

Abstract
Rationale:
Cancer-associated mutations have the potential to generate neoantigens and elicit CD8 positive T cell dependent adaptive immune responses. There are currently no reports of CD8 positive T cells with specificity for neoepitopes generated by EGFR mutations, which are driver oncogenes in a subset of patients with lung cancer.
Objectives:
To identify putative protective HLA class I allotypes that are predicted in silico to bind and present mutant EGFR generated peptides and associate the presence or absence of these alleles with clinical outcomes in lung adenocarcinoma.
Findings:
We identified 12 HLA class I alleles that fulfilled predefined criteria for being protective for EGFR p.L858R and 6 for EGFR p.E746_A750del, the two most common EGFR mutations in lung cancer. We validated the in silico predictions for peptide-HLA allele binding in vitro. A third (12 of 36) of mostly early stage lung adenocarcinoma patients in The Cancer Genome Atlas (TCGA) with either EGFR p.L858R or EGFR p.E746_A750del had at least one protective allele in their host genomes. Importantly, patients with protective alleles showed better disease-free (HR=0.20, 95% CI 0.05-0.78) and overall survival (HR=0.13, 95% CI 0.02-0.64) and this effect was independent from EGFR mutation type, stage, age and sex.
Conclusions:
Our data demonstrate that clinical outcomes were improved in patients with EGFR mutation positive lung adenocarcinoma who harbored protective HLA class I alleles. Thus, immunity with specificity for mutant EGFR is possible in a subset of patients with early stage lung cancer and portends a better prognosis.
Keywords: HLA, EGFR, adaptive immunity, neoepitopes
Introduction2
Non-small cell lung cancer (NSCLC) is the most common type of lung cancer and the leading cause of cancer related mortality in the US and globally.1 Tobacco products are responsible for most lung cancers; carcinogens like air pollutants, radon gas, arsenic and others account for some lung cancer cases with no tobacco exposure, but the etiology remains obscure for many other patients.2 Lung cancers that are not smoking related frequently harbor driver mutations in key oncogenes, including EGFR, ALK, ROS1 and others.3, 4 Available systemic treatments for lung cancer provide disease control but not cure in the metastatic setting and include chemotherapy, targeted therapies (in the presence of actionable mutations), and immune checkpoint inhibitors.5
Our understanding of the immune underpinnings of cancer has led to approval of a number of immune based therapies for patients with the disease.6, 7 Cancer-related mutations potentially generate immunogenic neoantigens and elicit adaptive immune responses.8, 9 T cell receptors specifically bind to peptides presented by human leucocyte antigen (HLA) molecules.9, 10 HLA class I genes are located in the major histocompatibility complex region I (MHC I) on chromosome 6 and are highly polymorphic.11, 12
HLA-A, -B and –C, which are the most polymorphic HLA class I molecules, are present at the cell surface of all nucleated cells. They form complexes with beta 2 microglobulin and present 8-11 amino acid long peptides (8-11mers) to CD8 positive T cells. Cancer cells themselves present mutation containing peptides loaded on MHC I complexes and activate adaptive CD8 positive T cell responses against the tumor.13
Noteworthy, most neoantigens recognized by the immune system in this fashion are derived by passenger mutations,14 whereas driver mutations are rarely identified as immunogenic.15 A possible reason is that the latter are very important for tumor development and evade the immune system early in the natural history of established tumors. Interestingly, the host MHC genotype restricts the landscape of cancer driver mutations that occur in a given tumor.16, 17 On this basis, we herein test the hypothesis that the capacity of HLA alleles in certain individuals to bind neoantigen peptides from EGFR is associated with improved outcomes in patients with EGFR mutant lung cancer.
Methods
Peptide-HLA affinity prediction
We used NETMHCpan 4.0,18–21 to predict the binding affinities of all 72 8-11 amino acid long EGFR derived peptides (8-11mer) containing the EGFR p.L858R or the EGFR p.E746_A750del (i.e., containing in succession both amino acids lysine and threonine that flank the p.E746_A750 in the wild type EGFR sequence). An IC50 value of 500 nM has previously been found to be associated with the vast majority of known cytotoxic T lymphocyte epitopes for their corresponding restricting allele, and thus represent a practical threshold to define binder vs. non-binder peptides with respect to a specific HLA allele in the MHC I region.22 On the basis of this threshold, for both in silico and in vitro assessments of binding affinity, peptides with predicted IC50 ≤ 500 nM are defined as high affinity binders, peptides with IC50 > 500-5000 nM are deemed intermediate binders, and affinities >5000 nM as non-binders, for the corresponding HLA allele. All wild type and mutant peptides used for predictions of binding affinity with various HLA alleles are shown in supplementary table 1.
Determination of protective and non-protective alleles
We followed a pre-defined strategy to classify a large number of common HLA-A, -B and -C alleles as either protective or non-protective with respect to EGFR p.L858R, based on differential binding of mutant and wild type EGFR derived peptides. We took into account the wild type peptide in predicting the immunogenicity of the mutant peptide because central tolerance processes that affect the former might spread to the latter if the two peptides are highly similar, as is the case with single amino acid change point mutations.23 An allele was deemed protective for EGFR p.L858R if an EGFR p.L858R containing 8-11mer binds to this allele (IC50≤500 nM), whereas the closest (1 amino acid difference) wild type peptide does not bind (IC50>500 nM) and there is a three or more-fold difference in binding affinity of the two peptides (mtIC50/wtIC50≤3). With regards to EGFR pE746_A750del, an HLA allele was classified as protective if there are EGFR p.E746_A750del containing 8-11mers that bind to this specific allele (IC50≤500 nM). In this case, we did not consider the wild type peptide binding because the closest such peptide would still be several amino acids different from the mutant peptide and was thus not expected to affect the immunogenicity of the latter. Any allele that was not considered protective with regards to a particular mutation, was classified as non-protective.
Patients
The Institutional Review Board of the University of Colorado, which waived the need to obtain written informed consent from the de-identified TCGA patients, has approved the study. We used the lung adenocarcinoma cohort in The Cancer Genome Atlas (TCGA, PanCancer Atlas).24 We obtained the “TCGA-CDR-SupplementalTableS1.xlsx” file from the Genomic Data Commons of the National Institute of Health. This file is recommended for extraction of clinical elements and survival data for the TCGA PanCancer Atlas cohort.24 We obtained the published HLA-A, -B and -C genotypes at 4 digit (which distinguishes HLA allotypes at the polypeptide sequence) level.6 We identified patients with EGFR mutations in the cBioPortal platform.25, 26 We classified patients with the EGFR p.L858R or the EGFR p.E746_A750del into the protective group if they had a protective allele and into the non-protective group otherwise.
HLA class I Purification and Peptide-Binding Assays
Purification of HLA class I molecules by affinity chromatography was performed as detailed elsewhere.27 Briefly, EBV transformed homozygous cell lines were utilized as sources of HLA class I molecules. HLA-A molecules were purified from cell pellet lysates by repeated passage over Protein A Sepharose beads conjugated with the W6/32 (anti-HLA -A, -B, -C) antibody following separation of HLA-B and -C molecules by pre-passage over a B1.23.2 (anti-HLA-B and -C) column. Protein purity, concentration, and the effectiveness of depletion steps are monitored by SDS-PAGE and BCA assay.
Assays to quantitatively measure peptide binding to HLA class I molecules are based on the inhibition of binding of a high affinity radiolabeled peptide to purified HLA class I molecules, and were performed as detailed elsewhere.27 Briefly, 0.1-1 nM of radiolabeled peptide was co-incubated at room temperature with 1 μM to 1 nM of purified HLA class I in the presence of a cocktail of protease inhibitors and 1 μM β2-microglobulin. Following a two-day incubation, HLA bound radioactivity was determined by capturing HLA/peptide complexes on W6/32 (anti-class I) antibody coated Lumitrac 600 plates (Greiner Bio-one, Frickenhausen, Germany), and measuring bound cpm using the TopCount (Packard Instrument Co., Meriden, CT) microscintillation counter. In the case of competitive assays, the concentration of peptide yielding 50% inhibition of the binding of the radiolabeled peptide was calculated. Under the conditions utilized, where [label]<[HLA] and IC50 ≥ [HLA], the measured IC50 values are reasonable approximations of the true Kd values.28, 29 Each competitor peptide was tested at six different concentrations covering a 100,000-fold dose range, and in three or more independent experiments. As a positive control, the unlabeled version of the radiolabeled probe was also tested in each experiment.
Statistical Analysis
We used the Pearson correlation coefficient (R) to evaluate the correlation of the IC50 values predicted in silico and measured in vitro. We used the 2-tailed Fisher’s exact and chi square tests to compare clinicopathologic variables among patients in the protective and non-protective groups. We estimated disease-fee survival (DFS), defined as the time in months from diagnosis to recurrence or death and overall survival (OS) defined as the time in months from diagnosis to death for the protective and non-protective groups using Kaplan Meier curves. We used the log-rank test to compare the curves and the Cox proportional hazards model to adjust for confounders. JMP (SAS Institute, Inc., Cary, NC) was used for all statistics.
Results
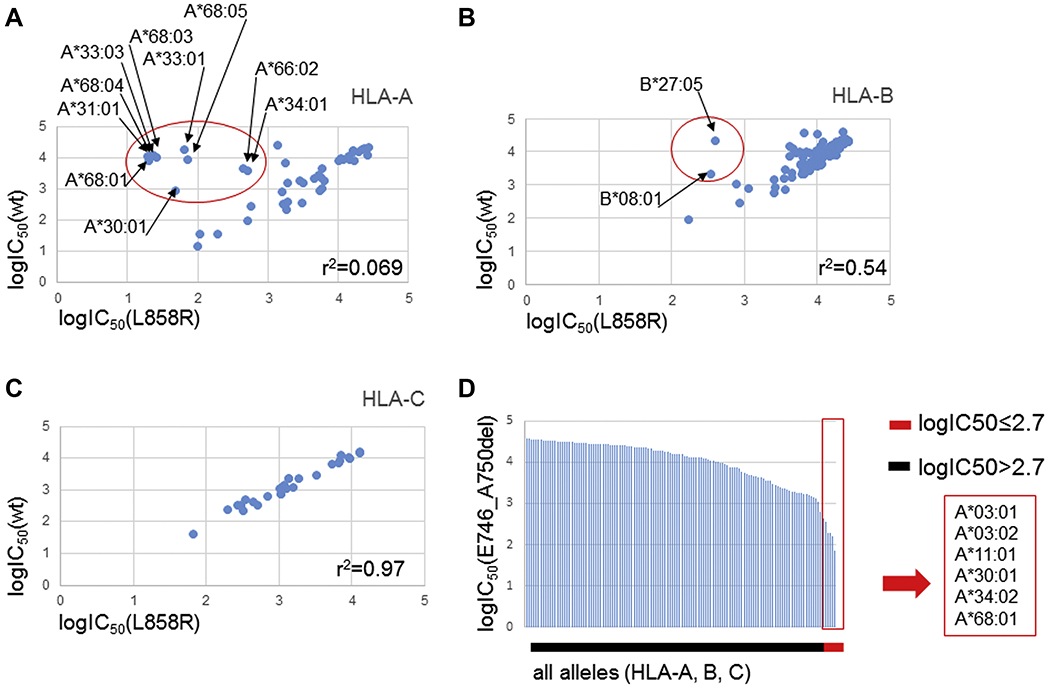
We predicted in silico the binding affinity of all possible 8-11mers containing the EGFR p.L858R with any given HLA-A, -B or -C allele from a list of 46 HLA-A, 98 HLA-B and 31 HLA-C alleles common in the contemporary USA.30 From these data, we identified HLA class I allotypes with predicted higher affinity to mutant peptides (potential neoantigen) than corresponding wildtype peptides, which were deemed protective. All other HLA class I allotypes were termed non-protective. We identified the following 10 HLA-A alleles that fulfill the predefined criteria for protective alleles: A*30:01, A*31:01, A*33:01, A*33:03, A*34:01, A*66:02, A*68:01, A*68:03, A*68:04 and A*68:05 (Fig. 1A). Similarly, we identified HLA-B*08:01 and HLA-B*27:05 as being protective for EGFR p.L858R among the HLA-B alleles (Fig. 1B) and none among the HLA-C alleles (Fig. 1C). We then predicted in silico the binding affinity of all possible 8-11mers containing the EGFR p.E746_A750del and identified HLA-A*03:01, HLA-A*03:02, HLA-A*11:01, HLA-A*30:01, HLA-A*34:02, HLA-A*68:01 as protective for EGFR p.E746_A750 (Fig. 1D). We used peptides with the lowest IC50 values per HLA allotype in the plots of figure 1. The specific peptides and IC50 values for EGFR p.L858R and EGFR p.E746_A750del that were used to construct the graphs in figure 1 are shown in Supplementary Tables 2 and 3, respectively.
Figure 1.
A, B, C: Correlation of IC50 values for 8-11mer peptides with lowest IC50 containing EGFR p.L858R and closest wild type peptide following logarithmic transformation (log10) for HLA-A, -B and -C alleles. Protective alleles are highlighted in red circle. D. IC50 values for 8-11mers with lowest IC50 containing EGFR p.E746_A750del following logarithmic transformation for HLA-A, -B and -C alleles. Protective alleles are shown in red rectangle.
We identified 21 patients with EGFR p.L858R and 15 patients with EGFR p.E746_A750 in the 511 patients from the TCGA lung adenocarcinoma PanCancer Atlas cohort with available HLA genotypes. The TCGA lung adenocarcinoma cohort includes mostly patients of European descent who have undergone surgical resection of their tumors and this is reflected in the EGFR mutation positive patients; only one of these patients was diagnosed with stage IV disease and there were only three patients with other than European ancestry (one African American and two Asians). We matched the patients with HLA genotypes from a publicly available source using the TCGA IDs 6 and classified the EGFR mutation positive patients in the protective group if they had protective alleles matching their specific mutation or the non-protective group if no such alleles were present. Table 1 summarizes the clinicopathologic characteristics for the patients with EGFR p.L858R or EGFR p.E746_A750del in the non-protective and protective groups as well as the patients with KRAS mutations and the entire TCGA lung adenocarcinoma cohort. We found that there was no difference between the protective and non-protective groups with regard to sex, age at diagnosis, ethnicity and type of EGFR mutation. However, stage distribution is different for patients with EGFR mutations in the protective group and non-protective groups. This difference is statistically significant (p=0.0038) and mainly driven by the difference in patients with stage III disease. Specifically, there are no patients with stage III disease in the protective group, whereas 8 of 23 patients in the non-protective group have stage III.
Table 1.
Clinicopathologic characteristics
| Variable | EGFR p.L858R or p.E746_A750del |
p value | Any KRAS mutation |
p value | All patients in TCGA Lung Adenocarcinoma PanCancer Atlas | ||
|---|---|---|---|---|---|---|---|
| Protective group | Non-protective group | Protective group | Non-protective group | ||||
| n | 12 | 24 | - | 96 | 57 | - | 511 |
| Sex | |||||||
| Male | 4 | 5 | 0.44 | 39 | 32 | 0.06 | 237 |
| Female | 8 | 19 | 57 | 25 | 274 | ||
| NA | - | - | - | - | - | ||
| Age | |||||||
| >65 | 5 | 11 | 1.0 | 51 | 26 | 0.39 | 239 |
| ≤65 | 6 | 12 | 41 | 28 | 253 | ||
| NA | 1 | 1 | 4 | 2 | 19 | ||
| Ethnicity | |||||||
| African American | 1 | 0 | 0.45 | 5 | 8 | 0.09 | 52 |
| European | 9 | 21 | 76 | 40 | 387 | ||
| Asian | 1 | 1 | 0 | 2 | 7 | ||
| Other | 0 | 0 | 0 | 0 | 1 | ||
| NA | 1 | 2 | 15 | 7 | 64 | ||
| Stage | |||||||
| I | 4 | 12 | 0.0038 | 46 | 29 | 0.81 | 273 |
| II | 7 | 4 | 25 | 14 | 122 | ||
| III | 0 | 8 | 19 | 9 | 84 | ||
| IV | 1 | 0 | 4 | 4 | 24 | ||
| NA | - | - | 2 | 1 | 8 | ||
| EGFR mutation | |||||||
| p.L858R | 5 | 16 | 0.17 | - | - | - | 21 |
| p.E746_A750del | 7 | 8 | 15 | ||||
Interestingly, at the population level, most of the protective alleles are more frequent in individuals of European compared to Asian ethnicity, with the notable exception of HLA A*11:01.30 In the TCGA cohort, we did not observe any difference in the frequency of protective HLA alleles between patients with EGFR mutations and the rest of the lung adenocarcinoma patients with the exception of HLA B*27:05. This allele was classified as protective for EGFR p.L858R and was under represented in patients with EGFR p.L858R (0/20 or 0%) compared to patients with EGFR p.E746_A750del (2/15 or 13.3%) and the rest of the TCGA lung adenocarcinoma cohort (non EGFR p.L858R, 33/491 or 6.7%). This difference however was not statistically significant reflecting low numbers of patients with EGFR mutation positive lung cancer in the TCGA. In the EGFR mutation positive patients in the TCGA cohort we found patients with EGFR p.L858R and at least one of HLA A*68:01, HLA A*31:01, HLA A*33:03 or HLA B*08:01. Likewise, we identified patients with EGFR p.E746_A750del and at least one of HLA A*03:01 or HLA A*11:01.
We then analyzed for differences in clinical outcomes for patients with EGFR p.L858R or EGFR p.E746_A750 in the protective and non-protective groups. One patient (TCGA-75-6207) in the non-protective group did not have available follow up data and was not included in the analysis. Median follow up was similar in the two groups at 21 months (0.4-58) in the protective and 19 months (2.6-29.6) in the non-protective group, as was the event rate for relapse and death (supplementary table 4). Interestingly, we found that the protective group had significantly longer DFS and OS compared to patients in the non-protective group. Median DFS for patients in the protective and non-protective groups is 49.3 and 18.8 months, respectively (HR=0.20, 0.05-0.78, p=0.012, Fig. 2A). Median OS for patients in the protective and non-protective groups is 59.2 and 38.2 months, respectively (HR 0.13, 0.02-0.64, p=0.005, Fig. 2B). Differences in OS are still significant after adjusting for mutation type, age at diagnosis, sex and stage for the 34 patients with available data, shown in table 2 (multivariate Cox proportional hazards model HR=0.04, 0.004-0.52).
Figure 2.
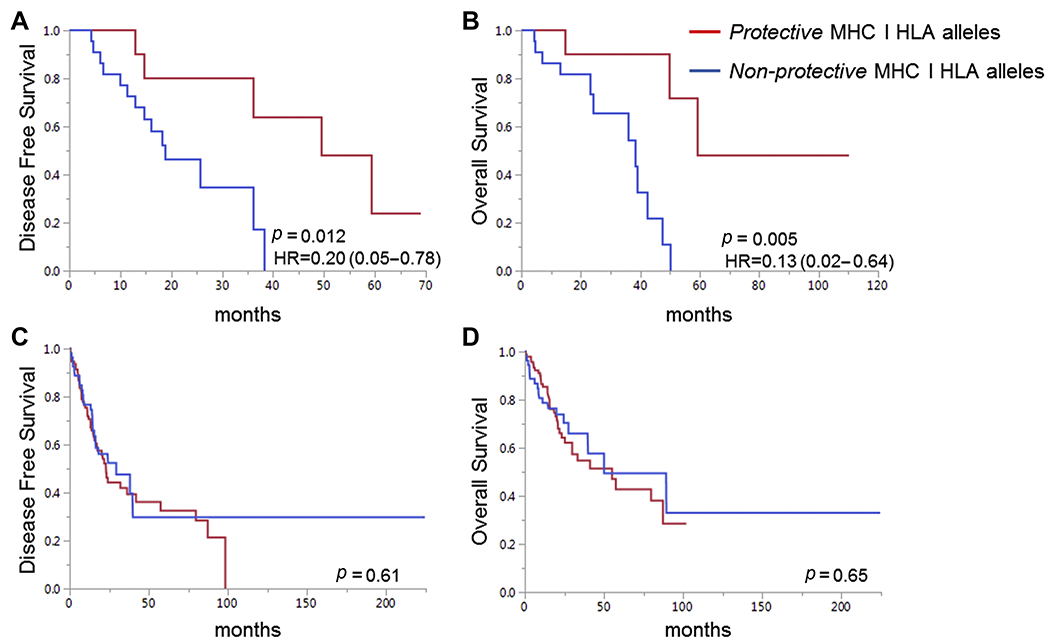
Disease-free (A) and overall survival (B) for patients with EGFR p.L858R or EGFR p.E746_A750del in the TCGA Lung Adenocarcinoma cohort for patients with non-protective only (n=23) and protective (n=12) HLA alleles in their host genome. Disease-free (C) and overall (D) survival in patients with any KRAS mutation in the TCGA Lung Adenocarcinoma cohort. KRAS mutation positive patients were classified in the protective group (n=94) if they had any protective allele specific for EGFR p.L858R or EGFR p.E746_A750del that was actually observed in patients with EGFR mutations in the same cohort and in the non-protective group (n=55) otherwise. Log rank p values displayed.
Table 2.
Cox Proportional Hazards Model, OS
| Variable | HR 95% CI, univariate | HR 95% CI, multivariate (n=34) |
|---|---|---|
| Age>65 | 0.77 (0.27-2.17) | 1.53 (0.41-5.75) |
| Male sex | 0.87 (0.26-2.84) | 1.34 (0.29-6.08) |
| Stage | ||
| I | 1 | 1 |
| II | 1.13 (0.32-4.01) | 2.17 (0.48-9.76) |
| III | 2.23 (0.64-7.79) | 1.02 (0.24-4.34) |
| IV | N/A | N/A |
| L858R vs. E746_A750del | 4.81 (1.33-17.33) | 7.4 (1.18-46.31) |
| Protective vs. Non-protective group | 0.13 (0.02-0.64) | 0.04 (0.004-0.52) |
To test if the protection from mutant EGFR-specific protective alleles is specific to EGFR or generally applicable to lung adenocarcinoma, we repeated the analysis on patients with KRAS mutations. We observed no difference in survival for patients with KRAS mutations and mutant EGFR specific protective or non-protective allotypes (Fig. 2C and 2D). This finding strongly suggests the protection we observed is specific to EGFR.
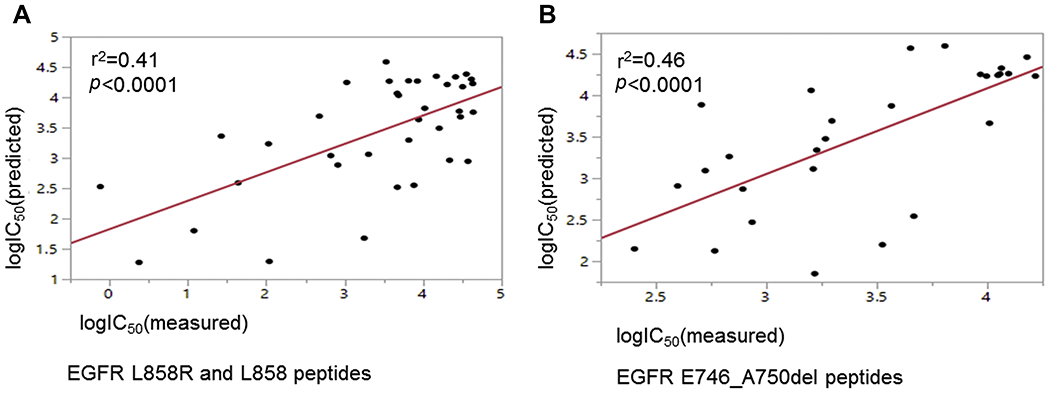
We validated the accuracy of in silico predictions for binding of peptides to HLA allotypes using in vitro binding assays. We tested the binding of selected HLA allotypes to peptides containing the EGFR L858R (mutant peptides) or EGFR L858 (wild type peptides) as well as peptides containing the EGFR E746_A750del (mutant peptides). Results are summarized in Supplementary Tables 5 and 6 for EGFR p.L858R and EGFR p.E746_A750del peptides, respectively. We found that mutant EGFR L858R but not wild type EGFR L858 peptides bound to A*31:01, A*33:01, A*68:01 and B*27:05 as predicted. Additionally, no mutant EGFR L858R peptides bound to A*01:01, A*02:01, A*03:01 and A*11:01, used as controls. B*08:01 bound to both the mutant EGFR L858R and the wild type EGFR L858 peptide, although the binding affinity for the former was 35 fold stronger than the latter. Neither the wild-type nor the mutant peptide had high affinity for the A*30:01 allele. We also validated the in silico predictions for the binding of EGFR E746_A750del peptides to A*11:01 and A*03:01. We found that multiple EGFR E746_A750del peptides bound to A*11:01 and A*03:01 with high (IC50 <500 nM) or intermediate affinity (IC50 500-5000 nM), whereas none bound to the control allele A*02:01 as shown in Supplementary table 6. Finally, we found significant level of correlation between the in silico predicted and the in vitro measured IC50 values following logarithmic transformation, for EGFR L858R, EGFR p.L858 (Fig. 3A) and EGFR p.E746_A750del peptides (Fig. 3B).
Figure 3.
Correlation between IC50 values for peptide-HLA allele binding following logarithmic transformation (log10) between in vitro measurements and in silico predictions. Correlation for EGFR p.L858R and EGFR p.L858 peptides shown in (A) and for EGFR p.E746_A750del peptides shown in (B).
Discussion
Collectively, our data indicate that certain host genome HLA genotypes exert protective functions in patients with EGFR p.L858R and EGFR p.E746_A750del positive lung cancer. Specifically, patients with these HLA genotypes have lower stage and better prognosis measured by DFS and OS compared with patients without protective alleles in the mostly surgical TCGA patient population. Although lower stage might account partially for better prognosis in this group, protective alleles remain prognostic after adjusting for disease stage. It is also possible that patients with protective alleles develop adaptive immunity with specificity for peptides derived from EGFR p.L858R and EGFR p.E746_A750, which restricts tumors at a lower stage. Our work herein is suggestive of the presence of effector T lymphocytes with specificity for EGFR mutation containing peptides as has been shown in at least one case report31. Further, our study maps the HLA allotypes in North American populations that could predict the presence of such immunity. Similar adaptive immunity with specificity for a driver oncogene rather than a passenger mutation is known to exist in rare case reports for other cancer types.15,32
Intriguingly, most of the protective alleles, with the exception of HLA A*11:01 are more common in individuals of European compared to Asian descent 30 indicating at the population level that the significantly lower frequency of EGFR mutation positive lung cancers in Europeans compared to Asians might be related to a protective immune system in the former. It is possible that adaptive immunity with specificity for EGFR mutation reduces the risk of development of clinical EGFR driven lung cancers, or that it prevents clinical progression of early stage tumors. Importantly, we found that HLA B*27:05, which is protective for EGFR p.L858R, to be under represented in patients with EGFR p.L858R compared to patients with EGFR p.E746_A750 or other driver mutations in the TCGA.
All identified protective HLA-A alleles belong to the HLA-A03 supertype 33 which share a preference for arginine or lysine at the carboxy-terminus of the binding peptide. It is possible that protection with respect to EGFR mutations is a more general feature of this supertype that could involve other less common alleles that have not been tested here. Likewise, both HLA-B*08:01 and HLA-B*27:05 have preference for arginine as main anchor specificity which implies that other alleles with the same binding specificity could have protective effects. Further studies are required to provide direct evidence for the existence of the EGFR mutation-specific T lymphocytes in patients and further clarify their role in the natural history of EGFR mutation positive lung cancer.
We validated the in silico predictions with in vitro measurements of HLA-peptide binding. In general, the majority of predicted binding vs. not binding using the IC50 500 nM threshold were validated in vitro. A*30:01 was an exception, where mutant EGFR p.L858R (but not wild type EGFR p.L858) was predicted to bind in silico, but was found to be a relatively weak binder in vitro; nevertheless, the mutant was found to bind with ~20-fold higher affinity than the wild type. This would not change the outcomes of the survival analysis in the TCGA dataset, as there were no patients classified in the protective group solely because of positive A*30:01 status. For B*08:01, although both mutant EGFR p.L858R and wild type EGFR p.L858 peptides had measured IC50 values below 500 nM in vitro, the IC50 for the former was 35 fold lower than the IC50 value for the latter. Therefore, there is preferential binding of the mutant peptide that could possibly trigger an adaptive immune reaction.
Applying stricter predefined criteria for differential binding predictions between EGFR p.L858R and wild type peptides to the same HLA class I allele, might exclude some alleles with potential to bind mutant peptides in vivo from the protective group and is a limitation in our study. However, stricter criteria also correctly exclude alleles that are predicted to bind mutant peptides in silico, or bind peptides in vitro, although they would not necessarily participate in immune responses in vivo because of central tolerance to wild type sequences that eliminate the corresponding T cell clones.23
Li et al31 show L858R peptides raised a T cell immune reaction in a patient with EGFR L858R positive lung cancer. Among the EGFR p.L858R containing peptide/allele combinations, HVKITDFGR/HLA-A*31:01 was associated with the most robust IFNγ response in vitro, as well as positive tetramer assay. On the contrary, the KITDFGRAK/HLA-A*11:01 was associated with much weaker IFNγ response in vitro and the tetramer assay was negative. Interestingly, both HVKITDFGR and KITDFGRAK bind to the corresponding HLA class I alleles (supplementary table 7). However, the wild type sequence for the former (HVKITDFGL) does not bind HLA-A*31:01, whereas the wild type sequence for the latter (KITDFGLAK) does bind HLA-A*11:01 with slightly higher affinity than the mutant peptide. To test the possibility that still the mutant peptide could raise a T cell immune reaction despite both the mutant and wild type peptides binding HLA-A*11:01, by different T cell receptor binding, we tested the binding for a number of substitutions at P7 to include non-conservative charged residues (i.e., D, E, and K), and several conservative/semi-conservative to probe different chemical specificities (e.g., hydrophobic, small polar etc.).34, 35 We found a wide range of binding affinities to HLA-A*11:01 (from weakest to strongest >30-fold) that follows chemical specificity, consistent with the varying residue P7 directly interacting with the HLA rather than the T cell receptor (supplementary table 7).
Our study has a number of limitations. First, the number of patients with either EGFR p.L858R or EGFR p.E746_A750 positive lung cancer in the TCGA is relatively small. Second, the TCGA includes mostly patients with early stage disease who have undergone surgical resection of their tumors and thus it is unclear whether the conclusions may also apply to patients with metastatic disease. Finally, we have based classification into protective and non-protective alleles on the HLA-peptide binding which does not guarantee immunogenicity on its own as it does not involve the peptide-TCR interaction. Nevertheless, we used predefined criteria to predict adaptive immunity for EGFR p.L858R and EGFR p.E746_A750 positive lung cancer and correlated with outcomes only in EGFR mutation positive patients, but not in patients with KRAS or other mutations. Our data suggest that patients with EGFR mutation specific adaptive immunity might be enriched in earlier stage patients. Overall, our study expands our understanding of the natural history of EGFR mutation positive lung cancer and the methodology can be expanded to other driver oncogenes.
Supplementary Material
Acknowledgements:
The results (published or shown) here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga.
Funding source: SPORE in Lung (P50 CA058187), 2018 AACR-Conquer Cancer Foundation of ASCO YIA for Translational Cancer Research (AD), NIH contract 75N93019C00001 (AS), R21 AI134127 (AS).
Disclosures: RCD has stock ownership in Rain Therapeutics; has income from advisory boards/consulting for Genentech/Roche, Takeda, and Rain Therapeutics; research funding from Ignyta; and patents licensed to Rain Therapeutics and Abbott Molecular. AD, PG, JS, AS, PJN have no disclosures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations: EGFR: Epidermal Growth Factor Receptor, HLA: Human Leucocyte Antigen, TCGA: The Cancer Genome Atlas, NSCLC: Non-small cell lung cancer, MHC: Major Histocompatibility Complex, EBV: Epstein Barr Virus, OS: Overall Survival, DFS: Disease-Free Survival
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin 2019;69:7–34. [DOI] [PubMed] [Google Scholar]
- 2.Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers--a different disease. Nat Rev Cancer 2007;7:778–790. [DOI] [PubMed] [Google Scholar]
- 3.Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell 2012;150:1121–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511:543–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pacheco JM, Dimou A, Bunn PA. Advances in lung cancer. Oncotarget 2017;8:78247–78248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorsson V, Gibbs DL, Brown SD, et al. The Immune Landscape of Cancer. Immunity 2018;48:812–830 e814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Serrano P, Hartmann M, Schmitt E, et al. Clinical Development and Initial Approval of Novel Immune Checkpoint Inhibitors in Oncology: Insights From a Global Regulatory Perspective. Clin Pharmacol Ther 2019;105:582–597. [DOI] [PubMed] [Google Scholar]
- 8.Yossef R, Tran E, Deniger DC, et al. Enhanced detection of neoantigen-reactive T cells targeting unique and shared oncogenes for personalized cancer immunotherapy. JCI Insight 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kalaora S, Wolf Y, Feferman T, et al. Combined Analysis of Antigen Presentation and T-cell Recognition Reveals Restricted Immune Responses in Melanoma. Cancer Discov 2018;8:1366–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wieczorek M, Abualrous ET, Sticht J, et al. Major Histocompatibility Complex (MHC) Class I and MHC Class II Proteins: Conformational Plasticity in Antigen Presentation. Front Immunol 2017;8:292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Horton R, Wilming L, Rand V, et al. Gene map of the extended human MHC. Nat Rev Genet 2004;5:889–899. [DOI] [PubMed] [Google Scholar]
- 12.Robinson J, Guethlein LA, Cereb N, et al. Distinguishing functional polymorphism from random variation in the sequences of >10,000 HLA-A, -B and -C alleles. PLoS Genet 2017;13:e1006862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chowell D, Morris LGT, Grigg CM, et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science 2018;359:582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Efremova M, Finotello F, Rieder D, et al. Neoantigens Generated by Individual Mutations and Their Role in Cancer Immunity and Immunotherapy. Front Immunol 2017;8:1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veatch JR, Lee SM, Fitzgibbon M, et al. Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma. J Clin Invest 2018;128:1563–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marty R, Kaabinejadian S, Rossell D, et al. MHC-I Genotype Restricts the Oncogenic Mutational Landscape. Cell 2017;171:1272–1283 e1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marty Pyke R, Thompson WK, Salem RM, et al. Evolutionary Pressure against MHC Class II Binding Cancer Mutations. Cell 2018;175:416–428 e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoof I, Peters B, Sidney J, et al. NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics 2009;61:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jurtz V, Paul S, Andreatta M, et al. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J Immunol 2017;199:3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nielsen M, Andreatta M. NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med 2016;8:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mei S, Li F, Leier A, et al. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction. Brief Bioinform 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Paul S, Weiskopf D, Angelo MA, et al. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. J Immunol 2013;191:5831–5839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duan F, Duitama J, Al Seesi S, et al. Genomic and bioinformatic profiling of mutational neoepitopes reveals new rules to predict anticancer immunogenicity. J Exp Med 2014;211:2231–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Lichtenberg T, Hoadley KA, et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018;173:400–416 e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cerami E, Gao J, Dogrusoz U, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data (vol 2, pg 401, 2012). Cancer Discovery 2012;2:960–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao JJ, Aksoy BA, Dogrusoz U, et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal 2013;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sidney J, Southwood S, Moore C, et al. Measurement of MHC/peptide interactions by gel filtration or monoclonal antibody capture. Curr Protoc Immunol 2013;Chapter 18:Unit 18 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 1973;22:3099–3108. [DOI] [PubMed] [Google Scholar]
- 29.Gulukota K, Sidney J, Sette A, et al. Two complementary methods for predicting peptides binding major histocompatibility complex molecules. J Mol Biol 1997;267:1258–1267. [DOI] [PubMed] [Google Scholar]
- 30.Cao K, Hollenbach J, Shi X, et al. Analysis of the frequencies of HLA-A, B, and C alleles and haplotypes in the five major ethnic groups of the United States reveals high levels of diversity in these loci and contrasting distribution patterns in these populations. Hum Immunol 2001;62:1009–1030. [DOI] [PubMed] [Google Scholar]
- 31.Li F, Chen C, Ju T, et al. Rapid tumor regression in an Asian lung cancer patient following personalized neoepitope peptide vaccination. Oncoimmunology 2016;5:e1238539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tran E, Robbins PF, Lu YC, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med 2016;375:2255–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sidney J, Peters B, Frahm N, et al. HLA class I supertypes: a revised and updated classification. BMC Immunol 2008;9:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madden DR. The three-dimensional structure of peptide-MHC complexes. Annu Rev Immunol 1995;13:587–622. [DOI] [PubMed] [Google Scholar]
- 35.Ruppert J, Sidney J, Celis E, et al. Prominent role of secondary anchor residues in peptide binding to HLA-A2.1 molecules. Cell 1993;74:929–937. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.