


Abstract
The XRCC1–DNA ligase IIIα complex (XL) is critical for DNA single-strand break repair, a key target for PARP inhibitors in cancer cells deficient in homologous recombination. Here, we combined biophysical approaches to gain insights into the shape and conformational flexibility of the XL as well as XRCC1 and DNA ligase IIIα (LigIIIα) alone. Structurally-guided mutational analyses based on the crystal structure of the human BRCT–BRCT heterodimer identified the network of salt bridges that together with the N-terminal extension of the XRCC1 C-terminal BRCT domain constitute the XL molecular interface. Coupling size exclusion chromatography with small angle X-ray scattering and multiangle light scattering (SEC-SAXS–MALS), we determined that the XL is more compact than either XRCC1 or LigIIIα, both of which form transient homodimers and are highly disordered. The reduced disorder and flexibility allowed us to build models of XL particles visualized by negative stain electron microscopy that predict close spatial organization between the LigIIIα catalytic core and both BRCT domains of XRCC1. Together our results identify an atypical BRCT–BRCT interaction as the stable nucleating core of the XL that links the flexible nick sensing and catalytic domains of LigIIIα to other protein partners of the flexible XRCC1 scaffold.
Graphical Abstract
Graphical Abstract.
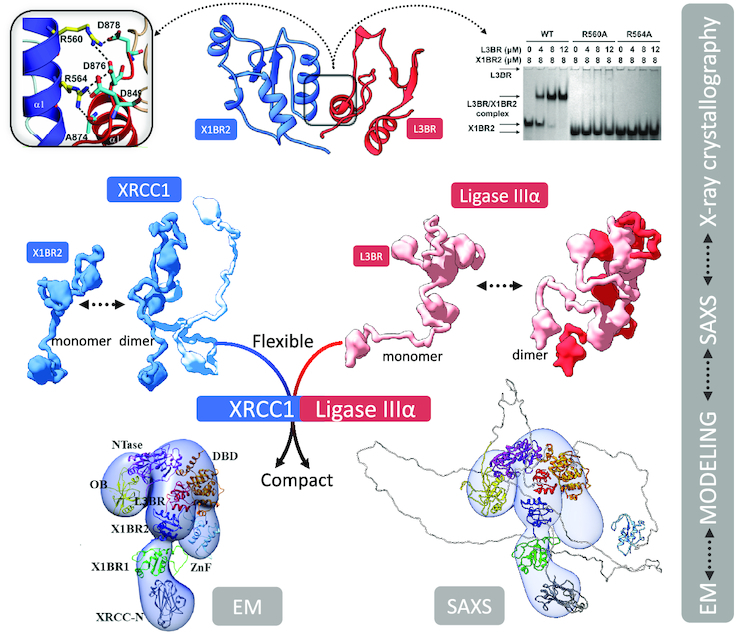
Integrative modeling from EM and SAXS with atomic resolution domain structures reveals the functional compaction of the XRCC1-DNA ligase IIIα complex that extends beyond the interacting C-terminal BRCT–BRCT domains to the catalytic region of DNA ligase IIIα.
INTRODUCTION
The DNA repair gene XRCC1 was first identified in a screen of human cDNAs that complemented the alkylation and X-ray sensitive phenotypes of the mutant Chinese Hamster Ovary cell line EM9 (1). Besides DNA damage sensitivity, a hallmark feature of these cells is a high spontaneous level of sister chromatid exchanges, indicative of hyperrecombination (1). XRCC1 protein lacks enzymatic activity; yet, it plays a key role in multiple DNA repair pathways by acting as a scaffold that binds to both DNA single strand breaks and gaps, poly (ADP-ribose) and to numerous DNA repair enzymes (2). DNA ligase IIIα (LigIIIα) was the first XRCC1-interacting protein to be identified (3), and is critical to bridge two DNA ends to promote specific intermolecular DNA end joining (4). This was followed by DNA glycosylases (5–10), AP endonuclease 1 (11), and DNA polymerase β (Polβ) (12,13), implicating XRCC1 in coordinating observed product complexes (14) for the short patch subpathway of base excision repair. An interaction with automodified PARP1 serves to recruit XRCC1 and other interacting proteins that also include the end-processing enzymes, polynucleotide kinase phosphorylase (PNKP) (15), aprataxin (APTX) (16) and APLF (17) to DNA single strand breaks (18–20). While these protein-protein interactions and the phenotype of xrcc1 cells show XRCC1 is a key factor in single-strand break repair (SSBR), other SSBR and base excision repair (BER) pathways involving DNA ligase I (LigI) and other DNA replication proteins are at least partially functionally redundant (21–24). This redundancy between LigI- and LigIIIα-dependent subpathways also occurs in nucleotide excision repair and in back-up pathways of DNA double-strand break repair, also known as alternative end joining (25–27).
XRCC1-deficient cells have reduced steady-state levels of both Polβ and LigIIIα, indicating that XRCC1 interactions promote the stability of these DNA repair enzymes along with their recruitment to DNA damage sites (28,29). In contrast, the steady-state levels of the other XRCC1-interacting proteins appear unaffected by XRCC1 absence. Interestingly, the binding sites for XRCC1-interacting proteins appear to be arranged linearly along the XRCC1 polypeptide with Polβ interacting with the N-terminal region and LigIIIα with the C-terminal region (2). While most binding regions appear non-overlapping, enabling XRCC1 to assemble multiprotein complexes, PNKP, MRE11, aprataxin and APLF binding sites are mutually exclusive and dependent upon XRCC1 phosphorylation, explaining the detection of distinct XRCC1 complexes containing either PNKP or aprataxin (2,15,17,30–34). Interestingly, mutations in the genes encoding XRCC1, PNKP and aprataxin are the causative factor in inherited neurodegenerative diseases, indicating that XRCC1-dependent repair of single strand breaks with non-ligatable ends plays a key role in neuronal cells (35–37).
Three folded domains have been identified within XRCC1 (Figure 1A); an N-terminal domain that interacts with Polβ (38), a central BRCT domain (X1BR1) that interacts with poly(ADP-ribosyl)ated (PARylated) PARP1 (18,39) and a C-terminal BRCT domain (X1BR2) that interacts with the C-terminal BRCT domain of LigIIIα (L3BR) (40–42). While inactivation of the central X1BR1 domain results in DNA damage hypersensitivity due to a defect in S phase-dependent DNA repair, disruption of the interaction between the C-terminal BRCTs of XRCC1 and LigIIIα, which results in destabilization of LigIIIα, does not, indicating that XRCC1 has nuclear DNA repair functions that are independent of LigIIIα (43–45). In contrast, LigIIIα is essential for mitochondrial DNA metabolism but XRCC1 is not present in these organelles (21,23,46,47).
Figure 1.
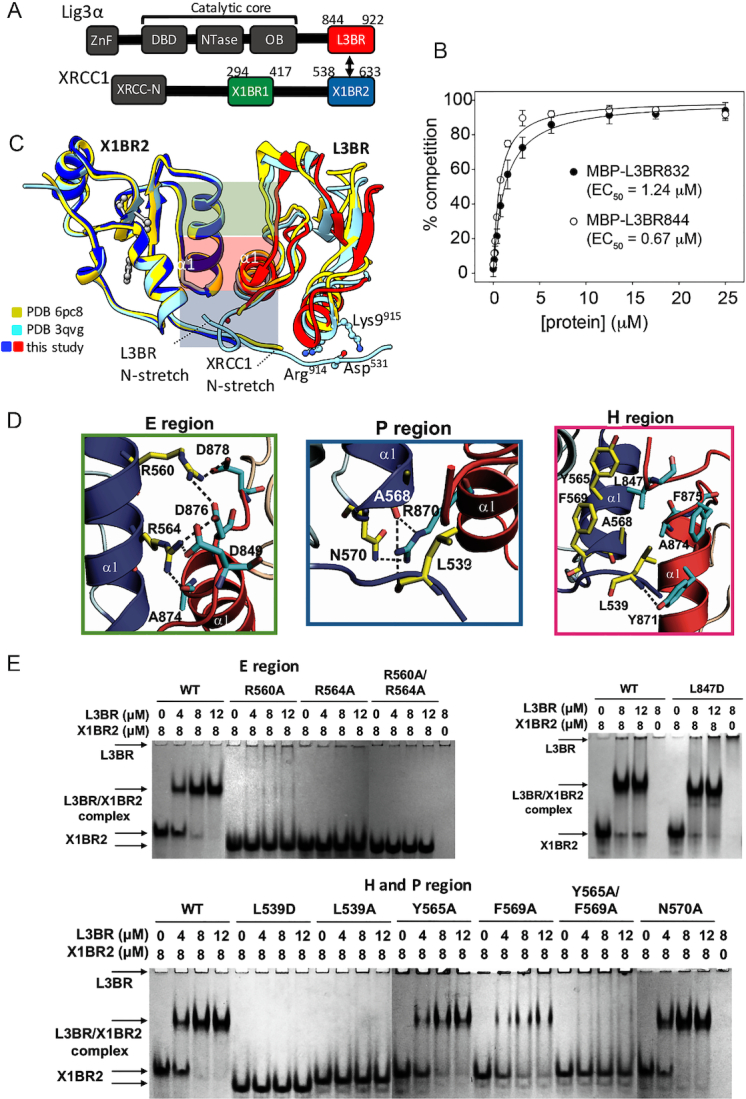
Human BRCT–BRCT heterodimer structure stably links XRCC1 and LigIIIα via a mutationally verified interface. (A) Domain organization of human XRCC1 and LigIIIα with the C-terminal BRCT domains used for crystallization (red and blue-boxes) (B) GFP-fluorescence-based competition assay (see Supplementary Figure S3) measuring specific binding affinity of L3BR832 and L3BR844 for X1BR2. The data shown represent the mean values and standard deviations from three independent experiments. (C) Structure of human XLBR-BR complex (X1BR2, blue; L3BR, red) is superimposed on to previously reported structures of heterodimers between mouse X1BR2 and human L3BR with two different lengths of N-stretch region. The different lengths of N-stretch regions of X1BR2 and L3BR constructs are highlighted. The XLBR-BR interfaces, which are classified based on the location and main type of interaction [electrostatic (E region, top, green), hydrophobic (H region, middle, purple) and polar (P region, bottom, blue) interactions], are indicated. (D) Panels show close-up of three main binding interfaces between X1BR2 and L3BR. (E) Effects of substituting residues that are located at the BRCT–BRCT interface on X1BR2–L3BR complex formation measured by native gel analysis; upper left panel, substitution of amino acids in the E region; upper right panel, substitution of Leu847 of L3BR, an equivalent of Leu539 of X1BR2; lower panel, substitution of amino acids in the H and P regions of the binding interface. The X1BR-only and L3BR-only control reactions are shown in the leftmost and rightmost lane of each gel, respectively. For residues in E region (upper left panel) or H and P regions (lower panel), gels were run with negative control (X1BR2 or L3BR alone) and positive control (with wild-type X1BR2, WT) reactions, and then combined in a single panel to compare their effects on L3BR binding. Representative gels from two independent experiments are shown. Since the theoretical pI for L3BR is 9.22 compared with 4.90 for X1BR2, L3BR does not enter the gels, which are run at pH 7.5, unless it is complexed with X1BR2.
For these various XRCC1 biological functions, it is important to understand the XRCC1 architectural structure and its relationship to its scaffolding functions. Since the C-terminal BRCT domains of XRCC1 and LigIIIα can form homodimers (48,49), we reasoned that these proteins may function as homodimers as well as heterodimers. Yet, although the structure of a hybrid complex between human L3BR and mouse X1BR2 has been determined (48), the structure of full-length complexes of dimeric XRCC1 and XRCC1-LigIIIα (XL) are unknown. Here, we solved the crystal structure of human L3BR complexed with human X1BR2 and furthermore define the architectures and structural flexibility of human XRCC1, LigIIIα and the XL heterodimer by combining small-angle X-ray scattering (SAXS) and electron microscopy (EM).
MATERIALS AND METHODS
Protein expression and purification
Full-length XRCC1 and LigIIIα [in pRSFduet (Novagen)] and XRCC1ΔN [residues 294–633 in pET32a (Novagen)] were expressed in Escherichia coli Rosetta cells, and purified using the same protocol as LigIIIβ described previously (50). L3BR (residues 825–922) and X1BR2 (residues 538–633) domains were expressed from pET28a with N-terminal His-tag in E. coli Rosetta cells. After Ni-NTA (Qiagen) affinity chromatography, proteins were purified by Superdex 200 (GE Healthcare) size exclusion column. For pull-down assay, L3BR, X1BR2 and X1BR1 (residues 294–417) were cloned into pGEX-6p1 (GE Healthcare). GST-tagged L3BR, X1BR2 and X1BR1 were expressed in E. coli Rosetta cells. After glutathione-sepharose chromatography (GE Healthcare), proteins were further purified by Superdex 200 column. X1BR1 was further purified by Sephadex 100 (GE Healthcare) after GST-X1BR1 was cleaved with precission protease. GFP- and MBP-tagged X1BR1 and L3BR were expressed from pET28a with N-terminal His-tag in E. coli Rosetta cells and purified using Ni-NTA affinity chromatography, followed by Superdex 200 size exclusion chromatography.
For crystallization, L3BR (residues 844–922 with C922S mutation) and X1BR2 (residues 538–633) were fused by a 22-amino acid flexible linker containing a His-tag and precission protease cleavage site, and the fusion protein was expressed in E. coli Rosetta cells. The L3BR-X1BR2 fusion protein was cleaved by purified precission protease after Ni-NTA column, and then further purified by Superdex 200 column. Selenomethionine labeled L3BR-X1BR2 fusion protein was expressed in Rosetta cells using amino acids to suppress methionine biosynthesis, as described (51), and was purified by the same protocol as the native protein. Protein crystallization conditions and structure determination methods are described in Supplementary Material.
Full-length XRCC1-LigIIIα and XRCC1 were purified from insect cells after infection with baculoviruses encoding streptactin-tagged LigIIIα and His-tagged XRCC1 (52,53). The XL complex was purified sequentially by HisTrap HP (GE Healthcare), Superdex 200 16/60 (GE Healthcare) and dsDNA cellulose (Sigma Aldrich) column chromatography. 6His-XRCC1 was purified sequentially by HisTrap HP and Superdex 200 16/60 column chromatography.
Protein–protein interaction assays
In a fluorescence-based competition assay, GST-L3BR (7 μM) and X1BR2-GFP (8 μM) were pre-incubated in a buffer containing 50 mM Tris–HCl pH 7.5, 150 mM NaCl, 10% glycerol and 2 mM DTT for 20 min on ice, and complexes were immobilized to GSH-coated magnetic beads (Promega). The beads were washed three times with the same buffer. The pre-bound complexes were competed off by adding MBP, MBP-L3BR832 and MBP-L3BR844 (0–25 μM). After the beads were pelleted using a magnet, the release of the X1BR2-GFP was monitored by the increased GFP fluorescence of the supernatant in 96-well plates using Synergy-2 microplate reader (BioTek).
To confirm the predicted key role of specific amino acids in the heterodimer interface, mutant versions of X1BR2 (residues 538–633) and L3BR (residues 825–922) were made using the Quickchange site-directed mutagenesis kit (Stratagene). Due to the high tendency of X1BR2 to aggregate in the absence of L3BR, we changed Leu626 in the C-terminus of X1BR2, which contributes to crystal packing interactions in both X1BR2 and the XLBR–BR complex structure (data not shown) to Glu. Since this substitution did not affect L3BR binding and it also significantly reduced aggregation, it was designated as wild type (WT) in Figure 1E and used as the template for further mutational analyses. Mutant proteins were expressed and purified by the same protocol as the native protein. Purified X1BR2 (8 μM) and L3BR proteins (0–12 μM) were incubated in 20 μl binding buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 2 mM DTT and 10% glycerol) for 30 min at 22°C. Samples were separated by electrophoresis on a native 10% polyacrylamide gel as described by Kang et al. (54).
Multi-angle light scattering coupled to size-exclusion chromatography
SEC-MALS was carried out either in parallel with SAXS (see below) or stand-alone. For stand-alone SEC-MALS, XRCC1 and the XL complex were injected at 0.35 ml/min with an AKTAmicro (GE Healthcare) onto a Superdex 200 Increase 10/300 GL column equilibrated in 40 mM HEPES pH 7.5, 200 mM NaCl, 10% glycerol and 0.1 mM TCEP. Molecular masses were determined using the Dawn HELEOS II MALS and OptiLab T-rEX online refractive index detectors (Wyatt Technology). The detectors were calibrated with the BSA monomer and data processed with ASTRA Version 6.1.6.5 (Wyatt Technology). The size-exclusion column was calibrated using protein standards (Bio-Rad).
Small angle X-ray scattering
SAXS was performed at the SIBLYS beamline at the Advanced Light Source (55,56). For SAXS coupled with multi-angle light scattering in line with size-exclusion chromatography (SEC-SAXS-MALS) experiments, 60 μl containing either 4.3 mg/ml 6His-XRCC1 in 200 mM NaCl, 20 mM Tris–HCl pH 7.5 , 2% glycerol (Figure 2); 5.96 mg/ml Strp-LigIIIα/6His-Flag-XRCC1 in 50 mM Tris–HCl pH 7.5, 100 mM NaCl, 5 mM MgCl2, 0.2 mM PMSF, 1 mM benzamidine (Figure 5). SEC-SAXS-MALS were collected at the ALS beamline 12.3.1 LBNL Berkeley, California. X-ray wavelength was set at λ = 1.127 Å and the sample-to-detector distance was 2100 mm resulting in scattering vectors, q, ranging from 0.01 to 0.4 Å–1. The scattering vector is defined as q = 4πsin θ/λ, where 2θ is the scattering angle. All experiments were performed at 20°C and data was processed as described (57). Briefly, a SAXS flow cell was directly coupled with an online Agilent 1260 Infinity HPLC system using a Shodex KW804 or Superdex 200 300/10 column giving similar SAXS results for Strep-LigIIIα/6His-Flag-XRCC1. The column was equilibrated with running buffer as indicated above with a flow rate of 0.5 ml/min. 55 μl of each sample was run through the SEC and three second X-ray exposures were collected continuously during a 30-min elution. The SAXS frames recorded prior to the protein elution peak were used to subtract all other frames. The subtracted frames were investigated by radius of gyration (Rg) derived by the Guinier approximation I(q) = I(0) exp(–q2Rg2/3) with the limits qRg < 1.5 (58). The elution peak was mapped by comparing the integral of ratios to background and Rg relative to the recorded frame using the program SCÅTTER. Non-uniform Rg values across an elution peak represent a heterogeneous assembly. Final merged SAXS profiles, derived by integrating multiple frames at the peak of the elution peak, where used for further analysis including Guinier plot which determined aggregation free state. The program SCÅTTER was used to compute the pair distribution function (P(r)). The distance r where P(r) approach zero intensity identifies the maximal dimension of the macromolecule (Dmax). P(r) functions were normalized based on the molecular weight of the assemblies as determined by SCÅTTER using volume of correlation Vc (59). Eluent was subsequently split 3 to 1 between the SAXS line and a series of UV at 280 and 260 nm, multi-angle light scattering (MALS), quasi-elastic light scattering (QELS) and refractometer detectors. MALS experiments were performed using an 18-angle DAWN HELEOS II light scattering detector connected in tandem to an Optilab refractive index concentration detector (Wyatt Technology). System normalization and calibration was performed with BSA monomer using a 45 μl sample at 10 mg/ml in the same SEC running buffer and a dn/dc value of 0.19. The light scattering experiments were used to perform analytical scale chromatographic separations for Mw determination of the principle peaks in the SEC analysis. UV, MALS, and differential refractive index data was analyzed using Wyatt Astra 7 software to monitor the homogeneity of the sample across the elution peak complementary to the above-mentioned SEC-SAXS signal validation.
Figure 5.
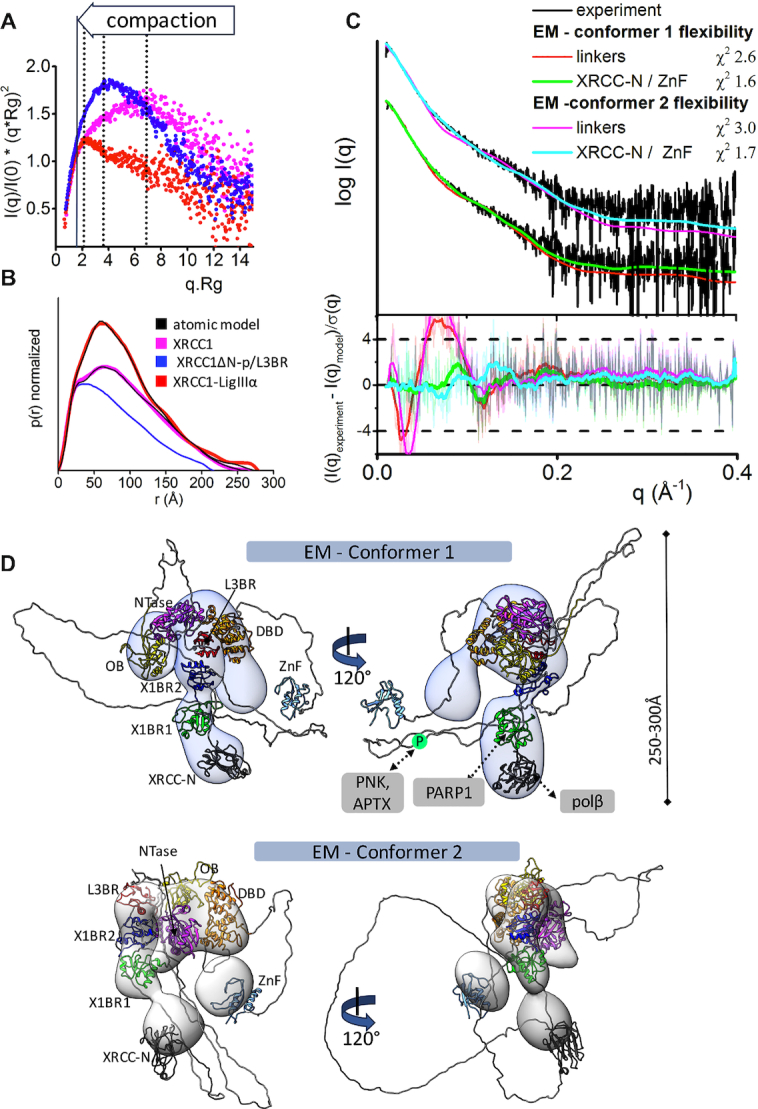
Increased XL compaction from comparison of EM and SAXS results. (A) Normalized Kratky plot of XRCC1 (magenta) in comparison with XRCC1ΔN-p/L3BR (blue) and XL complex (red) revealed compaction of larger XL complexes. The SAXS profiles are representative of two independent experiments. (B) Normalized P(r) functions obtained for experimental SAXS shown in panel A fitted to the theoretical P(r) functions (black) of atomistic models of XRCC1 (Figure 2E) and the multistate model of XL complex derived from the EM conformer 1 shown in panel D. P(r) function of XRCC1ΔN-p/L3BR complex reveal that the XRCC1ΔN has an elongated, flexible conformation, even when bound to L3BR domain. (C) Experimental (black) and theoretical (colored as indicated) SAXS profiles for the two XL EM conformers with optimized conformations of the linker regions (red and magenta) and optimized locations of the N-terminal domain of XRCC1 and the LigIII N-terminal ZnF domain (XRCC-N, cyan and ZnF, green). SAXS fits are shown together with the fit residuals in the lower panel and χ2 values indicating goodness of fit. (D) The top weighted model from the multistate SAXS-model of XL complex was modelled using EM conformer 1 (top) and conformer 2 (bottom) as initial model. Both conformers of multistate models are shown in Supplementary Figure S12. Models are superimposed on to the 3D EM maps. Corresponding SAXS fits for the atomistic models are shown in panel C and further shown as a theoretical P(r) functions in panel B. XRCC1 is constitutively phosphorylated by CK2 (30), and the phosphorylation site is indicated by P within a green circle. Interacting regions of XRCC1 with partner proteins are indicated.
Full-length LigIIIα, XRCC1ΔN, phosphorylated XRCC1ΔN (XRCC1ΔN-p), L3BR, X1BR2 and XRCC1ΔN-p/L3BR complex samples were dialyzed with a buffer containing 25 mM Tris–HCl pH 7.5, 150 mM NaCl, 10% glycerol and 2 mM DTT. SAXS data were collected at three different concentrations on the ALS beamline 12.3.1 LBNL Berkeley, California in high-throughput (HT) mode (60). Incident X-rays were tuned to a wavelength of λ = 1.0 Å at a sample-to-detector distance of 1.5 m, resulting in scattering vectors (q) ranging from 0.01 to 0.31 Å−1. All experiments were performed at 10°C and data was processed as described (55). Briefly, the data were acquired at short and long-time exposures (0.5 s, 5 s) then scaled and merged to obtained interference free SAXS profile. Further data processing was identical with process described for SEC-SAXS merged profiles.
Solution structure modeling
An initial atomistic model of XRCC1ΔN was built by connecting the NMR structure of X1BR1 (PDB ID: 2D8M) and our crystal structure of X1BR2 by MODELLER (61). A full length atomistic model of XRCC1 was built by connecting NMR structure of XRCC1 N-terminal domain (PDB ID: 1XNA) (62) to the XRCC1ΔN model. An initial homology model of full length LigIIIα was built based on the crystal structure of LigIIIβ (PDB ID: 3L2P) (63), the NMR structure of the N-terminal zinc finger (ZnF; PDB ID: 3OD8) (64) and our crystal structure of the C-terminal L3BR domain by connecting the regions with linkers modeled by MODELLER (61). Two initial atomistic models of dimeric XL complexes were built by adding missing linkers into two EM-derived conformers (see below). Minimal molecular dynamics (MD) simulations were performed on flexible regions in the models by the rigid body modeling strategy BILBOMD in order to optimize the conformational space of domain linkers alone (XL complex) or individual domains (XRCC1ΔN, XRCC1, LigIIIα and XL complex) (65). The experimental SAXS profiles were then compared to theoretical scattering curves generated from atomistic models using FOXS (66,67) followed by multistate model selection by MultiFOXS (68).
Crosslinking, negative-stain electron microscopy and image analysis
Chemical crosslinking of XRCC1 and the XL complex (dsDNA-cellulose-purified) was carried out in the dark, on ice, for 5 min with 0.05% (v/v) glutaraldehyde in 40 mM HEPES pH 7.5, 200 mM NaCl, 10% glycerol. Reactions were quenched with 1 M Tris pH 7.5 and buffer exchanged to EM buffer (40 mM HEPES pH 7.5, 50 mM NaCl, 0.1 mM TCEP). Crosslinked and non-crosslinked samples were diluted in EM buffer and deposited onto glow-discharged (Agar Scientific) carbon-coated copper grids (Electron Microscopy Sciences) for 1 min before blotting excess liquid with Whatman No. 1 filter paper. Freshly-prepared uranyl formate (1.5%; Electron Microscopy Sciences) was immediately applied for 1 min, followed by blotting excess liquid and air drying. Micrographs were collected using a FEI Tecnai T12 transmission electron microscopy fitted with a LaB6 filament and operated at 120 keV at room temperature. Low-dose mode was implemented in SerialEM (69) to acquire images at 67 000× magnification (corresponding to a pixel size of 1.64 Å) using a FEI Eagle 4k × 4k CCD camera with defocus values between 0.5 and 2 μm.
Image processing was carried out with the Scipion framework (70). None of the processing steps corrected for contrast transfer function nor applied symmetry. Automatic particle-picking using a reference-free algorithm (71) resulted in 78,400 coordinates of XRCC1 (non-crosslinked) and 12,700 coordinates of the crosslinked XL complex. Particles were extracted with two-fold downsampling in 264 Å boxes. Bad picks were discarded through iterative 2D classification using CL2D from Xmipp (72).The XL dataset was pruned to remove non-XL particles, resulting in 7648 particles that were used to generate an initial model through a stochastic gradient descent algorithm (73). The initial model was low-pass filtered to 60 Å and used for 3D classification (74) into three classes using a 200 Å circular mask. Classes 1 and 3 (31% and 28%, respectively) were grouped based on their overall similarity and the resulting merged class 1 (low-pass filtered to 60 Å) was refined using the grouped particles without applying a mask (EM conformer 1). Class 2 (41% of particles) was refined separately (EM conformer 2). The resolutions of the XL complex conformers were determined using the gold-standard Fourier shell correlation at the 0.5 criterion (75). Crystal and solution structures were docked into the EM maps using UCSF Chimera (76).
RESULTS
Human XRCC1 and DNA ligase IIIα C-terminal BRCT domains determine homodimer and heterodimer formation
XRCC1 and LigIIIα are elongated, multidomain proteins that form a stable complex through interactions between their C-terminal BRCT domains (48,49,63) (Figure 1A). While there is significant evidence that the XL complex is a key factor in BER and SSBR, the ability of these proteins to also form homodimers that may have specific functions (21,23,43,44,48,49), raises questions about the mechanisms that underlie formation of heterodimers and homodimers. In initial size exclusion experiments with the purified BRCT domains of human XRCC1 and LigIIIα, L3BR832 (residues 832–922) expressed either as an N-terminal MBP-fusion protein (Supplementary Figure S1A) or fused to C-terminal GFP (Supplementary Figure S1B) forms a dimer, whereas the dimerization of X1BR2 fused to C-terminal GFP appears to be transient under these conditions, consistent with the reported concentration-dependent dimerization of XRCC1 (49). Notably, deletion of 12 N-terminal residues from L3BR832 results in the MBP-fusion protein behaving as a monomer (Supplementary Figure S2A). Despite efficient homodimerization, untagged L3BR832 readily formed a 1:1 complex with MBP or GFP-tagged X1BR2 (Supplementary Figure S2). This finding suggests that the N-terminal linker region of L3BR (residues 832–843) is important for homodimerization but is dispensable for heterodimerization. Notably, in all size-exclusion chromatography experiments, the formation of heterodimers of X1BR2 and L3BR was considerably more favorable than homodimerization (Supplementary Figure S2).
To measure the apparent dimerization affinity of BRCT–BRCT interactions, a GFP-fluorescence-based, competition binding assay was developed (see Materials and Methods and Supplementary Figure S3). For this assay, GST-tagged L3BR832 was immobilized on glutathione-coated magnetic beads in complex with a GFP-labeled X1BR2 domain. The release of the X1BR2–GFP protein into the supernatant was monitored by the increase in GFP fluorescence as MBP-tagged L3BR832 protein was added to the binding reaction for competition. In this competition binding assay (Figure 1B), L3BR844 shows a slightly higher X1BR2-binding affinity than L3BR832 (EC50 values of 0.67 ± 0.08 and 1.24 ± 0.06 μM for L3BR844 and L3BR832, respectively), whereas MBP alone showed no interaction (Supplementary Figure S3). Together, we conclude that LigIIIα and XRCC1 specifically heterodimerize with sub- to low micromolar affinity through their C-terminal BRCT domains, and the N-terminal linker of L3BR contributes to the determination of homodimer and heterodimer formation, prompting us to further investigate the interface of human XL heterodimer and to identify key determinants of the specific XL complex formation.
BRCT heterodimer atomic resolution structure
We solved and refined the human X1BR2–L3BR844 complex (XLBR–BR) structure at 2.4 Å resolution (Supplementary Table S1, Figure 1C and Supplementary Figure S4). Overall, the structure of this complex resembles the previously reported hybrid complex between human L3BR and mouse X1BR2 (48) (Figure 1C). In the human XLBR–BR complex, there is a V-shaped ‘head-to-head’ arrangement of interacting L3BR–X1BR2 domains that buries 1164 Å2 of solvent accessible surface. Complementary surfaces of the BRCT domains are mostly contributed by helix α1 and the proximal ‘α1-β2 loop’, together with a short N-terminal extension (N-stretch) from each BRCT domain (Figure 1C). In comparison with the hybrid XLBR–BR structure, there is an increase in the tilt angle between L3BR and X1BR2 domains in the human structure. Notably, the structure of the hybrid XLBR–BR heterodimer with a longer X1BR2 N-stretch (Figure 1C, cyan, PDB ID: 3QVG) is more compact than the human XLBR–BR complex with a shorter N-stretch region (Figure 1C, red/blue), suggesting that an additional electrostatic interaction between the X1BR2 N-stretch residue Asp531 and L3BR residues Arg914 and Lys915 contributes to compaction of the complex (Figure 1C and Supplementary Figure S5). However, there are also differences in the main interaction interface between the similar-length hybrid XLBR–BR (PDB ID: 3PC8) (48) and the human complex, presumably reflecting differences in the amino acids of mouse and human X1BR that contribute to the interface.
In the human complex, three protruding side chains of X1BR (Leu539 from the N-stretch and Arg560 and Arg564 from helix α1) contact the hydrophobic C-terminal half of α1 in L3BR (Figure 1D). Together they make a network of electrostatic interactions with nearby residues Asp849, Asp876 and Asp878 of L3BR. Similar, but not strictly equivalent, reciprocal interactions are made by analogous residues of L3BR. Leu847 within the N-stretch of L3BR and Arg870 from α1 contact the hydrophobic C-terminal half of α1 and its proximal surfaces of X1BR2 (Figure 1D). Overall, the two BRCT domains are interlinked by protruding Leu and Arg residues from each subunit that stabilize the human XLBR-BR heterodimer by a combination of electrostatic (E region), hydrophobic (H region) and polar interactions (P region) (see Figure 1C and D). In the E region, Arg560 and Arg564 from α1 of X1BR2 form a network of salt bridges with Asp849, Asp876 and Asp878 residues from L3BR (Figure 1D). In addition, Arg564 from α1 of X1BR2 makes a hydrogen bond with the main chain carbonyl oxygen of Ala874 of L3BR. The center of the BRCT–BRCT interface (H region) is contributed by hydrophobic side chains from each subunit that form an interdomain hydrophobic core (Figure 1D). X1BR2 Leu539 appears critical for this hydrophobic core by virtue of extensive hydrophobic interactions with Leu847, Tyr871, Ala874 and Phe875 of L3BR. In addition, Leu539 main-chain amide hydrogen bonds with the Tyr871 side-chain of L3BR. Similarly, L3BR Leu847 makes hydrophobic interactions with XRCC1 residues Leu539, Tyr565 and Phe569. Finally, in the P region, Arg870 of L3BR makes hydrogen bonds with the side-chain amide of Asn570 and the main-chain carbonyl oxygens of Leu539 and Ala568 in X1BR2 (Figure 1D, P region).
X1BR2 N-stretch and α1 are critical for XL heterodimerization
To test the structurally-implicated critical roles of the E, H and P regions in XL heterodimerization, a series of amino acid substitutions were introduced into all three regions of the XLBR-BR interface (Figure 1E). Substitutions of Leu539 from the N-stretch of X1BR2 with Ala or Glu completely disrupted complex formation with L3BR as detected by native gel electrophoresis (Figure 1E). This reveals the functional importance of this region extending from the BRCT core and also supports and extends the previous finding that the N-terminal segment of X1BR2 significantly expands the buried surface area upon binding to L3BR (48). In contrast, L3BR Leu847, a structural equivalent of Leu539 in the N-stretch of XRCC1, appears to be dispensable for XL complex formation (Figure 1E), despite the similar appearance of both Leu residues and their local interactions with either side of the dimer interface. Although contradictory to a previous report (48), this is consistent with the dispensable role of the N-terminal linker of L3BR for XRCC1 binding and is indicative of functional asymmetry in this heterotypic BRCT–BRCT interaction. Given the dispensable role of L3BR Leu847, the severe defect in the X1BR2 Y565A/F569A double mutant (Figure 1E) is likely to be explained by some other interaction, perhaps a change in packing between XRCC1 helices α1 and α3 due to the replacement of bulky aromatic side chains. While X1BR2 Y565A, F569A and N570A mutants exhibited little or no effect on complex formation with L3BR (Figure 1E), the R560A, R564A and R560A/R564A mutants either failed to or had greatly diminished ability to form complexes with L3BR. Taken together, these mutational analyses defined specific hydrophobic interactions involving the N-stretch (Leu539) of XRCC1 and corresponding surfaces of LigIIIα, along with electrostatic interactions between α1s from XRCC1 (Arg560 and Arg564) and LigIIIα, as key determinants of XRCC1–LigIIIα heterodimer formation.
Atypical asymmetric BRCT–BRCT interaction promotes XL heterodimerization
The α1 helices of X1BR2 and L3BR domains share a remarkable sequence similarity (50% identity in α1 and surrounding residues, versus 29% overall sequence identity for the whole BRCT domains; Supplementary Figure S6), including similar patterns of charged and hydrophobic patches of surface residues around the dimer interface. Given the pseudo two-fold symmetry of interacting subunits in the XLBR-BR heterodimer (Supplementary Figure S6), this raises the question of why the XL heterodimer is preferred over the L3BR homodimer (Supplementary Figure S2). To address this, we compared structures of our XLBR–BR heterodimer to the X1BR2 homodimer (PDB ID: 1CDZ) (48) [2.4 Å resolution, r.m.s.d. of 0.8 Å for Cα atoms]. The XLBR–BR complex deviates significantly from strict 2-fold symmetry. Notably, the α1 of the L3BR subunit in the XL heterodimer is rotated ∼16° relative to the corresponding α1 of X1BR2 in the homodimer, as a result of different inter-subunit contacts in the two complexes with L3BR Arg870 acting as a pivot point for this conformational rotation (Supplementary Figure S6).
In the E region, α1 rotation causes a 3 Å displacement of L3BR Asp849 side chain with respect to Asp541 in the X1BR2 homodimer. Notably, this change results in a new salt bridge between X1BR2 Arg564 and L3BR Asp849 in the XL heterodimer (Figure 1D) that is essential for heterodimerization. In addition, L3BR Asp876 stands in for X1BR2 Asn570, completing the salt bridge network in the XL heterodimer (Figure 1D, E region). In the H region, the rotated BRCT conformation in the XL dimer appears to strengthen the X1BR2 Leu539-L3BR Tyr871 interaction, based on the closer distance (3.3 Å) in the XL heterodimer compared to the Leu539–Tyr565 interaction in the X1BR2 homodimer (3.9 Å) (Figure 1D). In contrast to the E and H regions, interactions in the P region of the XL heterodimer are diminished compared to the P region of the X1BR2 homodimer. For example, unlike Arg560 in the X1BR2 homodimer, L3BR Ser866 in the XL complex is unable to make interactions with Asn570 and Glu572 of the X1BR2 subunit (Figure 1D and E, and Supplementary Figure S6).
The asymmetric XL structure explains the functional asymmetry between the essential role of X1BR2 Leu539 and the dispensable role of L3BR Leu847 (Figure 1E). This asymmetry may provide flexibility in the L3BR N-stretch to allow the rotated conformation of L3BR upon X1BR2 binding. Collectively, the asymmetry induced by conformational rotation of L3BR contributes to favorable XL heterodimerization by strengthening the two crucial interactions mediated by Leu539 and Arg564 of X1BR2 in the XL heterodimer (Figure 1D and E).
Full-length XRCC1 and LigIIIα form transient homodimers with a largely unstructured multi-domain architecture
Apart from ordered X1BR1, X1BR2 and N-terminal domains (Figure 1A), XRCC1 appears unstructured. To experimentally examine the intrinsic flexibility of the full-length human XRCC1, we used SAXS (77,78) and SEC-SAXS-MALS. SEC-SAXS analysis of individual SAXS profiles across the elution peak revealed a large decrease of Rg (65–45 Å) and molecular mass (110–70 kDa) (Figure 2A). This indicates heterogeneity within the sample that presumably reflects transient dimerization of XRCC1 (49). The Kratky plot (Figure 2B) from the merged SAXS curve (Supplementary Figure S7A), together with the P(r) functions (Figure 2C), demonstrate that XRCC1 is a partially unfolded, multidomain scaffold-like protein with an estimated maximal elongation of ∼230 Å that matches the value estimated from analytic sedimentation equilibrium studies (49).
Figure 2.

Human XRCC1 is an elongated, disordered protein that transiently forms homodimers. (A) SEC-SAXS-MALS chromatographs for XRCC1. Solid lines represent the UV 280 nm (light magenta) or SAXS signal (magenta) in arbitrary units, while symbols represent molecular mass (light magenta) and Rg values for each collected SAXS frame (magenta) versus elution time. The SEC-SAXS-MALS results, which show full-length XRCC1 is a mixture of monomer/dimer, are representative of at least two independent preparations of XRCC1. (B) Normalized Kratky plot of XRCC1 (magenta) in comparison with XRCC1ΔN-p (dark green) and XRCC1ΔN (light green). (C) Normalized P(r) functions of XRCC1 (magenta) in comparison with XRCC1ΔN-p (dark green) matching theoretical P(r) functions of atomistic models (black) shown in panels D and E. Weighted ensemble atomistic model shown in molecular surface representation were used to fit experimental SAXS curves for XRCC1ΔN-p (D) and XRCC1 (E) shown in Supplementary Figure S7A and represented as theoretical P(r) functions in panel C.
As phosphorylation of the region between X1BR1–X1BR2 by casein kinase II (CK2) stabilizes the protein and is required for localization to DNA damage foci and efficient SSBR activity (30,34,79) we purified XRCC1ΔN after co-expression with CK2α in E. coli (80). In comparison with unmodified XRCC1ΔN, we found that phosphorylation of XRCC1ΔN does not alter its binding activity towards L3BR (Supplementary Figure S8) or its solution conformation, based on the nearly identical SAXS profiles of phosphorylated and unphosphorylated XRCC1ΔN (Figure 2B). The P(r) curve for XRCC1ΔN-p exhibits a gradual fall off at high R-values that is indicative of a large Dmax (∼200 Å). The radius of gyration (Rg = 53 Å) is also unexpectedly large for the 340-residue XRCC1ΔN-p protein (Figure 2C) and, similar to full length XRCC1, XRCC1ΔN-p is a mixture of monomers and dimers in solution as judged by estimated molecular mass (59) (Supplementary Table S2). Collectively, these experimental data are consistent with a transient dimerization of XRCC1 via the X1BR2 domains.
To explore the conformational space that can be occupied by XRCC1, we performed rigid body modeling that was validated by SAXS fitting. Initially we determined the atomistic model of XRCC1ΔN by connecting the NMR structure of X1BR1 (PDB ID: 2D8M) and the crystal structure of X1BR2 (see Materials and Methods). A selected minimal ensemble model (Figure 2D), that includes 40% extended monomer, 35% extended dimer and 25% compact dimer, was the best fit for the experimental SAXS profile and P(r) function (Figure 2C and Supplementary Figure S7A). Next, we modeled full length XRCC1 by connecting the NMR structure of XRCC1 N-terminal domain (PDB ID: 1XNA) (62) to the XRCC1ΔN model. An ensemble model (Figure 2E) that includes 61% extended monomer, 14% extended dimer and 25% compact dimer closely matched the experimental SAXS profile and P(r) function (Figure 2B and C, and Supplementary Figure S7A). While these ensembles do not provide a unique model, they validate the intrinsically disordered nature and transient dimerization of XRCC1.
To gain insights into the structure of full-length human LigIIIα, we initially determined a solution state of the L3BR832. Based on the estimated molecular mass and SAXS fitting of rigid body modeling, the solution structure model for L3BR832 closely matches the two-fold symmetric homodimer of L3BR (PDB ID: 3PC7) (Figure 3A and Supplementary Figure S7C, Table S2). The results of the Kratky plot (Figure 3B) and the P(r) function (Figure 3A) analyses of full-length LigIIIα indicate that, like LigIIIβ (63), LigIIIα has a disordered multidomain architecture. Unlike the L3BR domain that is exclusively dimeric in solution (Supplementary Figure S7C, Table S2), full-length LigIIIα is a mixture of monomer and dimers in solution, with maximal elongation of ∼200 Å (Figure 3C and Supplementary Figure S7B, Table S2). A comparison of normalized Kratky plots of LigIIIβ with LigIIIα indicates a similar level of protein disorder (Figure 3B) despite Lig IIIβ behaving as a monomer due to the absence of the L3BR domain (63). As expected (63), a comparison of LigIIIβ (LigIIIβ 1–755) missing the disordered C-terminal 107 residues and LigIIIβ missing the N-terminal ZnF domain (LigIIIβ 1–755 ΔZnF) (Figure 3B) confirmed the significant contribution of the ZnF region to the disordered character of LigIIIβ (63). Together, our rigid body modeling analysis indicates that the N- terminal ZnF extension and the C-terminal BRCT domain of LigIIIα, contribute significantly to the elongated shape of the DNA ligase III polypeptides.
Figure 3.
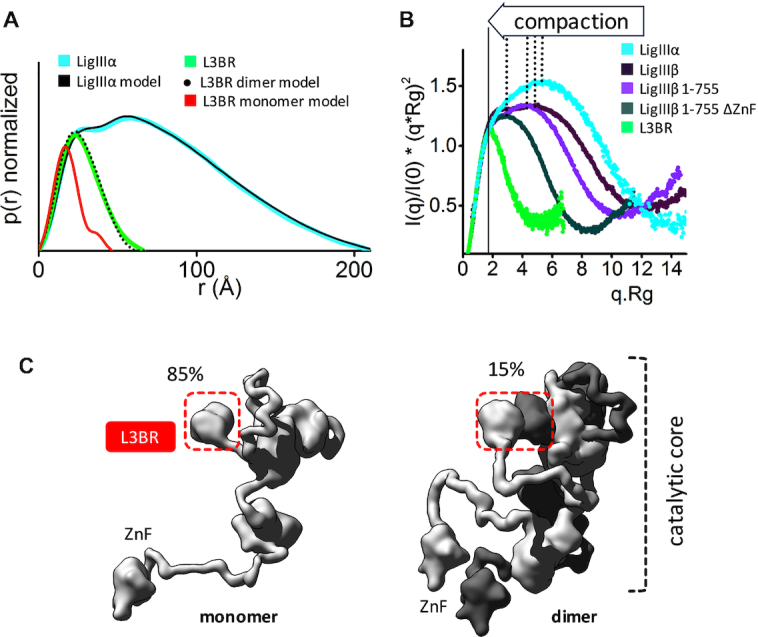
Human LigIIIα is an elongated, disordered protein that transiently forms homodimers. (A) Normalized P(r) functions for experimental SAXS curves for full length LigIIIα (cyan) and L3BR (green) are fitted to the theoretical P(r) functions (black) of atomistic models of full length LigIIIα (panel C) and the two-fold symmetric crystal structure of the L3BR homodimer (PDBID: 3PC7, black dots) and L3BR monomer (red). (B) Comparison of normalized Kratky plots of LigIIIα (cyan), L3BR dimer (green) with the SAXS curves of LigIIIβ (dark violet), LigIIIβ 1–755 (violet), and LigIIIβ 1–755 ΔZnF (dark green) (taken from (63)). Kratky plots show persistent disorder of full length LigIIIα and LigIIIβ but significant less disorder in the LigIIIβ construct with truncated ZnF domain. (C) Weighted ensemble atomistic models of LigIIIα monomer (left) and homodimer (right) are shown in surface representation. Corresponding fits of the SAXS curves for the atomistic models are shown in the Supplementary Figure S7D and as a theoretical P(r) function in panel A.
Visualization of XL complex by negative stain electron microscopy reveals a loosely packed domain arrangement
To gain insights into the possible domain organization of the XL complex, we co-purified the full-length proteins and confirmed that they form a ∼180 kDa heterodimer by SEC-MALS despite eluting earlier than expected from the gel filtration column (Figure 4A and B). XRCC1 and the XL complex were each stabilized with chemical crosslinking (Figure 4C), buffer exchanged and visualized using negative-stain EM. Micrograph quality was greatly improved by the crosslinking and subsequent buffer exchange steps (Supplementary Figure S9). Yet, 2D classification of the single particles from the XRCC1 sample revealed a conformationally heterogenous population of XRCC1 (Figure 4D) that is, based on our SAXS data (Figure 2A), likely to reflect a mixture of monomers and dimers. Crosslinking did not reduce this heterogeneity (data not shown). Efforts to generate an initial 3D model of XRCC1 failed, probably due to its intrinsic structural and oligomeric heterogeneity. Despite efficient crosslinking, classes of particles representing XRCC1-only particles were detected in the population of particles from the XL sample (Figure 4E). After removal of the XRCC1-only particles, the uniformly-sized XL particles (Figure 4F) were subjected to 3D classification to identify conformationally different forms of the XL complex (Supplementary Figure S10). Particles belonging to two visually similar classes were pooled and used to refine EM conformer 1. Particles from a third class were used to create EM conformer 2 (both conformers shown in Figure 4G).
Figure 4.

Visualization of XL particles by electron microscopy reveals a dynamic domain arrangement. (A) SEC-MALS analysis shows that XRCC1 either alone or in complex with full-length LigIIIα elutes earlier than globular protein standards despite molar masses of ∼77 and ∼180 kDa, respectively. (B) The XL peak seen in (A) is composed of a heterodimer. (C) The XL dimer was chemically crosslinked prior to negative-stain EM imaging. (D) 2D classification reveals oligomeric and conformational heterogeneity in non-crosslinked XRCC1. (E) Particles belonging to non-XL 2D classes were removed from the cross-linked XL dataset. (F) The final dataset contained uniformly-sized 2D classes of particles. 3D classification and subsequent refinement resulted in two conformers of the XL complex. (G) The 3D EM maps are rotated to each display a linear protrusion from a larger area of density. The linear extension potentially represents XRCC1 domains X1BR2 (PDB ID: 3PC8 chain A), X1BR1 (PDB ID: 2D8M) and the N-terminus (PDB ID: 3K77). The LigIIIα ZnF (PDB ID: 1UW0), three domain catalytic fragment (DBD, NTase and OBD, PDB ID: 3L2P) and L3BR (PDB ID: 3PC8 chain C) domains were docked into the larger area of density of each conformer.
Some structural heterogeneity for the XL complex was expected given the flexible and dynamic nature of both proteins. The conformers presented here do not represent definitive models of domain organization, but rather present a framework for modelling potential domain arrangements. Although other conceivable domain configurations might also be consistent with the EM results, our domain docking EM map interpretation was guided by known crystal structures of LigIIIα domains and our insights into the conformation of XRCC1 alone from 2D classes (Figure 4D). The XRCC1 monomer often adopts a linear arrangement that can be observed in both EM conformers (Figure 4G). The published atomic coordinates of individual XRCC1 domains were docked within this linear protrusion so as to have the N-terminal domain (XRCC-N) distal to the rest of the complex and the L3BR–X1BR2 heterodimer at the interface with the remaining volume. At the other side of the complex, we interpreted the isolated spherical density as the LigIIIα ZnF. Linking the two sides of the complex would be the LigIIIα catalytic fragment composed of the DNA binding, adenylation and OB-fold domains (63). These conserved domains are known to adopt an open conformation, as seen for the Saccharolobus solfataricus DNA Ligase (81) as well as a closed conformation with nicked DNA (63). The EM map for conformer 1 accommodates the catalytic domains in an approximately open conformation whereas a more compact LigIIIα is a better fit for the wider volume seen in conformer 2. Overall, our interpretation of two EM maps suggest a dynamic LigIIIα catalytic fragment that in turn orients XRCC1 at different positions within the complex. The full dynamics of XRCC1 are probably underestimated due to limitations in visualizing unstructured linkers by EM. However, the observed conformers allowed us to build a framework of domains upon which we added quantitative assessment of flexibility by SAXS as described below.
The XL complex is more structured and compact than XRCC1 and LigIIIα
Prior to investigating the solution structure of full-length XL, we determined that the interaction of L3BR832 with XRCC1ΔN-p did not change the extended shape of XRCC1ΔN as judged by the Kratky plot, P(r) function and SAXS parameters of the complex (Dmax of ∼200 Å and Rg of 54 Å (Figure 5A and B, Supplementary Table S2). We then examined the conformational flexibility of the full-length XL complex by performing SEC-SAXS-MALS experiments (Supplementary Figure S7D). Consistent with Figure 4A, the major peak eluting from the gel filtration column corresponded to the XL heterodimer, as judged by molecular estimates from MALS and SAXS (Supplementary Figure S7D, Table S2). The decrease of Rg values determined for individual SAXS frames across the elution peak (Figure 5C) suggests XL conformational heterogeneity through transient interactions between multiple XL domains. However, the Kratky plots revealed significant compaction of the XL heterodimer relative to the XRCC1 dimer (Figure 5A), consistent with the reduced heterogeneity the XL particles compared with XRCC1 particles seen by EM. To determine whether the compaction extends beyond the interacting C-terminal BRCT domains in the XL complex, we examined the protease-sensitivity of labeled, adenylated LigIII. Notably, the labeled catalytic fragment of LigIII was more resistant to proteolysis when complexed with XRCC1 (Supplementary Figure S11), suggesting that the BRCT–BRCT heterodimer interacts with and influences the conformation of the LigIII catalytic region.
To test how the EM conformers 1 and 2 (Figure 4G) match the solution state, we compared models based on the available atomic structures that do not include the linker regions between domains to the SAXS curve. Without linker regions that are missing from the atomic resolution structures, fits of the EM models were poor (χ2conformer1 = 11.6 and χ2conformer2 = 15.1, see Supplementary Figure S7E). To determine whether this discrepancy was due to the flexibility of the missing regions, these sections were built using MODELLER (61) and conformational sampling was applied using BILBOMD (65) to mimic the flexibility of the added regions (Supplementary Figure S12). While the SAXS fit was improved by including linker flexibility (χ2conformer1 = 2.6 and χ2conformer2 = 3.0), the remaining discrepancy between theoretical and experimental SAXS curves in the low resolution range of SAXS (q 0.05–0.15 Å−1, see Figure 5C) suggested larger conformational rearrangements of the XL domains in solution. Closer inspection of the EM-derived models showed that the N-terminal domain of XRCC1 and the LigIII N-terminal ZnF domain are spatially separated from the XL core (X1BR1, X1BR2, L3BR and LigIII catalytic domains) (Figure 4G). Thus, we performed rigid body modeling that included flexibility of the N-terminal domains of XRCC1 and LigIII using EM conformers 1 and 2, respectively as initial models. From approximately 10,000 conformers derived by conformational sampling (Supplementary Figure S7F), a multistate model containing two conformers was selected by MultiFOXS (68) (Figure 5D and Supplementary Figure S12). Displacement of the N-terminal domain of XRCC1 and the LigIII N-terminal ZnF domain from their initial locations (Figure 5D and Supplementary Figure S12) significantly improved the SAXS fit (χ2conformer1 = 1.6 and χ2conformer2 = 1.7, Figure 5C) in the low-resolution range, in accord with the flexibility of these regions inferred by our EM studies and shown by others (49,50) (Figure 5D and Supplementary Figure S12). Notably, the most dominant conformers in both conformational samplings (Figure 5D) adopted a similar arrangement to the EM-derived conformers. Taken together, the SAXS-derived model of full-length XL supports the domain arrangement predicted by EM in which the BRCT–BRCT heterodimer is the stable core of the XL complex that increases the compaction of the LigIII catalytic region and links the flexible N-terminal regions of XRCC1 that are accessible for additional protein-protein interactions with the flexible N-terminal nick sensing region of LigIIIα.
DISCUSSION
Based on previous studies, both XRCC1 and LigIIIα are elongated proteins composed of some well-folded domains connected by flexible, unstructured regions (2,49,63). While there is atomic resolution structural information for the well-folded domains (39,48,62,63,82,83), less is known about the functional architecture of the full length proteins, particularly the XL complex that plays a key role in coordinating base excision and single strand break repair (84,85). Accurate structural analyses of large and flexible complexes can require combined approaches to assess both flexibility (by SAXS), shape (by SAXS and EM) and detail (by X-ray crystallography and NMR) (86,87). Here we integrated results of crystallographic, EM and SAXS experiments for LigIIIα and XRCC1 with existing structural information (39,48,62,63,82,83) to build a complete data-based model of the highly flexible XL heterodimer that is anchored by a unique BRCT–BRCT interaction (Figures 4 and 5).
XRCC1 and LigIIIα BRCT domains are critical for the stability and function of nuclear LigIIIα (29,42,44). Yet, XRCC1 has LigIIIα-independent functions in nuclear DNA repair (42,44,45). Furthermore, the essential function of LigIIIα in mitochondrial DNA metabolism occurs in the absence of XRCC1 and is not dependent upon its C-terminal BRCT domain (21,23,47). In accord with published studies concluding that the C-terminal BRCT domains also homodimerize (48,49), we observed formation of XRCC1 and LigIIIα homodimers albeit to a lesser extent than XL heterodimers. Overall, our atomic resolution structure of the human BRCT–BRCT heterodimer closely resembles that formed by mouse X1BR2 with human L3BR and supports and extends the notion that interactions with the region N-terminal to X1BR2 contribute to the preferred formation of hetero- versus homodimers (48). However, differences between the human and mouse-human hybrid structures including an increase in the tilt angle between the X1BR2 with L3BR domains in the human structure evidently reflect differences in the amino acids contributing to the interaction interface. Residue substitutions confirmed the key role of specific amino acids in the structurally-defined interface. They furthermore highlighted the structural and functional asymmetry, in particular rotation of L3BR α1 helix in the XL heterodimer relative to the corresponding α1 helix of X1BR2 in the homodimer (48), that seems to favor heterodimer formation.
While the XL complex is flexible, it is significantly more compact than either XRCC1 or LigIIIα (Figure 5A). The linker region between the X1BR1 and X1BR2 domains is extensively phosphorylated by CK2, enhancing interactions with PNKP and APTX via their FHA domains (30,33,80,88). However, CK2 phosphorylation had no detectable effect on XRCC1 conformation. Since the catalytic region of LigIII undergoes a large conformational change when it engages nicked DNA (50), we examined the effect of complex formation with XRCC1 on the LigIII catalytic region. Notably, it was much more resistant to proteolysis within the XL heterodimer. Thus, the BRCT–BRCT heterodimer not only anchors these proteins but also presumably initiates secondary interactions that reduce the flexibility of the LigIII catalytic region, an effect that may contribute to the ability of XRCC1 to stimulate ligation by and maintain the steady state levels of LigIIIα (29,42).
During SSBR, the stable heterodimer core of XL defined here effectively tethers the flexible XRCC1 scaffold that mediates recruitment of LigIIIα, as well as other XRCC1-interacting proteins, including Polβ, PNKP and APTX (12,13,15,16) to sites of DNA damage via an interaction between X1BR1 and PARylated PARP1 (18,20,39) (Figure 5D). The N-terminal ZnF of LigIIIα is part of a flexible SSB sensing module that may facilitate location of SSBs in the context of local high concentrations of poly(ADP-ribose) (50,89,90). If the SSB is non-ligatable, we envision that the flexibility of XRCC1 and LigIIIα will enable dynamic sensing and processing of the SSB termini by the LigIIIα ZnF and XRCC1-associated proteins within the XL complex, culminating in the circularization of the LigIIIα catalytic fragment around a ligatable nick to complete repair (63). Future structural and biochemical studies are needed to address how the binding of other XRCC1 partner proteins impact the flexibility of the XRCC1 scaffold and the repair of base damage as well as both single- and double-strand breaks. For example, in BRCA2-defective cells, there are increased interactions between XRCC1 and both MRE11 and DNA polymerase theta that may contribute to the observed genome rearrangements in BRCA-deficient cancers (91), so these structural results may inform ongoing investigations of existing and novel chemotherapeutic strategies.
Here, we have used crystal structures combined with negative stain EM and SAXS to obtain complementary insights into the structure and conformational flexibility of an intact highly dynamic protein complex that could not be obtained by either X-ray crystallography or NMR alone. The combined results show that accurate determination of XL structures must include its dynamic and functional flexibility as was recently found for the DNA PK complex (92). Our integrative modeling from EM and SAXS plus atomic resolution domain structures revealed the functional compaction of the XL complex that extended beyond the interacting C-terminal BRCT–BRCT domains to the catalytic region of LigIIIα. The consensus models of the XL complex suggest that the interconversions of the flexible N-terminal extensions of LigIIIα and XRCC1 facilitate functional interactions with diverse protein partners during BER and strand break repair.
DATA AVAILABILITY
Coordinates and structure factors have been deposited in the PDB with accession codes 6WH1 for the XLBR-BR complex and 6WH2 for X1BR2. The EM maps have been deposited in the EMDB with accession codes 22130 and 22307 for EM conformer 1 and EM conformer 2, respectively. SAXS data and atomistic models of XRCC1ΔN-p, XRCC1, LigIIIα and XL complex have been deposited in the SASBDB with accession codes SASDJ52, SASDJ62, SASDJ72 and SASDJ82.
Supplementary Material
ACKNOWLEDGEMENTS
Mark Glover kindly provided us with pGEX-CK2α plasmid. Drs Leslie Hicks and Sophie Alvarez provided expert mass spectrometry analysis of proteins at the Proteomics & Mass Spectrometry Facility of the Donald Danforth Plant Science Center (Saint Louis, MO).
Author contributions: M.H., T.E., J.M.P., I-K.K., J.A.T. and A.E.T. designed the overall research. M-S.T., I-K.K, A.S. and I.R. were responsible for protein expression and purification; I-K.K, I.R. and A.S. performed the biochemistry and mutagenesis studies, I-K.K., Y.P. and T.E., the X-ray crystallography and analysis, M.H. the small angle X-ray scattering and analysis and A.S. and J.M.P. the electron microscopy and analysis. The manuscript was written by I-K.K., M.H., T.E., J.M.P., J.A.T. and A.E.T. with contributions from A.S. and I.R. All authors have given approval to the final version of the manuscript.
Contributor Information
Michal Hammel, Molecular Biophysics & Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
Ishtiaque Rashid, Departments of Internal Medicine, Molecular Genetics & Microbiology and the University of New Mexico Comprehensive Cancer Center, University of New Mexico, Albuquerque, NM 87131, USA.
Aleksandr Sverzhinsky, Department of Biochemistry and Molecular Medicine, Université de Montréal, Montréal, Québec, Canada.
Yasin Pourfarjam, Department of Chemistry, University of Cincinnati, 301 Clifton Ct, Cincinnati, OH 45221, USA.
Miaw-Sheue Tsai, Molecular Biophysics & Integrated Bioimaging, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA.
Tom Ellenberger, Department of Biochemistry, Washington University, St. Louis, MO, USA.
John M Pascal, Department of Biochemistry and Molecular Medicine, Université de Montréal, Montréal, Québec, Canada.
In-Kwon Kim, Department of Chemistry, University of Cincinnati, 301 Clifton Ct, Cincinnati, OH 45221, USA.
John A Tainer, Departments of Cancer Biology and Molecular & Cellular Oncology, University of Texas, MD Anderson Cancer Center, Houston, TX 77030, USA.
Alan E Tomkinson, Departments of Internal Medicine, Molecular Genetics & Microbiology and the University of New Mexico Comprehensive Cancer Center, University of New Mexico, Albuquerque, NM 87131, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01 ES012512 to A.E.T., R35 CA220430 to J.A.T., in part]; The Structural Cell Biology of DNA Repair Program [P01 CA092584 to A.E.T., T.E., J.A.T.]; V Foundation V Scholar Grant [V2018-25 to I.K.K.]; J.A.T. acknowledges the Cancer Prevention Research Institute of Texas (CPRIT) [RP180813]; Robert A. Welch Chemistry Chair; National Science and Engineering Research Council of Canada [RGPIN-2015-05776 to J.M.P.]; SIBYLS beamline at the Advanced Light Source was provided in part by the Offices of Science and Biological and Environmental Research, U.S. Department of Energy through DOE BER Integrated Diffraction Analysis Technologies (IDAT) program; NIGMS [P30 GM124169-01 ALS-ENABLE]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Thompson L.H., Brookman K.W., Jones N.J., Allen S.A., Carrano A.V.. Molecular cloning of the human XRCC1 gene, which corrects defective DNA strand break repair and sister chromatid exchange. Mol. Cell. Biol. 1990; 10:6160–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Caldecott K.W. XRCC1 protein; form and function. DNA Repair (Amst.). 2019; 81:102664. [DOI] [PubMed] [Google Scholar]
- 3. Caldecott K.W., McKeown C.K., Tucker J.D., Ljungquist S., Thompson L.H.. An interaction between the mammalian DNA repair protein XRCC1 and DNA ligase III. Mol. Cell. Biol. 1994; 14:68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kukshal V., Kim I.K., Hura G.L., Tomkinson A.E., Tainer J.A., Ellenberger T.. Human DNA ligase III bridges two DNA ends to promote specific intermolecular DNA end joining. Nucleic. Acids. Res. 2015; 43:7021–7031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marsin S., Vidal A.E., Sossou M., Menissier-de Murcia J., Le Page F., Boiteux S., de Murcia G., Radicella J.P.. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J. Biol. Chem. 2003; 278:44068–44074. [DOI] [PubMed] [Google Scholar]
- 6. Das A., Wiederhold L., Leppard J.B., Kedar P., Prasad R., Wang H., Boldogh I., Karimi-Busheri F., Weinfeld M., Tomkinson A.E. et al.. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair (Amst.). 2006; 5:1439–1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wiederhold L., Leppard J.B., Kedar P., Karimi-Busheri F., Rasouli-Nia A., Weinfeld M., Tomkinson A.E., Izumi T., Prasad R., Wilson S.H. et al.. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell. 2004; 15:209–220. [DOI] [PubMed] [Google Scholar]
- 8. Akbari M., Solvang-Garten K., Hanssen-Bauer A., Lieske N.V., Pettersen H.S., Pettersen G.K., Wilson D.M. 3rd, Krokan H.E., Otterlei M.. Direct interaction between XRCC1 and UNG2 facilitates rapid repair of uracil in DNA by XRCC1 complexes. DNA Repair (Amst.). 2010; 9:785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Campalans A., Marsin S., Nakabeppu Y., O’Connor TR., Boiteux S., Radicella J.P.. XRCC1 interactions with multiple DNA glycosylases: a model for its recruitment to base excision repair. DNA Repair (Amst.). 2005; 4:826–835. [DOI] [PubMed] [Google Scholar]
- 10. Steinacher R., Barekati Z., Botev P., Kusnierczyk A., Slupphaug G., Schar P.. SUMOylation coordinates BERosome assembly in active DNA demethylation during cell differentiation. EMBO J. 2019; 38:e99242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vidal A.E., Boiteux S., Hickson I.D., Radicella J.P.. XRCC1 coordinates the initial and late stages of DNA abasic site repair through protein-protein interactions. EMBO J. 2001; 20:6530–6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kubota Y., Nash R.A., Klungland A., Schar P., Barnes D.E., Lindahl T.. Reconstitution of DNA base excision-repair with purified human proteins: interaction between DNA polymerase beta and the XRCC1 protein. EMBO J. 1996; 15:6662–6670. [PMC free article] [PubMed] [Google Scholar]
- 13. Caldecott K.W., Aoufouchi S., Johnson P., Shall S.. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular 'nick-sensor' in vitro. Nucleic Acids Res. 1996; 24:4387–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mol C.D., Izumi T., Mitra S., Tainer J.A.. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination [corrected]. Nature. 2000; 403:451–456. [DOI] [PubMed] [Google Scholar]
- 15. Whitehouse C.J., Taylor R.M., Thistlethwaite A., Zhang H., Karimi-Busheri F., Lasko D.D., Weinfeld M., Caldecott K.W.. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell. 2001; 104:107–117. [DOI] [PubMed] [Google Scholar]
- 16. Date H., Igarashi S., Sano Y., Takahashi T., Takano H., Tsuji S., Nishizawa M., Onodera O.. The FHA domain of aprataxin interacts with the C-terminal region of XRCC1. Biochem. Biophys. Res. Commun. 2004; 325:1279–1285. [DOI] [PubMed] [Google Scholar]
- 17. Iles N., Rulten S., El-Khamisy S.F., Caldecott K.W.. APLF (C2orf13) is a novel human protein involved in the cellular response to chromosomal DNA strand breaks. Mol. Cell. Biol. 2007; 27:3793–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Masson M., Niedergang C., Schreiber V., Muller S., Menissier-de Murcia J., de Murcia G.. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol. Cell. Biol. 1998; 18:3563–3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lan L., Nakajima S., Oohata Y., Takao M., Okano S., Masutani M., Wilson S.H., Yasui A.. In situ analysis of repair processes for oxidative DNA damage in mammalian cells. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:13738–13743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Okano S., Lan L., Caldecott K.W., Mori T., Yasui A.. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol. Cell. Biol. 2003; 23:3974–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gao Y., Katyal S., Lee Y., Zhao J., Rehg J.E., Russell H.R., McKinnon P.J.. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature. 2011; 471:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Katyal S., McKinnon P.J.. Disconnecting XRCC1 and DNA ligase III. Cell Cycle. 2011; 10:2269–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Simsek D., Furda A., Gao Y., Artus J., Brunet E., Hadjantonakis A.K., Van Houten B., Shuman S., McKinnon P.J., Jasin M.. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature. 2011; 471:245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Frosina G., Fortini P., Rossi O., Carrozzino F., Raspaglio G., Cox L.S., Lane D.P., Abbondandolo A., Dogliotti E.. Two pathways for base excision repair in mammalian cells. J. Biol. Chem. 1996; 271:9573–9578. [DOI] [PubMed] [Google Scholar]
- 25. Moser J., Kool H., Giakzidis I., Caldecott K., Mullenders L.H., Fousteri M.I.. Sealing of chromosomal DNA nicks during nucleotide excision repair requires XRCC1 and DNA ligase III alpha in a cell-cycle-specific manner. Mol. Cell. 2007; 27:311–323. [DOI] [PubMed] [Google Scholar]
- 26. Simsek D., Brunet E., Wong S.Y., Katyal S., Gao Y., McKinnon P.J., Lou J., Zhang L., Li J., Rebar E.J. et al.. DNA ligase III promotes alternative nonhomologous end-joining during chromosomal translocation formation. PLos Genet. 2011; 7:e1002080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liang L., Deng L., Nguyen S.C., Zhao X., Maulion C.D., Shao C., Tischfield J.A.. Human DNA ligases I and III, but not ligase IV, are required for microhomology-mediated end joining of DNA double-strand breaks. Nucleic Acids Res. 2008; 36:3297–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Parsons J.L., Tait P.S., Finch D., Dianova I.I., Allinson S.L., Dianov G.L.. CHIP-mediated degradation and DNA damage-dependent stabilization regulate base excision repair proteins. Mol. Cell. 2008; 29:477–487. [DOI] [PubMed] [Google Scholar]
- 29. Caldecott K.W., Tucker J.D., Stanker L.H., Thompson L.H.. Characterization of the XRCC1–DNA ligase III complex in vitro and its absence from mutant hamster cells. Nucleic Acids Res. 1995; 23:4836–4843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Loizou J.I., El-Khamisy S.F., Zlatanou A., Moore D.J., Chan D.W., Qin J., Sarno S., Meggio F., Pinna L.A., Caldecott K.W.. The protein kinase CK2 facilitates repair of chromosomal DNA single-strand breaks. Cell. 2004; 117:17–28. [DOI] [PubMed] [Google Scholar]
- 31. Mani R.S., Fanta M., Karimi-Busheri F., Silver E., Virgen C.A., Caldecott K.W., Cass C.E., Weinfeld M.. XRCC1 stimulates polynucleotide kinase by enhancing its damage discrimination and displacement from DNA repair intermediates. J. Biol. Chem. 2007; 282:28004–28013. [DOI] [PubMed] [Google Scholar]
- 32. Kanno S., Kuzuoka H., Sasao S., Hong Z., Lan L., Nakajima S., Yasui A.. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. EMBO J. 2007; 26:2094–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luo H., Chan D.W., Yang T., Rodriguez M., Chen B.P., Leng M., Mu J.J., Chen D., Songyang Z., Wang Y. et al.. A new XRCC1-containing complex and its role in cellular survival of methyl methanesulfonate treatment. Mol. Cell. Biol. 2004; 24:8356–8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dutta A., Eckelmann B., Adhikari S., Ahmed K.M., Sengupta S., Pandey A., Hegde P.M., Tsai M.S., Tainer J.A., Weinfeld M. et al.. Microhomology-mediated end joining is activated in irradiated human cells due to phosphorylation-dependent formation of the XRCC1 repair complex. Nucleic Acids Res. 2017; 45:2585–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ahel I., Rass U., El-Khamisy S.F., Katyal S., Clements P.M., McKinnon P.J., Caldecott K.W., West S.C.. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 2006; 443:713–716. [DOI] [PubMed] [Google Scholar]
- 36. Hoch N.C., Hanzlikova H., Rulten S.L., Tetreault M., Komulainen E., Ju L., Hornyak P., Zeng Z., Gittens W., Rey S.A. et al.. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature. 2017; 541:87–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen J., Gilmore E.C., Marshall C.A., Haddadin M., Reynolds J.J., Eyaid W., Bodell A., Barry B., Gleason D., Allen K. et al.. Mutations in PNKP cause microcephaly, seizures and defects in DNA repair. Nat. Genet. 2010; 42:245–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marintchev A., Robertson A., Dimitriadis E.K., Prasad R., Wilson S.H., Mullen G.P.. Domain specific interaction in the XRCC1–DNA polymerase beta complex. Nucleic Acids Res. 2000; 28:2049–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Polo L.M., Xu Y., Hornyak P., Garces F., Zeng Z., Hailstone R., Matthews S.J., Caldecott K.W., Oliver A.W., Pearl L.H.. Efficient single-strand break repair requires binding to both poly(ADP-ribose) and DNA by the central BRCT domain of XRCC1. Cell Rep. 2019; 26:573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nash R.A., Caldecott K.W., Barnes D.E., Lindahl T.. XRCC1 protein interacts with one of two distinct forms of DNA ligase III. Biochemistry. 1997; 36:5207–5211. [DOI] [PubMed] [Google Scholar]
- 41. Mackey Z.B., Ramos W., Levin D.S., Walter C.A., McCarrey J.R., Tomkinson A.E.. An alternative splicing event which occurs in mouse pachytene spermatocytes generates a form of DNA ligase III with distinct biochemical properties that may function in meiotic recombination. Mol. Cell. Biol. 1997; 17:989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Taylor R.M., Wickstead B., Cronin S., Caldecott K.W.. Role of a BRCT domain in the interaction of DNA ligase III-alpha with the DNA repair protein XRCC1. Curr. Biol. 1998; 8:877–880. [DOI] [PubMed] [Google Scholar]
- 43. Moore D.J., Taylor R.M., Clements P., Caldecott K.W.. Mutation of a BRCT domain selectively disrupts DNA single-strand break repair in noncycling Chinese hamster ovary cells. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:13649–13654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Taylor R.M., Moore D.J., Whitehouse J., Johnson P., Caldecott K.W.. A cell cycle-specific requirement for the XRCC1 BRCT II domain during mammalian DNA strand break repair. Mol. Cell. Biol. 2000; 20:735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Taylor R.M., Thistlethwaite A., Caldecott K.W.. Central role for the XRCC1 BRCT I domain in mammalian DNA single-strand break repair. Mol. Cell. Biol. 2002; 22:2556–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lakshmipathy U., Campbell C.. The human DNA ligase III gene encodes nuclear and mitochondrial proteins. Mol. Cell. Biol. 1999; 19:3869–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lakshmipathy U., Campbell C.. Mitochondrial DNA ligase III function is independent of Xrcc1. Nucleic. Acids. Res. 2000; 28:3880–3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cuneo M.J., Gabel S.A., Krahn J.M., Ricker M.A., London R.E.. The structural basis for partitioning of the XRCC1/DNA ligase III-{alpha} BRCT-mediated dimer complexes. Nucleic Acids Res. 2011; 39:7816–7827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mani R.S., Karimi-Busheri F., Fanta M., Caldecott K.W., Cass C.E., Weinfeld M.. Biophysical characterization of human XRCC1 and its binding to damaged and undamaged DNA. Biochemistry. 2004; 43:16505–16514. [DOI] [PubMed] [Google Scholar]
- 50. Cotner-Gohara E., Kim I.K., Tomkinson A.E., Ellenberger T.. Two DNA-binding and nick recognition modules in human DNA ligase III. J. Biol. Chem. 2008; 283:10764–10772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Van Duyne G.D., Standaert R.F., Karplus P.A., Schreiber S.L., Clardy J.. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 1993; 229:105–124. [DOI] [PubMed] [Google Scholar]
- 52. Della-Maria J., Hegde M.L., McNeill D.R., Matsumoto Y., Tsai M.S., Ellenberger T., Wilson D.M. 3rd, Mitra S., Tomkinson A.E.. The interaction between polynucleotide kinase phosphatase and the DNA repair protein XRCC1 is critical for repair of DNA alkylation damage and stable association at DNA damage sites. J. Biol. Chem. 2012; 287:39233–39244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Della-Maria J., Zhou Y., Tsai M.S., Kuhnlein J., Carney J.P., Paull T.T., Tomkinson A.E.. Human Mre11/human Rad50/Nbs1 and DNA ligase III{alpha}/XRCC1 protein complexes act together in an alternative nonhomologous end joining pathway. J. Biol. Chem. 2011; 286:33845–33853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kang J.G., Paget M.S., Seok Y.J., Hahn M.Y., Bae J.B., Hahn J.S., Kleanthous C., Buttner M.J., Roe J.H.. RsrA, an anti-sigma factor regulated by redox change. EMBO J. 1999; 18:4292–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hura G.L., Menon A.L., Hammel M., Rambo R.P., Poole F.L. 2nd, Tsutakawa S.E., Jenney F.E. Jr., Classen S., Frankel K.A., Hopkins R.C. et al.. Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nat. Methods. 2009; 6:606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Classen S., Hura G.L., Holton J.M., Rambo R.P., Rodic I., McGuire P.J., Dyer K., Hammel M., Meigs G., Frankel K.A. et al.. Implementation and performance of SIBYLS: a dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the advanced light source. J. Appl. Crystallogr. 2013; 46:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Knott G.J., Cress B.F., Liu J.J., Thornton B.W., Lew R.J., Al-Shayeb B., Rosenberg D.J., Hammel M., Adler B.A., Lobba M.J. et al.. Structural basis for AcrVA4 inhibition of specific CRISPR-Cas12a. Elife. 2019; 8:e49110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Guinier A., Fournet F.. Small Angle Scattering of X-rays. 1955; NY: Wiley Interscience. [Google Scholar]
- 59. Rambo R.P., Tainer J.A.. Accurate assessment of mass, models and resolution by small-angle scattering. Nature. 2013; 496:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Dyer K.N., Hammel M., Rambo R.P., Tsutakawa S.E., Rodic I., Classen S., Tainer J.A., Hura G.L.. High-throughput SAXS for the characterization of biomolecules in solution: a practical approach. Methods Mol. Biol. 2014; 1091:245–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sali A., Blundell T.L.. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993; 234:779–815. [DOI] [PubMed] [Google Scholar]
- 62. Marintchev A., Mullen M.A., Maciejewski M.W., Pan B., Gryk M.R., Mullen G.P.. Solution structure of the single-strand break repair protein XRCC1 N-terminal domain. Nat. Struct. Biol. 1999; 6:884–893. [DOI] [PubMed] [Google Scholar]
- 63. Cotner-Gohara E., Kim I.K., Hammel M., Tainer J.A., Tomkinson A.E., Ellenberger T.. Human DNA ligase III recognizes DNA ends by dynamic switching between two DNA-bound states. Biochemistry. 2010; 49:6165–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Langelier M.F., Planck J.L., Roy S., Pascal J.M.. Crystal structures of poly(ADP-ribose) polymerase-1 (PARP-1) zinc fingers bound to DNA: structural and functional insights into DNA-dependent PARP-1 activity. J. Biol. Chem. 2011; 286:10690–10701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Pelikan M., Hura G.L., Hammel M.. Structure and flexibility within proteins as identified through small angle X-ray scattering. Gen. Physiol. Biophys. 2009; 28:174–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schneidman-Duhovny D., Hammel M., Sali A.. FoXS: a web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010; 38:W540–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Schneidman-Duhovny D., Hammel M., Tainer J.A., Sali A.. Accurate SAXS profile computation and its assessment by contrast variation experiments. Biophys. J. 2013; 105:962–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schneidman-Duhovny D., Hammel M., Tainer J.A., Sali A.. FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles. Nucleic Acids Res. 2016; 44:W424–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Mastronarde D.N. Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 2005; 152:36–51. [DOI] [PubMed] [Google Scholar]
- 70. Marabini R., Ludtke S.J., Murray S.C., Chiu W., de la Rosa-Trevin J.M., Patwardhan A., Heymann J.B., Carazo J.M.. The Electron Microscopy eXchange (EMX) initiative. J. Struct. Biol. 2016; 194:156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kimanius D., Forsberg B.O., Scheres S.H., Lindahl E.. Accelerated cryo-EM structure determination with parallelisation using GPUs in RELION-2. Elife. 2016; 5:e18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sorzano C.O., Martin-Ramos A., Prieto F., Melero R., Martin-Benito J., Jonic S., Navas-Calvente J., Vargas J., Oton J., Abrishami V. et al.. Local analysis of strains and rotations for macromolecular electron microscopy maps. J. Struct. Biol. 2016; 195:123–128. [DOI] [PubMed] [Google Scholar]
- 73. Punjani A., Rubinstein J.L., Fleet D.J., Brubaker M.A.. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods. 2017; 14:290–296. [DOI] [PubMed] [Google Scholar]
- 74. Scheres S.H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012; 180:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Scheres S.H., Chen S.. Prevention of overfitting in cryo-EM structure determination. Nat. Methods. 2012; 9:853–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 77. Putnam C.D., Hammel M., Hura G.L., Tainer J.A.. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007; 40:191–285. [DOI] [PubMed] [Google Scholar]
- 78. Rambo R.P., Tainer J.A.. Characterizing flexible and intrinsically unstructured biological macromolecules by SAS using the Porod-Debye law. Biopolymers. 2011; 95:559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Parsons J.L., Dianova I.I., Finch D., Tait P.S., Strom C.E., Helleday T., Dianov G.L.. XRCC1 phosphorylation by CK2 is required for its stability and efficient DNA repair. DNA Repair (Amst.). 2010; 9:835–841. [DOI] [PubMed] [Google Scholar]
- 80. Kim I.K., Stegeman R.A., Brosey C.A., Ellenberger T.. A quantitative assay reveals ligand specificity of the DNA scaffold repair protein XRCC1 and efficient disassembly of complexes of XRCC1 and the poly(ADP-ribose) polymerase 1 by poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 2015; 290:3775–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Pascal J.M., Tsodikov O.V., Hura G.L., Song W., Cotner E.A., Classen S., Tomkinson A.E., Tainer J.A., Ellenberger T.. A flexible interface between DNA ligase and PCNA supports conformational switching and efficient ligation of DNA. Mol. Cell. 2006; 24:279–291. [DOI] [PubMed] [Google Scholar]
- 82. Zhang X., Morera S., Bates P.A., Whitehead P.C., Coffer A.I., Hainbucher K., Nash R.A., Sternberg M.J., Lindahl T., Freemont P.S.. Structure of an XRCC1 BRCT domain: a new protein-protein interaction module. EMBO J. 1998; 17:6404–6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kulcyzk A.W., Yang J.-C., Neuhas D.. Solution structure and DNA binding of the zinc-finger domain from DNA ligase IIIα. J. Mol. Biol. 2004; 341:723–738. [DOI] [PubMed] [Google Scholar]
- 84. Caldecott K.W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 2008; 9:619–631. [DOI] [PubMed] [Google Scholar]
- 85. Tomkinson A.E., Chen L., Dong Z., Leppard J.B., Levin D.S., Mackey Z.B., Motycka T.A.. Completion of base excision repair by mammalian DNA ligases. Prog. Nucleic Acid Res. Mol. Biol. 2001; 68:151–164. [DOI] [PubMed] [Google Scholar]
- 86. Rambo R.P., Tainer J.A.. Bridging the solution divide: comprehensive structural analyses of dynamic RNA, DNA, and protein assemblies by small-angle X-ray scattering. Curr. Opin. Struct. Biol. 2010; 20:128–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Brosey C.A., Tainer J.A.. Evolving SAXS versatility: solution X-ray scattering for macromolecular architecture, functional landscapes, and integrative structural biology. Curr. Opin. Struct. Biol. 2019; 58:197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Bernstein N.K., Hammel M., Mani R.S., Weinfeld M., Pelikan M., Tainer J.A., Glover J.N.. Mechanism of DNA substrate recognition by the mammalian DNA repair enzyme, Polynucleotide Kinase. Nucleic Acids Res. 2009; 37:6161–6173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Leppard J.B., Dong Z., Mackey Z.B., Tomkinson A.E.. Physical and functional interaction between DNA ligase IIIalpha and poly(ADP-Ribose) polymerase 1 in DNA single-strand break repair. Mol. Cell. Biol. 2003; 23:5919–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mackey Z.B., Niedergang C., Murcia J.M., Leppard J., Au K., Chen J., de Murcia G., Tomkinson A.E.. DNA ligase III is recruited to DNA strand breaks by a zinc finger motif homologous to that of poly(ADP-ribose) polymerase. Identification of two functionally distinct DNA binding regions within DNA ligase III. J. Biol. Chem. 1999; 274:21679–21687. [DOI] [PubMed] [Google Scholar]
- 91. Eckelmann B.J., Bacolla A., Wang H., Ye Z., Guerrero E.N., Jiang W., El-Zein R., Hegde M.L., Tomkinson A.E., Tainer J.A. et al.. XRCC1 promotes replication restart, nascent fork degradation and mutagenic DNA repair in BRCA2-deficient cells. NAR Cancer. 2020; 2:zcaa013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hammel M., Rosenberg D.J., Bierma J., Hura G.L., Thapar R., Lees-Miller S.P., Tainer J.A.. Visualizing functional dynamicity in the DNA-dependent protein kinase holoenzyme DNA-PK complex by integrating SAXS with cryo-EM. Prog. Biophys. Mol. Biol. 2020; S0079-6107:30091–30092. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors have been deposited in the PDB with accession codes 6WH1 for the XLBR-BR complex and 6WH2 for X1BR2. The EM maps have been deposited in the EMDB with accession codes 22130 and 22307 for EM conformer 1 and EM conformer 2, respectively. SAXS data and atomistic models of XRCC1ΔN-p, XRCC1, LigIIIα and XL complex have been deposited in the SASBDB with accession codes SASDJ52, SASDJ62, SASDJ72 and SASDJ82.