


Abstract
Nucleic acid therapeutics (NATs) have proven useful in promoting the degradation of specific transcripts, modifying gene expression, and regulating mRNA splicing. In each situation, efficient delivery of nucleic acids to cells, tissues and intracellular compartments is crucial—both for optimizing efficacy and reducing side effects. Despite successes in NATs, our understanding of their cellular uptake and distribution in tissues is limited. Current methods have yielded insights into distribution of NATs within cells and tissues, but the sensitivity and resolution of these approaches are limited. Here, we show that nanoscale secondary ion mass spectrometry (NanoSIMS) imaging can be used to define the distribution of 5-bromo-2′-deoxythymidine (5-BrdT) modified antisense oligonucleotides (ASO) in cells and tissues with high sensitivity and spatial resolution. This approach makes it possible to define ASO uptake and distribution in different subcellular compartments and to quantify the impact of targeting ligands designed to promote ASO uptake by cells. Our studies showed that phosphorothioate ASOs are associated with filopodia and the inner nuclear membrane in cultured cells, and also revealed substantial cellular and subcellular heterogeneity of ASO uptake in mouse tissues. NanoSIMS imaging represents a significant advance in visualizing uptake and distribution of NATs; this approach will be useful in optimizing efficacy and delivery of NATs for treating human disease.
INTRODUCTION
Nucleic acid–based therapeutics (NATs) represent an important platform for drug discovery within the pharmaceutical industry. Multiple classes of NATs, including antisense oligonucleotides (ASOs), small interfering RNA (siRNA), mRNA and CRISPR-based therapeutics are being investigated for the treatment of human disease, ranging from cardiovascular diseases that affect millions of patients to diseases that affect a single individual (1–4). The idea that NATs could be useful therapeutic agents was attractive because they could be designed against RNA targets by applying Watson–Crick base-pairing rules. Over the past four decades, documenting efficacy of ASOs and siRNA in altering gene expression has become straightforward, but the creation of useful drugs has been more difficult, largely due to challenges in delivering nucleic acids to specific cells and tissues (5,6). Fortunately, advances in NAT design, chemistry, and delivery strategies have risen to these challenges, making NATs a growth industry (7).
Despite steady progress in NATs, our understanding of their uptake and distribution within cells and tissues remains limited. Studies with radiolabeled oligonucleotides and immunohistochemical studies with a phosphorothioate ASO–specific antibody have yielded insights into the ASO pharmacokinetics (8–10), but insights into ASO distribution in different cell types and subcellular compartments is typically lost. Fluorescence microscopy studies involving ASOs and siRNAs conjugated to fluorescent tags have made it possible to visualize ASOs (11–13), but those studies also have limitations. First, modifying NATs with fluorescent dyes (e.g. Cy3) can alter their pharmacokinetic properties, such that they may not provide a faithful representation of their cell-uptake properties (14,15). Second, the resolution and sensitivity of confocal microscopy is limited. For example, capillary endothelial cells—which are a potential barrier to NAT transport—are as thin as ∼70 nm, making those cells very difficult to image by confocal microscopy. Even with advances in super-resolution microscopy, it is difficult to resolve the basolateral and luminal plasma membranes of endothelial cells or to visualize the organelles of those cells. Third, most confocal fluorescence microscopy studies have involved the delivery of NATs to cells with transfection reagents rather than by ‘free uptake’ (16). Studies with transfected cells facilitate the detection of fluorescently labeled NATs within cells, but they are not relevant to NAT uptake in animals and may not recapitulate the cellular processes involved in NAT uptake.
Recently, nanoscale secondary ion mass spectrometry (NanoSIMS) imaging has been useful for generating high-resolution images of drugs and metabolites within cells (17–20). The NanoSIMS instrument bombards cells (or tissue sections) with a focused primary 133Cs+ beam, resulting in the release of millions of negatively-charged secondary ions (e.g. 1H–, 12C14N–, 16O–, 32S–, 79Br–) that are sized by mass spectrometry and quantified (21). Using the secondary ion data, it is possible to create images of cells and tissues based solely on distribution of a single isotope—or a ratio of different isotopes (e.g. the ratio of 32S– ions to 34S– ions). The NanoSIMS instrument is extremely sensitive, making it possible to generate images of cells and tissues based solely on the content of rare stable isotopes (e.g. 2H, with a natural abundance of 0.015%; 13C, with a natural abundance of 1.1%; 34S, with a natural abundance of 4.25%) (19,22,23). Thus, after administering a drug (or metabolite) containing a stable isotope, NanoSIMS imaging can examine the distribution of the stable isotope and define localized regions of stable isotope enrichment (24). NanoSIMS imaging is also useful for visualizing elements that are absent or are present only at low levels in cells and tissues (e.g. lanthanide metals, platinum, bromine, gold) (20,25–27). When drugs contain those elements, the background in NanoSIMS images is low. Specifically, bromine labeling has been utilized successfully for nucleotide imaging by SIMS techniques (28,29). A low background allows detection of isotopes and elements with a high dynamic range, making it possible to analyze features with different levels of isotope enrichment and to analyze both rare isotopes and abundant isotopes at the same time (30,31).
NanoSIMS imaging can provide images with up to 40-nm lateral resolution, greater than what is typically provided by confocal microscopy but lower than transmission electron microscopy or backscattered electron (BSE) imaging with a scanning electron microscope. Additionally, we have demonstrated by performing BSE imaging and NanoSIMS imaging sequentially on the very same section, it is possible to correlate the chemical information of NanoSIMS imaging (isotopic content of cells and subcellular structures) with the greater morphological detail provided by BSE images (19,20,32,33).
In the current study, we explored the utility of NanoSIMS imaging, alone or in combination with correlative BSE imaging, for defining the distribution of ASOs in cells and tissues. We labeled ASOs with both bromine and 34S and analyzed the utility of both labeling strategies for generating informative NanoSIMS images. In combination with BSE imaging, we used bromine-labeled ASOs to assess the distribution of ASOs in cellular and subcellular compartments in liver, kidney, and heart of wild-type mice. Our studies show that NanoSIMS imaging reveals, with both high sensitivity and high spatial resolution, the distribution of ASOs in both cultured cells and in the tissues of mice.
MATERIALS AND METHODS
Mouse strains and cell lines
Wild-type mice were purchased from The Jackson Laboratory (strain C57BL/6) and they were housed in a barrier facility at UCLA. HeLa, wild-type HEK293, and 3T3-L1 cell lines were purchased from ATCC, and were grown in Eagle's Minimum Essential Medium (EMEM; ATCC) containing 10% fetal bovine serum at 37°C with 5% CO2. Asialoglycoprotein receptor 1 (AGSR1)–expressing HEK293 (1A4) cells were created and characterized by Tanowitz et al. (34). 1A4 cells were grown in Eagle's Minimum Essential Medium (EMEM; ATCC) containing 10% FBS at 37°C with 5% CO2.
Antisense oligonucleotide (ASO) synthesis and labeling
The parental ASO and bromine-labeled ASO, (targeting Malat1) were synthesized on an ABI 394 DNA/RNA synthesizer (2 μmol scale) or an AKTA oligopilot 10 (20 μmol scale) with polystyrene-based Primer Support 5G UnyLinker or NittoPhase UnyLinker Solid Support (300–400 μmol/g). GalNAc-conjugated bromine-labeled ASOs (GalNAc-Br-ASOs) and the GalNAc-conjugated ASO (GalNAc-ASO) were prepared using GalNAc-loaded Primer Support 5G which was prepared as described (35). The fully protected nucleoside phosphoramidites were purchased from commercial sources (Glen Research) and incorporated using standard solid-phase ASO synthesis conditions (i.e. 3% dichloroacetic acid in dichloromethane for deblocking, 1 M 4,5-dicyanoimidazole and 0.1 M N-methylimidazole in acetonitrile (MeCN) as activator, 10% acetic anhydride in tetrahydrofuran (THF) and 10% N-methylimidazole in THF/pyridine for capping, and 0.1 M xanthane hydride in pyridine:MeCN 3:2 (v:v) for thiolation.
In the case of 34S labeling on the backbone of ASOs, 34S-labeled phenylacetyl disulfide (34S-PADS) was used as the sulfurizing reagent (36). DNA amidites were dissolved at 0.1 M MeCN:toluene 1:1 (v:v) and incorporated with 6-min coupling times. After the conclusion of the synthesis, the protecting groups were removed, and the ASOs were cleaved from the resin by treating with 15% diethylamine in toluene (v:v) for 20 min, followed by suspending the solid support in aqueous concentrated ammonia at room temperature for 48 h. The support was then removed by filtration and the crude mixture purified by high-performance liquid chromatography on a strong anion-exchange column (source Q support) using buffer A [50 mM NaOH (aq)] and buffer B [1.5 M NaCl 50 mM NaOH (aq)]. ASOs were desalted with C18 reverse-phase chromatography and lyophilized to a solid. A fluorescent ASO (Cy3-ASO) targeting Malat1 was purchased from Integrated DNA Technologies (IDT).
Antisense activities of oligonucleotides
3T3-L1 cells were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) and 10% fetal bovine serum (FBS) in a 37°C incubator with 5% CO2. One day before ASO transfection, the cells were seeded in 96-well plates (8000 cells/well). On the following day, ASOs (parental ASO, 34S-ASO and Br-ASO) were diluted in Opti-MEM reduced serum media (Gibco) and combined with an equal volume of Opti-MEM containing 12 μl/ml RNAiMax (Thermo Fisher Scientific). Complexes were incubated for 20 min then applied to cells at 100 μl/well for 8 h. Total RNA from cells grown in 96-well plates was prepared with an RNeasy mini kit (Qiagen). qRT-PCR was performed with TaqMan primer probe sets (Thermo Fisher Scientific). Briefly, ∼50 ng of total RNA in 5 μl water was mixed with 0.3 μl primer probe sets containing forward and reverse primers (10 μM each) and fluorescently labeled probe (3 μM), 0.3 μl RT enzyme mix (Qiagen), 4.4 μl RNase-free water and 10 μl of 2× PCR reaction buffer (20-μl reaction volume). Reverse transcription was performed at 48°C for 10 min; 40 PCR cycles were performed (94°C for 20 s and 60°C for 20 s) with the StepOne Plus RT-PCR system (Applied Biosystems). The transcript levels were normalized to the amount of total RNA in each reaction, determined in triplicate by the Ribogreen assay (Life Technologies). To measure receptor-mediated ASO gapmer activity by free uptake, ASGR1-expressing HEK293 (1A4) cells were seeded into 96-well plates (10 000 cells/well) and allowed to attach for at least 16 h. ASOs (parental ASO, GalNAc-ASO and GalNAc-Br-ASO) were diluted into complete growth media over a 12-point 3-fold dilution series at 10× final concentration. Diluted ASOs were then applied to triplicate treatment wells at 0.1× final volume and plates were returned to the incubator for 20–24 h. Total RNA was prepared using an RNeasy mini kit and analyzed for transcript levels as described above.
Exposure of ASOs to cells for imaging
Hela cells were cultured in DMEM containing 10% FBS. For studies examining the uptake of ASOs in the absence of transfection reagents (free uptake), cells were plated at 30% confluency on glass coverslips (for fluorescence studies) or Thermanox plastic cell culture coverslips (Thermo Fisher Scientific; for electron microscopy and NanoSIMS). Coverslips were placed in 24-well plates, and the cells were allowed to grow for 16 h. Next, cells were incubated in fresh medium containing 5 μM ASOs for 24 h (Cy3-ASO for fluorescence studies and 34S-ASO or Br-ASO for NanoSIMS analyses). For ASO transfection studies, cells were grown to 80% confluency and then treated with 100 μl Opti-MEM containing the ASOs and 1 μl lipofectamine (Lipofectamine LTX reagent, Thermo Fisher Scientific) in 500 μl of medium for 4 h at 37°C (final ASO concentration, 50 nM). The cells were washed three times with warm PBS/Ca2+/Mg2+ (3 min each).
Wild-type HEK293 and 1A4 cells were grown in Eagle's minimum essential medium (EMEM; ATCC) containing 10% FBS. Cells were plated at 50% confluency on Thermanox plastic cell culture coverslips (Thermo Fisher Scientific) and grown for 16 h. Cells were incubated with or without 150 nM Br-ASO or GalNAc-Br-ASO in growth medium for 24 h at 37°C. Cells were washed three times with warm PBS/Ca2+/Mg2+ (3 min each).
Visualization of a Cy3-ASO by fluorescence microscopy
For studies investigating the ‘free uptake’ of Cy3-ASO by HeLa cells, the cells were incubated in growth media with 5 μM ASOs for 24 h at 37°C as described earlier. The cells were fixed with 4% paraformaldehyde (PFA) for 15 min at room temperature. Cells were stained with DAPI for 5 min and rinse three times with PBS/Ca2+/Mg2+ before they were mounted. In some experiments, we examined the distribution of both Cy3-ASO and transferrin. Following the 24-h-incubation with Cy3-ASO, the cells were placed on ice for 10 min and then incubated for 20 min at 37 °C with 25 μg/ml of Alexa 488-transferrin (Thermo Fisher Scientific) in PBS/Ca2+/Mg2+ containing 0.2% bovine serum albumin (BSA). Cells were rinsed three times with PBS/Ca2+/Mg2+ containing 0.2% BSA and then fixed with 4% PFA for 10 min at room temperature. Cells were stained with DAPI for 5 min, rinsed three times with PBS/Ca2+/Mg2+, and then mounted. Images were recorded with an LSM880 microscope (Zeiss) and processed with Zen 2.3 software system (Zeiss).
Administration of ASOs to mice
A 6-week-old wild-type male mouse was injected subcutaneously with Br-ASO (32 mg/kg) in 1 ml saline solution (filtered with a 0.22-μm filter). As a control, a 6-week-old wild-type male mouse was injected subcutaneously with 1 ml of the saline solution without an ASO. After 72 h, the mice were euthanized and perfused with 10 ml of PBS, followed by perfusion with 10 ml of 3.7% paraformaldehyde (PFA; Electron Microscopy Sciences) in PBS. Heart, liver, and kidney were harvested and prepared for BSE and NanoSIMS imaging.
Preparation of cultured cells for BSE and NanoSIMS imaging
Cells that had been treated with Br-ASO (as described earlier) were processed with a protocol modified from the serial blockface scanning electron microscopy protocol from National Center for Microscopy and Imaging Research (37). First, the cells were washed once with warm fixative solution containing 2.5% glutaraldehyde in 0.1 M sodium cacodylate and then incubated in a fresh fixative solution for 10 min on the bench followed by 1 h on ice. After fixation, the cells were washed 5 times (3 min each) with cold 0.1 M sodium cacodylate and then post-fixed with 2% osmium tetroxide in 0.1 M sodium cacodylate for 1 h on ice. Next, cells were washed five times (3 min each) with cold distilled water, followed by a 20-min incubation in freshly prepared 1% thiocarbohydrazide solution at room temperature. The coverslips were transferred to a clean well and washed five times (3 min each) with distilled water at room temperature. The coverslips were then incubated for 30 min in 2% osmium tetroxide at room temperature, followed by washing with cold distilled water (five times, 3 min each). Next, the coverslips were incubated with 2% aqueous uranyl acetate overnight at 4°C. On the next day, the cells were washed five times for 3 min each with cold distilled water and dehydrated with increasing amounts of ethanol (30%, 50%, 70%, 85%, 95%, 100%) for 2 min each, followed by two 2-min incubations with 100% ethanol. Next, the samples were infiltrated with Embed812 resin (Electron Microscopy Sciences) by incubating samples in 50% resin (diluted in anhydrous acetone) for 1 h, 66% resin overnight, and 100% resin for 2 h. The backside of coverslips were wiped of excess resin; the coverslips were then inverted (cell side down) onto individual BEEM capsules (Electron Microscopy Sciences) which had been filled with resin. Capsules with coverslips were polymerized in a vacuum oven for 48 h at 65°C. To prepare samples for sectioning, the BEEM capsules were cut away from the polymerized resin with a razor blade. Next, the resin block, with the coverslip still attached, was loaded into a sample chuck and inverted (coverslip down) onto a 65°C hotplate for 30 sec, making it possible to remove the coverslip from the block. In this way, the cells were transferred from the coverslip onto the resin block. The block face was trimmed, and 500-nm sections were cut with a Leica UC6 ultramicrotome with a Diatome diamond knife. Cells that had been incubated with 34S-ASO were processed in a similar fashion, but without the thiocarbohydrazide staining step and the second osmium tetroxide staining step (thereby avoiding 34S background from thiocarbohydrazide staining).
Preparation of mouse tissues for electron microscopy and NanoSIMS analysis
Tissues were also processed with a protocol modified from the serial blockface scanning electron microscopy protocol from National Center for Microscopy and Imaging Research (37). After tissues were excised, they were fixed at 4°C overnight in a solution containing 4% paraformaldehyde, 2.5% glutaraldehyde, and 2.1% sucrose in 0.1 M sodium cacodylate. On the next day, the samples were rinsed 5 times for 3 min each with 0.1 M sodium cacodylate and then incubated in a solution containing 2% osmium tetroxide and 1.5% potassium ferricyanide in 0.1 M sodium cacodylate for 1 h at 4°C. Next, samples were rinsed five times for 3 min each with distilled water and incubated with 1% thiocarbohydrazide for 20 min at room temperature. Samples were rinsed five times for 3 min each with distilled water and then incubated with 2% aqueous osmium tetroxide for 30 min at room temperature. The samples were then rinsed five times for 3 min each with distilled water and incubated with 2% uranyl acetate at 4°C overnight. On the following day, the samples were rinsed with distilled water and dehydrated with a series of increasing amounts of ethanol (30%, 50%, 70%, 85%, 95%, 100%) for 10 min each followed by two additional 10-min incubations with 100% ethanol. Next, the samples were infiltrated with Embed812 resin (Electron Microscopy Sciences) by incubating samples in 33% resin (diluted in anhydrous acetone) for 2 h, 66% resin overnight, and 100% resin for 4 h. Samples were embedded in fresh resin with polypropylene molds (Electron Microscopy Sciences) and polymerized in a vacuum oven for 48 h at 65°C. After polymerization, samples were removed from molds; block faces were trimmed; and 500-nm sections were cut with a Leica UC6 ultramicrotome equipped with a Diatome diamond knife.
Scanning electron microscopy
500-nm sections of resin-embedded cells and mouse tissues were mounted onto 1-mm2 silicon wafers. Backscattered electron (BSE) images were obtained with an FEI Verios scanning electron microscope (Thermo Fisher Scientific).
NanoSIMS analyses
Sections were coated with 5 nm of gold and analyzed with a NanoSIMS instrument (NanoSIMS 50 or NanoSIMS 50L) (CAMECA, France). Samples were scanned with a focused 16-keV 133Cs+ beam, and secondary ions (12C– or 13C–,12C14N–, 31P–, 32S–, 79Br– and/or 81Br–) were collected.
For visualizing Br-ASO in Figures 2–4, Supplementary Figures S3–S4, and S7–S11, 30 × 30-μm regions of selected cells were scanned with a primary ion beam current of ∼1 nA (primary aperture D1 = 1) to reach a dose of ∼1 × 1017 ions/cm2 (‘presputtering,’ implanting 133Cs+ and ensuring a steady state of secondary ion release). Regions of 20 × 20-μm or 25 × 25-μm were imaged with an 8-pA beam current (primary aperture D1 = 2) and a total dwell time of 60 ms/pixel. 256 × 256-pixel images were obtained. Some cells were imaged by BSE before NanoSIMS imaging. Very small differences in organelle morphology can be detected in the BSE and NanoSIMS images as a result of presputtering with the 133Cs+ beam (which removes a small amount of the surface of the section), but in general the correlation between BSE and NanoSIMS images is extremely high.
Figure 2.
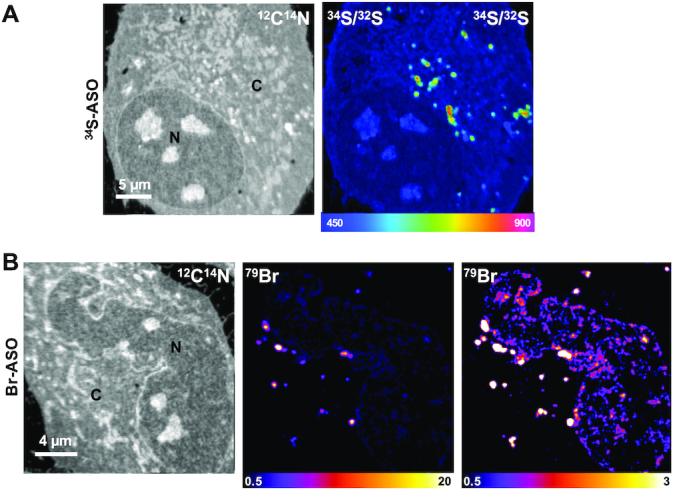
NanoSIMS imaging of 34S-labeled and Br-labeled ASOs in HeLa cells. HeLa cells were incubated in medium containing (A) 34S-ASO or (B) Br-ASO for 24 h and then processed for NanoSIMS analyses. 12C14N images were useful for defining cell morphology; 34S/32S and 79Br NanoSIMS images show distributions of 34S-ASO and Br-ASO, respectively. Two 79Br images, with different scales, are shown to depict the relative distributions of the Br-ASO in the cytoplasm and nucleus. The 34S/32S ratio scale is multiplied by 10 000. N, nucleus; C, cytoplasm.
Figure 3.
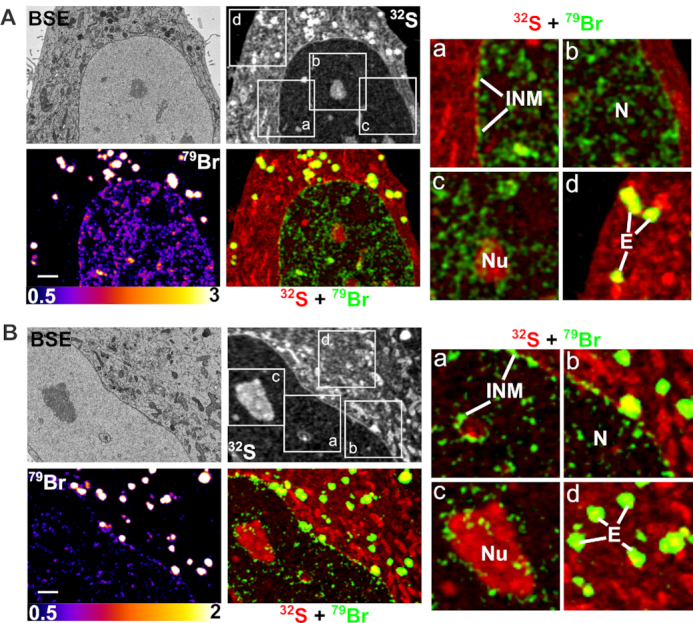
Correlative backscattered electron (BSE) and NanoSIMS imaging of Br-ASO taken up into cells by ‘free uptake.’ HeLa cells were incubated in medium containing 5 μM Br-ASO for 24 h and then processed for BSE/NanoSIMS analyses. BSE and 32S NanoSIMS images are useful for cell morphology; 79Br and composite 32S (red) + 79Br (green) NanoSIMS images were generated to show distributions of Br-ASO within cells. Two cells (panels A and B) are shown. In each panel, boxed regions (a, b, c, d) are shown on the right in higher-magnification 32S (red) + 79Br (green) composite images; these images reveal the Br-ASO along the inner nuclear membrane (INM) and in the nucleoplasm (N), nucleolus (Nu) and endosomes (E). Scale bars, 2 μm.
Figure 4.
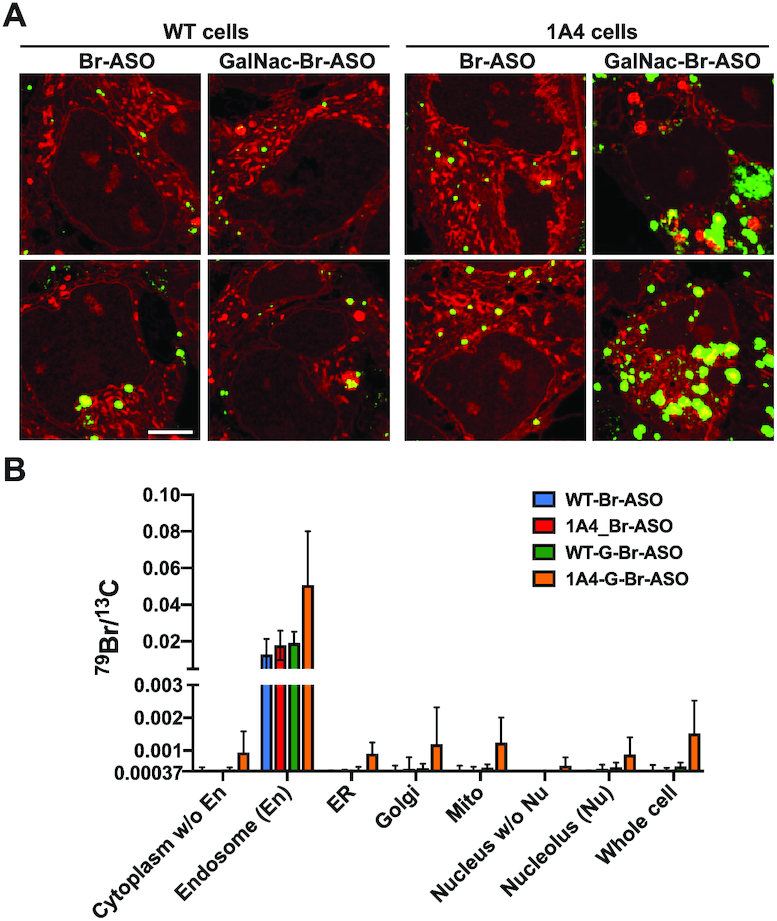
Examining the impact of a targeted ligand (GalNAc) and expressing a receptor for GalNAc (asialoglycoprotein receptor 1 (AGSR1) on ASO uptake and subcellular distribution of ASOs. Wild-type (WT) HEK293 cells or HEK293 cells stably expressing AGSR1 (1A4 cells) were incubated in medium containing 150 nM Br-ASO for 24 h (‘free uptake’) and then processed for NanoSIMS analyses. (A) Composite 32S (red) + 79Br (green) NanoSIMS images reveal the distribution of Br-ASO and GalNAc-Br-ASO in WT and 1A4 cells. Scale bars, 5 μm. Additional NanoSIMS images are shown in Supplementary Figures S8–S9. (B) Quantification of 79Br, normalized to 13C, in WT and 1A4 cells that had been incubated with Br-ASO or GalNAc-Br-ASO (mean ± SD). Signals were quantified in nucleolus (Nu), endosomes (En), nucleus, mitochondria (Mito), endoplasmic reticulum (ER), Golgi apparatus (Golgi), cytoplasm excluding endosomes, nucleoplasm, and whole cells. Numbers of cells evaluated: n = 6 for Br-ASO–treated WT cells; n = 9 for GalNAc-Br-ASO–treated WT cells; n = 8 for Br-ASO–treated 1A4 cells; n = 11 for GalNAc-Br-ASO–treated 1A4 cells. The 79Br/13C threshold was set at 0.00037 (corresponding to the 79Br/13C ratio in cells that were never incubated with an ASO). Data, presented as mean ± SD, were analyzed with one-way ANOVA with multiple comparisons. The 79Br/13C ratio in GalNAc-Br-ASO–treated 1A4 cells was higher than in the WT cells and higher than in Br-ASO–treated WT or 1A4 cells. P = 0.0031 for cytoplasm without endosomes; P = 0.0003 for endosomes; P <0.0001 for ER; P = 0.0961 for Golgi apparatus; P = 0.0006 for mitochondria; P = 0.0006 for nucleus without nucleolus; P = 0.0069 for nucleolus; P = 0.0004 for the whole cell.
For visualizing 34S-ASO in Figure 2, 35 × 35-μm regions of selected cells were scanned with a primary ion beam current of ∼1 nA (primary aperture D1 = 1) to reach a dose of ∼1 × 1017 ions/cm2. Regions of 30 × 30-μm were imaged with an 8-pA beam current (primary aperture D1 = 2) and a total dwell time of 35 ms/pixel. 256 × 256-pixel images were obtained.
For visualizing Br-ASO in the liver in Figure 5, a 20 × 20-μm region was scanned with a primary ion beam current of ∼1.1 nA (primary aperture D1 = 1) to reach a dose of ∼5 × 1016 ions/cm2. A region of 15 × 15-μm was imaged with an ∼1.5-pA beam current (primary aperture D1 = 3) and a total dwell time of 60 ms/pixel. For the heart images in Figure 5, a region was scanned with a primary ion beam current of ∼1.1 nA (primary aperture D1 = 1) to reach a dose of ∼1 × 1017 ions/cm2, and a region of 16 × 16-μm was imaged with a ∼1.5-pA beam current (primary aperture D1 = 3) and a total dwell time of 60 ms/pixel. For the other liver, heart, and kidney images in Figures 6–8 and Supplementary Figures S12–S14, regions of interest were scanned with a primary ion beam current of ∼1.1 nA (primary aperture D1 = 1) to reach a dose of ∼1 × 1017 ions/cm2. Regions of 38 × 38-μm were imaged with an 8-pA beam current (primary aperture D1 = 2) and a total dwell time of 40 ms/pixel. 256 × 256-pixel images were obtained.
Figure 5.
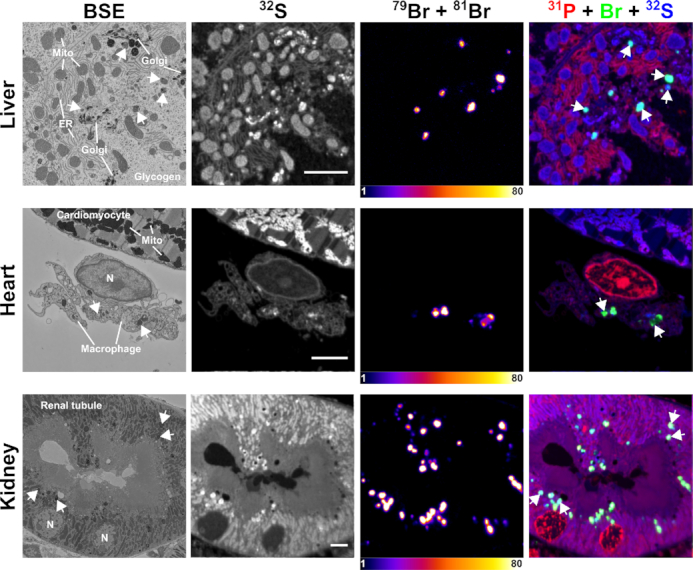
Correlative backscattered electron (BSE) and NanoSIMS images of mouse tissues following treatment with Br-ASO. A wild-type mouse was given a subcutaneous injection of Br-ASO (32 mg/kg). After 72 h, tissues were harvested and processed for BSE and NanoSIMS imaging. 32S NanoSIMS images were useful for visualizing morphology; bromine images (79Br + 81Br) and composite bromine (green) + 31P (red) + 32S (blue) NanoSIMS images were created to show the distribution of Br-ASO in the liver, heart, and kidney. Arrows point to endolysosomes. Mito, mitochondria; Golgi, Golgi apparatus; ER, endoplasmic reticulum. Scale bars, 4 μm.
Figure 6.

Correlative backscattered electron (BSE) and NanoSIMS mosaic images showing the distribution of Br-ASO in the liver. NanoSIMS imaging on a liver section from a mouse that had been injected subcutaneously with Br-ASO. After 72 h, liver was harvested and processed for NanoSIMS imaging. (A) BSE image; 32S and bromine (sum of 79Br and 81Br NanoSIMS images; and composite 31P (red) + Br (sum of 79Br and 81Br; green) and 32S (blue) NanoSIMS images of the liver. (B) Higher-magnification images of three boxed regions (Area a, Area b, Area c) from the NanoSIMS mosaic shown in panel A. 32S images were useful for morphology; the bromine and composite NanoSIMS images revealed an uneven distribution of Br-ASO in the liver. Br-ASO accumulation was observed in endolysosomes of hepatocytes and scattered cells within the liver parenchyma (Kupffer cells). Bromine levels were higher in hepatocytes in selected regions of the field. High levels of bromine enrichment were also observed in nuclei of Kupffer cells (K), cytoplasmic endolysosomes within Kupffer cells (arrows), capillary endothelial cells (arrowheads), and the cytoplasm (C) and nucleus (N) of hepatocytes. *capillary lumen; H, hepatocyte; K, Kupffer cell; N, cell nucleus. Scale bars: A, 20 μm; B, 4 μm. (C) Quantification of bromine signals (sum of 79Br and 81Br), normalized to 13C, in hepatocytes, Kupffer cells and capillary endothelial cells in the liver from a mouse that had been injected with Br-ASO. Numbers of cells evaluated: n = 68 for hepatocytes; n = 9 for Kupffer cells; n = 18 for capillary endothelial cells. The (79Br + 81Br)/13C threshold was set at 0.0025 (corresponding to slightly higher than the 79Br/13C ratio in liver cells from a control mouse that did not receive Br-ASO). Data is presented as mean ± SD. (D) Heterogeneity of bromine signals (sum of 79Br and 81Br), normalized to 13C, in hepatocytes in a liver section from a mouse had been injected with Br-ASO (treated) and a liver section from a mouse had not been injected with Br-ASO (control). Numbers of cells presented: n = 68 for hepatocytes from a treated mouse; n = 34 for hepatocytes from a control mouse. Each point represent data from one hepatocyte. The bromine background in the liver that had not exposed to Br-ASO is relatively homogeneous. (E) Quantification of bromine enrichment (sum of 79Br and 81Br), normalized to 13C, in subcellular features in hepatocytes and Kupffer cells from a mouse had been injected with Br-ASO. Numbers of features evaluated: n = 26 for hepatocyte nucleus; n = 44 for hepatocyte cytoplasm (excluding endolysosomes); n = 159 for hepatocyte foci; n = 8 for Kupffer cell nucleus; n = 9 for Kupffer cell cytoplasm (excluding endolysosomes); n = 7 for Kupffer cell foci. The (79Br + 81Br)/13C threshold was set at 0.0025 (corresponding to slightly higher than the 79Br/13C ratio in the liver cells from a mouse that were never injected with Br-ASO). Data is presented as mean ± SD.
Figure 7.
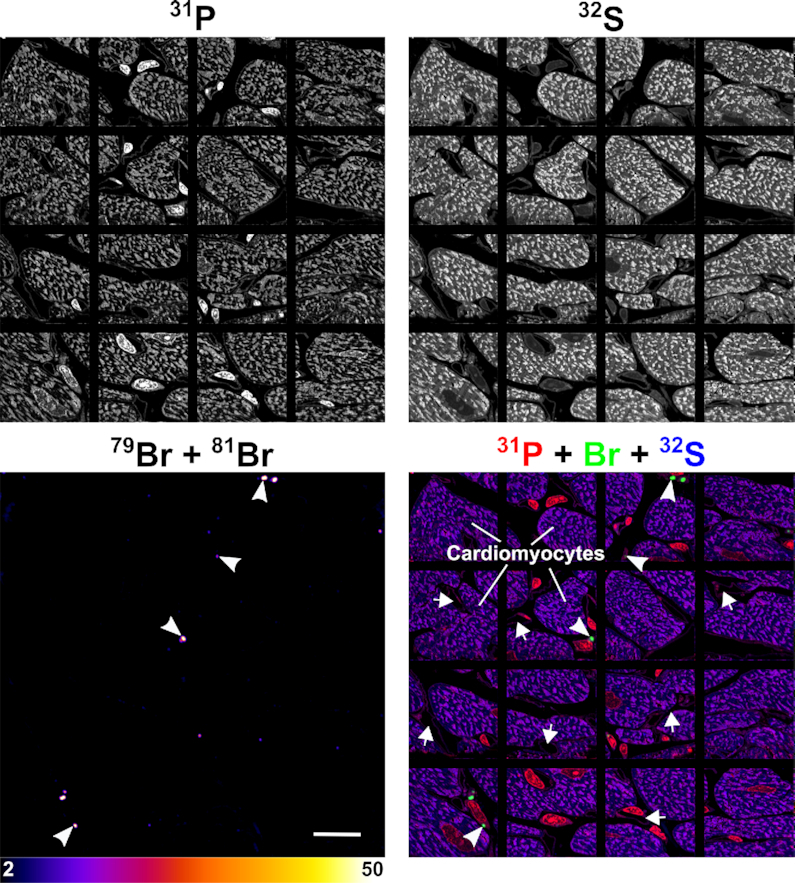
NanoSIMS mosaic showing the distribution of Br-ASO in the heart. Additional NanoSIMS images from the same heart section shown in Figure 4. 31P and 32S NanoSIMS images were useful for defining cell morphology; bromine (79Br + 81Br) NanoSIMS images and composite bromine (green) + 31P (red) + 32S (blue) NanoSIMS images were generated to show the distribution of Br-ASO in the heart. Arrows, capillaries; Arrowheads, bromine-enriched endolysosomes in interstitial cells [likely macrophages based on studies by Pinto et al. (44)] between cardiomyocytes. Scale bar, 20 μm.
Figure 8.
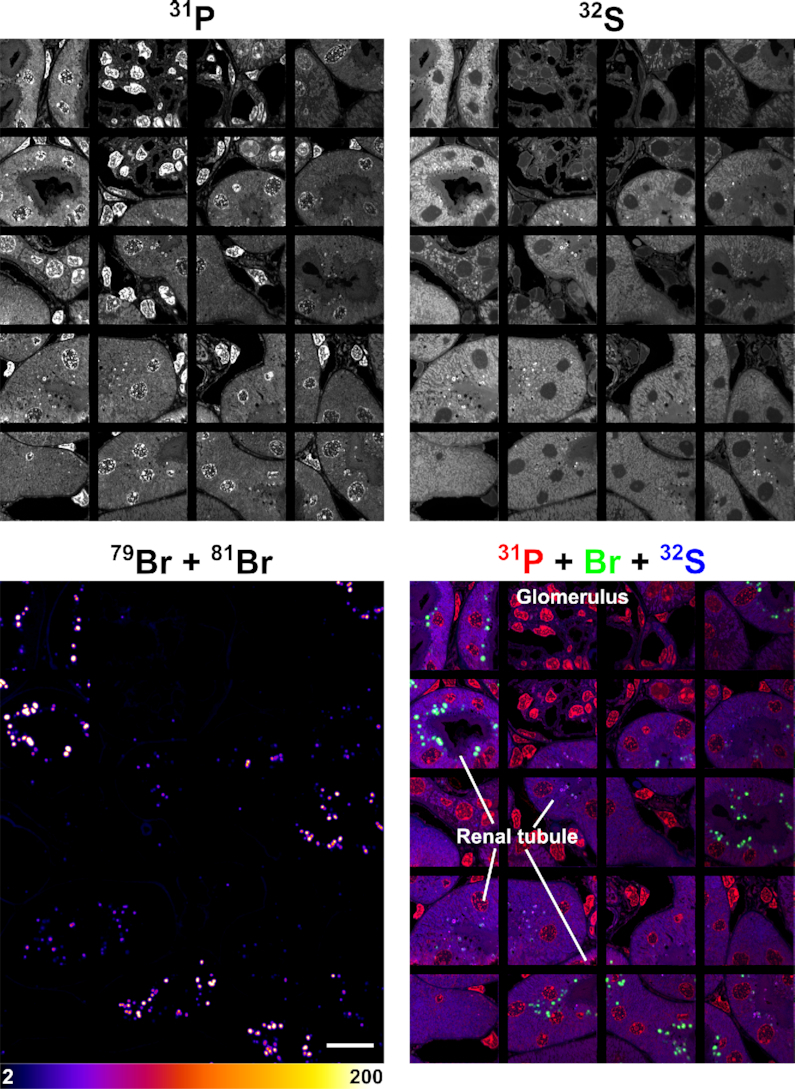
NanoSIMS images of the same kidney section shown in Figure 4, revealing the distribution of Br-ASO in the kidney. Shown here are 31P, 32S, 79Br, and composite 31P (red) + 32S (blue) + 79Br (green) NanoSIMS images of kidney from a mouse that had been injected subcutaneously with Br-ASO. Images show localized areas of Br-ASO enrichment (endolysosomes) in renal tubule cells. Scale bars, 20 μm.
Images were processed and prepared with the OpenMIMS plugin in ImageJ. A Gaussian Blur filter was applied to 79Br and to the (79Br + 81Br) images to reduce noise. The threshold of 79Br images of cells treated with bromine-labeled ASO were set based on the 79Br background from the control cells in each set of experiments. The threshold and scale for (79Br + 81Br) images of Br-ASO–treated mice were established based on the (79Br + 81Br) signal background from tissues of untreated mice. For image quantification, regions-of-interests were drawn manually based on the morphological features in NanoSIMS and BSE images. Bromine signals in regions-of-interests, normalized to 13C, were extracted using the OpenMIMS plugin and processed by GraphPad Prism 7.0.
Institutional approvals
Animal protocols were approved by UCLA’s Animal Research Committee. Mice were fed a chow diet and housed in AAALAC (Association for Assessment and Accreditation of Laboratory Animal Care International)-approved facilities with a 12-h light–dark cycle.
Statistics
Statistical analyses were performed with GraphPad Prism 7.0 software. Differences were assessed using one-way ANOVA with multiple comparisons.
RESULTS
Synthesis of ASOs for NanoSIMS imaging
We synthesized Malat1-specific ASOs that were labeled with bromine (Br-ASO) (Figure 1A) or 34S (34S-ASO) (Figure 1B). The Br-ASO contained both 79Br and 81Br (natural abundance, 51% and 49%, respectively) (Figure 1A). Both 34S-ASO and Br-ASO contained constrained ethyl modifications (Figure 1), a modification that enhances affinity for complementary RNA and stability from nuclease-mediated degradation (38). In 34S-ASO, the 34S (natural abundance, 4.5%) was incorporated into the backbone, replacing 32S—the most abundant sulfur isotope in nature. Enriching the ASO with 34S does not alter the properties of the ASO (36). We chose a bromine-labeling strategy because oligonucleotides containing bromine-modified uridines have activities similar to those of unmodified oligonucleotides (39). We found that the activities of 34S-ASO and Br-ASO (Figure 1) in reducing Malat1 expression in cultured cells were similar to the activity of the unmodified parental ASO, with IC50s ranging from 12 to 18 nM (Figure 1C and D).
Figure 1.

ASOs for NanoSIMS imaging. (A) Labeling ASOs with bromine by incorporating 5-bromo-2′-deoxyuridine (5-BrdU) residues. (B) 34S labeling of ASOs by incorporating 34S into the ASO backbone. (C and D) Design, chemistry, and antisense activities of unlabeled ASO (ASO 1), Br-labeled ASO (ASO 2), and 34S-labeled ASO (ASO 3). Blue, constrained ethyl (cEt) modifications; green, 5-BrdU-labeled nucleotides; red, 34S-labeled nucleotides. (D) Effect of the ASOs on MALAT1 transcript levels in 3T3-L1 cells. Transcript levels were measured after treating cells overnight with different ASO concentrations; data were normalized to MALAT1 transcript levels in untreated cells.
Imaging ASO distribution within cells by NanoSIMS
We incubated HeLa cells in medium containing 34S-ASO or Br-ASO (5 μM) in the absence of transfection reagents (‘free uptake’). After 24 h, the cells were fixed and prepared for NanoSIMS imaging (Figure 2). 34S/32S and 79Br NanoSIMS images revealed an accumulation of 34S-ASO and Br-ASO, respectively, in cytoplasmic organelles, many of which were visible in the 12C14N images. 12C14N images depict the nitrogen distribution in the cell and are helpful for defining cell morphology. Because fluorescently labeled ASOs have been reported to accumulate in endosomes (16), we suspected that the cytoplasmic features with increased amount of 34S and 79Br enrichment corresponded to endosomes. To explore this issue, we first incubated HeLa cells in medium containing a fluorescently labeled ASO (Cy3-ASO). As expected, we observed intensely fluorescent foci in the cytoplasm (Supplementary Figure S1). Additional studies revealed the fluorescent ASO largely (but not completely) co-localized with a fluorescently labeled transferrin (a marker of endosomes) (Supplementary Figure S2). The Cy3-ASO could be detected in the nucleus of HeLa cells, but only in small amounts and only in a subset of cells. In contrast, Br-ASO was easily detectable in HeLa cell nuclei by NanoSIMS imaging (Figure 2B). The greater ability of NanoSIMS to detect Br-ASO in the nucleus of HeLa cells was likely due, at least in part, to relatively low amounts of bromine in cells (which results in a lower background in NanoSIMS images). Our ability to visualize 34S-ASO in HeLa cells was less robust, likely because there are large amounts of sulfur in cells and because 4.5% of that sulfur is 34S (resulting in a high background and a reduced ability to detect localized regions of 34S enrichment). We were able to detect localized accumulations of 34S-ASO in endosomes, but the level of 34S enrichment was only 38% above 34S natural abundance. Because the Br images were superior to the 34S images, we focused the remainder of our efforts on imaging Br-ASO in cells and tissues.
Visualizing Br-ASO in cells by correlative backscattered electron (BSE) and NanoSIMS imaging
HeLa cells that had been incubated in medium containing Br-ASO (‘free uptake’) or had been transfected with Br-ASO with lipofectamine were processed for BSE and NanoSIMS imaging. The ultrastructural features of 500-nm-thick HeLa cell sections were mapped by BSE imaging with a scanning electron microscope (SEM) and then analyzed by NanoSIMS. This combined imaging approach allowed us to correlate the chemical information of NanoSIMS imaging with the greater ultrastructural detail provided by BSE imaging. In HeLa cells that were incubated in medium containing Br-ASO (free uptake studies), Br-ASO was readily detectable in endosomes, along the inner nuclear membrane, and in the nucleoplasm and nucleolus (Figure 3 and Supplementary Figure S3). In HeLa cells that had been transfected with Br-ASO, bromine enrichment was observed in the nucleoplasm and in granular and filamentous features of the nucleus (40,41), but not along the inner nuclear membrane (Supplementary Figure S4). When HeLa cells were transfected with Cy3-ASO and imaged by confocal microscopy, it was possible to detect the ASO in endosomes and in the nucleus (Supplementary Figure S5), but the resolution of the confocal images was far lower than the NanoSIMS images.
Assessing the effect of a targeting ligand (N-acetylgalactosamine; GalNAc) on the distribution of an ASOs
We synthesized a Br-ASO conjugated to GalNAc (Supplementary Figure S6A), a ligand for the asialoglycoprotein receptor (ASGR). Conjugation of ASOs to GalNAc has been shown to increase ASO activity by 20–30-fold in the liver (42,43). We found that the activity of GalNAc-conjugated ASO (GalNAc-ASO) in reducing Malat1 expression in cultured ASGR1-expressing HEK293 cells (1A4 cells) (34) increased >60-fold compared to the activity of the parental ASO without GalNAc conjugation (Supplementary Figure S6B). Bromine-labeling of GalNAc-conjugated ASOs (GalNAc-Br-ASO) did not alter the activity, with IC50 of 30 nM for GalNAc-Br-ASO and 33 nM for GalNAc-ASO (Supplementary Figure S6B). Wild-type HEK293 cells or 1A4 cells were incubated in medium containing the Br-ASO or GalNAc-Br-ASO and then processed for BSE and NanoSIMS imaging. In 1A4 cells, the uptake of GalNAc-Br-ASO was much greater than uptake of the Br-ASO, but there was no effect of GalNAc conjugation on ASO uptake in wild-type HEK293 cells (Figure 4A, Supplementary Figures S7 and S8). Correlative BSE imaging was helpful for identifying subcellular organelles (Supplementary Figure S9). In 1A4 cells, we found increased amounts of the GalNAc-conjugated ASO in all subcellular structures, including endosomes, mitochondria, endoplasmic reticulum (ER), Golgi apparatus (Golgi), nucleoplasm, and nucleolus (Figure 4B). We also noted increased Br-ASO and GalNAc-Br-ASO binding to filopodia, even when ASO binding to adjacent segments of the plasma membrane was negligible (Supplementary Figure S10). As controls, we imaged cells that were not incubated with an ASO. As expected, bromine enrichment in the nontreated cells was extremely low (Supplementary Figure S11). The control studies were important for quantifying ‘bromine background’ in cells. In studies with ASO-Br–treated cells, the bromine enrichment was corrected for background levels of bromine.
NanoSIMS analyses of Br-ASO distribution in mice
We administered, by subcutaneous injection, Br-ASO (32 mg/kg) to a wild-type mouse. After 72 h, the liver, kidney, and heart were harvested and processed for correlative BSE and NanoSIMS imaging. Subcellular organelles (e.g. mitochondria, endolysosomes, Golgi apparatus, endoplasmic reticulum, and nucleus) were identified in BSE images, and the distribution of Br-ASO in those structures was assessed by NanoSIMS (Figure 5). In preliminary studies, we were able to detect Br-ASO in endolysosomes of hepatocytes, renal tubular cells, and solitary cells between cardiomyocytes [likely macrophages based on studies by Pinto et al. (44)] (Figure 5).
To further explore the distribution of Br-ASO, we mapped large regions of the liver, heart, and kidney by NanoSIMS (Figures 6, 7, 8, and Supplementary Figures S12–S13). We observed uptake of Br-ASOs in hepatocytes, Kupffer cells, and sinusoidal capillary endothelial cells—with the highest levels of uptake in Kupffer cells (Figure 6A–C). The bromine signal (normalized to 13C) in Kupffer cells and capillary endothelial cells was 5.6-fold and 2.2-fold higher, respectively, than in hepatocytes (Figure 6C). We observed substantial heterogeneity in Br-ASO accumulation in different hepatocytes (Figure 6D)—even in immediately adjacent hepatocytes (Figure 6B, Area b and Area c). In some hepatocytes of Br-ASO–treated mice, bromine enrichment was barely above background (i.e. levels observed in hepatocytes of untreated mice), but in other hepatocytes the bromine enrichment was ∼6.5-fold higher than in untreated mice (Figure 6D). Br-ASO could be visualized in endolysosomes of most hepatocytes (Figure 6 and Supplementary Figure S12). The bromine signals in endolysosomes of hepatocytes were ∼20-fold higher than in the remainder of the cytoplasm and ∼26-fold higher than in the hepatocyte nucleus (Figure 6E). We observed Br-ASO accumulation in the nucleus of Kupffer calls and in endolysosomes of the cytoplasm (Figure 6B, Area a; Figure 6E and Supplementary Figure S12). Bromine enrichment in the nucleus, cytoplasm, and endolysosomes of Kupffer cells was ∼4.9-, ∼2.4- and ∼3.2-fold greater, respectively, than in hepatocytes (Figure 6E). Br-ASO was also observed in discrete cytoplasmic foci within the capillary endothelial cells that line liver sinusoids (Figure 6B; Area a and Area b).
NanoSIMS imaging of a larger region of the heart (Figure 7) (encompassing the cardiac macrophage in Figure 4) revealed bromine accumulation in most macrophages between cardiomyocytes, but the levels of bromine enrichment in cardiomyocytes were negligible (no greater than background). In contrast, bromine enrichment was high in the heart macrophages (Figure 7), with levels of bromine enrichment in endolysosomes being ∼33 times higher than in control mice.
A larger region of kidney was also mapped (Figure 8), including cells shown in Figure 4. Br-ASO accumulation was observed in endolysosomes of renal tubule cells. When the bromine scale was altered, it was possible to visualize bromine enrichment in the basement membranes of the kidney (Supplementary Figure S13) (45). Basement membrane proteins are normally brominated by the HOBr generated by peroxidasin, a peroxidase within the extracellular matrix (45). Bromine enrichment in endolysosomes of renal tubule cells in Br-ASO–treated mice was ∼70-fold higher than in the basement membranes and ∼178-fold higher than bromine enrichment in endolysosomes of renal tubule cells from control mice that had not been treated with Br-ASO (Supplementary Figure S13). Levels of bromine enrichment in tissues of mice given an injection of normal saline alone were extremely low (Supplementary Figure S14). Once again, the control studies were important for quantifying ‘bromine background.’ In our studies of tissues from ASO-Br–treated mice, bromine enrichment was corrected for background levels observed in wild-type control mice.
DISCUSSION
Over the past five years, we have used NanoSIMS imaging and correlative electron microscopy to visualize the trafficking of lipoprotein–derived fatty acids into cells and tissues (19,22); to define the distribution of cholesterol within the plasma membrane (46,47); and to define the cellular and subcellular distributions of pharmaceutical agents (20,27,32). Based on these studies, we suspected that it might be possible to use similar experimental approaches to refine our understanding of the trafficking and distribution of NATs in cells and tissues. As noted earlier, published studies on the distribution of fluorescently labeled NATs in cultured cells have provided useful information (48–50), but most of those studies were confined to transfected cells, and they were always subject to the caveat that the fluorescent tag could alter the properties of the NAT. In the current studies, we synthesized ASOs with two different elemental alterations (34S or Br) that do not significantly alter the biological activity of the ASO and would not be expected to change the physicochemical properties of the ASO. When we incubated cells with the 34S-labeled ASO (34S-ASO), we had no difficulty in identifying, by NanoSIMS, 34S enrichment in endosomes, but the large amounts of 34S in nature (4.25% of sulfur atoms) limited our ability to document 34S enrichment in additional organelles. NanoSIMS images of Br-ASO were superior. Bromine can be detected in mammalian cells and tissues and is enriched in basement membranes (45), but the background of bromine NanoSIMS analyses is far lower than with 34S. 34S levels in the endosomes of 34S-ASO–treated cells were only ∼40% higher than the natural abundance of 34S, whereas bromine levels in endosomes of Br-ASO–treated cells and tissues were as much as 170-fold higher than in endosomes of untreated control mice. Also, Br-ASO contained both 79Br and 81Br, and the ability to detect and quantify both isotopes improved image quality. Our ability to interpret NanoSIMS images of bromine enrichment was enhanced by BSE imaging, allowing us match regions of bromine enrichment in NanoSIMS images to the greater ultrastructural detail of BSE images.
We focused our efforts on Br-ASO distribution because our ability to detect bromine enrichment was robust. We emphasize, however, that there are other potential strategies for visualizing ASO distribution by NanoSIMS. For example, if the expense of stable isotope reagents was of no concern, it would be possible to minimize NanoSIMS background by synthesizing ASOs containing 33S (natural abundance, 0.75%) or 36S (natural abundance, 0.01%). One could also generate ASOs containing exclusively 79Br or 81Br. In the latter case, it would be possible to define ASO distribution by visualizing deviations from the 79Br/81Br ratio found in nature (which is 1.0279). The availability of two bromine isotopes would also make it possible to image two different ASOs simultaneously. Sulfur- and bromine-labeled ASOs are not the only strategy. It would be possible to prepare ASOs labeled with iodine or generate ASOs uniformly labeled with 15N (natural abundance, 0.37%). One could even produce ASOs that were uniformly labeled with both 15N and 13C (natural abundance, 1.1%) and then produce images from the cluster secondary ion 13C15N–, which is formed only when 13C and 15N atoms are located a few nanometers apart (51). That approach would likely yield images with a very low background.
In the current studies, we imaged cultured cells in which the Br-ASO was delivered to HeLa cells by ‘free uptake’ and by lipofectamine-mediated transfection. In the free uptake studies, we observed an accumulation of Br-ASO along the inner nuclear membrane of HeLa cells, raising the possibility that binding sites along inner nuclear membrane influence ASO trafficking and distribution. We did not observe Br-ASO accumulation along the inner nuclear membrane in lipofectamine-transfected cells, where the import of ASOs into the nucleus would differ. We also did not observe inner nuclear membrane accumulation of Br-ASO in HEK293 cells, where the concentration of Br-ASO in the cell culture medium was lower (∼3% of concentrations in the HeLa experiments). Differences in ASO distribution in HeLa and HEK293 cells might also reflect differences in the expression of cell-surface receptors and/or nuclear membrane proteins (50).
Our NanoSIMS studies revealed that a targeting ligand, GalNAc, augments ASO uptake, but only in cells expressing ASGR1 (1A4 cells). In 1A4 cells, the uptake of GalNAc-Br-ASO was ∼3.8-fold higher than the uptake of Br-ASO. In GalNAc-Br-ASO–treated 1A4 cells, we observed very high levels of bromine enrichment in endosomes, but greater bromine enrichment was also observed in each of the subcellular features that we analyzed. An unexpected finding—and one that was independent of GalNAc conjugation or ASGR1 expression—was an accumulation of ASOs on filopodia of HEK293 cells. We do not understand this finding, but we suspect that it could relate to increased amounts of cholesterol in the plasma membrane covering filopodia. In earlier studies, we found, by NanoSIMS analyses, higher levels of ‘accessible cholesterol’ in the filopodia of macrophages (46). We also observed higher levels of accessible cholesterol in the microvilli of Chinese Hamster ovary cells (18). Of note, phosphorothioate ASOs, such as those used in the current studies, bind preferentially to membranes containing high levels of cholesterol, and cholesterol supplementation increases phosphorothioate ASO uptake and activity (52,53).
NanoSIMS imaging was also useful for visualizing bromine enrichment in mouse tissues three days after a subcutaneous injection of Br-ASO. Given that ASOs are quite effective in reducing apolipoprotein expression in the liver (54,55), the fact that we were able to detect Br-ASO in hepatocytes was not a surprise. However, NanoSIMS analyses provided a more refined understanding of ASO distribution in the liver. For example, we observed intense bromine enrichment in the nucleus and endolysosomes of Kupffer cells and in localized foci of sinusoidal endothelial cells. Of note, we encountered considerable heterogeneity in the uptake of Br-ASO in hepatocytes—even in immediately adjacent hepatocytes. At this point, we do not understand the heterogeneity in Br-ASO uptake and distribution. Many metabolic functions in the liver are separated in hepatocytes located at different sites along liver sinusoids (e.g. periportal hepatocytes, centrilobular hepatocytes). This phenomenon (‘liver zonation’) could contribute to the heterogeneity in Br-ASO distribution in hepatocytes. However, we recommend caution in drawing firm conclusions about Br-ASO distribution based on tissues harvested after a single Br-ASO injection. It is possible that the heterogeneity would be far less impressive after several months of weekly Br-ASO injections.
ASOs are known to reach high concentrations in the kidney (6), and for that reason we were not surprised to find bromine enrichment in the kidney in Br-ASO-treated mice. Once again, however, NanoSIMS images provided more refined insights. First, we found that virtually all of the Br-ASO was located in renal tubule epithelial cells—with nearly all of the Br-ASO in endolysosomes. The bromine enrichment of endolysosomes in the kidney was ∼6.5-fold and ∼6.2-fold higher, respectively, than in liver and heart. Outside of endolysosomes of renal tubular cells, the bromine enrichment in renal tubular cells was negligible and in general was no greater than in renal tubular cells of a control mouse that had been injected with normal saline. The amount of bromine enrichment in the glomerulus of the Br-ASO–treated mouse was extremely low. Once again, however, we caution against overinterpreting that observation, given that our studies provided only a snapshot of bromine distribution three days after a single dose of Br-ASO.
Bromine enrichment in the cardiomyocytes in a Br-ASO–injected mouse was no greater than in a control mouse that had been given normal saline. Interestingly, however, we observed intense bromine enrichment in the macrophages scattered among the cardiomyocytes. Bromine enrichment in heart macrophages (and in the endolysosomes of those cells) was comparable to levels of enrichment in hepatocytes. Insights such as these could be relevant to human disease. For example, it is conceivable that ASO therapeutics could be useful for treating rejection of cardiac allografts—a process in which macrophages are assumed to be effectors of tissue injury (56).
In summary, we show that NanoSIMS imaging, particularly when combined with correlative BSE imaging, provides novel insights into ASO distribution in cultured cells and mouse tissues. We showed that phosphorothioate ASOs associate with filopodia and the inner nuclear membrane of cells, and we documented substantial cellular and subcellular heterogeneity in ASO distribution in the liver, heart, and kidney. While the current studies focused on ASOs, we anticipate that the same experimental approaches will be useful for characterizing the uptake, trafficking of other classes of NATs (e.g. siRNA, sgRNA, mRNAs). NanoSIMS imaging will make it possible to address key issues in NATs, for example the efficiency of NAT transport across endothelial cells, the uptake of NATs by different cell types and different subcellular compartments, how and when ligands separate from their NAT cargo in endo-lysosomal vesicles, how lysosomal accumulation contributes to the duration of NAT activity, and how the uptake of siRNAs by cells and tissues differ from single-stranded ASOs. Insights such as these will be useful in guiding ongoing efforts to optimize the design and delivery of NATs for the treatment of human disease. Moreover, optimization of methods will maximize the potential of NanoSIMS imaging of NATs, allowing for imaging of multiple NATs in the same cells with increased sensitivity and a higher signal-to-noise ratio. One limiting factor is that NanoSIMS instruments are not currently widely available. However, it is feasible to collaborate with NanoSIMS facilities from afar, both because the samples are stable for storage and shipping and because it is feasible to operate NanoSIMS instruments remotely via internet connections.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Jeremy Bougoure and Dr Alexandra Suvorova (Centre for Microscopy, Characterisation and Analysis, University of Western Australia) for their assistance on NanoSIMS analyses and scanning electron microscopy. The authors are grateful to the Centre for Microscopy, Characterisation & Analysis at the University of Western Australia, which is funded by the University and both the State and Commonwealth Governments.
Contributor Information
Cuiwen He, Department of Medicine, University of California, Los Angeles, CA 90095, USA.
Michael T Migawa, Ionis Pharmaceuticals, Inc., Carlsbad, CA 92010, USA.
Kai Chen, School of Molecular Sciences, University of Western Australia, Perth 6009, Australia.
Thomas A Weston, Department of Medicine, University of California, Los Angeles, CA 90095, USA.
Michael Tanowitz, Ionis Pharmaceuticals, Inc., Carlsbad, CA 92010, USA.
Wenxin Song, Department of Medicine, University of California, Los Angeles, CA 90095, USA.
Paul Guagliardo, Centre for Microscopy, Characterisation and Analysis, University of Western Australia, Perth 6009, Australia.
K Swaminathan Iyer, School of Molecular Sciences, University of Western Australia, Perth 6009, Australia.
C Frank Bennett, Ionis Pharmaceuticals, Inc., Carlsbad, CA 92010, USA.
Loren G Fong, Department of Medicine, University of California, Los Angeles, CA 90095, USA.
Punit P Seth, Ionis Pharmaceuticals, Inc., Carlsbad, CA 92010, USA.
Stephen G Young, Department of Medicine, University of California, Los Angeles, CA 90095, USA; Department of Human Genetics, University of California, Los Angeles, CA 90095, USA.
Haibo Jiang, School of Molecular Sciences, University of Western Australia, Perth 6009, Australia; Department of Chemistry, The University of Hong Kong, Hong Kong, China.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Australian Research Council Discovery Early Career Researcher Award; Rebecca L Cooper Medical Research Foundation Project Grants (to H.J.); UWA Research Income Growth Grant (to H.J. and F.B.); NHLBI [HL090553, HL087228, HL125335 to S.G.Y.]. Funding for open access charge: Ionis Pharmaceuticals, Inc.
Conflict of interest statement. The authors have no conflicts of interest or financial interests to declare aside from the fact that several of the authors are employees of Ionis Pharmaceuticals.
REFERENCES
- 1. Crooke S.T., Witztum J.L., Bennett C.F., Baker B.F.. RNA-targeted therapeutics. Cell Metab. 2018; 27:714–739. [DOI] [PubMed] [Google Scholar]
- 2. Setten R.L., Rossi J.J., Han S.-p.. The current state and future directions of RNAi-based therapeutics. Nat. Rev. Drug Discov. 2019; 18:421–446. [DOI] [PubMed] [Google Scholar]
- 3. Shen X., Corey D.R.. Chemistry, mechanism and clinical status of antisense oligonucleotides and duplex RNAs. Nucleic Acids Res. 2018; 46:1584–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim J., Hu C., Moufawad El Achkar C., Black L.E., Douville J., Larson A., Pendergast M.K., Goldkind S.F., Lee E.A., Kuniholm A.. Patient-customized oligonucleotide therapy for a rare genetic disease. N. Engl. J. Med. 2019; 381:1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Roberts T.C., Langer R., Wood M.J.. Advances in oligonucleotide drug delivery. Nat. Rev. Drug Discov. 2020; 19:673–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seth P.P., Tanowitz M., Bennett C.F.. Selective tissue targeting of synthetic nucleic acid drugs. J. Clin. Invest. 2019; 129:915–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bennett C.F. Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 2019; 70:307–321. [DOI] [PubMed] [Google Scholar]
- 8. Geary R.S., Norris D., Yu R., Bennett C.F.. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv. Drug. Deliv. Rev. 2015; 87:46–51. [DOI] [PubMed] [Google Scholar]
- 9. Prakash T.P., Mullick A.E., Lee R.G., Yu J., Yeh S.T., Low A., Chappell A.E., Østergaard M.E., Murray S., Gaus H.J.. Fatty acid conjugation enhances potency of antisense oligonucleotides in muscle. Nucleic Acids Res. 2019; 47:6029–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chappell A.E., Gaus H.J., Berdeja A., Gupta R., Jo M., Prakash T.P., Oestergaard M., Swayze E.E., Seth P.P.. Mechanisms of palmitic acid-conjugated antisense oligonucleotide distribution in mice. Nucleic Acids Res. 2020; 48:4382–4395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mazur C., Powers B., Zasadny K., Sullivan J.M., Dimant H., Kamme F., Hesterman J., Matson J., Oestergaard M., Seaman M.. Brain pharmacology of intrathecal antisense oligonucleotides revealed through multimodal imaging. JCI insight. 2019; 4:e129240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Deprey K., Batistatou N., Kritzer J.A.. A critical analysis of methods used to investigate the cellular uptake and subcellular localization of RNA therapeutics. Nucleic Acids Res. 2020; 48:7623–7639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alterman J.F., Godinho B.M., Hassler M.R., Ferguson C.M., Echeverria D., Sapp E., Haraszti R.A., Coles A.H., Conroy F., Miller R.. A divalent siRNA chemical scaffold for potent and sustained modulation of gene expression throughout the central nervous system. Nat. Biotechnol. 2019; 37:884–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Anderson B.J., Larkin C., Guja K., Schildbach J.F.. Using fluorophore-labeled oligonucleotides to measure affinities of protein–DNA interactions. Methods Enzymol. 2008; 450:253–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Moreira B.G., You Y., Owczarzy R.. Cy3 and Cy5 dyes attached to oligonucleotide terminus stabilize DNA duplexes: predictive thermodynamic model. Biophys. Chem. 2015; 198:36–44. [DOI] [PubMed] [Google Scholar]
- 16. Crooke S.T., Wang S., Vickers T.A., Shen W., Liang X.H.. Cellular uptake and trafficking of antisense oligonucleotides. Nat. Biotechnol. 2017; 35:230–237. [DOI] [PubMed] [Google Scholar]
- 17. Jiang H., Favaro E., Goulbourne C., Rakowska P., Hughes G., Ryadnov M., Fong L., Young S., Ferguson D., Harris A.. Stable isotope imaging of biological samples with high resolution secondary ion mass spectrometry and complementary techniques. Methods. 2014; 68:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He C., Hu X., Jung R.S., Weston T.A., Sandoval N.P., Tontonoz P., Kilburn M., Fong L.G., Young S.G., Jiang H.. High-resolution imaging and quantification of plasma membrane cholesterol by NanoSIMS. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:2000–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. He C., Weston T.A., Jung R.S., Heizer P., Larsson M., Hu X., Allan C.M., Tontonoz P., Reue K., Beigneux A.P. et al.. NanoSIMS analysis of intravascular lipolysis and lipid movement across capillaries and into cardiomyocytes. Cell Metab. 2018; 27:1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Greenwood D.J., Dos Santos M.S., Huang S., Russell M.R., Collinson L.M., MacRae J.I., West A., Jiang H., Gutierrez M.G.. Subcellular antibiotic visualization reveals a dynamic drug reservoir in infected macrophages. Science. 2019; 364:1279–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nuñez J., Renslow R., Cliff J.B. III, Anderton C.R.. NanoSIMS for biological applications: current practices and analyses. Biointerphases. 2018; 13:03B301. [DOI] [PubMed] [Google Scholar]
- 22. Jiang H., Goulbourne C.N., Tatar A., Turlo K., Wu D., Beigneux A.P., Grovenor C.R., Fong L.G., Young S.G.. High-resolution imaging of dietary lipids in cells and tissues by NanoSIMS analysis. J. Lipid Res. 2014; 55:2156–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Raina J.-B., Clode P.L., Cheong S., Bougoure J., Kilburn M.R., Reeder A., Forêt S., Stat M., Beltran V., Thomas-Hall P.. Subcellular tracking reveals the location of dimethylsulfoniopropionate in microalgae and visualises its uptake by marine bacteria. Elife. 2017; 6:e23008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jiang H., Kilburn M.R., Decelle J., Musat N.. NanoSIMS chemical imaging combined with correlative microscopy for biological sample analysis. Curr. Opin. Biotechnol. 2016; 41:130–135. [DOI] [PubMed] [Google Scholar]
- 25. Angelo M., Bendall S.C., Finck R., Hale M.B., Hitzman C., Borowsky A.D., Levenson R.M., Lowe J.B., Liu S.D., Zhao S.. Multiplexed ion beam imaging of human breast tumors. Nat. Med. 2014; 20:436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hu D., Yang C., Lok C.N., Xing F., Lee P.Y., Fung Y.M.E., Jiang H., Che C.M.. An antitumor bis (N‐heterocyclic carbene) platinum (II) complex that engages asparagine synthetase as an anticancer target. Angew. Chem. Int. Ed. 2019; 58:10914–10918. [DOI] [PubMed] [Google Scholar]
- 27. Tong K.-C., Lok C.-N., Wan P.-K., Hu D., Fung Y.M.E., Chang X.-Y., Huang S., Jiang H., Che C.-M.. An anticancer gold (III)-activated porphyrin scaffold that covalently modifies protein cysteine thiols. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Levi‐Setti R., Chabala J.M., Gavrilov K., Espinosa R. III, Le Beau M.M.. Imaging of BrdU labeled human metaphase chromosomes with a high resolution scanning ion microprobe. Microsc. Res. Tech. 1997; 36:301–312. [DOI] [PubMed] [Google Scholar]
- 29. Berry J.-P., Galle P., Chassoux D., Escaig F., Linarez-Cruz L.G., Lespinats G.. Mapping of intracellular halogenous molecules by low and high resolution SIMS microscopy. Biol. Cell. 1992; 74:93–98. [DOI] [PubMed] [Google Scholar]
- 30. Kilburn M.R., Wacey D.. Grice K., Arrigan D., Reddy S.M.. Nanoscale secondary ion mass spectrometry (NanoSIMS) as an analytical tool in the geosciences. Principles and Practice of Analytical Techniques in Geosciences. 2014; The Royal Society of Chemistry. [Google Scholar]
- 31. Mueller C.W., Weber P.K., Kilburn M.R., Hoeschen C., Kleber M., Pett-Ridge J.. Advances in Agronomy. 2013; 121:Elsevier; 1–46. [Google Scholar]
- 32. Jiang H., Passarelli M.K., Munro P.M., Kilburn M.R., West A., Dollery C.T., Gilmore I.S., Rakowska P.D.. High-resolution sub-cellular imaging by correlative NanoSIMS and electron microscopy of amiodarone internalisation by lung macrophages as evidence for drug-induced phospholipidosis. Chem. Commun. 2017; 53:1506–1509. [DOI] [PubMed] [Google Scholar]
- 33. Proetto M.T., Callmann C.E., Cliff J., Szymanski C.J., Hu D., Howell S.B., Evans J.E., Orr G., Gianneschi N.C.. Tumor retention of enzyme-responsive Pt (II) drug-loaded nanoparticles imaged by nanoscale secondary ion mass spectrometry and fluorescence microscopy. ACS Central Science. 2018; 4:1477–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tanowitz M., Hettrick L., Revenko A., Kinberger G.A., Prakash T.P., Seth P.P.. Asialoglycoprotein receptor 1 mediates productive uptake of N-acetylgalactosamine-conjugated and unconjugated phosphorothioate antisense oligonucleotides into liver hepatocytes. Nucleic Acids Res. 2017; 45:12388–12400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Prakash T.P., Yu J., Migawa M.T., Kinberger G.A., Wan W.B., Østergaard M.E., Carty R.L., Vasquez G., Low A., Chappell A.. Comprehensive structure–activity relationship of triantennary N-acetylgalactosamine conjugated antisense oligonucleotides for targeted delivery to hepatocytes. J. Med. Chem. 2016; 59:2718–2733. [DOI] [PubMed] [Google Scholar]
- 36. Stulz R., Meuller J., Baždarević D., Wennberg Huldt C., Strömberg R., Andersson S., Dahlén A.. A versatile and convenient synthesis of 34S‐labeled phosphorothioate oligonucleotides. Chem. BioChem. 2018; 19:2114–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deerinck T.J., Bushong E.A., Thor A., Ellisman M.H.. NCMIR methods for 3D EM: a new protocol for preparation of biological specimens for serial block face scanning electron microscopy. National Center forMicroscopyand Imaging Research. 2010; https://ncmir.ucsd.edu/sbem-protocol. [Google Scholar]
- 38. Seth P.P., Vasquez G., Allerson C.A., Berdeja A., Gaus H., Kinberger G.A., Prakash T.P., Migawa M.T., Bhat B., Swayze E.E.. Synthesis and biophysical evaluation of 2', 4'-constrained 2' O-methoxyethyl and 2', 4'-constrained 2' O-ethyl nucleic acid analogues. J. Org. Chem. 2010; 75:1569–1581. [DOI] [PubMed] [Google Scholar]
- 39. Chiu Y.-L., Rana T.M.. siRNA function in RNAi: a chemical modification analysis. RNA. 2003; 9:1034–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lorenz P., Baker B.F., Bennett C.F., Spector D.L.. Phosphorothioate antisense oligonucleotides induce the formation of nuclear bodies. Mol. Biol. Cell. 1998; 9:1007–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liang X.H., Shen W., Sun H., Prakash T.P., Crooke S.T.. TCP1 complex proteins interact with phosphorothioate oligonucleotides and can co-localize in oligonucleotide-induced nuclear bodies in mammalian cells. Nucleic Acids Res. 2014; 42:7819–7832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Prakash T.P., Graham M.J., Yu J., Carty R., Low A., Chappell A., Schmidt K., Zhao C., Aghajan M., Murray H.F.. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic Acids Res. 2014; 42:8796–8807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt K., Prakash T.P., Donner A.J., Kinberger G.A., Gaus H.J., Low A., Østergaard M.E., Bell M., Swayze E.E., Seth P.P.. Characterizing the effect of GalNAc and phosphorothioate backbone on binding of antisense oligonucleotides to the asialoglycoprotein receptor. Nucleic Acids Res. 2017; 45:2294–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pinto A.R., Paolicelli R., Salimova E., Gospocic J., Slonimsky E., Bilbao-Cortes D., Godwin J.W., Rosenthal N.A.. An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS One. 2012; 7:e36814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cuiwen H., Wenxin S., Thomas A.W., Caitlyn T., Ira K., Jonathan E.Z., Paul G., Jeffrey H.M., Sergey V.I., Jeremy B.. Peroxidasin-mediated Bromine Enrichment of Basement Membranes. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:15827–15836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. He C., Hu X., Weston T.A., Jung R.S., Sandhu J., Huang S., Heizer P., Kim J., Ellison R., Xu J. et al.. Macrophages release plasma membrane-derived particles rich in accessible cholesterol. Proc. Natl. Acad. Sci. U.S.A. 2018; 115:E8499–E8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hu X., Weston T.A., He C., Jung R.S., Heizer P.J., Young B.D., Tu Y., Tontonoz P., Wohlschlegel J.A., Jiang H.. Release of cholesterol-rich particles from the macrophage plasma membrane during movement of filopodia and lamellipodia. Elife. 2019; 8:e50231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang S., Sun H., Tanowitz M., Liang X.H., Crooke S.T.. Intra-endosomal trafficking mediated by lysobisphosphatidic acid contributes to intracellular release of phosphorothioate-modified antisense oligonucleotides. Nucleic Acids Res. 2017; 45:5309–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liang X.H., Sun H., Nichols J.G., Allen N., Wang S., Vickers T.A., Shen W., Hsu C.-W., Crooke S.T.. COPII vesicles can affect the activity of antisense oligonucleotides by facilitating the release of oligonucleotides from endocytic pathways. Nucleic Acids Res. 2018; 46:10225–10245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang S., Allen N., Vickers T.A., Revenko A.S., Sun H., Liang X.H., Crooke S.T.. Cellular uptake mediated by epidermal growth factor receptor facilitates the intracellular activity of phosphorothioate-modified antisense oligonucleotides. Nucleic Acids Res. 2018; 46:3579–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moss F.R. III, Boxer S.G.. Atomic recombination in dynamic secondary ion mass spectrometry probes distance in lipid assemblies: A nanometer chemical ruler. J. Am. Chem. Soc. 2016; 138:16737–16744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lu D., Rhodes D.G.. Binding of phosphorothioate oligonucleotides to zwitterionic liposomes. Bioch. Biophys. Acta (BBA)-Biomembranes. 2002; 1563:45–52. [DOI] [PubMed] [Google Scholar]
- 53. Wang S., Allen N., Liang X.H., Crooke S.T.. Membrane destabilization induced by lipid species increases activity of phosphorothioate-antisense oligonucleotides. Mol. Ther.-Nucleic Acids. 2018; 13:686–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Crooke R.M., Graham M.J., Lemonidis K.M., Whipple C.P., Koo S., Perera R.J.. An apolipoprotein B antisense oligonucleotide lowers LDL cholesterol in hyperlipidemic mice without causing hepatic steatosis. J. Lipid Res. 2005; 46:872–884. [DOI] [PubMed] [Google Scholar]
- 55. Graham M.J., Lee R.G., Bell T.A. III, Fu W., Mullick A.E., Alexander V.J., Singleton W., Viney N., Geary R., Su J.. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ. Res. 2013; 112:1479–1490. [DOI] [PubMed] [Google Scholar]
- 56. Liu Y., Kloc M., Li X.C.. Macrophages as effectors of acute and chronic allograft injury. Curr. Transplant. Rep. 2016; 3:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.