

Abstract
Aims
Cyclophilin-D is a well-known regulator of the mitochondrial permeability transition pore (PTP), the main effector of cardiac ischaemia/reperfusion injury. However, the binding of CypD to the PTP is poorly understood. Cysteine 202 (C202) of CypD is highly conserved among species and can undergo redox-sensitive post-translational modifications. We investigated whether C202 regulates the opening of PTP.
Methods and results
We developed a knock-in mouse model using CRISPR where CypD-C202 was mutated to a serine (C202S). Infarct size is reduced in CypD-C202S Langendorff perfused hearts compared to wild type (WT). Cardiac mitochondria from CypD-C202S mice also have higher calcium retention capacity compared to WT. Therefore, we hypothesized that oxidation of C202 might target CypD to the PTP. Indeed, isolated cardiac mitochondria subjected to oxidative stress exhibit less binding of CypD-C202S to the proposed PTP component F1F0-ATP-synthase. We previously found C202 to be S-nitrosylated in ischaemic preconditioning. Cysteine residues can also undergo S-acylation, and C202 matched an S-acylation motif. S-acylation of CypD-C202 was assessed using a resin-assisted capture (Acyl-RAC). WT hearts are abundantly S-acylated on CypD C202 under baseline conditions indicating that S-acylation on C202 per se does not lead to PTP opening. CypD C202S knock-in hearts are protected from ischaemia/reperfusion injury suggesting further that lack of CypD S-acylation at C202 is not detrimental (when C is mutated to S) and does not induce PTP opening. However, we find that ischaemia leads to de-acylation of C202 and that calcium overload in isolated mitochondria promotes de-acylation of CypD. Furthermore, a high bolus of calcium in WT cardiac mitochondria displaces CypD from its physiological binding partners and possibly renders it available for interaction with the PTP.
Conclusions
Taken together the data suggest that with ischaemia CypD is de-acylated at C202 allowing the free cysteine residue to undergo oxidation during the first minutes of reperfusion which in turn targets it to the PTP.
Keywords: Ischaemia/reperfusion, Cyclophilin-D, Permeability transition, S-acylation, Oxidation
Graphical Abstract
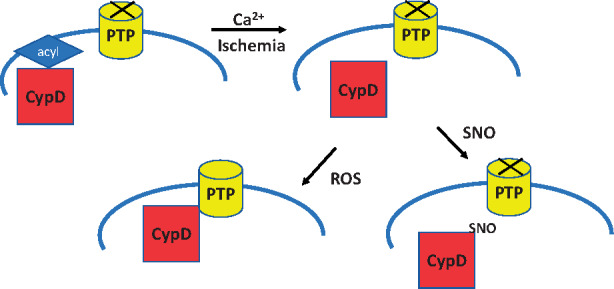
1. Introduction
Cell death following ischaemia/reperfusion (I/R) is thought to be due to opening of the mitochondrial permeability transition pore (PTP), a large conductance channel in the inner mitochondrial membrane.1–4 Calcium and reactive oxygen species are well-established triggers of PTP opening and both are elevated during I/R. Opening of the PTP5–8 leads to loss of mitochondrial membrane potential, loss of ATP-production, and ultimately cell-death. Although the molecular identity of PTP is debated, the F1F0-ATP-synthase has been proposed as the pore-forming component.9–11 In contrast to the uncertainty regarding the identity of the pore-forming unit of the PTP, genetic knockdown and inhibitor studies have documented that cyclophilin-D (CypD), a peptidyl-prolyl cis-trans isomerase localized in the mitochondrial matrix, is a regulator of PTP.12–14 In I/R injury, Ca+2 overload and oxidative stress promote the translocation of CypD to the inner mitochondrial membrane which in turn sensitizes PTP and triggers cell-death.15,16 PTP has been shown to be redox-sensitive, which makes thiols on cysteines of CypD an interesting target.17–20 Cysteine 202 (C202) in mouse CypD is highly conserved among species, and previous studies from our group have shown that it is oxidized during I/R injury and S-nitrosylated in ischaemic preconditioning.21 Another common post-translational modification of cysteines is S-acylation. S-acylation is the covalent attachment of a fatty acid to a protein thiol group and has been shown to affect the localization of proteins or their catalytic activity.22,23 CypD C202 matches an S-acylation site motif found commonly in soluble proteins.24 We have previously shown that the mutation of C202 to a serine (C202S) which cannot undergo redox-sensitive post-translational modifications desensitized PTP opening in mouse embryonic fibroblasts, suggesting that C202 is important for PTP opening.25
The goal of the present work is to elucidate the role of C202 of CypD in mediating PTP opening. We developed a CypD C202S knock-in mouse model using CRISPR in a C57BL/6N background. We find that C202 in addition to being a site of S-nitrosylation,21 is a major site of S-acylation. We report that CypD C202S mice exhibit reduced I/R injury and desensitized PTP opening. Furthermore, CypD C202S exhibits diminished binding to the proposed PTP component ATP-synthase under oxidative stress.
2. Methods
2.1 Animals
All animals were treated and cared for in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health), and protocols were approved by the Institutional Animal Care and Use Committee of the National Heart, Lung, and Blood Institute. Global CypD C202S mice were maintained on a C57BL/6N background. Genotyping was performed as described elsewhere.25 Mice were anaesthetized with an intraperitoneal injection of sodium pentobarbital (200 mg/kg, P3761, Sigma). Heparin (3000 U/kg, Sigma) was co-administered to prevent blood clotting during the isolation procedure for the perfused hearts. Induction of anaesthesia was confirmed within 5 min of injection by the absence of physical responses, including pedal withdrawal and corneal reflexes.
2.2 Langendorff perfused hearts
Hearts were excised quickly from mice and placed into ice-cold Krebs–Henseleit buffer of the following composition (mmol/L): 120 NaCl, 11 d-glucose, 25 NaHCO3, 1.75 CaCl2, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4 (pH 7.4). Aortas were cannulated and connected to a non-recirculating Langendorff perfusion apparatus, through which the heart was perfused retrogradely with Krebs buffer (37°C, aerated with 95% O2/5% CO2) at a constant pressure of 100 cm H2O. For all experiments, hearts were first equilibrated for 20 min and then subjected to global ischaemia for 20 min followed by reperfusion for 90 min. Where indicated hearts were perfused with cyclosporine A 5 min before the onset of ischaemia to 5 min after onset of reperfusion. To measure left ventricular developed pressure (LVDP), a latex balloon connected to a pressure transducer was inserted in the left ventricle and recordings were made using PowerLab (ADInstruments). Rate pressure product (RPP), a measure of functional recovery, was calculated as the post-ischaemic LVDP multiplied by heart rate divided by the pre-ischaemic values. After reperfusion hearts were perfused with 1% 2,3,5-triphenyltetrazolium chloride (TTC) and then incubated in TTC for 30 min at 37°C. Hearts were fixed in 10% formaldehyde, and cross-sections were imaged. Infarct size was calculated as a percentage of total cross-sectional area, and for each heart, the mean value was determined from six separate images.
2.3 Western blotting
Heart was homogenized and lysates prepared as previously described.26 Proteins were resolved by SDS-PAGE, transferred to nitrocellulose membranes (Invitrogen) and incubated overnight with antibodies against cyclophilin D (ab110324, Abcam), ATP5A (ab14748, Abcam), ATP5O (sc-365162, Santa Cruz), VDAC1 (sc-8828, Santa Cruz). Membranes were then incubated with secondary antibodies conjugated with HRP (1:10 000, Santa Cruz) for 1 h at room temperature, ECL reagents were added (RPN2232, Amersham ECL Prime, GE Healthcare Life Sciences), and chemiluminescence was measured using an Amersham Imager 600 (GE Healthcare Life Sciences).
2.4 Cardiac mitochondria isolation
Cardiac mitochondria were isolated by standard differential centrifugation. First, hearts were minced in ice-cold mitochondria isolation buffer of the following composition (mmol/L): 225 mannitol, 75 sucrose, 5 MOPS, 0.5 EGTA, 2 taurine (pH 7.25), and homogenized briefly on ice by a Polytron homogenizer (Ultra Turrax T25, IKA Labortechnik). To digest contractile proteins, trypsin was added (0.1 mg per 100 mg wet tissue, T1426, Sigma) to the homogenate for 5 min on ice. Trypsin digestion was stopped upon addition of 0.5 mL of mitochondria isolation buffer supplemented with 0.2% fatty-acid free bovine serum albumin. Any remaining tissue chunks were briefly homogenized by hand with a Teflon pestle. The homogenate was centrifuged at 500 g for 5 min and the resulting supernatant was then spun at 11 000 g for 5 min to pellet the mitochondria. All procedures were performed at 4°C. Protein was quantified using the Bradford assay (23200, Thermo Scientific).
2.5 Calcium retention capacity and swelling assays
Calcium-induced mitochondrial swelling was measured as a decrease in absorbance at 540 nm using a microplate reader (FLUOstar Omega, BMG Labtech). Isolated cardiac mitochondria were resuspended (100 µg in 200 µL) in physiological buffer of the following composition (mmol/L): 120 KCl, 10 Tris–HCl, 5 MOPS, 5 KH2PO4, 10 glutamate, 5 malate, and 0.01 EGTA (pH 7.4). A bolus of 250 μmol/L CaCl2 was added, and absorbance at 540 nm was recorded continuously. Calcium retention capacity was examined by using the fluorescent Ca2+ indicator Calcium Green-5N (1 µmol/L, C3737, Molecular Probes), which measures extramitochondrial calcium. Mitochondria were first depleted of their endogenous calcium with a 5 min incubation in depletion buffer of the following composition (mmol/L): 125 KCl, 20 HEPES, 15 NaCl, 5 MgCl2, 1 K2EDTA, 1 EGTA, 2 KH2PO4, 0.1 malate (pH 7.1).27 Subsequently, mitochondria were resuspended in physiological buffer (100 µg in 200 µL, see above) and 10–25 µmol/L CaCl2 were repeatedly added. In some experiments, mitochondria were treated with Cyclosporine-A (1 µmol/L, 30024, Sigma).
2.6 In vitro oxidation of cardiac mitochondria and immunoprecipitation of the ATP synthase
After isolation of cardiac mitochondria, 250–500 µg were pelleted and resuspended in physiological buffer of the following composition (mmol/L): 120 KCl, 10 Tris–HCl, 5 MOPS, 5 KH2PO4 (pH 7.4) without glutamate/malate at 3–5 mg/mL. Phenylarsenide (P3075, Sigma) was added at final concentration of 100 µmol/L and an incubation followed for 10 min at room temperature. Then the mitochondria were pelleted and resuspended in IP buffer of the following composition (mmol/L): 150 NaCl, 250 HEPES (pH 7.4), supplemented with protease/phosphatase inhibitors (HALT cocktail 78440, Thermo Fisher Scientific). To solubilize mitochondrial membranes 1.6 g n-dodecyl-β-d-maltopyranoside (D4641, Sigma) per g protein was added, and an incubation on ice for 30 min followed. Samples were then centrifuged at 21 000 g for 10 min at 4°C, and supernatants were incubated overnight at 4°C with an antibody against the ATP subunit O (1:200, sc-365162, Santa Cruz). Samples were incubated the following day with pre-washed magnetic beads coupled to protein G (50–100 μL Dynabeads Protein G, Invitrogen) for 1 h at room temperature. The beads were washed five times with IP buffer supplemented with 0.05% n-dodecyl-β-D-maltopyranoside, and eluted with 2× LDS sample buffer supplemented with 10% β-mercaptoethanol (Invitrogen NuPAGE) by incubating at room temperature for 10 min. The eluates were then analysed by western blot and probing with anti-cyclophilin D (ab110324, Abcam) and anti-ATP5O (sc-365162, Santa Cruz), or anti-ATP5A (ab14748, Abcam). CypD signal was normalized to the signal of ATP5O or ATP5A in each lane.
2.7 Acyl-resin-assisted-capture (Acyl-RAC) of S-acylated cysteines
To assess acylation of C202, we employed a resin-assisted capture.28 Murine hearts were perfused for 20 min. Subsequently, mitochondria were isolated as described above. Mitochondrial pellets of 150–500 µg were resuspended into HE-buffer of the following composition (mmol/L): 150 NaCl, 250 HEPES, 1 EDTA, pH 7.5 with NaOH, protease and phosphatase inhibitors (HALT cocktail 78440, Thermo Fisher Scientific), supplemented with 1% SDS and 40 mmol/L N-ethylmaleimide (04259, Sigma). N-ethylmaleimide was used to block free cysteines, and an incubation at 50°C for 30 min followed. The samples were then subjected to a de-salting column (Zeba spin desalting columns, 7K MWCO 89883, Thermo Fisher Scientific) to remove excess SDS and N-ethylmaleimide. Thiopropyl-Sepharose 6B (GE Healthcare Life Sciences) was prepared according to the instructions of the manufacturer. The samples were then transferred to the resin (8 mg of dry resin per sample) and fresh 250 mmol/L hydroxylamine-HCl (26103, Thermo Scientific, 2.5 mol/L stock, pH adjusted to 7 with NaOH) or 250 mmol/L NaCl (negative control) were added. An incubation at room temperature for 2 h followed. Hydroxylamine removes fatty acids from cysteines, which can be subsequently captured by the sepharose resin. After the incubation period, the resin was washed four times with HE-buffer supplemented with 1% SDS, and four times with HE-buffer. Elution of proteins captured by the resin was performed with 60 μL of 2× LDS sample buffer supplemented with 10% β-mercaptoethanol (Invitrogen NuPAGE) at 70°C for 10 min. The eluates were then analysed by western blotting with anti-cyclophilin D (ab110324, Abcam).
2.8 In vitro S-palmitoylation of cyclophilin D
The assay was performed as described previously.29 Purified wild-type (WT) human cyclophilin D was obtained from Abnova, CA, USA. Aliquots of 250 nmol/L were made into buffer of the following composition (mmol/L): 120 NaCl, 40 Tris, 8 HEPES, pH 8.0. Subsequently, 2 μmol/L palmitoyl-CoA (P9716, Sigma) was added and the samples were incubated at 30°C for 1 h. DMSO 0.1% served as vehicle control.
2.9 Cyclophilin D peptidyl-prolyl isomerase activity
The assay was performed as described previously.18,25 In glass cuvettes (10 × 10 mm) were added 10 nmol/L purified cyclophilin D (Abnova, stock 250 nmol/L) and 6 mg/mL α-chymotrypsin (C4129, Sigma, stock 60 mg/mL in 0.001% HCl) at a final volume of 1 mL. The cuvette was placed in a Nanodrop 2000C (Thermo Fisher Scientific) with stirring, and 75 nmol/L of N-Succinyl-Ala-Ala-Pro-Phe p-nitroanilide (S7388, Sigma, stock in trifluoroethanol supplemented with 0.47 mol/L LiCl) were added. Absorbance was measured at 380 nm every 2 s over a period of 3 min. Data were fitted to a first-order equation (A = A0 − A1e−kt, with k as rate constant) and rate constants (kobs) were obtained. The kcat/km values were calculated according to the equation kcat/km = (kobs − k0)/[CypD], where k0 is the first-order rate constant for spontaneous cis-trans isomerization.
2.10 In vitro treatment of cardiac mitochondria and assessment of S-acylation of CypD
Cardiac mitochondria were isolated from WT C57BL6/N mice as previously described and 150–250 μg were incubated in physiological buffer with phenylarsenide (100 μmol/L), CCCP (2 μmol/L), or CaCl2 (500 μmol/g mitochondria) for 5 min at room temperature. At the end of the incubation, the samples were centrifuged at 11 000 g for 2 min and S-acylation was assessed with Acyl-RAC, as described above.
2.11 In vitro crosslinking of cardiac mitochondria and assessment of binding partners of CypD
Cardiac mitochondria were isolated from WT C57BL6/N mice as previously described and 80 µg were incubated in physiological buffer with CaCl2 (500 μmol/g mitochondria) for 10 min at room temperature. At the end of the incubation fresh prepared disuccinimidyl suberate (DSS 21555, Thermo Scientific) was added at a final concentration of 3 mmol/L and the samples incubated for 30 min at room temperature. The crosslinking reaction was then quenched with 20 mmol/L Tris incubated for 15 min at room temperature. An aliquot of each sample (10 µg) was then resolved by SDS-PAGE and western blotting as described above.
2.12 Statistical analysis
Data are reported as mean ± SEM. Student’s t-test and two-way ANOVA with Sidak’s correction for multiple comparisons were used for statistical analysis. Differences were considered significant at the level of P < 0.05. All statistical analyses and curve-fitting were performed with Prism, version 7.02 (GraphPad).
3. Results
3.1 Cypd C202S protects against I/R injury
We first investigated whether C202 of CypD plays a role in I/R injury. To accomplish this, we mutated C202 to a serine and subjected Langendorff perfused hearts to global ischaemia for 20 min followed by 90 min of reperfusion. The recovery of contractile function (RPP) at reperfusion was improved and infarct size was attenuated in CypD C202S mice compared to their WT littermates (Figure 1A), suggesting that C202 of CypD plays a role in regulating I/R injury.
Figure 1.

CypD C202S mice are cardioprotected. (A) Infarct size assessment by TTC; unpaired two-tailed Student’s t-test, n = 6 mice per group. (B) Representative microphotograph of infarcted heart tissue appearing white after TTC staining. (C) Rate pressure product recovery at reperfusion; unpaired two-tailed Student’s t-test, n = 6 mice per group. (D) CypD C202S expression level was similar to WT in heart lysates; unpaired two-tailed Student’s t-test, n = 8 mice per group. (E) Representative western blot of CypD expression level in heart lysates, VDAC1 was used as loading control. CypD, cyclophilin D; KI, CypD C202S; LVDP, left ventricular developed pressure; VDAC, voltage dependent anion channel; WT, wild type.
3.2 Cypd C202S desensitizes the permeability transition pore to calcium
The protection observed in CypD C202S hearts could be due to a reduction in the level of CypD in these hearts. To test this, we assessed CypD C202S expression levels in heart lysates using western blot and found that the expression level did not differ in comparison to the WT CypD (Figure 1B and C).
We next tested whether cardiac mitochondria from CypD C202S-KI mice exhibited altered sensitivity to the PTP. We assessed calcium retention capacity, a measure of PTP opening in mitochondria from WT and CypD C202S hearts. Consistent with the protection against I/R injury, isolated cardiac mitochondria from CypD C202S mice exhibited higher calcium retention capacity compared to WT (Figure 2A and C). We further evaluated whether mutant CypD conferred additional protection to the known PTP desensitizer cyclosporine A. In the presence of 1 μmol/L cyclosporine A, CypD C202S showed calcium retention capacity similar to WT (Figure 2B and C). Notably, the calcium retention capacity was increased when mitochondria from CypD C202S mice were treated with cyclosporine A. To test whether this is mirrored in an increased protection against infarction we treated WT and CypD C202S mice with 0.2 μmol/L cyclosporine A for 5 min before the onset of ischaemia to 5 min after onset of reperfusion (Figure 2D and E). In contrast with isolated mitochondria and WT hearts, CypD C202S hearts did not exhibit additional cardioprotection when treated with cyclosporine A. This might suggest that PTP regulation differs between isolated mitochondria and an ex vivo perfused heart or that there is not enough dynamic range to resolve the additional protection by cyclosporine A in these hearts.
Figure 2.

Cardiac mitochondria from CypD C202S mice exhibit higher calcium retention capacity compared to wild type; representative traces of calcium retention capacity assays in cardiac mitochondria without (A) and with (B) 1 μmol/L cyclosporine A; calcium retention capacity was measured using calcium-sensitive probe calcium green-5N. (C) Summary of calcium retention capacity per genotype and treatment. Infarct size assessment with TTC (D) and rate pressure product recovery at reperfusion (E) in hearts perfused with 0.2 μmol/L cyclosporine A or DMSO control. Two-way ANOVA with Sidak’s post hoc test, n = 5 mice per group for panels C–E. CsA, cyclosporine A; CypD, cyclophilin D; RFU, relative fluorescence units; WT, wild type.
Previous studies showed that mutation of C202 did not alter the isomerase activity of CypD,18 thus it was unlikely that the reduced PTP observed in the CypD mutant mitochondria was due to loss of isomerase activity.30 We therefore investigated whether mutation of CypD altered the targeting of CypD to the F1F0-ATP-synthase, which has been proposed as the PTP.
3.3 Cypd C202S binds less to proposed PTP component ATP-synthase under oxidative stress
Previous studies have shown that cysteine 202 is the primary residue of CypD that undergoes oxidation during I/R injury.18,21 Oxidative stress has been reported to enhance CypD binding to the mitochondrial membrane12,13 where it activates PTP. To test if C202 is important in targeting CypD, cardiac mitochondria were subjected to the oxidizing agent phenylarsenide and the binding of CypD to the proposed PTP component, the F1F0-ATP-synthase was evaluated. Following phenylarsenide addition, CypD C202S exhibited less association with the ATP synthase compared to WT CypD (Figure 3). The data are consistent with the hypothesis that oxidation of cysteine 202 promotes its binding to proposed PTP components. Mutating the cysteine to a serine which cannot undergo oxidation attenuates its binding to PTP and confers protection against I/R injury. These data are consistent with the hypothesis that oxidation of C202 plays a role in targeting CypD to the PTP.
Figure 3.
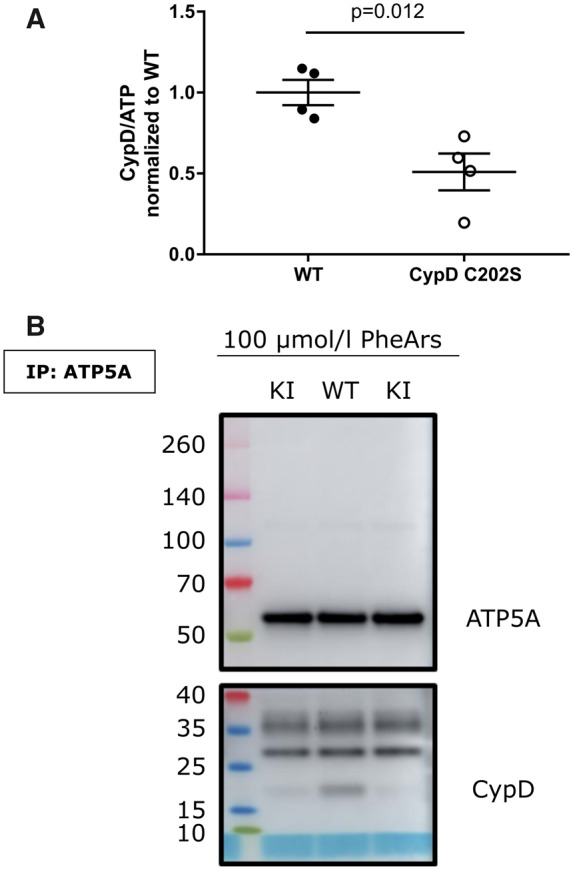
CypD C202S exhibits reduced binding to the proposed PTP component ATP-synthase under oxidative stress. (A) Immunoprecipitation of ATP-synthase in de-energized and oxidized mitochondria; the outputs were assessed for cyclophilin D and the ATP-synthase subunits alpha and O; unpaired two-tailed Student’s t-test, n = 4 mice per group. (B) Representative western blot of the outputs, one mouse for WT and two mice for KI. ATP5A, ATP-synthase subunit alpha; CypD, cyclophilin D; KI, CypD C202S; PheArs, phenylarsenide; WT, wild type.
3.4 Cysteine 202 is the main S-acylation site of CypD
In a previous study, we found CypD to be S-nitrosylated.21 A number of studies have shown cross-talk between S-nitrosylation and S-acylation.31–33 Cysteine 202 matches an S-acylation site motif found commonly in soluble proteins (-C–L-).24 To assess S-acylation of cysteine 202 we employed a resin-assisted capture. We compared S-acylation of WT hearts immediately following removal from the mouse to S-acylation in Langendorff perfused mouse hearts. We found that S-acylation is not altered by 60 min of perfusion. We also compared S-acylation between WT and CypD-KI mice and found that at baseline, CypD C202S hearts exhibits significantly less acylation compared to WT (Figure 4A). WT CypD shows two-fold more S-acylation compared to CypD C202S, suggesting that C202 is a major site of acylation.
Figure 4.

Assessment of CypD S-acylation in perfused murine hearts under baseline conditions and at the end of ischaemia. S-acylation of CypD was assessed by a resin-assisted capture (Acyl-RAC) that is based on the capture of S-acylated cysteines by a sepharose resin. Inputs represent samples before the Acyl-RAC was run and outputs represent the sample following the Acyl-RAC which is enriched in S-acylated proteins. (A) CypD C202S mice exhibit less S-acylation of CypD under baseline perfusion compared to wild type; unpaired two-tailed Student’s t-test, n = 5 mice per group; representative western blots. (B) CypD S-acylation decreases after 20 min of global ischaemia in wild-type hearts; unpaired two-tailed Student’s t-test, n = 4 mice per group; representative western blots. CypD, cyclophilin D; HAM, hydroxylamine; Isch, ischaemia; Perf, perfusion; pos.ctrl, positive control for the western blot; WT, wild type.
Previous studies suggest that S-acylation can affect enzyme activity34–37 and there is evidence that the opening of PTP depends on the isomerase activity of CypD.3 To determine whether S-acylation affects the cis-trans isomerase activity of CypD, purified WT CypD was S-palmitoylated non-enzymatically in vitro, then its catalytic activity was measured. The effectiveness of S-palmitoylation was assessed using Acyl-RAC. Incubation of CypD with palmitoyl-CoA resulted in a 6.7-fold increase of S-acylation (Figure 5A). The catalytic activity of S-palmitoylated CypD was measured and did not differ from that of non-palmitoylated CypD (Figure 5B).
Figure 5.

(A) Purified CypD (obtained from Abnova) can be S-palmitoylated non-enzymatically in-vitro; unpaired two-tailed Student’s t-test, n = 8 independent replicates per group (each replicate represents an independent reaction) using two different stocks of purified CypD, performed on four different days; representative western blot. S-acylation of CypD was assessed by a resin-assisted capture (Acyl-RAC) that is based on the capture of S-acylated cysteines by a sepharose resin. Inputs represent samples before the Acyl-RAC was run and outputs represent the sample following the Acyl-RAC which is enriched in S-acylated proteins. (B) S-palmitoylation of CypD does not affect its isomerase activity; representative trace and summary of data. CypD isomerase activity was assessed spectrophotometrically; two-way ANOVA with Sidak’s post hoc test, n = 5 independent replicates per group (each replicate represents an independent reaction of in vitro palmitoylation). CsA, cyclosporine A; DMSO blank, spontaneous cis-trans isomerization without CypD; PCoA, palmitoyl-CoA.
3.5 Cypd C202 is de-acylated during ischaemia
To assess whether CypD acylation is altered during I/R, Langendorff perfused murine hearts were subjected to baseline perfusion or I/R and CypD S-acylation was assessed. In WT hearts CypD acylation was significantly reduced following ischaemia compared to baseline perfusion (Figure 4B). Notably, CypD C202S mice cannot undergo oxidation or S-acylation at cysteine 202, and there was no change in acylation in the hearts during I/R.
We were interested in examining the mechanism responsible for the loss of S-acylation of C202 during ischaemia in the WT hearts. We considered that perhaps the increase in calcium or uncoupling during I/R might enhance de-acylation at C202. To test this hypothesis, we incubated isolated mitochondria with high calcium or the uncoupler carbonyl cyanide m-chlorophenyl hydrazine (CCCP). Addition of 500 μmol calcium per g of mitochondria which exceeds the calcium retention capacity for WT mitochondria (Figure 2) and triggers PTP opening resulted in a 37% decrease in S-acylation. Thus, the addition of calcium leads to de-acylation of CypD to levels similar to those observed following ischaemia (Figure 6A). We also tested whether a decrease in mitochondrial membrane potential might stimulate de-acylation. CCCP addition did not alter levels of S-acylation of C202 (Figure 6B). To further test the hypothesis that calcium regulates S-acylation we measured S-acylation of CypD in total non-perfused heart lysates from global germline mitochondrial calcium uniporter knock-out (MCU-KO) mice, which have reduced mitochondrial calcium,38 and we find an increase in S-acylation of CypD (Figure 7A). The data would be consistent with the hypothesis that a free C202 is needed for CypD to activate PTP. We hypothesized that with ischaemia the increase in mitochondrial calcium leads to de-acylation of C202 of CypD making it available for interaction with the PTP. To further test this hypothesis, we tested whether addition of 500 μmol calcium per g mitochondria would result in a change in CypD binding partners. We show that if we add a crosslinker to WT cardiac mitochondria there is association of CypD with many high molecular weight proteins. If we add a bolus of calcium that triggers PTP opening (500 μmol/g mitochondria) and then add the crosslinker we see that calcium reduces the association of CypD with these proteins as indicated by an increase in CypD at 20 kDa (Figure 7B).
Figure 6.

(A) Calcium resulted in a 37% decrease in S-acylation of CypD; unpaired two-tailed Student’s t-test, n = 9 mice per group, representative western blot. (B) CCCP addition did not alter levels of S-acylation of CypD; unpaired two-tailed Student’s t-test, n = 7 mice per group, representative western blot. For (A) and (B) S-acylation of CypD was assessed by a resin-assisted capture (Acyl-RAC) that is based on the capture of S-acylated cysteines by a sepharose resin. Inputs represent samples before the Acyl-RAC was run and outputs represent the sample following the Acyl-RAC which is enriched in S-acylated proteins. CCCP, Carbonyl cyanide m-chlorophenyl hydrazone; Ctrl, controls; CypD, cyclophilin D; HAM, hydroxylamine; WT, wild type.
Figure 7.

(A) Global germline MCU-KO mice exhibit higher levels of CypD S-acylation compared to WT in non-perfused total heart lysates; unpaired two-tailed Student’s t-test, n = 6 mice per group; representative western blot. S-acylation of CypD was assessed by a resin-assisted capture (Acyl-RAC) that is based on the capture of S-acylated cysteines by a sepharose resin. Outputs represent the sample following the Acyl-RAC which is enriched in S-acylated proteins. (B) Isolated cardiac wild-type mitochondria were treated without/with calcium, crosslinked with DSS and subjected to western blot. Treatment of WT cardiac mitochondria with calcium results in disassociation from its binding partners; unpaired two-tailed Student’s t-test, n = 6 mice per group. AU, arbitrary units; CypD, cyclophilin D; HAM, hydroxylamine; MCU, mitochondrial calcium uniporter; WT, wild type.
4. Discussion
CypD is a well-established regulator of PTP. In the present study, we show that C202 acts as an integrator of cell-death signalling; its post-translational modifications such as S-nitrosylation, oxidation, and S-acylation are modulators of the PTP. The mutation of C202 to a serine is cardioprotective, desensitizes PTP, and leads to reduced CypD binding to the proposed PTP component ATP-synthase following oxidative stress. CypD C202 has been identified as a redox-sensitive residue.18 We have previously demonstrated that CypD C202 undergoes S-nitrosylation in ischaemic preconditioning, a cardioprotective manoeuvre which consists of short non-lethal I/R cycles before prolonged ischaemia. We propose that S-nitrosylation contributes to cardioprotection by shielding C202 of CypD from oxidation.25
To further study the importance of CypD C202 in I/R injury we developed a global C202S CypD knock-in mouse in a C57BL6/N background using CRISPR. Mice with this KI mutation (CypD C202S) exhibit reduced I/R injury in the isolated perfused heart model. The protection observed is not due to attenuated levels of the mutant CypD, since the expression of CypD C202S is similar to the expression of the WT CypD. Isolated cardiac mitochondria from CypD C202S mice exhibit a higher calcium retention capacity compared to WT consistent with desensitization of PTP opening by calcium, suggesting a role for C202 in modulating PTP opening.
The F1F0-ATP-synthase has recently been proposed as a core PTP component.9,11 Oxidative stress has been reported to promote the translocation of CypD to the inner mitochondrial membrane and proposed PTP components.9,12,39 Consistent with this hypothesis we found that oxidation of C202 promotes the binding of CypD to the ATP-synthase. In CypD C202S mice, the serine at residue 202 cannot undergo oxidation, possibly limiting the translocation of CypD to PTP, thus de-sensitizing it and conferring protection.
Cysteine residues can also undergo S-acylation and CypD C202 matches an S-acylation motif.24,40 Previous studies have shown that S-nitrosylation and S-acylation can reciprocally regulate the targeting of proteins31,32 and thus we were interested in assessing the role of S-acylation of CypD in I/R injury. The assessment of S-acylation requires enrichment which is typically achieved with the Acyl-RAC or an acyl-biotin-switch method.40 Both methods are based on the use of hydroxylamine to release fatty acids from cysteine residues which can then bind to the resin (Acyl-RAC) or to biotin (biotin switch). WT hearts are abundantly S-acylated on CypD C202 under baseline conditions indicating that S-acylation on C202 per se does not lead to PTP opening. CypD C202S knock-in hearts are protected from I/R injury suggesting further that lack of CypD S-acylation at C202 is not detrimental if the C is mutated to a S, and does not induce PTP opening. However, we find that ischaemia leads to de-acylation of C202 and it appears that an increase in calcium promotes de-acylation.
Consistent with a role for mitochondrial calcium in regulating levels of CypD acylation, global germline CD1 MCU-KO mice which have reduced mitochondrial calcium compared to WT38 exhibit higher amount of S-acylated CypD. However, these MCU-KO mice are not protected from I/R injury likely due to other adaptations that occur in these mice. For example, we have shown previously that their CypD-mediated regulation of PTP is altered because of increased phosphorylation at serine 42 of CypD.26
These findings are reminiscent of the role of palmitoylation in regulating eNOS function. Palmitoylation targets eNOS to caveolae where interaction with caveolin which inhibits eNOS.41 However, eNOS targeting to caveolae is needed for proper functioning of eNOS.42,43 Loss of eNOS palmitoylation and targeting to caveolae alters the availability of substrates and cofactors and thereby alters eNOS generation of NO. Agonists activation of calcium-calmodulin leads to activation and depalmitoylation of eNOS.44 Thus, although palmitoylation targets eNOS to caveolin which inhibits its activity, it also appears to target it to the proper location to undergo activation by calcium-calmodulin which results in depalmitoylation.
Taken together the data suggest that mutation of C202 to a serine reduced PTP opening and is cardioprotective. These data are consistent with the hypothesis that a free cysteine is needed to target CypD to the PTP. It is possible that further oxidation of this cysteine is involved in targeting CypD to the PTP. During ischaemia, the rise in mitochondrial calcium leads to depalmitoylation which following oxidative stress leads to CypD oxidation and activation of PTP. If an NO donor is present at the time of depalmitoylation, S-nitrosylation of the C202 can occur which blocks oxidation and targeting to the PTP. Consistent with this hypothesis Brookes et al.45 showed that addition of an NO donor blocks or attenuates calcium-induced PTP opening. Thus, calcium activated PTP opening is blocked in the presence of an NO donor which can S-nitrosylate C202 as it becomes deacylated. Overall, our findings suggest that C202 of CypD plays an important role in mediating cell-death as its post-translational modifications are potent regulators of I/R injury.
Authors’ contributions
G.A.: acquisition, analysis, and interpretation of data, drafting of manuscript. J.S.: acquisition, analysis, and interpretation of data. M.F.: acquisition, analysis, and interpretation of data. S.M.: acquisition, analysis of data. C.L.: analysis and interpretation of data. J.M.: interpretation of data. E.M.: study design, interpretation of data, drafting of manuscript.
Conflict of interest: none declared.
Funding
This work was supported by intramural funding from the National Heart Lung and Blood Institute at the National Institutes of Health [ZIA HL002066 and ZIA HL006059 to E.M.]; and Foundation Leducq [6CVD04 to E.M. and J.D.M.].
Time for primary review: 28 days
Translational perspective
In this study, we demonstrated cysteine 202 of CypD undergoes multiple post-translational modifications that regulate its ability to activate permeability transition pore (PTP). We provide novel data demonstrating acylation of CypD and show that acylation and SNO compete for modification of C202. Together these novel data suggest that CypD integrates signals to regulate the PTP and cell death.
References
- 1. Griffiths EJ, Halestrap AP.. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 1995;307:93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Petronilli V, Penzo D, Scorrano L, Bernardi P, Di Lisa F.. The mitochondrial permeability transition, release of cytochrome c and cell death—correlation with the duration of pore openings in situ. J Biol Chem 2001;276:12030–12034. [DOI] [PubMed] [Google Scholar]
- 3. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD.. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005;434:658–662. [DOI] [PubMed] [Google Scholar]
- 4. Baines CP. The mitochondrial permeability transition pore as a target of cardioprotective signaling. Am J Physiol Heart Circ Physiol 2007;293:H903–H904. [DOI] [PubMed] [Google Scholar]
- 5. Bernardi P, Petronilli V.. The permeability transition pore as a mitochondrial calcium release channel: a critical appraisal. J Bioenerg Biomembr 1996;28:131–138. [DOI] [PubMed] [Google Scholar]
- 6. Halestrap AP. What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 2009;46:821–831. [DOI] [PubMed] [Google Scholar]
- 7. Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- 8. Dhingra R, Guberman M, Rabinovich-Nikitin I, Gerstein J, Margulets V, Gang H, Madden N, Thliveris J, Kirshenbaum LA.. Impaired NF-kappaB signalling underlies cyclophilin D-mediated mitochondrial permeability transition pore opening in doxorubicin cardiomyopathy. Cardiovasc Res 2020;116:1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Giorgio V, Bisetto E, Soriano ME, Dabbeni-Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G.. Cyclophilin D modulates mitochondrial F0F1-ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem 2009;284:33982–33988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bernardi P, Rasola A, Forte M, Lippe G.. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev 2015;95:1111–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bonora M, Morganti C, Morciano G, Pedriali G, Lebiedzinska-Arciszewska M, Aquila G, Giorgi C, Rizzo P, Campo G, Ferrari R, Kroemer G, Wieckowski MR, Galluzzi L, Pinton P.. Mitochondrial permeability transition involves dissociation of F1FO ATP synthase dimers and C-ring conformation. EMBO Rep 2017;18:1077–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Connern CP, Halestrap AP.. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem J 1994;302:321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nicolli A, Basso E, Petronilli V, Wenger RM, Bernardi P.. Interactions of cyclophilin with the mitochondrial inner membrane and regulation of the permeability transition pore, a cyclosporin A-sensitive channel. J Biol Chem 1996;271:2185–2192. [DOI] [PubMed] [Google Scholar]
- 14. Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX.. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 1989;337:476–478. [DOI] [PubMed] [Google Scholar]
- 15. Bochaton T, Crola-Da-Silva C, Pillot B, Villedieu C, Ferreras L, Alam MR, Thibault H, Strina M, Gharib A, Ovize M, Baetz D.. Inhibition of myocardial reperfusion injury by ischemic postconditioning requires sirtuin 3-mediated deacetylation of cyclophilin D. J Mol Cell Cardiol 2015;84:61–69. [DOI] [PubMed] [Google Scholar]
- 16. Garcia-Dorado D, Ruiz-Meana M, Inserte J, Rodriguez-Sinovas A, Piper HM.. Calcium-mediated cell death during myocardial reperfusion. Cardiovasc Res 2012;94:168–180. [DOI] [PubMed] [Google Scholar]
- 17. Chernyak BV. Redox regulation of the mitochondrial permeability transition pore. Biosci Rep 1997;17:293–302. [DOI] [PubMed] [Google Scholar]
- 18. Linard D, Kandlbinder A, Degand H, Morsomme P, Dietz K-J, Knoops B.. Redox characterization of human cyclophilin D: identification of a new mammalian mitochondrial redox sensor? Arch Biochem Biophys 2009;491:39–45. [DOI] [PubMed] [Google Scholar]
- 19. Kowaltowski AJ, Castilho RF, Vercesi AE.. Opening of the mitochondrial permeability transition pore by uncoupling or inorganic phosphate in the presence of Ca2+ is dependent on mitochondrial-generated reactive oxygen species. FEBS Lett 1996;378:150–152. [DOI] [PubMed] [Google Scholar]
- 20. Bibli SI, Papapetropoulos A, Iliodromitis EK, Daiber A, Randriamboavonjy V, Steven S, Brouckaert P, Chatzianastasiou A, Kypreos KE, Hausenloy DJ, Fleming I, Andreadou I.. Nitroglycerine limits infarct size through S-nitrosation of cyclophilin D: a novel mechanism for an old drug. Cardiovasc Res 2019;115:625–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kohr MJ, Sun J, Aponte A, Wang G, Gucek M, Murphy E, Steenbergen C.. Simultaneous measurement of protein oxidation and S-nitrosylation during preconditioning and ischemia/reperfusion injury with resin-assisted capture. Circ Res 2011;108:418–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Roy S, Plowman S, Rotblat B, Prior IA, Muncke C, Grainger S, Parton RG, Henis YI, Kloog Y, Hancock JF.. Individual palmitoyl residues serve distinct roles in H-ras trafficking, microlocalization, and signaling. Mol Cell Biol 2005;25:6722–6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee H, Woodman SE, Engelman JA, Volonte D, Galbiati F, Kaufman HL, Lublin DM, Lisanti MP.. Palmitoylation of caveolin-1 at a single site (Cys-156) controls its coupling to the c-Src tyrosine kinase: targeting of dually acylated molecules (GPI-linked, transmembrane, or cytoplasmic) to caveolae effectively uncouples c-Src and caveolin-1 (TYR-14). J Biol Chem 2001;276:35150–35158. [DOI] [PubMed] [Google Scholar]
- 24. Collins MO, Woodley KT, Choudhary JS.. Global, site-specific analysis of neuronal protein S-acylation. Sci Rep 2017;7:4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nguyen TT, Stevens MV, Kohr M, Steenbergen C, Sack MN, Murphy E.. Cysteine 203 of cyclophilin D is critical for cyclophilin D activation of the mitochondrial permeability transition pore. J Biol Chem 2011;286:40184–40192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parks RJ, Menazza S, Holmstrom KM, Amanakis G, Fergusson M, Ma H, Aponte AM, Bernardi P, Finkel T, Murphy E.. Cyclophilin D-mediated regulation of the permeability transition pore is altered in mice lacking the mitochondrial calcium uniporter. Cardiovasc Res 2019;115:385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Territo PR, French SA, Dunleavy MC, Evans FJ, Balaban RS.. Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, NADH, AND light scattering. J Biol Chem 2001;276:2586–2599. [DOI] [PubMed] [Google Scholar]
- 28. Forrester MT, Hess DT, Thompson JW, Hultman R, Moseley MA, Stamler JS, Casey PJ.. Site-specific analysis of protein S-acylation by resin-assisted capture. J Lipid Res 2011;52:393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Scholich K, Yigzaw Y, Patel TB.. Cysteine 3 is not the site of in vitro palmitoylation on G(salpha). Biochem Biophys Res Commun 2000;270:131–136. [DOI] [PubMed] [Google Scholar]
- 30. Scorrano L, Nicolli A, Basso E, Petronilli V, Bernardi P.. Two modes of activation of the permeability transition pore: the role of mitochondrial cyclophilin. Mol Cell Biochem 1997;174:181–184. [PubMed] [Google Scholar]
- 31. Hess DT, Patterson SI, Smith DS, Skene JH.. Neuronal growth cone collapse and inhibition of protein fatty acylation by nitric oxide. Nature 1993;366:562–565. [DOI] [PubMed] [Google Scholar]
- 32. Ho GP, Selvakumar B, Mukai J, Hester LD, Wang Y, Gogos JA, Snyder SH.. S-nitrosylation and S-palmitoylation reciprocally regulate synaptic targeting of PSD-95. Neuron 2011;71:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zareba-Koziol M, Bartkowiak-Kaczmarek A, Figiel I, Krzystyniak A, Wojtowicz T, Bijata M, Wlodarczyk J.. Stress-induced changes in the S-palmitoylation and S-nitrosylation of synaptic proteins. Mol Cell Proteomics 2019;18:1916–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chaube R, Hess DT, Wang YJ, Plummer B, Sun QA, Laurita K, Stamler JS.. Regulation of the skeletal muscle ryanodine receptor/Ca2+-release channel RyR1 by S-palmitoylation. J Biol Chem 2014;289:8612–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Holland SM, Collura KM, Ketschek A, Noma K, Ferguson TA, Jin Y, Gallo G, Thomas GM.. Palmitoylation controls DLK localization, interactions and activity to ensure effective axonal injury signaling. Proc Natl Acad Sci USA 2016;113:763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McMichael TM, Zhang L, Chemudupati M, Hach JC, Kenney AD, Hang HC, Yount JS.. The palmitoyltransferase ZDHHC20 enhances interferon-induced transmembrane protein 3 (IFITM3) palmitoylation and antiviral activity. J Biol Chem 2017;292:21517–21526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ni J, Qu L, Yang H, Wang M, Huang Y.. Palmitoylation and its effect on the GTPase-activating activity and conformation of RGS2. Int J Biochem Cell Biol 2006;38:2209–2218. [DOI] [PubMed] [Google Scholar]
- 38. Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T.. The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 2013;15:1464–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Connern CP, Halestrap AP.. Chaotropic agents and increased matrix volume enhance binding of mitochondrial cyclophilin to the inner mitochondrial membrane and sensitize the mitochondrial permeability transition to [Ca2+]. Biochemistry 1996;35:8172–8180. [DOI] [PubMed] [Google Scholar]
- 40. Edmonds MJ, Geary B, Doherty MK, Morgan A.. Analysis of the brain palmitoyl-proteome using both acyl-biotin exchange and acyl-resin-assisted capture methods. Sci Rep 2017;7:3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Michel JB, Feron O, Sacks D, Michel T.. Reciprocal regulation of endothelial nitric-oxide synthase by Ca2+-calmodulin and caveolin. J Biol Chem 1997;272:15583–15586. [DOI] [PubMed] [Google Scholar]
- 42. Liu J, García-Cardeña G, Sessa WC.. Palmitoylation of endothelial nitric oxide synthase is necessary for optimal stimulated release of nitric oxide: implications for caveolae localization. Biochemistry 1996;35:13277–13281. [DOI] [PubMed] [Google Scholar]
- 43. Shaul PW, Smart EJ, Robinson LJ, German Z, Yuhanna IS, Ying Y, Anderson RG, Michel T.. Acylation targets emdothelial nitric-oxide synthase to plasmalemmal caveolae. J Biol Chem 1996;271:6518–6522. [DOI] [PubMed] [Google Scholar]
- 44. Yeh DC, Duncan JA, Yamashita S, Michel T.. Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca(2+)-calmodulin. J Biol Chem 1999;274:33148–33154. [DOI] [PubMed] [Google Scholar]
- 45. Brookes PS, Salinas EP, Darley-Usmar K, Eiserich JP, Freeman BA, Darley-Usmar VM, Anderson PG.. Concentration-dependent effects of nitric oxide on mitochondrial permeability transition and cytochrome c release. J Biol Chem 2000;275:20474–20479. [DOI] [PubMed] [Google Scholar]