

Abstract
Aims
Heart failure is a major complication in cancer treatment due to the cardiotoxic effects of anticancer drugs, especially from the anthracyclines such as doxorubicin (DXR). DXR enhances oxidative stress and stimulates matrix metalloproteinase-2 (MMP-2) in cardiomyocytes. We investigated whether MMP inhibitors protect against DXR cardiotoxicity given the role of MMP-2 in proteolyzing sarcomeric proteins in the heart and remodelling the extracellular matrix.
Methods and results
Eight-week-old male C57BL/6J mice were treated with DXR weekly with or without MMP inhibitors doxycycline or ONO-4817 by daily oral gavage for 4 weeks. Echocardiography was used to determine cardiac function and left ventricular remodelling before and after treatment. MMP inhibitors ameliorated DXR-induced systolic and diastolic dysfunction by reducing the loss in left ventricular ejection fraction, fractional shortening, and E′/A′. MMP inhibitors attenuated adverse left ventricular remodelling, reduced cardiomyocyte dropout, and prevented myocardial fibrosis. DXR increased myocardial MMP-2 activity in part also by upregulating N-terminal truncated MMP-2. Immunogold transmission electron microscopy showed that DXR elevated MMP-2 levels within the sarcomere and mitochondria which were associated with myofilament lysis, mitochondrial degeneration, and T-tubule distention. DXR-induced myofilament lysis was associated with increased titin proteolysis in the heart which was prevented by ONO-4817. DXR also increased the level and activity of MMP-2 in human embryonic stem cell-derived cardiomyocytes, which was reduced by ONO-4817.
Conclusions
MMP-2 activation is an early event in DXR cardiotoxicity and contributes to myofilament lysis by proteolyzing cardiac titin. Two orally available MMP inhibitors ameliorated DXR cardiotoxicity by attenuating intracellular and extracellular matrix remodelling, suggesting their use may be a potential prophylactic strategy to prevent heart injury during chemotherapy.
Keywords: Matrix metalloproteinases, Cardiotoxicity, Cardiac remodelling, Sarcomere, Doxycycline, Titin
Graphical Abstract
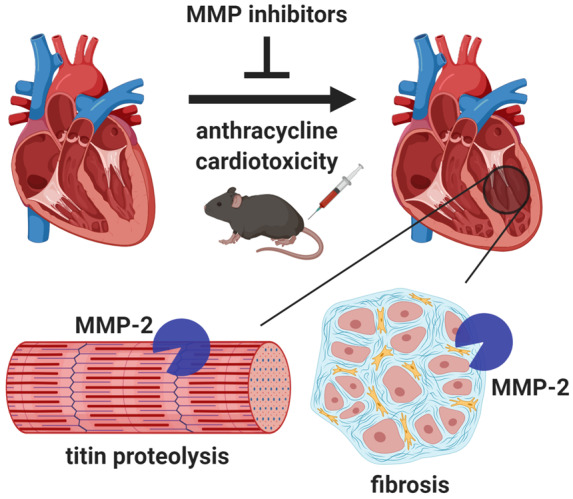
1. Introduction
Anthracyclines including doxorubicin (DXR) are still used to treat over 50% of childhood and adult cancers.1 Paradoxically, improvements in cancer survival have dramatically increased cardiovascular complications as a result of the severe, irreversible cardiotoxic side effects of anthracyclines.2 One in four patients develop asymptomatic left ventricular dysfunction, and 1 in 20 patients develop overt heart failure when taking anthracyclines.3 Anthracycline cardiotoxicity is associated with increased oxidative stress,4 which results in extracellular matrix remodelling5 and degradation of sarcomeric proteins.6 Unfortunately, antioxidant therapy failed to prevent cardiotoxicity.7,8 Prophylactic treatment with β-blockers, angiotensin-converting enzyme inhibitors, or angiotensin II receptor antagonists have shown modest, short-term improvements in ejection fraction, but long-term cardiac remodelling is unabated.9,10 Hence, a better adjuvant therapy is needed to prevent anthracycline cardiotoxicity.
Matrix metalloproteinases (MMPs) are zinc-dependent proteases best recognized for proteolyzing extracellular matrix proteins in angiogenesis, wound healing, cellular proliferation, cancer metastasis, atherosclerosis, and myocardial infarction. It is now known that MMPs also target several non-extracellular matrix substrates both inside and outside the cell. MMP-2 is abundant in cardiomyocytes and localized in discrete subcellular compartments including the sarcomere, cytoskeleton, mitochondria, and nuclei.11 Oxidative stress activates intracellular MMP-2 through distinct mechanisms by: (i) its direct post-translational modification via peroxynitrite-induced S-glutathiolation12 and, (ii) enhancing its transcription13 as well as that of an N-terminal truncated (NTT)-MMP-2.14 Recently, we found that DXR stimulates MMP-2 in isolated cardiomyocytes by enhancing its protein levels and activity and also by inducing de novo expression of NTT-MMP-2.15 When activated, MMP-2 impairs cardiac contractile function by proteolyzing sarcomeric proteins including troponin I,16 myosin light chain-1,17 and titin.18
Titin is a giant ∼3000 kDa myofilament protein which functions as a scaffold for sarcomeric assembly and as a molecular spring in striated muscle cells to regulate both systolic and diastolic function.19 Titin molecules anchor to the Z-disc and M-line of the sarcomere. Titin is composed of both an extensible and highly conserved inextensible regions, which secure the thin and thick filaments near the Z-disc and in the A-band region, respectively. In cardiomyocytes, the extensible I-band region is spliced by ribonucleic acid (RNA) binding motif protein 20 to produce either the N2B or N2BA titin isoforms.20 Alterations in titin isoform expression and titin proteolysis contribute to contractile dysfunction in dilated cardiomyopathy and ischaemic heart injury.18,21,22 In isolated cardiomyocytes, titin proteolysis6 and increased intracellular MMP-2 activity15 are implicated in DXR-induced injury. However, the role of MMP-2 in titin proteolysis in anthracycline cardiotoxicity in vivo is unknown.
MMP inhibitors were first developed as anticancer drugs and have since been shown to have beneficial effects on the heart. MMP inhibitors attenuate myocardial ischaemia-reperfusion injury, which causes marked oxidative stress, by preventing the degradation of sarcomeric proteins including titin.11 Doxycycline (Doxy), used clinically as an MMP inhibitor at sub-antimicrobial dosing,23 reduced adverse left ventricular remodelling in patients with acute myocardial infarction.24 Given the roles of MMP-2 in cardiac remodelling, we determined whether titin is a target of MMP-2 and comparatively evaluated the effect of two distinct MMP inhibitors on cardiac dysfunction and left ventricular intracellular (titin) and extracellular (collagen) remodelling in DXR cardiotoxicity.
2. Methods
All animal experiments were approved by the University of Alberta Institutional Animal Care and Use Committee, in accordance to the Guide to the Care and Use of Experimental Animals published by the Canadian Council on Animal Care (CCAC), and the Guide for Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH, 8th edition, revised 2011).
For any procedures not mentioned below see Supplementary material online.
2.1. DXR cardiotoxicity
DXR (Sigma-Aldrich, St. Louis, MO, USA) was administered to male C57BL/6J mice (Charles River Laboratories, Saint-Constant, QC, Canada) at 8 weeks of age once a week for 4 weeks (6 mg/kg/wk, i.p., cumulative dose of 24 mg/kg). This dose of DXR is clinically relevant as it reaches a therapeutic plasma concentration of 0.05 μM by 24 h.25,26 Beginning on Day 1, control and DXR groups were treated daily with saline or MMP inhibitors 15 mg/kg Doxy (Sigma-Aldrich, St. Louis, MO, USA) or 60 mg/kg ONO-4817 (Ono Pharmaceutical Co., Osaka, Japan), prepared in 2% carboxymethyl cellulose, by oral gavage for 28 days. These MMP inhibitors have been tested at similar doses in different rodent models of disease for 2 weeks without any adverse effects.27–30 The experimental protocol is shown in Supplementary material online, Figure S1. Mice were randomly assigned to control, Doxy, ONO-4817, DXR, DXR + Doxy, or DXR + ONO-4817 groups (n = 10/group).
2.2. Human embryonic stem cell-derived cardiomyocytes
Human embryonic stem cells (hESCs) were purchased from WiCell (H7; WA07) and experiments were approved by the University of Arizona stem cell oversight committee. hESC-derived cardiomyocytes (hESC-CMs) were differentiated using a monolayer method as previously described.31 In brief, Matrigel™-coated 15 cm culture plates (Corning, Corning, NY, USA) were seeded with hESCs at 1.2 × 106 cells per plate and grown for 4 days prior to differentiation. RPMI medium supplemented with B27 minus insulin (A1895601, Thermo Fisher Scientific, Waltham, MA, USA) and 4 µM CHIR-99021 (CT99021, Selleckchem, Houston, TX, USA) was added to the hESCs for 2 days to initiate differentiation. CHIR-99021 was then removed and cells were cultured for 1 day in RPMI supplemented with B27 minus insulin. RPMI medium supplemented with B27 minus insulin and 5 µM IWR-1 (I0161, Sigma-Aldrich) was introduced for 2 days and then cultured with RPMI containing B27 minus insulin for another 2 days. Cultures were maintained in RPMI with B27 plus insulin (17504-044, Thermo Fisher Scientific). hESC-CMs were dissociated with Accutase (STEMCELL Technologies, Vancouver, BC, Canada) and seeded on Matrigel™-coated 3.5 cm culture wells at 2 × 106 cells per well and cultured for 4 days prior to experimentation. hESC-CMs were treated with 1 µM DXR (Sigma-Aldrich) in the presence or absence of 1 µM ONO-4817 for 24 h. The conditioned media and cell lysates were collected as previously described15 for biochemical analysis.
2.3. Echocardiography
Mice were anesthetized with 1.5% isoflurane and in vivo cardiac function was assessed, in a blinded fashion, by transthoracic 2D/M-mode echocardiography using a Vevo 770 high-resolution imaging system with a 30 MHz transducer (VisualSonics, Toronto, ON, Canada). M-mode images were obtained to measure left posterior ventricular wall and interventricular septum thickness. Complete systolic and diastolic parameters were measured at baseline (Day 1) and Day 28 following treatment. On Day 28 following echocardiography, the mice were anesthetized with 240 mg/kg sodium pentobarbital (i.p., Bimeda-MTC Animal Health Inc., Cambridge, ON, Canada). The mice were euthanized by bilateral thoracotomy and the hearts were rapidly excised and exsanguinated with phosphate buffered saline (PBS).
2.4. Histology
Paraffin-embedded ventricular tissue was cut into 4 µm thick sections and mounted onto Superfrost Plus slides. All procedures were performed at room temperature. Haematoxylin and eosin staining of rehydrated ventricular sections was performed using Weigert’s iron haematoxylin set (Sigma-Aldrich). Collagen content in the ventricular sections was stained using picrosirius red solution (Direct Red 80, Sigma-Aldrich). Sections were dipped in 0.2% phosphomolybdic acid for 30 min, then stained in picrosirius red solution for 1 h, rinsed in 0.2% acetic acid, then dehydrated in ethanol before mounting with Permount (Thermo Fisher Scientific). Collagen birefringence from stained ventricular sections was imaged on an Axio Imager M1 polarized light microscope (Zeiss, Oberkochen, Germany). Collagen area fraction was determined by binarizing the images before analysing using ImageJ. Collagen area fraction was determined by dividing the area of collagen by the total tissue area of each image.
2.5. Western blot
The protein concentration of ventricular extracts was measured using the bicinchoninic acid assay (Sigma-Aldrich) using bovine serum albumin (BSA, Pierce Life Technologies, Rockford, IL, USA) as a standard. Thirty micrograms of total extract protein from mouse ventricular extracts, hESC-CM cell lysates, and conditioned media were separated on 8% polyacrylamide gels and then wet transferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA). Membranes were immunoblotted with primary monoclonal antibodies against MMP-2 (ab92536, Abcam, Cambridge, UK), tissue inhibitor of metalloproteinase-3 (TIMP-3, sc-30075, Santa Cruz Biotechnology, Santa Cruz, CA, USA), TIMP-4 (ab58425, Abcam), α-tubulin (ab7291, Abcam), and GAPDH (2118S, Cell Signaling Technology, Danvers, MA, USA). α-Tubulin and GAPDH were used as loading controls for mouse ventricular extracts and hESC-CM lysates, respectively. Protein bands were detected using horseradish peroxidase-conjugated secondary antibodies using chemiluminescent detection reagent (Clarity Western ECL Substrate; Bio-Rad, Hercules, CA, USA).
2.6. Gelatin zymography
Gelatin zymography was performed to determine MMP-2/-9 activities in mouse ventricular extracts, hESC-CM cell lysates, and conditioned media. Thirty micrograms of protein per sample, mixed in non-reducing loading buffer, were separated on 8% polyacrylamide gels co-polymerized with 2 mg/mL porcine gelatin (G8150, Sigma-Aldrich). The gels were rinsed in 2.5% Triton X-100 (20 min) and incubated in zymographic activity buffer (50 mM Tris–HCl, 150 mM NaCl, 5 mM CaCl2·2H2O, and 0.05% NaN3, pH 7.6) for 18 h at 37°C. Gels were stained with 0.05% Coomassie brilliant blue G-250 for 3 h and destained in 4% methanol and 8% acetic acid solution.
2.7. qPCR
Total cellular RNA was extracted from ventricular tissue using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), 5 mm stainless steel beads, and the TissueLyzer II system (Qiagen, Hilden, Germany). Total cellular RNA was quantified using a NanoDrop ND8000 spectrophotometer. RNA integrity was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), which determines the ratio of 28S and 18S ribosomal RNA. To eliminate genomic deoxyribonucleic acid (DNA) contamination, RNA was digested with DNase I (Invitrogen) at room temperature for 15 min and then at 65°C for 10 min. Reverse transcription was performed using the qScript cDNA SuperMix (Quanta Biosciences, Beverly, MA, USA). qPCR primers for B2M, Mmp2, NTT-Mmp2, and Rbm20 are shown in Supplementary material online, Table S1. Each primer pair was optimized in silico (Primer-BLAST, NCBI) and verified directly by PCR product sequencing. Samples were analysed in triplicate by qPCR using the SYBR Green I Master Mix (Roche Diagnostics, Risch-Rotkreuz, Switzerland) for 40 cycles (15 s denaturation, 45 s annealing, 60 s extension) in a LightCycler 480 System (Roche Diagnostics). Cq values and melt curves were analysed with the LightCycler 480 software v1.5.1. (Roche Diagnostics). A standard curve was generated to calculate copy numbers in the samples by loading increasing amounts of corresponding purified DNA standards of known concentration for each gene during each assay. Messenger RNA (mRNA) expression of each gene was calculated by first normalizing the copy numbers of each sample to the reference gene B2M and then normalizing to the control group.
2.8. Titin isolation and isoform expression
Titin was solubilized as described18 to determine the level of titin isoforms and degradation products in ventricular extracts. Extracts were loaded on a 1% agarose gel and electrophoresed at 15 mA (300–400 V) for 3 h at 4°C on a Hoefer SE600 gel unit. After electrophoresis, the gel was stained with Neuhoff’s optimized Coomassie blue. The optical density of T1 titin (total of N2BA and N2B titin), T2 titin (degradation product), and myosin heavy chain (MHC) were determined as a function of the sample volume loaded. The slope of the linear range of the relation between integrated optical density and loading volume was obtained for each sample. Titin proteolysis was determined by the ratio of titin degradation product T2 to T1 titin.
2.9. Transmission electron microscopy
For immuno transmission electron microscopic study of the left ventricles, the fixed tissue was dehydrated in an ethanol series, embedded in London Resin White (Ted Pella Inc., Redding, CA, USA), and polymerized under ultraviolet light. The ultraviolet polymerized tissue was longitudinally sectioned along the myofilaments and 80 nm ultrathin sections were transferred to a 300 mesh bare nickel grid for immunolabelling.
The grids were first incubated with a mixture of 5% BSA and 2% cold fish skin gelatin (G7765, Sigma-Aldrich) in 0.01 M PBS for 30 min at room temperature. Then they were incubated with monoclonal mouse anti-MMP-2 immunoglobulin G (IgG) (1:50, Cat. #MAB3008, Millipore, Burlington, MA, USA) overnight at 4°C followed by incubation with 18 nm colloidal gold-conjugated donkey anti-mouse secondary IgG (1:10, Cat. #715-215-150, Jackson ImmunoResearch Laboratories, West Grove, PA, USA) for 90 min at room temperature. Contrast was enhanced with 0.25× UranyLess and coating with carbon (7 nm thick) using a high vacuum carbon evaporator (Leica EM ACE600, Leica Microsystems, Concord Ontario, Canada). The grids were visualized using a Hitachi H-7650 transmission electron microscope. Quantitative analysis of intracellular MMP-2 level is detailed in the Supplementary material online.
2.10. Statistical analysis
Data are expressed as mean ± SEM of n independent experiments. Echocardiography, qPCR, western blot, gelatin zymography, and titin proteolysis experiments were analysed by one-way ANOVA followed by the Sidak’s post hoc test (GraphPad Prism 7 Software, La Jolla, CA, USA). Baseline (Day 1) echocardiography was analysed followed by one-way ANOVA using the Tukey’s post hoc test. P-values <0.05 were considered significant.
3. Results
3.1. DXR-induced cardiac contractile dysfunction and remodelling are attenuated by MMP inhibition
DXR impaired both systolic and diastolic function marked by a significant reduction in left ventricular fractional shortening (Figure 1A) and ejection fraction (Figure 1B) relative to control. DXR also caused mild diastolic dysfunction marked by a reduction in E′/A′ (Figure 1C) compared to control. Both MMP inhibitors attenuated DXR-impaired systolic and diastolic dysfunction by improving fractional shortening, ejection fraction, and E′/A′ (Figure 1A–C). DXR reduced cardiac output, an effect that was prevented by ONO-4817 but not Doxy (P = 0.076) (Figure 1D).
Figure 1.

Cardiac function and morphology in control (C) and DXR-administered mice treated with MMP inhibitors Doxy or ONO-4817 (ONO). (A–D) DXR impaired both systolic and diastolic function and reduced cardiac output. Doxy and ONO rescued DXR-reduced fractional shortening and ejection fraction. (E–H) DXR caused adverse cardiac remodelling marked by the thinning of the posterior wall and interventricular septum and expansion of the left ventricle end-systole, which were attenuated by Doxy. n = 10 mice per group. *P < 0.05 by one-way ANOVA followed by the Sidak’s post hoc test.
DXR caused adverse remodelling of the heart including thinning of the left ventricular posterior wall (Figure 1E) and interventricular septum (Figure 1F) during systole. DXR also increased the left ventricular internal diameter during systole (Figure 1G) and expanded the left ventricular end-systolic volume (Figure 1H). These adverse changes in cardiac morphology were prevented by Doxy but not ONO-4817 (Figure 1E–H). Full cardiac morphology and functional parameters measured at the end of the experiment (Day 28) are summarized in Supplementary material online, Table S2.
Baseline echocardiography data (Day 1) from all mice showed no significant differences in these parameters between all groups (Supplementary material online, Table S3). All echocardiographic parameters at Day 28 were unaffected by Doxy or ONO-4817 given without DXR, with the exception of a small reduction in systolic and diastolic interventricular septum thickness with ONO-4817 (Supplementary material online, Table S4). Intra-individual comparison of Day 1 and Day 28 ejection fraction and fractional shortening data for all groups are shown in Supplementary material online, Figure S2A and B.
3.2. MMP inhibitors prevent cellular remodelling in DXR cardiotoxicity
Haematoxylin and eosin staining of left ventricular free wall sections revealed increased nuclear size and crowding of cardiomyocytes in DXR mice (Figure 2A and B). DXR + Doxy and DXR + ONO-4817 attenuated nuclear size compared to DXR hearts (Figure 2C and D). DXR hearts exhibit increased cardiomyocyte paucity or dropout with increased interstitial oedema and myofibrillar disorganization compared to control hearts (Figure 2A and B). DXR + Doxy and DXR + ONO-4817 hearts exhibited similar myofibrillar morphology to control hearts and reduced cardiomyocyte dropout compared to DXR hearts (Figure 2C and D). Collagen staining with picrosirius red visualized with polarized light revealed that DXR caused interstitial fibrosis (Figure 2E–H). Quantification showed that DXR increased collagen deposition by ∼350%, and this was prevented with Doxy or ONO-4817 (Figure 2I).
Figure 2.

MMP inhibitors ameliorate cardiac remodelling and fibrosis in DXR cardiotoxicity. (A–D) Haematoxylin and eosin stained sections of the left ventricular free wall from control, DXR, DXR + Doxy, and DXR + ONO groups (representative of n = 5 hearts per group). Doxy and ONO attenuated DXR-induced cardiomyocyte dropout (white arrows) and crowding of the cardiomyocytes. The myocardium of DXR mice exhibited increased nuclear size of the cardiomyocytes when compared to control. DXR + Doxy and DXR + ONO hearts show an attenuation in nuclear size compared to DXR hearts. (E–H) Fibrosis of the left ventricular free wall was evaluated by staining collagen with picrosirius red using polarized light microscopy. Scale bar = 50 µm. (I) Doxy or ONO prevented DXR-induced left ventricular interstitial fibrosis (n = 5). *P < 0.05 by one-way ANOVA followed by the Sidak’s post hoc test.
3.3. DXR cardiotoxicity is associated with enhanced MMP-2 level and activity
DXR significantly increased MMP-2 protein levels in the heart to ∼250% of control, which was prevented with Doxy or ONO-4817 (Figure 3A). Changes in MMP-2 protein were accompanied by a two-fold increase in myocardial 72 kDa MMP-2 activity by gelatin zymography with no evidence of other gelatinolytic activities, including 64 kDa MMP-2 or MMP-9 (Figure 3B).
Figure 3.

MMP-2 levels and activity in the ventricle and plasma from DXR mice treated with MMP inhibitors. (A and B) DXR increased MMP-2 protein levels (n = 8) and activity (n = 10) in the heart, which were prevented by Doxy or ONO. (C and D) The plasma from DXR-treated mice exhibited increased levels of MMP-2 protein (n = 7–8) and activity (n = 9–10), which were reduced by ONO. *P < 0.05 by one-way ANOVA followed by the Sidak’s post hoc test.
Plasma MMP-2 protein and activity were also measured to determine changes in its circulating levels as a biomarker of DXR cardiotoxicity. DXR increased MMP-2 protein which was attenuated by ONO-4817 but not Doxy (Figure 3C). These changes in MMP-2 protein were accompanied by increased MMP-2 gelatinolytic activity which was abolished by ONO-4817 (Figure 3D).
MMP-2 activity is modulated by TIMPs, namely TIMP-3 in the extracellular matrix and TIMP-4, the most abundant endogenous inhibitor of intracellular MMP activity in cardiac myocytes.32 TIMP-3 and TIMP-4 protein levels in the heart were unaffected by DXR in the presence or absence of Doxy or ONO-4817 (Supplementary material online, Figure S3A and B).
Reactive oxygen–nitrogen species in combination with glutathione can activate MMP-2 by its S-glutathiolation.12S-glutathiolated MMP-2 in ventricular extracts was determined following immunoprecipitation of MMP-2. S-glutathiolated MMP-2 was detected in both control and DXR groups (Supplementary material online, Figure S4). DXR-treated mice exhibited increased oxidative stress in the heart indicated by reduced aconitase activity (Supplementary material online, Figure S5). Doxy, but not ONO-4817, normalized aconitase activity to that observed in control hearts. As oxidative stress can also transcriptionally up-regulate MMP-2, the mRNA levels of Mmp2 and NTT-Mmp2 were measured. DXR increased NTT-Mmp2 expression by two-fold (Supplementary material online, Figure S6A), but did not change that of 72 kDa Mmp2 (Supplementary material online, Figure S6B). Doxy and ONO-4817 significantly reduced DXR-elevated levels of NTT-Mmp2 mRNA to 105% and 74% of control, respectively (Supplementary material online, Figure S6A).
NTT-MMP-2 is involved in the innate immune response and is associated with elevated levels of interleukin-6 (IL6) and chemokine (C-X-C) motif ligand 1 (CXCL1).33 The DXR-induced de novo expression of NTT-Mmp2 was accompanied by an increase in IL6 (Supplementary material online, Figure S7A), but not CXCL1 (Supplementary material online, Figure S7B). ONO-4817 prevented DXR-induced increased IL6 expression.
3.4. Ultrastructure of the left ventricle by transmission electron microscopy
Transmission electron micrographs of the myofilaments from longitudinal left ventricular sections revealed significant differences in the ultrastructure of the sarcomere (Figure 4A–D) and mitochondria (Figure 4E–H) between control- and DXR-treated groups. We observed marked sarcomeric degeneration which included reduced sarcomere lengths, more diffuse and less prominent Z-discs, M-line disassembly, and significant myofilament disorganization compared to control (Figure 4A and B). Doxy and, in particular, ONO-4817, showed protection against DXR-induced damage to the sarcomere. Doxy hearts showed minor myofilament disorganization compared to DXR hearts while ONO-4817 hearts exhibited similar myofilament morphology to control hearts (Figure 4C and D). The mitochondria in DXR hearts appeared fragmented and the cristae were disrupted (Figure 4E and F), an effect that was prevented by Doxy or ONO-4817 (Figure 4G and H).
Figure 4.

Transmission electron micrographs of cardiac myocytes from left ventricular sections of control- and DXR-treated mice. (A) Control hearts exhibit normal myofilament morphology. (B) The myocardium of DXR mice exhibits extensive myofilament disorganization, diffuse Z-discs (Z), and disassembly of M-line (M), shown by black arrows, compared to control. (C and D) Doxy or ONO protected against DXR-induced sarcomeric damage in the heart. (E and F) DXR hearts exhibit mitochondrial degeneration (white arrows) and increased glycogen particles (arrowheads) compared to control. (G and H) Doxy or ONO hearts exhibited similar mitochondrial morphology to control. Representative of more than 50 images captured from 4 hearts from each group. Scale bar = 500 nm.
3.5. DXR increases MMP-2 levels in the sarcomere and mitochondria
Immunogold transmission electron microscopy was performed on left ventricular sections of control and DXR hearts to visualize the subcellular localization of MMP-2. Consistent with previous reports,16,34,35 MMP-2 localized to the sarcomere and mitochondria (Figure 5A and B). MMP-2 was localized to the Z-disc, I-band, A-band, and M-line of the sarcomere. MMP-2 was localized within the mitochondria and along the mitochondrial-associated membrane in both control and DXR hearts (Figure 5A and B). We determined the number of MMP-2 molecules/µm2 within the mitochondria or sarcomere as the colloidal gold-conjugated secondary antibodies bind to the primary antibodies with 1:1 stoichiometry. There was nearly twice the density of MMP-2 molecules in the mitochondria than in the sarcomere in both control and DXR hearts (Figure 5C and D). DXR doubled the density of MMP-2 molecules in both the sarcomere and mitochondria (Figure 5C and D).
Figure 5.

Determination of mitochondrial and sarcomeric MMP-2 in the left ventricle of control and DXR mice by immuno transmission electron microscopy. (A and B) Representative localization of MMP-2 in the sarcomere (black arrows) and mitochondria (white arrows) from control and DXR hearts. MMP-2 is localized to the Z-disc (Z), M-line (M), I-band, and A-band region of the sarcomere. Scale bar = 500 nm. (C and D) Quantification of colloidal gold particles, conjugated to anti-MMP-2 antibody that were localized within the sarcomere and mitochondria. A total of 24 images were captured and analysed from 4 hearts from each group (n = 4). *P < 0.05 by paired two-tailed Student’s t-test.
3.6. DXR cardiotoxicity induces titin proteolysis which is prevented by MMP inhibition
We then determined whether DXR-induced myofilament lysis is associated with cardiac titin proteolysis and/or altered titin isoform expression. The levels of titin isoforms (N2BA and N2B), intact titin (T1 = N2BA + N2B), and its major degradation product (T2) were measured in ventricular extracts. The ventricles from DXR-treated mice revealed three titin bands, including two full-length titin bands (>3 MDa) corresponding to N2BA and N2B titin, and a lower molecular weight degradation product, T2 (Figure 6A). The ratio of T2 to T1 titin determines the degree of titin degradation. DXR significantly increased cardiac titin proteolysis, which was attenuated by ONO-4817 but not Doxy (Figure 6B). DXR did not significantly change the ratio of titin (T1 + T2) to MHC compared to control hearts (Figure 6C). Heart failure is associated with changes in titin isoform expression.21 However, we observed no changes in the N2BA:N2B titin ratio across all groups (Figure 6D). This was confirmed by the absence of alterations in the expression of Rbm20 (Figure 6E), the titin splice factor which dictates isoform expression.
Figure 6.

Cardiac titin proteolysis and isoform expression in DXR cardiotoxicity. (A) Representative Coomassie blue-stained agarose gel showing titin levels in ventricular extracts. The ratio of titin degradation product (T2) to N2BA and N2B titin (T1) was determined in left ventricular extracts from control, DXR, DXR + Doxy, and DXR + ONO groups. MHC was used as a loading control. (B) Cardiac titin proteolysis, represented by the T2 to T1 titin ratio, was significantly increased in DXR and DXR + Doxy mice. DXR-induced titin proteolysis was prevented with ONO (n = 7–8). (C) Ratio of total titin (T1 + T2) to MHC content was unchanged between groups (n = 7–8). (D and E) DXR did not alter cardiac titin isoform expression, seen by the absence of changes in N2BA to N2B titin (n = 7–8) and Rbm20 mRNA expression (n = 6–8). STD is a standard ventricular extract from a control mouse that was not part of this study. *P < 0.05 by one-way ANOVA followed by the Sidak’s post hoc test.
3.7. DXR increases MMP-2 expression in hESC-CM
We next determined whether acute DXR treatment affects MMP-2 in human cardiomyocytes. hESC-CM were treated with a clinically relevant concentration of DXR that is observed in the plasma of patients within an hour after administering the drug.36 In cell lysates, DXR increased MMP-2 protein levels two-fold which was not affected by ONO-4817 (Figure 7A). DXR did not significantly enhance MMP-2 gelatinolytic activity in hESC-CM lysates (Figure 7B) but increased the level and activity of secreted MMP-2 by approximately two-fold (Figure 7C and D). ONO-4817 did not affect the DXR-induced increase in MMP-2 protein but significantly reduced its activity (Figure 7C and D).
Figure 7.

DXR-induced effects on MMP-2 in hESC-CMs. (A) MMP-2 protein levels were elevated in lysates from hESC-CM treated with DXR (1 µM, 24 h, n = 4). (B) DXR did not affect MMP-2 activity in hESC-CM lysates (n = 4). (C) Secreted MMP-2 protein levels and (D) activity were elevated in the conditioned media of DXR-treated hESC-CM (n = 4). Elevated MMP-2 activity in the conditioned media was blocked by ONO. *P < 0.05 by one-way ANOVA followed by the Sidak’s post hoc test.
4. Discussion
We demonstrated here for the first time that two orally available MMP inhibitors, Doxy or ONO-4817, attenuate DXR cardiotoxicity in vivo in mice. We established that DXR increased MMP-2 in the heart by increasing its transcription, protein levels, and activity. DXR also elevated the level of secreted MMP-2 activity in human cardiomyocytes, which was reduced by ONO-4817. Blocking the activity of MMP-2 prevented myofilament lysis, titin proteolysis, and interstitial fibrosis in mouse hearts. The resultant protective actions of MMP inhibitors on both the intracellular and extracellular matrices improved both systolic and diastolic function and remodelling impaired by DXR.
Prophylactic administration of drugs used to treat heart failure is the current treatment recommendation to alleviate chemotherapy-induced cardiotoxicity.37 However, these drugs only modestly improve ejection fraction without preventing left ventricular remodelling.10 In patients with acute myocardial infarction, Doxy reduced adverse left ventricular remodelling and improved ejection fraction.24 In mice, Doxy reduced DXR-induced myocardial oxidative stress by stimulating the activity of superoxide dismutase and glutathione peroxidase and diminished apoptosis.38 Tocchetti et al.39 demonstrated in mice that DXR-induced upregulation of MMP-2 was associated with increased deposition of interstitial collagen. After 1 week of treatment, cardioprotection with the sodium channel blocker ranolazine prevented early upregulation of MMP-2, interstitial fibrosis, and acute cardiac contractile dysfunction.39 A recent study by this group showed that phenylalanine-butyramide also attenuated DXR-induced MMP-2 upregulation, which was associated with improved cardiac contractile function and reduced left ventricular remodelling.40 We show here that Doxy or ONO-4817 diminished DXR cardiotoxicity by attenuating both cardiac contractile dysfunction and remodelling, suggesting that the detrimental effects of DXR are attributed, in part, to enhanced MMP activity.
MMP-2 is stimulated by oxidative stress at both transcriptional and post-translational levels. First, oxidative stress enhances MMP-2 transcription13 including the de novo expression of NTT-MMP-2 through an alternative promoter within the first intron.14 Second, intracellular MMP-2 is directly activated by peroxynitrite via S-glutathiolation to expose its catalytic site.12 We revealed that DXR increases MMP-2 levels and activity in the heart, also in part, by upregulating NTT-MMP-2 expression, consistent with our previous findings in isolated cardiomyocytes that DXR elevates MMP-2 protein and activity and de novo expression of NTT-MMP-2.15 We verified that a clinically relevant concentration of DXR also upregulated MMP-2 in human cardiomyocytes.
Cardiomyocyte dropout, interstitial fibrosis, and inflammation are characteristic of DXR cardiotoxicity. These dynamic changes take place in the extracellular matrix, which plays an important role in tissue architecture and cell signalling. Oxidative stress and inflammation increase MMP-2 activity in the injured myocardium, driving extracellular matrix remodelling, myofibroblast proliferation, and collagen deposition.41 NTT-MMP-2 initiates pro-inflammatory and pro-apoptotic innate immune responses in myocardial injury.14,42 NTT-MMP-2 triggers the activation of NFAT and NF-κB signalling cascades and the expression of a highly defined innate immunity transcriptome including IL6.42 Our results show that DXR cardiotoxicity is associated with increased expression of NTT-Mmp2 and IL6, and interstitial fibrosis, which were prevented by Doxy or ONO-4817. Consistent with a previous study,38 we demonstrated that Doxy normalized enhanced oxidative stress in hearts from DXR-treated mice to control levels. By preventing oxidative stress and degradation of the intracellular and extracellular matrices, MMP inhibitors likely attenuate the pernicious cycle of inflammatory processes and prevent the upregulation and activation of MMP-2 in the heart.
MMP-2 is elevated within cardiomyocytes in cardiomyopathy and oxidative stress injury.11,43 Intracellular MMP-2 activity directly affects cardiomyocyte structure and function by cleaving contractile proteins in the sarcomere.44 Degradation of titin or alteration in its splicing causes detrimental effects on cardiac contractile function.18,21 Here DXR increased MMP-2 levels directly in the sarcomere to contribute to titin proteolysis, which was associated with reduced sarcomere length, myofilament lysis, and cardiac contractile dysfunction. These effects are likely attributed to increased MMP-2 activity given that titin proteolysis was markedly reduced when MMP activity was blocked by ONO-4817. Data suggesting that DXR stimulated titin proteolysis in isolated cardiomyocytes is calpain-dependent6 was based on the protease inhibitor used, which later was revealed to also potently inhibit MMP-2 activity.45 In addition, the lysine methyltransferase SMYD2 dissociates from the near Z-disc region of titin during oxidative stress and renders titin susceptible to in vitro proteolysis by MMP-2.46 Although altered titin isoform expression can occur in patients with dilated cardiomyopathy21 we found that DXR did not affect titin splicing as there were no effects on Rbm20 expression nor on the N2BA:N2B titin ratio. Clinical studies have associated the abundance of urinary N-terminal titin fragments and MMP-2 degraded titin fragments in sera from patients with dilated cardiomyopathy47 and cardiac injury,48,49 respectively. Titin fragments should be investigated in future studies as a potential biomarker of DXR cardiotoxicity.
Our study demonstrated that two different MMP inhibitors protect against DXR cardiotoxicity by preventing adverse cardiac remodelling or titin proteolysis. While these inhibitors both target MMP-2, the intracellular concentration of Doxy relative to ONO-4817 within cardiomyocytes is unknown. Doxy preferentially accumulates in heart tissue over plasma50 and we have shown that ONO-4817 can inhibit MMP-2 inside cardiomyocytes.45 The relative selectivity of Doxy or ONO-4817 for intracellular vs. extracellular MMP-2 isoforms is also unknown. However, ONO-4817 is the more potent MMP inhibitor and shows greater selectivity for gelatinases (MMP-2 and MMP-9) than Doxy. While ONO-4817 inhibits MMP-2 from proteolyzing titin, the cardioprotective effects of Doxy may be a result of inhibiting MMP-2 and other MMPs involved in ventricular remodelling.
Clinical and pre-clinical studies have reported sex-related differences in DXR-induced cardiotoxicity.51,52 We only used male mice which limits the interpretation of our results. Determining the effects of DXR and MMP inhibitors in females may reveal novel mechanisms underlying the sexual dimorphism of anthracycline cardiotoxicity and will be very important to test in future studies. Additionally, it remains an open question whether MMP-2 contributes to titin turnover under physiological conditions by helping dismantle damaged titin for further processing by the ubiquitin-proteasome pathway. Furthermore, although we observed differences in MMP-2 activity in hESC-CMs, we do not know whether DXR elevates MMP-2 or increases titin proteolysis in mature human myocardium.
Together, our results elucidate the role of MMP-2 in intracellular and extracellular matrix remodelling in DXR cardiotoxicity. We demonstrate that two orally available, selective MMP inhibitors ameliorate DXR-induced cardiac dysfunction and remodelling by attenuating titin proteolysis, myofilament lysis, and interstitial fibrosis. Prophylactic inhibition of MMP activity could be an effective means to prevent heart injury caused by anthracyclines in the treatment of cancer.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
The authors thank Donna Beker for performing echocardiography, Chandrasekhar Saripalli and Nils Moser for technical assistance, and Mohammad A.M. Ali for his helpful comments on the manuscript.
Conflict of interest: None declared.
Funding
This work was supported by the Canadian Institutes of Health Research [Foundation #143299 to R.S.], the Heart and Stroke Foundation of Canada (to R.S.), and the National Institutes of Health [1R35HL144998 to H.G.]. B.Y.H.C. was supported by a graduate studentship from the Women and Children’s Health Research Institute, through the generous support of the Stollery Children’s Hospital Foundation and the Royal Alexandra Hospital Foundation. Some experiments were performed at the University of Alberta Faculty of Medicine & Dentistry Cell Imaging Centre, which receives financial support from the Faculty of Medicine & Dentistry, the Department of Medical Microbiology and Immunology, and Canada Foundation for Innovation awards to contributing investigators.
Time for primary review: 28 days
Translational perspective
Heart failure is the primary chronic toxicity of anthracycline chemotherapy. Anthracyclines such as doxorubicin cause left ventricular remodelling and loss of myofilament proteins. We determined in mice whether matrix metalloproteinase-2, an intracellular and extracellular protease in the heart, contributes to doxorubicin cardiotoxicity. Doxorubicin activated myocardial MMP-2 in mouse hearts and human cardiomyocytes, including de novo expression of an N-terminal truncated MMP-2 isoform which is exclusively expressed by increased oxidative stress. Increased MMP-2 levels and activity in the heart contributed to left ventricular remodelling, interstitial fibrosis, and titin proteolysis in doxorubicin cardiotoxicity. These adverse effects on the heart were prevented by two orally available MMP inhibitors, demonstrating the potential benefits of MMP inhibition in the prophylaxis of doxorubicin cardiotoxicity.
References
- 1. Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, Jones A.. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer 2010;10:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bloom MW, Hamo CE, Cardinale D, Ky B, Nohria A, Baer L, Skopicki H, Lenihan DJ, Gheorghiade M, Lyon AR, Butler J.. Cancer therapy-related cardiac dysfunction and heart failure: part 1: definitions, pathophysiology, risk factors, and imaging. Circ Heart Fail 2016;9:e002661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yeh ET, Bickford CL.. Cardiovascular complications of cancer therapy: incidence, pathogenesis, diagnosis, and management. J Am Coll Cardiol 2009;53:2231–2247. [DOI] [PubMed] [Google Scholar]
- 4. Singal P, Iliskovic N.. Doxorubicin-induced cardiomyopathy. N Engl J Med 1998;339:900–905. [DOI] [PubMed] [Google Scholar]
- 5. Goetzenich A, Hatam N, Zernecke A, Weber C, Czarnotta T, Autschbach R, Christiansen S.. Alteration of matrix metalloproteinases in selective left ventricular adriamycin-induced cardiomyopathy in the pig. J Heart Lung Transplant 2009;28:1087–1093. [DOI] [PubMed] [Google Scholar]
- 6. Lim CC, Zuppinger C, Guo X, Kuster GM, Helmes M, Eppenberger HM, Suter TM, Liao R, Sawyer DB.. Anthracyclines induce calpain-dependent titin proteolysis and necrosis in cardiomyocytes. J Biol Chem 2004;279:8290–8299. [DOI] [PubMed] [Google Scholar]
- 7. Myers C, Bonow R, Palmeri S, Jenkins J, Corden B, Locker G, Doroshow J, Epstein S.. A randomized controlled trial assessing the prevention of doxorubicin cardiomyopathy by N-acetylcysteine. Semin Oncol 1983;10:53–55. [PubMed] [Google Scholar]
- 8. Martin E, Thougaard AV, Grauslund M, Jensen PB, Bjorkling F, Hasinoff BB, Tjornelund J, Sehested M, Jensen LH.. Evaluation of the topoisomerase II-inactive bisdioxopiperazine ICRF-161 as a protectant against doxorubicin-induced cardiomyopathy. Toxicology 2009;255:72–79. [DOI] [PubMed] [Google Scholar]
- 9. Bosch X, Rovira M, Sitges M, Domenech A, Ortiz-Perez JT, de Caralt TM, Morales-Ruiz M, Perea RJ, Monzo M, Esteve J.. Enalapril and carvedilol for preventing chemotherapy-induced left ventricular systolic dysfunction in patients with malignant hemopathies: the OVERCOME trial (preventiOn of left Ventricular dysfunction with Enalapril and caRvedilol in patients submitted to intensive ChemOtherapy for the treatment of Malignant hEmopathies). J Am Coll Cardiol 2013;61:2355–2362. [DOI] [PubMed] [Google Scholar]
- 10. Pituskin E, Mackey JR, Koshman S, Jassal D, Pitz M, Haykowsky MJ, Pagano JJ, Chow K, Thompson RB, Vos LJ, Ghosh S, Oudit GY, Ezekowitz JA, Paterson DI.. Multidisciplinary approach to novel therapies in cardio-oncology research (MANTICORE 101-Breast): a randomized trial for the prevention of trastuzumab-associated cardiotoxicity. J Clin Oncol 2017;35:870–877. [DOI] [PubMed] [Google Scholar]
- 11. Hughes BG, Schulz R.. Targeting MMP-2 to treat ischemic heart injury. Basic Res Cardiol 2014;109:424. [DOI] [PubMed] [Google Scholar]
- 12. Viappiani S, Nicolescu AC, Holt A, Sawicki G, Crawford BD, Leon H, van Mulligen T, Schulz R.. Activation and modulation of 72kDa matrix metalloproteinase-2 by peroxynitrite and glutathione. Biochem Pharmacol 2009;77:826–834. [DOI] [PubMed] [Google Scholar]
- 13. Alfonso-Jaume MA, Bergman MR, Mahimkar R, Cheng S, Jin ZQ, Karliner JS, Lovett DH.. Cardiac ischemia-reperfusion injury induces matrix metalloproteinase-2 expression through the AP-1 components FosB and JunB. Am J Physiol Heart Circ Physiol 2006;291:H1838–H1846. [DOI] [PubMed] [Google Scholar]
- 14. Lovett DH, Mahimkar R, Raffai RL, Cape L, Maklashina E, Cecchini G, Karliner JS.. A novel intracellular isoform of matrix metalloproteinase-2 induced by oxidative stress activates innate immunity. PLoS One 2012;7:e34177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chan BYH, Roczkowsky A, Moser N, Poirier M, Hughes BG, Ilarraza R, Schulz R.. Doxorubicin induces de novo expression of N-terminal-truncated matrix metalloproteinase-2 in cardiac myocytes. Can J Physiol Pharmacol 2018;96:1238–1245. [DOI] [PubMed] [Google Scholar]
- 16. Wang W, Schulze CJ, Suarez-Pinzon W, Dyck J, Sawicki S, Schulz R.. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation 2002;106:1543–1549. [DOI] [PubMed] [Google Scholar]
- 17. Sawicki G, Leon H, Sawicka J, Sariahmetoglu M, Schulze CJ, Scott PG, Szczesna-Cordary D, Schulz R.. Degradation of myosin light chain in isolated rat hearts subjected to ischemia-reperfusion injury: a new intracellular target for matrix metalloproteinase-2. Circulation 2005;112:544–552. [DOI] [PubMed] [Google Scholar]
- 18. Ali MA, Cho WJ, Hudson B, Kassiri Z, Granzier H, Schulz R.. Titin is a target of matrix metalloproteinase-2: implications in myocardial ischemia/reperfusion injury. Circulation 2010;122:2039–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. LeWinter MM, Granzier H.. Cardiac titin: a multifunctional giant. Circulation 2010;121:2137–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, Dauksaite V, Vakeel P, Klaassen S, Gerull B, Thierfelder L, Regitz-Zagrosek V, Hacker TA, Saupe KW, Dec GW, Ellinor PT, MacRae CA, Spallek B, Fischer R, Perrot A, Ozcelik C, Saar K, Hubner N, Gotthardt M.. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med 2012;18:766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL.. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation 2004;110:155–162. [DOI] [PubMed] [Google Scholar]
- 22. Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA.. Titin isoform switch in ischemic human heart disease. Circulation 2002;106:1333–1341. [DOI] [PubMed] [Google Scholar]
- 23. Lee HM, Ciancio SG, Tuter G, Ryan ME, Komaroff E, Golub LM.. Subantimicrobial dose doxycycline efficacy as a matrix metalloproteinase inhibitor in chronic periodontitis patients is enhanced when combined with a non-steroidal anti-inflammatory drug. J Periodontol 2004;75:453–463. [DOI] [PubMed] [Google Scholar]
- 24. Cerisano G, Buonamici P, Valenti R, Sciagrà R, Raspanti S, Santini A, Carrabba N, Dovellini EV, Romito R, Pupi A, Colonna P, Antoniucci D.. Early short-term doxycycline therapy in patients with acute myocardial infarction and left ventricular dysfunction to prevent the ominous progression to adverse remodelling: the TIPTOP trial. Eur Heart J 2014;35:184–191. [DOI] [PubMed] [Google Scholar]
- 25. Barpe DR, Rosa DD, Froehlich PE.. Pharmacokinetic evaluation of doxorubicin plasma levels in normal and overweight patients with breast cancer and simulation of dose adjustment by different indexes of body mass. Eur J Pharm Sci 2010;41:458–463. [DOI] [PubMed] [Google Scholar]
- 26. Johansen PB. Doxorubicin pharmacokinetics after intravenous and intraperitoneal administration in the nude mouse. Cancer Chemother Pharmacol 1981;5:267–270. [DOI] [PubMed] [Google Scholar]
- 27. Castro MM, Rizzi E, Figueiredo-Lopes L, Fernandes K, Bendhack LM, Pitol DL, Gerlach RF, Tanus-Santos JE.. Metalloproteinase inhibition ameliorates hypertension and prevents vascular dysfunction and remodeling in renovascular hypertensive rats. Atherosclerosis 2008;198:320–331. [DOI] [PubMed] [Google Scholar]
- 28. Hariya A, Takazawa K, Yamamoto T, Amano A.. ONO-4817, a novel matrix metalloproteinase inhibitor, attenuates allograft vasculopathy in a rat cardiac transplant. J Heart Lung Transplant 2004;23:1163–1169. [DOI] [PubMed] [Google Scholar]
- 29. Naito Y, Takagi T, Kuroda M, Katada K, Ichikawa H, Kokura S, Yoshida N, Okanoue T, Yoshikawa T.. An orally active matrix metalloproteinase inhibitor, ONO-4817, reduces dextran sulfate sodium-induced colitis in mice. Inflamm Res 2004;53:462–468. [DOI] [PubMed] [Google Scholar]
- 30. Yaras N, Sariahmetoglu M, Bilginoglu A, Aydemir-Koksoy A, Onay-Besikci A, Turan B, Schulz R.. Protective action of doxycycline against diabetic cardiomyopathy in rats. Br J Pharmacol 2008;155:1174–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Churko JM, Garg P, Treutlein B, Venkatasubramanian M, Wu H, Lee J, Wessells QN, Chen SY, Chen WY, Chetal K, Mantalas G, Neff N, Jabart E, Sharma A, Nolan GP, Salomonis N, Wu JC.. Defining human cardiac transcription factor hierarchies using integrated single-cell heterogeneity analysis. Nat Commun 2018;9:4906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schulze CJ, Wang W, Suarez-Pinzon WL, Sawicka J, Sawicki G, Schulz R.. Imbalance between tissue inhibitor of betalloproteinase-4 and matrix metalloproteinases during acute myoctardial ischemia-reperfusion injury. Circulation 2003;107:2487–2492. [DOI] [PubMed] [Google Scholar]
- 33. Joshi SK, Lee L, Lovett DH, Kang H, Kim HT, Delgado C, Liu X.. Novel intracellular N-terminal truncated matrix metalloproteinase-2 isoform in skeletal muscle ischemia-reperfusion injury. J Orthop Res 2016;34:502–509. [DOI] [PubMed] [Google Scholar]
- 34. Hughes BG, Fan X, Cho WJ, Schulz R.. MMP-2 is localized to the mitochondria-associated membrane of the heart. Am J Physiol Heart Circ Physiol 2014;306:H764–H770. [DOI] [PubMed] [Google Scholar]
- 35. Lovett DH, Chu C, Wang G, Ratcliffe MB, Baker AJ.. A N-terminal truncated intracellular isoform of matrix metalloproteinase-2 impairs contractility of mouse myocardium. Front Physiol 2014;5:363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Greene RF, Collins JM, Jenkins JF, Speyer JL, Myers CE.. Plasma pharmacokinetics of adriamycin and adriamycinol: implications for the design of in vitro experiments and treatment protocols. Cancer Res 1983;43:3417–3421. [PubMed] [Google Scholar]
- 37. Curigliano G, Cardinale D, Suter T, Plataniotis G, de Azambuja E, Sandri MT, Criscitiello C, Goldhirsch A, Cipolla C, Roila F.. Cardiovascular toxicity induced by chemotherapy, targeted agents and radiotherapy: ESMO Clinical Practice Guidelines. Ann Oncol 2012;23(Suppl. 7):vii155–vii166. [DOI] [PubMed] [Google Scholar]
- 38. Lai HC, Yeh YC, Ting CT, Lee WL, Lee HW, Wang LC, Wang KY, Lai HC, Wu A, Liu TJ.. Doxycycline suppresses doxorubicin-induced oxidative stress and cellular apoptosis in mouse hearts. Eur J Pharmacol 2010;644:176–187. [DOI] [PubMed] [Google Scholar]
- 39. Tocchetti CG, Carpi A, Coppola C, Quintavalle C, Rea D, Campesan M, Arcari A, Piscopo G, Cipresso C, Monti MG, De Lorenzo C, Arra C, Condorelli G, Di Lisa F, Maurea N.. Ranolazine protects from doxorubicin-induced oxidative stress and cardiac dysfunction. Eur J Heart Fail 2014;16:358–366. [DOI] [PubMed] [Google Scholar]
- 40. Russo M, Guida F, Paparo L, Trinchese G, Aitoro R, Avagliano C, Fiordelisi A, Napolitano F, Mercurio V, Sala V, Li M, Sorriento D, Ciccarelli M, Ghigo A, Hirsch E, Bianco R, Iaccarino G, Abete P, Bonaduce D, Calignano A, Berni Canani R, Tocchetti CG.. The novel butyrate derivative phenylalanine-butyramide protects from doxorubicin-induced cardiotoxicity. Eur J Heart Fail 2019;21:519–528. [DOI] [PubMed] [Google Scholar]
- 41. Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev 2007;87:1285–1342. [DOI] [PubMed] [Google Scholar]
- 42. Lovett DH, Mahimkar R, Raffai RL, Cape L, Zhu BQ, Jin ZQ, Baker AJ, Karliner JS.. N-terminal truncated intracellular matrix metalloproteinase-2 induces cardiomyocyte hypertrophy, inflammation and systolic heart failure. PLoS One 2013;8:e68154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rouet-Benzineb P, Buhler JM, Dreyfus P, Delcourt A, Dorent R, Perennec J, Crozatier B, Harf A, Lafuma C.. Altered balance between matrix gelatinases (MMP-2 and MMP-9) and their tissue inhibitors in human dilated cardiomyopathy: potential role of MMP-9 in myosin-heavy chain degradation. Eur J Heart Fail 1999;1:337–352. [DOI] [PubMed] [Google Scholar]
- 44. Ali MAM, Fan F, Schulz R.. Cardiac sarcomeric proteins: novel intracellular targets of matrix metalloproteinase-2 in heart disease. Trends Cardiovasc Med 2011;21:112–118. [DOI] [PubMed] [Google Scholar]
- 45. Ali MA, Stepanko A, Fan X, Holt A, Schulz R.. Calpain inhibitors exhibit matrix metalloproteinase-2 inhibitory activity. Biochem Biophys Res Commun 2012;423:1–5. [DOI] [PubMed] [Google Scholar]
- 46. Munkanatta Godage DNP, VanHecke GC, Samarasinghe KTG, Feng HZ, Hiske M, Holcomb J, Yang Z, Jin JP, Chung CS, Ahn YH.. SMYD2 glutathionylation contributes to degradation of sarcomeric proteins. Nat Commun 2018;9:4341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yoshihisa A, Kimishima Y, Kiko T, Sato Y, Watanabe S, Kanno Y, Abe S, Miyata M, Sato T, Suzuki S, Oikawa M, Kobayashi A, Yamaki T, Kunii H, Nakazato K, Ishida T, Takeishi Y.. Usefulness of urinary N-terminal fragment of titin to predict mortality in dilated cardiomyopathy. Am J Cardiol 2018;121:1260–1265. [DOI] [PubMed] [Google Scholar]
- 48. Bogomolovas J, Gasch A, Bajoras V, Karčiauskaitė D, Šerpytis P, Grabauskienė V, Labeit D, Labeit S.. Cardiac specific titin N2B exon is a novel sensitive serological marker for cardiac injury. Int J Cardiol 2016;212:232–234. [DOI] [PubMed] [Google Scholar]
- 49. Rahim MAA, Rahim ZHA, Ahmad WAW, Bakri MM, Ismail MD, Hashim OH.. Inverse changes in plasma tetranectin and titin levels in patients with type 2 diabetes mellitus: a potential predictor of acute myocardial infarction? Acta Pharmacol Sin 2018;39:1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Romero-Perez D, Fricovsky E, Yamasaki KG, Griffin M, Barraza-Hidalgo M, Dillmann W, Villarreal F.. Cardiac uptake of minocycline and mechanisms for in vivo cardioprotection. J Am Coll Cardiol 2008;52:1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Moulin M, Piquereau J, Mateo P, Fortin D, Rucker-Martin C, Gressette M, Lefebvre F, Gresikova M, Solgadi A, Veksler V, Garnier A, Ventura-Clapier R.. Sexual dimorphism of doxorubicin-mediated cardiotoxicity: potential role of energy metabolism remodeling. Circ Heart Fail 2015;8:98–108. [DOI] [PubMed] [Google Scholar]
- 52. Lipshultz SE, Lipsitz SR, Mone SM, Goorin AM, Sallan SE, Sanders SP, Orav EJ, Gelber RD, Colan SD.. Female sex and higher drug dose as risk factors for late cardiotoxic effects of doxorubicin therapy for childhood cancer. N Engl J Med 1995;332:1738–1743. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.