


Abstract
Human genome stability requires efficient repair of oxidized bases, which is initiated via damage recognition and excision by NEIL1 and other base excision repair (BER) pathway DNA glycosylases (DGs). However, the biological mechanisms underlying detection of damaged bases among the million-fold excess of undamaged bases remain enigmatic. Indeed, mutation rates vary greatly within individual genomes, and lesion recognition by purified DGs in the chromatin context is inefficient. Employing super-resolution microscopy and co-immunoprecipitation assays, we find that acetylated NEIL1 (AcNEIL1), but not its non-acetylated form, is predominantly localized in the nucleus in association with epigenetic marks of uncondensed chromatin. Furthermore, chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) revealed non-random AcNEIL1 binding near transcription start sites of weakly transcribed genes and along highly transcribed chromatin domains. Bioinformatic analyses revealed a striking correspondence between AcNEIL1 occupancy along the genome and mutation rates, with AcNEIL1-occupied sites exhibiting fewer mutations compared to AcNEIL1-free domains, both in cancer genomes and in population variation. Intriguingly, from the evolutionarily conserved unstructured domain that targets NEIL1 to open chromatin, its damage surveillance of highly oxidation-susceptible sites to preserve essential gene function and to limit instability and cancer likely originated ∼500 million years ago during the buildup of free atmospheric oxygen.
Graphical Abstract
Graphical Abstract.
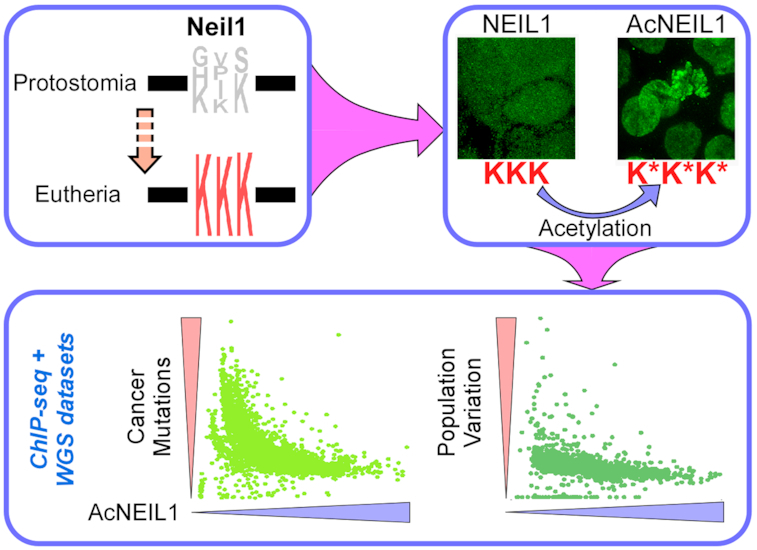
DNA glycosylase NEIL1 has acquired a 3-Lys acetylation center in eutherians, which upon acetylation confers stabilization to the enzyme on chromatin, thereby enabling efficient protection against oxidative DNA damage.
INTRODUCTION
It is estimated that ∼70 000 lesions occur in genomic DNA in each human cell per day, most of which (75%) originate from oxidation reactions with endogenous byproducts of metabolism and base hydrolysis (1). To counteract the continuous threat these lesions pose to genome stability, cells have evolved a wide-ranging arsenal of repair programs, including DNA base excision repair (BER), mismatch repair (MMR), nucleotide excision repair (NER), translesion synthesis (TLS) and strand break repair (homologous recombination and various non-homologous end joining pathways), which together act upon particular types of lesion or at specific phases of the cell cycle to prevent mutations in DNA and cell death. Oxidative stress induces a plethora of small base modifications, including the stable 2′-deoxycytidine derivatives 5-hydroxy-2′-deoxycytidine, 5-hydroxy-2′-deoxyuridine and 5,6-dihydroxy-5,6-dihydro-2′-deoxyuridine, and the 2′-deoxyguanosine base 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxoG), which accumulate at sufficiently high rates in human tissues to be readily detected (2). If not removed, these lesions are potentially mutagenic since replicative DNA polymerases, although possessing high fidelity, are not able to discriminate between a canonical and a noncanonical incoming base during DNA synthesis, thereby giving rise to mismatches that are then fixed into mutations at the next round of replication. Oxidized nucleotide pools are also mutagenic as they are readily incorporated into nascent DNA.
Oxidative lesions and selected mismatches are primarily repaired by BER, which is initiated by recognition and cleavage by one of several DNA glycosylases (DGs) (monofunctional: MBD4, MYH, MPG, SMUG1, TDG, UDG; bifunctional: OGG1, NTH1 and NEIL1/2/3) to generate an apurinic/apyrimidinic (AP) site processed, in the case of monofunctional enzymes, by AP endonuclease 1 (APE1), yielding a single-strand break (3–6). Full reconstitution of the original DNA sequence then proceeds through either short or long patch repair, which minimally requires DNA synthesis (Pol β, Pol λ and Pol ϵ) across the break followed by ligation (Lig1 and Lig3) (7–10)).
Structural analyses of BER initiation regions in the genome have revealed detection of local base damage by active surveillance of intact DNA helices by DGs (5,11–12) followed by concerted steps, whereby one enzyme channels its product to the next enzyme in the pathway (13,14). However, the efficient global mechanisms through which damaged bases are robustly detected within the vast excess of unmodified bases, particularly in the context of chromatin, have remained largely enigmatic. Indeed, whereas on naked DNA DGs are capable of interrogating helix integrity via effective mechanisms, including sliding, hopping and jumping, damage recognition in the context of reconstituted nucleosomal DNA is highly inefficient (reviewed in (7,9,15–16)).
Recent findings have implicated posttranslational modification (PTM) of BER factors in the assembly of active ‘BERosome’ complexes onto chromatin, thereby promoting the concept that reversible PTM may be critical for mediating and regulating protein-protein interactions among DNA repair factors, modified histones and other chromatin remodeling factors in the cell (17–22). However, as only very few ChIP-seq analyses of BER components are available, the extent to which these factors share genomic space has not been addressed. This question is intriguing given that the composition and PTM landscape differ in different regions of the genome, so would BERosome assembly along the genome, potentially leading to local variations in BER and hence mutation rates. Indeed, despite the fact that replication-associated mutations have been linked to high frequencies of single base-pair substitutions (SBSs) in human cancers (23), mutational landscapes in cancer genomes are highly heterogeneous (24,25). In fact, it has been noted that both in cancer and in the context of human population variation, mutation rates are lower in early replicating euchromatic regions than in late replicating compact heterochromatic regions (reviewed in (26)), raising questions as to the underlying mechanisms.
Here we examined human Nei-like-1 (NEIL1), a prototypic DG that initiates BER by excising oxidized base lesions in both double-stranded and single-stranded DNA (27,28). Motivated by our recent study showing significantly higher BER activity of acetylated (AcNEIL1) compared to nonacetylated NEIL1 (17), we determined their localization. We found that contrary to total NEIL1, which punctuates uniformly both nuclei and cytoplasm, AcNEIL1 is mostly confined to nuclei from where it can be readily isolated in complex with RNA pol II and other components of the active chromatin. We showed that NEIL1 becomes stabilized on chromatin after site-specific (Lys296–298) acetylation, and accumulates almost exclusively at highly transcribed genomic regions as well as the transcription start sites (TSS) of weakly expressed genes, some of which are associated with poor prognosis when overexpressed in cancer. Bioinformatic analyses using cancer genome datasets and human germline population mutation datasets provide, with unprecedented resolution, information on the relationship between local variations in single base substitution (SBS) rates and ChIP-seq AcNEIL1 occupancy, both in cancer genomes and the germline. The AcNEIL1 acetylation center appears to have consolidated from variable amino acids in protostomes ∼540 million years ago, during a transition from sulfur to oxygen as an energy source. By combining our results with available data, we conclude that AcNEIL1-containing BERosomes are stabilized onto chromatin through PTM, where they are poised for lesion recognition and repair in areas of the genome which, because of high transcription, are particularly vulnerable to oxidative damage.
MATERIALS AND METHODS
Cell lines and treatments
The human colorectal adenocarcinoma HCT116 (ATCC # CCL-247), human ovarian cancer SKOV3 (ATCC # HTB-77), and human osteosarcoma U2OS (ATCC # HTB-96) cell lines were grown in McCoy's 5A medium (Gibco/Life Technologies). The human cervical adenocarcinoma HeLa (ATCC #CCL2) and human embryonic kidney epithelial HEK293 (ATCC # CRL-1573) cell lines were maintained in DMEM-high glucose medium (Hyclone). Human breast cancer MDA-MB-231 (ATCC # HTB-26), human acute leukemia Jurkat (ATCC # TIB-152) and human lung cancer H1563 (ATCC # CRL-5875) cells were maintained in RPMI 1640 medium (Corning). Human prostate cancer PC3 (ATCC # CRL-1435) cells were cultured in F-12K Medium (Corning). The human lymphoblast K562 (ATCC # CCL-243) and the near-haploid human HAP1 (Horizon) cells were maintained in Iscove's Modified Dulbecco's Medium (Corning). All culture media were supplemented with 10% fetal bovine serum (FBS, Sigma-Aldrich) and 1% (v/v) penicillin/streptomycin (Life Technologies). The cells were maintained in a humidified incubator at 37°C with 5% CO2. Exponentially growing cells were treated with 1 μM retinoic acid (RA; Sigma) according to experiments for mRNA expression and ChIP assays. Downregulation of endogenous NEIL1 was carried out by Lipofectamine RNAimax-mediated transfection of HCT116 and HeLa cells with NEIL1-specific siRNA (Sigma; sense: 5′CCGUGAUGAUGUUUGUUUAUU3′; antisense: 5′UAAACAAACAUCAUCACGGUU3′) or the universal negative control siRNA (Sigma; # SIC001) following the manufacturer's protocol. The inhibition of NEIL1 acetylation and deacetylation was achieved by treating HCT116 cells with DMSO (control), the histone acetyltransferases (HAT, such as p300 and PCAF) inhibitor Garcinol (50 and 10 μM; Santa Cruz Biotech), and the histone deacetylase (HDAC) inhibitor nicotinamide (NAM) (2 and 0.2 mM; Sigma), respectively.
Chromatin fractionation
Subcellular fractionation for chromatin extracts from HCT116 cells was performed following our previously published protocol with slight modifications (17). Briefly, 80–90% confluent HCT116 cells grown in 150 cm plates were washed twice in DPBS, and then lysed in ice-cold cytoplasmic lysis buffer (10 mM Tris–HCl pH 7.9, 0.34 M sucrose, 3 mM CaCl2, 2 mM MgCl2, 0.1 mM ethylenediaminetetraacetic acid (EDTA), 1 mM 1,4-dithiothreitol (DTT), 0.1% NP-40 and cocktail protease inhibitors (Thermo Scientific); 750 μl lysis buffer per 150 cm plate). After pelleting the nuclei by centrifugation at 3500 g for 15 min at 4°C, pellets were lysed in ice-cold nuclear lysis buffer (20 mM HEPES pH 7.9, 1.5 mM MgCl2, 3 mM EDTA, 150 mM K-acetate, 10% glycerol, 0.5% NP-40 and cocktail protease inhibitors; 200 μl lysis buffer per 150 cm plate), vortexed for 15 min at 4°C, followed by centrifugation at 14 000 rpm for 15 min at 4°C to pellet the chromatin. The supernatant was labeled as the soluble nuclear extract. The chromatin pellet was dissolved in chilled chromatin lysis buffer (150 mM HEPES pH 7.9, 1.5 mM MgCl2, 150 mM K-acetate, 10% glycerol and cocktail protease inhibitors; 150 μl lysis buffer per 150 cm plate), and incubated with 0.15 unit/μl of Benzonase (Novagen) at 37°C for 30 min, followed by centrifugation at 14 000 rpm for 15 min at 4°C. The supernatants (chromatin extracts) were collected for western blotting.
Antibodies
The following antibodies were used: α-H3 (Cell Signaling; #4499), α-H3K27Ac (Active Motif; #39685), α-H3K27Ac (Cell Signaling; 8173), α-H4K16Ac (Active Motif; #61529), α-H4K16Ac (Cell Signaling; #13534), anti-G-quadruplex DNA 1H6 (Millipore; #MABE1126), α-MeCP2 (AbCam; #ab2828), α-Pan-Methyl-H3K9 (Cell Signaling; #4473), α-methyl-histone H3K9 (Cell Signaling; #5327), α-RNA Pol II (Santa Cruz Biotechnology; #sc-899), α-APE1 (Novus Biologicals; #NB100-116). The α-NEIL1 antibody was as in (28); the α-AcNEIL1 antibody was custom-generated through EZBiolab using a chemically synthesized NEIL1 peptide (288APKGRKSRK*K*K*SKA301) with acetylated Lys (*) as hapten to induce IgG antibody in rabbit, as previously described (17).
Cytospin
Cells were harvested and washed twice with ice-cold phosphate-buffered saline (PBS); 1 × 105 cells were resuspended in 200 μL cold PBS containing 10% FBS. The cell suspension was added to the cytospin apparatus, followed by centrifugation at 800 rpm for 5 min (Shandon Cytospin 4 cytocentrifuge; Thermo Scientific). After removal from centrifugation, slides were dried at room temperature and fixed with 4% paraformaldehyde (PFA) for immunofluorescence staining.
Immunofluorescence and imaging
HCT116 cells were cultured onto glass coverslips and grown for 24 h (75% confluence), washed twice with PBS and fixed for 20 min with 4% paraformaldehyde at room temperature. Then cells were washed with PBS and permeabilized with 0.1% saponin for 30 min in PBS, and subsequently blocked in 2% BSA blocking buffer for 1 h. Next, cells were stained with primary antibodies overnight at 4°C. Coverslips were rinsed five times with PBS and subsequently incubated with anti-Rabbit IgG Atto 488 (Sigma-Aldrich; #18772) and anti-Mouse IgG Atto 555 (Sigma-Aldrich; #43394) (1:200) for 1 h at room temperature followed by further five washes in PBS. Cells were then incubated with or without 100 nM Acti-stain™670 phalloidin (Cytoskeleton, Inc) for another 30 min. After washing with PBS five times, coverslips were mounted on glass slides by using DAPI-containing mounting media (Invitrogen) and analyzed by an LSM710 confocal microscope (Carl Zeiss AG).
ChIP assays
Direct ChIP assays
ChIP assays were performed on exponentially growing cells (one 70–80%-confluent 10-cm plate for one ChIP reaction) with Magna ChIP Protein A/G magnetic beads (Millipore, # 16-663) using the custom-generated rabbit α-AcNEIL1 antibody, rabbit α-H3K27Ac (Cell Signaling; 8173) antibody, or control IgG as described below. After washing in PBS, cells were crosslinked with 1% formaldehyde (15 min at room temperature) in PBS. Crosslinked cells were washed three times in PBS and scraped off the plates into PBS containing a protease inhibitor (PI) cocktail (Thermo Scientific/Pierce; # 88666) and pelleted at 900 RPM (10 min, 4°C). Pelleted cells were lysed in sodium dodecyl sulphate (SDS) lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl pH 8, and PI), incubated on ice for 10 min and subjected to sonication (XL-2000 QSonica LLC) on ice, setting the pulse at 4 for 4–5 times with a 1 min interval in between, followed by centrifugation (14,000 RPM, 15 min, 4°C) to collect the sheared chromatin lysate. IP was performed in this chromatin lysate with 30 μl Protein A/G magnetic beads, the corresponding antibody (5 μg; control IgG was included in a separate IP) in a total volume of 2 ml (diluted 1:10 with ChIP dilution buffer: 0.01% SDS, 1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM Tris–HCl, pH 8.1, 167 mM NaCl, PI) overnight (4°C) with constant shaking. The next day, the IPs were washed sequentially with low salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8, 150 mM NaCl), high salt immune complex wash buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8, 500 mM NaCl), LiCl immune complex wash buffer (0.25 M LiCl, 1% NP40, 1% Na-deoxycholate, 1 mM EDTA, 10 mM Tris–HCl, pH 8) and TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8). The protein–DNA complexes were eluted in ChIP elution buffer (1% SDS, 0.1 M NaHCO3), de-crosslinked in 200 mM NaCl for 4 h at 65°C. ChIP DNA was purified by proteinase K digestion, RNAse treatment, phenol chloroform extraction and ethanol precipitation using standard protocols. The ChIP-purified DNA was finally dissolved in 10 mM Tris–HCl pH 8. The ChIP and 10% input DNA were subjected to SYBR GREEN-based quantitative polymerase chain reaction (qPCR) (7500 real-time PCR system; Applied Biosystems) with primers (Supplementary Table S2) and SYBR Premix Ex Taq (TaKaRa). Data are represented as percentage input according to http://www.lifetechnologies.com/us/en/home/life-science/epigenetics-noncoding-rna-research/chromatin-remodeling/chromatin-immunoprecipitation-chip/chip-analysis.html.
ChIP-seq assays
For ChIP-seq assays, the direct ChIP protocol above was followed with the following modifications: about 70%-confluent exponentially growing HCT116 cells in 10-cm plates (10–15 plates) were pelleted after crosslinking and PBS wash. Hypotonic lysis buffer (20 mM HEPES pH 7.9. 10 mM KCl, 1 mM EDTA, 10% glycerol, 1 mM DTT, PI) was added to the cell pellet, followed by incubation on ice for 15 min. Cells were then centrifuged at 900 RPM (10 min, 4°C), and RIPA buffer (10 mM Tris–HCl pH 8, 140 mM NaCl, 1% Triton-X 100, 0.1% SDS, 1% sodium deoxycholate, 1 mM DTT, PI) was added to the pellet, incubated on ice for 10 min to extract the nuclear lysate, which was sonicated 12 times. IP was carried out in the sheared chromatin lysate with 80 μl protein A/G magnetic beads, the corresponding antibody (10 μg; control IgG was included in a separate IP) in a total volume of 3–4 ml (diluted with ChIP dilution buffer) overnight at 4°C with constant shaking. Next day, the IPs were washed three times with RIPA buffer, then with PBS and finally with TE buffer. The protein-DNA complexes were eluted in ChIP elution buffer, de-crosslinked and finally the ChIP-ed DNA was purified by proteinase K digestion, RNAse treatment, phenol chloroform extraction and ethanol precipitation. The ChIP-purified DNA was finally dissolved in 10 mM Tris–HCl pH 8 and subjected to next-generation sequencing (NGS) on an Illumina HiSeq 4000 sequencer performing a 76-nt paired-end sequencing read format. ChIP DNA was quantified by Nanodrop Spectrophotometer and then used to create an NGS library following the TruSeq v2 (Illumina) protocol.
Co-IP assays
Co-IP assays were performed using nuclear extracts from HCT116 cells. Exponentially growing cells were first lysed in cytoplasmic lysis buffer (10 mM Tris–HCl pH 8, 0.34 M sucrose, 3 mM CaCl2, 2 mM MgCl2, 0.1 mM EDTA, 1 mM DTT, 0.1% Nonidet P-40 and PI) and nuclei were pelleted by centrifugation at 4000 RPM for 15 min at 4°C. Nuclear pellets were lysed in cell lysis buffer (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton-X, PI), followed by vortexing for 15 min at 4°C, and centrifugation at 14 000 RPM for 30 min at 4°C. The supernatant was the nuclear extract. The nuclear lysates were precleared by incubating with IgG and protein A (Millipore; 16-661) or protein A/G magnetic beads for 1 h at 4°C with constant shaking. The precleared lysates after separating from the magnetic beads were incubated overnight with the corresponding antibody (or IgG) and fresh protein A or A/G magnetic beads at 4°C with constant shaking. Next day, the beads were washed sequentially three times with Tris buffered saline (TBS) containing 0.05% Tween 20, followed by sequentially low salt, high salt, immune complex and TE wash buffers. The washed beads were eluted in Laemmli buffer followed by SDS-polyacrylaminde gel electrophoresis (PAGE) for Western blot analysis with the appropriate antibodies.
Real time RT-PCR assays
Total RNA from control and experimental cells was isolated with Qiagen RNeasy mini kit including on-column DNase1 digestion and processed for cDNA synthesis using the Superscript III first-strand synthesis kit (Invitrogen), following the manufacturer's protocol. RARβ2 expression levels were analyzed in samples with SYBR GREEN-based Real Time PCR and specific primers (Supplementary Table S2), using HPRT1 as an internal control. Data were represented as relative quantitation with respect to the reference samples set at 1 based on the 2−ΔΔCT method.
Cell imaging
HCT116 cells were grown on coverslip, fixed by the addition of 4% (w/vol) paraformaldehyde (PFA) pH 8.0 and washed six to seven times with PBS, pH 8.0. Cells were permeabilized with 0.5% Triton X-100 on ice for 5 min followed by three PBS washes, blocked with blocking buffer (PBS, 3% BSA, 5% FBS and 0.5% Triton X-100) for 2 h at room temperature or overnight at 4°C. Following three additional washes with PBS, cells were incubated with primary antibody overnight in PBS, 3% BSA and 0.5% Triton X-100. Cells were washed five to six times with PBS and then incubated with Atto488-conjugated secondary antibody. After 2–3 h incubation, cells were washed six to seven times with PBS and fixed with 1% PFA for 30 min. Following another five to six washes, coverslips were mounted onto a slide with mounting medium (0.1% p-phenylenediamine and 75% glycerol in PBS at pH 7.5–8.0) for FLIM or in Prolong Diamond for STED nanoscopy.
FLIM images were captured using a Leica SP5 II confocal microscope with internal FLIM detector or a Leica SP8 FALCON. Atto488 was excited at 900 nm with titanium–sapphire pumped laser (Mai Tai BB, Spectral Physics) with 710–920 nm tunability and 70 femtosecond pulse width. A Becker & Hickl SPC830 data and image acquisition card was used for time-correlated single photon counting (TCSPC); electrical time resolution was 8 picoseconds with a pixel resolution of 256 × 256. Data processing and analysis were performed using a B&H SPC FLIM analysis software. The fluorescence decays were fitted to a single exponential decay model. STED images were captured with a Leica TCS SP8 STED 3× microscope capable of continuous-wave Stimulated Emission Depletion (cwSTED) and gated STED imaging, equipped with a 405 nm diode laser and a tuneable super-continuum White Light Laser (WLL, 470–670 nm) for excitation, as well as a 592, 660 and 775 nm continuous wave depletion laser. An oil immersion objective (100×, NA 1.4) with optimal color correction was used for imaging fixed samples at ∼23°C embedded in refractive index-matched media. Raw data acquired using the Leica LAS X software were imported into the integrated Huygens deconvolution software and processed.
ChIP-seq analysis and quality control
ChIP sequencing was performed in the Advanced Technology Genomics Core (ATGC) Facility at MD Anderson Cancer Center. Briefly, Illumina compatible indexed libraries were prepared from 10–20 ng of Diagenode Biorupter Pico sheared ChIP DNA using the KAPA Hyper Library Preparation Kit (KAPA Biosystems, Inc.). Libraries were amplified by 15 cycles of PCR, then assessed for size distribution using the 4200 TapeStation High Sensitivity D1000 ScreenTape (Agilent Technologies) and quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher.). Equimolar quantities of the indexed libraries were multiplexed, nine libraries per pool. The pool was quantified by qPCR using the KAPA Library Quantification Kit (KAPA Biosystems) then sequenced in one lane of the Illumina HiSeq4000 sequencer using the 76 nt paired-end run format. Triplicate samples were subjected to NGS for AcNEIL1, Input and IgG controls. Fastq files were preprocessed with the Trim Galore wrapper (trim_galore –fastqc –length 20 –paired run1, run2) for adapter trimming (cutadapt) and reads quality control (FastQC). On average, 1.9 ± 1.6% (mean ± SD) sequence pairs were discarded because their length was below the threshold and 37 345 085 ± 2 279 420 (values are given as mean ± SD unless otherwise specified) reads passed the filters. Trimmed reads were mapped to both GRCh37/hg19 and GRCh38/hg38 human genome assemblies with Bowtie2 using the -q –very-sensitive parameter. The overall alignment rate was ∼1–2% higher for the hg38 than for the hg19 assembly. The alignment rate for IgG, 47.0 ± 6.0%, was low compared to Input (98.4 ± 0.1%) and AcNEIL1 (98.0 ± 0.9%). Thus, Input was used as control. Sorted bam files and bam indices were obtained with samtools. Sorted bam files were processed with ‘bedtools intersect’ to filter out a blacklist (file wgEncodeDacMapabilityConsensusExcludable.bed.gz from (http://hgdownload.cse.ucsc.edu/goldenpath/hg19/encodeDCC/wgEncodeMapability/ lifted to hg38 coordinates for the hg38 mapping) of genomic regions identified by the ENCODE consortium as yielding artifactually high signals. The utilities ‘macs2 filterdup’ and ‘macs2 predictd’ were then used to remove duplicates and to extract fragment length to build a shifting model for peak detection. Narrow peaks were called with ‘macs2 callpeak’ using both a Poisson-distributed shifting model or a ‘noModel’ process with the fragment length obtained from the ‘predictd’ parameter. The average number of peaks obtained from the two methods was similar (49 608 ± 10 741 for noModel and 49 955 ± 10 169 for model for hg38; p = NS). Therefore, we chose the peak collection from the model for subsequent analyses. ChIPQC was used to assess the quality of called peaks.
ChIP-seq downstream analyses
We used ChIPpeakAnno with the TxDb.Hsapiens.UCSC.hg38.knownGene library to annotate the peaks to genomic features using a proximal promoter cutoff of 2 kb (from −2 kb to TSS) and an ‘immediate downstream’ region cutoff of 1 kb (from TSS to 1 kb). The percentage values obtained were normalized to the number of bases comprising each genomic feature by running the python script extract_transcript_regions.py (https://github.com/stephenfloor/extract-transcript-regions) on the UCSC knownGenes.txt file (http://hgdownload.cse.ucsc.edu/goldenpath/hg38/database/). The number of bases comprising promoters was obtained from the UCSC refGene.txt file and a custom C++ script that provided a list of nonredundant genomic coordinates. The total number of bases covering genomic features were 63 159 536 for promoters, 32 933 397 for immediate downstream, 11 328 275 for 5′ UTRs, 46 852 188 for 3′ UTRs, 146 342 265 for exons, 1 642 804 337 for introns and 1 235 963 694 for intergenic regions.
We used two methods to map reads near TSSs. In the first, we used seqMiner. Specifically, we loaded the summit coordinates of the first AcNEIL1 replicate (A1.1) along with its bam file, set a seed value to 15 642 605, selected the hg38_refSeq file as reference, chose the four most prominent peak clusters (Figure 1B) and used the annotation file to plot the number of peaks relative to ±20 kb of TSSs in intervals of 250 bp. In the second method, we used an in-house script to provide a bp-resolution map near TSSs. To this end, we extended the summits’ coordinates by ±100 positions and then integrated these genomic coordinates within ±5 kb of TSS (from file refGene.txt) for all three replicates. This was performed either without any filtering or by selecting only non-redundant coordinates from the reads. In cases where there were common TSSs for multiple gene names (like FAM138A, C, F), only one TSS instance was selected.
Figure 1.

AcNEIL1 is predominantly localized in the nucleus. (A) 3D super-resolution STED nanoscopy of HCT116 cells comparing localization between AcNEIL1 (top; green) and total NEIL1 (TtNEIL1) (bottom; green) with H3K27Ac (magenta) and DNA (blue); scale bar, 10 μm. (B) Confocal microscopy images of HCT116 cells stained with DAPI for nuclear DNA (blue), anti-AcNEIL1 specific antibody (green) and F-actin for cytoplasmic cytoskeleton (yellow), and merged images, showing the efficient knockdown of NEIL1 by siRNA; scale bar, 10 μm. (C) Immunoblotting (IB) of AcNEIL1 and total NEIL1 after siRNA treatment in HCT116 cells. (D) Confocal microscopy and with fluorescence lifetime measurements and values displayed as false color lifetime images mapped with pixel-by-pixel corresponding lifetime values (histograms). Fluorescence lifetime imaging microscopy (FLIM) was used to measure the lifetime of Atto488-labeled AcNEIl1 as donor. FRET between Atto488-labeled AcNEIL1 and SiR-stained DNA in the presence of H3K27Ac (ii) or total H3 (iii). Reference lifetime with donor alone (i) and donor without the acceptor (SiR Hoechst) but with Atto594 (iv). Control for FRET between Atto488 and SiR Hoechst without Atto594-H3 (v). (vi) Control measuring the reference lifetime of APE1. (E) Immunoblotting (IB) of NEIL1, AcNEIL1 and histone H3 from the chromatin fractions of HCT116 cells. Histone H3 served as the loading control. (F) Co-immunoprecipitation (Co-IP) blots probing the association between AcNEIL1 and RNA Pol II, H4K16Ac, H3K27Ac, MeCP2 or H3K9me2 in IP complexes from HCT116 cells. The detection of NEIL1 in AcNEIL1-pulled down fractions is expected since anti-NEIL1 antibody recognizes both forms. (G) Co-IP blots showing total NEIL1 and AcNEIL1 in H3K27Ac-containing IP complexes in HCT116 cells.
We used ChIPpeakAnno to find peaks in common (PICs) among the three AcNEIL1 replicates and the genes containing these peaks. The P-values associated with the probability of any two peak clusters between two replicates overlapping were obtained by both omitting the parameter ‘totalTest’, in which case the estimated total number of binding sites was ∼113 900 and by specifying totalTest and then varying the estimated total number of AcNEIL1 binding sites.
Gene set enrichment analyses (GSEA) were performed with DAVID (https://david.ncifcrf.gov/) and IPA (https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/) for genes that contained PICs within 1, 2, 4, 6, 8, 10 kb of their TSSs (PIC genes) and, for IPA, five additional control gene sets, each containing 1000 random genes. P-values for DAVID’s analyses were corrected for multiple testing employing Benjamini's correction. Pathway analyses were from IPA.
External ChIP-seq data and analyses
The ChIP-seq data for modified histones (H3K27me3, H3K36me3, H3K9m3 and Input control in HCT116 cells were obtained from http://hgdownload.cse.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeSydhHistone/ and consisted of the bigWig files. The annotation file for the TSSs coordinates was gencode.v17.annotation.gtf from ftp://ftp.sanger.ac.uk/pub/gencode/release_17/. For XRCC1, we downloaded the original fastq file from the GEO (https://www.ncbi.nlm.nih.gov/geo/) repository (GSE95302, file SRR5282040) and processed it according to the pipeline we used here. For Pol β, we obtained the bedGraph file from GEO, accession number GSM2137770 and converted it to the bigWig format using the bedGraphToBigWig utility from UCSC. This file contained fewer reads than expected. The file for OGG1 was GSM2357433_CP-Sample_Flag-OGG1-Con-2.tdf from the GEO record GSM2357433; the file was first converted to a bedGraph format using the ‘igvtools tdftobedgraph’and then to bigWig. All these ChIP-seq data were derived from the MCF7 breast cancer cell line. When needed, files were converted to the appropriate assembly using ‘bwtool lift’ (29). For mapping the ChIP-seq peaks to TSSs, we used the ‘bwtool agg’ utility on the bigWig files and the hg19 assembly.
The H3K27ac and H3K9me3 ChIP-seq files used to map the peaks overlapping with AcNEIL1 genome-wide were ENCFF450AGJ and ENCFF077NZX, respectively, mapped to the hg38 assembly from https://www.encodeproject.org/reference-epigenomes/ENCSR361KMF/, which corresponded to the narrow-peaks replicates 1 and 2 obtained in the HCT116 cancer cell line in bed format. The intersection with AcNEIL1 peaks was performed by extending the center-peak positions by 100 bases on either side and retrieving the identical base positions from both input files.
Expression of genes with PICs
We used TCGA-Assembler to download gene-normalized RNA-seq expression data from TCGA available as rsem-transformed data (.rsem.genes.normalized_results files). Genes were separated into AcNEIL1-negative and AcNEIL1-positive according to whether or not they contained PICs, and log2(normalized rsem + 1) values were computed in both groups for all aggregated tumor types and for each individual tumor type for PIC genes. RNA-seq data for normal tissues were from (30). The resulting P-values (Welch's t-tests) were then used to build a 2D Euclidean hierarchical-clustering heat map with the R libraries ‘gplots’ and ‘RColorBrewer’. A similar heat map with the same color-range in P-values was used to aggregate the gene expression values for the five sets of control genes relative to all other genes. Expression for genes with PICs within 1 kb of TSS and for CBX8 was also compared between TCGA tumors and matched controls for those tumor types in which at least 10 control samples were available (15 total). For the hierarchical clustering of the RNA-seq data in normal tissues, AcNEIL1-positive and AcNEIL1-negative genes were highlighted as separate clusters on the y-axis. Hox gene expression data in normal tissues were obtained from the GTEx Portal at https://gtexportal.org/home/.
Survival analyses
For the Hox gene-related analyses, we first used the TCGA RNA-seq gene expression data and clinical information to conduct a comprehensive analysis of patient survival in all 33 available TCGA tumor types. To this end we divided patients with each tumor type into two groups: group 1, with expression of gene x above the mean; and group 2, with expression of gene x below or at the mean value. We then used groups 1 and 2 to compute the Kaplan–Meier survival curves and hazard ratios from the Cox proportional hazards regression model using the R libraries ‘dplyr‘, ‘survival’, ‘survminer’ and the function ‘coxph’. P-values were from log rank tests. For the gene correlation expression analyses (GCEA), accurate P-values were obtained through the c++ boost F- and t-distribution functions (https://www.boost.org), which enable the computation of values approaching the numeric limit of 2.2e-306. Tumor abbreviations are as follows. ACC, adrenocortical carcinoma; BLCA, bladder urothelial carcinoma; BRCA, breast invasive carcinoma; CESC, cervical squamous cell carcinoma and endocervical adenocarcinoma; CHOL, cholangiocarcinoma; COAD, colon adenocarcinoma; DLBC, lymphoid neoplasm diffuse large B-cell lymphoma; ESCA, esophageal carcinoma; GBM, glioblastoma multiforme; HNSC, head and neck squamous cell carcinoma; KICH, kidney chromophobe; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LAML, acute myeloid leukemia; LGG, brain lower grade glioma; LIHC, liver hepatocellular carcinoma; LUAD, lung adenocarcinoma; LUSC, lung squamous cell carcinoma; MESO, mesothelioma; OV, ovarian serous cystadenocarcinoma; PAAD, pancreatic adenocarcinoma; PCPG, pheochromocytoma and paraganglioma; PRAD, prostate adenocarcinoma; READ, rectum adenocarcinoma; SARC, sarcoma; SKCM, skin cutaneous melanoma; STAD, stomach adenocarcinoma; TGCT, testicular germ cell tumors, THCA, thyroid carcinoma; THYM, thymoma, UCEC, uterine corpus endometrial carcinoma; UCS, uterine carcinosarcoma; UVM, uveal melanoma. Somatic mutations in cancer were obtained from the Catalogue Of Somatic Mutations In Cancer (COSMIC, https://cancer.sanger.ac.uk/cosmic/), release v91, 7 April 2020, file CosmicGenomeScreensMutantExport.tsv.gz.
UCSC track visualization
To visualize the AcNEIL1 ChIP-seq data on the UCSC browser, we used macs2 with the ‘bdgcmp’ option to generate fold enrichment (FE) and logLR peak enrichment files in bedGraph format, which were then transformed into BigWig format with the bedtools ‘slop’ option, followed by sorting with bedClip and the UCSC utility bedGraphToBigWig. For correlations involving ChIP signals, we used the data from the FE bedGraph files and integrated the signals at each bp over 1 MB intervals. Intervals at the end of chromosomes or over sequence gaps containing <1 MB intervals were discarded. P-values for the correlations between ChIP-signals and transcription or gene density were derived from Welch's t-tests using custom c++ scripts.
Mutation data
A dataset of 25 472 007 SBSs in cancer genomes was obtained from COSMIC (https://cancer.sanger.ac.uk/cosmic/download) release v89 (19 May 2019) files CosmicGenomeScreensMutantExport.tsv and CosmicNCV.tsv, comprising coding and noncoding variants, respectively. The dataset was used after 2 763 747 (10.8%) duplicate entries were filtered out. The list of SNPs (single nucleotide polymorphisms) occurring in the general population was obtained from file snp151.txt (UCSC), which contained 683 635 300 entries. The file was filtered first by selecting field ‘genomic single’ and excluding fields ‘SingleClassTriAllelic’ and ‘SingleClassQuadAllelic’, which left 555 714 904 entries. The file was further filtered by selecting the data from the SweGen study, which yielded a total of 34 956 631 SNPs present in the Swedish population (31). Data from the Human Gene Mutation Database (HGMD Professional, version 2017.3; (32)) were downloaded and only variants for which genomic coordinates and TSS information provided by HGMD were available were used for the analysis (10 377 variants). All HGMD variant classes were included (i.e. DM, DM?, FP, DP and DFP; see Supplementary Table S3 for definitions).
Intrinsic protein disorder predictions
The Neil1 protein sequences for Homo sapiens (Q96FI4), Mus musculus (Q8K4Q6), Rattus norvegicus (Q4KLM0), Danio rerio (Q6TGW9), Bos taurus (F1MC42), Gallus gallus (F1NLM0), Ovis aries (W5NWT2), Gorilla gorilla (G3RFS6), Xenopus tropicalis (B1H3L2), Crassostrea gigas (K1R5G2), Xenopus laevis (Q6GLL8), Capitella teleta (R7VAR0) and Takifugu rubripes (H2UBV7) and their phylogenetic tree were obtained from UniProtKB at http://www.uniprot.org/uniprot/. Protein disorder predictions were evaluated using the online GeneSilico MetaDisorder Service at http://genesilico.pl/metadisorder/. The results selected for plotting were those obtained with MetaDisorderMD2, a method based on 13 disorder predictors.
Assembly of Alvinella pompejana genome and identification of Neil1 gene
To identify the Neil1 gene in the extreme thermophile Alvinella pompejana, we assembled the genome of this annelid from ∼43 gigabases (Gb) of short WGS reads from samples collected from hydrothermal vent sites (9°N, 50/104°W17) during a past expedition (33). For contig and scaffold assembly, we used SOAPdenovo (http://soap.genomics.org.cn/soapdenovo.html) with k-mer settings of 23–93 in steps of 10. Other variable settings were 100 and 151 for maximum read length, 436 for average insert, 100 and 143 for read length cutoff and 32 for map length, respectively. The best setting combination (k-mer 63, max read length 100 and read length cutoff 143) yielded a contig N50 of 3657 bp and a scaffold N50 of 22.13 kb. Next we performed a genome-wide ab-initio prediction of genes using Augustus (http://bioinf.uni-greifswald.de/augustus/) after training the software on an A. pompejana EST library merged from three laboratory sources (34). The combined genome and EST libraries yielded a proteome comprising >60 000 full-length proteins and partial peptides. A. pompejana Neil1 was identified in this annelid's proteome from a blast search (e-value, ∼5 × 10−105) using human NEIL1 as bait, which returned both full-length genomic and an EST entry from (34).
Human NEIL1 homologues
Protein sequence data for 241 human NEIL1 homologues were obtained with blastp (https://blast.ncbi.nlm.gov/Blast.cgi?PAGE=Proteins) and aligned using Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/). In Sauropsida, protein alignments with human NEIL1 were manually adjusted to account for a 4-aa insertion in Passeriformes after S295. In Supplementary Figure S6C, the lengths of lines do not imply divergence times.
Statistical information
Pairwise Student's t-tests and Welch's tests used two-tailed distributions. P-values for DAVID’s gene enrichment were corrected for multiple testing using the Benjamini–Hochberg procedure.
RESULTS
We recently showed that p300 acetylates NEIL1 (herein referred to as AcNEIL1) at residues Lys296, Lys297 and Lys298, which serves to increase DG activity and stabilize the enzyme on chromatin-bound complexes (17). We reasoned that defining the extent to which the cell uses this PTM to achieve genome-wide repair could provide insights into efficient damage recognition and the contribution of AcNEIL1 to modulating mutational loads in the context of both evolution and cancer genomes.
AcNEIL1 localization
Stimulated emission depletion (STED) nanoscopy provides super-resolution images through the selective deactivation of fluorophores. STED of human colorectal adenocarcinoma HCT116 cells labeled with anti-AcNEIL1 or anti-total-NEIL1 antibodies showed strong AcNEIL1 nuclear localization, as opposed to diffuse staining in nuclei and cytoplasm for non-acetylated NEIL1. AcNEIL1 staining superimposed upon histone H3 acetylated at lysine 27 (H3K27Ac), a marker of active enhancers and transcriptionally-active chromatin (Figure 1A). We therefore used confocal microscopy to examine AcNEIL1 localization in several human-derived cell lines with various degrees of ploidy, from hypodiploid (Skov3) to near-diploid (HCT116, Jurkat), aneuploid (HeLa, MDAMB231), near-triploid (PC3, K562) and hypertriploid (U2OS); in all cases, AcNEIL1 displayed preferential localization to the nuclei (Supplementary Figure S1A).
We also assessed AcNEIL1 localization in a chronic myelogenous leukemia cell line (HAP1), which has a distinctive near-haploid genotype; in these cells, we detected AcNEIL1 throughout the cell bodies (Supplementary Figure S1B). We validated the specificity of the anti-AcNEIL1 antibody using siRNA-mediated NEIL1 knockdown, which abolished both cellular NEIL1 staining (Figure 1B and Supplementary Figure S1C) and protein levels (Figure 1C and Supplementary Figure S1D). Thus, except for the near-haploid leukemia HAP1 cell line, the data shows AcNEIL1 staining appears to be confined to nuclei in cancer cells.
Next, we used fluorescence lifetime imaging microscopy (FLIM) to detect fluorescence resonance energy transfer (FRET) between donor, Atto488-labeled AcNEIL1 and acceptor, SiR-stained DNA. FRET occurs if donor and acceptor are within 10 nm of each other, which causes a reduction in donor fluorescence lifetime. The Atto488-labeled AcNEIL1 fluorescence lifetime centered around 2.35 nanoseconds (ns) (Figure 1Di), but left-shifted to 2.2 ns in the presence of labeled DNA (Figure 1Dii). This significant 150 picoseconds (ps) reduction in fluorescence lifetime indicates that AcNEIL1 localized to within 10 nm of chromosomal DNA. Cells were also counter stained with H3K27Ac and histone H3 with an Atto594-coupled secondary antibody (Figure 1Dii and iii). Despite histone proteins being wrapped around DNA, no FRET between Atto594-histone H3 and SiR-Hoechst was observed, perhaps reflecting a greater distance of the histone tails from DNA than the one observed for AcNEIL1. The specific nature of FRET between Atto488-labeled AcNEIL1 and SiR-Hoechst was confirmed with cells lacking SiR-Hoechst (Figure 1Div versus v).
H3K27Ac (Figure 1Dii) colocalized with AcNEIL1 on condensed chromosomes to a greater extent than total histone H3 (Figure 1Diii) and, likewise, AcNEIL1 yielded stronger fluorescent signal than total NEIL1 in nuclei (Supplementary Figure S2A, green trace). We did not observe FRET between SiR-DNA and Atto488-labeled total apurinic/apyrimidinic (AP) endonuclease 1 (APE1), a component of the BER pathway that is also stabilized on chromatin upon acetylation (35). We also co-stained AcNEIL1 with the heterochromatin marker HeK9me3 in HCT116 cells and used FLIM to determine their distance. Unlike with AcNEIL1 and H3K27ac, we found only a small portion of co-localization between AcNEIL1 and HeK9me3 (Supplementary Figure S2B). Likewise, with FLIM we did not see a shortening of the average fluorescence lifetime for the AcNEIL1/HeK9me3 pair relative to AcNEIL1 alone, suggesting negligible interaction between them (Supplementary Figure S2B).
We therefore performed western blotting to further assess the comparative recruitment of total NEIL1 versus AcNEIL1 at the chromatin in HCT116 cells. Immunoblotting showed that 50 μM of the HAT inhibitor garcinol reduced the level of AcNEIL1 compared to 10 μM. Hence the level of total NEIL1 was also proportionally decreased in the chromatin fractions compared to that of mock treatment. Likewise, treating the cells with the HDAC inhibitor NAM at varying doses increased the chromatin contents of both total NEIL1 and AcNEIL1 compared to the mock treatment (Figure 1E and Supplementary Figure S2C). These results, which support and extend our previous findings (17), suggest that recruitment of NEIL1 on the chromatin depends on its level of acetylation.
Co-immunoprecipitation (co-IP) experiments, in which either AcNEIL1 (Figure 1F) or H3K27Ac (Figure 1G) were pulled-down from cellular fractions confirmed close association between AcNEIL1 and H3K27Ac. In addition, histone H4 acetylated on lysine 16 (H4K16Ac) and RNA polymerase II were also associated with AcNEIL1, supporting the colocalization of AcNEIL1 within euchromatin domains. By contrast, no coprecipitation was observed for either methyl CpG binding protein 2 (MECP2) or histone H3 methylated on lysine 9 (H3K9me), both of which are markers of heterochromatin. We conclude that AcNEIL1 is recruited to nuclear foci in near-diploid and aneuploid cancer cells, where it forms chromosomal complexes within regions of transcriptionally active chromatin.
AcNEIL1 punctuates TSSs of genes involved in cancer and development
To obtain a genome-wide map of AcNEIL1 localization, we employed chromatin immunoprecipitation-sequencing (ChIP-seq) assays. From three independent experiments in HCT116 cells, anti-AcNEIL1 antibody achieved an ∼3-fold enrichment of reads within peaks (6.3 ± 1.1%; mean ± SD) relative to input controls (2.0 ± 0.1%, P = 0.002 from Student's t-test), with few reads in false-positive hit regions (blacklist regions, Supplementary Figure S3A and Table S1). A clustering heatmap on the co-occurrence of peaks also showed a net distinction between the AcNEIL1 samples and input controls (Supplementary Figure S3B), increasing our confidence in the interpretation of downstream analyses. Mapping of reads within genomic features, using either narrow- or broad-peak methods, revealed the highest AcNEIL1 densities within the coding portions of genes and their promoters (∼0.1% peaks/Mb), followed by introns (∼0.038% peaks/Mb) and lastly intergenic regions (∼0.01% peaks/Mb) (Figure 2A). Peak enrichment relative to controls varied along the PCR-amplified fragments (Figure 2B); however, AcNEIL1 binding occurred most frequently within 1 kb downstream of TSSs (Figure 2C and Supplementary Figure S3C).
Figure 2.

AcNEIL1 punctuates TSSs of genes associated with poor survival in cancer. (A) Annotation of AcNEIL1 peaks associated with genomic features for the 3 replicates. Pairwise P-values from Student's t-test between TSS to 1kb downstream (ImmDown) and other features. Results represent the mean ± SD. ** 0.001; *** < 0.001; n = 3. (B) Clusters of peaks enriched for AcNEIL1 (A1.1) relative to Input (I1.1) for the first replicate extended ±200 bp from the summits as obtained from seqMiner. (C) Annotation of peaks from (B) to TSSs using the seqMiner annotation file to display the number of peaks relative to ±20 kb of TSSs in intervals of 250 bp. (D) Venn diagram of PICs for the three AcNEIL1 replicates (A1.1, A1.2, A1.3) obtained with ChIPpeakAnno. (E) Main DAVID-enriched gene ontology (GO) categories for genes containing AcNEIL1 PICs obtained with ChIPpeakAnno. Gene sets, distance of PICs from TSSs. IPRO, INTERPRO; SM, SMART; USF, UP_SEQ_FEATURE. (F) Heat map of log rank P-values from Kaplan–Meier survival curves where expression of a Hox gene either above (blue) or below (pink) the mean value was a risk factor in cancer patients. (G) Bar plot of significantly enriched GO terms containing one or more Hox genes from GSEA using all genes associated with poor survival (log rank P-value < 0.001) in LGG and KIRC when expressed above mean levels.
Given our confidence in the specific PCR amplification of chromatin-bound AcNEIL1, we examined peaks that overlapped among the three replicates, which we termed ‘peaks in common’ (PICs) and which represented high-confidence AcNEIL1 binding sites. A total of 13 647 PICs (Figure 2D) were identified through the narrow-peak method (21 173 PICs were identified through the broad-peak method), which we assessed for statistical significance. The P-value, obtained from the hypergeometric test (using an estimate of 113 900 for the total number of binding sites, the amount of coding sequence and regulatory DNA and average peak width), was highly significant (Supplementary Figure S3D).
The functional annotation of genes containing PICs located from 1 to 10 kb from TSSs revealed the strong overrepresentation of homeodomain-containing Hox genes (Figure 2E), both for the narrow- and broad-peak methods. In addition, Ingenuity Pathway Analysis (IPA) identified a variety of ‘terms’ associated with genes implicated in cancer, whose P-values vastly exceeded those obtained from randomly chosen gene sets (Supplementary Figure S4A). IPA also revealed the enrichment of genes related to ‘gene expression’ and ‘cell movement’ (Supplementary Figure S4B). For cancer- and development-related genes, causal network analysis identified CBX8 (for cancer, Supplementary Figure S4C) and Hox genes (for development, Supplementary Figure S4D) as target nodes, suggesting that AcNEIL1 preferentially targets the TSSs of genes that are relevant to cancer and organ development.
High expression of PICs-containing Hox genes is associated with poor survival in cancer
To test the possible relevance of AcNEIL1-bound Hox genes to cancer, we performed a survey across the 33 TCGA tumor types to assess association between differential expression of Hox genes and patient survival by using the Kaplan–Meier estimator and Cox-derived hazard ratios. Of the 39 Hox genes, 12 were associated with poor prognosis in 17 types of tumor for a total of 43 cases; 40 cases in which high expression (above the mean in tumor samples) was a risk factor, and 3 in which low expression (below the mean in tumor samples) was a risk factor (Figure 2F). Of the 12 Hox genes, 7 (58%) contained AcNEIL1 PICs out of the 9 with PICs, more than expected by chance alone (P = 0.002, Fisher exact test). We therefore extended the survival analysis to all genes in all TCGA tumor types to perform a comprehensive gene set enrichment analysis (GSEA) and extracted gene ontology (GO) terms enriched in Hox genes. At a log rank P-value < 0.001 from the Kaplan–Meier estimator, for the association between high expression and poor survival, Hox genes were enriched in eight GO terms in low grade glioma (LGG), which were related to embryonic pattern specification, cell-matrix adhesion and transcription (Figure 2G). Two Hox-containing GO terms were also observed in kidney renal clear cell carcinoma (KIRC), both of which were related to transcriptional regulation (Figure 2G).
The association of Hox overexpression with poor survival raised the question whether other genes might have been responsible for the outome, rather than Hox. Therefore, we performed a comprehensive GSEA on all ∼20 000 genes in all 33 TCGA tumor types; in both LGG and KIRC, poor survival was dominated by the overexpression of genes related to the activation of cell cycle and mitosis (FDR values down to 1 × 10−15 in both cases) and, in LGG, to a strong activation of the innate immune response (FDR of 4.7 × 10−21). GO terms linked to mitotic genes also led the association with poor survival in other tumors, including adrenocortical carcinoma (ACC), mesothelioma (MESO), lung adenocarcinoma (LUAD), pancreatic adenocarcinoma (PAAD), kidney renal papillary cell carcinoma (KIRP) and breast invasive carcinoma (BRCA). Thus, cell hyperproliferation was the most common prognostic marker for poor survival in cancer. Of the tumor types in which a Hox overexpression-survival relationship was noted, ∼10 – 25 Hox genes were expected to be expressed, in some cases at high levels, in 8/12 of their putative adult tissues of origin (Supplementary Figure S5A). Therefore, we sought to clarify the relationship between cell hyperproliferation and Hox overexpression by selecting the top genes that were enriched in GO terms in at least 6 tumor types in the comprehensive survival analysis, and which would also be overexpressed in all tumor types when compared to matched controls (15 tumor types with matched controls were available). Thirty genes fulfilled our criteria, including two prominent transcription factors (TFs) with critical roles in cell prolideration, FOXM1 and MYBL2. This enabled us to conduct a gene coexpression analysis (GCEA) to assess the linkage between Hox overexpression and the activation of cell proliferation in LGG.
At a Bonferroni-corrected -log10 P-value threshold of 5.40 (regression coefficient r = ∼|0.200|), ∼80% of all Hox genes displayed significant co-expression with MYBL2 and FOXM1. Yet, only about half of the PICs-containing Hox genes did; moreover, 7/9 PICs-containing Hox genes showed coexpression levels below the median value, with HOXB1, HOXC12 and HOXD12 being expressed in <100/532 LGG cases (Supplementary Figures S5A and B). A list of all genes ranked by the strength of coexpression with FOXM1 and MYBL2 indicated -log10 P-values down to ∼10−250 with r-values up to 0.94 for genes such as HJURP and KIFC1, which play critical roles in mitosis. The ranking order was biphasic, with an estimated ∼200 genes being prime proliferative targets or co-amplified with MYBL2 and FOXM1, as assessed from initial slope extrapolation (Supplementary Figure S5B, insets), and genes with -log10 P-values below 18–20 likely representing weak, indirect transactivation and possibly non-casual relationships. In sum, analysis of the available data indicates that independently of other key drivers of cellular hyperproliferation, such as FOXM1 or MYBL2 (36), in LGG activation of Hox gene expression, particularly for PICs-containing Hox genes, leads to poor outcome.
Transcription spreads AcNEIL1 along gene bodies
Based on the co-IP results (Figure 1F and G), we anticipated that AcNEIL1 would localize to highly transcribed genes; thus, the colocalization between PIC-containing genes and the Hox family was surprising given that these transcription factors exhibit broadly restricted expression in adult tissues (37) (Supplementary Figure S5A). Indeed, quantitative transcriptomic data from 32 types of adult normal tissue (30) revealed that the 1148 PIC-containing genes with AcNEIL1 within 1kb of TSSs (A1_1kb) clustered mostly with weakly transcribed genes (Supplementary Figure S6A) and were expressed at lower levels than the remaining 19 196 genes (Figure 3A). The same pattern was observed in cancer tissues, where A1_1kb-associated genes were expressed at lower levels than the remainder of the genes for all 33 types of tumor from TCGA (Figure 3B, upper row). Despite these commonalities, A1_1kb-associated genes were expressed at higher levels in the tumor masses than in matched normal tissues for 7/15 tumor types, particularly in liver hepatocellular carcinoma and non-small cell lung carcinoma (Figure 3C); CBX8, in particular, was upregulated in all tumor types except chromophobe renal cell carcinoma (KICH, Supplementary Figure S6B).
Figure 3.

Transcription spreads AcNEIL1 along gene bodies. (A) Geometric box plot of aggregate transcript levels for genes with and without AcNEIL1 PICs within 1 kb of TSS in normal tissues. Dot, mean; bar, median; P-value from Wilcoxon test. (B) Left, 2D hierarchical clustering of P-values from Wilcoxon tests for transcript levels for genes with and without AcNEIL1 PICs within 1, 2, 4, 6, 8, 10 kb of TSSs in 33 TCGA tumor types (black labels) and in normal tissues (red label). Green, lower expression in genes with AcNEIL1 than in genes without AcNEIL1; red, higher expression in genes with AcNEIL1 than in genes without AcNEIL1. Scale, -log P-values. Note that unsupervised clustering ordered the P-values according to AcNEIL1 distances from TSSs (left label). Right, geometric box plots for transcript levels in 33 TCGA datasets for genes with AcNEIL1 at various distance from TSS; box widths scaled to the squared root of the number of observations: 37 392 (1 kb), 51 153 (2 kb), 69 568 (4 kb), 81 778 (6 kb), 91 645 (8 kb) and 99 367 (10 kb). P-values from Welch's t-tests. (C) Box plots of gene expression for genes with AcNEIL1 PICs within 1 kb of TSS in TCGA tumors and matched controls. Only control datasets with >10 samples were assessed. P-values from Wilcoxon tests. (D) Bottom, percent genomic positions within the immediate downstream 1 kb of TSS (ImmDown) for the three AcNEIL1 replicates at different fold enrichment values: 4.3 (≥4.2 and ≤4.31), 3.3 (≥3.26 and ≤3.31), 2.3 (≥2.25 and ≤2.48). Positions analyzed were from summits ±100 bases. The ensemble of genomic features included promoters (−2 kb to TSS), 5′-UTRs, ImmDown, exons and 3′-UTRs. Top, box plots of gene expression in normal tissues for genes with and without AcNEIL1 containing peaks at various fold enrichments (Peak enrichment) from the three replicates. P-values from Welch's t-tests. (E) Length of AcNEIL1 PICs identified with ChIPpeakAnno as a function of distance from TSSs; data show mean and SD; P-values from Welch's t-tests.
Next, we analyzed the expression profiles of PIC-containing genes when AcNEIL1 was located at increasing distances from TSSs. Transcription levels increased, in both TCGA (with the sole exception of acute myeloid leukemia) and in normal tissues, often exceeding those of AcNEIL1-negative genes (Figure 3B). This differential transcription between AcNEIL1-positive and AcNEIL1-negative genes was not due to sampling bias, as confirmed by a parallel analysis of five sets of 1000 randomly chosen genes each, which showed no differences in transcription levels between the random genes and the AcNEIL1-negative genes for 162/165 cases, and weak P-values for the remaining three cases (mesothelioma 0.03, testicular germ cell tumor 0.01 and uterine carcinosarcoma 0.04, Supplementary Figure S6C).
We then analyzed PICs in more detail, expecting PICs to contain the upper (strongest) end of P-values from the list of all called peaks from the three replicates, since these P-values would imply high-confidence AcNEIL1 mapping. Surprisingly, PIC analysis missed 1938/149 498 peaks (1.3%) with the strongest P-values and the highest enrichment ratios (>5.3, Supplementary Figure S6D). Indeed, the percentage of peaks mapping just downstream of TSSs were at their highest for both the most- and least-enriched peaks (Figure 3D), and the genes containing these peaks were expressed at low levels when located within 1 kb of TSSs. These apparent anomalies imply that peak enrichment per se is not a prime metric for AcNEIL1 binding. Rather, high-confidence AcNEIL1 binding appears to originate from the overlap of adjacent genomic binding areas, as deduced from the longer peak lengths near TSSs (Supplementary Figure S6E) and the smooth decrease in peak lengths that were observed when AcNEIL1 was found further away from TSSs (Figure 3E). These composite data show that AcNEIL1 binding to genomic DNA is fluid, in accordance with its lack of sequence-binding specificity and DNA-tracking properties. In sum, AcNEIL1 localizes to the TSS region in weakly transcribed genes; however, an increase in transcriptional activity may provide a larger or more long-lived open chromatin environment to enable wider genomic segments to be scanned by AcNEIL1.
Gene-dense and highly transcribed domains are enriched in AcNEIL1
To further examine NEIL1 locailzation, we tested whether AcNEIL1 mapping could be confirmed by quantitative assays. Therefore, we conducted AcNEIL1 ChIP at selected loci followed by quantitative PCR amplification (ChIP qPCR) (Supplementary Table S2) and compared the results using Chip-seq peaks visualized on the University of California, Santa Cruz (UCSC) genome browser. At the highly transcribed MYC locus, ChIP qPCR yielded a signal, ∼20% relative to the input, while ChIP-seq identified several strong peaks over broad regions (Figure 4A, left). By contrast, at the poorly transcribed MYL1 locus, the ChIP qPCR signal was ∼10-fold lower than at MYC and ChIP-seq displayed sparse peaks (Figure 4A, right). The ChIP qPCR signals for both AcNEIL1 and H3K27Ac at three high-transcription loci and three low-transcription loci revealed preferential AcNEIL1 occupancy at high-transcription loci (Figure 4B), confirming our observation that AcNEIL1 maps throughout transcribed gene bodies.
Figure 4.

Gene-dense and highly transcribed domains are enriched in AcNEIL1. (A) ChIP-seq enriched peaks for the AcNEIL1 replicates visualized on the UCSC genome browser in ln(logLR + 1) units (range = 0–3, top) with splicing isoforms (Genes) and direction of transcription (arrows); transcriptional profiling (RNA-seq) and histone H3 acetylated at Lys27 (H3K27Ac,) from ENCODE; AcNEIL1 ChIP qPCR quantitation (% relative to Input) at a strongly (MYC, left) and weakly (MYL1, right) transcribed gene. (B) AcNEIL1 and H3K27Ac ChIP qPCR quantitation (from A). Results represent the mean ± SEM; n = 4. P-values are from Welch's t-tests for the aggregate data at high (orange, 14.1 ± 3.2 mean ± SD) versus low transcription (blue, 3.2 ± 1.7 mean ± SD). (C–E) Modulation of promoter-specific AcNEIL1 levels during RA-induced transcriptional activation of RARB in HEK293 cells. (C) As in A for the RARB gene in HCT116 cells showing the paucity of AcNEIL1 peaks at TSSs. (D) RARB expression by RT-PCR normalized to HPRT1 expression; mean ± SEM, n = 3. (E) AcNEIL1 ChIP qPCR for the −165 to +82 region containing RARE on the RARB promoter versus a non-specific (ns) control region on chromosome 17 with no RARE after RA treatment; mean ±SEM, n = 3. (F) UCSC genome browser custom tracks for AcNEIL1 ICS peaks, as in A, for chromosome 1, showing all TSSs (maroon ticks) and gene density in TSS/Mb (top gray). (G) Plot of AcNEIL1 ICS (x-axis) for A1.2 versus gene density. Each point represents the integration over 1 Mb of ICS with FE (fold enrichment) output and the number of annotated TSSs over the same genomic region (y-axis). (H) As in G, the y-axis represents the RNA-seq fpkm data from averaged normal tissues from (22). (I) Profile of aggregate Chip-seq read depth near TSSs in HCT116 cells for AcNEIL1, XRCC1 and Pol β. Signals were normalized by the total amount of signal within the −5–5 kb interval.
We next explored whether AcNEIL1 is dislodged from its placement at TSSs upon transcriptional activation. The promoter for the retinoic acid (RA) receptor β gene (RARB) contains an RA-responsive element (RARE) which, when activated, stimulates gene expression and BER activity in response to a transient surge in ROS (38); however, in HCT116 cells RARE is not inducible by RA (39,40), and the RARB promoter is poorly occupied by AcNEIL1 (Figure 4C). By contrast, in the embryonic cell line HEK293, RARE is readily activated by RA (Figure 4D). Prior to activation, the RARB promoter was fully occupied by AcNEIL1 and full occupancy continued to be observed 30 min after RA activation, while transcription occurred at background levels. After 2 h, when transcriptional activation was detectable, the amount of AcNEIL1 had decreased by 5-fold (Figure 4E). Thus, this RA-inducible system provided proof-of-principle that AcNEIL1 found at TSSs is poised for transcription-induced repair.
On a chromosome-wide scale, the AcNEIL1 landscape comprises densely populated ‘cities’ interspersed with ‘deserts’, and the sex chromosomes were generally found to be devoid of cities (Figure 4F and Supplementary Figure S7). Close inspection of ChIP-seq peaks suggested that AcNEIL1 acted as a marker for high-transcription areas in closely spaced gene domains (Figure 4F); at the 1-Mb scale, the AcNEIL1 integrated ChIP-signal (ICS) correlated with both gene density (Figure 4G) and transcription level (Figure 4H). The strength of the correlation between AcNEIL1 and the transcription level was also strong for the different TCGA tumor types, with the sole exception of acute myeloid leukemia (Supplementary Figure S8A), where AcNEIL1 PIC-containing genes failed to display TSS distance-dependent increases in transcription (Figure 3B).
To assess the extent to which AcNEIL1 might be part of BERosomes preloaded onto chromatin, we conducted a comparison between AcNEIL1 and available ChIP-seq data for other BER components. Despite differences in methods used, the peak profiles for AcNEIL1, XRCC1 and Pol β near TSSs were remarkably similar, with a ‘gully’ at TSSs flanked by ridges (Figure 4I). By contrast, OGG1 displayed a prominent maximum at TSSs (Supplementary Figure S8B), as previously reported (41). The profiles for the modified histone H3K27ac were also qualitatively similar to that of AcNEIL1 close to TSSs (Supplementary Figure S8B), but contrasted with the marks of more condensed chromatin: H3K9me3, H3K36me3 and H3K27me3 (Supplementary Figure S8C). On a genome-wide scale, the overlap of AcNEIL1 peaks with those of H3K27ac was 11-fold greater than for those with H3K9me3 and 36-fold greater than the overlap between H3K27ac and H3K9me3 (Supplementary Figure S8D), in accordance with our IP, immunofluorescence and FLIM analyses. Visualization of the ChIP-seq peaks on the UCSC genome browser for a portion of chromosome 1 also indicated close correspondence between the BER components AcNEIL1, XRCC1 and the more limited data for Pol β (Supplementary Figure S8E). Taken together, these data provide a foundation for the concept that AcNEIL1 acts as a sentinel for surveilling DNA damage in open chromatin in the context of active BERosome complexes.
AcNEIL1 occupancy correlates with low mutation rates
If AcNEIL1 were primarily responsible for detecting and correcting DNA damage, we reasoned that mutational loads ought to decrease with increasing AcNEIL1 occupancy. We therefore analyzed ∼25.4 million SBSs specific to tumor samples, including 5.6 million coding region mutations and 19.8 million non-coding variants. When analyzing 20 1-Mb genomic bin pairs, where AcNEIL1 ICS was low (<350 000) in one member of the pair but high (>950 000) in the other member of the pair, and where each pair shared near-equal transcription levels, AcNEIL1-high bins exhibited significantly fewer SBSs than AcNEIL1-low bins (Supplementary Figure S9A). When SBSs were decomposed into the six contributing spectra (i.e. G>A plus C>T etc., simplified to G>A), all types of substitution occurred less frequently in AcNEIL1-high bins than in AcNEIL1-low bins, with the exception of G>A transitions, whose numbers remained unchanged (Supplementary Figure S9B).
To explore these observations further, we separately addressed the contributions of transcription and AcNEIL1 to mutagenesis, with the goal of capturing zeroth-order kinetic behaviors, where the rate, k, is derived from linear equation slopes, such that k = Δ[P]/Δ[X]. In this case, P was the number of SBSs per 1-Mb bins and X was either the AcNEIL1 concentration or mRNA levels, as a proxy for the amount of single-stranded DNA exposed to damage or modification by enzymes, such as apolipoprotein B mRNA-editing enzyme, catalytic polypeptide-like (APOBEC) (42,43). No AcNEIL1-low bins were associated with high transcription levels; therefore, we compiled six sets of 10 1-Mb bins containing AcNEIL1-high regions (>950 000 ICS), with varying levels of transcription. We excluded the bin at chr17:7000000 associated with TP53, which contained an abnormally high number of SBSs, 45 787/63 999 of which occurred solely at the TP53 locus. This bin also displayed the highest level of transcription (350.6 units). A plot of SBSs versus transcription levels revealed a positive and linear relationship (Figure 5A), consistent with a zeroth-order kinetics model. A total of five to six mutation spectra also revealed positive slopes, with G>A displaying an ∼10-fold higher rate than those for G>C, G>T, A>C and A>G, whereas A>T exhibited no changes (Figure 5B). Conversely, the plot for 351 1-Mb bins with variable amounts of AcNEIL1 ICS but no transcription (<5 units) followed zeroth-order behavior but the rate was negative (Figure 5C). Importantly, G>A transitions remained constant, whereas those for the other five types of substitution decreased, particularly A>T transversions (Figure 5D).
Figure 5.

AcNEIL1 occupancy correlates with low mutation rates. (A) Mutation loads in cancer genomes increase with transcription. Mutations were computed for 6 different types of genomic region each containing 10 1-Mb bins with high AcNEIL1 ICS (∼1 million counts/bin) but increasing transcription. Transcription data were from normal tissues. Data represent mean and SD (vertical bars for mutations and horizontal bars for transcription). The bin containing TP53 with 45 787 mutations was excluded. (B) Mutation loads in cancer genomes decrease with increasing AcNEIL1 density. Mutations were computed for 322 1-Mb bins each containing low transcription (0.5–5) as a function of AcNEIL1 ICS. (C) Deconvolution of mutation loads in cancer genomes into six mutation spectra as a function of transcription at steady AcNEIL1 concentration, from A. Data were fitted to linear regressions with rates representing dy/dx changes. P-values were from the regression coefficients. (D) Deconvolution of mutation loads in cancer genomes into six mutation spectra as a function of AcNEIL1 ICS and constant transcription from C. Data were fitted to linear regression with rates representing dy/dx changes. P-values were from the regression coefficients. (E–G) Genome-wide number of mutations as a function of AcNEIL1 ICS in cancer genomes. (E) G>A; (F) A>T; (G) A>C. Orange highlight, low AcNEIL1-containing bins enriched in olfactory receptor (OR) genes. (H–J) Genome-wide number of SNPs as a function of AcNEIL1 ICS in the Swedish population. (H) G>A; (I) A>T; (J) A>C. Orange highlight, low AcNEIL1-containing bins enriched in olfactory receptor (OR) genes. (K) Relative difference in base changes between cancer genomes and the germline. The percentage base changes occurring in all bins with low (≤5 × 105) versus high (>5 × 105) AcNEIL1 ICS in cancer genomes subtracted from those in the Swedish population.
On a genome-wide scale, the rates of G>A transitions increased steadily (Figure 5E) whereas those for the other five types of substitution displayed negative initial velocities, followed by near-constant SBS accumulations (Figure 5F and G), as expected. We conclude that transcription is an intrinsically mutagenic process, and that chromatin-bound AcNEIL1 is the active DG form of NElL1 in the context of BERosomes, which is responsible for the repair of oxidative DNA damage and the prevention of base-pair change accumulations, particularly transversion mutations at A:T base pairs.
Segmental patterns of DNA repair by AcNEIL1 are heritable
After establishing that the local variations in mutation rates observed in cancer genomes coincide with segmental AcNEIL1 occupancy, we next addressed whether similar patterns exist for genetic variations between populations. We selected SweGen, a database of single nucleotide polymorphisms (SNPs) in the Swedish population, which is comparable in size (∼23 million SNPs) to the cancer dataset. No qualitative differences were observed in the genome-wide distribution of SNPs as a function of AcNEIL1 ICS compared with those observed for cancer. Specifically, G>A transitions displayed a linear dependence on AcNEIL1 (Figure 5H), whereas the remaining five mutational spectra exhibited two distinct rates (Figure 5I and J), a higher rate at low ICS and a lower rate at high ICS. No qualitative differences were observed among different types of bins containing sparse AcNEIL1 ICS, which were associated with olfactory receptor (OR) genes (Figure 5F and I). Quantitatively, the frequency of incurring base changes at low AcNEIL1 (<500 000 ICS) was greater in cancer than in the germline, particularly for A>T and A>C transversions (Supplementary Figure S9C). Plots of SBSs or SNPs as a function of transcription level failed to distinguish two rates of base substitution (Supplementary Figure S9D and E), either on a linear or a logarithmic scale, since bins with and without AcNEIL1 were no longer distinguished.
Using custom scripts (36,44), we searched for DNA motifs that can fold into stable G4 structures and detected their enrichment at TSS-flanking regions (Supplementary Figure S9F), where some are expected to be incorporated into 5′-untranslated regions (UTRs). G4-DNA and its complementary C-rich strand contain unpaired bases, which are susceptible to DNA damage (45); AcNEIL1 colocalization at TSSs is suggestive of protection at these vulnerable sites. We surveyed the Human Gene Mutation Database (HGMD) to assess the frequency of variants at G4 DNA motifs in 5′-UTRs known to cause or predispose toward inherited disease. A total of 13 loci were identified (Supplementary Table S3); however, two of them, located respectively within the DNA repair genes RAD51 and XRCC1 with pivotal roles at the replication-repair interface (46,47), may confer an increased burden of morbidity upon the human population by increasing genome instability and hence susceptibility to cancer.
Evolutionary origin of the AcNEIL1 acetylation center
Given the key and prototypical role of AcNEIL1 in genome repair, we examined the evolutionary conservation of Neil1 and its acetylation center in the two main lineages of bilateria (560–0 Ma), deuterostomes and protostomes. We included the sequence of the polychaete worm A. pompejana, an ancestral extremophile residing in deep-sea hydrothermal vents (48), which we obtained from partial genome assembly from specimens collected during an expedition on the East Pacific Rise (Supplementary Figure S10A). A total of 241/260 species comprising all deuterostomes (540–0 Ma) and Lophotrochozoa (536–0 Ma), and part of the Ecdysozoa clade displayed strong evolutionary conservation (blastp values between 7e−64 and 33−112), although no significant hits were found in Nematoda and Hexapoda, which include the model genomes of Caenorhabditis elegans and Drosophila melanogaster (Figure 6A).
Figure 6.

Evolutionary origin of the AcNEIL1 acetylation center. (A) Phylogenetic tree of invertebrate species with (red) and without (black) human NEIL1 gene homologs, as assessed by blastp. (B) Sequence conservation at the acetylation center in NEIL1 C-ter flexible region. Black, >93% sequence homology among 241 species; blue, highly conserved lineage-specific amino acids; red, conserved Lys residues carrying the acetylation mark in human NEIL1.
The AcNEIL1 acetyl acceptors, Lys296, Lys297 and Lys298, are embedded within an intrinsically unstructured carboxyl-terminal (C-ter) domain (49) (Supplementary Figure S10B), whose disordered nature has been well-conserved (Supplementary Figure S10B–D) despite the high degree of ordered protein folds expected among thermophilic lifeforms. Notably, amino acids Gly283 and Gly286, which reside beyond the last structured portion of human NEIL1 (Phe281 in β10) and are not required for either glycosylase/lyase or DNA binding activity (50,51), have been extremely well conserved in all 241 species examined over the last 500 million years (Figure 6B). Likewise, the acetyl-acceptor center, weakly discernible in Protostomia and located just downstream of another highly conserved residue (Pro290, 93%), evidences strong evolutionary conservation across most of the deuterostome lineage. Thus, the evolutionary conservation of the disordered NEIL1 domain, which supports its putative fundamental role in reducing the number of oxidative mutations in cell genomes, is consistent with an acquired critical role in orchestrating oxidative DNA damage repair in bilateria, originating ∼500 million years ago during the buildup of free oxygen in the atmosphere.
DISCUSSION
We find that the map of local variations in mutation rates among cancer genomes coincides spatially with the map of chromatin-bound DG AcNEIL1. A fraction of NEIL1 is acetylated, resulting in its stable integration at selected genomic loci which, in turn, display lower mutation rates than loci without the integrated enzyme. This finding implies that the integrated AcNEIL1 performs local scanning for DNA damage and repair, likely in the context of functional BERosomes. The observed loading and stabilization of AcNEIL1 on chromatin is critically dependent upon PTM at three consecutive lysine residues (Lys297–299), an acetylation center that appears to be the result of consolidation among variable amino acids in protostomes, probably occurring during the Cambrian explosion, ∼540 million years ago, during a transition from sulfur to oxygen as an energy source. The nesting of the acetylation center within a conserved intrinsically disordered protein domain is a recurrent theme for the PTM-mediated regulation of signaling pathways and chromatin architecture (52), enabling supramolecular assemblies and liquid–liquid phase transition structures (53–55), which can organize euchromatin into large transcriptional hubs (56). Preloading of BER enzymes to chromatin may be required to access base lesions hidden within nucleosome-bound DNA (57,58).
Uneven AcNEIL1 mapping along chromosomes defines three portions of the genome that are differentially targeted for oxidative damage repair: the active transcriptome, which is heavily protected; a portion of the genome that is weakly transcribed and partially protected; and a portion that is not at all targeted for repair. This repair pattern appears deeply engraved into the cellular genetic program and is retained upon oncogenic transformation. G>A transitions occur independently of AcNEIL1 localization, and their increase in incidence as transcription levels increase demonstrates the vulnerability of the active transcriptome to mutations and the necessity of an effective repair program to protect its integrity. C>T (G>A) substitutions occur frequently at methylated CpG dinucleotides (59), particularly in single-stranded DNA where deamination of 5-methylcytosine to thymine, which produces G:T mismatches resulting in mutations (i.e. 5mC:G>T:G>T:A), is accelerated relative to duplex DNA (60). C>T transitions also arise from stable cytosine oxidation products, such as 5-hydroxycytosine (61). In addition, germline homozygous deletions in NTHL1 have been found to predispose to adenomatous polyposis and colorectal cancer and to cause an accumulation of C>T transitions (62), thereby supporting a role for NTHL1 in the global genome repair of oxidized G:C base pairs (63). 5-hydroxycytosine is a substrate for NEIL1 in vitro (64); however, it does not accumulate in Neil1−/− mice (65), in line with our observation that G>A substitutions do not correlate with NEIL1 occupancy. NEIL2 has been shown to stably interact with the transcriptional complex (66), and Neil2−/− mice display increased oxidative DNA damage in actively transcribed regions relative to wild-type littermates (57). Thus, although ChIP-seq data for NEIL2 are not yet available, it is conceivable that AcNEIL1 and NEIL2 share portions of the genomic surveillance space. Indeed, even an intersection of base and nucleotide excision repair has been seen for oxidative alkylations such as cigarette-smoke-derived O(6)-4-(3-pyridyl)-4-oxobutylguanine (67), implying that the pervasive nature of oxidative base damage may select for DG’s to have evolved over-lapping specificities that allow them to back each other up. Regarding transcription-associated mutagenesis, the tumor suppressor TP53, the most commonly mutated gene associated with cancer (68,69), strikingly resides within the most highly transcribed 1-Mb domain in the human genome, raising the intriguing possibility that susceptibility to human cancer may stem, in part, from intrinsic genome architecture.
In a separate Neil1−/− mouse model, 4,6-diamino-5-formamidopyrimidine (FapyA) and thymine glycol (TG) base-derivatives displayed the strongest increase relative to wild-type littermates, followed by 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) (70), all of which are validated substrates for NEIL1 (28,71–72). Studies on the mutagenic potential of formamidopyrimidines have suggested that direct base misincorporation can generate both transition and transversion mutations (73–75). TG blocks DNA replication and its bypass, aided by translesion synthesis polymerases, can yield mutations when Polθ is utilized (76). Nei-like DNA glycosylases exhibit distinct DNA substrate specificities as compared to the Endonuclease III/Nth family of DNA glycosylases. NEIL1 can excise bulky DNA adducts, such as aflatoxin-Fapy-dG (77), peptide-DNA crosslinks (78) and cyclo-deoxyadenosines (79). Such bulky DNA lesions occurring at A:T (leading to A>T,C mutations) and G:C (leading to G>T mutations) base pairs in transcribed DNA could be repaired specifically by NEIL1, but not by NTHL1, which would account for the mutator phenotype observed in NEIL1-deficient mammalian cells (80). These composite observations are in agreement with our kinetic rates and genome-wide mutation loads, which support DNA damage at A:T and G:C base pairs as preferred substrates for NEIL1 and the antimutagenic role of AcNEIL1 in human cells.
Besides the active transcriptome, NEIL1 marks the transcriptional origin of ∼1000 genes, including developmentally related genes of the Hox family and members of gene regulatory networks, such as the polycomb repressive complex 1-like members (CBX8, CBX4 and PCGF3), which repress Hox expression outside of target tissues (81). Our analysis that Hox overexpression, and especially AcNEIL1-containing Hox gene reactivation in low grade glioma, contributes to poor survival strengthens the growing support for their key role in tumorigenesis (82–84). Neil1−/− (or Neil2−/−) mouse embryoid bodies display neural defects, the downregulation of key developmental genes, including Hox genes, elevated levels of reactive oxygen species (ROS) and a pro-apoptotic TP53-associated DNA damage response (85). Neil1−/− mice that are challenged with ionizing radiation also show behavioral and neurological defects (86). ROS, which generate the formamidopyrimidines NEIL1 substrates (87), increase during neurogenesis, a process that is dependent on oxidative phosphorylation (88). These composite observations support an essential role for NEIL1 and other BER enzymes in the protection of the developing embryo from the harmful effects of endogenous DNA damage, a side effect of tissue differentiation during development. The confinement of AcNEIL1 near the TSSs of weakly expressed genes would, therefore, be consistent with transcriptional repression and the enforcement of Pol II promoter-proximal pausing (89,90), which would prevent AcNEIL1 from leaving the promoter area. The altered expression and mutations associated with polycomb complexes have also been linked to cancer (91,92), and it will be of interest to determine the extent to which escape from NEIL1 damage repair beyond the early developmental stages may contribute to tumorigenesis. The genome-wide base substitution patterns that are observed between population variation and cancer are similar; therefore, notwithstanding the potential roles played by carcinogens and other factors during cancer development, endogenous oxidative DNA damage may represent the strongest source of base changes for both cancer and population variation, with the two processes differing quantitatively due to the higher rates of oxidative stress that occurs in cancer.
In almost one third of the genome, AcNEIL1 peak density is <25% of that in gene-coding regions, and in ∼80 million bp this density is <10%. The genomic domains containing olfactory receptor (OR) gene family members, a region to which we mapped the highest mutation rates, is particularly poor in AcNEIL1 peaks. The OR gene family comprises ∼1000 members, in both mouse and human; however, >50% of these genes have been inactivated in the human lineage as compared to ∼20% of inactivated genes in the mouse (93,94). Evolutionary comparisons suggest that the loss of OR gene expression in primates may have been driven by anatomical (nose shape and size) and behavioral (transition from fruit-rich to leaf-rich diet) changes that alleviated the dependency on smell for survival (95). However, the high frequencies of SNPs in these genomic regions supports the view that the loss of OR genes is an ongoing process in humans and that the lack of oxidative DNA damage repair by NEIL1 and perhaps other BER enzymes has contributed to this loss.
Besides clarifying a key question in cancer biology related to the mechanisms underlying the variation in mutation rates in cancer, our work raises several points for future investigation. Whether PTM-dependent NEIL1 chromatin-stabilization also enhances its diffusion rates along DNA (96,97) remains to be explored. Whether the coelution of NEIL1 with RNA Pol II observed here reflects a functional or casual association also merits further validation, since NEIL1 maps to both within and outside transcriptional units. In similar vein, the association between early replication timing and low mutation loads (98) may underscore an association between BERosomes and replisomes in euchromatin, where they are abundant and preloaded on chromatin, but not in heterochromatin (late replication). This is particularly relevant in the context of NEIL1 given its recognized association with PCNA, FEN1 and other DNA replication factors (99–102). It will be of interest to assess the contribution of selection pressure in the context of population variation and differential mutational loads in transcribed versus non-transcribed regions, as it relates to uneven BERosome occupancy. That endogenous non-acetylated NEIL1 was found both in the cytoplasm and the nuclei contrasts with earlier work showing discrete nuclear localization of C-terminal fluorescently-tagged NEIL1 in HeLa and U2OS cells (103); an improved antibody for total-NEIL1, such as one designed against the key flexible C-terminal region (49) expected to induce monoclonal antibodies that recognize the intract protein (104), might help resolve such discrepancies. Notably, this same NEIL1 C-terminal acetylated region is reported to bind Poly(ADP-Ribose) Polymerase 1 (PARP1) (105), suggesting future NEIL1 localization studies might test the role of PARylation using PARP1 and poly(ADP-ribose) glycohydrolase (PARG) inhibitors (106,107). Because mouse models support shared substrate specificity for BER enzymes (108,109), despite distinct physical genomic targets (110), it will also be useful to determine the relative contribution of other BER and DNA repair enzymes, such as those associated with mismatch repair (111) or with the multi-repair pathway nuclease XPG associated with both nucleotide and base excision repair (112), to genome stability. More generally and illustrating how DG’s may overcome the pervasive nature of oxidative DNA base damage by over-lapping specificities that allow them to back each other up, the intersection of base and nucleotide excision repair has been seen for oxidative alkylations such as cigarette-smoke-derived O(6)-4-(3-pyridyl)-4-oxobutylguanine (67). This notwithstanding, the results presented here show that the prototypic BER DG NEIL1 is targeted to open chromatin sites for efficient surveillance and repair of oxidative DNA damage occurring during transcription. Thus, although transcription is DNA damaging due to opening DNA bases to increased oxidation and deanimation, mutational load from transcription depends upon repair efficiency, as directly shown here for NEIL1.
DATA AVAILABILITY
The datasets generated during the current study are available in the GEO repository at http://www.ncbi.nlm.nih.gov/geo/ with accession number GSE142324 and in GenBank at http://www.ncbi.nlm.nih.gov/genbank/ with accession number MT380562.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Katharina Schlacher and Chris Brosey for suggestions; Aloke Sarkar for cell lines; David Shin, Craig Cary and Jill Fuss for key efforts in obtaining A. pompejana samples for sequencing; and Stanislav Vitha for assistance with FLIM. The use of the Microscopy and Imaging Center facility at Texas A&M University is acknowledged. The Leica SP8 confocal microscope acquisition was supported by the Office of the Vice President for Research at Texas A&M University.
Author Contributions: S.S., C.Y., Z.Y., J.M. and M.L.H. performed ChIP, IP and IB experiments; A.B. and R.B.D. conducted bioinformatics analyses; Z.A., Z.Y. and S.S. performed STED nanoscopy, FLIM and nuclear staining; M.M. and D.N.C. analyzed HGMD data; A.B., J.A.T. and S.M. conceived the experiments and wrote the paper.
Notes
Present address: Shiladitya Sengupta, Texas Heart Institute, Houston, TX 77030, USA.
Present address: Chunying Yang, School of Biomedical Informatics, The University of Texas Health Science Center, Houston, TX 77030, USA.
Contributor Information
Albino Bacolla, Departments of Cancer Biology and of Molecular and Cellular Oncology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Shiladitya Sengupta, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA; Weill Cornell Medical College, Cornell University, New York, NY 10065, USA.
Zu Ye, Departments of Cancer Biology and of Molecular and Cellular Oncology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Chunying Yang, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA.
Joy Mitra, Department of Neurosurgery, Center for Neuroregeneration, Houston Methodist Research Institute, Houston, TX 77030, USA.
Ruth B De-Paula, Departments of Cancer Biology and of Molecular and Cellular Oncology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Muralidhar L Hegde, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA; Weill Cornell Medical College, Cornell University, New York, NY 10065, USA; Department of Neurosurgery, Center for Neuroregeneration, Houston Methodist Research Institute, Houston, TX 77030, USA.
Zamal Ahmed, Departments of Cancer Biology and of Molecular and Cellular Oncology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
Matthew Mort, Institute of Medical Genetics, School of Medicine, Cardiff University, Heath Park, Cardiff CF14 4XN, UK.
David N Cooper, Institute of Medical Genetics, School of Medicine, Cardiff University, Heath Park, Cardiff CF14 4XN, UK.
Sankar Mitra, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, TX 77030, USA; Weill Cornell Medical College, Cornell University, New York, NY 10065, USA; Houston Methodist Cancer Center, Houston Methodist Research Institute, Houston, TX 77030, USA.
John A Tainer, Departments of Cancer Biology and of Molecular and Cellular Oncology, University of Texas MD Anderson Cancer Center, Houston, TX 77030, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
MATERIALS AND CORRESPONDENCE
All requests for materials should be addressed to Prof. John A. Tainer.
FUNDING
National Institutes of Health (NIH) [CA158910, GM105090 to S.M., P01 CA092584 to S.M., J.A.T, R35 CA220430 to J.A.T, NS088645 to M.L.H.]; Cancer Prevention and Research Institute of Texas [RP180813 to Z.A., J.A.T.]; Robert A. Welch Chair in Chemistry (to J.A.T.); National Science Foundation [ACI-1548562, ACI-1134872]; Qiagen Inc (to M.M., D.N.C.). Funding for open access charge: NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1. Tubbs A., Nussenzweig A.. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017; 168:644–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. De Bont R., van Larebeke N.. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004; 19:169–185. [DOI] [PubMed] [Google Scholar]
- 3. Mol C.D., Izumi T., Mitra S., Tainer J.A.. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000; 403:451–456. [DOI] [PubMed] [Google Scholar]
- 4. Guan Y., Manuel R.C., Arvai A.S., Parikh S.S., Mol C.D., Miller J.H., Lloyd S., Tainer J.A.. MutY catalytic core, mutant and bound adenine structures define specificity for DNA repair enzyme superfamily. Nat. Struct. Biol. 1998; 5:1058–1064. [DOI] [PubMed] [Google Scholar]
- 5. Slupphaug G., Mol C.D., Kavli B., Arvai A.S., Krokan H.E., Tainer J.A.. A nucleotide-flipping mechanism from the structure of human uracil-DNA glycosylase bound to DNA. Nature. 1996; 384:87–92. [DOI] [PubMed] [Google Scholar]
- 6. Huffman J.L., Sundheim O., Tainer J.A.. DNA base damage recognition and removal: new twists and grooves. Mutat. Res. 2005; 577:55–76. [DOI] [PubMed] [Google Scholar]
- 7. Beard W.A., Horton J.K., Prasad R., Wilson S.H.. Eukaryotic base excision repair: new approaches shine light on mechanism. Annu. Rev. Biochem. 2019; 88:137–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dutta A., Yang C., Sengupta S., Mitra S., Hegde M.L.. New paradigms in the repair of oxidative damage in human genome: mechanisms ensuring repair of mutagenic base lesions during replication and involvement of accessory proteins. Cell. Mol. Life Sci. 2015; 72:1679–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wallace S.S. Base excision repair: a critical player in many games. DNA Repair. 2014; 19:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Barnes D.E., Lindahl T.. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu. Rev. Genet. 2004; 38:445–476. [DOI] [PubMed] [Google Scholar]
- 11. Banerjee A., Santos W.L., Verdine G.L.. Structure of a DNA glycosylase searching for lesions. Science. 2006; 311:1153–1157. [DOI] [PubMed] [Google Scholar]
- 12. Mol C.D., Parikh S.S., Putnam C.D., Lo T.P., Tainer J.A.. DNA repair mechanisms for the recognition and removal of damaged DNA bases. Annu. Rev. Biophys. Biomol. Struct. 1999; 28:101–128. [DOI] [PubMed] [Google Scholar]
- 13. Prasad R., Beard W.A., Batra V.K., Liu Y., Shock D.D., Wilson S.H.. A review of recent experiments on step-to-step “hand-off” of the DNA intermediates in mammalian base excision repair pathways. Mol. Biol. 2011; 45:586–600. [PMC free article] [PubMed] [Google Scholar]
- 14. Hitomi K., Iwai S., Tainer J.A.. The intricate structural chemistry of base excision repair machinery: implications for DNA damage recognition, removal, and repair. DNA Repair. 2007; 6:410–428. [DOI] [PubMed] [Google Scholar]
- 15. Howard M.J., Wilson S.H.. DNA scanning by base excision repair enzymes and implications for pathway coordination. DNA Repair. 2018; 71:101–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Parikh S.S., Putnam C.D., Tainer J.A.. Lessons learned from structural results on uracil-DNA glycosylase. Mutat. Res. 2000; 460:183–199. [DOI] [PubMed] [Google Scholar]
- 17. Sengupta S., Yang C., Hegde M.L., Hegde P.M., Mitra J., Pandey A., Dutta A., Datarwala A.T., Bhakat K.K., Mitra S.. Acetylation of oxidized base repair-initiating NEIL1 DNA glycosylase required for chromatin bound repair complex formation in human genome increases cellular resistance to oxidative stress. DNA Repair. 2018; 66-67:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moor N.A., Lavrik O.I.. Protein-protein interactions in DNA base excision repair. Biochemistry (Mosc). 2018; 83:411–422. [DOI] [PubMed] [Google Scholar]
- 19. Carter R.J., Parsons J.L.. Base excision repair, a pathway regulated by posttranslational modifications. Mol. Cell. Biol. 2016; 36:1426–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Prakash A., Cao V.B., Doublie S.. Phosphorylation sites identified in the NEIL1 DNA glycosylase are potential targets for the JNK1 kinase. PLoS One. 2016; 11:e0157860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bhakat K.K., Hazra T.K., Mitra S.. Acetylation of the human DNA glycosylase NEIL2 and inhibition of its activity. Nucleic Acids Res. 2004; 32:3033–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bhakat K.K., Mokkapati S.K., Boldogh I., Hazra T.K., Mitra S.. Acetylation of human 8-oxoguanine-DNA glycosylase by p300 and its role in 8-oxoguanine repair in vivo. Mol. Cell. Biol. 2006; 26:1654–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tomasetti C., Li L., Vogelstein B.. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017; 355:1330–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A. et al.. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013; 499:214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Helleday T., Eshtad S., Nik-Zainal S.. Mechanisms underlying mutational signatures in human cancers. Nat. Rev. Genet. 2014; 15:585–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gonzalez-Perez A., Sabarinathan R., Lopez-Bigas N.. Local determinants of the mutational landscape of the human genome. Cell. 2019; 177:101–114. [DOI] [PubMed] [Google Scholar]
- 27. Hegde M.L., Hazra T.K., Mitra S.. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008; 18:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hazra T.K., Izumi T., Boldogh I., Imhoff B., Kow Y.W., Jaruga P., Dizdaroglu M., Mitra S.. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2002; 99:3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pohl A., Beato M.. bwtool: a tool for bigWig files. Bioinformatics. 2014; 30:1618–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A. et al.. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347:1260419. [DOI] [PubMed] [Google Scholar]
- 31. Ameur A., Dahlberg J., Olason P., Vezzi F., Karlsson R., Martin M., Viklund J., Kahari A.K., Lundin P., Che H. et al.. SweGen: a whole-genome data resource of genetic variability in a cross-section of the Swedish population. Eur. J. Hum. Genet. 2017; 25:1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Stenson P.D., Mort M., Ball E.V., Chapman M., Evans K., Azevedo L., Hayden M., Heywood S., Millar D.S., Phillips A.D. et al.. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum. Genet. 2020; 139:1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shin D.S., Didonato M., Barondeau D.P., Hura G.L., Hitomi C., Berglund J.A., Getzoff E.D., Cary S.C., Tainer J.A.. Superoxide dismutase from the eukaryotic thermophile Alvinella pompejana: structures, stability, mechanism, and insights into amyotrophic lateral sclerosis. J. Mol. Biol. 2009; 385:1534–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holder T., Basquin C., Ebert J., Randel N., Jollivet D., Conti E., Jekely G., Bono F.. Deep transcriptome-sequencing and proteome analysis of the hydrothermal vent annelid Alvinella pompejana identifies the CvP-bias as a robust measure of eukaryotic thermostability. Biol. Direct. 2013; 8:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Roychoudhury S., Nath S., Song H., Hegde M.L., Bellot L.J., Mantha A.K., Sengupta S., Ray S., Natarajan A., Bhakat K.K.. Human apurinic/apyrimidinic endonuclease (APE1) is acetylated at DNA damage sites in chromatin, and acetylation modulates its DNA repair activity. Mol. Cell. Biol. 2017; 37:e00401-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bacolla A., Ye Z., Ahmed Z., Tainer J.A.. Cancer mutational burden is shaped by G4 DNA, replication stress and mitochondrial dysfunction. Prog. Biophys. Mol. Biol. 2019; 147:47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dunwell T.L., Holland P.W.. Diversity of human and mouse homeobox gene expression in development and adult tissues. BMC Dev. Biol. 2016; 16:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sengupta S., Wang H., Yang C., Szczesny B., Hegde M.L., Mitra S.. Ligand-induced gene activation is associated with oxidative genome damage whose repair is required for transcription. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:22183–22192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van der Leede B.M., van den Brink C.E., van der Saag P.T.. Retinoic acid receptor and retinoid X receptor expression in retinoic acid-resistant human tumor cell lines. Mol. Carcinog. 1993; 8:112–122. [DOI] [PubMed] [Google Scholar]
- 40. Moison C., Senamaud-Beaufort C., Fourriere L., Champion C., Ceccaldi A., Lacomme S., Daunay A., Tost J., Arimondo P.B., Guieysse-Peugeot A.L.. DNA methylation associated with polycomb repression in retinoic acid receptor beta silencing. FASEB J. 2013; 27:1468–1478. [DOI] [PubMed] [Google Scholar]
- 41. Hao W., Qi T., Pan L., Wang R., Zhu B., Aguilera-Aguirre L., Radak Z., Hazra T.K., Vlahopoulos S.A., Bacsi A. et al.. Effects of the stimuli-dependent enrichment of 8-oxoguanine DNA glycosylase1 on chromatinized DNA. Redox Biol. 2018; 18:43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chan K., Sterling J.F., Roberts S.A., Bhagwat A.S., Resnick M.A., Gordenin D.A.. Base damage within single-strand DNA underlies in vivo hypermutability induced by a ubiquitous environmental agent. PLoS Genet. 2012; 8:e1003149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Buisson R., Langenbucher A., Bowen D., Kwan E.E., Benes C.H., Zou L., Lawrence M.S.. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science. 2019; 364:eaaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bacolla A., Tainer J.A., Vasquez K.M., Cooper D.N.. Translocation and deletion breakpoints in cancer genomes are associated with potential non-B DNA-forming sequences. Nucleic Acids Res. 2016; 44:5673–5688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Fleming A.M., Burrows C.J.. Formation and processing of DNA damage substrates for the hNEIL enzymes. Free Radic. Biol. Med. 2017; 107:35–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bhat K.P., Cortez D.. RPA and RAD51: fork reversal, fork protection, and genome stability. Nat. Struct. Mol. Biol. 2018; 25:446–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Eckelmann B.J., Bacolla A., Wang H., Ye Z., Guerrero E.N., Jiang W., El-Zein R., Hegde M.L., Tomkinson A.E., Tainer J.A. et al.. XRCC1 promotes replication restart, nascent fork degradation and mutagenic DNA repair in BRCA2-deficient cells. NAR Cancer. 2020; 2:zcaa013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fontanillas E., Galzitskaya O.V., Lecompte O., Lobanov M.Y., Tanguy A., Mary J., Girguis P.R., Hourdez S., Jollivet D.. Proteome evolution of deep-sea hydrothermal vent alvinellid polychaetes supports the ancestry of thermophily and subsequent adaptation to cold in some lineages. Genome Biol. Evol. 2017; 9:279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Hegde M.L., Tsutakawa S.E., Hegde P.M., Holthauzen L.M., Li J., Oezguen N., Hilser V.J., Tainer J.A., Mitra S.. The disordered C-terminal domain of human DNA glycosylase NEIL1 contributes to its stability via intramolecular interactions. J. Mol. Biol. 2013; 425:2359–2371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Doublie S., Bandaru V., Bond J.P., Wallace S.S.. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc. Natl. Acad. Sci. U.S.A. 2004; 101:10284–10289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu C., Lu L., Zhang J., Yue Z., Song J., Zong S., Liu M., Stovicek O., Gao Y.Q., Yi C.. Tautomerization-dependent recognition and excision of oxidation damage in base-excision DNA repair. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:7792–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gibson B.A., Doolittle L.K., Schneider M.W.G., Jensen L.E., Gamarra N., Henry L., Gerlich D.W., Redding S., Rosen M.K.. Organization of chromatin by intrinsic and regulated phase separation. Cell. 2019; 179:470–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Danilenko N., Lercher L., Kirkpatrick J., Gabel F., Codutti L., Carlomagno T.. Histone chaperone exploits intrinsic disorder to switch acetylation specificity. Nat. Commun. 2019; 10:3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhou J., Zhao S., Dunker A.K.. Intrinsically disordered proteins link alternative splicing and post-translational modifications to complex cell signaling and regulation. J. Mol. Biol. 2018; 430:2342–2359. [DOI] [PubMed] [Google Scholar]
- 55. Uversky V.N. Intrinsically disordered proteins in overcrowded milieu: Membrane-less organelles, phase separation, and intrinsic disorder. Curr. Opin. Struct. Biol. 2017; 44:18–30. [DOI] [PubMed] [Google Scholar]
- 56. Furlong E.E.M., Levine M.. Developmental enhancers and chromosome topology. Science. 2018; 361:1341–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chakraborty A., Wakamiya M., Venkova-Canova T., Pandita R.K., Aguilera-Aguirre L., Sarker A.H., Singh D.K., Hosoki K., Wood T.G., Sharma G. et al.. Neil2-null mice accumulate oxidized DNA bases in the transcriptionally active sequences of the genome and are susceptible to innate inflammation. J. Biol. Chem. 2015; 290:24636–24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Montaldo N.P., Bordin D.L., Brambilla A., Rosinger M., Fordyce Martin S.L., Bjoras K.O., Bradamante S., Aas P.A., Furrer A., Olsen L.C. et al.. Alkyladenine DNA glycosylase associates with transcription elongation to coordinate DNA repair with gene expression. Nat. Commun. 2019; 10:5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Petljak M., Alexandrov L.B., Brammeld J.S., Price S., Wedge D.C., Grossmann S., Dawson K.J., Ju Y.S., Iorio F., Tubio J.M.C. et al.. Characterizing mutational signatures in human cancer cell lines reveals episodic APOBEC mutagenesis. Cell. 2019; 176:1282–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Frederico L.A., Kunkel T.A., Shaw B.R.. A sensitive genetic assay for the detection of cytosine deamination: determination of rate constants and the activation energy. Biochemistry. 1990; 29:2532–2537. [DOI] [PubMed] [Google Scholar]
- 61. Zahn K.E., Averill A., Wallace S.S., Doublie S.. The miscoding potential of 5-hydroxycytosine arises due to template instability in the replicative polymerase active site. Biochemistry. 2011; 50:10350–10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Weren R.D., Ligtenberg M.J., Kets C.M., de Voer R.M., Verwiel E.T., Spruijt L., van Zelst-Stams W.A., Jongmans M.C., Gilissen C., Hehir-Kwa J.Y. et al.. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015; 47:668–671. [DOI] [PubMed] [Google Scholar]
- 63. Galick H.A., Kathe S., Liu M., Robey-Bond S., Kidane D., Wallace S.S., Sweasy J.B.. Germ-line variant of human NTH1 DNA glycosylase induces genomic instability and cellular transformation. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:14314–14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Vik E.S., Alseth I., Forsbring M., Helle I.H., Morland I., Luna L., Bjoras M., Dalhus B.. Biochemical mapping of human NEIL1 DNA glycosylase and AP lyase activities. DNA Repair. 2012; 11:766–773. [DOI] [PubMed] [Google Scholar]
- 65. Rolseth V., Luna L., Olsen A.K., Suganthan R., Scheffler K., Neurauter C.G., Esbensen Y., Kusnierczyk A., Hildrestrand G.A., Graupner A. et al.. No cancer predisposition or increased spontaneous mutation frequencies in NEIL DNA glycosylases-deficient mice. Sci. Rep. 2017; 7:4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Banerjee D., Mandal S.M., Das A., Hegde M.L., Das S., Bhakat K.K., Boldogh I., Sarkar P.S., Mitra S., Hazra T.K.. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 2011; 286:6006–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Tubbs J.L., Latypov V., Kanugula S., Butt A., Melikishvili M., Kraehenbuehl R., Fleck O., Marriott A., Watson A.J., Verbeek B. et al.. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009; 459:808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Knijnenburg T.A., Wang L., Zimmermann M.T., Chambwe N., Gao G.F., Cherniack A.D., Fan H., Shen H., Way G.P., Greene C.S. et al.. Genomic and molecular landscape of DNA damage repair deficiency across The Cancer Genome Atlas. Cell Rep. 2018; 23:239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bailey M.H., Tokheim C., Porta-Pardo E., Sengupta S., Bertrand D., Weerasinghe A., Colaprico A., Wendl M.C., Kim J., Reardon B. et al.. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018; 173:371–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Calkins M.J., Vartanian V., Owen N., Kirkali G., Jaruga P., Dizdaroglu M., McCullough A.K., Lloyd R.S.. Enhanced sensitivity of Neil1(-/-) mice to chronic UVB exposure. DNA Repair. 2016; 48:43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Jaruga P., Birincioglu M., Rosenquist T.A., Dizdaroglu M.. Mouse NEIL1 protein is specific for excision of 2,6-diamino-4-hydroxy-5-formamidopyrimidine and 4,6-diamino-5-formamidopyrimidine from oxidatively damaged DNA. Biochemistry. 2004; 43:15909–15914. [DOI] [PubMed] [Google Scholar]
- 72. Rosenquist T.A., Zaika E., Fernandes A.S., Zharkov D.O., Miller H., Grollman A.P.. The novel DNA glycosylase, NEIL1, protects mammalian cells from radiation-mediated cell death. DNA Repair. 2003; 2:581–591. [DOI] [PubMed] [Google Scholar]
- 73. Gehrke T.H., Lischke U., Gasteiger K.L., Schneider S., Arnold S., Muller H.C., Stephenson D.S., Zipse H., Carell T.. Unexpected non-Hoogsteen-based mutagenicity mechanism of FaPy-DNA lesions. Nat. Chem. Biol. 2013; 9:455–461. [DOI] [PubMed] [Google Scholar]
- 74. Greenberg M.M. The formamidopyrimidines: purine lesions formed in competition with 8-oxopurines from oxidative stress. Acc. Chem. Res. 2012; 45:588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kalam M.A., Haraguchi K., Chandani S., Loechler E.L., Moriya M., Greenberg M.M., Basu A.K.. Genetic effects of oxidative DNA damages: comparative mutagenesis of the imidazole ring-opened formamidopyrimidines (Fapy lesions) and 8-oxo-purines in simian kidney cells. Nucleic Acids Res. 2006; 34:2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yoon J.H., Roy Choudhury J., Park J., Prakash S., Prakash L.. A role for DNA polymerase theta in promoting replication through oxidative DNA lesion, thymine glycol, in human cells. J. Biol. Chem. 2014; 289:13177–13185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Vartanian V., Minko I.G., Chawanthayatham S., Egner P.A., Lin Y.C., Earley L.F., Makar R., Eng J.R., Camp M.T., Li L. et al.. NEIL1 protects against aflatoxin-induced hepatocellular carcinoma in mice. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:4207–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McKibbin P.L., Fleming A.M., Towheed M.A., Van Houten B., Burrows C.J., David S.S.. Repair of hydantoin lesions and their amine adducts in DNA by base and nucleotide excision repair. J. Am. Chem. Soc. 2013; 135:13851–13861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Jaruga P., Xiao Y., Vartanian V., Lloyd R.S., Dizdaroglu M.. Evidence for the involvement of DNA repair enzyme NEIL1 in nucleotide excision repair of (5'R)- and (5'S)-8,5'-cyclo-2'-deoxyadenosines. Biochemistry. 2010; 49:1053–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Maiti A.K., Boldogh I., Spratt H., Mitra S., Hazra T.K.. Mutator phenotype of mammalian cells due to deficiency of NEIL1 DNA glycosylase, an oxidized base-specific repair enzyme. DNA Repair. 2008; 7:1213–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cheutin T., Cavalli G.. Loss of PRC1 induces higher-order opening of Hox loci independently of transcription during Drosophila embryogenesis. Nat. Commun. 2018; 9:3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Brotto D.B., Siena A.D.D., de B. II, Carvalho S., Muys B.R., Goedert L., Cardoso C., Placa J.R., Ramao A., Squire J.A. et al.. Contributions of HOX genes to cancer hallmarks: enrichment pathway analysis and review. Tumour Biol. 2020; 42:1010428320918050. [DOI] [PubMed] [Google Scholar]
- 83. Pomerantz M.M., Qiu X., Zhu Y., Takeda D.Y., Pan W., Baca S.C., Gusev A., Korthauer K.D., Severson T.M., Ha G. et al.. Prostate cancer reactivates developmental epigenomic programs during metastatic progression. Nat. Genet. 2020; 52:790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cimino P.J., Kim Y., Wu H.J., Alexander J., Wirsching H.G., Szulzewsky F., Pitter K., Ozawa T., Wang J., Vazquez J. et al.. Increased HOXA5 expression provides a selective advantage for gain of whole chromosome 7 in IDH wild-type glioblastoma. Genes Dev. 2018; 32:512–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Han D., Schomacher L., Schule K.M., Mallick M., Musheev M.U., Karaulanov E., Krebs L., von Seggern A., Niehrs C.. NEIL1 and NEIL2 DNA glycosylases protect neural crest development against mitochondrial oxidative stress. Elife. 2019; 8:e49044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Yang B., Figueroa D.M., Hou Y., Babbar M., Baringer S.L., Croteau D.L., Bohr V.A.. NEIL1 stimulates neurogenesis and suppresses neuroinflammation after stress. Free Radic. Biol. Med. 2019; 141:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cadet J., Douki T., Ravanat J.L.. Oxidatively generated base damage to cellular DNA. Free Radic. Biol. Med. 2010; 49:9–21. [DOI] [PubMed] [Google Scholar]
- 88. van den Ameele J., Brand A.H.. Neural stem cell temporal patterning and brain tumour growth rely on oxidative phosphorylation. Elife. 2019; 8:e47887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Pherson M., Misulovin Z., Gause M., Mihindukulasuriya K., Swain A., Dorsett D.. Polycomb repressive complex 1 modifies transcription of active genes. Sci. Adv. 2017; 3:e1700944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Vos S.M., Farnung L., Urlaub H., Cramer P.. Structure of paused transcription complex Pol II-DSIF-NELF. Nature. 2018; 560:601–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Schuettengruber B., Bourbon H.M., Di Croce L., Cavalli G.. Genome regulation by Polycomb and Trithorax: 70 years and counting. Cell. 2017; 171:34–57. [DOI] [PubMed] [Google Scholar]
- 92. Sashida G., Oshima M., Iwama A.. Deregulated Polycomb functions in myeloproliferative neoplasms. Int. J. Hematol. 2019; 110:170–178. [DOI] [PubMed] [Google Scholar]
- 93. Gilad Y., Man O., Paabo S., Lancet D.. Human specific loss of olfactory receptor genes. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:3324–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dong D., He G., Zhang S., Zhang Z.. Evolution of olfactory receptor genes in primates dominated by birth-and-death process. Genome Biol. Evol. 2009; 1:258–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Niimura Y., Matsui A., Touhara K.. Acceleration of olfactory receptor gene loss in primate evolution: possible link to anatomical change in sensory systems and dietary transition. Mol. Biol. Evol. 2018; 35:1437–1450. [DOI] [PubMed] [Google Scholar]
- 96. Ahmadi A., Rosnes I., Blicher P., Diekmann R., Schuttpelz M., Glette K., Torresen J., Bjoras M., Dalhus B., Rowe A.D.. Breaking the speed limit with multimode fast scanning of DNA by Endonuclease V. Nat. Commun. 2018; 9:5381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lee A.J., Wallace S.S.. Hide and seek: How do DNA glycosylases locate oxidatively damaged DNA bases amidst a sea of undamaged bases. Free Radic. Biol. Med. 2017; 107:170–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Koren A., Polak P., Nemesh J., Michaelson J.J., Sebat J., Sunyaev S.R., McCarroll S.A.. Differential relationship of DNA replication timing to different forms of human mutation and variation. Am. J. Hum. Genet. 2012; 91:1033–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Hegde P.M., Dutta A., Sengupta S., Mitra J., Adhikari S., Tomkinson A.E., Li G.M., Boldogh I., Hazra T.K., Mitra S. et al.. The C-terminal domain (CTD) of human DNA glycosylase NEIL1 is required for forming BERosome repair complex with DNA replication proteins at the replicating genome: dominant negative functin of the CTD. J. Biol. Chem. 2015; 290:20919–20933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hegde M.L., Hegde P.M., Bellot L.J., Mandal S.M., Hazra T.K., Li G.M., Boldogh I., Tomkinson A.E., Mitra S.. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:E3090–E3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dou H., Theriot C.A., Das A., Hegde M.L., Matsumoto Y., Boldogh I., Hazra T.K., Bhakat K.K., Mitra S.. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 2008; 283:3130–3140. [DOI] [PubMed] [Google Scholar]
- 102. Hegde M.L., Theriot C.A., Das A., Hegde P.M., Guo Z., Gary R.K., Hazra T.K., Shen B., Mitra S.. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 2008; 283:27028–27037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. McNeill D.R., Paramasivam M., Baldwin J., Huang J., Vyjayanti V.N., Seidman M.M., Wilson D.M. 3rd.. NEIL1 responds and binds to psoralen-induced DNA interstrand crosslinks. J. Biol. Chem. 2013; 288:12426–12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Fieser T.M., Tainer J.A., Geysen H.M., Houghten R.A., Lerner R.A.. Influence of protein flexibility and peptide conformation on reactivity of monoclonal anti-peptide antibodies with a protein alpha-helix. Proc. Natl. Acad. Sci. U.S.A. 1987; 84:8568–8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Noren Hooten N., Fitzpatrick M., Kompaniez K., Jacob K.D., Moore B.R., Nagle J., Barnes J., Lohani A., Evans M.K.. Coordination of DNA repair by NEIL1 and PARP-1: a possible link to aging. Aging. 2012; 4:674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Bryant H.E., Schultz N., Thomas H.D., Parker K.M., Flower D., Lopez E., Kyle S., Meuth M., Curtin N.J., Helleday T.. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005; 434:913–917. [DOI] [PubMed] [Google Scholar]
- 107. Houl J.H., Ye Z., Brosey C.A., Balapiti-Modarage L.P.F., Namjoshi S., Bacolla A., Laverty D., Walker B.L., Pourfarjam Y., Warden L.S. et al.. Selective small molecule PARG inhibitor causes replication fork stalling and cancer cell death. Nat. Commun. 2019; 10:5654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Chimienti G., Pesce V., Fracasso F., Russo F., de Souza-Pinto N.C., Bohr V.A., Lezza A.M.S.. Deletion of OGG1 results in a differential signature of oxidized purine base damage in mtDNA regions. Int. J. Mol. Sci. 2019; 20:3302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Hu J., de Souza-Pinto N.C., Haraguchi K., Hogue B.A., Jaruga P., Greenberg M.M., Dizdaroglu M., Bohr V.A.. Repair of formamidopyrimidines in DNA involves different glycosylases: role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem. 2005; 280:40544–40551. [DOI] [PubMed] [Google Scholar]
- 110. Le Page F., Klungland A., Barnes D.E., Sarasin A., Boiteux S.. Transcription coupled repair of 8-oxoguanine in murine cells: the ogg1 protein is required for repair in nontranscribed sequences but not in transcribed sequences. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:8397–8402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Frigola J., Sabarinathan R., Mularoni L., Muinos F., Gonzalez-Perez A., Lopez-Bigas N.. Reduced mutation rate in exons due to differential mismatch repair. Nat. Genet. 2017; 49:1684–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Tsutakawa S.E., Sarker A.H., Ng C., Arvai A.S., Shin D.S., Shih B., Jiang S., Thwin A.C., Tsai M.S., Willcox A. et al.. Human XPG nuclease structure, assembly, and activities with insights for neurodegeneration and cancer from pathogenic mutations. Proc. Natl. Acad. Sci. U.S.A. 2020; 117:14127–14138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during the current study are available in the GEO repository at http://www.ncbi.nlm.nih.gov/geo/ with accession number GSE142324 and in GenBank at http://www.ncbi.nlm.nih.gov/genbank/ with accession number MT380562.