


Abstract
RNA modifications can regulate the stability of RNAs, mRNA–protein interactions, and translation efficiency. Pseudouridine is a prevalent RNA modification, and its metabolic fate after RNA turnover was recently characterized in eukaryotes, in the plant Arabidopsis thaliana. Here, we present structural and biochemical analyses of PSEUDOURIDINE KINASE from Arabidopsis (AtPUKI), the enzyme catalyzing the first step in pseudouridine degradation. AtPUKI, a member of the PfkB family of carbohydrate kinases, is a homodimeric α/β protein with a protruding small β-strand domain, which serves simultaneously as dimerization interface and dynamic substrate specificity determinant. AtPUKI has a unique nucleoside binding site specifying the binding of pseudourine, in particular at the nucleobase, by multiple hydrophilic interactions, of which one is mediated by a loop from the small β-strand domain of the adjacent monomer. Conformational transition of the dimerized small β-strand domains containing active site residues is required for substrate specificity. These dynamic features explain the higher catalytic efficiency for pseudouridine over uridine. Both substrates bind well (similar Km), but only pseudouridine is turned over efficiently. Our studies provide an example for structural and functional divergence in the PfkB family and highlight how AtPUKI avoids futile uridine phosphorylation which in vivo would disturb pyrimidine homeostasis.
INTRODUCTION
RNA modifications play an important role in regulating the stability of various coding and non-coding RNAs, and influence gene expression (1,2). Across all organisms, there are presently more than 140 known post-transcriptional modifications in RNAs. Specific enzymes are required for RNA modification. Enzymes introducing and removing modifications are called writers and erasers, respectively, and the information stored in the modifications is interpreted via reader proteins (1–3). N6-Methylated adenine (m6A) and pseudouridine (Ψ), a C5-glycoside isomer of uridine (Figure 1A), are the most prevalent RNA modifications (3,4). N6-Methylated adenine occurs mainly in messenger RNAs (mRNAs) and regulates for example mRNAs stability, accessibility to RNA-binding proteins, and translation efficiency (3). By contrast, Ψ was first found in non-coding RNA, including transfer RNA (tRNA) and ribosomal RNA (rRNA) (5), and only recently in mRNAs (6,7). In Arabidopsis thaliana, there are 187 Ψ sites in rRNAs, 232 Ψ sites in tRNAs, and more than 450 Ψ sites in mRNAs (8). Ψ plays a role in the folding of rRNAs and in the stabilization of tRNA structures (9,10). In mRNA from yeast and human cells, pseudouridylation is regulated in response to changing cellular environments (6,7), and its presence alters translation (11). In plant mRNA, the role of pseudouridylation is not yet clear.
Figure 1.

Reaction scheme of pseudouridine catabolism and sequence comparison of PUKI from different sources. (A) In Arabidopsis thaliana, pseudouridine degradation requires two enzymatic reactions occurring in the peroxisome. PUKI phosphorylates pseudouridine with ATP as a phosphate donor to 5′-ΨMP, which is hydrolyzed into uracil and ribose 5-phosphate by PUMY. (B) The amino acid sequence of AtPUKI (At1g49350) is compared to that of other PUKI homologs from soybean (Glycine max), pepper (Capsicum annuum) rice (Oryza sativa), and a tobacco species (Nicotiana sylvestris). Highly conserved residues are shown in red and are boxed in blue, while strictly conserved residues are shown with a red background. Secondary structural elements defined in unliganded AtPUKI are shown for the corresponding sequences. Residues or structural elements are indicated using the following color code: residues in the substrate pocket for pseudouridine, black circles; ADP-binding residues, gray triangles; putative catalytic residues, black asterisks; the nucleoside substrate-binding loop, green bar; the small ATP-binding loop (Gly239–Asn241), magenta bar; the large ATP-binding loop (Pro297–Gly308), light blue bar. This figure was prepared using ESPript (48).
Compared to the knowledge regarding the biogenesis and regulatory functions of RNA modifications, little is known about the metabolic fate of non-canonical nucleotides derived from the degradation of modified RNAs. Metabolic turnover of m6A- or Ψ-containing RNAs produces the non-canonical nucleotides N6-methyl-adenosine monophosphate (N6-mAMP) or pseudouridine monophosphate (ΨMP) as products. Enzymes for the degradation of these modified nucleotides in eukaryotes have only recently been discovered (12,13). In Arabidopsis plants and human cells, N6-mAMP deaminase hydrolyzes N6-mAMP, generating inosine monophosphate (IMP) (12), which is an intermediate of either purine nucleotide biosynthesis or catabolism (14,15). The N6-mAMP deaminase reaction thus converts a non-canonical nucleotide into a nucleotide occurring in general nucleotide metabolism. The elucidation of the structure of N6-mAMP deaminase from Arabidopsis showed that this enzyme undergoes a ligand-induced conformational change and identified the amino acids in the active site that mediate the substrate specificity (16,17). The discovery of the in-vivo role of N6-mAMP deaminase has shown for the first time that eukaryotic cells can catabolize non-canonical nucleotides released from modified nucleic acids.
The catabolic pathway for ΨMP has recently been described in Arabidopsis (13). In this plant, a significant proportion of RNA degradation seems to occur in the vacuole (18,19), where breakdown of Ψ-containing RNA generates ΨMP (presumably 3′-ΨMP), which is then dephosphorylated to pseudouridine and exported to the cytosol (13). For degradation, pseudouridine must reach the peroxisome. The catabolic pathway for pseudouridine in the peroxisome consists of two enzymes: PSEUDOURIDINE KINASE (PUKI), which phosphorylates pseudouridine to 5′-ΨMP, and PSEUDOURIDINE MONOPHOSPHATE GLYCOSYLASE (PUMY), which hydrolyzes 5′-ΨMP producing uracil and ribose 5-phosphate (Figure 1A) (13). Both reaction products are intermediates of main metabolic pathways. Uracil may be reincorporated into uridine monophosphate in a so-called salvage reaction or may enter pyrimidine ring catabolism (20, 21). Ribose 5-phosphate may be activated to 5-phosphoribosyl 1-pyrophosphate needed for phosphoribosyl transfer reactions or may enter the pentose phosphate pathway. PUKI and PUMY are required for the removal of pseudouridine. Malfunction of these enzymes causes massive accumulation of pseudouridine, resulting in spurious formation of 5′-ΨMP catalyzed by cytosolic UMP kinases. Cytosolic 5′-ΨMP appears to be particularly toxic and causes delayed seed germination and growth inhibition (13).
Degradation of ΨMP was initially observed for pyrimidine auxotrophic Escherichia coli mutants (22). Later, detailed investigations of these mutants led to the discovery of PUKI and PUMY, called YeiC and YeiN in E. coli (23). Bioinformatic analyses suggested that PUKI and PUMY are present in many organisms including eukaryotes, but not in mammals (13,23,24). Interestingly, the kinase and the glycosidase reside on a single polypeptide in animals, but these putatively bifunctional enzymes have not been characterized so far. PUKI is a member of the phosphofructokinase B (PfkB) family of carbohydrate kinases (13,25). PUKI from Arabidopsis (AtPUKI; encoded at the locus At1g49350) has putative orthologs of highly similar sequence in other plants (Figure 1B) (13). PUKI sequences formed a well-supported clade when all PfkB kinases from five evolutionary distant plant species were phylogenetically analyzed (26). This indicates that PUKI has conserved sequence elements of functional relevance that are distinct from other PfkB family kinases. One intriguing biochemical observation for AtPUKI is its high specificity towards the non-canonical nucleoside pseudouridine (13), leading to the question of how AtPUKI discriminates its authentic substrate from other chemically and/or structurally similar pyrimidine nucleosides.
In this study, we determined crystal structures of AtPUKI in the absence of a ligand, its binary complex with adenosine triphosphate (ATP), and a ternary complex with pseudouridine and adenosine diphosphate (ADP). We also carried out mutational and kinetic analyses to investigate the substrate specificity of the enzyme. Our results provide a structural and catalytic rationale for the high preference of AtPUKI for the non-canonical nucleoside pseudouridine.
MATERIALS AND METHODS
Cloning and purification
A synthetic cDNA of AtPUKI (At1g49350) (Bioneer, Daejeon, South Korea) codon-optimized for expression in Escherichia coli was amplified by PCR. The resulting PCR product was cloned into a modified pET-28a vector (Merck, Kenilworth, NJ, USA) containing a tobacco etch virus (TEV) protease cleavage site at the junction between the coding sequence for an N-terminal (His)6-maltose binding protein (MBP) and the multiple cloning site. Later, we found that the (His)6-MBP tag could not be cleaved by the TEV protease during purification. For a facile cleavage of the tag, which is required for crystallization, additional linkers coding for Gly-Gly-Gly-Ser (GGGS) were inserted, resulting in the N-terminal region of the AtPUKI fusion protein containing the sequence (H)6-MBP-GGGS-GGGS-ENLYFQS (TEV protease cleavage site)–GGGS. Escherichia coli BL21 (DE3) cells (Merck) transformed with the resulting construct were cultured at 37°C in Luria-Bertani medium until the absorbance at 600 nm reached 0.6. Expression of AtPUKI was induced with 0.5 mM isopropyl-β-d-1-thiogalactopyranoside, followed by incubation at 12°C for 40 h. Cells were collected by centrifugation and sonicated in buffer A (50 mM Tris–HCl at pH 8.0, 300 mM NaCl, 2 mM dithiothreitol, and 5% [w/v] glycerol), and the supernatant was obtained by centrifugation at 30 000 × g for 1 h at 4°C. The fusion protein was purified using a BabyBio Ni-NTA column (Bio-Works Technologies AB, Uppsala, Sweden) equilibrated with buffer A, and was then eluted with buffer A containing an additional 0.5 M imidazole. The (His)6-MBP tag of AtPUKI was subsequently removed by a TEV protease treatment overnight at 4°C using a 20:1 molar ratio of AtPUKI to protease. The N-terminal His-tagged fragment and the TEV protease were removed by a BabyBio Ni-NTA column, and the tag-free AtPUKI was further purified by size-exclusion chromatography using a Superdex 200 column (GE Healthcare, Chicago, IL, USA) with buffer A.
For structural analyses of AtPUKI, seleno-l-methionine (SeMet)-substituted AtPUKI was expressed in E. coli BL21 (DE3) in M9 minimal medium containing the required amino acids according to a published protocol (27). The purification of SeMet-substituted AtPUKI was carried out as described before for the native AtPUKI.
Crystallization and structure determination
Crystallization was carried out by the sitting-drop vapor-diffusion method at 22°C using native and SeMet-substituted AtPUKI (8 mg/ml). Crystals of both native and SeMet-substituted AtPUKI were produced in crystallization conditions containing 0.1 M tri-sodium citrate and 22% PEG3350. Subsequently, co-crystallization of a binary complex with 2 mM ATP and 4 mM MgCl2, and a ternary complex with 4 mM ADP and 4 mM pseudouridine were also achieved under crystallization conditions identical to those for native AtPUKI. Prior to data collection, crystals for the binary and ternary complexes were further soaked in the crystallization solution plus 2 mM ATP and 4 mM MgCl2 for the binary complex or 4 mM ADP and 4 mM pseudouridine for the ternary complex. In all cases, 20% glycerol was used as a cryoprotectant for data collection at 100 K.
Diffraction data were collected with a 0.5° oscillation angle at beamline 7A of the Pohang Accelerator Laboratory (Pohang, South Korea). The diffraction images were indexed, integrated, and scaled with HKL2000 (28), and a CC1/2 statistical value of ∼0.5 was used for the high-resolution cut-off (29,30). All crystals belong to the space group P21, with two monomers in the asymmetric unit. Specifically, sub-structure determination and the initial phase calculations for the structure of SeMet-substituted AtPUKI were carried out with the single-wavelength anomalous dispersion method using the program PHENIX (31). The refined model of the SeMet-substituted AtPUKI structure was used as search model for a molecular replacement of its binary and ternary complexes, which was conducted using the PHENIX AutoMR program (31). The models were manually rebuilt by COOT (32) and refined with PHENIX (31). After several cycles of refinement, most residues of AtPUKI were located from Met1 to Leu378, except for highly disordered residues, particularly between Phe200 and Pro300. Details on data collection and refinement statistics are shown in Table 1.
Table 1.
Data collection and refinement statistics
| Data set | Unliganded AtPUKI | AtPUKI-pseudouridine-ADP | AtPUKI-ATP |
|---|---|---|---|
| PDB ID | 7C1X | 7C1Y | 7C1Z |
| Data collection | |||
| Crystal | SeMet | Native | Native |
| Wavelength (Å) | 0.97935 | 0.97935 | 0.97935 |
| Resolution (Å) | 50.0–2.4 | 50.0–2.1 | 50.0–2.1 |
| (2.49–2.40)a | (2.18–2.10)a | (2.18–2.10)a | |
| Unique reflections | 7,873 | 11,095 | 11,212 |
| Multiplicity | 3.4 (3.4) | 3.6 (3.7) | 3.5 (3.5) |
| Completeness (%) | 99.2 (99.3) | 99.6 (100.0) | 99.5 (99.7) |
| Mean I/sigma(I) | 13.4 (1.7) | 13.1 (1.1) | 15.3 (1.4) |
| Wilson B-factors (Å2) | 48.3 | 43.1 | 36.4 |
| R-mergeb | 0.13 (0.90) | 0.12 (1.13) | 0.10 (0.93) |
| CC1/2c | 0.97 (0.73) | 0.98 (0.52) | 0.99 (0.61) |
| Space group | P21 | P21 | P21 |
| Unit cell a, b, c (Å) | 80.7, 48.4, 92.8 | 79.3, 48.9, 91.8 | 80.6, 47.9, 91.9 |
| α, β, γ (°) | 90.0, 109.1, 90.0 | 90.0, 107.5, 90.0 | 90.0 109.0 90.0 |
| Refinement | |||
| R-workd (%) | 20.8 | 19.1 | 18.6 |
| R-freee (%) | 24.2 | 23.5 | 23.7 |
| No. of atoms | 5225 | 5380 | 5465 |
| Macromolecules | 5198 | 5209 | 5290 |
| Ligands | - | 171 | 66 |
| Water | 27 | 83 | 109 |
| RMS (bonds) (Å) | 0.003 | 0.008 | 0.008 |
| RMS (angles) (°) | 0.89 | 1.23 | 1.25 |
| Ramachandran | |||
| favored (%) | 97.4 | 98.0 | 96.7 |
| outliers (%) | 0.2 | 1.9 | 0.0 |
| Average B-factor (Å2) | 57.8 | 54.7 | 45.2 |
| Macromolecules | 57.8 | 54.8 | 45.0 |
| Ligands | - | 49.5 | 60.9 |
| Water | 57.4 | 49.0 | 47.6 |
aNumbers in parentheses refer to data in the highest resolution shell.
b R merge = Σ|Ih – <Ih>|/Σ Ih, where Ih is the observed intensity and <Ih> is the average intensity.
cThe CC1/2 is the Pearson correlation coefficient (CC) calculated from each subset containing a random half of the measurements of unique reflection.
d R work = Σ ||Fobs| – |Fcal||/ Σ|Fobs|
e R free is the same as Robs for a selected subset (5%) of the reflections that was not included in prior refinement calculations.
Activity assays
We employed two different assay protocols for AtPUKI: an enzyme-coupled assay (33,34) and a direct assay (35) not requiring coupling enzymes. The wild-type and various mutants of AtPUKI were purified as described above.
Given that the enzyme-coupled assay requires K+ and Mg2+ ions as co-factors for one of the coupling enzymes (pyruvate kinase, see below), a direct assay protocol was also needed to determine the effects of different monovalent and divalent cations on AtPUKI activity, and to determine the substrate preferences of AtPUKI toward various nucleotide triphosphates including ATP, guanosine triphosphate (GTP), cytidine triphosphate (CTP), and uridine triphosphate (UTP). Typically, the assay mixture included 40 mM Tris–HCl pH 7.5, 20 mM divalent cation, 50 mM monovalent cation, 0.003% phenol red, 4 mM ATP, and the wild-type AtPUKI. After incubation at 25°C for 2 min, 1.25 mM pseudouridine was added to initiate the enzyme reaction, and changes in the absorbance at 430 nm were monitored for 30 s. For the monovalent cation dependency of AtPUKI, its activity was measured in the absence or presence of 50 mM LiCl, NaCl, KCl, RbCl or CsCl. For the effects of divalent cations, the AtPUKI activity was also assayed in the absence or presence of 20 mM MgCl2, MnCl2 or CaCl2. For the substrate preference regarding the phosphate donor, the direct assay was performed in the presence of 1 mM ATP, GTP, CTP or UTP. All assays were conducted in triplicate.
The enzyme-coupled assay was employed for steady-state kinetic measurements using K+ and Mg2+ as mono- and di-valent cations, because these were the most stimulating ions for AtPUKI activity in the direct assay. Briefly, ADP released from an AtPUKI-dependent reaction was detected in the coupled assay, with concurrent consumption of NADH, via two sequential reactions by pyruvate kinase and the NADH-dependent lactate dehydrogenase. Specifically, the pre-reaction mixture consisted of 40 mM Tris–HCl pH 7.5, 20 mM MgCl2, 50 mM KCl, 0.2 mM NADH, 2 mM phosphoenolpyruvate, indicated concentrations of AtPUKI, 18–28 units of lactate dehydrogenase and 12–20 units of pyruvate kinase/ml, and one of the substrates (either pseudouridine or ATP). Following incubation at 25°C for 2 min, the other substrate was added to initiate the enzyme reaction. Then we monitored the decrease in absorbance at 340 nm using a UV–vis light spectrophotometer (Jasco, Tokyo, Japan). We verified that the concentrations of the coupling enzymes were saturating in our assay and that the initial velocity was dependent on the concentration of AtPUKI. The initial velocity was determined between 15 and 45 s and expressed as the corresponding ADP concentration change per min, using an extinction coefficient of 6220 M–1cm–1 at 340 nm for NADH. Sigmaplot (Systat Software, San Jose, CA, USA) was used to calculate the Km and Vmax.
RESULTS AND DISCUSSION
Structure of unliganded AtPUKI
The calculated molecular weight of monomeric AtPUKI is 40.3 kDa. The native protein had a molecular weight of ∼80 kDa determined by size-exclusion chromatography during protein purification, indicating that it is a dimer (Supplementary Figure S1). Consistent with the chromatographic result for the enzyme in solution, crystalline AtPUKI was also a dimer in the asymmetric unit, with a buried surface area of 2420 Å2 calculated from the PISA analyses (36). Therefore, the dimer observed in the crystal represents the biologically relevant dimeric state.
Monomeric AtPUKI (Met1−Leu378) is folded into an α/β domain, with a protruding small β-stranded domain (Figures 1B and 2A). The α/β domain is reminiscent of a Rossmann fold employing the β−α unit as a basic structural motif. In the α/β domain, juxtaposition of the β−α units results in a central β-sheet, with seven α-helices (α3–α9) on the convex side of the β-sheet and the remaining α-helices (α1, α2 and α10–α12) on the other side. These structural arrangements result in a crevice along the central β-sheet (Figure 2B, area in magenta). Specifically, the central β-sheet contains 10 β-strands that are positioned in the order of β5–β4–β1–β8–β9–β10–β11–β12–β13–β14 with a parallel orientation, except for β12 and β14. Among the β−α units, the β1−α1 and β5−α3 units on the edge of the central β-sheet contain ∼30-residue-long insertions in the loop region that are folded into β-strands: β2 and β3 in β1−α1, and β6 and β7 in β5−α3 (Figures 1B and 2A). The two consecutive β-strands in each insertion are folded in an antiparallel manner and protrude from the main domain. In the small domain, the antiparallel β-strands from the two β−α units are packed in an edge-to-edge orientation, resulting in a continuous four β-stranded domain (Figure 2A). The small domain is bent over the crevice on the top of the central β-sheet, leaving a cleft just underneath the small domain.
Figure 2.
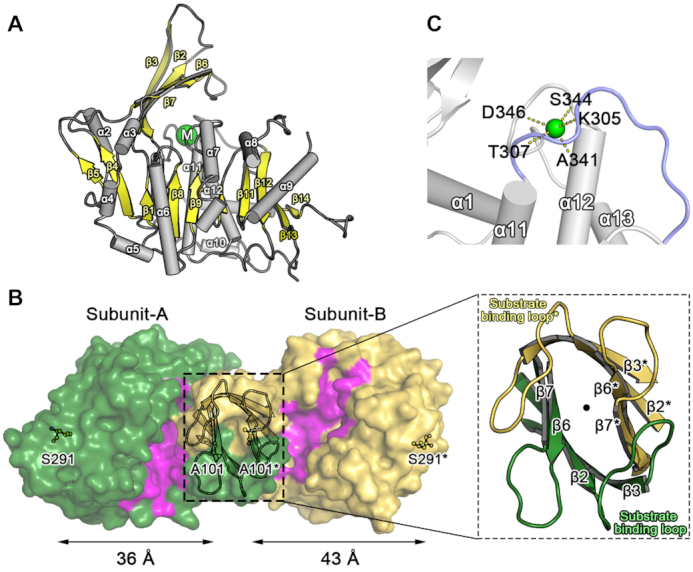
Monomeric and dimeric structures of AtPUKI. (A) The monomeric structure of unliganded AtPUKI is shown in a side view along the central β-sheet (yellow), with the protruding small β-stranded domain on the top (yellow). The monovalent ion binding site is indicated with a green circle containing the letter M. (B) Top view of dimeric AtPUKI is displayed in surface representation, with a zoom-in view of the β-clasp motif for dimerization. Rotation of the side view in (A) horizontally by 90° towards the reader would orient AtPUKI almost in the position of subunit-B shown in yellow. The surface in magenta forms a crevice comprising the binding sites for pseudouridine and ADP (see the characterization of the ternary complex). In subunit-A, the crevice surface is more extensive than in subunit-B. In a zoom-in view, the substrate-binding loops between β2 and β3 from both subunits are labeled and positioned almost in a two-fold symmetric manner around the perpendicular 2-fold axis running through the center, as indicated by the dot. (C) The monovalent cation-binding site in AtPUKI. The cation represented by the green sphere connects and stabilizes two loops: the loop between α12 and α13 (gray) and the large ATP-binding loop (light blue) preceding α11. The dashed lines represent coordination between the monovalent cation and the main chain carbonyl oxygen atoms of the indicated residues.
Dimerization of AtPUKI is mediated mainly by two elements: the small β-strand domain (Figure 2B) and α2 on the edge of the central β-sheet (Figure 2A). The face-to-face orientation with an almost orthogonal manner of the four β-strands in the small domains from both subunits results in a β-clasp motif for dimerization (37). In the β-clasp motif, there are two loops protruding from each subunit that are present in β2−β3 and β6−β7. These two loops are oriented in a two-fold symmetric way with the two equivalent loops from the adjacent subunit (Figure 2B, zoom-in view). In detail, the loop present in the β2−β3 strand of one subunit crosses over the β-clasp motif and interacts with the loop in the β6*−β7* strand of the adjacent subunit (*indicates residues or elements from the adjacent monomer) and vice versa. Later we found that the loop in β2−β3 (i.e., Ser18–Pro28) is involved in the active site of the adjacent subunit (see below). Therefore we refer to this loop as the substrate-binding loop in the following.
The two subunits of the dimer adopt different conformations. In one monomer, referred to as subunit-A, the area of the crevice along the central β-sheet is less exposed compared to the other monomer (i.e. subunit-B) (Figure 2B). These features originate from differences in the bending angle of the small β-strand domains toward the crevice on the large α/β domain (Supplementary Figure S2). Structural superposition showed that the large α/β domains of the two subunits maintain an essentially identical conformation, with a root-mean-square-deviation (RMSD) of 0.63 Å for the 298 Cα atoms in the large domain. However, due to differences in orientation, the small domains of both subunits showed large positional deviations, up to 10 Å, for their corresponding Cα atoms (Supplementary Figure S2). In fact, the small domain in subunit-A, specifically the β6 and β7 regions, is bent by 15 degrees more compared to that in subunit-B, leaving the crevice in subunit-A less accessible from the surface. Accordingly, the inter-atomic Cα distance between Ala101 in the small domain β7 region and Ser291, the residue most distant from the small domain, is different for the respective subunits: ∼36 Å for subunit-A and ∼43 Å for subunit-B. This asymmetry indicates that the small domains, particularly the β6 and β7 regions, have dynamic features in AtPUKI.
Structural homologs of AtPUKI and the structure of the monovalent cation-binding site
Sequence analyses indicated that AtPUKI is a member of the PfkB family (25). Accordingly, structural features of monomeric and dimeric AtPUKI are also highly homologous to those of this protein family. A structure similarity search using the program DALI (38) indicated that the AtPUKI monomer exhibits high structural homology with the PfkB family kinases catalyzing the phosphorylation of ribose, other carbohydrates, and adenosine, with a Z-score > 20. Among those structures, E. coli ribokinase (EcRBSK; Protein Data Bank [PDB] ID, 1RK2) (39) is the closest homolog with a Z-score of 37.7, along with other ribokinases from A. thaliana (AtRBSK; PDB ID, 6ILR; Z-score, 35.0) (34) and Vibrio cholerae (PDB ID, 4XDA; Z-score, 34.8) (40). Human adenosine kinase (PDB ID, 1BX4; Z-score, 30.6) is also homologous to the AtPUKI monomer. Due to these structural similarities, we adopted the structural nomenclature from ribokinase, particularly for the ATP binding features (34,37).
A direct (i.e. coupling enzyme-independent) activity assay showed that AtPUKI reaches maximum activity in the presence of the monovalent cation K+, the divalent cation Mg2+, and with ATP as substrate (Figure 3A–C). These functional features are essentially identical to those of ribokinase. Inorganic phosphate is not necessary for AtPUKI activity (Figure 3D). This is different from ribokinase of E. coli and from human adenosine kinase, which require inorganic phosphate as an activator (41,42). The binding sites for the cations and ligands in ribokinases from various sources are well characterized (34,37,40). In AtPUKI, the monovalent cation-binding site was present at the location corresponding to that in ribokinases. It is formed by two adjacent loops: one between α12 and α13, and the other preceding α11, which was defined as the large ATP loop in ribokinase for the ATP-binding element (Figure 2C). Most likely a sodium ion occupies the position of the monovalent cation in our structure, based on the results of metal and refinement analyses (Supplementary Figure S3), although more direct evidence is required to fully ascertain the identity of the metal. It is coordinated by the main chain carbonyl oxygens of Lys305, Thr307, Ala341, Ser344 and Asp346, and the coordination shell differs from the square bipyramidal shell in AtRBSK with six ligands (34). The binding environment of the divalent cation Mg2+ is unique to AtPUKI. It will be discussed below in the description of the binary complex with ATP (see the section ‘Mg2+-binding site and catalysis in AtPUKI’).
Figure 3.
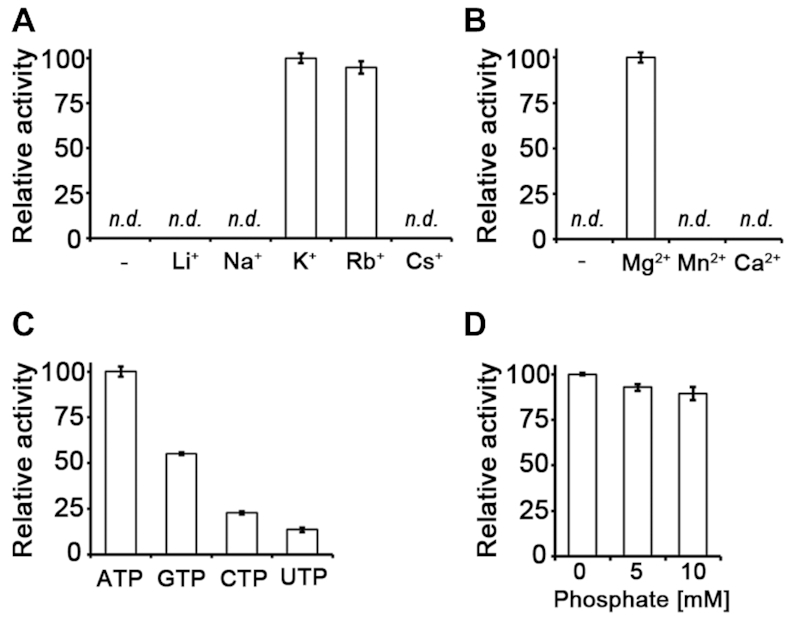
Effects of metal ions, different nucleoside triphosphates, and phosphate on AtPUKI activity. The relative activity depending on (A) monovalent cations (each at 50 mM), (B) divalent cations (each at 20 mM) and (C) different nucleoside triphosphates as phosphate donors (each at 1 mM) was assessed with a direct activity assay. (D) Using an enzyme-coupled assay, the effects of phosphate on AtPUKI activity were measured. The mean value of the highest measurement was set to 100% in each panel. Error bars are SD (n = 3). n.d., not detected.
The ternary complex of AtPUKI with pseudouridine and ADP
We determined the crystal structure of AtPUKI complexed with pseudouridine and ADP. By co-crystallization and further soaking of the crystal with both ligands, we achieved unequivocal electron densities for pseudouridine and ADP (Figure 4A). The structure of the ternary complex displays conformational differences compared to the unliganded AtPUKI (Supplementary Figure S4). However, there are virtually no changes in the tertiary structures of both subunits, with RMSD values of 0.67 Å for the 351 Cα atoms of subunit-A and of 0.48 Å for the 342 Cα atoms of subunit-B between the ternary complex and the unliganded AtPUKI. The changes occur mainly in the quaternary structure by a rigid-body rotation of one subunit. As a result, the inter-atomic Cα distances between reference points on both subunits are altered. For example, the distance between the two equivalent Pro202 residues in the two subunits is reduced by ∼3 Å due to substrate binding (Supplementary Figure S4). In the ternary complex, the substrate pseudouridine and the product ADP are bound to both subunits.
Figure 4.

Structural features for the ternary complex of AtPUKI with pseudouridine and ADP. (A) The binding sites of pseudouridine and ADP in subunit-A are displayed, with an overlaid omitted Fo-Fc electron density map contoured at 2.5 σ. This enlarged surface representation is a top view of the crevice (magenta) from subunit-A of the ternary complex. The orientation of the ternary complex is similar to the one shown in Figure 2B, illustrating that the small domain (green) partly covers pseudouridine and ADP in the crevice. The proposed catalytic base Asp311 indicated as blue surface is located in the vicinity of the O5′ ribosyl moiety of pseudouridine. (B) The binding environment of pseudouridine is shown for subunit-A of the ternary complex. Residues are classified in the following color code: small domain residues, green; residues from subunit-B, yellow and asterisk; catalytic base Asp311, blue label; other residues, gray. The dashed lines indicate possible hydrogen bonds within 3.0 Å. Notably, the substrate-binding loop from subunit-B is part of the subunit-A active site, and Thr26* therefore interacts specifically with the N1 of the nucleobase of pseudouridine. (C) The binding environment of pseudouridine in subunit-B is shown. For comparison with that of subunit-A, the orientation of the view in subunit-B is adjusted to be almost identical to that of (B). Residues in subunit-B are indicated with an asterisk and classified using the following color code: small domain residues, yellow; residues from subunit-A, green; catalytic base Asp311, blue label; other residues, gray. Notably, the hydrophobic residues in the β6* and β7* region of the small domain and the substrate-binding loop from subunit-A are farther distant from the nucleobase (see text for details) in subunit-B than in subunit-A. (D) The ADP-binding mode is shown in subunit-B of the ternary complex, with the small ATP-binding loop (Gly239–Asn241, magenta) and large ATP-binding loop (Pro297–Gly308, light blue) as defined in Figure 1(B). The adenine base is enclosed by hydrophobic residues and the monovalent cation-binding site is indicated as a green circle. These features are identical in both subunits.
In subunit-A, pseudouridine and ADP, which are separated by ∼6 Å between the O5′ of the pseudouridine ribosyl moiety and the β-phosphate atom of ADP (Figure 4A), are bound in an extended mode along the crevice in the concave side of the central β-sheet. The small β-strand domain is located in an overhanging manner above the two ligands. Pseudouridine is located at the cleft underneath the small β-strand domain, with its planar nucleobase (hereafter referred to as uracil-Ψ) almost perpendicular to the ribose (Figure 4 A and B). Overall, pseudouridine is oriented such that the hydroxyl O5′ of the ribosyl moiety is positioned toward the β-phosphate group of ADP, whereas its nucleobase points into the opposite direction and is closer to the dimerization interface. The uracil-Ψ ring is embedded into the pocket lined with the hydrophobic residues Ile10, Val90, Ala105, Val107 and Val162, and the hydrophilic residues Thr26*, Asn137 and Lys166 (Figure 4B). The extended side chain of Lys166 likely serves to limit the depth of the pocket. The hydrophobic residues in the pocket are in stacking orientations against the planar uracil-Ψ ring, with Val162 on one side of the ring and the remaining residues, mainly from β6 and β7 of the small domain, on the other side. The hydrophilic residues are located within 3.0 Å of uracil-Ψ ring. In particular, Asn137 mediates bidendate interactions with the N3 and O4 of the base (Figure 4B), and Lys166 interacts with the O2 of the base. Another key residue for the pseudouridine specificity is Thr26*, a residue in the substrate-binding loop of the adjacent subunit-B involved in forming the binding pocket of subunit-A. Its side chain hydroxyl group is located within ∼2.6 Å of the N1 of uracil-Ψ, the hallmark atom for differentiating uracil-Ψ from uracil in uridine.
However, the possible involvement of Thr26* in determining pseudouridine specificity was observed only in subunit-A. In subunit-B, the side chain hydroxyl group of Thr26 is more than ∼7 Å away from the N1 of uracil-Ψ, mainly due to the different conformation of the substrate-binding loop in β2−β3 (Figure 4C). Additionally, subunit-B also shows noticeable differences in the position of the small β-strand domain relative to that in subunit-A (Figure 4C and Supplementary Figure S2). The β6 and β7 region, containing Val90, Ala105, and Val107 for hydophobic interactions with uracil-Ψ, is about 1.5–3.0 Å more distant from uracil-Ψ in subunit-B than in subunit-A. As a consequence, the relative positions of pseudouridine in the two subunits show some differences. In subunit-B, pseudouridine is positioned further away from ADP compared to subunit-A, but positional displacements are minor, ranging only from 0.3 to 1.3 Å depending on the respective atoms (Supplementary Figure S5). For example, the pseudouridine ribosyl moiety is found almost at the same location in the two subunits with displacements of only 0.5–0.7 Å, but larger displacements of 0.3–1.3 Å are observed for the planar uracil-Ψ ring located near the hydrophobic residues in β6 and β7 region of the small β-strand domain. Therefore, the hydrophobic interactions are likely to be weaker in the active site of subunit-B and the specificity for uracil-Ψ is partially lost. These observations suggest that the small β-strand domain, which contains the substrate-binding loop and the hydrophobic residues interacting with uracil-Ψ, is able to undergo structural transitions.
Unlike the nucleobase, the pseudouridine ribosyl moiety and ADP are bound almost at the same location in both subunits (Figure 4 B and C). In particular, the planar ribosyl moiety sits equally on the bed of β2 and β3 in the small domain. Its O2′ and O3′ form bidendate interactions with Asp12, and the O5′ is within ∼2.8 Å of Asp311, a putative catalytic residue as suggested for ribokinase (34). ADP is bound in an extended configuration to the crevice along the central β-sheet (Figure 4A), analogous to the binding of ADP in EcRBSK (37) and AtRBSK (34). Its adenine base is embedded into the cavity between the two loops, the large and small ATP loop defined as the ATP-binding elements in ribokinase (34,37), and is involved in extensive hydrophobic interactions (Figure 4D). Its associated ribosyl and di-phosphate groups are extended towards the ribosyl moiety of pseudouridine (Figure 4A), fulfilling the geometrical requirement for a phosphorylation reaction occurring on the ribosyl O5′ of the substrate. When the ATP in subunit-A of the AtPUKI-ATP binary complex is overlaid on the ADP in the corresponding subunit of the ternary complex, the distance between the O5′ of the ribose in pseudouridine and the γ-phosphate atom of ATP is ∼4.4 Å (Supplementary Figure S6), a distance proximal enough for a kinase reaction.
Functional features of the AtPUKI active site residues
By employing an enzyme-coupled assay, we first carried out steady-state kinetic analyses of the wild-type AtPUKI (Figure 5A). Under our assay conditions, the Km and kcat values are 39.6 μM and 5.28 s–1 for pseudouridine, and 761 μM and 5.80 s–1 for ATP. Under saturation conditions for ATP (i.e. 4 mM ATP), we evaluated the substrate selectivity of AtPUKI towards various nucleosides at a concentration of 1.25 mM each (Figure 5B). Consistent with a previous study (13), AtPUKI was highly specific for pseudouridine, with relative activities towards cytidine and uridine of 15.4% and 1.6%, respectively, and negligible activities for other potential substrates. Thus, AtPUKI exhibits a narrow selectivity towards pyrimidine nucleosides and likely possesses structural determinants distinguishing pseudouridine from the chemically similar uridine or cytidine.
Figure 5.

Kinetic analyses of AtPUKI and activity comparisons using several substrates and a variety of mutants. (A) Steady-state kinetic analyses of AtPUKI with pseudouridine and ATP as substrates. Error bars are SD (n = 3). Our measurements did not show any recognizable deviations among the measured values; thus, the error bars are not clearly visible. Details of the reaction conditions for (A), (B) and (C) can be found in the ‘Activity assays’ section. (B) Specific activity of AtPUKI with different substrate candidates at 1.25 mM, including ribose (blue), pyrimidine nucleosides (green) and purine nucleosides (black). The resulting activity was compared to that with pseudouridine (red, 8.54 μmol min–1 mg–1 protein set to 100%). Specific activities are given in parentheses. Error bars are SD (n = 3). n.d., not detected. (C) Specific activity of AtPUKI variants with pseudouridine. Color code: mutants of the substrate pocket (blue), the ADP-binding site (black), and catalysis (yellow; see the section ‘Mg2+-binding site and catalysis in AtPUKI’). The activities were compared to that of the wild-type AtPUKI (set to 100%); the wild-type enzyme data is identical to that shown in (B). Specific activities are given in parentheses. Error bars are SD (n = 3). n.d., not detected.
We further performed activity assays and kinetic analyses of AtPUKI mutants. Residues for site-directed mutagenesis were selected based on the binding site of pseudouridine and ADP in subunit-A (Figure 4B and D). ADP production was monitored for 30 s in these assays using 4 mM ATP and 1.25 mM pseudouridine as substrates. Mutations in the nucleoside binding pocket reduced the activity to a greater extent than those in the ADP-binding site (Figure 5C). Mutation of the three residues that interact likely via hydrogen bonds with the base of pseudouridine (T26A, T26V, N137A, K166A) led to activities in the range of 2.5–16.3% relative to the wild-type. The T26S mutant exhibited an activity of ∼53%, suggesting a role of the identified hydrogen bond in catalysis. The D12A and D12N mutations that probably interfere with proper ribose binding abolished the activity almost completely, whereas a mutation of the hydrophobic residue proximal to uracil-Ψ (I10A) maintained ∼50% activity.
To further validate the structure-based functional assignments of the active site residues, the kinetic parameters of the various mutants were determined. The analysis confirmed the proposed functional roles for active site residues (Figure 6A). For example, I10A increased the apparent Km value by ∼2-fold but not the kcat value, as would be expected for a nucleobase-interacting residue. Asn137 plays a central role in recognizing uracil-Ψ via a bidentate interaction with the base. Accordingly, a ∼114-fold increase in the Km value and a ∼4.3-fold decrease in the kcat value were observed for the N137A mutant. The K166A mutation resulted in a negative effect on Km (2.5-fold higher) and kcat (5.7-fold lower), because the long side chain of Lys166 is located at the bottom end of the binding pocket and its terminal NZ recognizes the O2 of uracil-Ψ (Figure 4B). The most intriguing residue was Thr26 in the substrate-binding loop donated from the other subunit that recognizes the N1 of the nucleobase (Figure 4B). Whereas the T26A and T26V mutants lose the ability to form the proposed hydrogen bond to N1 of the base, the T26S variant maintains this interaction. Accordingly, two groups can be discerned based on kinetic features: (1) the T26A and T26V mutants both with an about 4-fold higher Km and a 6-fold lower kcat value relative to the wild-type AtPUKI and (2) the T26S variant with an almost equal Km to the wild-type and a kcat value reaching 62% of that of the wild-type enzyme. These data clearly show that Thr26 plays an important role in recognizing uracil-Ψ and demonstrate that the structure of subunit-A in the ternary complex represents the catalytically active conformation of the substrate-binding loop in AtPUKI (Figure 4B). Consistently, the active site residues described are highly conserved in PUKI orthologs of plants (Figure 1B). Especially, the hydrophilic residues Thr26, Asn137 and Lys166 interacting with the uracil-Ψ are also highly conserved in PUKI (YeiC) from bacteria and in the metazoan bifunctional enzymes containing both PUKI and PUMY domains (13). In some enzymes, the equivalent position to Thr26 is replaced by serine (Figure 1B). Therefore, it is probable that the binding mode of uracil-Ψ to the active site of AtPUKI is representative for PUKI in general.
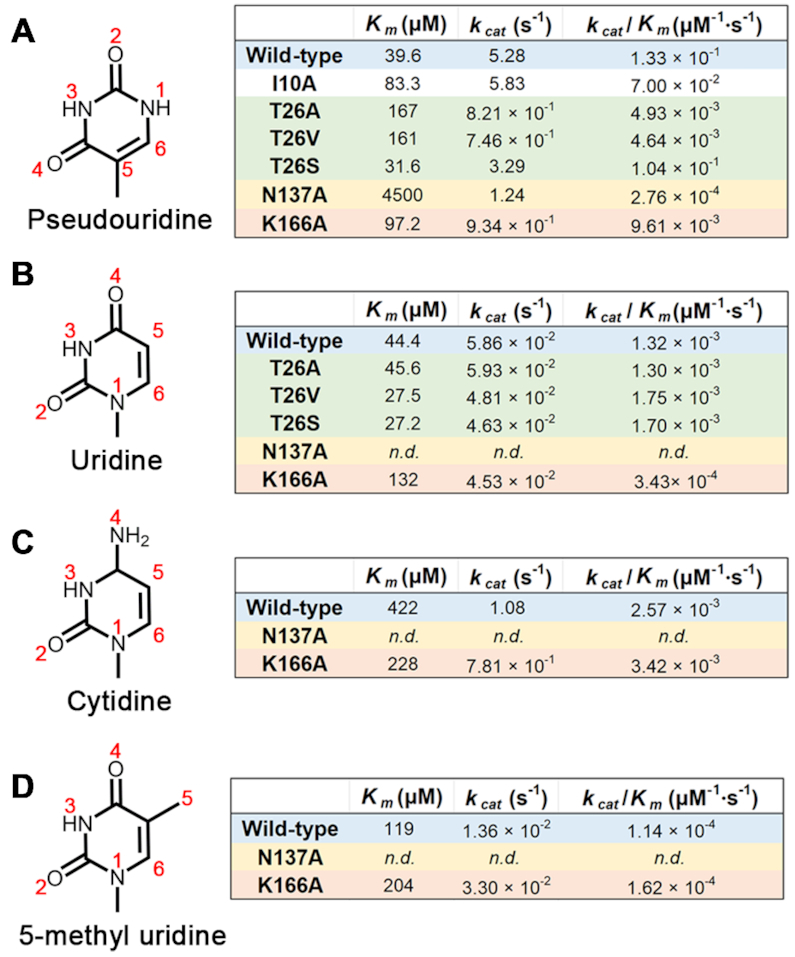
Figure 6.
Kinetic analyses of AtPUKI variants with pseudouridine and structurally and chemically similar nucleosides. For clarity, only the nucleobases of the nucleosides are shown. In (A)–(D), steady-state kinetic analyses in the presence of 4 mM ATP as described in the section ‘Activity assays.’ Atom numbers of the pyrimidine ring are given for each nucleoside; n.d., not detected.
High fidelity of AtPUKI for pseudouridine
The question of how AtPUKI achieves its high fidelity, distinguishing pseudouridine from other pyrimidine nucleosides, was addressed by further kinetic analyses employing the substrates uridine, cytidine, and 5-methyl uridine carrying a methylated uracil base (Figure 6B–D).
Comparing the kinetic parameters of wild-type and mutant enzymes for chemically similar substrates confirmed the previous notion that the formation of several specific hydrogen bonds between AtPUKI and pseudouridine is the key factor for achieving this high substrate specificity. This is reminiscent to the so-called ‘direct readout’ for sequence-specific interactions characterized in protein–DNA recognitions (43,44). There are three crucial amino acids for the fidelity of AtPUKI: Thr26, Asn137 and Lys166. In this study, we noticed that Asn137 is essential for recognizing pseudouridine, but it is also absolutely required for binding the base of pyrimidine nucleosides in general, because the wild-type AtPUKI exhibited activity (although with large variation) to all substrates, whereas the N137A variant was essentially inactive. Specifically, the side chain carbonyl and amino groups of Asn137 fulfill the requirement for hydrogen bond formation by accepting and donating a hydrogen, respectively, from/to the hydrogen donor N3 and acceptor O4 of uracil-Ψ (or the equivalent N3 and O2 atoms of the other three nucleosides, Figure 6B–D). Lys166 likely recognizes the hydrogen acceptor O2 of uracil-Ψ that is equivalent to O4 in uridine but chemically differs from the hydrogen donor N4 in cytidine, belonging to the amino group distinguishing cytidine from uridine. Accordingly, the Km values of AtPUKI for pseudouridine and uridine are almost equal, but that for cytidine is ∼9.5-fold higher. However, the kcat value for cytidine is higher than for uridine (Figure 6C). Although AtPUKI is a better kinase for cytidine than uridine at high substrate concentrations, this will be of little relevance in vivo, because the content of cytidine is generally lower than that of uridine in Arabidopsis (45) and uridine is not an in vivo substrate of AtPUKI (13). Thr26 in the substrate-binding loop probably forms a hydrogen bond to N1 of pseudouridine. The formation of this bond is not possible when 5-methyl uridine is the substrate (Figure 6D), resulting in an ∼3-fold higher Km value and a 390-fold reduction of the kcat value compared to pseudouridine as substrate.
It is quite unusual that the Km values for pseudouridine and uridine are almost identical for the wild-type AtPUKI, whereas there is an ∼90-fold difference in the kcat values (Figure 6 A and B). Apparently, AtPUKI discriminates its substrates not by binding affinity but by differential turnover. AtPUKI and its mutant variants T26A, T26V, and T26S have similar Km and kcat values for uridine (Figure 6B). This is not the case for the authentic substrate, pseudouridine, for which the kinetic parameters of the T26S variant, in contrast to the T26A and T26V mutants, are almost like those of the wild-type. It appears that the interactions mediated via Thr26 in the substrate-binding loop facilitate catalysis. These observations together with the dynamic features of the β6 and β7 regions of the small domain and the substrate-binding loop, which were implied by comparing the structures of subunits-A and -B (Figure 4 B and C, and Supplementary Figure S2), lead us to propose that AtPUKI undergoes a structural transition between a catalytically active form and an inactive form. The transition likely affects the catalytic efficiency, mainly through increasing the turnover. Probably only pseudouridine can induce the structural transition effectively recruiting the essential elements of the enzyme into the active site. Pseudouridine is therefore the by far best substrate for AtPUKI.
The substrate pocket of AtPUKI in comparison to ribokinase and adenosine kinase
Structure comparisons indicate that among structurally homologous proteins from the PfkB family, the nucleobase binding pocket of AtPUKI is unique in particular for the high number of specific interactions with the base involving also inter-subunit interactions and conformational changes (Figure 7A).
Figure 7.
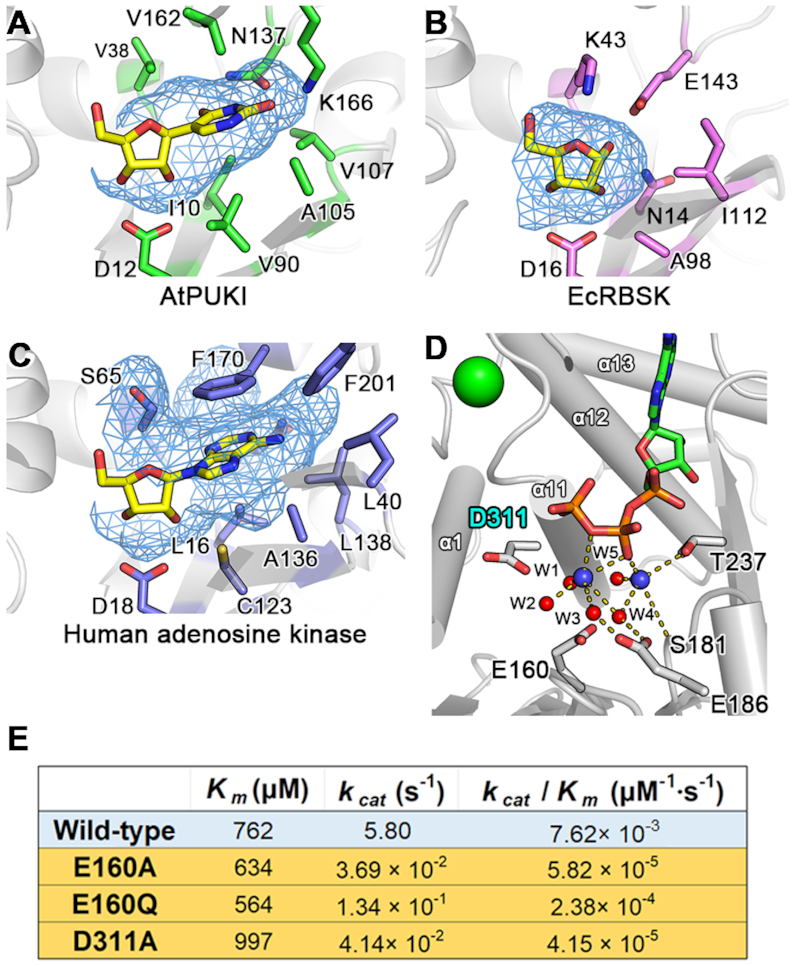
The substrate pocket of AtPUKI, ribokinase, and adenosine kinase, and the Mg2+-binding site of AtPUKI. (A) The mesh in blue calculated by PyMol (www.pymol.org) represents the molecular surface of the protein in the absence of ligand(s), and therefore displays the pseudouridine binding pocket of AtPUKI. This view is almost identical to that of Figure 4(B). (B) Substrate pocket of E. coli ribokinase (PDB id, 1RK2) (37) showing a region of the kinase that is equivalent to that shown for AtPUKI in (A). Note that a nucleobase cannot be accommodated, mainly because Lys43 and Glu143 occupy the space. (C) The binding environment for adenosine in human adenosine kinase (PDB ID: 1BX4) shown in an identical manner as AtPUKI in (A). (D) The Mg2+-binding site in the vicinity of ADP in the ternary complex of AtPUKI. The following symbols are used: monovalent cation, green circle; magnesium ions, blue circles; water molecule, red circles. The Mg2+-coordinating shell is indicated by the dashed lines, and for clarity, possible hydrogen bonds are not shown between Glu160 and water molecules including W4 and W5. (E) Steady-state kinetic analyses employing 1.25 mM pseudouridine and different concentrations of ATP, as described in the section ‘Activity assays.’ The D311A variant was produced to analyze a mutant of the catalytic base Asp311.
In dimeric EcRBSK, each monomer has a vacancy suitable only for ribose, without any involvements of structural elements from the adjacent monomer. The region corresponding to the nucleobase pocket in AtPUKI is effectively occupied, mainly by Lys43 and Glu143, which are strictly conserved in the family of ribokinases (Figure 7B). The monomeric human adenosine kinase contains a nucleoside binding pocket essentially at an equivalent location to where it is found in AtPUKI (Figure 7C). However, the pocket is larger than that of AtPUKI to allow accommodation of a nucleoside with a purine base, and it consists mainly of hydrophobic residues which are also conserved in other adenosine kinases. In contrast to AtPUKI, the nucleobase binding part of the pocket lacks hydrophilic residues that could mediate specific hydrogen bonds to the adenine moiety. These structural features explain the more pronounced promiscuity of adenosine kinases regarding the substrate (46,47). It appears that the nucleoside binding pocket of PUKI is especially equipped for a thorough identity control of the nucleobase. This might be required to avoid the in vivo phosphorylation of similar nucleosides like uridine, which is more abundant than pseudouridine in Arabidopsis (13).
The Mg2+-binding site and catalysis in AtPUKI
A recent study on AtRBSK suggested a possible reaction mechanism by which ribose is converted into ribose-5′-phosphate involving the catalytic base Asp325 and catalytic acid Lys291 (34). In detail, Asp325AtRBSK (i.e. residue Asp325 of AtRBSK) in the vicinity of the ribose catalyzes the de-protonation of the ribose 5′-OH group, and Lys291AtRBSK near the β-phosphate group of ATP is responsible for the protonation of the ADP leaving group after a nucleophilic attack of the deprotonated O5′ of ribose on the γ-phosphate group of ATP. Structural comparisons show that the binding sites for ribose, ADP, and ATP are almost identical between AtPUKI and members of the ribokinase family (34,37,40). From a structural perspective, the catalytic base Asp325AtRBSK corresponds to Asp311AtPUKI (Figure 7D), but the catalytic acid Lys291AtRBSK does not have a functional counterpart in AtPUKI. Using the binary complex of AtPUKI with ATP, we observed that the Cα position of Lys291AtRBSK is superimposable to that of Thr237AtPUKI, but the side chain of Thr237AtPUKI is more than 6 Å away from the β-phosphate group of ATP and unable to carry out a role as a catalytic acid (Figure 7D).
In the binary complex, we identified two Mg2+-binding sites in the vicinity of the β- and γ-phosphate groups of ATP (Figure 7D), based on the number of ligands and the coordination geometry. These features are identical in both monomers. One magnesium ion (MgA) between the β- and γ-phosphate groups of ATP, has six ligands: two from the oxygen atoms of ATP and the remaining four with water molecules (W1 to W4). The second magnesium ion (MgB) near the β-phosphate group of ATP, has five coordinating ligands, including two water molecules (W4 and W5), and three oxygen atoms: one from the β-phosphate group of ATP, one from the side chain hydroxyl group of Thr237, and one from the main chain carbonyl group of Ser181. Therefore, there are no candidates for the catalytic acid in close vicinity of the β-phosphate group of ATP. In the Mg2+ shell of the binary complex, water molecules are coordinated within 3.0 Å with the nearby residues Glu160 and Glu186 (Figure 7D): Glu160 to W4 and W5 and Glu186 to W3 and W4. The two glutamates are located more than 4 Å distant from the two magnesium ions, making a direct coordination with the magnesium ions impossible. Mutagenesis of Glu160 as well as a proposed catalytic base Asp311 essentially abolishes enzyme activity (Figure 5C). Mutants E160A, E160Q and D311A only had 0.5%, 2.0% and 0.6% of the specific activity of wild-type AtPUKI, respectively. Kinetic measurements varying the ATP concentration at a saturating concentration of pseudouridine (1.25 mM) indicated that the mutations caused minor effects on the Km value for ATP but greatly reduced the enzyme efficiencies (kcat/Km) to less than 3% compared to wild-type AtPUKI (Figure 7E). These data and the structural environment around Glu160 suggest that a water molecule in the Mg2+-coordinating shell likely plays a role in the formation of the ADP leaving group during the AtPUKI kinase reaction. Consistent with such a pivotal function, Glu160 and Glu186 are strictly conserved in the PUKI sequences from plants (Figure 1B) and also in the bifunctional enzymes from animals. In bacterial PUKI (YeiC) enzymes, a Glu or an Asp are found at the position equivalent to Glu160, and Glu186 is also here highly conserved (13).
CONCLUSIONS
AtPUKI exhibits several features, which are unique among the PfkB family proteins. The substrate binding pocket is mainly formed by hydrophobic residues from the small domain, and hydrophilic residues in the pocket, in part stemming from the adjacent subunit, mediate several specific hydrogen bonds between AtPUKI and the nucleobase of pseudouridine (Figure 4B). By contrast, in the binding pocket of adenosine kinase, the base is recognized less specifically (Figure 7C). Second, the dynamic properties of the pocket-forming elements are closely related to the catalytic function of AtPUKI. Upon structural transition (Figure 2B and Supplementary Figure S2) hydrophobic residues, mainly from β6 and β7 of the small domain, move toward one face of the nucleobase. Simultaneously, the substrate binding loop between β2* and β3* of the adjacent subunit closes the lateral flank of the binding pocket mediating a hydrogen bond with the characteristic N1 of the substrate (Figure 4B and C). Because N1 clearly distinguishes pseudouridine from uridine, this dynamic inter-subunit interaction is decisive for pseudouridine specificity and catalytic efficiency (Figure 6). Such dynamic features are not observed in other PfkB kinases. Third, AtPUKI lacks the catalytic acid identified in AtRBSK (34). Instead, a water molecule in the Mg2+-coordinating shell might take over that role (Figure 7D and E). Our analyses reveal an example of structural and functional divergence in the PfkB protein family and explain how pseudouridine can be phosphorylated in vivo without interfering with uridine and cytosine homeostasis.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession number 7C1X, 7C1Y and 7C1Z.
Supplementary Material
Contributor Information
Sang-Hoon Kim, Department of Agricultural Biotechnology, Seoul National University, Seoul, Korea.
Claus-Peter Witte, Department of Molecular Nutrition and Biochemistry of Plants, Leibniz University Hannover, Hannover, Germany.
Sangkee Rhee, Department of Agricultural Biotechnology, Seoul National University, Seoul, Korea; Research Institute of Agriculture and Life Sciences, Seoul National University, Seoul, Korea.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) [2020R1A4A1018890 to S.R.]; Deutsche Forschungsgemeinschaft [DFG grant CH2292/1-1 to C.-P.W.]. Funding for open access charge: Seoul National University, Korea.
Conflict of interest statement. None declared.
REFERENCES
- 1. Roundtree I.A., Evans M.E., Pan T., He C.. Dynamic RNA modifications in gene expression regulation. Cell. 2017; 169:1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Frye M., Harada B.T., Behm M., He C.. RNA modifications modulate gene expression during development. Science. 2018; 361:1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zaccara S., Ries R.J., Jaffrey S.R.. Reading, writing and erasing mRNA methylation. Nat. Rev. Mol. Cell Biol. 2019; 20:608–624. [DOI] [PubMed] [Google Scholar]
- 4. Gilbert W.V., Bell T.A., Schaening C.. Messenger RNA modifications: form, distribution, and function. Science. 2016; 352:1408–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cohn W.E. Pseudouridine, a carbon-carbon linked ribonucleoside in ribonucleic acids: isolation, structure, and chemical characteristics. J. Biol. Chem. 1960; 235:1488–1498. [PubMed] [Google Scholar]
- 6. Carlile T.M., Rojas-Duran M.F., Zinshteyn B., Shin H., Bartoli K.M., Gilbert W.V.. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature. 2014; 515:143–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schwartz S., Bernstein D.A., Mumbach M.R., Jovanovic M., Herbst R.H., León-Ricardo B.X., Engreitz J.M., Guttman M., Satija R., Lander E.S. et al.. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell. 2014; 159:148–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun L., Xu Y., Bai S., Bai X., Zhu H., Dong H., Wang W., Zhu X., Hao F., Song C.-P.. Transcriptome-wide analysis of pseudouridylation of mRNA and non-coding RNAs in Arabidopsis. J. Exp. Bot. 2019; 70:5089–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Charette M., Gray M.W.. Pseudouridine in RNA: what, where, how, and why. IUBMB Life. 2000; 49:341–351. [DOI] [PubMed] [Google Scholar]
- 10. Helm M. Post-transcriptional nucleotide modification and alternative folding of RNA. Nucleic Acids Res. 2006; 34:721–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eyler D.E., Franco M.K., Batool Z., Wu M.Z., Dubuke M.L., Dobosz-Bartoszek M., Jones J.D., Polikanov Y.S., Roy B., Koutmou K.S.. Pseudouridinylation of mRNA coding sequences alters translation. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:23068–23074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen M., Urs M.J., Sánchez-González I., Olayioye M.A., Herde M., Witte C.-P.. M6A RNA degradation products are catabolized by an evolutionarily conserved N6-methyl-AMP deaminase in plant and mammalian cells. Plant Cell. 2018; 30:1511–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen M., Witte C.-P.. A kinase and a glycosylase catabolize pseudouridine in the peroxisome to prevent toxic pseudouridine monophosphate accumulation. Plant Cell. 2020; 32:722–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Witte C.-P., Herde M.. Nucleotide metabolism in plants. Plant Physiol. 2020; 182:63–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Baccolini C., Witte C.-P.. AMP and GMP catabolism in Arabidopsis converge on xanthosine, which Is degraded by a nucleoside hydrolase heterocomplex. Plant Cell. 2019; 31:734–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jia Q., Xie W.. Alternative conformation induced by substrate binding for Arabidopsis thaliana N6-methyl-AMP deaminase. Nucleic Acids Res. 2019; 47:3233–3243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wu B., Zhang D., Nie H., Shen S., Li Y., Li S.. Structure of Arabidopsis thaliana N6-methyl-AMP deaminase ADAL with bound GMP and IMP and implications for N6-methyl-AMP recognition and processing. RNA Biol. 2019; 16:1504–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Floyd B.E., Morriss S.C., MacIntosh G.C., Bassham D.C.. Evidence for autophagy-dependent pathways of rRNA turnover in Arabidopsis. Autophagy. 2015; 11:2199–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hickl D., Drews F., Girke C., Zimmer D., Mühlhaus T., Hauth J., Nordström K., Trentmann O., Neuhaus H.E., Fehlmann T. et al.. Differential degradation of RNA species by autophagy related pathways in plants. 2019; bioRxiv doi:17 October 2019, preprint: not peer reviewed 10.1101/793950. [DOI] [PubMed]
- 20. Loh K.D., Gyaneshwar P., Markenscoff Papadimitriou E., Fong R., Kim K.S., Parales R., Zhou Z., Inwood W, Kustu S.. A previously undescribed pathway for pyrimidine catabolism. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:5114–5119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zrenner R., Riegler H., Marquard C.R., Lange P.R., Geserick C., Bartosz C.E., Chen C.T., Slocum R.D.. A functional analysis of the pyrimidine catabolic pathway in Arabidopsis. New Phytol. 2009; 183:117–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Breitman T.R. Pseudouridulate synthetase of Escherichia coli: correlation of its activity with utilization of pseudouridine for growth. J. Bacteriol. 1970; 103:263–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Preumont A., Snoussi K., Stroobant V., Collet J.-F., Van Schaftingen E.. Molecular identification of pseudouridine metabolizing enzymes. J. Biol. Chem. 2008; 283:25238–25246. [DOI] [PubMed] [Google Scholar]
- 24. Reumann S. Toward a definition of the complete proteome of plant peroxisomes: where experimental proteomics must be complemented by bioinformatics. Proteomics. 2011; 11:1764–1779. [DOI] [PubMed] [Google Scholar]
- 25. Park J., Gupta R.S.. Adenosine kinase and ribokinase: the RK family of proteins. Cell Mol. Life Sci. 2008; 65:2875–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schroeder R.Y., Zhu A., Eubel H., Dahncke K., Witte C.-P.. The ribokinases of Arabidopsis thaliana and Saccharomyces cerevisiae are required for ribose recycling from nucleotide catabolism, which in plants is not essential to survive prolonged dark stress. New Phytol. 2018; 217:233–244. [DOI] [PubMed] [Google Scholar]
- 27. Van Duyne G.D., Standaert R.F., Karplus P.A., Schreiber S.L., Clardy J.. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 1993; 229:105–124. [DOI] [PubMed] [Google Scholar]
- 28. Otwinowski Z., Minor W.. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997; 276:307–326. [DOI] [PubMed] [Google Scholar]
- 29. Karplus P.A., Diederichs K.. Linking crystallographic model and data quality. Science. 2012; 336:1030–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Diederichs K., Karplus P.A.. Better models by discarding data?. Acta Crystallogr. D Biol. Crystallogra. 2013; 69:1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grossekunstleve R.W.. PHENIX: a comprehensive python-based system for macromolecular structure solution. Acta. Crystallogr. D. Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta. Crystallogr. D. Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mallette E., Kimber M.S.. Structural and kinetic characterization of (S)-1-amino-2-propanol kinase from the aminoacetone utilization microcompartment of Mycobacterium smegmatis. J. Biol. Chem. 2018; 293:19909–19918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kang P.-A., Oh J., Lee H., Witte C.-P., Rhee S.. Crystal structure and mutational analyses of ribokinase from Arabidopsis thaliana. J. Struct. Biol. 2019; 206:110–118. [DOI] [PubMed] [Google Scholar]
- 35. Andersson C.E., Mowbray S.L.. Activation of ribokinase by monovalent cations. J. Mol. Biol. 2002; 315:409–419. [DOI] [PubMed] [Google Scholar]
- 36. Krissinel E., Henrick K.. Inference of macromolecular assemblies from crystalline state. J Mol Biol. 2007; 372:774–797. [DOI] [PubMed] [Google Scholar]
- 37. Sigrell J.A., Cameron A.D., Jones T.A., Mowbray S.L.. Structure of Escherichia coli ribokinase in complex with ribose and dinucleotide determined to 1.8 å resolution: insights into a new family of kinase structures. Structure. 1998; 6:183–193. [DOI] [PubMed] [Google Scholar]
- 38. Holm L., Laakso L.M.. Dali server update. Nucleic Acids Res. 2016; 44:W351–W355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sigrell J.A., Cameron A.D., Mowbray S.L.. Induced fit on sugar binding activates ribokinase. J. Mol. Biol. 1999; 290:1009–1018. [DOI] [PubMed] [Google Scholar]
- 40. Paul R., Patra M.D., Sen U.. Crystal structure of apo and ligand bound Vibrio cholerae ribokinase (Vc-RK): role of monovalent cation induced activation and structural flexibility in sugar phosphorylation. Adv. Exp. Med. Biol. 2015; 842:293–307. [DOI] [PubMed] [Google Scholar]
- 41. Maj M.C., Gupta R.S.. The effect of inorganic phosphate on the activity of bacterial ribokinase. J Protein Chem. 2001; 20:139–144. [DOI] [PubMed] [Google Scholar]
- 42. Park J., Singh B., Maj M.C., Gupta R.S.. Phosphorylated derivatives that activate or inhibit mammalian adenosine kinase provide insights into the role of pentavalent ions in AK catalysis. Protein J. 2004; 23:167–177. [DOI] [PubMed] [Google Scholar]
- 43. Pabo C.O., Sauer R.T.. Transcription factors: structural families and principles of DNA recognition. Annu. Rev. Biochem. 1992; 61:1053–1095. [DOI] [PubMed] [Google Scholar]
- 44. Steitz T.A. Structural studies of protein-nucleic acid interaction: the sources of sequence-specific binding. Q. Rev. Biophys. 1990; 23:205–280. [DOI] [PubMed] [Google Scholar]
- 45. Chen M., Herde M., Witte C.-P.. Of the nine cytidine deaminase-like genes in Arabidopsis, eight are pseudogenes and only one is required to maintain pyrimidine homeostasis in vivo. Plant Physiol. 2016; 171:799–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bennett L.L Jr, Hill D.L.. Structural requirements for activity of nucleosides as substrates for adenosine kinase: orientation of substituents on the pentofuranosyl ring. Mol. Pharmacol. 1975; 11:803–808. [PubMed] [Google Scholar]
- 47. Romanello L., Bachega J.F., Cassago A., Brandão-Neto J., DeMarco R., Garratt R.C., Pereira H.D.. Adenosine kinase from Schistosoma mansoni: structural basis for the differential incorporation of nucleoside analogues. Acta. Crystallogr. D. Biol. Crystallogr. 2013; 69:126–136. [DOI] [PubMed] [Google Scholar]
- 48. Robert X., Gouet P.. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014; 42:W320–W324. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited with the Protein Data bank under accession number 7C1X, 7C1Y and 7C1Z.