


Abstract
Short-chain acylations of lysine residues in eukaryotic proteins are recognized as essential posttranslational chemical modifications (PTMs) that regulate cellular processes from transcription, cell cycle, metabolism, to signal transduction. Lysine butyrylation was initially discovered as a normal straight chain butyrylation (Knbu). Here we report its structural isomer, branched chain butyrylation, i.e. lysine isobutyrylation (Kibu), existing as a new PTM on nuclear histones. Uniquely, isobutyryl-CoA is derived from valine catabolism and branched chain fatty acid oxidation which is distinct from the metabolism of n-butyryl-CoA. Several histone acetyltransferases were found to possess lysine isobutyryltransferase activity in vitro, especially p300 and HAT1. Transfection and western blot experiments showed that p300 regulated histone isobutyrylation levels in the cell. We resolved the X-ray crystal structures of HAT1 in complex with isobutyryl-CoA that gleaned an atomic level insight into HAT-catalyzed isobutyrylation. RNA-Seq profiling revealed that isobutyrate greatly affected the expression of genes associated with many pivotal biological pathways. Together, our findings identify Kibu as a novel chemical modification mark in histones and suggest its extensive role in regulating epigenetics and cellular physiology.
INTRODUCTION
Protein posttranslational modifications (PTMs) fundamentally impact on cellular physiology and phenotype in eukaryotic organisms (1). Fatty acylation on the side-chain amino group of lysine residues has been recognized as an important type of reversible PTM marks that impart various regulatory functions on key cellular processes such as gene transcription, metabolism, protein homeostasis and signal transduction (2–4). So far, ∼20 different lysine acylations have been discovered including acetylation, propionylation, butyrylation, crotonylation, succinylation, malonylation, and glutarylation (5–8). Lysine acetyltransferases (KATs) are the writer enzymes that introduce acylation marks on specific lysine residues using acyl-CoA molecules as the acyl donor (9). Acyllysines are recognized by downstream reader proteins, and enzymatically reversed by eraser proteins, histone deacylases (HDAC) (10,11). Deregulation of lysine acylation dynamics caused by aberrant expression or mutation of either writer, reader or eraser proteins is broadly associated with various disease phenotypes including inflammation, neurodegeneration, cancer, etc. (12–14). Even occurring on the same residues, different acylations could result in distinct biological outcomes. For instance, butyrylation competes with acetylation on H4K5/K8 and reduces the binding of the reader protein BRDT on these loci, which causes delayed histone removal and gene expression in spermatogenic cells (15). It remains an imperative task to map out cellular substrates and modification sites of different lysine acylations and investigate their functional impacts in different physiological and pathological pathways.
In the last decade, the development of high resolution mass spectrometry has greatly facilitated discovery of many novel lysine acylation marks (16,17). Lysine butyrylation (Kbu) was first discovered by Zhao et al. as a normal straight chain n-butyrylation (Knbu) which is biochemically dependent on n-butyryl-CoA (18). Later studies demonstrated that KAT members p300/CBP possess lysine n-butyryltransferase activity (19). Etiologically, n-butyryl-CoA is a metabolic intermediate from the fatty acid oxidation pathway and therefore, serving as an ample donor source for Knbu. From a chemical perspective, butyrylation mark may also exist in its isomeric structure, i.e. isobutyrylation (2-methylpropionylation) (Figure 1). Our proposition is further inspired by the natural existence of isobutyryl-CoA which is endogenously generated as a result of valine catabolism and the oxidation of branched chain fatty acids (20). Taking into account the physiological existence of isobutyryl-CoA and its structural relevance with n-butyryl-CoA, we posit that isobutyryl-CoA may also function as an acyl group donor and lead to an unexplored acylation mark in proteins, lysine isobutyrylation. Pathologically, dysregulation of isobutyryl-CoA level caused by genetic deficiency of isobutyryl-CoA dehydrogenase (IBD) is associated with multiple disease symptoms including speech delay, anemia, and dilated cardiomyopathy in newborn patients while mechanisms underlying these disorders are still poorly defined (21,22). Hence, understanding the catabolism of isobutyryl-CoA in relationship with dynamic regulation of protein function through the PTM mechanism has a profound pathophysiological significance. In the present study, we report and validate lysine isobutyrylation (Kibu) as a bona fide PTM in nuclear histones through a combined suite of biochemical, biophysical, and cellular studies.
Figure 1.

Distinct pathways for the formation of butyryl-CoA and isobutyl-CoA and their consequences in leading to protein lysine butyrylation. Lysine butyrylation was discovered in 2007 (18) and was recognized as n-butyrylation. In this study, we found that lysine butyrylation also exists in the form of isobutyrylation in histones. Endogenous n-butyryl-CoA is derived from fatty acid metabolism while isobutyryl-CoA is derived from valine metabolic pathway. Exogenous butyrate and isobutyrate can be converted to their acyl-CoA by the action of cellular short-chain acyl-CoA synthetases.
MATERIALS AND METHODS
Quantification of butyryl-CoA level in HEK 293T cells
Human embryonic kidney 293T (HEK 293T) cells were purchased from ATCC and cultured to 90% confluence in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% streptomycin–penicillin. Sodium d7-isobutyrate was prepared by adding concentrated NaOH solution to d7-isobutyric acid (Sigma-Aldrich, Cat# 632007), lyophilized and quantified by reverse phase (RP)-HPLC. Cells were treated with varied concentrations of sodium d7-isobutyrate and valine (Sigma-Aldrich, Cat#V0500) for 24 h, and washed with ice-cold PBS buffer followed by fixing in methanol at −80°C for 15 min. Cells were collected in 50% of methanol with gentle scrape, and centrifuged at 16 000 g at 4°C. The supernatant was collected for HPLC–MS/MS analysis.
An Atlantis® T3 (4.6 × 150 mm, 3 μm) column with a Phenomenex SecurityGuard C-18 guard column (4.0 mm × 2.0 mm) was applied to separate analytes. The column temperature was constant at 32°C. The mobile phase A was 10 mM ammonium acetate, and mobile phase B was acetonitrile. A gradient method was applied for separation, with a 0.4 ml/min flow rate, (time/minute, % mobile phase B): (0, 6), (15, 30), (15.01, 100), (22.50, 100), (22.51, 6). The injection volume was 30 μl, and the autosampler injection needle was washed with methanol after each injection. Nitrogen was used as the desolvation gas at a flow rate of 500 l/h. The desolvation temperature was 500°C and the source temperature was 120°C. Argon was used as the collision gas, and the collision cell pressure was 3.5 × 10−3 mbar. Samples were analyzed in the positive ion mode. The capillary voltage was 3.2 kV, the cone voltage was 42 V and the collision energy was 22 eV. A multiple reaction monitoring (MRM) function was applied for the detection of analytes. The ion transition 838→331 was monitored for iso- and n-butyryl-CoA, and 845→338 for d7-isobutyryl CoA.
Expression and purification of p300 and HAT1
The expression of p300 HAT domain (1287–1666) was done using the semisynthetic method developed by the Cole lab (23). A C-terminal 14-residue peptide (sequence 1653–1666: CMLVELHTQSQDRF) was synthesized by solid-phase peptide synthesis and purified by C18 RP-HPLC. The pTYB2 plasmid encoding the inactive p300 HAT domain (region 1287–1652) fused to intein-chitin-binding domain was transformed into Escherichia coli BL21(DE3)/RIL competent cells through heat-shock method and then grown on lysogeny broth (LB)-agar plates containing both ampicillin (final concentration 100 μg/ml) and chloramphenicol (final concentration 50 μg/ml). Colonies were harvested and grown at 37°C in 8 ml then 1 l cultures of terrific broth media containing both ampicillin and chloramphenicol. Protein expression was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) at 1.0 mM final concentration and shaken for 16 h at 16°C. The cells were collected by centrifugation at 4000 rpm for 25 min, suspended in lysis buffer (25 mM Na-HEPES (pH 8.0), 500 mM NaCl, 1 mM MgSO4, 10% glycerol and 2 mM phenylmethanesulfonyl fluoride (PMSF)), and lysed by a microfluidic cell disruptor. The lysates were cleared by centrifugation to obtain the supernatant. Chitin resin were equilibrated with column buffer (25 mM Na-HEPES (pH 8.0), 250 mM NaCl, 1 mM EDTA, 0.1% trition X-100 and 1 mM PMSF) before they were used to purify the supernatant. The column was thoroughly washed with column buffer and wash buffer (25 mM Na-HEPES (pH 8.0), 500 mM NaCl, 1 mM EDTA, 0.1% triton X-100 and 1 mM PMSF) before being ligated to the CT14 peptide to obtain active p300. CT14 peptide was dissolved in cleavage buffer (25 mM Na-HEPES (pH 8.0), 250 mM NaCl, 1 mM EDTA and 200 mM 2-mercaptoethanesulfonic acid (MESNA)) and added to the column. After the addition of the CT14 peptide in cleavage buffer, the column was shaken at room temperature for 16 h. The protein was then eluted from the column, and several volumes of cleavage buffer were added to ensure the complete elution. The eluted protein was further purified by cation exchange chromatography using the NGC fast protein liquid chromatography (FPLC) system, Bio-Rad. The ligated p300 HAT domain was characterized using a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Millipore centrifugal filtration was used to concentrate protein solution and the Bradford assay was used to determine final protein concentration. Lastly, the protein was aliquoted, flash frozen by liquid nitrogen and stored at −80°C.
The expression and purification of human HAT1 (20–341) was done following the method described Hong Wu et al. (24). The pET28a-LIC-HAT1 plasmid (plasmid #25239, Addgene) was transformed into BL21(DE3)-RIL competent cells through heat-shock and then spread on agar plates containing antibiotics kanamycin and chloramphenicol. Protein expression was induced by the addition of 1 mM IPTG and shaken for 16 h at 16°C. The cells were collected and suspended in lysis buffer (50 mM Na-phosphate (pH 7.4), 250 mM NaCl, 5 mM imidazole, 5% glycerol, 2 mM β-mercaptoethanol and 1 mM PMSF) and then disrupted using the cell disruptor. The supernatant was passed through a column containing Ni-NTA resin equilibrated with column washing buffer (20 mM Tris–HCl (pH 8.0), 250 mM NaCl, 5% glycerol, 30 mM imidazole and 1 mM PMSF) and the resin was washed with the same column washing buffer for twice. Next, the resin was washed with buffer containing a higher concentration of imidazole (20 mM Tris–HCl (pH 8.0), 250 mM NaCl, 5% glycerol, 50 mM imidazole and 1 mM PMSF) for three times. The proteins on the resin were then eluted with elution buffer (20 mM Tris–HCl (pH 8.0), 250 mM NaCl, 5% glycerol, 500 mM imidazole and 1 mM PMSF), and dialyzed in the dialysis buffer (25 mM Tris–HCl (pH 8.0), 150 mM NaCl, 10% glycerol, 1 mM DTT) at 4°C overnight. Thrombin was added to the dialyzed protein containing HAT1 and dialyzed in thrombin cleavage buffer (20 mM Tris–HCl (pH 8.0), 100 mM NaCl, 2.5 mM CaCl2, 5% glycerol, 1 mM DTT) for 20 h at 4°C to remove the His6x-tag. The resultant protein was concentrated and purified by anion exchange chromatography using the NGC FPLC system. HAT1 purity was checked using SDS-PAGE. Millipore centrifugal filter and Bradford assay were used to concentrate and determine protein concentration, respectively. The protein was aliquoted, flash frozen by liquid nitrogen, and stored at −80°C.
Synthesis of isobutyryl-CoA
2 mmol of isobutyric acid (176.2 mg) was dissolved in 5 ml of freshly distilled CH2Cl2. To this solution was added 1 mmol of N,N’-dicyclohexylcarbodiimide (DCC) (206.4 mg), and the reaction was allowed to proceed at room temperature for 4 h. The reaction mixture was filtered to remove dicyclohexylurea (DCU), and then CH2Cl2 was removed using rotary evaporation. The dried crude material was used in the next step without further purification. 0.013 mmol of CoA hydrate (10 mg) was dissolved in 1.5 ml of 0.5 M NaHCO3 (pH 8.0) and cooled down on ice bath. Then the crude isobutyric anhydride (10.3 mg, 0.065 mmol) in 1 ml of CH3CN/acetone (1:1 v/v) was added dropwise to the CoA solution. The reaction solution was stirred at 4°C overnight and quenched by adjusting pH to 4 with 1 M HCl. The reaction mixture was subjected to RP-HPLC purification with gradient 5─45% acetonitrile over 30 min at flow rate 5 ml/min; UV detection wavelength was fixed at 214 and 254 nm. HPLC buffer was 0.05% TFA in water (solution A) and 0.05% TFA in acetonitrile (solution B). The fractions were collected, rotary evaporated and lyophilized to yield 6.34 mg white solid. The purity of final product was checked by analytical RP-HPLC and molecular weight was confirmed by MALDI MS (found [M+H]+ 838.6).
Biochemical assays of lysine acyltransferase activities of KATs
Acetyl-CoA (Sigma-Aldrich, Cat#A2181), propionyl-CoA (Chem Impex Intl Inc, Cat# 01895) and n-butyryl-CoA (Sigma-Aldrich, Cat#B1508) salts were purchased from commercial suppliers. Synthetic histone peptides H3(1–20) or H4(1–20) (20 amino acids from the N-terminal of histone H3 and H4 (the sequence of H3(1–20) is Ac-ARTKQTARKSTGGKAPRKQL, the sequence of H4(1–20) is Ac-SGRGKGGKGLGKGGAKRHRK) were used as acyl acceptor substrates. For single-point quantification assays, 30 μM of each acyl-CoA molecule was incubated with individual KAT enzymes and 100 μM histone peptides. The enzymatic reactions were conducted in a KAT reaction buffer containing 50 mM HEPES-Na and 0.1 mM EDTA-Na at pH 8.0. KAT enzymes were mixed with individual acyl-CoA molecules and peptide substrates, followed by incubation at 30°C for 15 min to allow enzymatic transfer of acyl groups to lysine substrates and release of by-product CoASH. The fluorogenic probe 7-diethylamino-3-(4′-maleimidylphenyl)-4-methylcoumarin (abbr. CPM, ThermoFisher, Cat# D346) in 100% DMSO was then added to both quench the enzymatic reaction and react with CoASH to yield the fluorescent CPM–SCoA complex for fluorescence quantification (25). The fluorescence intensities were measured with excitation and emission wavelengths at 392 and 482 nm, respectively, with a FlexStation®3 microplate reader. Duplicated experiments were performed and the results were shown in a bar graph.
For kinetic characterization of p300 and HAT1 with acyl-CoA molecules, varied concentration of acyl-CoA was incubated with p300 or HAT1 and 200 μM of H4(1–20) peptide. All the reactions were conducted in the same KAT reaction buffer as the single-point assay. The fluorescence intensity was measured with the same method as the single-point assay and catalytic rate was determined based on fluorescence intensities. Kinetic constants including Michaelis constant (Km) and catalytic turnover (kcat) were determined by fitting the acyl-CoA concentration-catalytic velocity curves to the Michaelis–Menten equation using KaleidaGraph software.
Western blot analysis of HAT1 mediated in vitro histone H4 isobutyrylation
1 μg of human recombinant histone H4 (New England BioLabs, Cat# M2504S) was incubated with 50 μM n-butyryl- or isobutyryl-CoA and 0.2 μM of HAT1 at 30°C for 1 h. The reaction mixture was boiled in SDS-PAGE gel loading buffer and resolved on a 15% polyacrylamide gel followed by wet membrane transfer to a nitrocellulose (NC) membrane. The NC membrane was blocked with 5% non-fat milk in Tris-buffered saline + 0.1% Tween-20 (TBST) for 1 h at room temperature. Anti-butyryllysine antibody (PTM BioLabs, PTM#301) at 1:2000 dilution was incubated with the membrane overnight at 4°C. The membrane was washed with TBST buffer for three times and incubated with the goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology, Cat# sc-2004) with 1:3000 dilution at room temperature for 1 h. The membrane was then washed and subjected to chemiluminescence detection with the ECL substrate (ThermoFisher, Cat# 32209) on a LI-COR Odyssey system (LI-COR Biosciences).
Western blot analysis of in cellulo lysine isobutyrylation and lysine acetylation in response to sodium isobutyrate treatment
HEK 293T cells were cultured to 90% confluence in DMEM medium supplemented with 10% FBS and 1% streptomycin–penicillin antibiotics. For in-cell Kibu level analysis, cells were treated with 10 mM d7-isobutyrate for 16 h followed by cellular protein extraction. Whole cell lysate was extracted in M-PER™ Mammalian Protein Extraction Reagent (ThermoFisher Scientific, Cat# 78501) with gentle sonication and core histone proteins were extracted with the EpiQuik Total Histone Extraction Kit (Epigentek, Cat# OP-0006-100). The extracted lysates and histone proteins were resolved on a 4–20% gradient gel (BioRad) and a 15% polyacrylamide gel, respectively. Kibu levels were detected by western blot using the same procedure aforementioned. For in-cell Kac level analysis, cells were treated with 20 mM d7-isobutyrate for 24 h followed by core histone extraction. Histone extracts were then resolved on a 15% polyacrylamide gel and Kac levels were determined with western blot using anti-acetyllysine antibody (PTM Biolabs, Cat# PTM-101), with anti-Knbu/Kibu as the positive control using anti-butyryllysine antibody (PTM Biolabs, Cat# PTM-301) and histone H3 as the loading control using histone H3 antibody (Santa Cruz Biotechnology, Cat# sc-517576).
In cellulo analysis of lysine isobutyrylation in response to p300 and HAT1 overexpression
HEK 293T cells were cultured to 90% confluence in DMEM medium supplemented with 10% FBS and 1% streptomycin–penicillin antibiotics. Plasmids flag-HAT1 (GeneCopoeia, Cat# EX-I0105-M13-11) and pCMVβ-p300-myc (Addgene, Cat# 30489) were transfected into cells with Lipofectamine 3000™ Transfection Reagent (ThermoFisher, Cat# L3000008). The cells were then incubated with 20 mM sodium d7-isobutyrate for 24 h followed by core histone extraction. Kibu levels were analyzed by western blot using the same procedure and antibodies aforementioned.
HAT1 crystallization and structural determination
HAT1 was expressed and purified as described previously (24). HAT1 at 8 mg/ml was incubated with isobutyryl coenzyme A (IBuCoA) and histone H4 K12A mutant peptide (amino acids 1–20 of H4) at a 1:10:5 molar ratio of HAT1:IbuCoA:H4(K12A) and crystallized using the sitting drop vapor diffusion method by mixing 2 μl of protein solution with 1 μl of the reservoir solution containing 2 M sodium dihydrogen phosphate and 0.1 M MES, pH 6.5. Crystals were cryo-protected in the corresponding mother liquor supplemented with 30% glycerol and cryo-cooled in liquid nitrogen. X-ray diffraction data were collected at the Advanced Photon Source (APS) BEAMLINE 19-ID. Data were processed using XDS (26) and merged with Aimless (27). PDB ID: 2P0W was used in Fourier transform using Refmac5 (28). Model building and visualization was performed in COOT (29) and the structure was validated with Molprobity (30). Data collection and refinement statistics are summarized in Supplementary Table S1. The HAT1-IbuCoA-H4(K12A) structure factors and coordinates have been deposited in the Protein Data Bank with the PDB ID: 6VO5.
HPLC-MS/MS analysis of histone isobutyrylation
Core histones (∼4 μg) extracted from d7-sodium isobutyrate treated 293T cells were resolved in SDS-PAGE. Histones were excised from the gel, and subjected to in-gel digestion by trypsin (31). The digested peptide was dissolved in 2.5 μl water containing 0.1% formic acid (v/v), and then loaded onto a home-packed capillary column (10 cm length × 75 mm ID, 3 μm particle size, Dr Maisch GmbH, Germany) which was connected to an EASY-nLC 1000 system (Thermo Fisher.). The mobile phase A was water containing 0.1% formic acid (v/v), and mobile phase B was acetonitrile containing 0.1% formic acid (v/v). A 60-min gradient of 2–90% mobile phase B at a flow rate of 200 nl/min was used for the peptide separation. The eluted peptides were analyzed by a Q-Exactive mass spectrometer (Thermo Fisher). A Full mass scan was conducted in the Orbitrap mass analyzer in the range m/z 300–1400 with a resolution of 70 000 at m/z 200. The top 15 ions were fragmented with normalized collision energy of 27 and tandem mass spectra were acquired with a mass resolution of 17 500 at m/z 200.
The obtained MS/MS spectra were searched with Mascot (Matrix Science, London, UK) against UniProt Human protein database. Mono-methylation and di-methylation on lysine and arginine, tri-methylation on lysine, acetylation on lysine and protein N-terminal, oxidation on methionine, and d7-isobutyrylation on lysine were specified as variable modifications. Maximum missing cleavage was set at 4, and mass tolerance was set at 10 ppm for precursor ions and ±0.05 Da for MS/MS.
RNA-seq
Cellular total RNA was extracted using TRIzol reagent (Thermo Fisher). Indexed libraries were constructed using the Illumina TruSeq Stranded mRNA library prep kit. Samples were then sequenced on NovaSeq (PE50) with paired-end reading. Raw reads in FASTQ files were submitted for differential expression analysis using DEseq2. Gene set enrichment analysis (GSEA) was performed against KEGG pathway. Significantly altered gene sets were defined with the False Discovery Rate (FDR) and P value less than 0.25 and 0.05, respectively.
RESULTS
Isobutyryl-CoA is an abundant metabolite in the mammalian cell
Upon its discovery with mass spectrometry, lysine butyrylation (Kbu) was defined as a straight chain n-butyrylation (18). Nevertheless, two isomeric structures may contribute to the same molecular mass of the butyryl mark (+70 au): either normal linear butyryl or branched isobutyryl group. Lysine acylations rely on acyl-CoA molecules as the reactive acyl group donor (Figure 1); therefore, an ample pool of acyl-CoA is a prerequisite for the incidence of cognate lysine acylation. To determine cellular butyryl-CoA composition, we measured the levels of n- and iso-butyryl-CoA in HEK 293T cells with high-performance liquid chromatography tandem mass spectrometry (HPLC–MS/MS). Iso- and n-butyryl-CoA standards were resolved on the chromatogram at retention times of ∼13.85 and 14.05 min respectively, and monitored at the same ion transition (838→331) on the mass spectrometer (Figure 2A). The chromatographic peak area of n- or iso-butyryl-CoA molecules was integrated as the quantitation reference. Then 293T cell's acyl-CoA extracts were subjected to the same HPLC–MS/MS analysis and the abundance of n- and iso-butyryl-CoAs was compared. Duplicate experiments were conducted and showed that the ratios of iso- and n-butyryl-CoA were 2–3:1 (Figure 2B). This data revealed that the cellular butyryl-CoA is a mixture of two isomers, both of which may act as acyl donors leading to lysine butyrylation on cellular proteins.
Figure 2.

Detection and quantification of n- and iso-butyryl-CoA in 293T cells using LC–MS/MS. (A) Control experiments showing that n- and iso-butyryl-CoA were separated with the LC–MS/MS system with the retention times at 14.06 and 13.86 min and were detected at the same ion transition 810 → 313. (B) n- and iso-Butyryl-CoA from the 293T cell extracts were detected under the same LC–MS/MS condition. Their levels were compared by using the integrated chromatographic peak areas.
Cellular metabolism of isobutyrate and valine lead to isobutyryl-CoA production
Acyl-CoA molecules are metabolic intermediates mostly from glucose catabolism, fatty acid β-oxidation, and amino acid degradation (32). Also short chain fatty acids such as propionate and butyrate taken up from surrounding environment can be converted to cognate acyl-CoA molecules with the catalysis by short chain acyl-CoA synthetases (ACS) (33,34). It is thus highly possible that isobutyryl-CoA can be synthesized from this pathway with isobutyrate as the source agent. Importantly, isobutyryl-CoA is a key intermediate of valine metabolism pathway so valine may also stimulate biosynthesis of isobutyryl-CoA (Figure 1) (35). These isobutyryl-CoA synthetic pathways would further strengthen the possibility that isobutyryl-CoA acts as an important regulator in physiological processes. Herein, we studied the effect of isobutyrate and valine treatment on the change of cellular isobutyryl-CoA level. 293T cells were treated with varied concentrations of deuterated sodium isobutyrate (d7-isobutyrate), and acyl-CoA molecules were extracted and analyzed with the HPLC–MS/MS method aforementioned. The levels of butyryl-CoA molecules were measured and normalized to total cellular proteins (Supplementary Figure S1A). With the increase of d7-isobutyrate doses, d7-isobutyryl-CoA level increased drastically until a plateau was reached at 5 mM of d7-isobutyrate. The level of endogenous, non-isotopic isobutyryl-CoA remained barely changed. We also tested how isobutyryl-CoA level would change in response to valine feeding. The data showed that treatment of 293T cells with valine significantly increased the level of isobutyryl-CoA until a plateau was reached at the concentration of 5 mM valine (Supplementary Figure S1B). In contrast, the level of n-butyryl-CoA was not affected by valine treatment. This observation is consistent with the aforementioned valine metabolic pathway, in which isobutyryl-CoA is an important intermediate (35). Taken together, these data demonstrated that isobutyryl-CoA can be biosynthesized from the acyl-CoA synthetase pathway and the valine metabolism, which not only defines the anabolism of isobutyryl-CoA in mammalian cells but also provides us with an amenable approach to study dynamic changes of lysine isobutyrylation levels in cellular proteins.
KAT enzymes catalyze lysine isobutyrylation
Site-specific lysine acylations are enzymatically driven by KATs, which covalently deposit acyl groups from the cosubstrate acyl-CoA to lysine residues in protein substrates. Identification of isobutyryl-CoA as an ample acyl-CoA donor points out the high probability for the existence of lysine isobutyrylation in cellular proteins. In this regard, it is necessary to examine whether and which KATs can catalyze this acylation reaction. We quantitatively measured the acylation activities of nine human KATs from three major KAT families, MYST, GCN5/PCAF/HAT1, and p300/CBP, using a fluorogenic assay that was designed to quantify the byproduct CoA (25). Histone peptides H3(1–20) or H4(1–20) was used as the acyl acceptor to characterize the acetyl-, propionyl-, n-butyryl- and isobutyrylation activities of individual KAT enzymes (Supplementary Figure S2). Consistent with our recent study (36), all the tested KAT enzymes showed remarkable lysine propionylation activity, >20% of their native acetyl transfer activity, which demonstrated that almost all eukaryotic KATs may possess this intrinsic activity. Nevertheless, further increase of acyl chain length to four carbon units lead to a drastic decrease of butyrylation activity (Kbu) of KAT enzymes. Among the tested KAT enzymes, HAT1 showed an outstanding isobutyrylation (Kibu) activity with ∼25% of its acetylation activity, followed by HBO1 while the other KAT enzymes exhibited much lower or even barely detectable Kibu activity. p300 is known for its great cofactor promiscuity, functioning as lysine acetyl-, propionyl-, butyryl- and crotonyltransferase (2,18,19). Its unique acyl-CoA binding pocket enables p300 to bind larger acyl-CoA molecules without need of protein engineering (37,38). Therefore, we carried out steady-state kinetic characterization of p300 and HAT1 with various acyl-CoAs to determine their acyltransferase activities (Figure 3A and B). The results showed that the catalytic specificity constants kcat/Km of p300 with n- and iso-butyrylation activities were 0.25 min−1.μM−1 and 0.13 min−1.μM−1 respectively, about 7% and 13% of p300 acetylation activity (Figure 3C). In comparison, Knbu and Kibu activity of HAT1 was about 13% and 31% of its acetylation activity, respectively. Thus, both HAT1 and p300 possessed appreciable activities of lysine n-butyrylation and iso-butyrylation.
Figure 3.

Kinetic characterization of p300 and HAT1 acylation activities. p300 or HAT1 was incubated with individual acyl-CoA molecule at varied concentrations and H4(1–20) peptide substrates. The enzymatic reaction was quantified using the CPM assay. The reaction velocities as a function of acyl-CoA concentration were fit with the Michaelis-Menten equation to get the kinetic constants Km and kcat. (A) Initial velocity curves of p300 with acetyl-, propionyl-, n-butyryl- and isobutyryl-CoA. (B) Initial velocity curves of HAT1 with acetyl-, propionyl-, n-butyryl- and isobutyryl-CoA. (C) Kinetic parameters obtained from the velocity data fitting. Experimental conditions: 200 μM H4(1–20) peptide, 0–50 μM acyl-CoA, 40 nM or 100 nM HAT1, 20 nM or 100 nM p300, 15 min reaction time, temperature at 30°C.
HAT1 and p300 isobutyryltransferase activity on synthetic H3 and H4 peptides was further confirmed by MALDI-MS analysis of the reaction mixtures (Supplementary Figure S3). The recombinant p300 and HAT1 were incubated with isobutyryl-CoA and H3(1-20) (for p300) or H4(1-20) peptides (for HAT1) for 1 h. Next, the reaction mixture was subjected to MALDI-MS test. Product peaks (M+70) were observed in both p300 and HAT1 catalytic reactions, demonstrating that both enzymes can catalyze isobutyrylation on peptide substrates. We further tested if the isobutyrylation activity of HAT1 on histone H4 substrate can be detected with Western blot using the commercially available anti-butyryllysine antibody (PTM Biolab, Cat# PTM-301). Although this antibody was designed for detection of Knbu (15,39), we found that incubation of n- or iso-butyryl-CoA, histone H4 and HAT1 drastically increased the band intensity of both n-butyrylated H4 and iso-butyrylated H4, whereas lack of HAT1 induced little histone labeling (Supplementary Figure S4). Hence, this anti-Knbu antibody was also able to recognize Kibu mark. Our finding on the promiscuous specificity of the anti-butyryllysine antibody indicates that some butyrylated lysines identified in previous work could be a mixture of n- and iso-butyrylated lysines (40). Overall, the biochemical measurements and western blot data validated the Kibu activity of HAT1 and p300. Importantly, the capability of PTM-301 antibody in recognizing Kibu mark allows for a technical means to study protein Kibu signals from the cellular contexts.
Lysine isobutyrylation is a bona fide PTM mark on nucleosomal histones
Next we focused on the detection of lysine isobutyrylation (Kibu) on cellular proteins. 293T cells were treated with sodium d7-isobutyrate for 16 h, followed by extraction of the nuclear histone proteins or whole cellular proteome. The extracted proteins were resolved on a SDS-PAGE gel and analyzed with Western blot using the anti-Kbu antibody PTM-301. The chemiluminescent protein bands on the Western blot membrane are collective signals corresponding to Knbu and Kibu levels because of the promiscuity of this antibody. Nevertheless, any intensity change upon d7-isobutyrate treatment will reflect changes in the Kibu level because isobutyrate treatment induced synthesis of isobutyryl-CoA rather than n-butyryl-CoA. As shown in Figure 4, both whole proteome extracts and histone extracts showed increased Kibu levels on histones H3 and H4 as a result of isobutyrate treatment. Thus, Kibu is a bona fide histone PTM and is driven by isobutyryl-CoA. Surprisingly, under our condition, no appreciable change of chemiluminescence intensity was observed on non-histone proteins upon isobutyrate treatment.
Figure 4.
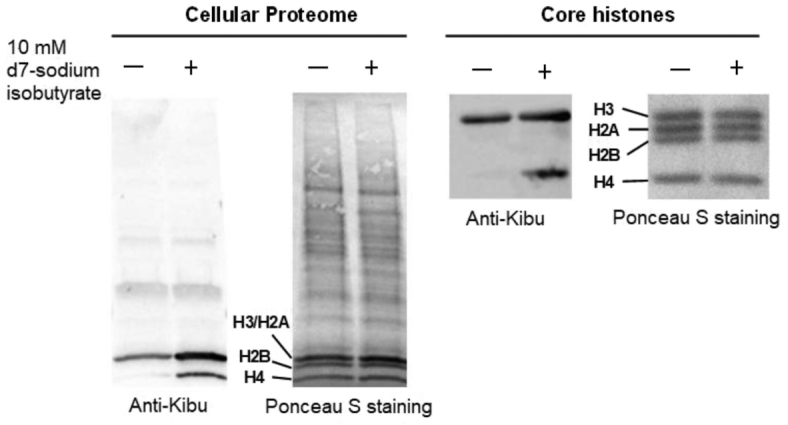
Detection of lysine isobutyrylation on protein lysines in 293T cells. Cellular proteomes and core histone proteins were extracted and tested with anti-butyryllysine antibody (PTM Biolabs, Cat#PTM-301). Treatment of cells with isobutyrate induced increase of lysine isobutyrylation level on core histone proteins while no appreciable change was observed on non-histone protein isobutyrylation upon isobutyrate treatment.
To confirm histone Kibu in cells, we performed HPLC-MS/MS analysis of the histone extract (Figure 5). The core histones were extracted from the sodium d7-isobutyrate treated 293T cells and subsequently digested with trypsin. The resulting tryptic peptides were subjected to nano-HPLC/MS/MS analysis and protein sequence alignment with Mascot algorithm. Our analysis led to the identification of two modified H3 peptides, KSTGGKAPR and KQLATKAAR, which contain a mass shift of +77.0858 Da at sites H3K14 and H3K23, respectively. This mass shift is the same as that caused by the addition of a d7-isobutyralation, demonstrating the existence of two novel isobutyralation sites on histone H3.
Figure 5.

MS/MS spectra showing isobutyrylation of H3K14 (A) and H3K23 (B) sites in the histones extracted from 293T cells. The b and y ions refer to peptide backbone fragment ions containing N- and C-terminus, respectively.
The p300 acetyltransferase possesses histone isobutyrylation activity in the cell
We moved on to test which KAT can mediate Kibu levels in the cell. We focused on p300 and HAT1 as they both showed clear isobutyrylation activity on peptide substrates in vitro (Figure 3 and Supplementary Figure S2). To determine if they could catalyze in cellulo lysine isobutyrylation, we performed transient transfection of 293T cells with p300 and HAT1 plasmids and then treated the cells with sodium d7-isobutyrate. Consistent with the above observations (Figure 4), isobutyrate significantly increased Kibu levels in histones, suggesting isobutyrate is the source of isobutyryl group to promote isobutyrylation (Supplementary Figure S5, lanes 2 and 6). In the presence of p300 overexpression and isobutyrate treatment together, Kibu levels were further boosted (Supplementary Figure S5, lane 4) especially on histone H3 region. These data suggest that histone H3 may be a major Kibu target mediated by p300. The result is consistent with previous studies that p300 acetylates multiple sites in histone H3 (41,42). Surprisingly, we did not observe any significant change in the Kibu level with HAT1 overexpression (Supplementary Figure S5, lane 8). Such a discrepancy between the biochemical and in cellulo isobutyrylation activities might reflect the context dependence of HAT1 activity.
Structural insights of the HAT1 and p300 interactions with isobutyryl-CoA
To elucidate the structural basis of KATs’ isobutyryltransferase activity, we first generated the 1.6 Å crystal structure of the ternary complex of HAT1 with isobutyryl-CoA (IbuCoA) and histone H4(K12A) mutant peptide, referred to here as HAT1-IbuCoA-H4(K12A) (PDB ID: 6VO5) (Supplementary Table S1). We used the H4(K12A) mutant peptide instead of the wild type counterpart to avoid any reaction intermediates between the ϵ-amino group of lysine12 (K12) of H4 and IbuCoA, and eventually to better resolve the electron density of the isobutyryl moiety of IbuCoA in HAT1-IbuCoA-H4(K12A). For comparison, the wild type H4K12 side chain inserts into the HAT1 active site tunnel in the crystal structure of HAT1 bound to acetyl-CoA and H4, referred to here as HAT1-AcCoA-H4 (PDB ID: 2P0W). The overall IbuCoA-HAT1 interaction is similar to that of AcCoA-HAT1 interaction observed in HAT1-AcCoA-H4 (Figure 6A, B), except for the adenosine ring of IbuCoA in HAT1-IbuCoA-H4(K12A), which adopts a pi-stacking interaction with Phe288 side chain of HAT1 instead of Lys284 (Supplementary Figure S6A). The electron density omit-map for IbuCoA in the crystal structure of HAT1-IbuCoA-H4(K12A) contained clear density for the extra isobutyryl moiety of IbuCoA (Supplementary Figure S6B), thus revealing that HAT1 also accommodates IbuCoA in its active site without any structural rearrangements.
Figure 6.

Crystal structure of HAT1 in complex with isobutyryl-CoA. (A) The overall fold of HAT1-IbuCoA-H4(K12A) (PDB ID: 6VO5) shown in cyan, superposed on HAT1-AcCoA-H4 (PBD ID: 2P0W) shown in grey. IbuCoA and histone H4(K12A) peptide are shown in sticks representation in magenta in yellow, respectively. Acetyl-CoA and histone H4 from HAT1-AcCoA-H4 structure (PBD ID: 2P0W) is shown in stick representation in grey. (B) Close-up view of the isobutyryl moiety of IbuCoA binding site. (C) Comparison of the acyl-CoA binding pockets in HAT1 (PDB ID: 2P0W) and p300 (PDB ID: 5LKT). Acetyl-CoA and H4 peptide are shown in sticks representation in magenta in yellow, respectively. Butyryl-CoA is shown in green. (D) Model of an isobutyryl-CoA (in magenta) superposed onto butyryl-CoA (in cyan) in the crystal structure of p300 (PDB ID: 5LKT). Model was generated by fitting the IbuCoA into the butyryl-CoA electron density map in the crystal structure of the p300-butyryl-CoA complex.
Compared to HAT1, the p300 HAT domain has a larger acyl-CoA binding pocket and thus can accommodate a diverse array of acyl-CoAs including bulky acyl-groups (Figure 6C). Several p300 structures in complex with acyl-CoA variants including acetyl-CoA and butyryl-CoA were reported (19,43–45), providing the structural basis for its promiscuous acyl-transferase activities. To further corroborate the p300 isobutyryltransferase activity observed in our experiments, we modeled IbuCoA in the crystal structure of p300 bound to n-butyryl-CoA (PDB ID: 5LKT). Clearly, p300 accommodates well isobutyryl-CoA in its acyl-CoA binding pocket, accounting for its observed Kibu activity (Figure 6D).
Isobutyrate globally affects the transcriptional profile of 293T cells
To systemically understand epigenetic changes affected by lysine isobutyrylation, we performed RNA-seq profiling on isobutyrate-treated 293T cells. Gene set enrichment analysis (GSEA) was used to compare the rank-ordered dataset of the transcripts from isobutyrate-treated cells versus untreated cells (control) with respect to the KEGG pathways, which offered molecular information on transcription alterations induced by isobutyrate treatment (Figure 7 and supplementary Figure S7). The results revealed that isobutyrate treatment caused upregulation of a number of genes associated with such important biological pathways as diabetes-related signaling, calcium signaling, hedgehog signaling, and JAK/STAT signaling. On the other hand, there were also a few genes downregulated by isobutyrate treatment, which included aminoacyl-tRNA biosynthesis, mRNA splicing, DNA replication and DNA repair.
Figure 7.

Enrichment analysis of the top gene sets altered by isobutyrate treatment in 293T cells. Significantly altered gene sets were defined with the False Discovery Rate (FDR) and P value less than 0.25 and 0.05, respectively. These genes were analyzed against the KEGG pathway for upregulation (red) and downregulation (blue).
DISCUSSION
Histone modifications are key players in the epigenetic regulation of chromatin dynamics and transcriptional programing. Various histone PTM marks in combination create epigenetic patterns that are associated with transcription regulatory changes (46). Lysine butyrylation was recognized as n-butyrylation upon its discovery (18). Later studies showed that butyrylation competes with acetylation on the same lysine residues and leads to different biological outcomes (15). Herein, we identified lysine isobutyrylation, the structural isomer of n-butyrylation, as a new PTM in histones. Although our mass spectrometry experiment only identified two isobutyrylated sites in histones, i.e. H3K18 and H3K23, the Western blot data showed that histone H4 was also isobutyrylated (Figure 4). In the future, it is needed to develop isobutyryllysine-specific antibody or chemical probes and apply them to more precisely map out lysine isobutyrylation sites in the chromatin histones as well as in the whole proteome. The integration of the chromatographic peak area showed that isobutyryl-CoA abundance in our cellular system was at the comparative level as its isomeric form butyryl-CoA (Figure 2), which provides the etiological basis that lysine isobutyrylation is driven by acyl-CoA. Both n-butyryl-CoA and isobutyryl-CoA are metabolites in mammalian cells but differ in their biosynthetic pathways. While n-butyryl-CoA is derived from fatty acid metabolism, isobutyryl-CoA comes from the valine metabolism (Figure 1). Dairy and ruminant foods are rich in branched chain fatty acids whose degradation provides another rich source of isobutyryl-CoA (47). Although butyryl-CoA mutase enzyme is known in prokaryotic organisms that interconverts n- and iso-butyryl-CoA (48), our blast search did not yield any possible orthologs existing in eukaryotic organisms. Indeed, treatment of 293T cells with d7-isobutyryate or valine only boosted isobutryl-CoA level, but not n-butyryl-CoA (Supplementary Figure S1), supporting that an isobutyryl-CoA mutase likely is not present in higher organisms.
The biochemical assays clearly showed that KAT members p300 and HAT1 catalyze lysine isobutyrylation activity in vitro. This is further substantiated by structural determination that both KATs are able to bind isobutyryl-CoA in their acetyl-CoA binding pocket. However only p300 showed cellular Kibu activity in our tested cell model. The discrepancy of HAT1 isobutyrylation activity in the biochemical assays from its lack of cellular activity could be a consequence of cellular context. HAT1 has been shown to form a protein complex with RIP1/3 (49), which may change the molecular environment of HAT1 and contribute to alterations in isobutyrylation substrate specificity in cells. It is necessary in the future to investigate the isobutyrylation activity of p300, HAT1 and other KATs (e.g. HBO1) in a broader scope and in different biological systems. These functional studies of new KAT activities can be more effectively executed by joint use of selective HAT small molecule inhibitors as chemical genetic tools (50,51).
RNA-seq profiling showed that sodium isobutyrate led to an extensive change in the expression levels of multiple genes in 293T cells. This exemplifies the intricate connection between metabolites and epigenetics: isobutyrate increases cellular levels of isobutyryl-CoA and thereby leads to isobutyrylation of nuclear histones and transcriptional changes. In terms of biophysical properties, lysine isobutyrylation may have similar effects as acetylation on chromatin structure and gene transcription: both acylations neutralize the positive charge on the lysine side chain and disrupt or weaken the electrostatic interaction between histones and DNA. As a result, the chromatin architecture becomes more loosely stacked and transcriptionally active gene sets are enhanced. At this point, it remains unknown whether any reader protein modules exist to recognize isobutyryllysine and distinguish it from other lysine acylation marks. Previously, a few human bromodomain proteins (i.e. BRD9 and CECR2 and the second bromodomain of TAF1) are reported to bind n-butyryllysine marks on histone peptides with affinities comparable to acetyllysine (52). It would be interesting to test if any of these and other acetyllysine binding domains interact with isobutyryllysine containing peptides. Given that isobutyryl group has a branched-chain structure, its binding property with reader modules may differ from those linear-chain acyl groups. New structural elucidation will be particularly valuable in understanding isobutyryl mark recognition by reader proteins. Moreover, isobutyryl-CoA may compete with acetyl-CoA for HAT active site binding, thereby affecting acetylation levels in HAT substrates. A few examples of acyl-CoAs in HAT acetyltransferase activity inhibition through competitive binding have been reported earlier (53,54). Another possibility is that isobutyrylation may compete for acetylation on the same lysine sites in histones, thus impeding bromodomain-dependent regulation of transcription. Indeed, our RNA-seq data showed that certain genes are downregulated upon isobutyrate treatment (Figure 7). The gene-repressive effect of isobutyrate, furthermore, could also be attributed by its pharmacological impact on cellular HDAC activities. Previous studies showed that n-butyrate, β-hydroxybutyrate and 4-phenylbutyrate all have inhibitory effects on the HDAC enzymes (55), which would positively influence gene expression by increasing the chromatin accessibility of transcriptional factors. In line with these studies, our initial biochemical results indeed showed that isobutyrate also acted as an inhibitor of HDACs, albeit with a weaker potency than n-butyrate (Supplementary Figure S8). Together, isobutyrate may contribute to gene transcription regulation via pleiotropic effects including nucleosomal histone isobutyrylation, isobutyryl-CoA competition for acetyl-CoA binding with KATs, and small molecule inhibition of HDAC activities, etc. In the future, it would be necessary to define gene expression changes directly mediated by histone isobutyrylation with ChIP-seq experiments. Our efforts are undergoing to develop selective anti-isobutyryllysine antibodies for further understanding of the biological functions of lysine isobutyrylation. Overall, this study calls for a comprehensive assessment of transcriptional and pharmacological effects of different short chain fatty acid molecules and their acyl-CoA metabolites in cells.
Acetate, propionate and n-butyrate, produced by human gut microbiota directly impact on host physiology (56). For instance, these short chain fatty acids exhibit anti-inflammatory and anti-proliferative properties in the gastrointestinal tract, which provides novel strategies to develop anti-inflammation or anti-tumor therapeutics (57). It will be intriguing to examine the percentage of isobutyrate molecules produced by luminal microbiota and how it affects the physiology of the intestinal epithelial cells. Also, protein acylations are influenced by cellular concentrations of fatty acy-CoAs which are greatly influenced by diet conditions (54,58,59). During valine catabolism, degradation of isobutyryl-CoA is catalyzed by isobutyryl-CoA dehydrogenase (IBD) encoded by the ACAD8 gene (60). Mutations in ACAD8 cause a rare inborn metabolic disorder, IBD deficiency, and lead to impaired isobutyryl-CoA breakdown and reduced energy production (60). Although most IBD deficiency patients are asymptomatic, some develop severe features of dilated cardiomyopathy, hypotonia and anemia (61). The range of symptoms associated with IBD deficiency remain unclear, and biomarkers for IBD deficiency are being investigated to allow efficient and effective diagnosis (62). It is important to examine cellular isobutyryl-CoA abundance and histone isobutyrylation levels in patients with IBD deficiency. We anticipate that isobutyryl-CoA level would increase and hence protein lysine isobutyrylation may be upregulated in those disease models. These are biological mysteries that warrant in-depth investigation in the future.
DATA AVAILABILITY
The HAT1-IbuCoA-H4(K12A) structure factors and coordinates have been deposited in the Protein Data Bank with the PDB ID: 6VO5.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Aiping Dong for reviewing and depositing the HAT1 structure. Structural results shown in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center (SBC) at the Advanced Photon Source. SBC-CAT is operated by UChicago Argonne, LLC, for the U.S. Department of Energy, Office of Biological and Environmental Research under contract DE-AC02-06CH11357. The Structural Genomics Consortium is a registered charity (no.: 1097737) that receives funds from AbbVie; Bayer Pharma AG; Boehringer Ingelheim; Canada Foundation for Innovation; Eshelman Institute for Innovation; Genome Canada through Ontario Genomics Institute [OGI-055]; Innovative Medicines Initiative (EU/EFPIA) [ULTRA-DD: 115766]; Janssen, Merck & Co.; Novartis Pharma AG; Ontario Ministry of Research Innovation and Science (MRIS); Pfizer, São Paulo Research Foundation-FAPESP, Takeda and the Wellcome Trust.
Contributor Information
Zhesi Zhu, Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of Georgia, Athens, GA 30602, USA.
Zhen Han, Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of Georgia, Athens, GA 30602, USA.
Levon Halabelian, Structural Genomics Consortium, University of Toronto, Toronto, ON M5G 1L7, Canada.
Xiangkun Yang, Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of Georgia, Athens, GA 30602, USA.
Jun Ding, Ben May Department for Cancer Research, The University of Chicago, Chicago, IL 60637, USA.
Nawei Zhang, Chemical Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA.
Liza Ngo, Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of Georgia, Athens, GA 30602, USA.
Jiabao Song, Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of Georgia, Athens, GA 30602, USA.
Hong Zeng, Structural Genomics Consortium, University of Toronto, Toronto, ON M5G 1L7, Canada.
Maomao He, Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of Georgia, Athens, GA 30602, USA.
Yingming Zhao, Ben May Department for Cancer Research, The University of Chicago, Chicago, IL 60637, USA.
Cheryl H Arrowsmith, Structural Genomics Consortium, University of Toronto, Toronto, ON M5G 1L7, Canada; Department of Medical Biophysics, University of Toronto, Toronto, ON M5G 1L7, Canada; Princess Margaret Cancer Centre, University Health Network, Toronto, ON M5G 2M9, Canada.
Minkui Luo, Chemical Biology Program, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA; Program of Pharmacology, Weill Cornell Medical College of Cornell University, New York, NY 20021, USA.
Michael G Bartlett, Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of Georgia, Athens, GA 30602, USA.
Y George Zheng, Department of Pharmaceutical and Biomedical Sciences, College of Pharmacy, University of Georgia, Athens, GA 30602, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Y.G.Z. acknowledges the support of the National Science Foundation [1507741, 1808087]; acknowledge the Proteomics and Mass Spectrometry facility (PAMS) at UGA for the peptide MS support; Y.Z. is supported by supported by the University of Chicago, Nancy and Leonard Florsheim family fund (to Y.Z.); NIH grants [R01GM115961, R01DK118266 to Y.Z.]; Y.Z. is a founder, board member, advisor to, and inventor on patents licensed to PTM Biolabs Inc. (Chicago, IL) and Maponos Therapeutics Inc. (Chicago, IL); M.L. is supported by NIH grants [R35GM131858, 5P30CA0087]; M.L. is a member of the Scientific Advisory Board for Epi One Inc.. Funding for open access charge: NSF.
Conflict of interest statement. None declared.
REFERENCES
- 1. Aebersold R., Agar J.N., Amster I.J., Baker M.S., Bertozzi C.R., Boja E.S., Costello C.E., Cravatt B.F., Fenselau C., Garcia B.A. et al.. How many human proteoforms are there. Nat. Chem. Biol. 2018; 14:206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sabari B.R., Tang Z., Huang H., Yong-Gonzalez V., Molina H., Kong H.E., Dai L., Shimada M., Cross J.R., Zhao Y. et al.. Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell. 2015; 58:203–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Verdin E., Ott M.. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015; 16:258–264. [DOI] [PubMed] [Google Scholar]
- 4. Barnes C.E., English D.M., Cowley S.M.. Acetylation & Co: an expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem. 2019; 63:97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hirschey M.D., Zhao Y.. Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol. Cell. Proteomics. 2015; 14:2308–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sabari B.R., Zhang D., Allis C.D., Zhao Y.. Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 2017; 18:90–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lin H., Su X., He B.. Protein lysine acylation and cysteine succination by intermediates of energy metabolism. ACS Chem. Biol. 2012; 7:947–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bos J., Muir T.W.. A chemical probe for protein crotonylation. J. Am. Chem. Soc. 2018; 140:4757–4760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. He M., Han Z., Liu L., Zheng Y.G.. Chemical biology approaches for investigating the functions of lysine acetyltransferases. Angew. Chem. Int. Ed. 2018; 57:1162–1184. [DOI] [PubMed] [Google Scholar]
- 10. Marmorstein R., Zhou M.M.. Writers and readers of histone acetylation: structure, mechanism, and inhibition. Cold Spring Harb. Perspect. Biol. 2014; 6:a018762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bheda P., Jing H., Wolberger C., Lin H.. The substrate specificity of sirtuins. Annu. Rev. Biochem. 2016; 85:405–429. [DOI] [PubMed] [Google Scholar]
- 12. Schneider A., Chatterjee S., Bousiges O., Selvi B.R., Swaminathan A., Cassel R., Blanc F., Kundu T.K., Boutillier A.L.. Acetyltransferases (HATs) as targets for neurological therapeutics. Neurotherapeutics. 2013; 10:568–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Di Cerbo V., Schneider R.. Cancers with wrong HATs: the impact of acetylation. Brief. Funct. Genomics. 2013; 12:231–243. [DOI] [PubMed] [Google Scholar]
- 14. Mohammad H.P., Barbash O., Creasy C.L.. Targeting epigenetic modifications in cancer therapy: erasing the roadmap to cancer. Nat. Med. 2019; 25:403–418. [DOI] [PubMed] [Google Scholar]
- 15. Goudarzi A., Zhang D., Huang H., Barral S., Kwon O.K., Qi S., Tang Z., Buchou T., Vitte A.L., He T. et al.. Dynamic competing histone H4 K5K8 acetylation and butyrylation are hallmarks of highly active gene promoters. Mol. Cell. 2016; 62:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wu Z., Cheng Z., Sun M., Wan X., Liu P., He T., Tan M., Zhao Y.. A chemical proteomics approach for global analysis of lysine monomethylome profiling. Mol. Cell. Proteomics. 2015; 14:329–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang X., Sidoli S., Garcia B.A.. Zheng Y.G. Application of mass spectrometry in translational epigenetics. Epigenetic Technological Applications. 2015; San Diego: Academic Press; 55–78. [Google Scholar]
- 18. Chen Y., Sprung R., Tang Y., Ball H., Sangras B., Kim S.C., Falck J.R., Peng J., Gu W., Zhao Y.. Lysine propionylation and butyrylation are novel post-translational modifications in histones. Mol. Cell. Proteomics. 2007; 6:812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kaczmarska Z., Ortega E., Goudarzi A., Huang H., Kim S., Marquez J.A., Zhao Y., Khochbin S., Panne D.. Structure of p300 in complex with acyl-CoA variants. Nat. Chem. Biol. 2017; 13:21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Robinson W.G., Nagle R., Bachhawat B.K., Kupiecki F.P., Coon M.J.. Coenzyme-a thiol esters of isobutyric, methacrylic, and beta-hydroxyisobutyric acids as intermediates in the enzymatic degradation of valine. J.Biol. Chem. 1957; 224:1–11. [PubMed] [Google Scholar]
- 21. Yun J.W., Jo K.I., Woo H.I., Lee S.Y., Ki C.S., Kim J.W., Song J., Lee D.H., Lee Y.W., Park H.D.. A novel ACAD8 mutation in asymptomatic patients with isobutyryl-CoA dehydrogenase deficiency and a review of the ACAD8 mutation spectrum. Clin. Genet. 2015; 87:196–198. [DOI] [PubMed] [Google Scholar]
- 22. Santra S., Macdonald A., Preece M.A., Olsen R.K., Andresen B.S.. Long-term outcome of isobutyryl-CoA dehydrogenase deficiency diagnosed following an episode of ketotic hypoglycaemia. Mol. Genet. Metab. Rep. 2017; 10:28–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Thompson P.R., Wang D., Wang L., Fulco M., Pediconi N., Zhang D., An W., Ge Q., Roeder R.G., Wong J. et al.. Regulation of the p300 HAT domain via a novel activation loop. Nat. Struct. Mol. Biol. 2004; 11:308–315. [DOI] [PubMed] [Google Scholar]
- 24. Wu H., Moshkina N., Min J., Zeng H., Joshua J., Zhou M.M., Plotnikov A.N.. Structural basis for substrate specificity and catalysis of human histone acetyltransferase 1. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:8925–8930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gao T., Yang C., Zheng Y.G.. Comparative studies of thiol-sensitive fluorogenic probes for HAT assays. Anal. Bioanal. Chem. 2013; 405:1361–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kabsch W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Winn M.D., Ballard C.C., Cowtan K.D., Dodson E.J., Emsley P., Evans P.R., Keegan R.M., Krissinel E.B., Leslie A.G., McCoy A. et al.. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011; 67:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Evans P.R., Murshudov G.N.. How good are my data and what is the resolution. Acta Crystallogr. D Biol. Crystallogr. 2013; 69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Williams C.J., Headd J.J., Moriarty N.W., Prisant M.G., Videau L.L., Deis L.N., Verma V., Keedy D.A., Hintze B.J., Chen V.B. et al.. MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 2018; 27:293–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan M., Luo H., Lee S., Jin F., Yang J.S., Montellier E., Buchou T., Cheng Z., Rousseaux S., Rajagopal N. et al.. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011; 146:1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Grevengoed T.J., Klett E.L., Coleman R.A.. Acyl-CoA metabolism and partitioning. Annu. Rev. Nutr. 2014; 34:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Abdinejad A., Fisher A.M., Kumar S.. Production and utilization of butyryl-CoA by fatty acid synthetase from mammalian tissues. Arch. Biochem. Biophys. 1981; 208:135–145. [DOI] [PubMed] [Google Scholar]
- 34. Webster L.T. Jr, Gerowin L.D., Rakita L.. Purification and characteristics of a butyryl coenzyme a synthetase from bovine heart mitochondria. J. Biol. Chem. 1965; 240:29–33. [PubMed] [Google Scholar]
- 35. Roe C.R., Cederbaum S.D., Roe D.S., Mardach R., Galindo A., Sweetman L.. Isolated isobutyryl-CoA dehydrogenase deficiency: an unrecognized defect in human valine metabolism. Mol. Genet. Metab. 1998; 65:264–271. [DOI] [PubMed] [Google Scholar]
- 36. Han Z., Wu H., Kim S., Yang X., Li Q., Huang H., Cai H., Bartlett M.G., Dong A., Zeng H. et al.. Revealing the protein propionylation activity of the histone acetyltransferase MOF (males absent on the first). J. Biol. Chem. 2018; 293:3410–3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang C., Mi J., Feng Y., Ngo L., Gao T., Yan L., Zheng Y.G.. Labeling lysine acetyltransferase substrates with engineered enzymes and functionalized cofactor surrogates. J. Am. Chem. Soc. 2013; 135:7791–7794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yang Y.Y., Ascano J.M., Hang H.C.. Bioorthogonal chemical reporters for monitoring protein acetylation. J. Am. Chem. Soc. 2010; 132:3640–3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pougovkina O., te Brinke H., Wanders R.J.A., Houten S.M., de Boer V.C.J.. Aberrant protein acylation is a common observation in inborn errors of acyl-CoA metabolism. J. Inherit. Metab. Dis. 2014; 37:709–714. [DOI] [PubMed] [Google Scholar]
- 40. Xu G., Wang J., Wu Z., Qian L., Dai L., Wan X., Tan M., Zhao Y., Wu Y.. SAHA regulates histone acetylation, butyrylation, and protein expression in neuroblastoma. J. Proteome Res. 2014; 13:4211–4219. [DOI] [PubMed] [Google Scholar]
- 41. Henry R.A., Kuo Y.M., Bhattacharjee V., Yen T.J., Andrews A.J.. Changing the selectivity of p300 by acetyl-CoA modulation of histone acetylation. ACS Chem. Biol. 2015; 10:146–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dancy B.M., Cole P.A.. Protein lysine acetylation by p300/CBP. Chem. Rev. 2015; 115:2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu X., Wang L., Zhao K., Thompson P.R., Hwang Y., Marmorstein R., Cole P.A.. The structural basis of protein acetylation by the p300/CBP transcriptional coactivator. Nature. 2008; 451:846–850. [DOI] [PubMed] [Google Scholar]
- 44. Delvecchio M., Gaucher J., Aguilar-Gurrieri C., Ortega E., Panne D.. Structure of the p300 catalytic core and implications for chromatin targeting and HAT regulation. Nat. Struct. Mol. Biol. 2013; 20:1040–1046. [DOI] [PubMed] [Google Scholar]
- 45. Maksimoska J., Segura-Pena D., Cole P.A., Marmorstein R.. Structure of the p300 histone acetyltransferase bound to acetyl-coenzyme A and its analogues. Biochemistry. 2014; 53:3415–3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Papait R., Cattaneo P., Kunderfranco P., Greco C., Carullo P., Guffanti A., Viganò V., Stirparo G.G., Latronico M.V.G., Hasenfuss G. et al.. Genome-wide analysis of histone marks identifying an epigenetic signature of promoters and enhancers underlying cardiac hypertrophy. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:20164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ran-Ressler R.R., Bae S., Lawrence P., Wang D.H., Brenna J.T.. Branched-chain fatty acid content of foods and estimated intake in the USA. Br. J. Nutr. 2014; 112:565–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Jost M., Born D.A., Cracan V., Banerjee R., Drennan C.L.. Structural basis for substrate specificity in adenosylcobalamin-dependent isobutyryl-CoA mutase and related Acyl-CoA mutases. J. Biol. Chem. 2015; 290:26882–26898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carafa V., Nebbioso A., Cuomo F., Rotili D., Cobellis G., Bontempo P., Baldi A., Spugnini E.P., Citro G., Chambery A. et al.. RIP1-HAT1-SIRT complex identification and targeting in treatment and prevention of cancer. Clin. Cancer Res. 2018; 24:2886–2900. [DOI] [PubMed] [Google Scholar]
- 50. Lasko L.M., Jakob C.G., Edalji R.P., Qiu W., Montgomery D., Digiammarino E.L., Hansen T.M., Risi R.M., Frey R., Manaves V. et al.. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature. 2017; 550:128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bowers E.M., Yan G., Mukherjee C., Orry A., Wang L., Holbert M.A., Crump N.T., Hazzalin C.A., Liszczak G., Yuan H. et al.. Virtual ligand screening of the p300/CBP histone acetyltransferase: identification of a selective small molecule inhibitor. Chem. Biol. 2010; 17:471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Flynn E.M., Huang O.W., Poy F., Oppikofer M., Bellon S.F., Tang Y., Cochran A.G.. A Subset of human bromodomains recognizes butyryllysine and crotonyllysine histone peptide modifications. Structure. 2015; 23:1801–1814. [DOI] [PubMed] [Google Scholar]
- 53. Montgomery D.C., Sorum A.W., Guasch L., Nicklaus M.C., Meier J.L.. Metabolic regulation of histone acetyltransferases by endogenous Acyl-CoA cofactors. Chem. Biol. 2015; 22:1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Carrer A., Parris J.L., Trefely S., Henry R.A., Montgomery D.C., Torres A., Viola J.M., Kuo Y.M., Blair I.A., Meier J.L. et al.. Impact of a High-fat diet on tissue Acyl-CoA and histone acetylation levels. J. Biol. Chem. 2017; 292:3312–3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Chriett S., Dąbek A., Wojtala M., Vidal H., Balcerczyk A., Pirola L.. Prominent action of butyrate over β-hydroxybutyrate as histone deacetylase inhibitor, transcriptional modulator and anti-inflammatory molecule. Sci. Rep. 2019; 9:742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Louis P., Hold G.L., Flint H.J.. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014; 12:661–672. [DOI] [PubMed] [Google Scholar]
- 57. Parada Venegas D., De la Fuente M.K., Landskron G., Gonzalez M.J., Quera R., Dijkstra G., Harmsen H.J.M., Faber K.N., Hermoso M.A.. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front Immunol. 2019; 10:277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Trefely S., Lovell C.D., Snyder N.W., Wellen K.E.. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab. 2020; 38:100941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lee J.V., Carrer A., Shah S., Snyder N.W., Wei S., Venneti S., Worth A.J., Yuan Z.F., Lim H.W., Liu S. et al.. Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab. 2014; 20:306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nguyen T.V., Andresen B.S., Corydon T.J., Ghisla S., Abd-El Razik N., Mohsen A.-W.A., Cederbaum S.D., Roe D.S., Roe C.R., Lench N.J. et al.. Identification of isobutyryl-CoA dehydrogenase and its deficiency in humans. Mol. Genet. Metab. 2002; 77:68–79. [DOI] [PubMed] [Google Scholar]
- 61. Santra S., Macdonald A., Preece M.A., Olsen R.K., Andresen B.S.. Long-term outcome of isobutyryl-CoA dehydrogenase deficiency diagnosed following an episode of ketotic hypoglycaemia. Mol. Genet. Metab. Rep. 2017; 10:28–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Oglesbee D., He M., Majumder N., Vockley J., Ahmad A., Angle B., Burton B., Charrow J., Ensenauer R., Ficicioglu C.H. et al.. Development of a newborn screening follow-up algorithm for the diagnosis of isobutyryl-CoA dehydrogenase deficiency. Genet. Med. 2007; 9:108–116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The HAT1-IbuCoA-H4(K12A) structure factors and coordinates have been deposited in the Protein Data Bank with the PDB ID: 6VO5.