

Abstract
A major thrust of our laboratory has been to identify how physiological stress is transduced into transcriptional responses that feed back to overcome the inciting stress or its consequences, thereby fostering survival and repair. To this end, we have adopted the use of an in vitro model of ferroptosis, a caspase-independent, but iron-dependent form of cell death (Dixon et al., 2012; Ratan, 2020). In this review, we highlight three distinct epigenetic targets that have evolved from our studies and which have been validated in in vivo studies. In the first section, we discuss our studies of broad, pan-selective histone deacetylase (HDAC) inhibitors in ferroptosis and how these studies led to the validation of HDAC inhibitors as candidate therapeutics in a host of disease models. In the second section, we discuss our studies that revealed a role for transglutaminase as an epigenetic modulator of proferroptotic pathways and how these studies set the stage for recent elucidation of monoamines as post-translation modifiers of histone function. In the final section, we discuss our studies of iron-, 2-oxoglutarate-, and oxygen-dependent dioxygenases and the role of one family of these enzymes, the HIF prolyl hydroxylases, in mediating transcriptional events necessary for ferroptosis in vitro and for dysfunction in a host of neurological conditions. Overall, our studies highlight the importance of epigenetic proteins in mediating prodeath and prosurvival responses to ferroptosis. Pharmacological agents that target these epigenetic proteins are showing robust beneficial effects in diverse rodent models of stroke, Parkinson’s disease, Huntington’s disease, and Alzheimer’s disease.
Keywords: Ferroptosis, epigenetics, HDAC, transglutaminase, HIF prolyl hydroxylase
1. Introduction
An in vitro model of oxidative stress-induced ferroptosis in neurons
Oxidative stress has been linked to almost all neurological conditions; however, the use of antioxidants as therapy has been disappointing in the clinic (Kumar and Ratan, 2016; Patel, 2016). One of the many reasons for the failure of antioxidant approaches has been the absence of in vitro models that allow us to determine the molecular targets of oxidants in neurons that lead to dysfunction, as well as the mechanisms of defense and how they can be augmented therapeutically. These models are also essential for determining the role of oxidants in normal cell function. Clearly, over the past two decades, we have come to recognize the important role that hydrogen peroxide and nitric oxide play as messengers (Rhee, 1999), and we have to take this role into account as we develop “antioxidant” therapeutics.
Our laboratory has attempted to address this lack of effective models by investigating an in vitro model of oxidative stress first elucidated in Joseph Coyle’s lab by Tim Murphy at Johns Hopkins (Murphy et al., 1989). Unsurprisingly, the model involves exposure of neurons to the excitotoxin, glutamate. However, the mechanisms by which glutamate kills immature cortical neurons (from E17 rat embryos cultured 2 days in vitro [DIV]) prior to the development of functional glutamate receptors (which occurs at 14 DIV) are unique and instructive. In this model, glutamate inhibits the transport of cystine from the extracellular bathing medium into neurons by competition for cystine at its plasma membrane transporting agency, the Xc− transporter (Bannai, 1986). This chloride-dependent transporter does not require energy, is non-electrogenic, and exchanges cystine for glutamate via facilitated diffusion. About ten percent of the extracellular glutamate at physiological pH is in the anionic form and is thus able to compete with cystine. Accordingly, as one drives up the concentration of glutamate to mM levels, cystine transport is competitively inhibited. Inhibition of cystine transport leads to depletion of intracellular cysteine, a rate-limiting substrate for the versatile tripeptide antioxidant glutathione (γ-glutamyl-cysteinyl-glycine). Glutathione represents an available reservoir of electrons (Dringen et al., 1999) that can be leveraged to maintain protein thiols in their reduced state (via glutathione reductase), to neutralize electrophilic lipid peroxides and hydrogen peroxides (via glutathione peroxidases), and to neutralize electrophilic xenobiotics (via glutathione-S-transferases).
2. What are the consequences of glutathione depletion in neurons?
Early studies from our laboratory showed that glutamate-induced glutathione depletion led to neuronal death that was highly regulated and involved the de novo synthesis of “prodeath” proteins (Ratan et al., 1994). Subsequent elegant studies from a number of laboratories have shown that, despite their programmed nature, glutathione depletion-induced transcriptional events do not involve classic prodeath apoptotic proteins (e.g., Bax or Bad) or caspases. Instead, they appear to depend on iron. Hence, this form of regulated cell death is distinct from apoptosis or necrosis and is now known as ferroptosis (Dixon et al., 2012; Zille et al., 2017).
3. HDAC inhibitors block ferroptosis and are neuroprotective in vivo
The observation that depletion of a cellular antioxidant, glutathione, could lead to ferroptotic cell death driven by the synthesis of proferroptotic proteins led us to speculate that this dysfunction could be counterbalanced by the transcriptional synthesis of antiferroptotic proteins. This speculation was stimulated in large measure by elegant work that had illuminated the yin and yang of proapoptotic and antiapoptotic BH3 proteins (e.g., Bax and Bcl-2, respectively). At the time we initiated these studies, we did not know the identity of the specific antioxidant enzyme that prevented ferroptosis (it is now known to be glutathione peroxidase 4 (Gpx4) (Seiler et al., 2008)). However, we hypothesized that execution of ferroptosis required an inadequate induction of compensatory gene responses that could nullify oxidative stress in response to ferroptotic stimuli.
To begin to understand what might drive these antiferroptotic responses in the nucleus, we screened several antioxidant enzymes to determine whether specific binding sites were enriched in their regulatory promoter regions. One response element that was highly enriched in these antioxidant gene promoters was the GC box (5’-(G/T)GGGCGG(G/A)(G/A)(C/T)-3’, the binding site for the transcription factor Sp1 (specificity protein 1) (Ryu et al., 2003a; Ryu et al., 2003b). Electrophoretic mobility shift assays confirmed a robust enhancement of Sp1 and Sp3 DNA binding to a canonical GC box; this binding occurred after glutathione depletion but prior to the commitment of neurons to death (Ryu et al., 2003b). A candidate search for post-translational modifications of the Sp1 and Sp3 proteins that could mediate a change in stability and DNA affinity revealed an increase in Sp1 acetylation (Ryu et al., 2003a).
As an initial test of the role for Sp1 acetylation in regulating ferroptotic death, we took advantage of a host of isoform pan-selective inhibitors of histone deacetylases (HDACs) (Ryu et al., 2003a). While HDACs are classically associated with acetylation of histones, another role that is now well appreciated is that they can modulate the acetylation of a host of proteins, including transcription factors. Chemically diverse pan-HDAC inhibitors (isoform non-selective) increased Sp1 acetylation, Sp1 DNA binding, and protection from oxidative stress-induced ferroptosis (Ryu et al., 2003a). As oxidative stress had been implicated in a number of neurological diseases, we were able to extend these results to models of Huntington’s disease (Ferrante et al., 2003), stroke (Langley et al., 2008), multiple sclerosis (Camelo et al., 2005), Parkinson’s disease (Aime et al., 2020; Liu et al., 2017), and hearing loss (Drottar et al., 2006).
Subsequent studies demonstrated that the beneficial effects of pan-selective HDAC inhibitors in preventing ferroptosis could be traced to class I HDACs (HDACs 1, 2, and 3) and one class II HDAC (HDAC6). As HDAC6 is localized to the axon, inhibiting HDAC6 activity is not directly epigenetic and so will not be discussed here. By contrast, in vitro inhibition of HDACs 1, 2, and 3 together leads to the induction of more than 2,000 genes in cultured neurons [Bourassa et al., unpublished observations, (Bourassa et al., 2016)]. Moreover, HDAC inhibitors can lead to neuroprotection independent of HDAC inhibition (Sleiman et al., 2014). Accordingly, defining the specific coordinated cassette of genes responsible for the beneficial effects of these agents has been difficult, both in vitro and in vivo. Despite this, certain genes that were most upregulated in mouse models of disease (e.g., Huntington’s) are now appreciated to reduce ferroptosis (e.g., Mkp-1 and Mkp-3) (Ferrante et al., 2003). These genes are sensitive to oxidation and undergo redox inactivation in neurons in response to ferroptotic stimuli. Inhibition of Mkp-1 or Mkp-3 leads to the persistent activation of Erk, its translocation to the nucleus, and the induction of prodeath transcription factors that include c-Myc (Sleiman et al., 2011).
Transcriptional upregulation of Mkp1 and Mkp3 by sodium butyrate in R6/2 HD mouse models provides a putative mechanism that explains how HDAC inhibitors counteract the unopposed activation of Erk that is known to lead to ferroptotic death in vitro (Yagoda et al., 2007; Zille et al., 2017) and that has also been implicated in neurodegeneration in vivo (Chung et al., 2005; Feld et al., 2014; Pei et al., 2002; Santini et al., 2010). Moreover, sodium butyrate treatment also decreased tau expression, although this appeared to be limited to the neurons in the striatum (Ferrante et al., 2003). As tau antibodies are showing promise in a host of neurological conditions, this reduction may also be relevant to the broad neuroprotective effects of HDAC inhibitors in multiple disease models (Bourassa et al., 2016; Langley et al., 2008; Yanamandra et al., 2017).
Independent of whether the protective effects of HDAC inhibition depend on Sp1 in vivo, insight into the environmental signals that might drive Sp1 acetylation in ferroptosis may come from recent studies showing that Sp1 cooperates with TFAP2c to initiate adaptive transcriptional responses to ferroptosis, especially transcription of Gpx4 (Alim et al., 2019). This transcriptional response is driven above a protective threshold by pharmacological selenium, leading to sequential activation of TFAP2c and subsequent sustained activation of Sp1. Accordingly, selenium may possibly drive the expression of Gpx4 via its ability inhibit an HDAC or to drive a histone acetyltransferase (HAT). Indeed, selenium derivatives of suberoyl anilide hydroxamic acid (SAHA) are more effective than the parent compound in inducing apoptosis in melanoma cells. However, their effect on ferroptosis in post-mitotic neurons is uncertain (Gowda et al., 2012).
In summary, recent studies show that ferroptotic stimuli (e.g., depletion of the antioxidant, glutathione) activate the epigenetic induction of prosurvival genes via the transcription factors TFAP2 and Sp1. Ferroptotic stimuli appear to achieve this homeostatic response by increasing Sp1 and Sp3 acetylation, resulting in enhanced Sp1 and Sp3 DNA occupancy at specific protective genes (e.g., Gpx4, Mkp1/Mkp3) and reduction of prodegeneration genes (e.g., Tau) that trigger neuroprotection. Selenium or class I HDAC inhibitors can drive this protective response above a threshold required for survival in vitro and in vivo (Alim et al., 2019). The precise signals that initiate these adaptive responses are unclear but are likely involve reactive lipid species.
4. Transglutaminases: novel epigenetic kids on the CNS block
The ultimate execution of cell death by ferroptotic stimuli appears to involve an inadequate homeostatic response. The reason for this inadequacy is complex and may reflect the ability of prodeath signals to turn on epigenetic repressors of prosurvival transcription. Transcription factors (TFs) bind to response elements, which are specific sites in promoters or enhancers in the DNA, through their DNA binding domains. To convert that binding into transcription of the coding region of the proximal gene, TFs must possess a transactivation domain (Arnold et al., 2018). This transactivation domain binds coregulatory factors that have a dual purpose. In one case, these factors facilitate the unwinding of DNA. In the other, they recruit RNA polymerase (the enzyme that synthesizes RNA).
While transactivation domains come in many shapes and sizes, a number of the transcription factors that have been linked to stress adaptation (Sp1, CREB, and Nrf1) have glutamine-rich transactivation domains. We speculated that these glutamine-rich domains can be post-translationally modified by a calcium- and GTP-dependent enzyme, transglutaminase, which appears to serve as an epigenetic modulator. Transglutaminase can crosslink a glutamine on one protein to an adjacent lysine on another protein or it can catalyze the addition of polyamines or monoamines to glutamines on transcription factors or histones (Folk and Finlayson, 1977; Lorand and Conrad, 1984).
Interest in transglutaminase emerged because pharmacological and molecular inhibition of the transglutaminase 2 (TG2) isoform enzyme was effective in improving the behavior and extending the lifespan in models of Huntington’s disease, Parkinson’s disease, and hemorrhagic stroke (Gibrat et al., 2010; Mastroberardino et al., 2002; McConoughey et al., 2010; Okauchi et al., 2009). Initial hypotheses focused on the ability of transglutaminase, a calcium-regulated enzyme, to promote the aggregation of mutant proteins in the cytoplasm and confer toxicity (Cooper et al., 1999; Zainelli et al., 2004). However, our studies in cellular and fly models showed that transcriptional repression of metabolic genes involved in Huntington’s disease could be reversed by a selective peptide inhibitor of transglutaminase but not by its inactive control (McConoughey et al., 2010). Moreover, forced expression of TG2 repressed metabolic transcription, whereas a modified form of TG that either lacked transamidating activity or did not enter the nucleus failed to repress transcription (McConoughey et al., 2010).
Chromatin immunoprecipitation assays localized TG2 to the promoters of those genes (PGC1α, cytochrome C) that it was shown to repress and provided resistance to 3-nitroproprionic acid in rodent and human cells. Indeed, inhibition of TG2 normalized the expression of more than 40 percent of genes by greater than 25 percent. Inhibition of TG led to derepression independent of changes in histone acetylation, suggesting that TGs were not acting as HDAC inhibitors. Co-immunoprecipitation studies showed that TG does not interact with the wild type or mutant huntingtin protein per se, but instead interacts with histone H3. Although our findings suggested that TG2 catalyzed a repressive activity (polyamination) via interactions at glutamine residues, recent studies have highlighted the role of TG2-catalyzed serotonylation of the histone H3 protein at glutamine 5 in the transcriptional expression of differentiation genes (Farrelly et al., 2019).
TG2 has the ability to repress transcription via local action at targeted promoters and thereby promote neuronal death independent of mutant huntingtin protein (McConoughey et al., 2010). This observation led us to consider the possibility that TG2 could negatively influence viability in response to stresses such as oxidative stress that may be relevant to many neurological conditions. Consistent with this notion, we found that TG1 and TG2 are upregulated after ischemic stroke and that germline deletion of TG2 prevented ferroptotic death in mouse embryo fibroblasts (Basso et al., 2012). A series of structurally diverse inhibitors of TG abrogated death, even when delivered up to 14 hours after exposure to glutamate as a ferroptotic stimulus. Inhibitors of TG that could not penetrate the plasma membrane were not protective, and TG activity was significantly increased before the cells were committed to die. This increase in TG activity was completely suppressed by inhibitors of MEK, suggesting that TG activity acts downstream of nuclear Erk activation, a well-established mediator of ferroptosis in neurons (Basso et al., 2012). In this context, TG may repress genes involved in survival or activate genes involved in death.
Recent studies have shown that the TG-catalyzed serotonylation of glutamine 5 of Histone H3 adjacent to lysine 4 trimethylation is a combinatorial mark that recruits TFIID to recruit RNA polymerase and drive the synthesis of differentiation genes (Farrelly et al., 2019). Moreover, dopaminylation of glutamine 5, also catalyzed by TG2, can have similar roles in enhancing transcription (Lepack et al., 2020). Taken together with our previous results, these findings suggest that TG2 can act to foster repression or activation of gene expression through its ability to polyaminate or monoaminylate specific residues in histones or transcription factors. Future studies will clarify the role of TG2 as an epigenetic facilitator of ferroptosis in neurons and cancer cells.
5. Oxygen sensors as epigenetic regulators of ferroptosis
A central, defining feature of ferroptotic death in multiple cell types is its inhibition by chelators of low-molecular-weight iron (Ratan, 2020). Classically, these chelators were hypothesized to protect via their ability to inhibit the generation of toxic hydroxyl or peroxyl radicals. During the past 25 years, our lab has assembled support for an alternative model of iron toxicity that appears to involve the loading of a family of metalloenzymes that were originally defined as oxygen sensors (Aminova et al., 2005; Aminova et al., 2008; Karuppagounder et al., 2016; Siddiq et al., 2009; Siddiq et al., 2005; Smirnova et al., 2010; Zaman et al., 1999). The specific metalloenzymes are the HIF prolyl hydroxylases (PHDs) and iron-, oxygen-, and 2-oxoglutarate-dependent dioxygenases (Bruick and McKnight, 2001; Kaelin and Ratcliffe, 2008; Karuppagounder and Ratan, 2012; Ratan, 2020). The canonical function of these enzymes is to modulate the stability of the regulated hypoxia inducible factor-1 alpha subunit of the heterodimeric transcription factor, HIF.
The HIF PHDs catalyze the oxygen-dependent transfer of a hydroxyl group to the proline 403 and proline 564 of the HIF-1α protein oxygen-dependent domain. Hydroxylation of HIF-1α at proline 403 and 564 enhances its affinity for its E3 ubiquitin ligase, the Von Hippel Lindau (VHL) protein. VHL binding to HIF-1α leads to HIF-1α polyubiquitination and degradation. Under conditions of hypoxia, the oxygen concentration drops below the Km of the VHL enzyme, and HIF is not hydroxylated, polyubiquitinated, or degraded. We showed that iron chelators inhibit the HIF prolyl hydroxylases presumably by stealing iron from the active site of the enzyme (Karuppagounder et al., 2016; Ratan, 2020; Siddiq et al., 2005; Smirnova et al., 2010; Zaman et al., 1999). In this way, iron chelators “fool” the neuron into thinking it is hypoxic when it is not, and this, in vitro, can activate a cassette of genes associated with hypoxic adaptation. Accordingly, our model for protection was that iron chelators inhibit the oxygen sensors, fool ferroptotic neurons into thinking they are hypoxic, and drive a cassette of genes that operate in an integrated way to prevent death. Indeed, molecular depletion of HIF PHDs, using selective tat-linked peptide inhibitors in vitro, siRNAs to individual isoforms in vitro, or conditional deletion of all three isoforms in vivo, leads to reductions in cell death (Karuppagounder et al., 2016; Siddiq et al., 2009; Siddiq et al., 2005).
How does inhibiting the prolyl hydroxylases inhibit ferroptotic death? Our initial model was that inhibition of HIF PHDs leads to upregulation of HIF-dependent genes, including those of glycolytic enzymes, PDK1, and BNIP3 (Siddiq et al., 2005; Zaman et al., 1999). These genes would enhance the ability of the neuron to generate ATP by glycolysis and reduce mitochondrial biosynthetic and catabolic functions. Reductions in these mitochondrial activities would reduce the free radical production that normally occurs as electrons are passed down the mitochondrial electron transport chain and interact with oxygen to generate superoxide. However, several observations argued against this model. First, forced expression of HIF-VP16, an oxygen-stabilized and constitutively active form of HIF, potentiated ferroptotic death rather than inhibiting it (Aminova et al., 2005). Second, siRNA-mediated reductions in HIF-1α and HIF-2α failed to alter the protective effects of HIF PHD inhibition by the iron chelator, deferoxamine (DFO) or other chemically diverse HIF PHD inhibitors. Of note, penetratin-linked siRNAs for PHD1, but not for PHD2 or PHD3, prevented ferroptotic death in HT22 cells and primary neurons (Siddiq et al., 2009).
Collectively, our studies showed that while HIF PHD1 inhibition can prevent ferroptotic death, it does so independently of HIF-1, HIF-2, or CREB. Our prior studies showed that HIF PHD1 can act in the nucleus to hydroxylate Rbp1 at proline 1465 (within a canonical LGQLAP proline hydroxylation motif) to enhance Rbp1 activity (Mikhaylova et al., 2008). Rbp1 is a subunit of RNA polymerase II, which carries the enzymatic activity required for making new RNA. Under this scheme, HIF PHD1 inhibition would protect against ferroptosis by preventing RNA Pol II activity and the synthesis of de novo proferroptotic proteins.
Unbiased transcriptomics identified a mechanism by which HIF PHD1 could target promoter specific transcription. Specifically, we found that the protective effects of pharmacological HIF PHD inhibition were not associated with the upregulation of HIF regulated genes but were unexpectedly associated with specific downregulation of ATF4-dependent genes (Karuppagounder et al., 2016). Like HIF-1, ATF4 is known to possess phylogenetically conserved prolines that can be hydroxylated. Forced expression of a mutant form of ATF4, with all 5 conserved prolines changed to alanine (5×P/A mutant ATF4), failed to induce ferroptotic death like its wild type ATF4 counterpart. Indeed, forced expression of the 5×P/A mutant blocked ferroptosis, suggesting that the mutant can function as a dominant negative. Wild type ATF4 can be localized to the promoters of putative proferroptotic genes (e.g., Trib3), but the 5×P/A mutant cannot. As expected, pharmacological inhibition of the HIF PHDs reduced the level of hydroxylation of ATF4. Additional selective mutations of proline are required to establish the effects of mutations that might not be expected to create such dramatic alterations in protein structure.
In unpublished studies, we found that iron loading of neurons using proferroptotic hemin (used to model brain hemorrhage in a dish) dramatically enhances the activity of the HIF prolyl hydroxylases. The results are consistent with the notion that the HIF prolyl hydroxylases function as both oxygen and iron sensors. The mechanisms by which classical ferroptosis paradigms result in increases in free iron above a homeostatic threshold are unclear; however, disruptions of iron sulfur clusters via glutathione depletion or loss of iron storage via ferritinophagy have been proposed. Whatever the mechanism, our data are consistent with a scheme whereby increases in iron lead to loading of a specific chaperone, called poly (RC) binding protein (PCBP1). PCPB1, or its paralog PCBP2, were originally defined as RNA or DNA binding proteins but were also shown to function as chaperones to load ferritin, the major iron-storage protein in the cell. More recently, they were found to deliver iron to the HIF prolyl hydroxylases.
During ferroptosis, ferritin may undergo selective autophagy (Nandal et al., 2011), leading PCPB1 or PCBP2 to preferentially load the HIF PHDs and causing increased hydroxylations of substrates including HIF-1 and ATF4. While increased hydroxylation of HIF-1 would lead to its VHL-mediated ubiquitination and degradation, increased hydroxylation of ATF4 appears to be associated with enhanced transcription of its targets, including Trib3, Chac1, and Chop (Karuppagounder et al., 2016; Lange et al., 2008). Trib3 is a pseudokinase inhibitor of AKT that can inhibit CREB-dependent transcription, but it also can decrease the stability of Parkin, a protein that mediates mitophagy. By contrast, Chac1 degrades glutathione to its constituent amino acids, thereby reducing the likely antioxidant capacity as cells are committed to die. Similarly, Chop is a transcription factor that has been implicated in executing cell death in a host of contexts.
We speculate that enhanced ATF4 hydroxylation, driven by increased HIF PHD activity, leads to transcriptional upregulation of these and other target genes in the integrated execution of ferroptosis. Accordingly, HIF PHDs, and most likely HIF PHD1, serve as epigenetic iron sensors to transduce a lethal stress into the execution of a regulated necrotic pathway via de novo transcription. These iron-dependent mechanisms of ferroptotic death appear to serve in a host of neurological conditions as selective inhibitors of HIF PHDs (which do not globally chelate iron) that are effective in models of Huntington’s disease, Parkinson’s disease, stroke, traumatic brain injury, and Alzheimer’s disease.
Deferoxamine (DFO), has been shown to activate other transcriptional pathways involved in neuronal survival, although the mechanism is not clearly epigenetic. For example, a study by Ryu et al., (2005) suggested that a mitochondrial signal transduction pathway was activated by DFO directly leading to neuronal survival (Ryu et al., 2005). Indeed, while establishing that nuclear CREB is stabilized by DFO, Ryu and colleagues excitingly discovered that CREB, which was thought to be a nuclear protein, is also stabilized within the mitochondria. Further, DFO was found to activate PKA, a cAMP dependent kinase responsible for site-specific phosphorylation and activation of CREB in vitro and in vivo. The data showed an increase of mitochondrial CREB-mediated transcriptional activity of mitochondrially encoded genes. The studies by Ryu et al. suggested that iron chelators activate a coordinate transcriptional response between the nucleus and mitochondria downstream of cAMP that can augment metabolism and cell survival by activating transcription of nuclear and mitochondrial genes. Whether these responses are downstream of oxygen sensing or some other pathway has yet to be established.
6. Conclusion
Oxidative stress has been implicated in almost every neurological condition, and yet antioxidants have been disappointing therapeutics in the clinic. Some of this failure has been attributable to the lack of good in vitro models of oxidative stress in neurons. Immature neurons exposed to high concentrations of glutamate or its analogs experience glutathione insufficiency and programmed necrosis that is now recognized as ferroptosis. Studies over the last 25 years have implicated HDACs, transglutaminases, and HIF prolyl hydroxylases as epigenetic regulators of ferroptotic death. Pharmacological or molecular inhibition of each of these targets leads not only to abrogation of ferroptosis but also to reduced cell death and improved recovery in diverse neurological disease models. These findings suggest that ferroptotic pathways may represent a final common pathway by which diverse acquired and inherited diseases of the nervous system lead to neuronal loss and impairment.
Figure 1. HDAC inhibitors augment oxidative stress-induced Sp1 acetylation to increase expression of genes that inhibit oxidative stress and cell death [Adapted from (Ryu et al., 2003a)].
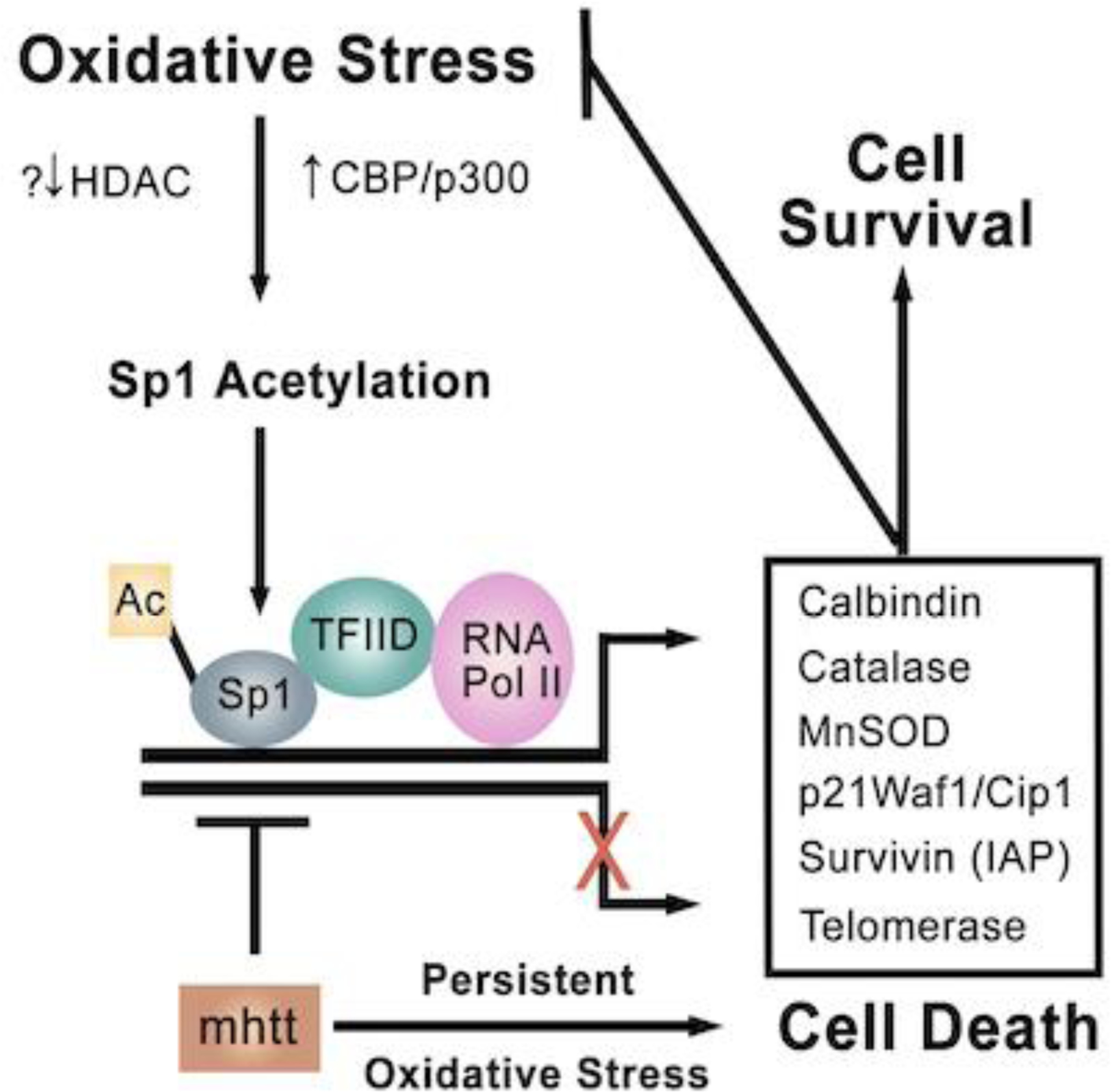
In response to hydrogen peroxide (a non ferroptotic stimulus) or glutathione depletion (a ferroptotic stimulus) acetylation of Sp1 is increased. Acetylated of Sp1 enhances its DNA binding and ability to bind TFIID, the factor that recruits RNA polymerase to produce mRNAs from target genes. A list of putative protective genes induced by Sp1 is listed. Some of these genes would be expected to inhibit the effector phase of cell death (e.g. surviving). Other genes would influence the affector phase of cell death (e.g. catalase, MnSOD). The ability of mutant huntingtin to inhibit Sp1 transcription (Dunah et al., 2002) provides a mechanism by which a gene mutation linked to a neurodegenerative disease could decreased the threshold for oxidative stress-induced death.
Figure 2. Selenium drives an adaptive transcriptional response to ferroptosis, excitotoxicity and ER stress via the induction of Gpx4 and other genes of the selenome [adapted from (Alim et al., 2019)].

Ferroptotic stimuli (e.g. inhibitors of the Xc− transporter or brain hemorrhage) drive a frustrated attempt at compensation which is mediated via the transient activation of the transcription factor TFAP2c and subsequent, more sustained activation of the transcription factor Sp1. This inadequate homeostatic response can be augmented to enhance survival via the exposure of neurons to selenium. Delivery of selenium occurs optimally via a peptide, Tat-Sel Pep that incorporates selenocysteines and can drive TFAP2c and Sp1 to enhance transcription of the selenome leading to broad cell protection in hemorrhagic or ischemic stroke.
Figure 3. Transglutaminase is an epigenetic modulator of transcription and ferroptosis [(Basso and Ratan, 2013)].

During ferroptosis, CaM and other calcium binding proteins are not sufficient to buffer calcium efficiently and other calcium-dependent proteins like TG2 are activated. 1.) Several putative mechanisms exist for how increased TG activity could repress prosurvival or activate prodeath gene expression. TG could polyaminate or monaminylate transcription factors (TFs) presenting a glutamine-rich sequence in their TF activation domain (TAD). 2) TG can crosslink actin and cofilin in nuclear rods, altering the subgeography of the nucleus and thus contributing to transcriptional activation or repression. 3.) TG can polyaminate or monaminylate histone proteins and favors chromatin in the close conformation to repress transcription (e.g. polyamination, shown in figure) or recruit TGIID to activate transcription (e.g., serotonylation or dopaminylation, not shown in figure).
Figure 4. HIF PHDs function as epigenetic iron sensors to drive proferroptotic transcription.
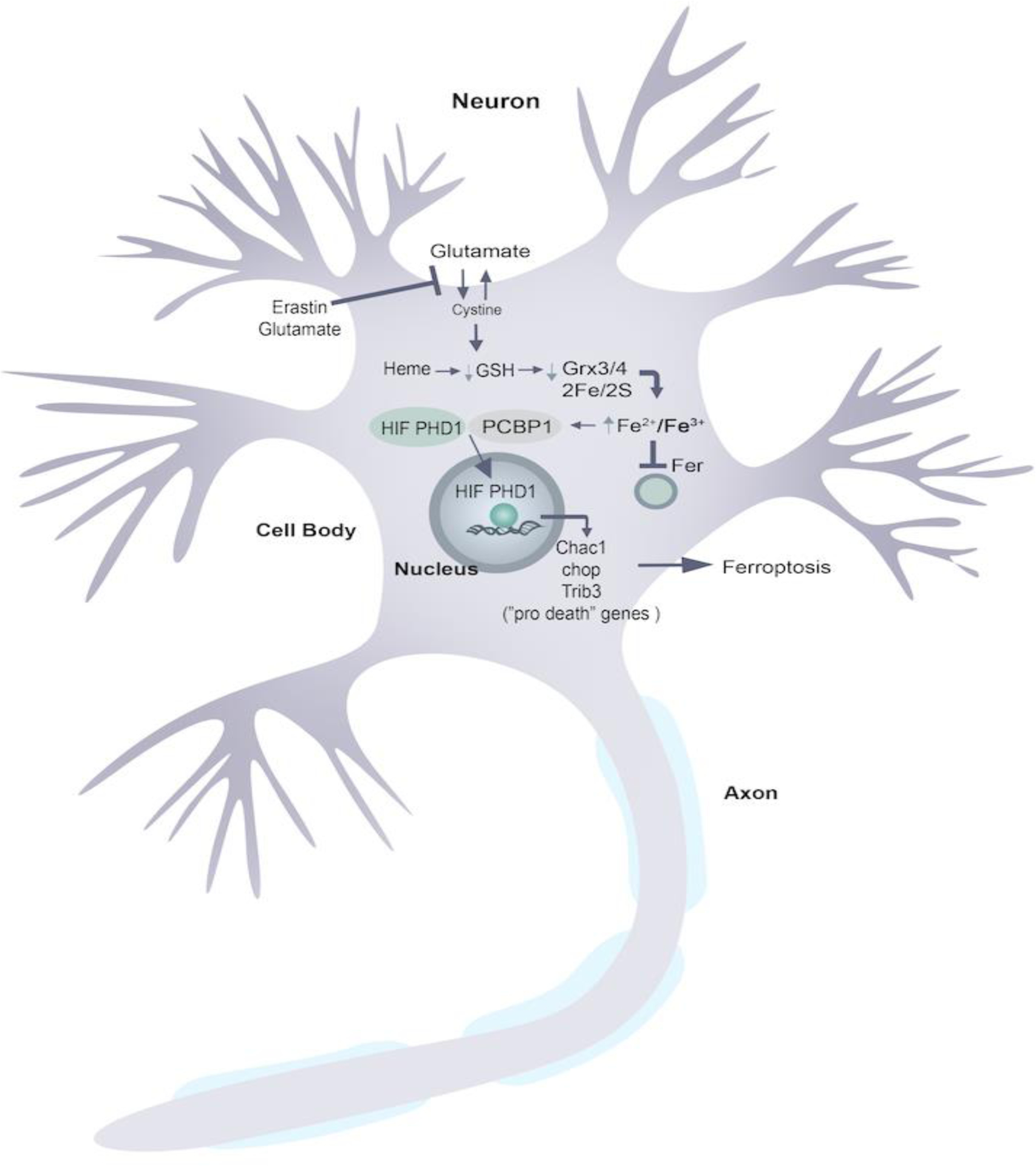
A working model for how glutathione depletion leads to activation of proferroptotic transcription. Erastin or glutamate inhibit the Xc− transporter leading to cysteine deprivation. As cysteine is the rate limiting precursor for glutathione synthesis, this leads to glutathione depletion. As a consequence of glutathione depletion iron is liberated from iron sulfur complexes and released to the iron chaperone PCBP1 or PCBP2. Through mechanisms that have yet to be worked out, PCPB1/2 load the HIF PHDs rather than ferritin leading to augmentation of HIF PHD activity, hydroxylation of ATF4 and induction of ATF4 dependent prodeath genes. Inhibitors of the HIF PHDs leads to epigenetic repression of ATF4 proferrotptic gene expression and protection from ferroptotic cell death.
Acknowledgements
R.R.R. is funded with generous support from the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation, the Burke Foundation, the Sperling Center for Hemorrhagic Stroke Recovery at the Burke Neurological Institute and the NIH (grant P01 NIA AG014930, project 1, to R.R.R.). We thank Laura Gilmartin with assistance with the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no significant financial conflicts of interest. R.R.R. holds patents (owned by Cornell University or Burke Neurological Institute) related to inhibitors of ferroptosis and their therapeutic applications in the nervous system. He is also on the Scientific Advisory Board for Neuronasal, Inc., which has licensed patents from the Ratan Laboratory. R.R.R. has an equity interest in Neuronasal and also serves in an advisory capacity to Biogen.
Reference:
- Aime P, et al. , 2020. The drug adaptaquin blocks ATF4/CHOP-dependent pro-death Trib3 induction and protects in cellular and mouse models of Parkinson’s disease. Neurobiol Dis. 136, 104725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alim I, et al. , 2019. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell. 177, 1262–1279 e25. [DOI] [PubMed] [Google Scholar]
- Aminova LR, et al. , 2005. Prosurvival and prodeath effects of hypoxia-inducible factor-1alpha stabilization in a murine hippocampal cell line. J Biol Chem. 280, 3996–4003. [DOI] [PubMed] [Google Scholar]
- Aminova LR, et al. , 2008. Antioxidants, HIF prolyl hydroxylase inhibitors or short interfering RNAs to BNIP3 or PUMA, can prevent prodeath effects of the transcriptional activator, HIF-1alpha, in a mouse hippocampal neuronal line. Antioxid Redox Signal. 10, 1989–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold CD, et al. , 2018. A high-throughput method to identify trans-activation domains within transcription factor sequences. EMBO J. 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai S, 1986. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 261, 2256–63. [PubMed] [Google Scholar]
- Basso M, et al. , 2012. Transglutaminase inhibition protects against oxidative stress-induced neuronal death downstream of pathological ERK activation. J Neurosci. 32, 6561–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basso M, Ratan RR, 2013. Transglutaminase is a therapeutic target for oxidative stress, excitotoxicity and stroke: a new epigenetic kid on the CNS block. J Cereb Blood Flow Metab. 33, 809–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilaqua LR, et al. , 1999. Experience-dependent increase in cAMP-responsive element binding protein in synaptic and nonsynaptic mitochondria of the rat hippocampus. Eur J Neurosci. 1999 October; 11(10):3753–6. Doi 10.1046/j.1460-9568.1999.00830.x. [DOI] [PubMed] [Google Scholar]
- Bourassa MW, et al. , 2016. Butyrate, neuroepigenetics and the gut microbiome: Can a high fiber diet improve brain health? Neurosci Lett. 625, 56–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruick RK, McKnight SL, 2001. A conserved family of prolyl-4-hydroxylases that modify HIF. Science. 294, 1337–40. [DOI] [PubMed] [Google Scholar]
- Camelo S, et al. , 2005. Transcriptional therapy with the histone deacetylase inhibitor trichostatin A ameliorates experimental autoimmune encephalomyelitis. J Neuroimmunol. 164, 10–21. [DOI] [PubMed] [Google Scholar]
- Chung YH, et al. , 2005. Immunohistochemical study on the distribution of phosphorylated extracellular signal-regulated kinase (ERK) in the central nervous system of SOD1G93A transgenic mice. Brain Res. 1050, 203–9. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, et al. , 1999. Pathogenesis of inclusion bodies in (CAG)n/Qn-expansion diseases with special reference to the role of tissue transglutaminase and to selective vulnerability. J Neurochem. 72, 889–99. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, et al. , 2012. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 149, 1060–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dringen R, et al. , 1999. Synthesis of the antioxidant glutathione in neurons: supply by astrocytes of CysGly as precursor for neuronal glutathione. J Neurosci. 19, 562–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drottar M, et al. , 2006. The histone deacetylase inhibitor sodium butyrate protects against cisplatin-induced hearing loss in guinea pigs. Laryngoscope. 116, 292–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunah AW, et al. , 2002. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 296, 2238–43. [DOI] [PubMed] [Google Scholar]
- Farrelly LA, et al. , 2019. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature. 567, 535–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feld M, et al. , 2014. Decrease of ERK/MAPK overactivation in prefrontal cortex reverses early memory deficit in a mouse model of Alzheimer’s disease. J Alzheimers Dis. 40, 69–82. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, et al. , 2003. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 23, 9418–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folk JE, Finlayson JS, 1977. The epsilon-(gamma-glutamyl)lysine crosslink and the catalytic role of transglutaminases. Adv Protein Chem. 31, 1–133. [DOI] [PubMed] [Google Scholar]
- Gibrat C, et al. , 2010. Cystamine prevents MPTP-induced toxicity in young adult mice via the up-regulation of the brain-derived neurotrophic factor. Prog Neuropsychopharmacol Biol Psychiatry. 34, 193–203. [DOI] [PubMed] [Google Scholar]
- Gowda R, et al. , 2012. Selenium-containing histone deacetylase inhibitors for melanoma management. Cancer Biol Ther. 13, 756–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada et al. , 1999. Phosphorylation and inactivation of BAD by mitochondria-anchored protein kinase A. Mol Cell Apr;3 (4):413–22. DOI 10.1016/s1097-2765(00)80469-4. [DOI] [PubMed] [Google Scholar]
- Kaelin WG Jr., Ratcliffe PJ, 2008. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 30, 393–402. [DOI] [PubMed] [Google Scholar]
- Karuppagounder SS, et al. , 2016. Therapeutic targeting of oxygen-sensing prolyl hydroxylases abrogates ATF4-dependent neuronal death and improves outcomes after brain hemorrhage in several rodent models. Sci Transl Med. 8, 328ra29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karuppagounder SS, Ratan RR, 2012. Hypoxia-inducible factor prolyl hydroxylase inhibition: robust new target or another big bust for stroke therapeutics? J Cereb Blood Flow Metab. 32, 1347–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Ratan RR, 2016. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J Huntingtons Dis. 5, 217–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange PS, et al. , 2008. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 205, 1227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley B, et al. , 2008. Pulse inhibition of histone deacetylases induces complete resistance to oxidative death in cortical neurons without toxicity and reveals a role for cytoplasmic p21(waf1/cip1) in cell cycle-independent neuroprotection. J Neurosci. 28, 163–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepack AE, et al. , 2020. Dopaminylation of histone H3 in ventral tegmental area regulates cocaine seeking. Science. 368, 197–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, et al. , 2017. Sodium butyrate exerts protective effect against Parkinson’s disease in mice via stimulation of glucagon like peptide-1. J Neurol Sci. 381, 176–181. [DOI] [PubMed] [Google Scholar]
- Lorand L, Conrad SM, 1984. Transglutaminases. Mol Cell Biochem. 58, 9–35. [DOI] [PubMed] [Google Scholar]
- Mastroberardino PG, et al. , 2002. ‘Tissue’ transglutaminase ablation reduces neuronal death and prolongs survival in a mouse model of Huntington’s disease. Cell Death Differ. 9, 873–80. [DOI] [PubMed] [Google Scholar]
- McConoughey SJ, et al. , 2010. Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol Med. 2, 349–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhaylova O, et al. , 2008. The von Hippel-Lindau tumor suppressor protein and Egl-9-Type proline hydroxylases regulate the large subunit of RNA polymerase II in response to oxidative stress. Mol Cell Biol. 28, 2701–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, et al. , 1989. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 2, 1547–58. [DOI] [PubMed] [Google Scholar]
- Nandal A, et al. , 2011. Activation of the HIF prolyl hydroxylase by the iron chaperones PCBP1 and PCBP2. Cell Metab. 14, 647–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okauchi M, et al. , 2009. Tissue-type transglutaminase and the effects of cystamine on intracerebral hemorrhage-induced brain edema and neurological deficits. Brain Res. 1249, 229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel M, 2016. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol Sci. 37, 768–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei JJ, et al. , 2002. Up-regulation of mitogen-activated protein kinases ERK1/2 and MEK1/2 is associated with the progression of neurofibrillary degeneration in Alzheimer’s disease. Brain Res Mol Brain Res. 109, 45–55. [DOI] [PubMed] [Google Scholar]
- Ratan RR, 2020. The Chemical Biology of Ferroptosis in the Central Nervous System. Cell Chem Biol. 27, 479–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratan RR, et al. , 1994. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 62, 376–9. [DOI] [PubMed] [Google Scholar]
- Rhee SG, 1999. Redox signaling: hydrogen peroxide as intracellular messenger. Exp Mol Med. 31, 53–9. [DOI] [PubMed] [Google Scholar]
- Ryu H, et al. , 2003a. Histone deacetylase inhibitors prevent oxidative neuronal death independent of expanded polyglutamine repeats via an Sp1-dependent pathway. Proc Natl Acad Sci U S A. 100, 4281–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, et al. , 2003b. Sp1 and Sp3 are oxidative stress-inducible, antideath transcription factors in cortical neurons. J Neurosci. 23, 3597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, et al. , 2005. Antioxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mitochondrial genome in neurons. PNAS September 27, 2005 102 (39) 13915–13920 doi: 10.1073/pnas.0502878102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, et al. , 2010. Distinct changes in cAMP and extracellular signal-regulated protein kinase signalling in L-DOPA-induced dyskinesia. PLoS One. 5, e12322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq A, et al. , 2009. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci. 29, 8828–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq A, et al. , 2005. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 280, 41732–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman SF, et al. , 2011. Histone Deacetylase Inhibitors and Mithramycin A Impact a Similar Neuroprotective Pathway at a Crossroad between Cancer and Neurodegeneration. Pharmaceuticals (Basel). 4, 1183–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleiman SF, et al. , 2014. Hydroxamic acid-based histone deacetylase (HDAC) inhibitors can mediate neuroprotection independent of HDAC inhibition. J Neurosci. 34, 14328–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smirnova NA, et al. , 2010. Utilization of an in vivo reporter for high throughput identification of branched small molecule regulators of hypoxic adaptation. Chem Biol. 17, 380–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takio K, et al. , 1982. Primary structure of the regulatory subunit of type II cAMP-dependent protein kinase from bovine cardiac muscle. Proc Natl Acad Sci USA. April;79(8):2544–8. Doi 10.1073/pnas.79.8.2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagoda N, et al. , 2007. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 447, 864–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, et al. , 2017. Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci Transl Med. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zainelli GM, et al. , 2004. Calmodulin regulates transglutaminase 2 cross-linking of huntingtin. J Neurosci. 24, 1954–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman K, et al. , 1999. Protection from oxidative stress-induced apoptosis in cortical neuronal cultures by iron chelators is associated with enhanced DNA binding of hypoxia-inducible factor-1 and ATF-1/CREB and increased expression of glycolytic enzymes, p21(waf1/cip1), and erythropoietin. J Neurosci. 19, 9821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zille M, et al. , 2017. Neuronal Death After Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke. 48, 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]