537 and C1538 (Figure 5E), Lys37 contacts U1541, and Phe50 contacts U1544 (Figure 5D). bS6 of the Bacteroidetes contains a phylum-specific C-terminal extension, which folds into an alpha helix in the F. johnsoniae ribosome (Supplementary Figure S14). Three residues within this helix (Arg103, Trp100 and Lys107) interact with C1543 and U1544, the last nucleotides of F. johnsoniae 16S rRNA (Figure 5D).
To evaluate whether the described protein-RNA interactions stabilizing the ASD sequence in F. johnsoniae could potentially occur in species of other phyla, we structurally aligned bS6, bS18 and bS21 of E. coli and T. thermophilus onto the structure of the F. johnsoniae ribosome. We found that critical interactions contributing to ASD occlusion in F. johnsoniae (including those established through Tyr54 of bS21; Phe50, Asn56 and Lys37 of bS18; and Arg103 and Trp100 of bS6) cannot occur with the residues occupying the structurally equivalent positions in the E. coli or T. thermophilus ribosome (Supplementary Figure S15).
Structural differences in the 50S subunit
The most notable differences in 23S rRNA were observed in helices H10, H16-H17, H28, H63 and H98 (Figure 7A and Supplementary Figure S16), which deviate in length, fold, and/or orientation compared to six other species. Helices H10 and H63 are the shortest in F. johnsoniae (Figure 7B and D, and Supplementary Figures S16A & S16B). Helix H28 is longer in F. johnsoniae and T. thermophilus than in E. coli and in all the other structures we compared (Figure 7C and Supplementary Figure S16C). Helices H16-H17 adopt a different conformation from the equivalent helices in E. coli (Figure 7E). The overall orientation of helices H16-H17 in F. johnsoniae was somewhat similar to that in the other four structures. However, obvious differences do exist. For example, P. aeruginosa contains the shortest and M. smegmatis the longest of these helices (Supplementary Figure S16D). Finally, the length of H98 in F. johnsoniae is much reduced, essentially eliminating the helix (Figure 7F & Supplementary Figure S16E).
Figure 7.

Comparison of structural differences in the 23S rRNA between the 50S subunit in F. johnsoniae and E. coli. (A) Solvent view of the 23S rRNA from F. johnsoniae with the regions distinct from E. coli (PDB ID: 6O9K) labelled and highlighted in different colors. (B–F) Comparison of helices H10, H28, H63, H16-H17, and H98, respectively from F. johnsoniae and E. coli along with their tertiary structure derived secondary structure diagram (except for helices H16-H17). Color codes for F. johnsoniae 23S rRNA elements are same as in (A), and E. coli’s elements are shown in grey. The red dashed lines in (E) indicate the segment which could not be modeled due to weaker densities. Comparisons with T. thermophilus, M. smegmatis, P. aeruginosa, B. subtilis, and S. aureus are shown in Supplementary Figure S16.
Discovery of a novel ribosomal protein: bL38
Besides the differences in the rRNA, we identified a region of protein density in the 50S subunit, which to our knowledge has not been assigned to any known protein in any ribosome. To identify the protein, purified ribosomes and 50S subunits were subjected to LC/MS-MS analysis. One uncharacterized protein (UniProt A5F9Y9; pfam 14128, DUF4295) was found in both samples (Supplementary Table S9). This small (5.6 kDa), basic (pI = 10.5) polypeptide is encoded by Fjoh_4981, which lies immediately downstream of rpmB (Fjoh_4983) and rpmG (Fjoh_4982), the genes for bL28 and bL33, respectively (Figure 8A and B). RNA-seq and ribo-seq read coverage is similar across these three genes (Figure 8B, (40)), suggesting that they belong to the same operon and are translated at comparable levels. While absent from other phyla, genes homologous to Fjoh_4981 are widely distributed across the Bacteroidetes (Supplementary Figure S17A). Among representative species of the phylum, the gene neighborhood of Fjoh_4981 looks well conserved (Supplementary Figure S17B). These observations suggest that Fjoh_4981 encodes a bona fide ribosomal protein, unique to the Bacteroidetes. Accordingly, we name the protein bL38 and the corresponding gene rpmL, in line with modern nomenclature (100,101).
Figure 8.

A novel ribosomal protein: bL38. (A) Sequence alignment of bL38 proteins from fifteen Bacteroidetes, three representatives of each class Flavobacteriia, Bacteroidia, Cytophagia, Sphingobacteriia, and Chitinophagia (top to bottom, species listed). (B) RNA-seq and ribo-seq read coverage across rpmB (bL28), rpmG (bL33), and rpmL (bL38) in F. johnsoniae. Ratios of average per-nucleotide coverage are 1.0:1.1:1.3 (RNA-seq) and 1.0:0.6:0.5 (ribo-seq), respectively. (C) Ribbon representation of the overall 50S subunit (side view) from F. johnsoniae, highlighting the location of the unassigned protein density with respect to uL6 (marine blue; left panel). The modeled amino acid sequence for bL38 (from Lys 14 to Lys 48) is shown fitted into the density of the cryo-EM map (right panel; top) along with a close-up view of the left panel (right panel; bottom). The close-up view shows a cartoon representation of the newly described r protein, bL38 folding into a β-hairpin that forms a mixed β-sheet with r protein uL6. It also shows its close association with helices H95 and H97 (labelled and highlighted in light blue).
The sequence of the protein encoded by Fjoh_4981 was used to build the molecular model using the unassigned density of the cryo-EM map (Figure 8C). The quality of density was sufficient to assign side chains with confidence. We found that the observed density represented the C-terminal region of the protein from Lys 14 to Lys 48. There was no observed density for the first thirteen residues of the N-terminus or the last two residues of the C-terminus (Figure 8C, right top panel). The newly described r protein bL38 folds into a β-hairpin that forms a mixed β-sheet with r protein uL6, located just above. bL38 also interacts with rRNA helices H95 and H97 (Figure 8C, right bottom panel).
Strong SD sequences lie upstream of Bacteroidetes genes encoding bS21 and/or bS18
We noticed that the bS21 gene (rpsU) of F. johnsoniae contains a ‘perfect’ SD sequence (Figure 9A), whereas all the other rps genes lack an obvious SD. To further investigate this, we analyzed the TIRs of all rps genes in 293 representative species of the Bacteroidetes. Using the program free_scan (91), the free energy of pairing between the canonical ASD (3′-UUCCUCCA-5′) and each 8-nt stretch of mRNA within the TIR was calculated and plotted (Supplementary Figuress S18-S22). For many members of class Flavobacteriia, including F. johnsoniae, a prominent trough was observed near position –8 of rpsU (Supplementary Figure S18), indicative of an authentic SD. Based on a stringent threshold of –7 kcal/mol, 60% of Flavobacteriia harbor a strong SD upstream of rpsU (Table 1). This value increases to 77% when only those organisms with a canonical ASD are considered. By contrast, other rps genes of Flavobacteriia lack SD sequences, with only a few potential exceptions. For class Bacteroidia, SDs were rarely seen for rpsU but commonly seen for rpsR (encoding S18) (Supplementary Figure S19). In fact, 66% of the Bacteroidia analyzed exhibit a strong SD (≤ -7 kcal/mol) upstream of rpsR (Table 1). Interestingly, the gene for uS11, rpsK, also contains a strong SD in a subset of the Bacteroidia (14%). For class Chitinophagia, SDs were found to be most prevalent upstream of rpsU and less common upstream of rpsR and/or rpsA (Table 1, Supplementary Figure S20). In the Cytophagia and Sphingobacteriia, strong SDs were commonly seen for rpsU and/or rpsR (Table 1, Supplementary Figures S21-S22). In Sphingobacteriia, strong SDs were also evident for rpsB (encoding uS2), rpsG (encoding uS7) and rpsS (encoding uS19). Thus, even though the vast majority of Bacteroidetes genes lack SD sequences (23–26), certain ribosomal genes clearly have them. Remarkably, overall in the phylum, rpsU and/or rpsR commonly contain a strong SD (Table 1), and the corresponding proteins bS21 and bS18 interact with the 16S rRNA in a way that occludes the ASD (Figure 5).
Figure 9.

Sequences near the start codon of rpsU in various Flavobacteriia (A) Potential base pairing between rRNA and rpsU mRNA in various Flavobacteriia that have the canonical ASD (as indicated). (B) Potential base pairing between rRNA and rpsU mRNA in various Flavobacteriia that have an alternative ASD (ASD2, as indicated). Bold, start codon. Red, mRNA nucleotides predicted to pair to 16S rRNA.
Table 1.
Occurrences of strong SD sequences in the Bacteroidetes
| Gene | Flavobacteriia | Bacteroidia | Chitinophagia | Cytophagia | Sphingobacteriia |
|---|---|---|---|---|---|
| Small subunit genes | |||||
| rpsA (uS1) | 0 | 0 | 0.182 | 0.022 | 0 |
| rpsB (uS2) | 0 | 0 | 0 | 0 | 0.769 |
| rpsC (uS3) | 0 | 0 | 0 | 0 | 0 |
| rpsD (uS4) | 0 | 0 | 0 | 0 | 0 |
| rpsE (uS5) | 0 | 0 | 0 | 0 | 0 |
| rpsF (bS6) | 0 | 0 | 0 | 0 | 0 |
| rpsG (uS7) | 0 | 0 | 0 | 0 | 0.462 |
| rpsH (uS8) | 0 | 0 | 0 | 0 | 0 |
| rpsI (uS9) | 0 | 0 | 0 | 0 | 0 |
| rpsJ (uS10) | 0 | 0 | 0 | 0 | 0 |
| rpsK (uS11) | 0 | 0.144 | 0 | 0 | 0 |
| rpsL (uS12) | 0 | 0 | 0 | 0 | 0 |
| rpsM (uS13) | 0 | 0 | 0 | 0 | 0 |
| rpsN (uS14) | 0 | 0 | 0 | 0 | 0 |
| rpsO (uS15) | 0 | 0 | 0 | 0 | 0 |
| rpsP (bS16) | 0 | 0 | 0 | 0 | 0 |
| rpsQ (uS17) | 0 | 0 | 0 | 0 | 0 |
| rpsR (bS18) | 0.008 (0.011)a | 0.660 | 0.091 | 0.696 | 0.615 |
| rpsS (uS19) | 0 | 0 | 0 | 0 | 0.154 |
| rpsT (bS20) | 0 | 0 | 0 | 0 | 0 |
| rpsU (bS21) | 0.597 (0.772)a | 0 | 0.455 | 0.478 | 0.846 |
| Large subunit genes | |||||
| rplA (uL1) | 0 | 0 | 0 | 0.261 | 0 |
| rplB (uL2) | 0 | 0 | 0 | 0 | 0 |
| rplC (uL3) | 0 | 0 | 0 | 0 | 0 |
| rplD (uL4) | 0 | 0.010 | 0 | 0 | 0 |
| rplE (uL5) | 0.008 (0.011)a | 0.481 | 0.364 | 0.413 | 0.769 |
| rplF (uL6) | 0 | 0.539 | 0 | 0.065 | 0.462 |
| rplI (bL9) | 0 | 0.825 | 0.182 | 0.152 | 0.692 |
| rplJ (uL10) | 0 | 0.126 | 0 | 0 | 0.077 |
| rplK (uL11) | 0 | 0 | 0 | 0 | 0 |
| rplL (uL7/12) | 0 | 0 | 0 | 0 | 0 |
| rplM (uL13) | 0 | 0 | 0 | 0 | 0 |
| rplN (uL14) | 0 | 0 | 0 | 0 | 0 |
| rplO (uL15) | 0 | 0 | 0 | 0.022 | 0 |
| rplP (uL16) | 0 | 0 | 0 | 0 | 0 |
| rplQ (bL17) | 0 | 0 | 0 | 0 | 0 |
| rplR (uL18) | 0 | 0 | 0 | 0 | 0 |
| rplS (bL19) | 0 | 0 | 0 | 0 | 0 |
| rplT (bL20) | 0 | 0 | 0 | 0 | 0 |
| rplU (bL21) | 0 | 0 | 0 | 0 | 0 |
| rplV (uL22) | 0.050 (0)a | 0 | 0 | 0 | 0 |
| rplW (uL23) | 0 | 0.385 | 0 | 0 | 0.692 |
| rplX (uL24) | 0 | 0 | 0.091 | 0.022 | 1 |
| rplY (bL25) | 0 | 0 | 0 | 0 | N |
| rpmA (bL27) | 0 | 0.327 | 0 | 0 | 0 |
| rpmB (bL28) | 0 | 0 | 0 | 0 | 0 |
| rpmC (uL29) | 0 | 0 | 0 | 0 | 0 |
| rpmD (uL30) | 0 | 0.385 | 0 | 0 | 0 |
| rpmE (bL31) | 0 | N | N | N | N |
| rpmF (bL32) | 0 | 0 | 0 | 0 | 0 |
| rpmG (bL33) | 0 | 0.087 | 0 | 0 | 0 |
| rpmH (bL34) | 0 | 0 | 0 | 0 | 0 |
| rpmI (bL35) | 0 | 0 | 0 | 0 | 0 |
| rpmJ (bL36) | 0 | 0 | 0 | 0 | 0 |
| All genesb | 0.002 (0.002)a | 0.017 | 0.006 | 0.007 | 0.011 |
Data represent the fraction of genes that contain a strong SD, defined by a free energy of pairing threshold of –7 kcal/mol. N, no such gene identified. Numbers of genomes (n) analyzed: Flavobacteriia, n = 119; Bacteroidia, n = 104; Chitinophagia, n = 11; Cytophagia, n = 46; Sphingobacteriia, n = 13.
aValues in parentheses derive from Flavobacteriia with the canonical ASD (n = 92).
bAll annotated protein coding genes, including ribosomal genes.
The fact that several genes of Sphingobacteriia besides rpsU and rpsR contain SD sequences prompted us to screen the large-subunit genes as well (Table 1). In Flavobacteriia, SDs are rarely seen in any rpl/rpm genes. By contrast, SDs are commonly seen for a subset of rpl/rpm genes in the other classes. In Bacteroidia, strong SDs occur with a frequency of > 0.1 upstream of genes encoding uL5, uL6, bL9, uL10, uL23, bL27 and uL30. A similar but smaller set of large-subunit genes (uL5, uL6, bL9, uL10, and uL23) exhibit SDs in the Sphingobacteriia. In Chitinophagia and Cytophagia, genes for uL5 and bL9 also exhibit SDs, albeit at lower frequency. SDs also lie upstream of rplA (uL1) in about one fourth of the Cytophagia species analyzed (Table 1).
We next applied the same screen to all TIRs in all genomes. This allowed us to compute the SD frequency for all genes per organism and per class (Supplementary Table S5). To assess the expected rate of false positives, we also screened an upstream control window (–25 to –40; too far from the start codon) and termed the resulting ‘hits’ mock SD (MSD) sequences. Three different free energy thresholds (–7, –6, and –5 kcal/mol) were used for these screens, and consistent trends emerged regardless of the threshold. The analysis revealed that Flavobacteriia exhibit the lowest average SD frequency (Table 1, bottom row; Supplementary Table S5). In most Flavobacteriia species, fewer SDs were identified than MSDs, suggesting that selective pressure has acted to largely eliminate SDs in these organisms (Supplementary Table S5). In Chitinophagia and Cytophagia, average SD frequencies are slightly higher, on par with MSD frequencies (Table 1, bottom row; Supplementary Table S5). Bacteroidia and Sphingobacteriia exhibit somewhat larger average SD frequencies. For most organisms in these classes, the number of SDs modestly exceeds the number of MSDs. These genome-wide metrics are in line with the ribosomal gene results (Table 1), and collectively the data reveal differences in SD usage among the Bacteroidetes classes.
Covariation between mRNA and rRNA in a subset of Flavobacteriia
Futcher and coworkers showed that 16S rRNA genes of Chryseobacteria, Riemerella and related genera encode an alternative ASD sequence, 5′-AUCUCAUU-3′ (henceforth termed ASD2), with two substitutions (C1535U and C1539A, underscored above) (28). We looked upstream of rpsU in these organisms and in all cases found a sequence fully complementary to ASD2 and able to form an extended mRNA-rRNA helix (Figure 9B). This prompted us to look more systematically in these organisms for ‘SD2’ sequences, predicted to pair with ASD2, using free_scan. No other ribosomal gene exhibits a convincing SD2 (Supplementary Figure S23). We then screened all genes and compared the frequencies of SD2 and mock SD2 (MSD2) sequences, using various free energy thresholds (Supplementary Table S5). We found that the SD2 sequences are generally rare in these organisms. At the -7 kcal/mol threshold, very few (if any) genes besides rpsU were identified. At the -6 and –5 kcal/mol thresholds, frequencies of SD2 sequences remained quite low, similar to MSD2 frequencies. Relative to MSD2, SD2 sequences seemed to be modestly underrepresented in the former case and modestly overrepresented in the latter case, although the biological relevance of this observation remains unclear. Regardless, it is evident that translation initiation rarely entails mRNA-rRNA pairing in the Flavobacteriia, whether their ribosomes harbor ASD or ASD2. Importantly, these data provide compelling evidence for natural covariation in the SD-ASD helix and further implicate SD-ASD pairing in translation of rpsU in the Flavobacteriia.
DISCUSSION
It has been known for a decade that Bacteroidetes lack SD sequences, even though ribosomes of most of these organisms retain the conserved ASD. Introduced SD elements cause no stimulation of translation in vivo, suggesting that pairing between 16S rRNA and mRNA is somehow prevented or made inconsequential. In this work, we uncover the basis of ASD inhibition in the Bacteroidetes. The 30S subunits purified from F. johnsoniae fail to recognize the SD in vitro, indicating that intra-subunit interactions are responsible for ASD occlusion. A high-resolution cryo-EM structure of the F. johnsoniae 70S ribosome reveals that the 3′ end of 16S rRNA interacts with bS21, bS18, and bS6 on the 30S platform, contacts which physically sequester the ASD bases. Many of these contacts involve amino acids uniquely conserved in the Bacteroidetes, suggesting that the same mechanism of ASD occlusion is operational across the phylum. The entire 3′ end of 16S rRNA is well resolved in the cryo-EM map. This differs from all other vacant bacterial ribosome structures (70,102,103), which exhibit weak density beyond nucleotide 1534. We infer that the 3′ tail of 16S rRNA binds stably to the 30S platform in the Bacteroidetes ribosome but is free and mobile in other bacterial ribosomes. Consequently, the Bacteroidetes ribosome is unable to recognize SD sequences as other ribosomes do.
Absent from the imaged 70S particles were ribosomal proteins bS1 and uS2. Based on SDS-PAGE analysis and concurrent cryo-EM work (94), we infer that bS1 was mainly lost during ribosome purification while uS2 was lost during purification and sample vitrification. Importantly, there is no indication that either protein affects ASD function. Levels of both proteins are higher in our 30S preparation (which contains stoichiometrically-bound uS2). Yet, the 30S and 70S particles similarly fail to recognize the SD in assays of IC formation (Figure 1). Moreover, occlusion of the ASD is directly observed in 70S particles lacking bS1 and uS2, indicating that neither protein is required for the mechanism. That being said, we do suspect that bS1 depletion contributes to the generally low efficiency of IC formation seen for F. johnsoniae ribosomes. Protein bS1 is known to interact with mRNA, and bS1-mRNA contacts may be particularly important for translation initiation in organisms like the Bacteroidetes, which generally lack SD sequences (23,40,104). Future work will be needed to clarify the role of bS1 in F. johnsoniae IC formation.
We also report that in the Bacteroidetes certain ribosomal genes often contain SD sequences. In many Flavobacteriia, rpsU is the only ribosomal gene with an obvious SD. Flavobacteriia with an alternative ASD (ASD2) also have the complementary sequence (SD2) upstream of rpsU, corroborating the functional importance of rRNA-mRNA pairing in translation of rpsU. The corresponding protein, bS21, makes specific contacts to the 3′ tail of 16S rRNA and orients it toward bS18 and bS6. These observations suggest a simple mechanism of translational autoregulation. We propose that replete and bS21-deficient ribosomes are both active in translation; however, the latter ribosomes initiate translation of rpsU mRNA at a higher rate because the ASD is functionally liberated (Figure 10). As levels of bS21 increase in the cell, so does the proportion of replete ribosomes, damping down further synthesis of bS21. Protein bS21 is one of the last proteins to be incorporated during 30S assembly (105,106). Thus, such feedback regulation may help ensure the completion of ribosome biogenesis, for example when cells rapidly enter an environment without nutrients. While future work will be needed to test this hypothesis, the basic concept is consistent with the following observations. First, bS21 is naturally absent from some bacteria (107) and nonessential in others (108–111), so there are ample examples of active bS21-deficient ribosomes. Second, homologs of bS21 are encoded by many marine bacteriophages (112). In tested cases, these viral proteins can assemble into host ribosomes, possibly to shunt those ribosomes to phage mRNA.
Figure 10.
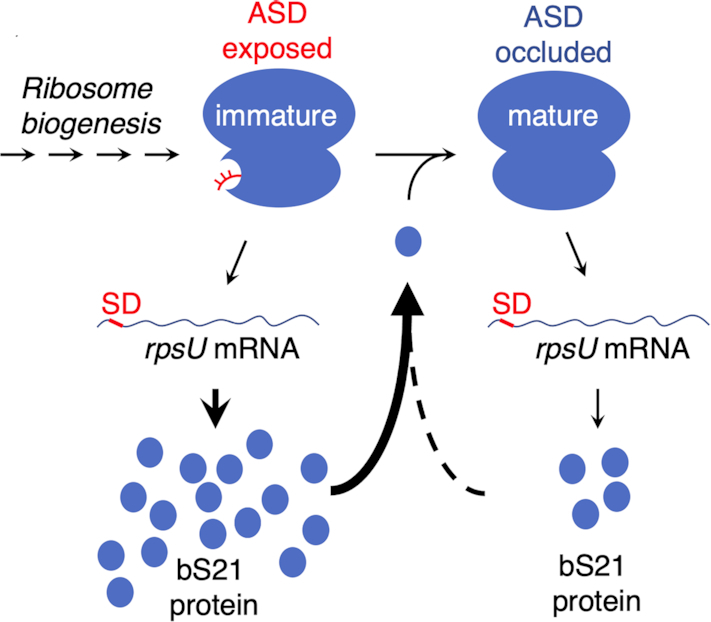
A potential mechanism of bS21 autoregulation. Incorporation of bS21 is one of the last steps in 30S biogenesis. Ribosomes lacking bS21 (immature) contain a liberated ASD, allowing for high-level translation of the rpsU mRNA. Product bS21 binds to generate the replete (mature) ribosome, in which the ASD is occluded. Replete ribosomes will also translate rpsU mRNA but at a reduced rate.
In Bacteroidia, SDs are absent from rpsU but frequently found upstream of rpsR, the gene encoding bS18. Like bS21, bS18 contributes to ASD occlusion and hence could regulate its own production through a mechanism analogous to that described above. Namely, we hypothesize that bS18-depleted subunits, which can readily engage SD sequences, increase the overall rate of translation of rpsR mRNA. The bS18 gene is nonessential in Porphyromonas gingivalis (108), supporting the plausibility of this hypothesis. Interestingly, SDs are prevalent in rpsU and/or rpsR in many Chitinophagia, Cytophagia, and Sphingobacteriia, raising the possibility that bS21- and/or bS18-depleted subunits contribute to translational control in these organisms.
In Bacteroidia, Chitinophagia, Cytophagia, and Sphingobacteriia, other ribosomal genes frequently contain SDs. While the purpose of these SDs remains unclear, they are likely involved in some form of translational control. Intriguingly, proteins of the 30S encoded by these genes (bS1, uS2, uS7, uS11) bind near bS21 and bS18 in the mature subunit, and there exist multiple functional associations between these proteins during 30S assembly (106,113,114). If our hypothesis above is correct, the functional difference between replete and bS21-depleted (or bS18-depleted) ribosomes could be exploited to regulate ribosome assembly. These depleted subunits, acting as proxies of assembly intermediates, might adjust translation of multiple mRNAs to ensure efficient ribosome biogenesis under various growth conditions. We envisage that this sort of feedback control has been elaborated to varying degrees, depending on the class of Bacteroidetes. A clear advantage gained by occlusion of the ASD in the vast majority of cellular ribosomes is an opportunity to use SDs as regulatory elements.
One of the most conserved features of the bacterial ribosome is the ASD. Recent genomic studies have identified the rare cases of ASD sequence variation, which occur almost exclusively in a subset of Flavobacteriia (27,28). These natural substitutions occur mainly at positions 1535 and/or 1539, as exemplified by ASD2 of Chyseobacteria and related organisms (Figure 9B). Notably, in the F. johnsoniae ribosome structure, the base of C1535 is oriented toward the solvent and neither C1535 nor C1539 makes specific contacts with the ribosome platform. Hence, substitutions at these positions are predicted to have little or no effect on the mechanism of ASD occlusion, in contrast to substitutions at neighboring positions. This hints that, in the Bacteroidetes, platform interactions impose stronger evolutionary constraints on the 3′ end of 16S rRNA than mRNA interactions do.
Finally, we show that F. johnsoniae initiation complexes can be reconstituted in vitro using purified components. Complex formation is strictly factor-dependent and occurs at the canonical start codon for all tested model mRNAs (from F. johnsoniae or E. coli). While 70S ICs were readily detected by toeprinting, 30S ICs were not (or gave weak toeprints) in parallel experiments. Either these 30S ICs were too labile to halt reverse transcriptase or they simply did not form under the conditions employed. It is possible that these 30S ICs fail to form due to loss of factor or subunit activity. However, efficient 70S IC formation requires all three F. johnsoniae initiation factors, suggesting that each purified factor exhibits activity. Moreover, replacement of F. johnsoniae IF3 with E. coli IF3 results in a heterologous 30S IC, readily detected by toeprinting, indicating that 30S subunits prepared from F. johnsoniae are capable of complex formation. Additionally, 70S ICs can be formed from isolated F. johnsoniae subunits (rather than tight-couple ribosomes), providing further evidence that these 30S subunits exhibit activity. Still, further work will be needed to determine whether the anomalous behavior of the F. johnsoniae 30S IC holds biological relevance or has a more trivial basis.
The ability of E. coli IF3 to stabilize F. johnsoniae 30S complexes came as a surprise, because studies of E. coli initiation have shown that IF3 has a net destabilizing effect on the 30S IC (60,115). IF3 increases both the binding rate (kon) and dissociation rate (koff) of fMet-tRNA, but the effect on koff is larger (115). Notably, the E. coli studies have employed SD-containing mRNA, and to our knowledge analogous studies using SD-lacking mRNA have yet to be performed. IF3 is a two-domain protein that exhibits multiple modes of binding during initiation (7,116). The C-terminal domain interacts with distinct sites on the 30S platform, and the N-terminal domain contacts the elbow region of fMet-tRNA. IF3 lies near the SD-ASD helix, and IF3 and IF1 affect the positioning of the SD-ASD helix (7). These observations raise the following questions, which remain open: (1) Does the role of IF3 differ in the two bacterial systems? (2) Does SD-ASD pairing influence IF3 dynamics and its interplay with fMet-tRNA? Additional experiments will be needed to address these questions and further advance our understanding of translation initiation in bacteria.
DATA AVAILABILITY
The electron microscopy map and the model for the F. johnsoniae 70S ribosome have been deposited in the Electron Microscopy Data Bank (EMDB) and in the Protein Data Bank (PDB) with accession codes EMD-22345 and PDB 7JIL, respectively.
Supplementary Material
ACKNOWLEDGEMENTS
We thank S. McNutt and F. Chu at the University of New Hampshire for performing the LC–MS/MS analysis; K. Sears, M. Strauss and other staff members of the Facility for Electron Microscopy Research (FEMR) at McGill University for help with microscope operation and data collection; and R. Green, A. Buskirk and T. Dever for feedback on the manuscript.
Contributor Information
Vikash Jha, Department of Anatomy and Cell Biology, McGill University, Montreal, Quebec H3A 0C7, Canada; Centre for Structural Biology, McGill University, Montreal, Quebec H3G 0B1, Canada.
Bappaditya Roy, Department of Microbiology, The Ohio State University, Columbus, OH 43210, USA; Center for RNA Biology, The Ohio State University, Columbus, OH 43210, USA.
Dushyant Jahagirdar, Department of Anatomy and Cell Biology, McGill University, Montreal, Quebec H3A 0C7, Canada; Centre for Structural Biology, McGill University, Montreal, Quebec H3G 0B1, Canada.
Zakkary A McNutt, Center for RNA Biology, The Ohio State University, Columbus, OH 43210, USA; Ohio State Biochemistry Program, The Ohio State University, Columbus, OH 43210, USA.
Elan A Shatoff, Center for RNA Biology, The Ohio State University, Columbus, OH 43210, USA; Department of Physics, The Ohio State University, Columbus, OH 43210, USA.
Bethany L Boleratz, Ohio State Biochemistry Program, The Ohio State University, Columbus, OH 43210, USA.
Dean E Watkins, Department of Microbiology, The Ohio State University, Columbus, OH 43210, USA.
Ralf Bundschuh, Center for RNA Biology, The Ohio State University, Columbus, OH 43210, USA; Department of Physics, The Ohio State University, Columbus, OH 43210, USA; Department of Chemistry & Biochemistry, Division of Hematology, The Ohio State University, Columbus, OH 43210, USA.
Kaustuv Basu, Department of Anatomy and Cell Biology, McGill University, Montreal, Quebec H3A 0C7, Canada; Centre for Structural Biology, McGill University, Montreal, Quebec H3G 0B1, Canada.
Joaquin Ortega, Department of Anatomy and Cell Biology, McGill University, Montreal, Quebec H3A 0C7, Canada; Centre for Structural Biology, McGill University, Montreal, Quebec H3G 0B1, Canada.
Kurt Fredrick, Department of Microbiology, The Ohio State University, Columbus, OH 43210, USA; Center for RNA Biology, The Ohio State University, Columbus, OH 43210, USA; Ohio State Biochemistry Program, The Ohio State University, Columbus, OH 43210, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [MCB-2029502 to K.F.]; NSERC Discovery Program [RGPIN-2019-05799 to J.O.]; Titan Krios cryo-EM data were collected at FEMR (McGill); FEMR is supported by the Canadian Foundation for Innovation, Quebec Government and McGill University. Funding for open access charge: National Natural Science Foundation of China [31870721].
Conflict of interest statement. None declared.
REFERENCES
- 1. Shine J., Dalgarno L.. The 3′-terminal sequence of Escherichia coli 16S ribosomal RNA: complementarity to nonsense triplets and ribosome binding sites. Proc. Natl. Acad. Sci. U.S.A. 1974; 71:1342–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shultzaberger R.K., Bucheimer R.E., Rudd K.E., Schneider T.D.. Anatomy of Escherichia coli ribosome binding sites. J. Mol. Biol. 2001; 313:215–228. [DOI] [PubMed] [Google Scholar]
- 3. Vellanoweth R.L., Rabinowitz J.C.. The influence of ribosome-binding-site elements on translational efficiency in Bacillus subtilis and Escherichia coli in vivo. Mol. Microbiol. 1992; 6:1105–1114. [DOI] [PubMed] [Google Scholar]
- 4. Steitz J.A., Jakes K.. How ribosomes select initiator regions in mRNA: base pair formation between the 3′ terminus of 16S rRNA and the mRNA during initiation of protein synthesis in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1975; 72:4734–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jenner L.B., Demeshkina N., Yusupova G., Yusupov M.. Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat. Struct. Mol. Biol. 2010; 17:555–560. [DOI] [PubMed] [Google Scholar]
- 6. Korostelev A., Trakhanov S., Asahara H., Laurberg M., Lancaster L., Noller H.F.. Interactions and dynamics of the Shine Dalgarno helix in the 70S ribosome. Proc. Natl. Acad. Sci. U.S.A. 2007; 104:16840–16843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hussain T., Llacer J.L., Wimberly B.T., Kieft J.S., Ramakrishnan V.. Large-Scale movements of IF3 and tRNA during bacterial translation initiation. Cell. 2016; 167:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kaminishi T., Wilson D.N., Takemoto C., Harms J.M., Kawazoe M., Schluenzen F., Hanawa-Suetsugu K., Shirouzu M., Fucini P., Yokoyama S.. A snapshot of the 30S ribosomal subunit capturing mRNA via the Shine-Dalgarno interaction. Structure. 2007; 15:289–297. [DOI] [PubMed] [Google Scholar]
- 9. Zhu H.Q., Hu G.Q., Ouyang Z.Q., Wang J., She Z.S.. Accuracy improvement for identifying translation initiation sites in microbial genomes. Bioinformatics. 2004; 20:3308–3317. [DOI] [PubMed] [Google Scholar]
- 10. Delcher A.L., Bratke K.A., Powers E.C., Salzberg S.L.. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007; 23:673–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Besemer J., Lomsadze A., Borodovsky M.. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001; 29:2607–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Suzek B.E., Ermolaeva M.D., Schreiber M., Salzberg S.L.. A probabilistic method for identifying start codons in bacterial genomes. Bioinformatics. 2001; 17:1123–1130. [DOI] [PubMed] [Google Scholar]
- 13. Ou H.Y., Guo F.B., Zhang C.T.. GS-Finder: a program to find bacterial gene start sites with a self-training method. Int. J. Biochem. Cell Biol. 2004; 36:535–544. [DOI] [PubMed] [Google Scholar]
- 14. Osada Y., Saito R., Tomita M.. Analysis of base-pairing potentials between 16S rRNA and 5′ UTR for translation initiation in various prokaryotes. Bioinformatics. 1999; 15:578–581. [DOI] [PubMed] [Google Scholar]
- 15. Ringquist S., Shinedling S., Barrick D., Green L., Binkley J., Stormo G.D., Gold L.. Translation initiation in Escherichia coli: sequences within the ribosome-binding site. Mol. Microbiol. 1992; 6:1219–1229. [DOI] [PubMed] [Google Scholar]
- 16. Schurr T., Nadir E., Margalit H.. Identification and characterization of E.coli ribosomal binding sites by free energy computation. Nucleic Acids Res. 1993; 21:4019–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. de Smit M.H., van Duin J.. Translational initiation on structured messengers. Another role for the Shine-Dalgarno interaction. J. Mol. Biol. 1994; 235:173–184. [DOI] [PubMed] [Google Scholar]
- 18. Hui A., de Boer H.A.. Specialized ribosome system: preferential translation of a single mRNA species by a subpopulation of mutated ribosomes in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:4762–4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jacob W.F., Santer M., Dahlberg A.E.. A single base change in the Shine-Dalgarno region of 16S rRNA of Escherichia coli affects translation of many proteins. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:4757–4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li G.W., Burkhardt D., Gross C., Weissman J.S.. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014; 157:624–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schrader J.M., Zhou B., Li G.W., Lasker K., Childers W.S., Williams B., Long T., Crosson S., McAdams H.H., Weissman J.S. et al.. The coding and noncoding architecture of the Caulobacter crescentus genome. PLoS Genet. 2014; 10:e1004463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Skorski P., Leroy P., Fayet O., Dreyfus M., Hermann-Le Denmat S.. The highly efficient translation initiation region from the Escherichia coli rpsA gene lacks a shine-dalgarno element. J. Bacteriol. 2006; 188:6277–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakagawa S., Niimura Y., Miura K., Gojobori T.. Dynamic evolution of translation initiation mechanisms in prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 2010; 107:6382–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nakagawa S., Niimura Y., Gojobori T.. Comparative genomic analysis of translation initiation mechanisms for genes lacking the Shine-Dalgarno sequence in prokaryotes. Nucleic Acids Res. 2017; 45:3922–3931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wegmann U., Horn N., Carding S.R.. Defining the bacteroides ribosomal binding site. Appl. Environ. Microbiol. 2013; 79:1980–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Accetto T., Avgustin G.. Inability of Prevotella bryantii to form a functional Shine-Dalgarno interaction reflects unique evolution of ribosome binding sites in Bacteroidetes. PLoS One. 2011; 6:e22914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lim K., Furuta Y., Kobayashi I.. Large variations in bacterial ribosomal RNA genes. Mol. Biol. Evol. 2012; 29:2937–2948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Amin M.R., Yurovsky A., Chen Y., Skiena S., Futcher B.. Re-annotation of 12,495 prokaryotic 16S rRNA 3′ ends and analysis of Shine-Dalgarno and anti-Shine-Dalgarno sequences. PLoS One. 2018; 13:e0202767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hahnke R.L., Meier-Kolthoff J.P., Garcia-Lopez M., Mukherjee S., Huntemann M., Ivanova N.N., Woyke T., Kyrpides N.C., Klenk H.P., Goker M.. Genome-Based taxonomic classification of bacteroidetes. Front Microbiol. 2016; 7:2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ley R.E., Hamady M., Lozupone C., Turnbaugh P.J., Ramey R.R., Bircher J.S., Schlegel M.L., Tucker T.A., Schrenzel M.D., Knight R. et al.. Evolution of mammals and their gut microbes. Science. 2008; 320:1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martens E.C., Roth R., Heuser J.E., Gordon J.I.. Coordinate regulation of glycan degradation and polysaccharide capsule biosynthesis by a prominent human gut symbiont. J. Biol. Chem. 2009; 284:18445–18457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martens E.C., Koropatkin N.M., Smith T.J., Gordon J.I.. Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J. Biol. Chem. 2009; 284:24673–24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martens E.C., Chiang H.C., Gordon J.I.. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008; 4:447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Johnson E.L., Heaver S.L., Walters W.A., Ley R.E.. Microbiome and metabolic disease: revisiting the bacterial phylum Bacteroidetes. J Mol Med (Berl). 2017; 95:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rinninella E., Raoul P., Cintoni M., Franceschi F., Miggiano G.A.D., Gasbarrini A., Mele M.C.. What is the healthy gut microbiota composition? A changing ecosystem across age, environment, diet, and diseases. Microorganisms. 2019; 7:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bayley D.P., Rocha E.R., Smith C.J.. Analysis of cepA and other Bacteroides fragilis genes reveals a unique promoter structure. FEMS Microbiol. Lett. 2000; 193:149–154. [DOI] [PubMed] [Google Scholar]
- 37. Chen S., Bagdasarian M., Kaufman M.G., Walker E.D.. Characterization of strong promoters from an environmental Flavobacterium hibernum strain by using a green fluorescent protein-based reporter system. Appl. Environ. Microbiol. 2007; 73:1089–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen S., Bagdasarian M., Kaufman M.G., Bates A.K., Walker E.D.. Mutational analysis of the ompA promoter from Flavobacterium johnsoniae. J. Bacteriol. 2007; 189:5108–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vingadassalom D., Kolb A., Mayer C., Rybkine T., Collatz E., Podglajen I.. An unusual primary sigma factor in the Bacteroidetes phylum. Mol. Microbiol. 2005; 56:888–902. [DOI] [PubMed] [Google Scholar]
- 40. Baez W.D., Roy B., McNutt Z.A., Shatoff E.A., Chen S., Bundschuh R., Fredrick K.. Global analysis of protein synthesis in Flavobacterium johnsoniae reveals the use of Kozak-like sequences in diverse bacteria. Nucleic Acids Res. 2019; 47:10477–10488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kozak M. Point mutations define a sequence flanking the AUG initiator codon that modulates translation by eukaryotic ribosomes. Cell. 1986; 44:283–292. [DOI] [PubMed] [Google Scholar]
- 42. Nakagawa S., Niimura Y., Gojobori T., Tanaka H., Miura K.. Diversity of preferred nucleotide sequences around the translation initiation codon in eukaryote genomes. Nucleic Acids Res. 2008; 36:861–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yamauchi K. The sequence flanking translational initiation site in protozoa. Nucleic Acids Res. 1991; 19:2715–2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. McBride M.J., Kempf M.J.. Development of techniques for the genetic manipulation of the gliding bacterium Cytophaga johnsonae. J. Bacteriol. 1996; 178:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sezonov G., Joseleau-Petit D., D’Ari R.. Escherichia coli physiology in Luria-Bertani broth. J. Bacteriol. 2007; 189:8746–8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Qin D., Abdi N.M., Fredrick K.. Characterization of 16S rRNA mutations that decrease the fidelity of translation initiation. RNA. 2007; 13:2348–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lancaster L., Kiel M.C., Kaji A., Noller H.F.. Orientation of ribosome recycling factor in the ribosome from directed hydroxyl radical probing. Cell. 2002; 111:129–140. [DOI] [PubMed] [Google Scholar]
- 48. Butler J.S., Springer M., Dondon J., Graffe M., Grunberg-Manago M.. Escherichia coli protein synthesis initiation factor IF3 controls its own gene expression at the translational level in vivo. J. Mol. Biol. 1986; 192:767–780. [DOI] [PubMed] [Google Scholar]
- 49. Gold L., Stormo G., Saunders R.. Escherichia coli translational initiation factor IF3: a unique case of translational regulation. Proc. Natl. Acad. Sci. U.S.A. 1984; 81:7061–7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sacerdot C., Chiaruttini C., Engst K., Graffe M., Milet M., Mathy N., Dondon J., Springer M.. The role of the AUU initiation codon in the negative feedback regulation of the gene for translation initiation factor IF3 in Escherichia coli. Mol. Microbiol. 1996; 21:331–346. [DOI] [PubMed] [Google Scholar]
- 51. Sussman J.K., Simons E.L., Simons R.W.. Escherichia coli translation initiation factor 3 discriminates the initiation codon in vivo. Mol. Microbiol. 1996; 21:347–360. [DOI] [PubMed] [Google Scholar]
- 52. McBride M.J., Xie G., Martens E.C., Lapidus A., Henrissat B., Rhodes R.G., Goltsman E., Wang W., Xu J., Hunnicutt D.W. et al.. Novel features of the polysaccharide-digesting gliding bacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl. Environ. Microbiol. 2009; 75:6864–6875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xie G., Bruce D.C., Challacombe J.F., Chertkov O., Detter J.C., Gilna P., Han C.S., Lucas S., Misra M., Myers G.L. et al.. Genome sequence of the cellulolytic gliding bacterium Cytophaga hutchinsonii. Appl. Environ. Microbiol. 2007; 73:3536–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Qin D., Fredrick K.. Control of translation initiation involves a factor-induced rearrangement of helix 44 of 16S ribosomal RNA. Mol. Microbiol. 2009; 71:1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dallas A., Noller H.F.. Interaction of translation initiation factor 3 with the 30S ribosomal subunit. Mol. Cell. 2001; 8:855–864. [DOI] [PubMed] [Google Scholar]
- 56. Walker S.E., Fredrick K.. Preparation and evaluation of acylated tRNAs. Methods. 2008; 44:81–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fredrick K., Noller H.F.. Accurate translocation of mRNA by the ribosome requires a peptidyl group or its analog on the tRNA moving into the 30S P site. Mol. Cell. 2002; 9:1125–1131. [DOI] [PubMed] [Google Scholar]
- 58. Shoji S., Walker S.E., Fredrick K.. Reverse translocation of tRNA in the ribosome. Mol. Cell. 2006; 24:931–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hartz D., McPheeters D.S., Gold L.. Selection of the initiator tRNA by Escherichia coli initiation factors. Genes Dev. 1989; 3:1899–1912. [DOI] [PubMed] [Google Scholar]
- 60. Qin D., Liu Q., Devaraj A., Fredrick K.. Role of helix 44 of 16S rRNA in the fidelity of translation initiation. RNA. 2012; 18:485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Roy B., Liu Q., Shoji S., Fredrick K.. IF2 and unique features of initiator tRNA(fMet) help establish the translational reading frame. RNA Biol. 2018; 15:604–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Beringer M., Bruell C., Xiong L., Pfister P., Bieling P., Katunin V.I., Mankin A.S., Bottger E.C., Rodnina M.V.. Essential mechanisms in the catalysis of peptide bond formation on the ribosome. J. Biol. Chem. 2005; 280:36065–36072. [DOI] [PubMed] [Google Scholar]
- 63. Fahlman R.P., Uhlenbeck O.C.. Contribution of the esterified amino acid to the binding of aminoacylated tRNAs to the ribosomal P- and A-sites. Biochemistry. 2004; 43:7575–7583. [DOI] [PubMed] [Google Scholar]
- 64. Shoji S., Abdi N.M., Bundschuh R., Fredrick K.. Contribution of ribosomal residues to P-site tRNA binding. Nucleic Acids Res. 2009; 37:4033–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lalanne J.B., Taggart J.C., Guo M.S., Herzel L., Schieler A., Li G.W.. Evolutionary convergence of pathway-specific enzyme expression stoichiometry. Cell. 2018; 173:749–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zeng-Elmore X., Gao X.Z., Pellarin R., Schneidman-Duhovny D., Zhang X.J., Kozacka K.A., Tang Y., Sali A., Chalkley R.J., Cote R.H. et al.. Molecular architecture of photoreceptor phosphodiesterase elucidated by chemical cross-linking and integrative modeling. J. Mol. Biol. 2014; 426:3713–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zheng S.Q., Palovcak E., Armache J.P., Verba K.A., Cheng Y., Agard D.A.. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods. 2017; 14:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zivanov J., Nakane T., Forsberg B.O., Kimanius D., Hagen W.J., Lindahl E., Scheres S.H.. New tools for automated high-resolution cryo-EM structure determination in RELION-3. Elife. 2018; 7:e42166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zhang K. Gctf: Real-time CTF determination and correction. J. Struct. Biol. 2016; 193:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Schuwirth B.S., Borovinskaya M.A., Hau C.W., Zhang W., Vila-Sanjurjo A., Holton J.M., Cate J.H.. Structures of the bacterial ribosome at 3.5 A resolution. Science. 2005; 310:827–834. [DOI] [PubMed] [Google Scholar]
- 71. de la Rosa-Trevin J.M., Oton J., Marabini R., Zaldivar A., Vargas J., Carazo J.M., Sorzano C.O.. Xmipp 3.0: an improved software suite for image processing in electron microscopy. J. Struct. Biol. 2013; 184:321–328. [DOI] [PubMed] [Google Scholar]
- 72. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W. et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Emsley P., Cowtan K.. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. Biol. Crystallogr. 2004; 60:2126–2132. [DOI] [PubMed] [Google Scholar]
- 74. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D. Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. UniProt C. UniProt: a worldwide hub of protein knowledge. Nucleic Acids Res. 2019; 47:D506–D515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Larkin M.A., Blackshields G., Brown N.P., Chenna R., McGettigan P.A., McWilliam H., Valentin F., Wallace I.M., Wilm A., Lopez R. et al.. Clustal W and Clustal X version 2.0. Bioinformatics. 2007; 23:2947–2948. [DOI] [PubMed] [Google Scholar]
- 77. Sohmen D., Chiba S., Shimokawa-Chiba N., Innis C.A., Berninghausen O., Beckmann R., Ito K., Wilson D.N.. Structure of the Bacillus subtilis 70S ribosome reveals the basis for species-specific stalling. Nat. Commun. 2015; 6:6941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Afonine P.V., Poon B.K., Read R.J., Sobolev O.V., Terwilliger T.C., Urzhumtsev A., Adams P.D.. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr. D Struct. Biol. 2018; 74:531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Afonine P.V., Klaholz B.P., Moriarty N.W., Poon B.K., Sobolev O.V., Terwilliger T.C., Adams P.D., Urzhumtsev A.. New tools for the analysis and validation of cryo-EM maps and atomic models. Acta Crystallogr. D Struct. Biol. 2018; 74:814–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B. et al.. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018; 27:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. DeLano W.L. The PyMOL Molecular Graphic Systems. 2002; Palo Alto, CA, USA: DeLano Scientific. [Google Scholar]
- 82. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 83. Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E.. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018; 27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lu X.J., Bussemaker H.J., Olson W.K.. DSSR: an integrated software tool for dissecting the spatial structure of RNA. Nucleic Acids Res. 2015; 43:e142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Darty K., Denise A., Ponty Y.. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics. 2009; 25:1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Okonechnikov K., Golosova O., Fursov M., team U.. Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics. 2012; 28:1166–1167. [DOI] [PubMed] [Google Scholar]
- 87. Katoh K., Misawa K., Kuma K., Miyata T.. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002; 30:3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Crooks G.E., Hon G., Chandonia J.M., Brenner S.E.. WebLogo: a sequence logo generator. Genome Res. 2004; 14:1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kozlowski L.P. IPC - Isoelectric Point Calculator. Biol. Direct. 2016; 11:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kitts P.A., Church D.M., Thibaud-Nissen F., Choi J., Hem V., Sapojnikov V., Smith R.G., Tatusova T., Xiang C., Zherikov A. et al.. Assembly: a resource for assembled genomes at NCBI. Nucleic Acids Res. 2016; 44:D73–D80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Starmer J., Stomp A., Vouk M., Bitzer D.. Predicting Shine-Dalgarno sequence locations exposes genome annotation errors. PLoS Comput. Biol. 2006; 2:e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Salis H.M., Mirsky E.A., Voigt C.A.. Automated design of synthetic ribosome binding sites to control protein expression. Nat. Biotechnol. 2009; 27:946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Subramanian A.R., van Duin J.. Exchange of individual ribosomal proteins between ribosomes as studied by heavy isotope-transfer experiments. Mol. Gen. Genet. 1977; 158:1–9. [DOI] [PubMed] [Google Scholar]
- 94. Jahagirdar D., Jha V., Basu B., Gomez-Blanco J., Vargas J., Ortega J.. Alternative conformations and motions adopted by 30S ribosomal subunits visualized by Cryo-Electron microscopy. RNA. 2020; 26:2017–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Kaledhonkar S., Fu Z., Caban K., Li W., Chen B., Sun M., Gonzalez R.L. Jr, Frank J.. Late steps in bacterial translation initiation visualized using time-resolved cryo-EM. Nature. 2019; 570:400–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Polikanov Y.S., Starosta A.L., Juette M.F., Altman R.B., Terry D.S., Lu W., Burnett B.J., Dinos G., Reynolds K.A., Blanchard S.C. et al.. Distinct tRNA accommodation intermediates observed on the ribosome with the antibiotics Hygromycin A and A201A. Mol. Cell. 2015; 58:832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hentschel J., Burnside C., Mignot I., Leibundgut M., Boehringer D., Ban N.. The complete structure of the Mycobacterium smegmatis 70S ribosome. Cell Rep. 2017; 20:149–160. [DOI] [PubMed] [Google Scholar]
- 98. Halfon Y., Jimenez-Fernandez A., La Rosa R., Espinosa Portero R., Krogh Johansen H., Matzov D., Eyal Z., Bashan A., Zimmerman E., Belousoff M. et al.. Structure of Pseudomonas aeruginosa ribosomes from an aminoglycoside-resistant clinical isolate. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:22275–22281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Khusainov I., Vicens Q., Bochler A., Grosse F., Myasnikov A., Menetret J.F., Chicher J., Marzi S., Romby P., Yusupova G. et al.. Structure of the 70S ribosome from human pathogen Staphylococcus aureus. Nucleic Acids Res. 2017; 45:1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ban N., Beckmann R., Cate J.H., Dinman J.D., Dragon F., Ellis S.R., Lafontaine D.L., Lindahl L., Liljas A., Lipton J.M. et al.. A new system for naming ribosomal proteins. Curr. Opin. Struct. Biol. 2014; 24:165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Li Z., Ge X., Zhang Y., Zheng L., Sanyal S., Gao N.. Cryo-EM structure of Mycobacterium smegmatis ribosome reveals two unidentified ribosomal proteins close to the functional centers. Protein Cell. 2018; 9:384–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Cocozaki A.I., Altman R.B., Huang J., Buurman E.T., Kazmirski S.L., Doig P., Prince D.B., Blanchard S.C., Cate J.H., Ferguson A.D.. Resistance mutations generate divergent antibiotic susceptibility profiles against translation inhibitors. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:8188–8193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Borovinskaya M.A., Pai R.D., Zhang W., Schuwirth B.S., Holton J.M., Hirokawa G., Kaji H., Kaji A., Cate J.H.. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat. Struct. Mol. Biol. 2007; 14:727–732. [DOI] [PubMed] [Google Scholar]
- 104. Salah P., Bisaglia M., Aliprandi P., Uzan M., Sizun C., Bontems F.. Probing the relationship between Gram-negative and Gram-positive S1 proteins by sequence analysis. Nucleic Acids Res. 2009; 37:5578–5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chen S.S., Williamson J.R.. Characterization of the ribosome biogenesis landscape in E. coli using quantitative mass spectrometry. J. Mol. Biol. 2013; 425:767–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Sashital D.G., Greeman C.A., Lyumkis D., Potter C.S., Carragher B., Williamson J.R.. A combined quantitative mass spectrometry and electron microscopy analysis of ribosomal 30S subunit assembly in E. coli. Elife. 2014; 3:e04491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Yutin N., Puigbo P., Koonin E.V., Wolf Y.I.. Phylogenomics of prokaryotic ribosomal proteins. PLoS One. 2012; 7:e36972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Klein B.A., Tenorio E.L., Lazinski D.W., Camilli A., Duncan M.J., Hu L.T.. Identification of essential genes of the periodontal pathogen Porphyromonas gingivalis. BMC Genomics. 2012; 13:578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Takada H., Morita M., Shiwa Y., Sugimoto R., Suzuki S., Kawamura F., Yoshikawa H.. Cell motility and biofilm formation in Bacillus subtilis are affected by the ribosomal proteins, S11 and S21. Biosci. Biotechnol. Biochem. 2014; 78:898–907. [DOI] [PubMed] [Google Scholar]
- 110. Akanuma G., Nanamiya H., Natori Y., Yano K., Suzuki S., Omata S., Ishizuka M., Sekine Y., Kawamura F.. Inactivation of ribosomal protein genes in Bacillus subtilis reveals importance of each ribosomal protein for cell proliferation and cell differentiation. J. Bacteriol. 2012; 194:6282–6291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Metselaar K.I., den Besten H.M., Boekhorst J., van Hijum S.A., Zwietering M.H., Abee T.. Diversity of acid stress resistant variants of Listeria monocytogenes and the potential role of ribosomal protein S21 encoded by rpsU. Front Microbiol. 2015; 6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Mizuno C.M., Guyomar C., Roux S., Lavigne R., Rodriguez-Valera F., Sullivan M.B., Gillet R., Forterre P., Krupovic M.. Numerous cultivated and uncultivated viruses encode ribosomal proteins. Nat. Commun. 2019; 10:752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Mizushima S., Nomura M.. Assembly mapping of 30S ribosomal proteins from E. coli. Nature. 1970; 226:1214. [DOI] [PubMed] [Google Scholar]
- 114. Mulder A.M., Yoshioka C., Beck A.H., Bunner A.E., Milligan R.A., Potter C.S., Carragher B., Williamson J.R.. Visualizing ribosome biogenesis: parallel assembly pathways for the 30S subunit. Science. 2010; 330:673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Antoun A., Pavlov M.Y., Lovmar M., Ehrenberg M.. How initiation factors tune the rate of initiation of protein synthesis in bacteria. EMBO J. 2006; 25:2539–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Elvekrog M.M., Gonzalez R.L. Jr. Conformational selection of translation initiation factor 3 signals proper substrate selection. Nat. Struct. Mol. Biol. 2013; 20:628–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The electron microscopy map and the model for the F. johnsoniae 70S ribosome have been deposited in the Electron Microscopy Data Bank (EMDB) and in the Protein Data Bank (PDB) with accession codes EMD-22345 and PDB 7JIL, respectively.