

Abstract
We review recent progress on natural products that target cytoskeletal components, including microtubules, actin, intermediate filaments, and septins and highlight their demonstrated and potential utility in the treatment of human disease. The anticancer efficacy of microtubule targeted agents identified from plants, microbes, and marine organisms is well documented. We highlight new microtubule targeted agents currently in clinical evaluations for the treatment of drug resistant cancers and the accumulating evidence that the anticancer efficacy of these agents is not solely due to their antimitotic effects. Indeed, the effects of microtubule targeted agents on interphase microtubules are leading to their potential for more mechanistically guided use in cancers as well as neurological disease. The discussion of these agents as more targeted drugs also prompts a reevaluation of our thinking about natural products that target other components of the cytoskeleton. For instance, actin active natural products are largely considered chemical probes and non-selective toxins. However, studies utilizing these probes have uncovered aspects of actin biology that can be more specifically targeted to potentially treat cancer, neurological disorders, and infectious disease. Compounds that target intermediate filaments and septins are understudied, but their continued discovery and mechanistic evaluations have implications for numerous therapeutic indications.
Introduction
The eukaryotic cytoskeleton is made up of at least four distinct classes of proteins that coordinate almost every aspect of cellular physiology, including structure, motility, propagation, and intracellular trafficking. The two most recognizable elements of the cytoskeleton are microtubules and actin microfilaments, which are polarized polymers consisting of tubulin heterodimers and actin monomers, respectively (Figure 1). The intermediate filaments are a more heterogeneous component of the cytoskeleton that can consist of different proteins, including vimentins, keratins, neurofilaments, lamins, or desmin, that are differentially expressed in distinct cell types. Septins are an often overlooked fourth cytoskeletal element that assemble into filaments and rings that can associate with microtubules and actin filaments to facilitate critical roles in cytokinesis and protein scaffolding. These proteins form distinct cellular structures (Figure 1) that are critical for signaling, intracellular transport, and cell motility. While the cytoskeleton is critical for many conserved cellular processes, there is a distinct coordination of cytoskeletal structure, localization, and dynamics in individual cell types. For instance, although actin filaments are found in all cells, the specific coordination of actin and myosin fibers into sarcomeres in muscle cells underlie their coordinated contractility whereas in the gastrointestinal track cross-linked actin filaments form specialized microvilli critical for nutrient absorption. Due to the diverse functionality of the cytoskeleton, it is often coopted to promote pathogenesis and can therefore serve as a target for therapeutic interventions. The most notable example of which is the prolific use of microtubule targeted drugs in the treatment of cancer. In this review, we will describe recent progress on natural products that target components of the eukaryotic cytoskeleton and describe how a more detailed understanding of the distinct biochemical and cell biological effects of these agents underlie their potential to be used as more targeted therapeutics in cancer as well as in other indications.
Figure 1.
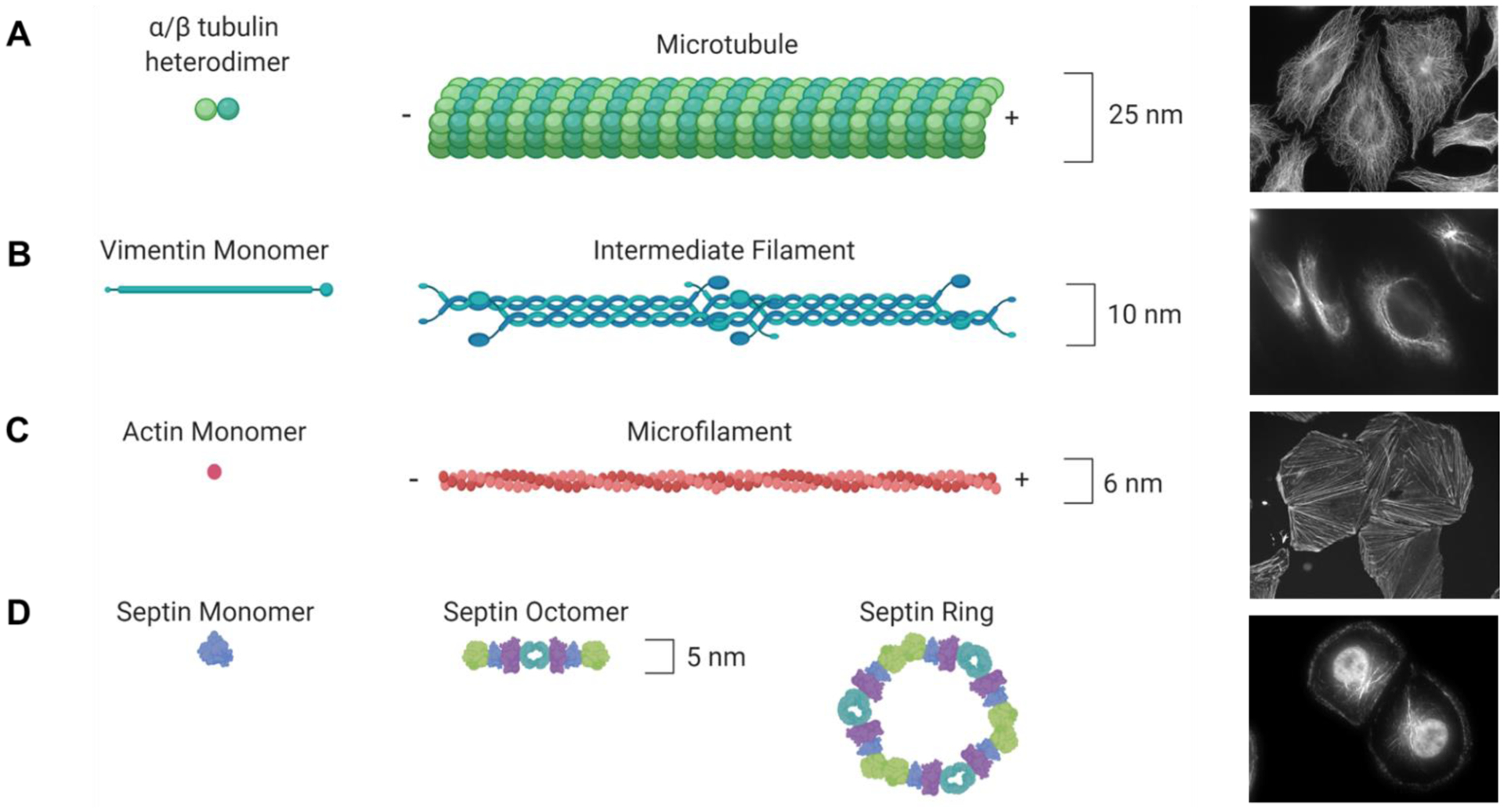
Schematic and actual representation of cytoskeletal proteins. (A) α/β-tubulin heterodimers are cotranslated to form the basic building blocks of the microtubule cytoskeleton. Cellular microtubule localization is visualized by β-tubulin immunofluorescence in HeLa cells. (B) Monomers of intermediate filament proteins, including vimentin, dimerize and then form head-to-tail tetramers that form intermediate filament structures. Cellular vimentin localization is visualized by immunofluorescence in MDA-MB-231 cells. (C) Actin monomers polymerize in a head-to-tail fashion to form microfilaments. Cellular F-actin localization is visualized by imaging TRITC-labeled phalloidin treated A10 cells. (D) Individual septin proteins come together to form linear and ringed cytoskeletal structures. Cellular localization of septin 9 is visualized by immunofluorescence in HCC1937 cells. Microtubule and microfilament structures have polarity depicted as + and − ends.
Microtubule targeted agents (MTAs) from diverse natural sources.
Natural products that bind to microtubules are some of the most effective drugs available for the treatment of solid tumors and hematological malignancies. MTAs continue to be used as single agents and in combination chemotherapy as well as in combination with molecular targeted therapies and immunotherapeutics to improve their anticancer efficacy. The first MTA identified, colchicine, was originally isolated from Colchicum autumnale (commonly known as autumn crocus) and used in its natural form as early as 1500 BC to treat joint pain and swelling. It is currently approved for the treatment of gout as a result of its anti-inflammatory effects. However, it does not have an acceptable therapeutic index for the treatment of cancer. Importantly, tubulin was first identified as the subunit of microtubules directly as a result of its interaction with colchicine,1 which binds within a deep pocket near the α/β-tubulin intradimer interface (Figure 2A).2 Vinblastine, was isolated from Catharanthus roseus (commonly known as Madagascar periwinkle) in 1958 for its purported effects against diabetes. Although no antidiabetic effects were observed, it was discovered that the vinca alkaloids elicit myelosuppressive effects that led to the approval of vinblastine as the first MTA for cancer treatment. Since these initial discoveries, there have been concerted efforts to identify new natural products that alter the microtubule cytoskeleton. This has resulted in the FDA approval of over a dozen distinct MTAs for the treatment of various cancers with numerous additional classes that have been described to have anticancer potential in preclinical evaluations. The first MTAs isolated, including vinblastine and colchicine, were classified as microtubule destabilizers because they inhibit microtubule polymerization and therefore decrease overall microtubule density. The identification of the mechanism of paclitaxel as an enhancer of microtubule polymerization by Dr. Susan Horwitz’s laboratory in 1979 introduced microtubule stabilizers as a second distinct class of MTAs.3 Although microtubule stabilizers and destabilizers have opposing roles on gross microtubule polymerization, they both initiate mitotic arrest as a result of a shared mechanism of disrupting microtubule dynamicity, which is required for formation of the microtubule spindle and was long thought to be their primary mechanism of anticancer activity. The interrogation of plant, marine, and microorganism-derived natural products led to the discovery of each of the six distinct binding sites on tubulin that can be exploited to alter microtubule polymerization and dynamics (Figures 2A and 3A).
Figure 2.

Binding sites of microtubule destabilizing agents. (A) Superimposition of the binding sites of colchicine (orange, PDB ID: 4O2B), maytansine (green, PDB ID: 4TV8), pironetin (pink, PDB ID: 5LA6), and vinblastine (purple, PDB ID: 1Z2B) on a single α/β tubulin heterodimer. (B) Superimposition of vinblastine (red, PDB ID: 1Z2B) and the peptide soblidotin (blue, PDB ID: 3E22) in their respective tubulin binding sites between two heterodimers, which together define the “vinca domain” (pink, α-tubulin; green, β-tubulin). GTP is depicted in purple.
Figure 3.

Binding sites of microtubule stabilizing agents. (A) Superimposition of paclitaxel (green, PDB ID: 1JFF) and laulimalide (orange, PDB ID: 4O4H) in their respective binding sites on β-tubulin. (B) Superimposition of the binding sites of covalent microtubule stabilizers zampanolide (blue, PDB ID: 4I4T), cyclostreptin (gold, PDB ID: 6QTN), and taccalonolide AJ (green, PDB ID: 5EZY) in the taxane binding pocket on β-tubulin. (C) Individual views of the covalent binding linkages of zampanolide (blue), cyclostreptin (gold), and taccalonolide AJ (green) with β-tubulin.

Vinca site microtubule destabilizers.
The vinca binding site, where vinblastine (1) and other vinca alkaloids bind, is the only microtubule destabilizer binding site that is currently represented by clinically approved cancer therapeutics. Vinca alkaloids currently approved for the treatment of cancer include vinblastine (1), vincristine (2), vinorelbine, vindesine, and vinflunine.4 The vinca alkaloids are particularly effective in the treatment of hematological malignancies, but each of the clinically approved agents of this class have distinct cancer indications and associated toxicity profiles highlighting the clinical diversity even among MTAs that bind to the same site on tubulin.5 The approval of eribulin (3) for the treatment of metastatic breast cancer and liposarcoma has been a major success story for marine-derived vinca-binding site MTAs. Eribulin, which is a simplified analog of the sponge-derived macrolide halichondrin B, binds within the vinca domain, although it has distinct mechanistic effects that are associated with stabilization of the tumor vasculature (which enhances drug delivery) and reversal of epithelial-to-mesenchymal transition (EMT) in breast cancer.6–8 Most strikingly, eribulin offers an overall survival advantage in breast cancer patients compared to other approved treatments9 and has superior efficacy compared to vinorelbine, which is the only other microtubule destabilizer approved for breast cancer.10 Currently, eribulin is being evaluated in clinical trials against pediatric and adult solid tumors, including sarcomas, cervical cancer, and brain metastases from breast cancer. An antibody-drug conjugate (ADC) of eribulin to the folate receptor α, MORAb-202, has demonstrated efficacy in preclinical cancer models of gastric and non-small cell lung cancers that overexpress this receptor11 and is currently in phase I clinical trials for patients with solid tumors.
There are several other classes of compounds identified from marine invertebrates that bind within the vinca site, including dolastatin 10 (4), hemiasterlin, arenastatin A, diazonamide A, and vitilevuamide.12 The vinca binding site consists of a primary core at the inter-dimer interface that can extend into what has been termed the “peptide site” that is occupied by peptide-based natural products, including hemiasterlin, dolastatin 10, and soblidotin13 (Figure 2B). The microtubule destabilization initiated by vinca-site ligands involves sequestration of tubulin dimers into ring-like oligomers and/or blocking the microtubule tip to prevent incorporation into microtubules. Several of these novel vinca-site binding agents and their synthetic analogs have been evaluated for clinical efficacy. A synthetic derivative of hemiasterlin, HTI-286, entered phase II trials for recurrent non-small-cell lung cancer but was terminated early with no further clinical testing initiated.14 Dolastatin 10 (4), a cyanobacterial metabolite originally discovered from a sea hare,15, 16 and related analogs soblidotin and tasidotin entered clinical evaluations in the late 1990s and early 2000s but toxicity and lack of efficacy inhibited their advancement.17 However, in 2003, Seattle Genetics published their work on the generation of an ADC using monomethyl auristatin E (MMAE, 5) as the cytotoxic payload,18 which resulted in the approval of the CD30-targeted MMAE conjugate bentuximab vedotin in 2011 for the treatment of lymphomas. ADCs that direct MMAE to other targets are in clinical evaluations for urothelial, cervical, ovarian, and breast cancers. The cryptophycins are vinca-site binding macrocyclic depsipeptides originally identified from cyanobacteria.19, 20 The synthetic cryptophycin, cryptophycin 52 (LY355703, 6), entered clinical trials for metastatic colorectal, ovarian, and non-small cell lung cancer, but development was halted due to lack of response and unacceptable toxicities.21 However, there has been a renewed interest in these exquisitely sensitive drugs as cytotoxic payloads for targeted therapeutics, similarly to the use of MMAE, in recent years.22, 23
Maytansine site microtubule destabilizers.
Several natural products have been identified that interfere with vinblastine (1) binding but are associated with a binding site adjacent but distinct from the vinca site, which has been classified as the maytansine site on tubulin (Figure 2A).24 Compounds that have demonstrated binding to this site include the marine-derived PM060184 and spongistatins, the plant-derived maytansine (7), and the bacterial-derived rhizoxin and disorazoles.25 Rhizoxin and maytansine entered clinical evaluations for cancer but did not show sufficient activity to progress toward further clinical development as single agents.26, 27 However, like the dolastatins, the clinical utility of maytansine (7) has been resurrected through the generation of ADCs. Trastuzumab emtansine (T-DM1), the first ADC that has been approved for the treatment of solid tumors, consists of the maytansinoid, emtansine (mertansine; DM1) conjugated to the HER2-targeted monoclonal antibody trastuzumab (Herceptin) for the treatment of HER2-positive breast cancer.
Colchicine-site microtubule destabilizers.
While colchicine (8) itself does not possess the therapeutic window for cancer treatment, there have been significant efforts over the last thirty years to develop a colchicine-site drug with sufficient anticancer efficacy for clinical use. This is best typified by the combretastatins (combretastatin A1, 9), first identified from the South African tree Combretum caffrum, which have been evaluated in clinical trials for over 16 years with no approval for any cancer indication. Given the synthetically tractable nature of colchicine-site binding drugs (as compared to the complex natural products typified as ligands to the other microtubule binding sites) and their general ability to circumvent clinically relevant mechanisms of drug resistance to approved MTAs, it is somewhat surprising that no agent of this class has entered the pharmacopeia of approved cancer drugs. Notable clinical limitations of colchicine-site drugs that have entered clinical evaluations include poor aqueous solubility, generation of toxic metabolites, and poor chemical or metabolic stability.28 While many colchicine site binding agents have entered and failed clinical trials over the last two decades, there are a few promising agents of this class currently in clinical evaluations. Plinabulin (NPI-2358, 10) is a synthetic analog of halimide, a colchicine-site MTA identified from an Aspergillus strain, which is currently in phase III clinical trials in combination with the microtubule stabilizer docetaxel for advanced non-small cell lung cancer based on the preclinical efficacy of this combination in KRAS-driven preclinical models.29 The drug BAL27862 (11) is a colchicine-site binding MTA and the active metabolite of the prodrug BAL101553, which is currently in clinical trials for glioblastoma. Brain cancers present an optimal opportunity for clinical development of a colchicine-site MTA as none of the clinically approved MTAs cross the blood-brain barrier. Although crystallographic evidence demonstrates that BAL27862 (11) resides within the colchicine pocket on tubulin, it has distinct effects on microtubule structure in biochemical and cellular assays, including distinct binding kinetics and effects on the mitotic spindle apparatus.30 The BAL27862 prodrug, BAL101553, has improved solubility, crosses the blood-brain barrier, and is orally bioavailable. It was found to be well tolerated in phase I clinical evaluations in 25 patients with recurrent glioblastoma and high-grade gliomas with one patient achieving a long-lasting partial response after progression on two lines of prior therapy and five patients experiencing stable disease.

VERU-111 (12) is an orally bioavailable colchicine-site MTA that is being developed as a third-line treatment against metastatic therapy resistant prostate cancer with a phase Ib/II trial currently recruiting patients. Colchicine-site agents as a class tend to retain efficacy in clinically relevant models of MTA resistance, including those expressing P-glycoprotein or the βIII isoform of tubulin. Therefore, there is a strong rationale that VERU-111 would have efficacy in prostate cancers and other cancers where MTAs have proven to be effective but are prone to development of resistance. As mentioned above, combretastatins are continuing to be evaluated clinically with OXi4503, a prodrug of combretastatin A1 (9), currently in phase I/II clinical trials for the treatment of relapsed or refractory AML and myelodysplastic syndromes. An intriguing effect of combretastatins and some other colchicine-site drugs is their ability to disrupt tumor vasculature to promote tumor necrosis.31, 32 OXi4503 and other combretastatins have demonstrated antitumor efficacy and vascular disruption in a variety of preclinical models, including MDA-MB-231 and MHEC5-T xenografts. However, the lack of clinical responses and presence of cerebrovascular toxicities, likely attributable to vascular disrupting activities of these agents, have so far inhibited the clinical development of this drug class. Ultimately, the colchicine-site MTAs for the treatment of cancers based on their efficacy in drug resistant models and the development of orally available inhibitors that cross the blood-brain barrier continues to have promise but has not been effectively translated to clinical efficacy.
Pironetin site microtubule destabilizer.
The fourth microtubule destabilizer binding site on tubulin was identified with pironetin (13), an α/β unsaturated lactone isolated from Streptomyces species. Pironetin is unique from all other microtubule destabilizing agents identified to date in that it covalently binds to Cys316 on α-tubulin, making it the only known MTA that exclusively interacts with α-tubulin.33 Pironetin has potent in vitro activity and retains efficacy in H69 and K562 taxane resistant in vitro models, likely due to its irreversible target binding.34, 35 It did not demonstrate antitumor effects in P388 leukemia bearing mice,36 although this may be due to a poor choice of model since this cell line is one of the least sensitive to pironetin in vitro.37
Taxane site microtubule stabilizers.
Paclitaxel (14), the first microtubule stabilizer identified from the bark of the Pacific Yew,3 remains one of the most widely used anticancer drugs for the treatment of solid tumors. Second and third generation taxanes, including docetaxel (15), cabazitaxel, and abraxane (nanoparticle albumin-bound paclitaxel) are semi-synthetic paclitaxel analogs that bind within the taxane pocket on β-tubulin (Figure 3A).38 Paclitaxel and docetaxel are approved for the treatment of a wide variety of solid tumors, including breast, ovarian, testicular, non-small cell lung, gastric, head and neck, and prostate cancers. Cabazitaxel was developed as a taxane that would retain efficacy in drug resistant tumors; however, it is only approved for drug-resistant prostate cancer. Abraxane (nab-paclitaxel) is an albumin-bound form of paclitaxel that is approved for pancreatic, breast, and non-small cell lung cancers. Although originally designed to overcome solvent associated problems with taxane treatment, the binding of albumin to the SPARC (secreted protein acidic and rich in cysteine) protein on breast, lung and prostate cancers also promotes selective tumor targeting and correlates with response to nab-paclitaxel.39 The epothilones are a class of microtubule stabilizers that were originally identified from myxobacterium. The semi-synthetic epothilone B (16) analog ixabepilone (17) was approved for the treatment of taxane resistant metastatic or locally advanced breast cancer.

Microtubule stabilizers are highly effective against solid tumors as single agents, in combination with chemotherapy regimens and with newer targeted drugs, including immunotherapy.40 All currently approved microtubule stabilizers bind within the same taxane binding pocket on β-tubulin, which resides within the microtubule lumen. However, there are noted differences among even drugs of this class with regard to their therapeutic use and toxicities that may be attributable to differences in their effects on microtubule dynamics and structure. Despite their efficacy, limitations of microtubule stabilizers, like destabilizers, include innate and acquired drug resistance as well as toxic side effects. New generation taxanes and other microtubule stabilizers are in various stages of clinical and preclinical development. Tesetaxel is an orally bioavailable taxane with nitrogen containing functional groups that dramatically increase solubility and plasma half-life as compared to other taxanes.41, 42 Tesetaxel is not efficiently effluxed by the p-glycoprotein drug transporter, allowing for efficacy in taxane resistant tumor models and for penetration of the blood-brain barrier, providing an opportunity for the treatment of primary brain tumors as well as brain metastases. It is currently in phase II and III clinical trials for the treatment of breast cancer including patients with brain metastases to evaluate their response as a secondary outcome. The natural product, epothilone B (16), was evaluated in ovarian cancer clinical trials but failed to demonstrate a survival advantage.43 Epothilone D and its derivative KOS-1584 did not progress past phase I/II clinical trials for cancer. The success of nab-paclitaxel has led to additional strategies to formulate taxanes to increase their tumor targeting while decreasing the need for the use of the toxic solvent cremophor, which is used for the formulation of paclitaxel. Genexol-PM is a cremophor-free micelle form of paclitaxel that demonstrated a modest response in locally advanced head and neck squamous cell carcinoma,44 efficacy and toxicity similar to standard of care in ovarian cancer,45 and is in phase II clinical evaluations for pancreatic adenocarcinoma and hepatocellular carcinoma. A polyglutamated nanoformulation of paclitaxel has not demonstrated efficacy in clinical evaluations46 although it remains in phase III clinical evaluations against late stage ovarian, peritoneal or fallopian tube cancer.
In addition to the taxanes, other classes of microtubule stabilizers have been identified from natural products. Discodermolide (18), a polyketide isolated from a marine sponge, is a potent microtubule stabilizer that can synergize with paclitaxel and retains efficacy in taxane-resistant tumor models due to a distinct mode of binding within the taxane pocket on β-tubulin.47–50 Although discodermolide entered clinical trials for cancer, its clinical development was halted due to pneumotoxicities.51 Dictyostatin (19) is another sponge-derived microtubule stabilizer that shows some structural similarity to discodermolide and retains efficacy in clinically relevant models of taxane resistance.52 Additionally, three microtubule stabilizers that bind covalently to β-tubulin within the taxane site have been identified; zampanolide from a marine sponge, cyclostreptin from Streptomyces cultures, and the taccalonolides (taccalonolides AF and AJ) from plants of the genus Tacca38 (Figure 3B, C). Each of these microtubule stabilizers retain efficacy in taxane-resistant models and exhibit distinct profiles of microtubule stability due to their irreversible binding.34, 53, 54 In particular, the taccalonolides have demonstrated highly persistent efficacy in taxane-resistant tumor models upon systemic administration or local tumor delivery.53, 55, 56 Additional classes of microtubule stabilizing drugs from natural products continue to be evaluated for their unique effects on microtubule structure, dynamics and efficacy in drug-resistant models, including the avocado toxin persin, and rhazinilam from plants of the Apocynaceae family among others.57, 58
Laulimalide site microtubule stabilizers.
The discovery of laulimalide (20) and peloruside microtubule stabilizers from marine sponges led to the identification of a second microtubule stabilizer binding site that is completely distinct from the taxane site (Figure 3A) although it appears to promote a common mechanism of M-loop stabilization.59 In addition to the distinct binding site on the β-tubulin protein, in the intact microtubule laulimalide is associated with the exterior of the microtubule in contrast to the taxanes, which are buried in the interior microtubule lumen.60 Peloruside demonstrated in vivo antitumor efficacy against H460 and A549 xenografts with recoverable weight loss but was found to be less effective than paclitaxel in a Pgp-expressing NCI/ADR-RES xenograft model.61 The in vivo efficacy of laulimalide/fijianolide has been mixed with slight inhibition of tumor growth in HCT-116 xenografts and a complete lack of antitumor activity in MDA-MB-435 and HT-1080 models over a range of doses and schedules.62, 63 This novel microtubule stabilizer binding site provides the possibility for the clinical development of a novel class of anticancer drugs.
Microtubule stabilizers in the treatment of neurological disorders.
While the focus on microtubule stabilizers has been on their efficacy as anticancer agents, there has been a somewhat recent surge in the interest in these drugs for the treatment of neurological disorders. The initial hypothesis for the use of microtubule stabilizers in the treatment of neurodegenerative disease stems from the fact that the pathology of many of these diseases is associated with defects in the folding and aggregation of the microtubule associated protein tau, which are commonly referred to as tauopathies. In neuronal axons, tau plays a critical role in the organization, dynamics, and overall stability of microtubules, which are used for axonal transport. In vitro studies have demonstrated that microtubule stabilizers, including taxanes, epothilones, and peloruside, can rescue tau-induced neuronal degeneration and axonal transport defects.64, 65
Consistent with the role of microtubule stabilization in counteracting tauopathy-associated phenotypes, the microtubule destabilizer colchicine is used as a method to induce an Alzheimer’s disease-like pathology in rats.66 However, the use of microtubule stabilizers to counteract tau-related neuropathologies are complicated by the poor brain penetration of clinically approved MSAs as well as their associated toxicities, including neuropathies. Dictyostatin (19), which is brain penetrant, improved microtubule density and axonal dystrophy in mice with tau pathologies, but was associated with unacceptable toxicities.67 More promising brain-penetrant microtubule stabilizers entered clinical evaluations for tau-associated neurodegenerative diseases, including epothilone D and the taxane TPI-287. The phase I study of epothilone D (KOS-862/BMS-241027) in patients with mild Alzheimer’s disease ended in 2013 and was subsequently discontinued while two phase I clinical trials of TPI-287 in patients with Alzheimer’s disease and other tau-associated neuropathologies are ongoing. Another hurdle in the use of MSAs in the treatment of tau-associated disease is the well-documented effect of these drugs on the promotion of peripheral neuropathies in cancer patients treated with these drugs even when they do not cross the blood brain barrier.68 While pharmacological microtubule stabilization can serve as a stand-in for tau-associated microtubule stabilization to some degree, it is important to recognize that these drugs promote distinct alterations in the structure and dynamics of microtubules.69 Microtubule hyperstabilization by MSAs alters axonal trafficking and can directly lead to neurotoxicity of the distal axon in cultured neurons.70, 71 Additionally, microtubule stabilizers can directly promote neuroinflamation.72 Intriguingly, microtubule stabilization by taxanes has a greater neurotoxicity than microtubule destabilization promoted by eribulin (3), even when eribulin showed greater penetration into peripheral nerve tissues.73 Therefore, the promise of developing more brain penetrant MSAs for the treatment of neurological disease and brain tumors and metastases should be tempered by an appreciation of their detrimental effects on normal neuronal function.
Non-mitotic effects of MTAs.
The observation that MTAs can have important clinical consequences in non-dividing neuronal cells leads to the important realization that the effects of these drugs on interphase microtubules are just as important as their ability to arrest cells in mitosis. Although scientists and clinicians often refer to microtubule binding drugs as antimitotics, accumulating studies demonstrate that the anticancer efficacy of these drugs goes beyond their antimitotic effects.74–76 The most critical evidence demonstrating that the antimitotic effects of MTAs are not sufficient for their anticancer efficacy is the finding that other classes of antimitotic chemotherapeutics that do not interact directly with microtubules are not effective in the treatment of solid tumors. At least 46 independent clinical trials of 20 different non-microtubule binding mitotic inhibitors resulted in an overall response rate of less than 2% against solid tumors even though side effects of mitotic inhibition, including neutropenia, were observed.76 This is consistent with preclinical data where complete responses were observed in only 2 of the 53 tumor models evaluated. The lack of clinical efficacy of mitotic inhibition is consistent with the fact that solid tumors double on average every 200 days, a much longer doubling time than what is achieved in preclinical in vitro and in vivo models wherein tumors divide once every 1 – 6 days. Additionally, in vivo imaging has provided direct visualization of the cytotoxic effects of MTA treatment independently of entry into mitosis.77, 78
The realization that the anticancer efficacy of MTAs is not solely attributable to their antimitotic effects leads to renewed interest in the mechanism of action of these drugs, which have been successfully used in the treatment of cancer for decades. In interphase, the microtubule cytoskeleton plays an important role in intracellular trafficking of proteins and organelles as well as in a number of signaling pathways. Known oncogenic drivers that traffic along microtubules include p53, c-myc, APC, BRCA1, Rb, ERα, HIF-1α, and Src among others.75 Elegant work has demonstrated that MTAs inhibit the nuclear trafficking of the androgen receptor in prostate cancer, providing a mechanism for the efficacy of these drugs in such a slow growing tumor.79 This renewed interest in the mechanism of MTAs led to the finding that clinically relevant splice variants that disrupt the interaction between the receptor and microtubules are no longer sensitive to MTAs, which provides the opportunity for more rational treatment of patients expressing these variants.80 Additionally, the finding that MTAs, but not other classes of mitotic inhibitors, disrupted the nuclear trafficking of DNA damage repair proteins provides a mechanistic rationale for the long-observed synergy of these drugs with DNA-damaging agents in cancer therapy.81 The realization that the anticancer efficacy of MTAs involves their disruption of interphase microtubules provides a rationale for previously unexplained clinical differences between MTAs. Even paclitaxel (14) and docetaxel (15), which are almost identical compounds, have distinct effects on microtubule structure including the formation of microtubules with different numbers of protofilaments and therefore different diameters that would be expected to have distinct cell biological consequences.82 These structural differences may underlie the different clinical utilities of these drugs as well as explain their differential toxicities, including a lower incidence of peripheral neuropathies for docetaxel as compared to paclitaxel. It is therefore likely that even more disparate classes of MTAs would have even larger differences. The unfortunate history of classifying all microtubule stabilizers and destabilizers together as a single drug class and focusing solely on their antimitotic effects has precluded a rigorous analysis of the mechanistic differences of these drugs on interphase microtubules. Intriguingly, the sponge-derived adociasulfates (particularly adociasulfates 2, 8, 13, and 14) bind to kinesin motor proteins effectively inhibiting their processive trafficking along the microtubule.83, 84 The specificity for these compounds against motor kinesins as compared to mitotic kinesis provides a valuable tool to more fully understand the consequences of distinct forms of pharmacological microtubule disruption. We propose that a deeper understanding of the cell biological consequences of different clinically approved MTAs and novel classes of preclinical agents on oncogenic driver pathways could lead to more personalized treatment of cancer patients with these drugs and provide the potential for the treatment of additional non-cancer indications in a targeted manner.
The Actin Cytoskeleton
The actin cytoskeleton is critical for many of the same cellular processes as the microtubule cytoskeleton and significant cross-talk occurs between these structures to organize core biological processes, including cell migration and division.85 The role of actin-based microfilaments in cancer, infection, neurodegenerative disease, and even substance abuse is well documented,86–89 suggesting that actin-binding drugs could have potential as therapeutics in a variety of disease indications. Although many actin-interacting compounds have been isolated from natural products, none of them is currently approved for the treatment of disease likely due to significant cardiac and respiratory toxicities as a result of the non-specific binding of these agents to actin. It is therefore unlikely that compounds promoting gross actin binding and disruption will be effective in the treatment of human disease. However, a detailed mechanistic understanding of actin dynamics and function has led to the development of more specific actin-disrupting agents that have increased clinical potential. Like microtubules, the formation of actin-based microfilaments can be modula ted by the binding of cellular proteins or small molecules that either promote polymerization of monomeric G-actin into filamentous F-actin, or promote microfilament depolymerization.

Actin depolymerizing agents from natural sources.
The sponge-derived latrunculins bind within the hydrophobic ATP pocket on actin monomers and promote allosteric changes that inhibit polymerization through monomer sequestration (Figure 4A). Although latrunculin A (21) has potent cytotoxic effects against cancer cells in vitro and minor antitumor efficacy in MKN45 and NUGC-4 in vivo models,90 a very narrow therapeutic window has halted any clinical development of this class. However, the latrunculins are highly valuable as molecular tools to study the role of actin in cellular processes. There are several other classes of natural product-derived microfilament depolymerizing compounds that bind to the growing, barbed ends of the actin microfilament blocking further polymerization and promoting net depolymerization. These include fungi-derived cytochalasins as well as several marine-derived macrolides, including trisoxazoles (kabiramide C, 22) and swinholides from sponges, tolytoxin from cyanobacterium, and lobophorolide from brown alga, among others.91 Marine-derived macrolides, including tolytoxin, misakinolide A, and kabiramide C (22) have been described to have antifungal activities, but it is unclear whether there is sufficient selectivity for any of these agents to be used clinically for the treatment of fungal infections.92 These diverse groups of compounds all have a similar mechanism of action of binding to the solvent-exposed barbed end of the microfilament at a site overlapping with endogenous actin binding proteins that sequester and sever actin, including gelsolin, CapG, and profilin. However, while all of these compounds promote microfilament depolymerization by sequestering monomeric actin, a subset can also intercalate into a hydrophobic patch on an intact microfilament to effectively sever the filament. Even minor affinity of these compounds for the cleft between monomers can have dramatic destabilizing effects due to the importance of cooperative binding for filament integrity. While there has long been concern that actin active agents will have significant cardiotoxicity that will preclude a therapeutic window for disease treatment,93 antitumor efficacy of cytochalasin B (23) has been observed in some tumor models with ip, iv, and sc injections, particularly when liposome encapsulated.94
Figure 4.

Binding sites of actin active agents. (A) Superimposition of the actin destabilizers latrunculin (PDB ID: 1ESV) and kabiramide C (PDB ID: 1QZ5) on actin. (B) Cryo-EM structure of three phalloidin molecules (green, PDB ID: 6D8C), each bridging three actin monomers, to stabilize the actin microfilament structure.
Actin polymerizing agents from natural sources.
Microfilament-binding natural products that increase microfilament polymerization have also been identified, the most notable being the cyclic depsipeptides phalloidin (24) from the death cap mushroom Amanita phalloides and jasplakinolide (25) from the marine sponge Jaspis johnstoni. Other structurally and functionally similar natural products include chondramide from the myxobacterim Chondromyces crocatus, dolastatin 11 and doliculide from the Indian Ocean sea hare Dolabella auricularia, and amphidinolide H from dinoflagellates of the Amphidinium genus. Hectochlorin and lyngbyabellin (26) represent a family of actin filament stabilizers are structurally distinct cyclic lipopeptides isolated from the cyanobacterium Lyngbya majuscula.95 Phalloidin (24) binding promotes microfilament stabilization through a coordinated interaction bridging 3 actin monomers via 3 distinct actin binding sites to enhance the innate cooperativity of microfilament polymerization (Figure 4B).96 A similar mechanism is employed by each of these microfilament stabilizers, although some like dolastatin 11 do not directly compete for phalloidin binding97 and amphidonolide H is unique in its ability to covalently bind actin.98 Phalloidin is non-cell permeable, but is taken up by the organic anion transporter Oatp1b2 in hepatocytes, which promotes acute liver injury.99 However, the high specificity of F-actin binding provided by 3 distinct binding sites across an intact microfilament has facilitated its use as a cell biological tool to specifically label F-actin structures in fixed cells.100 Jasplankinolide (25), which is cell permeable, serves as a complementary biological tool to evaluate the pharmacological effects of actin stabilization on a variety of cellular processes in diverse model systems.101–104 Although total synthesis of this rare, sponge-derived compound has been achieved,105 the significant cost of jasplakinolide (hundreds of dollars for μg quantities) has limited in vivo evaluations in models such as zebrafish where significant delayed toxicities were observed.106 Chondramide has been evaluated in murine tumor models where it had a minor effect on the growth of a MDA-MB-231 breast cancer xenograft tumor.107 Additionally, chondramide pretreatment slightly diminished, but notably did not inhibit, the colonization of intravenously injected 4T1 mammary cancer cells to the lung.108
Actin active agents with multiple mechanisms of action.
There are other classes of natural products that have demonstrated more complex interactions with cytoskeletal components that prohibit their strict inclusion in the previous categories of agents that stabilize or destabilize the microtubule or actin cytoskeleton. The cucurbitacins are a family of plant-derived triterpenes that have been reported to have diverse effects on the cytoskeleton, including promotion of actin aggregation and some slight microtubule depolymerizing effects.109 There is controversy with regard to the mechanism of the actin-associated effects of cucurbitacins, with roles attributed to VASP/RhoA pathway activation for cucurbitacin B,110 differential JAK/STAT3 pathway inhibition for cucurbitacins A, B, E, I, and Q,111 a direct interaction with the actin depolymerizing protein cofillin for cucurbitacins E and I,112 and a direct interaction with actin for cucurbitacin E.113 With regard to the direct interaction of cucurbitacin E (27) with actin, it inhibits actin depolymerization by forming a covalent bond with Cys257 of actin in the polymerized F-form, but not in the monomeric G-form, to promote irreversible net actin polymerization in the cell.113 The subsequent finding that cucurbitacins D, E, and I can covalently interact with multiple cysteine residues on cofilin as well as the reducing agent dithiothreitol (DTT), suggests that the reason so many different mechanisms have been attributed to their biological effects is that they interact non-specifically with many cellular targets.114
The marine macrolide aplyronine A (28) from the Pacific sea hare, Aplysia kurodai, appears to interact with both actin and tubulin.84, 115 Mechanistically, aplyronine A first binds to actin and this actin-aplyronine A complex is then able to interact with tubulin to effectively destabilize microtubules in biochemical experiments and in cells. Intriguingly, gross cellular disruption of microtubule structure and mitotic arrest occur at much lower concentrations of aplyronine A than cause gross cellular actin depolymerization. The structurally similar compound aplyronine C, which lacks a trimethylserine ester, retains actin-binding activity similar to aplyronine A but lacks its microtubule-associated effects. The identification of compounds that bind and depolymerize actin in a similar manner but that have differential effects on microtubule structure provides a distinct opportunity to comparatively evaluate the relative contributions of disruption of these two cytoskeletal structures. Indeed, the lack of microtubule destabilizing effects for aplyronine C is associated with 1000-fold less cytotoxic potency against HeLa S3 cells as compared to aplyronine A. In vivo evaluations demonstrate that i.p. dosing of 0.04 – 0.08 mg/kg aplyronine A can prolong survival in mice injected i.p. with human cancer cell lines.116
Natural products as modulators of actin associated protein interactions.
The compounds described above have, for the most part, been characterized as general disruptors of the microfilament cytoskeleton, which have diverse and multifaceted downstream effects on likely hundreds of actin-mediated processes. As the field has progressed, there have been efforts to more specifically target the actin cytoskeleton with the intent of identifying more tolerable actin-active compounds as therapeutic lead compounds. Although actin is a highly conserved target between all cells, there is diversity in the actin associated proteins that regulate the function of actin between distinct cell types. In particular, at least 40 distinct isoforms of tropomyosin can form copolymers with actin to define the intrinsic function of distinct cellular microfilaments. This diversity of the microfilament superstructure provides an opportunity to target distinct microfilament populations that are associated with disease. Indeed, synthetic compounds that target specific tropomyosin containing actin microfilaments have demonstrated in vivo efficacy for regulating glucose metabolism117 or antitumor efficacy118, 119 with limited side effects. Some actin-active natural products may also have the potential to exhibit specificity due to disruption of distinct microfilament interactions. The myxobacterial-derived compound miuraenamide A has similar actin stabilizing effects as jasplakinolide (25), however it competes with a different cohort of actin-associated proteins leading to distinct effects on cellular migration and transcription.120 Recently, the inhibition of actin nucleation and polymerization by myxobacterium derived chivosazole A has been attributed to disruption of interactions with multiple actin-associated regulatory factors, which promotes distinct biological effects compared to latrunculin B.121

Small molecules that disrupt actin independently of direct microfilament binding include Rho Kinase (ROCK) inhibitors, which have been developed for the treatment of a variety of disorders, including glaucoma, asthma, neuronal degeneration, and cancer among others.89, 122 While these efforts have been focused on the identification of small molecule kinase inhibitors, recent efforts have been initiated to identify selective ROCK inhibitors from natural products.123, 124 In addition to Rho, small molecule inhibitors of other actin regulatory G proteins, Rac and Cdc42, have been sought out to inhibit invasion and metastasis in cancer by regulating actomyosin contractility.125 Selective inhibitors of the Cdc42-binding kinase MRCK from fragment libraries have demonstrated efficacy in chemical carcinogen-induced skin cancer models, providing evidence of the potential for targeting actin at the level of these regulatory kinases.126 It is intriguing to consider that a more targeted approach to identify specific classes of actin-disrupting agents combined with a deeper detailed mechanistic understanding of actin active agents already identified could enhance our understanding of the specific functionality of disrupting distinct microfilament structures and aid in the potential to identify therapeutically relevant classes of natural products in the future.
Organism-specific actin disrupting agents.
While there has been a large focus on evaluating the anticancer potential of actin active drugs, particularly given the success of microtubule targeted drugs in this area, there are other disease indications where actin-based drugs may be more aptly suited. The glycolipopeptide occidiofungin (29) is an actin binding agent produced by the soil bacterium Burkholderia contaminans with a wide spectrum fungicidal activity with minimal toxicity to animal models.127 Actin disruption is a novel mechanism compared to clinically approved antifungal drugs and occidofungin displays a broad spectrum of activities against both filamentous and non-filamentous fungi through direct actin binding and the disruption of F-actin cables.128 While the mechanism of fungal selectivity is unclear and unexpected due to the conservation of the cellular target, differences in cellular uptake between fungal and mammalian cells have been proposed for the low toxicity observed in animals subjected to occidiofungin by intravenous or subcutaneous injection. Most recently, occidiofungin was found to be effective in the treatment of a vulvovaginal yeast infection in a murine model with no adverse effects noted.128 It is interesting to note that minor, 4-fold, resistance to occidofungin is conferred by the selective mutation of one tropomyosin isoform, but not other actin-regulatory proteins.128
Intermediate Filaments
The intermediate filaments (IFs) are cytoskeletal structures that are named due to their intermediate size (8 – 10 nm) as compared to thicker microtubules and thinner actin microfilaments (Figure 1). IFs are the largest and most diverse class of cytoskeletal structures and are made up from distinct proteins in different tissues or cellular organelles. Type I and II IFs are keratin-containing structures that reside in epithelial cells. Type III IFs are composed of vimentin in fibroblasts and endothelial cells, desmin in muscle, glial fibrillary acidic protein (GFAP) in astrocytes, and peripherin in peripheral nerve fibers. Type IV, V, and VI IFs are composed of neurofilaments, nuclear localized lamins, and nestin, respectively. In spite of their heterogeneity, all IF subunit proteins have a common domain structure that promotes formation of coiled-coil dimers that further tetramerize and pair into protofibrils that lack the polarity of the microtubule and microfilament cytoskeleton. IFs play vital roles in providing structural support to the cell and internal structures like the nucleus (lamin), epidermis (keratin) or sarcomeres in muscle (desmin). Over 80 human diseases are associated with mutations in IF proteins (IF-pathies) and there are no treatments available that act directly on IFs to correct these disorders.

Type I and II IFs.
Mutations in 15 of the 21 keratins are associated with disease and underlie a number of inherited skin fragility disorders as a result of decreased epithelial rigidity, including epidermolysis bullosa simplex (EBS). Natural products, including retinoids and sulforaphane can restore skin integrity by upregulating the expression of normal keratins to compensate for the mutant forms although additional skin irritation can occur as a side effect.129, 130 In small clinical studies of EBS patients, Oleogel-S10, a 10% triperpene dry extract from birch bark, accelerated wound healing131 and diacerein, an anti-inflammatory compound from rhubarb root, significantly reduced blistering.132 Additional clinical trials evaluating the efficacy of topical diacerein, rapamycin, or injected botulinum toxin for EBS treatment as modulators of inflammation and keratin expression are ongoing. Midostaurin (PKC412, 30), a semisynthetic derivative of staurosporine, demonstrated a distinct mechanism of normalizing the structure of IFs containing mutated keratins in hepatocytes through increasing their association with a non-muscle myosin heavy chain.133 This approach of identifying compounds that functionally normalize mutant IF structures in a cell-based assay that can be applied to the specific protein mutations associated with any IF-pathy provides an intriguing platform for screening natural products extracts, fractions, and pure compounds.134
Type III IFs.
Desmin-associated IF-pathies include a subgroup of progressive muscular diseases (myofibrillar myopathies) and cardiomyopathies that are associated with desmin mutations and desmin-positive protein aggregates.135, 136 Compounds that reduced aggregation of desmin mutants in cellular models include the geldanamycin derivative 17-DMAG, antioxidants alpha-lipoic acid, α-tocopherol, and acetyl-α-tocopherol, curcumin, colchicine, and the mTOR inhibitor pp242. Their mechanism of action has been attributed to antioxidant production and autophagy.137 Mutations in the IF protein GFAP are implicated in Alexander disease, which is associated with demyelination of neurons and progressive neurological side effects that often present within the first two years of life and poor prognosis. The antidepressant clomipramine was found in a screen for compounds that reduce GFAP expression and aggregation138 and there is an indication that compounds that alter proteasomal activity, the activation of stress pathways, and regulation of inflammation in astrocytes and their local environment could alter disease progression.139
The expression of vimentin in epithelial cells has an integral role in the initiation and progression of many solid tumors, including lung, prostate, breast, and colon cancers.140 Vimentin is normally expressed in fibroblast and endothelial cells, although vimentin homozygous knockout mice are viable with some arterial stiffening noted.141 The natural product withaferin A (31) from Withania somnifera, directly binds to vimentin through a covalent interaction with Cys327 to cause aggregation of vimentin filaments, antiangiogenic activity, and has demonstrated antitumor efficacy in over a dozen in vivo models.142, 143 However, over the years many additional cellular targets of withaferin A have been identified in addition to vimentin that likely contribute toward its antitumor efficacy, including covalent binding to cysteine residues on β-tubulin and annexin II, as well as disruption of the Hsp90 chaperone.142 Additionally, withaferin A has been demonstrated to inhibit NF-κB dependent inflammatory signaling in monocytes,144 have antidiabetic effects as a leptin sensitizer,145 and elicit numerous other biological activities likely due to its diverse cellular targets. Specific vimentin inhibitors identified in small molecule screens demonstrated a much higher degree of selectivity for mesenchymally transformed cells than withaferin A, likely as a result of these additional targets.146 Recently the garlic derived phytochemical ajoene was also found to covalently bind to vimentin at the same cysteine residue as withaferin A (with additional interacting partners also detected), disrupt the cellular vimentin network, and reduce cell migration.147 Other natural products identified to interact with vimentin include the statins simvastatin and mevastatin (32) that promote vimentin bundling and selective cytotoxicity of mesenchymal, vimentin-expressing breast cancer cells.148 The potential for clinical development of natural product vimentin inhibitors will likely rely on the reduction of or at least a more detailed understanding of their vimentin-independent effects.
Type IV, V, and VI IFs.
Nestin expression has also been implicated in cancer progression and angiogenesis.149 Although its role in normal endothelial cells may preclude its potential as a general therapeutic target, there does appear to be a case for nestin inhibition or targeting particularly in the treatment of glioblastoma.150–152 The natural products salvianolic acids, tetramethylpyrazine, and resveratrol can induce nestin expression, although the mechanisms are indirect and not fully elucidated.144 Several neuronal/axonal and neuropsychiatric disorders are caused by mutations in neurofilament IF proteins, which play essential roles in nerve conduction and synaptic function. Neurofilament disorders underlie forms of Charcot-Marie-Tooth disease, amyotrophic lateral sclerosis (ALS) susceptibility, Parkinson’s disease, and Alzheimer’s disease.153 Laminopathies, caused by alterations in nuclear localized lamin IF proteins, include the premature aging disease Hutchinson-Gilford Progeria syndrome (HGPS) as well as cardiomyopathies. In HGPS, a truncated toxic form of lamin A, called progerin, accumulates in nuclei and is also observed in normal aging.154 Metformin and farnesyltransferase inhibitors can disrupt that aberrant splicing that leads to progerin accumulation155, 156 and studies evaluating the efficacy of combinations of these agents are currently being evaluated in clinical trials with limited sucess.154 Autophagy induction by rapamycin, retainoids, proteosomal inhibition and sulforaphane promote progerin clearance leading to reversal of aging defects in HGPS patient skin fibroblasts and animal models although additional preclinical studies are needed to inform on optimal clinical evaluations of these agents.154
Septins
Although not traditionally considered one of the three major cytoskeletal components, septins are increasingly acknowledged to be the fourth component of the cytoskeleton. Septins are GTP-binding proteins that directly interact with and coordinate microtubule and actin microfilaments to regulate numerous cell biological processes, including localized signaling and vesicular trafficking.157 Four distinct septin subgroups, consisting of at least 13 genes with multiple isoforms have been identified in mammals that interact among one another to form filaments, rings and gauzes.158 Septin misregulation is associated with cancer,159 neurodegenerative disease,160 infectious disease,161 and a variety of organ-specific pathologies that underlie human disease.162 Septin proteins compete with other microtubule binding proteins to inhibit microtubule dynamicity, contribute to the resistance of cells to microtubule targeted chemotherapies in vitro, and misregulation of Septin 9 isoform expression is associated with poor prognosis in prostate and breast cancers.159 Septin targeting could also serve as an alternative entry point to the modulation of the actin cytoskeleton as septins 2, 6, and 7 coordinate actin remodeling during cancer cell migration and microbial infection.
Septin expression in general is high in neuronal tissues and is integral to the formation, growth and stability of axons and dendrites and synaptic vesical trafficking.160 Septin 4 is important for dopamine release and reuptake and has been associated with Parkinson’s disease due to a direct interaction with parkin. Septins 2 and 3 are increased in Alzheimer’s patients and a polymorphism of septin 3 was found to be significantly associated with the disease. Aberrant septin 5 expression is linked to a rare autosomal bleeding disorder, septin 2 is required for bacterial invasion, and septin 4 is critical for fertility due to its role in sperm morphology.162 The expression of distinct combinations of septin proteins in different tissues and disease states resulting in the formation of different macromolecular structures provides an opportunity for the targeted inhibition of pathological septin structures.
The only compound identified to date that disrupts septin assembly is the synthetic plant cytokine forchlorfenuron (33), which directly suppresses septin dynamics resulting in the formation of higher order structures that effectively deplete functional septin structures.163 Modeling studies indicate that forchlorfenuron binds to the nucleotide binding pocket of septins and locks them into a conformation that mimics the GTP bound state.164 This binding pocket is conserved among the septin 2, 3, and 7 paralogs, which results in cellular septin reorganization and inhibition of mitotic progression and cellular migration. There is significant untapped potential to discover more selective septin inhibitors that could be used to more fully understand septin regulation and the potential for the treatment of septin-associated disease.
Conclusions
The role of the eukaryotic cytoskeleton in human disease has long been appreciated. While direct targeting of the microtubule cytoskeleton has been an effective strategy for the treatment of cancer, inhibitors of other components of the eukaryotic cytoskeleton have so far not been translated to the clinic. Mounting evidence suggests that non-specific disruption of the actin microfilament cytoskeleton may be too toxic as a therapeutic strategy and that more targeted disruption of distinct actin structures or interacting proteins may be a more optimal approach. Additionally, the cell type specific expression and coordination of intermediate filaments and septins provides an opportunity for pharmacological disruption of more specific disease-associated cytoskeletal processes with a decreased likelihood of global toxic side-effects. As we gain additional structural and mechanistic insight into distinct cytoskeletal structures that are coopted in diseased states, more targeted strategies can be employed to identify selective inhibitors of these structures. Natural products have consistently provided novel and unanticipated chemical scaffolds that have revolutionized the treatment of human disease.165 The identification of novel natural products with the potential to impact the treatment of disease through cytoskeletal disruption/interaction will likely require the development of more sophisticated mechanistic screening efforts than what have been traditionally employed. There is also a strong possibility that natural products that have been historically classified as general inhibitors of cytoskeletal components could actually be more selective inhibitors of distinct cytoskeletal processes if more detailed mechanistic studies are undertaken.
Acknowledgements
We would like to acknowledge support from CA219948 to A.L.R and L.D.
Footnotes
Conflicts of interest
There are no conflicts to declare.
References
- 1.Shelanski ML and Taylor EW, J Cell Biol, 1967, 34, 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ravelli RB, Gigant B, Curmi PA, Jourdain I, Lachkar S, Sobel A and Knossow M, Nature, 2004, 428, 198–202. [DOI] [PubMed] [Google Scholar]
- 3.Schiff PB, Fant J and Horwitz SB, Nature, 1979, 277, 665–667. [DOI] [PubMed] [Google Scholar]
- 4.Moudi M, Go R, Yien CY and Nazre M, Int J Prev Med, 2013, 4, 1231–1235. [PMC free article] [PubMed] [Google Scholar]
- 5.Martino E, Casamassima G, Castiglione S, Cellupica E, Pantalone S, Papagni F, Rui M, Siciliano AM and Collina S, Bioorg Med Chem Lett, 2018, 28, 2816–2826. [DOI] [PubMed] [Google Scholar]
- 6.Funahashi Y, Okamoto K, Adachi Y, Semba T, Uesugi M, Ozawa Y, Tohyama O, Uehara T, Kimura T, Watanabe H, Asano M, Kawano S, Tizon X, McCracken PJ, Matsui J, Aoshima K, Nomoto K and Oda Y, Cancer Sci, 2014, 105, 1334–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshida T, Ozawa Y, Kimura T, Sato Y, Kuznetsov G, Xu S, Uesugi M, Agoulnik S, Taylor N, Funahashi Y and Matsui J, Br J Cancer, 2014, 110, 1497–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kashiwagi S, Asano Y, Goto W, Takada K, Takahashi K, Hatano T, Tanaka S, Takashima T, Tomita S, Motomura H, Ohsawa M, Hirakawa K and Ohira M, Anticancer Res, 2018, 38, 401–410. [DOI] [PubMed] [Google Scholar]
- 9.Cortes J, O’Shaughnessy J, Loesch D, Blum JL, Vahdat LT, Petrakova K, Chollet P, Manikas A, Dieras V, Delozier T, Vladimirov V, Cardoso F, Koh H, Bougnoux P, Dutcus CE, Seegobin S, Mir D, Meneses N, Wanders J, Twelves C and investigators E, Lancet, 2011, 377, 914–923. [DOI] [PubMed] [Google Scholar]
- 10.Yuan P, Hu X, Sun T, Li W, Zhang Q, Cui S, Cheng Y, Ouyang Q, Wang X, Chen Z, Hiraiwa M, Saito K, Funasaka S and Xu B, Eur J Cancer, 2019, 112, 57–65. [DOI] [PubMed] [Google Scholar]
- 11.Cheng X, Li J, Tanaka K, Majumder U, Milinichik AZ, Verdi AC, Maddage CJ, Rybinski KA, Fernando S, Fernando D, Kuc M, Furuuchi K, Fang F, Uenaka T, Grasso L and Albone EF, Mol Cancer Ther, 2018, 17, 2665–2675. [DOI] [PubMed] [Google Scholar]
- 12.Miller JH, Field JJ, Kanakkanthara A, Owen JG, Singh AJ and Northcote PT, J Nat Prod, 2018, 81, 691–702. [DOI] [PubMed] [Google Scholar]
- 13.Steinmetz MO and Prota AE, Trends Cell Biol, 2018, 28, 776–792. [DOI] [PubMed] [Google Scholar]
- 14.Andersen RJ, Biochem Pharmacol, 2017, 139, 3–14. [DOI] [PubMed] [Google Scholar]
- 15.Pettit GR, Kamano Y, Herald CL, Tuinman AA, Boettner FE, Kizu H, Schmidt JM, Baczynskyj L, Tomer KB and Bontems RJ, J Am Chem Soc, 1987, 109, 6883–6885. [Google Scholar]
- 16.Luesch H, Yoshida WY, Moore RE, Paul VJ, Mooberry SL and Corbett TH, J Nat Prod, 2002, 65, 16–20. [DOI] [PubMed] [Google Scholar]
- 17.Newman DJ and Cragg GM, Mar Drugs, 2017, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doronina SO, Toki BE, Torgov MY, Mendelsohn BA, Cerveny CG, Chace DF, DeBlanc RL, Gearing RP, Bovee TD, Siegall CB, Francisco JA, Wahl AF, Meyer DL and Senter PD, Nat Biotechnol, 2003, 21, 778–784. [DOI] [PubMed] [Google Scholar]
- 19.Smith CD, Zhang X, Mooberry SL, Patterson GM and Moore RE, Cancer Res, 1994, 54, 3779–3784. [PubMed] [Google Scholar]
- 20.Smith CD and Zhang X, J Biol Chem, 1996, 271, 6192–6198. [DOI] [PubMed] [Google Scholar]
- 21.Edelman MJ, Gandara DR, Hausner P, Israel V, Thornton D, DeSanto J and Doyle LA, Lung Cancer, 2003, 39, 197–199. [DOI] [PubMed] [Google Scholar]
- 22.Borbely A, Figueras E, Martins A, Bodero L, Raposo Moreira Dias A, Lopez Rivas P, Pina A, Arosio D, Gallinari P, Frese M, Steinkuhler C, Gennari C, Piarulli U and Sewald N, ChemistryOpen, 2019, 8, 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Verma VA, Pillow TH, DePalatis L, Li G, Phillips GL, Polson AG, Raab HE, Spencer S and Zheng B, Bioorg Med Chem Lett, 2015, 25, 864–868. [DOI] [PubMed] [Google Scholar]
- 24.Prota AE, Bargsten K, Diaz JF, Marsh M, Cuevas C, Liniger M, Neuhaus C, Andreu JM, Altmann KH and Steinmetz MO, Proc Natl Acad Sci U S A, 2014, 111, 13817–13821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Menchon G, Prota AE, Lucena-Agell D, Bucher P, Jansen R, Irschik H, Muller R, Paterson I, Diaz JF, Altmann KH and Steinmetz MO, Nat Commun, 2018, 9, 2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanauske AR, Catimel G, Aamdal S, ten Bokkel Huinink W, Paridaens R, Pavlidis N, Kaye SB, te Velde A, Wanders J and Verweij J, Br J Cancer, 1996, 73, 397–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ravry MJ, Omura GA and Birch R, Am J Clin Oncol, 1985, 8, 148–150. [DOI] [PubMed] [Google Scholar]
- 28.Wu X, Wang Q and Li W, Anticancer Agents Med Chem, 2016, 16, 1325–1338. [DOI] [PubMed] [Google Scholar]
- 29.Cimino PJ, Huang L, Du L, Wu Y, Bishop J, Dalsing-Hernandez J, Kotlarczyk K, Gonzales P, Carew J, Nawrocki S, Jordan MA, Wilson L, Lloyd GK and Wirsching HG, Biomed Rep, 2019, 10, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Prota AE, Danel F, Bachmann F, Bargsten K, Buey RM, Pohlmann J, Reinelt S, Lane H and Steinmetz MO, J Mol Biol, 2014, 426, 1848–1860. [DOI] [PubMed] [Google Scholar]
- 31.Hua J, Sheng Y, Pinney KG, Garner CM, Kane RR, Prezioso JA, Pettit GR, Chaplin DJ and Edvardsen K, Anticancer Res, 2003, 23, 1433–1440. [PubMed] [Google Scholar]
- 32.Ji YT, Liu YN and Liu ZP, Curr Med Chem, 2015, 22, 1348–1360. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Wang Y, Wang T, Jiang J, Botting CH, Liu H, Chen Q, Yang J, Naismith JH, Zhu X and Chen L, Nat Commun, 2016, 7, 12103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balaguer FA, Muhlethaler T, Estevez-Gallego J, Calvo E, Gimenez-Abian JF, Risinger AL, Sorensen EJ, Vanderwal CD, Altmann KH, Mooberry SL, Steinmetz MO, Oliva MA, Prota AE and Diaz JF, Int J Mol Sci, 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida M, Matsui Y, Ikarashi Y, Usui T, Osada H and Wakasugi H, Anticancer Res, 2007, 27, 729–736. [PubMed] [Google Scholar]
- 36.Kondoh M, Usui T, Kobayashi S, Tsuchiya K, Nishikawa K, Nishikiori T, Mayumi T and Osada H, Cancer Lett, 1998, 126, 29–32. [DOI] [PubMed] [Google Scholar]
- 37.Coulup SK and Georg GI, Bioorg Med Chem Lett, 2019, 29, 1865–1873. [DOI] [PubMed] [Google Scholar]
- 38.Rohena CC and Mooberry SL, Nat Prod Rep, 2014, 31, 335–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Desai N, Trieu V, Damascelli B and Soon-Shiong P, Transl Oncol, 2009, 2, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA and Investigators IMT, N Engl J Med, 2018, 379, 2108–2121. [DOI] [PubMed] [Google Scholar]
- 41.Flores JP and Saif MW, Clin Investig (Lond), 2013, 3, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.James J, Tang K and Wei T, Atlanta, 2019.
- 43.Colombo N, Kutarska E, Dimopoulos M, Bae DS, Rzepka-Gorska I, Bidzinski M, Scambia G, Engelholm SA, Joly F, Weber D, El-Hashimy M, Li J, Souami F, Wing P, Engelholm S, Bamias A and Schwartz P, J Clin Oncol, 2012, 30, 3841–3847. [DOI] [PubMed] [Google Scholar]
- 44.Keam B, Lee KW, Lee SH, Kim JS, Kim JH, Wu HG, Eom KY, Kim S, Ahn SH, Chung EJ, Kwon SK, Jeong WJ, Jung YH, Kim JW and Heo DS, Oncologist, 2019, 24, 751–e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee SW, Kim YM, Cho CH, Kim YT, Kim SM, Hur SY, Kim JH, Kim BG, Kim SC, Ryu HS and Kang SB, Cancer Res Treat, 2018, 50, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao J, Koay EJ, Li T, Wen X and Li C, Wiley Interdiscip Rev Nanomed Nanobiotechnol, 2018, 10, e1497. [DOI] [PubMed] [Google Scholar]
- 47.Prota AE, Bargsten K, Redondo-Horcajo M, Smith AB 3rd, Yang CH, McDaid HM, Paterson I, Horwitz SB, Fernando Diaz J and Steinmetz MO, Chembiochem, 2017, 18, 905–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Souza MV, ScientificWorldJournal, 2004, 4, 415–436.15243683 [Google Scholar]
- 49.Kowalski RJ, Giannakakou P, Gunasekera SP, Longley RE, Day BW and Hamel E, Mol Pharmacol, 1997, 52, 613–622. [PubMed] [Google Scholar]
- 50.Martello LA, McDaid HM, Regl DL, Yang CP, Meng D, Pettus TR, Kaufman MD, Arimoto H, Danishefsky SJ, Smith AB 3rd and Horwitz SB, Clin Cancer Res, 2000, 6, 1978–1987. [PubMed] [Google Scholar]
- 51.Mita A, Lockhart AC, Chen TL, Bochinski K, Curtright J, Cooper W, Hammond L, Rothenberg M, Rowinsky E and Sharma S, Journal of Clinical Oncology, 2004, 22, 2025. [Google Scholar]
- 52.Trigili C, Barasoain I, Sanchez-Murcia PA, Bargsten K, Redondo-Horcajo M, Nogales A, Gardner NM, Meyer A, Naylor GJ, Gomez-Rubio E, Gago F, Steinmetz MO, Paterson I, Prota AE and Diaz JF, ACS Omega, 2016, 1, 1192–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Risinger AL, Jackson EM, Polin LA, Helms GL, LeBoeuf DA, Joe PA, Hopper-Borge E, Luduena RF, Kruh GD and Mooberry SL, Cancer Res, 2008, 68, 8881–8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Field JJ, Northcote PT, Paterson I, Altmann KH, Diaz JF and Miller JH, Int J Mol Sci, 2017, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ola ARB, Risinger AL, Du L, Zammiello CL, Peng J, Cichewicz RH and Mooberry SL, J Nat Prod, 2018, 81, 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Risinger AL, Li J, Bennett MJ, Rohena CC, Peng J, Schriemer DC and Mooberry SL, Cancer Res, 2013, 73, 6780–6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bai R and Hamel E, Arch Biochem Biophys, 2016, 604, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Field JJ, Kanakkanthara A, Brooke DG, Sinha S, Pillai SD, Denny WA, Butt AJ and Miller JH, Invest New Drugs, 2016, 34, 277–289. [DOI] [PubMed] [Google Scholar]
- 59.Prota AE, Bargsten K, Northcote PT, Marsh M, Altmann KH, Miller JH, Diaz JF and Steinmetz MO, Angew Chem Int Ed Engl, 2014, 53, 1621–1625. [DOI] [PubMed] [Google Scholar]
- 60.Bennett MJ, Barakat K, Huzil JT, Tuszynski J and Schriemer DC, Chem Biol, 2010, 17, 725–734. [DOI] [PubMed] [Google Scholar]
- 61.Meyer CJ, Krauth M, Wick MJ, Shay JW, Gellert G, De Brabander JK, Northcote PT and Miller JH, Mol Cancer Ther, 2015, 14, 1816–1823. [DOI] [PubMed] [Google Scholar]
- 62.Liu J, Towle MJ, Cheng H, Saxton P, Reardon C, Wu J, Murphy EA, Kuznetsov G, Johannes CW, Tremblay MR, Zhao H, Pesant M, Fang FG, Vermeulen MW, Gallagher BM Jr. and Littlefield BA, Anticancer Res, 2007, 27, 1509–1518. [PubMed] [Google Scholar]
- 63.Johnson TA, Tenney K, Cichewicz RH, Morinaka BI, White KN, Amagata T, Subramanian B, Media J, Mooberry SL, Valeriote FA and Crews P, J Med Chem, 2007, 50, 3795–3803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang B, Maiti A, Shively S, Lakhani F, McDonald-Jones G, Bruce J, Lee EB, Xie SX, Joyce S, Li C, Toleikis PM, Lee VM and Trojanowski JQ, Proc Natl Acad Sci U S A, 2005, 102, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Das V and Miller JH, Eur J Neurosci, 2012, 35, 1705–1717. [DOI] [PubMed] [Google Scholar]
- 66.Pallavi D and Sidharth M, Journal of Alzheimer’s Disease Reports, 2019, 3, 179–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Makani V, Zhang B, Han H, Yao Y, Lassalas P, Lou K, Paterson I, Lee VM, Trojanowski JQ, Ballatore C, Smith AB 3rd and Brunden KR, Acta Neuropathol Commun, 2016, 4, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rivera E and Cianfrocca M, Cancer Chemother Pharmacol, 2015, 75, 659–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kellogg EH, Hejab NMA, Howes S, Northcote P, Miller JH, Diaz JF, Downing KH and Nogales E, J Mol Biol, 2017, 429, 633–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gornstein EL and Schwarz TL, Exp Neurol, 2017, 288, 153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Starobova H and Vetter I, Front Mol Neurosci, 2017, 10, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Makker PG, Duffy SS, Lees JG, Perera CJ, Tonkin RS, Butovsky O, Park SB, Goldstein D and Moalem-Taylor G, PLoS One, 2017, 12, e0170814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wozniak KM, Vornov JJ, Wu Y, Nomoto K, Littlefield BA, DesJardins C, Yu Y, Lai G, Reyderman L, Wong N and Slusher BS, Cancer Res, 2016, 76, 3332–3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Field JJ, Kanakkanthara A and Miller JH, Bioorg Med Chem, 2014, 22, 5050–5059. [DOI] [PubMed] [Google Scholar]
- 75.Komlodi-Pasztor E, Sackett D, Wilkerson J and Fojo T, Nat Rev Clin Oncol, 2011, 8, 244–250. [DOI] [PubMed] [Google Scholar]
- 76.Komlodi-Pasztor E, Sackett DL and Fojo AT, Clin Cancer Res, 2012, 18, 51–63. [DOI] [PubMed] [Google Scholar]
- 77.Orth JD, Kohler RH, Foijer F, Sorger PK, Weissleder R and Mitchison TJ, Cancer Res, 2011, 71, 4608–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Janssen A, Beerling E, Medema R and van Rheenen J, PLoS One, 2013, 8, e64029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Darshan MS, Loftus MS, Thadani-Mulero M, Levy BP, Escuin D, Zhou XK, Gjyrezi A, Chanel-Vos C, Shen R, Tagawa ST, Bander NH, Nanus DM and Giannakakou P, Cancer Res, 2011, 71, 6019–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Thadani-Mulero M, Portella L, Sun S, Sung M, Matov A, Vessella RL, Corey E, Nanus DM, Plymate SR and Giannakakou P, Cancer Res, 2014, 74, 2270–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Poruchynsky MS, Komlodi-Pasztor E, Trostel S, Wilkerson J, Regairaz M, Pommier Y, Zhang X, Kumar Maity T, Robey R, Burotto M, Sackett D, Guha U and Fojo AT, Proc Natl Acad Sci U S A, 2015, 112, 1571–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Matesanz R, Rodriguez-Salarichs J, Pera B, Canales A, Andreu JM, Jimenez-Barbero J, Bras W, Nogales A, Fang WS and Diaz JF, Biophys J, 2011, 101, 2970–2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Smith TE, Hong W, Zachariah MM, Harper MK, Matainaho TK, Van Wagoner RM, Ireland CM and Vershinin M, Proc Natl Acad Sci U S A, 2013, 110, 18880–18885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kita M and Kigoshi H, Nat Prod Rep, 2015, 32, 534–542. [DOI] [PubMed] [Google Scholar]
- 85.Dogterom M and Koenderink GH, Nat Rev Mol Cell Biol, 2019, 20, 38–54. [DOI] [PubMed] [Google Scholar]
- 86.Young EJ, Briggs SB and Miller CA, CNS Neurol Disord Drug Targets, 2015, 14, 731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brayford S, Schevzov G, Vos J and Gunning PW, in The cytoskeleton in health and disease, Springer, 2015. [Google Scholar]
- 88.Bugalhao JN, Mota LJ and Franco IS, Pathog Dis, 2015, 73, ftv078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eira J, Silva CS, Sousa MM and Liz MA, Prog Neurobiol, 2016, 141, 61–82. [DOI] [PubMed] [Google Scholar]
- 90.Konishi H, Kikuchi S, Ochiai T, Ikoma H, Kubota T, Ichikawa D, Fujiwara H, Okamoto K, Sakakura C, Sonoyama T, Kokuba Y, Sasaki H, Matsui T and Otsuji E, Anticancer Res, 2009, 29, 2091–2097. [PubMed] [Google Scholar]
- 91.Allingham JS, Klenchin VA and Rayment I, Cell Mol Life Sci, 2006, 63, 2119–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Karpinski TM, Mar Drugs, 2019, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Sachs HG, McDonald TF and Springer M, J Cell Sci, 1974, 14, 163–185. [DOI] [PubMed] [Google Scholar]
- 94.Trendowski M, Zoino JN, Christen TD, Acquafondata C and Fondy TP, Transl Oncol, 2015, 8, 308–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Luesch H, Yoshida WY, Moore RE, Paul VJ and Mooberry SL, J Nat Prod, 2000, 63, 611–615. [DOI] [PubMed] [Google Scholar]
- 96.Mentes A, Huehn A, Liu X, Zwolak A, Dominguez R, Shuman H, Ostap EM and Sindelar CV, Proc Natl Acad Sci U S A, 2018, 115, 1292–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Oda T, Crane ZD, Dicus CW, Sufi BA and Bates RB, J Mol Biol, 2003, 328, 319–324. [DOI] [PubMed] [Google Scholar]
- 98.Usui T, Kazami S, Dohmae N, Mashimo Y, Kondo H, Tsuda M, Terasaki AG, Ohashi K, Kobayashi J and Osada H, Chem Biol, 2004, 11, 1269–1277. [DOI] [PubMed] [Google Scholar]
- 99.Lu H, Choudhuri S, Ogura K, Csanaky IL, Lei X, Cheng X, Song PZ and Klaassen CD, Toxicol Sci, 2008, 103, 35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Melak M, Plessner M and Grosse R, J Cell Sci, 2017, 130, 525–530. [DOI] [PubMed] [Google Scholar]
- 101.Zhang X, Cui X, Cheng L, Guan X, Li H, Li X and Cheng M, PLoS One, 2012, 7, e50899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Childers RC, Sunyecz I, West TA, Cismowski MJ, Lucchesi PA and Gooch KJ, Am J Physiol Heart Circ Physiol, 2019, 316, H596–H608. [DOI] [PubMed] [Google Scholar]
- 103.Phillips JK, Sherman SA, Cotton KY, Heddleston JM, Taylor AB and Finan JD, J Neurosci Methods, 2019, 312, 154–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee S, Tan HY, Geneva II, Kruglov A and Calvert PD, J Cell Biol, 2018, 217, 2831–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ghosh AK and Moon DK, Org Lett, 2007, 9, 2425–2427. [DOI] [PubMed] [Google Scholar]
- 106.Trendowski M, Wong V, Wellington K, Hatfield S and Fondy TP, Org Lett, 2014, 28, 1021–1031. [PubMed] [Google Scholar]
- 107.Foerster F, Braig S, Moser C, Kubisch R, Busse J, Wagner E, Schmoeckel E, Mayr D, Schmitt S, Huettel S, Zischka H, Mueller R and Vollmar AM, Cell Death Dis, 2014, 5, e1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Menhofer MH, Kubisch R, Schreiner L, Zorn M, Foerster F, Mueller R, Raedler JO, Wagner E, Vollmar AM and Zahler S, PLoS One, 2014, 9, e112542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang X, Tanaka M, Peixoto HS and Wink M, PeerJ, 2017, 5, e3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhang YT, Xu LH, Lu Q, Liu KP, Liu PY, Ji F, Liu XM, Ouyang DY and He XH, PLoS One, 2014, 9, e93547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sun J, Blaskovich MA, Jove R, Livingston SK, Coppola D and Sebti SM, Oncogene, 2005, 24, 3236–3245. [DOI] [PubMed] [Google Scholar]
- 112.Nakashima S, Matsuda H, Kurume A, Oda Y, Nakamura S, Yamashita M and Yoshikawa M, Bioorg Med Chem Lett, 2010, 20, 2994–2997. [DOI] [PubMed] [Google Scholar]
- 113.Sorensen PM, Iacob RE, Fritzsche M, Engen JR, Brieher WM, Charras G and Eggert US, ACS Chem Biol, 2012, 7, 1502–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gabrielsen M, Schuldt M, Munro J, Borucka D, Cameron J, Baugh M, Mleczak A, Lilla S, Morrice N and Olson MF, Cell Commun Signal, 2013, 11, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kita M, Hirayama Y, Yoneda K, Yamagishi K, Chinen T, Usui T, Sumiya E, Uesugi M and Kigoshi H, J Am Chem Soc, 2013, 135, 18089–18095. [DOI] [PubMed] [Google Scholar]
- 116.Yamada K, Ojika M, Kigoshi H and Suenaga K, Proc Jpn Acad Ser B Phys Biol Sci, 2010, 86, 176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kee AJ, Chagan J, Chan JY, Bryce NS, Lucas CA, Zeng J, Hook J, Treutlein H, Laybutt DR, Stehn JR, Gunning PW and Hardeman EC, Sci Rep, 2018, 8, 4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Currier MA, Stehn JR, Swain A, Chen D, Hook J, Eiffe E, Heaton A, Brown D, Nartker BA, Eaves DW, Kloss N, Treutlein H, Zeng J, Alieva IB, Dugina VB, Hardeman EC, Gunning PW and Cripe TP, Mol Cancer Ther, 2017, 16, 1555–1565. [DOI] [PubMed] [Google Scholar]
- 119.Stehn JR, Haass NK, Bonello T, Desouza M, Kottyan G, Treutlein H, Zeng J, Nascimento PR, Sequeira VB, Butler TL, Allanson M, Fath T, Hill TA, McCluskey A, Schevzov G, Palmer SJ, Hardeman EC, Winlaw D, Reeve VE, Dixon I, Weninger W, Cripe TP and Gunning PW, Cancer Res, 2013, 73, 5169–5182. [DOI] [PubMed] [Google Scholar]
- 120.Wang S, Crevenna AH, Ugur I, Marion A, Antes I, Kazmaier U, Hoyer M, Lamb DC, Gegenfurtner F, Kliesmete Z, Ziegenhain C, Enard W, Vollmar A and Zahler S, Sci Rep, 2019, 9, 9731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wang S, Gegenfurtner FA, Crevenna AH, Ziegenhain C, Kliesmete Z, Enard W, Muller R, Vollmar AM, Schneider S and Zahler S, J Nat Prod, 2019, 82, 1961–1970. [DOI] [PubMed] [Google Scholar]
- 122.Feng Y, LoGrasso PV, Defert O and Li R, J Med Chem, 2016, 59, 2269–2300. [DOI] [PubMed] [Google Scholar]
- 123.Su H, Yan J, Xu J, Fan XZ, Sun XL and Chen KY, Pharm Biol, 2015, 53, 1201–1206. [DOI] [PubMed] [Google Scholar]
- 124.Amen Y, Zhu Q, Tran HB, Afifi MS, Halim AF, Ashour A, Fujimoto R, Goto T and Shimizu K, Nat Prod Res, 2018, 32, 1955–1959. [DOI] [PubMed] [Google Scholar]
- 125.Gandalovicova A, Rosel D, Fernandes M, Vesely P, Heneberg P, Cermak V, Petruzelka L, Kumar S, Sanz-Moreno V and Brabek J, Trends Cancer, 2017, 3, 391–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Unbekandt M, Belshaw S, Bower J, Clarke M, Cordes J, Crighton D, Croft DR, Drysdale MJ, Garnett MJ, Gill K, Gray C, Greenhalgh DA, Hall JAM, Konczal J, Lilla S, McArthur D, McConnell P, McDonald L, McGarry L, McKinnon H, McMenemy C, Mezna M, Morrice NA, Munro J, Naylor G, Rath N, Schuttelkopf AW, Sime M and Olson MF, Cancer Res, 2018, 78, 2096–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Tan W, Cooley J, Austin F, Lu SE, Pruett SB and Smith L, Int J Toxicol, 2012, 31, 326–336. [DOI] [PubMed] [Google Scholar]
- 128.Ravichandran A, Geng M, Hull KG, Li J, Romo D, Lu SE, Albee A, Nutter C, Gordon DM, Ghannoum MA, Lockless SW and Smith L, Antimicrob Agents Chemother, 2019, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Torma H, Dermatoendocrinol, 2011, 3, 136–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kerns ML, DePianto D, Dinkova-Kostova AT, Talalay P and Coulombe PA, Proc Natl Acad Sci U S A, 2007, 104, 14460–14465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schwieger-Briel A, Kiritsi D, Schempp C, Has C and Schumann H, Dermatol Res Pract, 2017, 2017, 5068969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wally V, Hovnanian A, Ly J, Buckova H, Brunner V, Lettner T, Ablinger M, Felder TK, Hofbauer P, Wolkersdorfer M, Lagler FB, Hitzl W, Laimer M, Kitzmuller S, Diem A and Bauer JW, J Am Acad Dermatol, 2018, 78, 892–901 e897. [DOI] [PubMed] [Google Scholar]
- 133.Kwan R, Chen L, Looi K, Tao GZ, Weerasinghe SV, Snider NT, Conti MA, Adelstein RS, Xie Q and Omary MB, Hepatology, 2015, 62, 1858–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sun J, Groppi VE, Gui H, Chen L, Xie Q, Liu L and Omary MB, Methods Enzymol, 2016, 568, 163–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lorenzon A, Beffagna G, Bauce B, De Bortoli M, Li Mura IE, Calore M, Dazzo E, Basso C, Nava A, Thiene G and Rampazzo A, Am J Cardiol, 2013, 111, 400–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Olive M, Kley RA and Goldfarb LG, Curr Opin Neurol, 2013, 26, 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Cabet E, Batonnet-Pichon S, Delort F, Gausseres B, Vicart P and Lilienbaum A, PLoS One, 2015, 10, e0137009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cho W, Brenner M, Peters N and Messing A, Hum Mol Genet, 2010, 19, 3169–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sosunov A, Olabarria M and Goldman JE, Brain Pathol, 2018, 28, 388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sharma P, Alsharif S, Fallatah A and Chung BM, Cells, 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Langlois B, Belozertseva E, Parlakian A, Bourhim M, Gao-Li J, Blanc J, Tian L, Coletti D, Labat C, Ramdame-Cherif Z, Challande P, Regnault V, Lacolley P and Li Z, Sci Rep, 2017, 7, 11628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hassannia B, Logie E, Vandenabeele P, Vanden Berghe T and Vanden Berghe W, Biochem Pharmacol, 2019, DOI: 10.1016/j.bcp.2019.08.004. [DOI] [PubMed] [Google Scholar]
- 143.Bargagna-Mohan P, Hamza A, Kim YE, Khuan Abby Ho Y, Mor-Vaknin N, Wendschlag N, Liu J, Evans RM, Markovitz DM, Zhan CG, Kim KB and Mohan R, Chem Biol, 2007, 14, 623–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dubey S, Yoon H, Cohen MS, Nagarkatti P, Nagarkatti M and Karan D, Front Immunol, 2018, 9, 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lee J, Liu J, Feng X, Salazar Hernandez MA, Mucka P, Ibi D, Choi JW and Ozcan U, Nat Med, 2016, 22, 1023–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Bollong MJ, Pietila M, Pearson AD, Sarkar TR, Ahmad I, Soundararajan R, Lyssiotis CA, Mani SA, Schultz PG and Lairson LL, Proc Natl Acad Sci U S A, 2017, 114, E9903–E9912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kaschula CH, Tuveri R, Ngarande E, Dzobo K, Barnett C, Kusza DA, Graham LM, Katz AA, Rafudeen MS, Parker MI, Hunter R and Schafer G, BMC Cancer, 2019, 19, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Trogden KP, Battaglia RA, Kabiraj P, Madden VJ, Herrmann H and Snider NT, FASEB J, 2018, 32, 2841–2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Nowak A and Dziegiel P, Int J Oncol, 2018, 53, 477–487. [DOI] [PubMed] [Google Scholar]
- 150.Prabhu S, Goda JS, Mutalik S, Mohanty BS, Chaudhari P, Rai S, Udupa N and Rao BSS, Nanoscale, 2017, 9, 10919–10932. [DOI] [PubMed] [Google Scholar]
- 151.Matsuda Y, Ishiwata T, Yoshimura H, Hagio M and Arai T, Cancer Lett, 2015, 357, 602–611. [DOI] [PubMed] [Google Scholar]
- 152.Dusart P, Fagerberg L, Perisic L, Civelek M, Struck E, Hedin U, Uhlen M, Tregouet DA, Renne T, Odeberg J and Butler LM, Sci Rep, 2018, 8, 14668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Yuan A, Rao MV, Veeranna and RA Nixon, Cold Spring Harb Perspect Biol, 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Harhouri K, Frankel D, Bartoli C, Roll P, De Sandre-Giovannoli A and Levy N, Nucleus, 2018, 9, 246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.DuBose AJ, Lichtenstein ST, Petrash NM, Erdos MR, Gordon LB and Collins FS, Proc Natl Acad Sci U S A, 2018, 115, 4206–4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Mattioli E, Andrenacci D, Mai A, Valente S, Robijns J, De Vos WH, Capanni C and Lattanzi G, Front Cell Dev Biol, 2019, 7, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Akhmetova KA, Chesnokov IN and Fedorova SA, Mol Biol (Mosk), 2018, 52, 155–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Neubauer K and Zieger B, Front Cell Dev Biol, 2017, 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Pous C, Klipfel L and Baillet A, Front Cell Dev Biol, 2016, 4, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Marttinen M, Kurkinen KM, Soininen H, Haapasalo A and Hiltunen M, Mol Neurodegener, 2015, 10, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Van Ngo H and Mostowy S, J Cell Sci, 2019, 132. [DOI] [PubMed] [Google Scholar]
- 162.Dolat L, Hu Q and Spiliotis ET, Biol Chem, 2014, 395, 123–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hu Q, Nelson WJ and Spiliotis ET, J Biol Chem, 2008, 283, 29563–29571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Angelis D, Karasmanis EP, Bai X and Spiliotis ET, PLoS One, 2014, 9, e96390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Newman DJ and Cragg GM, J Nat Prod, 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]