

Abstract
Natural killer (NK) cells are cytotoxic innate lymphocytes that protect against viral infection and tumor metastasis. Despite their inherent ability to kill a broad range of virally infected, stressed and transformed cells, low numbers of dysfunctional NK cells are often observed in many advanced solid human cancers. Here, we review the potential mechanisms that influence suboptimal mature NK cell recruitment and function in the tumor microenvironment (TME) of solid tumors. We further highlight current immunotherapy approaches aimed to circumvent NK cell dysfunction and discuss next‐generation strategies to enhance adoptive NK cell therapy through targeting intrinsic and extrinsic checkpoints the regulate NK cell functionality in the TME. Understanding the mechanisms that drive NK cell dysfunction in the TME will lead to novel immunotherapeutic approaches in the fight against cancer.
Keywords: adoptive NK cell therapy, CRISPR, dysfunction, genetic engineering, immunotherapy, natural killer cells, suppression, tumor microenvironment
In this review, we provide a summary of the current literature profiling intrinsic and extrinsic mechanisms of NK cells that promote dysfunctional responses in solid tumor microenvironments. We critically analyse the current literature and propose open questions as well as new directions for NK‐cell‐based immunotherapies in cancer.

Introduction
NK cells are cytotoxic innate lymphocytes (ILCs) which are capable of recognising stressed or transformed cells during viral infection and tumoigenesis. 1 Their endogenous anti‐tumor role in vivo has been demonstrated in mouse models of NK cell deficiency that develop more carcinogen‐induced tumors, oncogene‐driven tumors, and experimental lung and liver metastases. 2 , 3 , 4 Similarly, collective clinical evidence suggests that increased presence of NK cells correlates with better survival outcomes in multiple solid cancers and in response to checkpoint blockade immunotherapy in melanoma. 5 , 6 , 7 , 8 , 9 In mice, NK cells have been shown to be one of the earliest circulating lymphocytes recruited to distal sites of tumor metastasis and can mediate cancer regression through IFN‐γ production, rapid cytotoxicity, and recruitment and proliferation of type 1 conventional dendritic cells (cDC1) through production of XCL1 and FLT3 ligand. 10 , 11 NK cell activation is regulated by a balance of activating and inhibitory receptors such as natural cytotoxicity receptors (NCRs) 12 , 13 or killer cell lectin‐like receptor (NKG2) molecules NKG2D 14 and NKG2A, respectively 15 ; killer cell immunoglobulin‐like receptors (KIRs) in humans, and the mouse counterpart c‐type lectin‐like (Ly49) receptors. 1 Under homeostatic conditions, transient activating signals can be countered by potent inhibitory signals derived from MHC‐I molecules present on host cells. 16 During tumorigenesis, transformed cells can upregulate activating NKG2D ligands (aNKG2DL) as a result of replicative or genotoxic stress 14 , 17 and can mutate genes in the MHC‐I pathway to evade cytotoxic CD8+ T‐cell recognition. 1 These conditions shift the balance of activating to inhibitory signals in the NK cell–target cell synapse to lead to NK cell‐mediated lysis of tumor cells, which has been extensively reviewed previously. 7 , 12 , 13 , 18 In addition to membrane‐bound tumor ligands, NK cells have also been shown to recognise tumor cells through tumor shed soluble ligands capable of activating NK cells through NKG2D and NKp44. 19 , 20 However, MHC‐I‐deficient and aNKG2DLhi tumors can still progressively grow in mice and humans 17 , 21 suggesting that additional suppressive mechanisms exist within solid tumor microenvironments that inhibit endogenous NK cell anti‐tumor function. In this review, we discuss the potential mechanisms that influence suboptimal mature NK cell recruitment and function in the tumor microenvironment (TME) of solid tumors. We further highlight current immunotherapy approaches aimed to circumvent NK cell dysfunction and discuss next‐generation strategies to enhance adoptive NK cell therapy through targeting intrinsic and extrinsic checkpoints the regulate NK cell functionality in the TME.
NK Cell Maturation in the TME
NK cells consist of phenotypically and functionally diverse subsets that represent a developmental continuum during homeostasis (Figure 1). In mice, the surface markers CD27 and CD11b can be used to define NK cell developmental maturation stages. These maturation markers correlate with cytokine production and cytotoxic potential because evidence suggests that CD11b–CD27+ immature NK (iNK) cells are potent cytokine producers with heightened proliferative capacity, and CD11b+CD27− mature NK (mNK) cells have increased cytolytic capacity against tumor targets. 22 NK cells in the bone marrow (BM) are primarily iNK, whereas the majority of NK cells in the blood and most tissues are observed to be mNK during homeostasis. 22 However, multiple studies using preclinical mouse models have shown that this balance is disrupted during tumorigenesis. 23 , 24 , 25 For instance, transplanted EL4 thymoma tumors can block NK cell maturation in the bone marrow, spleen and TME. This is supported by experiments demonstrating that adoptively transferred CD11b– NK cells failed to upregulate CD11b when harvested from the spleen of EL4 tumor‐bearing mice 14 days later. 23 Tumor‐bearing mice displayed reduced levels of interleukin (IL)‐15R⍺+ in BM stromal cells, and transgenic IL‐15 expressing mice restored NK cell anti‐tumor function indicating that IL‐15R⍺ expression BM stromal cells can be suppressed during tumorigenesis in the BM niche. 23 In support of this hypothesis, a recent study using a mouse transgenic MYC‐driven T‐lymphoma model observed reduced mNK cells in peripheral organs due to suppressed NK cell development in the BM. 26 Type I interferon (IFN) signalling was suppressed in the BM microenvironment in the presence of MYC‐driven lymphomas, and type I IFN was found to be required for systemic mNK cell development. 26 Since type I IFN is known to induce IL‐15R⍺ on dendritic cells (DCs), 27 a potential decrease in IL‐15R⍺+ BM DCs could explain the block in NK cell development during lymphoma development in the BM. Additional studies have shown NK cells display an immature phenotype within the TME of B16 melanoma tumors and spontaneously forming PyMT breast tumors. 24 , 25 However, these studies did not find defects in NK maturation in splenic NK cells of tumor‐bearing mice, perhaps because these tumors had not yet seeded the BM to negatively impact systemic NK cell development and may suggest that iNK cells may also be preferentially recruited to the TME in certain solid cancers.
Figure 1.

Surface markers defining immature and mature NK Cells in mouse and human. Human and mouse NK cell subsets are simplified and divided into two main subsets: mature (mNK) and immature (iNK) cells, based off CD56 and CD16 in humans and CD11b and CD27 in mice. Important lineage markers, activation, inhibitory and chemokine receptors are depicted.
In humans, NK cells are classically divided into two major subsets: CD56dimCD16hi and CD56brightCD16lo (Figure 1). 16 , 28 Peripheral blood NK cells are composed of approximately 90% of CD56dimCD16hi NK cells and 10% of CD56brightCD16lo NK cells. 28 Similar to mouse NK cell subsets, human NK cells differ in maturation status and function. Human CD56brightCD16lo NK cells are considered terminally differentiated and predominantly exert cytolytic function, 16 while CD56brightCD16lo NK cells are less cytolytic, but have a higher propensity to produce effector cytokines. 16 A recent single‐cell RNA sequencing data set has unbiasedly identified two distinct NK cell subsets present in mouse and human samples, and concluded that mature CD56dimCD16hi human NK cells share similar gene signatures as CD11b+CD27− mouse mNK cells and human CD56brightCD16lo NK cell signatures overlap with CD11b–CD27+ iNK cells. 29 While these data suggest that CD56brightCD16lo human NK cells likely represent iNK cells (nomenclature we will use hereafter), it remains possible that these cells could represent an activation state or distinct lineage of human NK cells. Similar to mouse tumor models, human tumors are often are infiltrated with iNK cells with diminished cytotoxic function. Several studies looking at both lung and breast cancer have shown that a majority of tumor infiltrating NK cells are CD56brightperforinlo compared to matched healthy tissue. 30 , 31 , 32 Another study looking at breast tumors also found infiltrating NK cells were consistently CD56brightNKG2AhiCD16loKIRlo and had reduced functionality demonstrated by decreased interferon (IFN)‐γ and tumor necrosis factor (TNF)‐⍺ compared to peripheral blood NK cells. 33 This evidence suggests that either iNK cells have a higher propensity to reach the TME, or upon entering the TME, mNK cells revert to a poorly cytotoxic cell state. Further fate‐mapping studies will be helpful to understand whether developmental states of NK cells are preferentially recruited to the TME, or whether the TME can induce phenotypical and functional suppression of mNK cells.
NK Cell Trafficking to Tumors
The mechanisms that drive chemotaxis of innate lymphocytes to the TME is not well understood, and chemokine receptor expression can vary between iNK and mNK cells. 34 Human mNK cells express the chemokine receptors CXCR1, CXCR2, CX3CR1, ChemR23 and S1P5 while iNK cells typically express receptors CXCR3, CCR2, CCR3, CCR5 and CCR7 22 , 31 , 34 (Table 1). Because chemokine receptor expression on mouse NK cells is less well understood, we utilised RNA‐seq data from the ImmGen database 35 to examine chemokine receptor expression in iNK and mNK mouse subsets (Figure 2). Recent single‐cell sequencing data suggest that mouse and human iNK cells are enriched for Cxcr3 transcripts. 29 In addition, mouse iNK cells have increased Ccr2 transcripts and mNK cells show increased S1pr5 transcripts, 29 in agreement with the ImmGen database (Figure 2). Furthermore, recent evidence demonstrates that chemokine receptor heterogeneity exists within CD56bright and CD56dim subsets. 36 These data indicate that NK cell infiltration into different tissues may be more complex than current developmental classification suggest. In leukaemia and lymphoma mouse tumor models, CXCL9 and CXCL10 are produced at high levels and CXCL11 can be induced upon addition of exogenous IFN‐γ to the TME. 37 CXCL9 and CXCL10 expression in the TME can recruit iNK cells through expression of CXCR3. 38 However, another study found that while administration of CXCL9 and CXCL10 resulted in increased NK cell infiltration to the TME in a CXCR3 dependent manner, CXCR3 was dispensable for overall tumor burden and survival, suggesting potential redundancy in the chemokine receptors required for NK cell recruitment depending on the tumor model. 37 Furthermore, stress factors produced by tumor cells have been shown to be upstream of chemokine production in the TME because IL‐17D produced by MCA‐induced fibrosarcoma tumors can induce endothelial cell production of CCL2, which can recruit CCR2+ iNK cells to the tumor. 39 These studies collectively suggest that both CCR2 and CXCR3 are important for NK cell recruitment into solid tumors in mouse models, and suggest a preference for iNK over mNK recruitment into tumors. In support of this hypothesis, CD56brightperforinlo NK cells preferentially infiltrate human lung and breast tumors. 31 This accumulation is correlated with increased expression of CCL19, CXCL9, and CXCL10 in the TME. 31 In contrast, CX3CL1 (the ligand for CX3CR1 expressed on mNK cells), is a positive prognostic marker across 5 different human tumor types, 18 and CX3CR1+ mouse NK cells can better control experimental lung metastasis than CX3CR1– NK cells. 40 CX3CL1 was also shown to reduce tumor burden in a lymphoma mouse model through increased recruitment of activated NK cytotoxic cells. 41 Together, these results suggest that solid tumors may selectively recruit poorly cytotoxic iNK cells as a potential immune evasion mechanism, although future work will be necessary to test this hypothesis.
Table 1.
Chemokine receptor and ligands in NK cell subsets in mouse and human
| iNK (immature) | mNK (mature) | ||
|---|---|---|---|
| Human | CD56bright CD16− | CD56dim CD16+ | Ligand(s) |
| CCR5 | + | − | CCL3, CCL4, CCL5 |
| CCR7 | ++ | − | CCL19, CCL21 |
| CXCR1 | − | ++ | CXCL6, CXCL8 |
| CXCR2 | − | + | CXCL8, CXCL1, CXCL2, CXCL5 |
| CXCR3 | ++ | + | CXCL9, CXCL10, CXCL11 |
| CXCR4 | ++ | ++ | CXCL12 |
| CX3CR1 | − | ++ | CX3CL1 |
| S1P5 | − | + | S1P |
| CR23 (Cmklr1) | − | + | Chemerin |
| iNK (immature) | mNK (mature) | ||
|---|---|---|---|
| Mouse | CD27+CD11b– | CD27–CD11b+ | Ligand(s) |
| CCR5 | ND | ND | CCL3, CCL4, CCL5 |
| CCR7 | ND | ND | CCL19, CCL21 |
| CXCR1 | ND | ND | CXCL6, CXCL8 |
| CXCR2 | ND | ND | CXCL8, CXCL1, CXCL2, CXCL5 |
| CXCR3 | + | − | CXCL9, CXCL10, CXCL11 |
| CXCR4 | + | + | CXCL12 |
| CX3CR1 | ND | + | CX3CL1 |
| S1PR5 | − | + | S1P |
| CR23 (Cmklr1) | ND | ND | Chemerin |
References (human): Carrega et al. 31 , Lima et al. 34 , Berahovich et al. 36 , Kremer et al. 44 , Castriconi et al. 123 , Cheng et al. 124 , Li et al. 125 , Pachynski et al. 126 , Parodi et al. 127 , Pesce et al. 128 , Zhang et al. 129 References (mice): Marquardt et al. 38 , Bernardini et al. 130 , Mayol et al. 131 , Robinson et al. 132 , Susek et al. 133 ++, high expression; +, expression; −, no or low expression; ND, undetermined
Figure 2.

Chemokine expression patterns in murine NK cell subsets. RNA‐seq data from the ImmGen database (http://www.immgen.org/) show the differential expression of various chemokine receptors on mature CD27–CD11b+ , intermediate CD27+Cd11b+ and immature CD27+CD11b– NK cells. The expression value is plotted on the y‐axis.
Of course, chemokine receptor expression is not static, and can be modulated on NK cells during proliferation or by secreted factors enriched in the TME. Transforming growth factor (TGF)‐β, a well‐known immunosuppressive cytokine, can downregulate CX3CR1 on human NK cells. 42 Furthermore, CX3CR1 expression rapidly declines after 5 days in culture with IL‐15, 43 and ex vivo expanded NK cells decrease CXCR2 expression following 8 days of culture in vitro, 44 suggesting that proliferation and maturation of human NK cells may negatively impact their recruitment to the solid TME. However, CXCR2 and CXCR1 overexpression can improve NK cell migration, resulting in increased killing of tumor cells in vitro and in vivo human tumor xenograft models. 44 , 45 Thus, it will be important to genetically engineer NK cells to constitutively express chemokine receptors based on the chemokine expression profile present in individual cancer biopsies. Future studies will be needed to understand the specific chemokine gradients that preferentially recruit mNK cells to specific TMEs to enhance anti‐tumor responses.
Immunosuppressive Factors in the Tumor Microenvironment
Hypoxia
Solid tumors have been characterised by low oxygen tension which can result from rapid cancer cell proliferation, metabolism, and reduced nutrient and oxygen transport as a result of irregular solid tumor vasculature. 46 , 47 Lymphocytes can transcriptionally and metabolically adapt to low levels of oxygen (hypoxia) through hypoxia‐inducible factor 1A (HIF‐1α). 48 HIF‐1α regulates gene expression programs involved in many processes, such as cell proliferation, survival, glucose metabolism, and angiogenesis. 49 It has been demonstrated that NK cells are hyporesponsive under hypoxic conditions in vitro, displaying reduced expression of NKG2D, CD107a, and granzyme B with concomitant reductions in cytotoxicity. 49 , 50 , 51 , 52 Similarly, human peripheral NK cells cultured under hypoxic conditions in vitro displayed reduced IFN‐γ, granzyme B, and CD107a protein levels that could be restored using a HIF‐1α inhibitor. 53 Experiments using in vivo tumor models have proposed that HIF‐1α−/− NK cells promote tumor clearance by inhibiting vascular endothelial growth factor (VEGF)‐driven angiogenesis. 48 A recent study demonstrated that HIF‐1α−/− NK cells promoted tumor clearance in vivo through an increase in oxygen consumption rate and higher levels of granzyme B compared to WT NK cells. 54 HIF‐1α may regulate NK cell mitochondrial dynamics during periods of oxidative stress, because human NK cells in the liver TME and under ex vivo hypoxic conditions displayed small and fragmented mitochondria. 53 Mitochondrial fragmentation occurred through mTOR‐Drp1 signalling and silencing Drp1 gene expression using siRNA was sufficient to restore mitochondrial size and function, cytotoxicity, and in vivo efficacy of NK cells that were previously cultured under hypoxic conditions. 53 , 54 Together, this evidence suggests HIF‐1α and Drp1 may be candidates to enhance NK cell anti‐tumor responses and highlight the need for more mechanistic studies profiling mitochondrial dynamics in the regulation of NK cell effector functionality.
Transforming growth factor beta (TGFβ)
The TME is infiltrated with a preponderance of immunosuppressive cells such as tumor‐associated macrophages, myeloid‐derived suppressor cells (MDSCs), and regulatory T cells (Tregs). These cells can produce immunosuppressive signals like Prostaglandin E2 (PGE2), indoleamine 2,3‐dioxygenase (IDO), and TGF‐β which are capable of suppressing NK cell effector function 47 (Figure 3). Soluble modulators in the TME can negatively regulate NK cell activity by acting either directly on the NK cells or by acting on other immune cell types to produce additional suppressive molecules. 46 TGF‐β is a cytokine produced in the TME by tumor cells, Tregs, MDSCs, and other stromal cells. In addition to modifying the chemokine receptor repertoire of NK cells, TGF‐β can inhibit NK cell cytotoxicity and proliferation. TGF‐β impairs NK cell function directly by limiting NK cell antibody‐dependent cellular cytotoxicity (ADCC) and IFN‐γ production through inhibition of the transcription factor SMAD3. 55 TGF‐β is also capable of downregulating the expression of activating receptors NKG2D on peripheral NK cells derived from glioblastoma patients. 56 Antibody‐mediated depletion of TGF‐β can restore NKG2D expression on NK cells, indicating the therapeutic potential for targeting TGF‐β or its receptor. 57 Furthermore, TGF‐β is capable of upregulating fructose‐1,6‐bisphosphatase (FBP1) expression in NK cells which can lead to dysfunction by inhibiting glycolysis and impairing viability during tumor development. 58 While small molecule inhibition of FBP1 was observed to partially rescue glycolytic capability and viability of NK cells, cytotoxicity was independent of FPB1. Accordingly, in vivo tumor clearance was only partially improved following adoptive transfer of FBP1‐inhibited NK cells. 58 These results suggest that transient inhibition of FBP1 is not sufficient for maximal NK cell anti‐tumor activity and could benefit from a permanent reduction in FBP1 activity using genetic engineering approaches.
Figure 3.

The suppressive tumor microenvironment. Suppressive molecular signals are normally required to maintain tissue homeostasis and prevent damage after inflammation. However, when these signals (in red text) are expressed in the TME they can dampen NK cell responses and promote tumor escape. A majority of NK cells are often found in an immature, dysfunctional state in the TME indicated by blue iNK cells. Poorly structured tumor vasculature and rapidly proliferating tumor cells which consume much of the available oxygen result in an environment devoid of oxygen leaving the TME in a hypoxic state. Hypoxia is indicated by the lack of oxygen (O) molecules in the tissue. Highly glycolytic tumor cells also produce significant levels of lactate which acidifies the TME, further suppressing NK cell cytotoxic activity. In a series of enzymatic reactions, ATP is converted to AMP which is then converted to adenosine by tumor bound ectoenzymes CD39 and CD73, respectively. Adenosine suppresses NK cell cytotoxic function through receptor A2AR. Tumor cells with high COX2 activity can produce PGE2 which also suppresses NK cell cytotoxic function. Tumor‐derived enzyme IDO catabolises tryptophan in the TME thus starving NK cells of this essential amino acid. In additional to tumor cells, other cells present in the TME such as Tregs, MDSCs, and stromal cells are all capable of producing highly immunosuppressive molecule TGFβ which can directly by limit NK cell ADCC and IFN‐γ production.
Prostaglandin E2 (PGE2)
Tumor cells can produce high levels of PGE2 due to elevated cyclooxygenase‐2 (COX‐2) activity in tumor cells. 59 Early efforts using COX inhibitors showed therapeutic efficacy dependent on T cells or NK cells, but COX‐2 inhibitors exhibited safety concerns regarding toxicity. 59 PGE2 can signal through four G‐protein‐coupled receptors (EP1–4) expressed on cytotoxic lymphocytes, primarily EP4 in NK cells. 60 Experimental evidence suggests that a small molecule inhibitor of EP4 can restore IFN‐γ production, cell migration and anti‐tumor activity of mouse NK cells. 59 , 60 A recent study demonstrated that tumor‐derived PGE2 can suppress the production of CCL5 and XCL1 by mouse NK cells, which results in decreased recruitment of XCR1+ dendritic cells (cDC1s) and increased tumor burden. 10 Given the immunosuppressive role of PGE2 in the TME, it will be necessary for future experiments to determine whether EP4−/− NK cells display restored chemokine production and anti‐tumor function.
Extracellular metabolites
IDO is an enzyme overexpressed by cancer cells that catalyses the first step in the kynurenine pathway of tryptophan catabolism, thus depleting tryptophan from the TME and starving T and NK cells of this essential amino acid. 61 The by‐product of this reaction, L‐kynurenine, has additional immunosuppressive effects by directly downregulating expression of activating receptors NKG2D and NKp46 on NK cells. 61 Despite promising results from early phase clinical trials using IDO inhibitors, a phase III study in conjunction with programmed death‐1 (PD‐1) immune checkpoint blockade (ICB) ultimately showed no efficacy compared to ICB alone. 61 However, other approaches to inhibit this pathway continue to be considered. L‐kynurenine is imported into NK cells exclusively by transporter SLC7A5, making it a potentially attractive target to ablate during adoptive NK cell therapy. However, SLC7A5 is a primary L‐amino acid transporter in activated NK cells, and SLC7A5 is required for mTORC1‐driven cMYC activity which controls NK cell effector function. 62 A more complete understanding of L‐kynurenine‐driven mechanisms in NK cells will be required to design effective strategies to abrogate the immune suppressive effects in the TME without hindering NK cell effector function.
Rapidly proliferating tumors are recognised as massive consumers of ATP in order to fuel increased growth and proliferation during tumorigenesis. 46 Ectoenzymes CD39 and CD73 often found on tumor cells as well as some immune cells, catalyse ATP to AMP and finally to adenosine. 63 Adenosine is an important immunosuppressive metabolite critical for protecting tissues against damage during inflammatory responses. 64 Adenosine can signal through four G‐protein‐coupled adenosine receptors (A1, A2A, A2B and A3). 65 Adenosine receptor A2A (A2AR) is highly expressed on lymphocytes, including NK cells. Collective reports have demonstrated exogenous adenosine can reduce NK cell cytotoxicity and cytokine production in vitro. 65 , 66 This hypothesis is supported by experiments demonstrating A2AR −/− NK cells exhibit a mature (CD11b+CD27–) and cytotoxic phenotype capable of robust proliferation and increased tumor clearance in mouse tumor models. 67 Upon A2AR small molecule inhibition, human NK cells also display increased phenotypic maturation and cytotoxicity. 67 Further studies outlining the role of A2AR in human NK cells using preclinical humanised mouse models of cancer will be needed to validate whether A2AR is an attractive target for adoptive NK cell therapy.
Lactate is another immunosuppressive metabolite capable of limiting anti‐tumor responses. 47 The accumulation of lactate in the TME is mainly due to metabolic reprogramming of tumors, where instead of using oxidative phosphorylation, tumor cells primarily use glucose for glycolytic metabolism which produces lactate as a by‐product. 68 Lactate can directly decrease cytotoxic activity in mouse and human NK cells demonstrated by lower expression of granzyme B and perforin. 69 , 70 Lactate was capable of upregulating transcription factor NFAT in NK cells which led to a decrease in IFN‐γ and NKp46. 69 This cumulative evidence suggests lactate can directly inhibit NK cytolytic activity in the TME. Lactate also increases the number of MDSCs in the TME which inhibit NK cell cytotoxicity. 69 An additional study found liver‐resident NK cells migrating towards colorectal liver metastasis were unable to regulate intracellular pH levels resulting from lactate‐induced acidity of the TME, causing mitochondrial stress and apoptosis. 71 Further studies understanding how to improve NK cell responses in acidic environments will be essential to designing successful NK cell therapy in response to solid tumors.
NK cell intracellular checkpoints
IL‐15 pathway regulators
NK cells are reliant on IL‐15 signalling to maintain cellular homeostasis. 72 Recent studies have identified a number of intracellular negative regulators for NK cell activation, summarised in (Figure 4). One recent study has identified Otub1 as an important regulator of IL‐15 receptor signalling in NK cells and CD8+ T cells. 73 IL‐15 signalling recruits Otub1 to the membrane where it prevents the activation of AKT. Otub1 likely negatively regulates IL‐15 signalling in vivo because Otub1 −/− NK cells had significantly more CD11b+CD27– mNK cells capable of producing higher levels of granzyme B and CCL5. 73 Interestingly Otub1 −/− CD8 T cells had increased transcript levels of Cish, a known suppressor of IL‐15 signalling in NK cells. 74 Mice with inducible deletion of Otub1 during adulthood experienced reduced tumor burden and increased NK and CD8 T‐cell tumor infiltration. 73 Similarly, Cish −/− NK cells proliferate better in response to low levels of IL‐15 and show greater anti‐tumor efficacy in multiple mouse models. 74 , 75 CISH −/− human NK cells derived from induced pluripotent stem cells (iPSCs) demonstrate superior killing of tumor cells due to an improved metabolic profile compared to WT cells. 76 Several groups have already demonstrated successful genetic targeting of CISH in human NK cells resulting in enhanced anti‐tumor efficacy using humanised tumor models. 76 , 77 Together, these data suggest that targeting negative feedback mechanisms following IL‐15 signalling is a beneficial strategy to enhance NK cell anti‐tumor responses in vivo.
Figure 4.
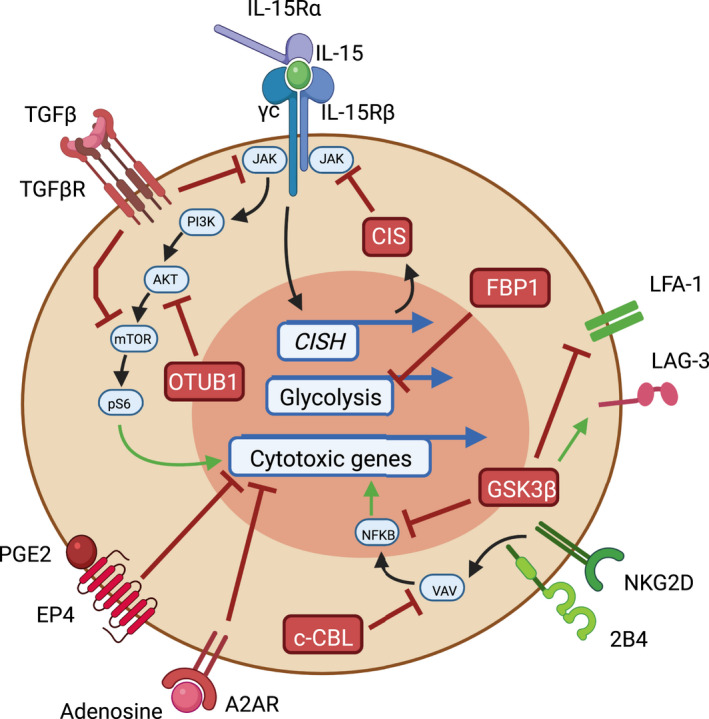
NK cell intrinsic checkpoints. Essential intracellular signalling pathways used by NK cells during an anti‐tumor response are depicted. Red targets and receptors are negative regulators while green arrows and receptors promote NK cell activation or cytotoxicity. IL‐15 signalling is required for NK cell proliferation, survival and anti‐tumor function in the TME. IL‐15 signalling drives expression of negative regulator CISH, producing CIS which acts as negative feedback loop towards IL‐15 signalling. OTUB1 can also suppress IL‐15 signalling by preventing activation of AKT. TGFβ can signal through its receptor TGFβR leading to suppression of multiple downstream targets of IL‐15 signalling. Furthermore, TGF‐β is capable of upregulating FBP1 expression in NK cells which can lead to dysfunction by inhibiting glycolysis. PGE2 through binding receptor EP4 and adenosine through binding A2AR can directly inhibit cytotoxic activity during anti‐tumor NK cell responses. c‐CBL can also inhibit VAV1 which is a common downstream signalling component of activating receptors NKG2D and 2B4 thereby suppressing NFκB‐driven cytotoxic programs. Additionally, GSK3β can inhibit NK cells at multiple points by suppressing NFκB and LFA‐1 expression. GSK3β is also capable of upregulating immunosuppressive surface receptor LAG‐3.
Glycogen synthase kinase‐3 (GSK3)
GSK3 is a serine‐threonine kinase which can influence gene expression directly by targeting transcription factors or indirectly by phosphorylating histones, histone deacetylases, and histone acetyltransferases. 78 15 years ago, small molecule inactivation of GSK‐3 increased the cytotoxicity and cytokine secretion of NK cells from individuals with XLP disease. 79 GSK‐3 inhibition in healthy donor NK cells was also shown to increase IFN‐γ, granzyme B and specific tumor cell lysis. 79 Furthermore, GSK‐3 inhibition increased the expression of integrin LFA‐1 on NK cells which can recognise ICAM1 present on AML cells resulting in improved specific cell lysis. 80 Recent work has further defined the benefit of GSK‐3 inhibition in NK cells during an adoptive transfer setting. CD57 is a maturation marker in human NK cells which is associated with increased cytotoxicity, TNF‐⍺ and IFN‐γ production. 16 The frequency of CD57+ NK cells is often higher in individuals who are seropositive for cytomegalovirus (CMV), and dimly expressed on CD56bright adult NK cells. 81 The CD57+NKG2C+ NK cell subset is considered to be the human equivalent of mouse Ly49H+ memory NK cells following MCMV infection, because they display memory‐like properties such as heightened effector function, persistence, and rapid recall responses. 82 GSK3 inhibition of NK cells from CMV seropositive donors during ex vivo NK cell expansion with IL‐15 significantly enhanced CD57 acquisition and maturation. Furthermore, GSK3‐inhibited NK cells displayed increased transcript levels of TBX21, ZEB2, and BLIMP‐1 which positively regulate NK cell maturation. 81 Additionally, GSK3‐inhibited NK cells demonstrated improved anti‐tumor efficacy in an adoptive transfer setting into humanised mice. 81 Another study suggested that GSK‐3 inhibition can downregulate the inhibitory receptor lymphocyte activation gene 3 (LAG‐3) on NK cells, further improving tumor clearance. 83 Thus, collective evidence suggests that approaches to therapeutically limit GSK3 function should improve NK cell anti‐tumor function in vivo.
Cbl Proto‐Oncogene (CBL)
CBL proteins, including c‐CBL and CBL‐b, are adaptor molecules with multiple functions, including E3 ubiquitin ligase activity. CBL proteins primarily repress signalling of lymphocyte receptors by promoting ubiquitylation, resulting in internalisation of surface receptors. 84 The TAM (Tyro3, Axl, and Mer) receptor family are receptor tyrosine kinases expressed by many immune cells including NK cells. TAM receptors directly phosphorylate E3 ubiquitin ligase CBL‐b which can then inhibit NK cell function by degrading important molecules such as SLC7A5, which is required for NK1.1‐ or NKG2D‐mediated activation and cytokine production. 85 Indeed, adoptive transfer of Cbl‐b −/− NK cells reduces tumor metastasis compared to transfer of WT NK cells. 86 c‐CBL can also inhibit VAV1 which is a common downstream signalling component of activating receptors NKG2D and 2B4. 87 A study demonstrated knockdown of c‐Cbl but not Cbl‐b enhanced NK cell cytotoxicity and cytokine secretion through a VAV1 dependent signalling cascade. 87 Given these conflicting results, future work will be necessary to understand the precise role of c‐CBL and CBL‐b in human NK cell anti‐tumor function.
Strategies to enhance endogenous NK cell function in the TME
Cytokine therapies
To initiate and sustain a proper immune response, pro‐inflammatory cytokines IL‐2, IL‐15, IL‐12, IL‐18 and IL‐21 are required to drive NK cell activation, proliferation and cytotoxicity. 1 Clinical efforts to stimulate NK cell anti‐tumor responses have used cytokines to increase NK cell activation and abundance within the TME. Early studies using IL‐2 proved to be unsuccessful due to activation of regulatory T (Treg) cells in addition to CD8+ T and NK cells. 88 Alternative IL‐2 molecules have been developed with decreased affinity for IL‐2R⍺ which is predominantly expressed on Treg cells. IL‐2Rβ‐biased agonist NKTR‐214 is shown to expand CD8+ T and NK cells with limited increase in Tregs. 89 In combination with anti‐PD‐1 immunotherapy, phase II clinical trial results indicate an objective response rate (ORR) of 53% (20/38), and a complete response rate (CR) of 34% (13/38) at a median follow up time of 18.6 months. 90 Phase III trials are currently underway using NKTR‐214 in combination with anti‐PD‐1 in renal cell carcinoma, melanoma and muscle‐invasive bladder cancer, 90 Alternatively, IL‐15 represents a preferred route of activating NK cells since IL15Rα expressed on antigen‐presenting cells can present IL‐15 in trans to IL‐2Rβγc receptors expressed on NK and CD8+ T cells without activating Tregs. 91 Recombinant human (rh)IL‐15 in combination with haploidentical NK cell infusions in refractory AML patients has shown a 35% CR, better than IL‐2 in previous trials, indicating rnIL‐15 is capable of augmenting adoptive NK cell therapy. In addition to rhIL‐15, IL‐15 superagonists such as ALT‐803 are capable of targeting both the IL‐2 and IL‐15 pathway in NK and CD8+ T cells. In phase I studies, ALT‐803 was shown to have some clinical response as a monotherapy and in combination with anti‐EGFR and anti‐PD‐1. 92 , 93 , 94 Additional studies have demonstrated an overnight activation with IL‐12, IL‐15, and IL‐18 generated superior NK cells with memory‐like capabilities exhibiting enhanced responses to cytokine or activating receptor restimulation for weeks to months after pre‐activation. 95 A phase 1 clinical trial using adoptive transfer of these cytokine‐induced memory‐like (CIML) NK cells with rhIL‐2 resulted in CR in four out of nine AML patients. 95 During the phase II trials, rhIL‐2 was replaced with IL‐15 superagonist ALT‐803 since studies have shown ALT‐803 resulted in better NK cell expansion than rhIL‐2. Interestingly a large expansion of host T cells resulted in the killing of donor CIML NK cells, suggesting that key caveats exist within systemic cytokine administration for tumor therapy. 95 Collectively, these studies indicate cytokine therapies may prove to be an effective and well‐tolerated treatment; however, these strategies are primarily efficacious in liquid tumors and it is not yet clear how effective these treatments will be for solid tumors.
Bispecific and trispecific killer cell engagers (BiKEs and TriKEs)
In addition to NK cell‐mediated tumor killing through reduced expression of MHC‐I and increased activating receptor ligands, NK cells also possess the Fc receptor CD16 which can initiate ADCC. 96 CD16 is primary expressed on CD56dim mNK cells which account for a majority of the NK cells in peripheral blood. CD16‐mediated ADCC is triggered by engagement with the Fc region of IgG Abs which signals through the immunoreceptor tyrosine‐based activation motif (ITAM) in association with adaptor molecules FcεR1γ and CD3ζ. 96 In order to further optimise this process, bispecific killer cell engager (BiKE) molecules possessing an anti‐CD16 scFv linked to an scFv specific to a tumor‐expressed antigen have been developed to bring CD16 and tumor epitopes in close contact. Preclinical models have suggested anti‐HER2 x anti‐CD16 BiKE molecules allow for superior ADCC compared to the Fc region alone on anti‐HER2 monoclonal antibody treatment. 97 To further improve this treatment, trispecific killer cell engager TriKE molecules can possess a third arm containing a cytokine such as IL‐15. This has resulted in even more robust NK cell ADCC compared to BiKE therapy, demonstrated by better human NK cell expansion and tumor control in a preclinical AML xenograft mouse model. 98 BiKE and TriKE molecules are relatively small modalities, much easier to manufacture than adoptive cell therapies, and can be used off‐the‐shelf. 99 In addition, the utilisation of multiple arms and the possibility to add even a fourth arm in TetraKEs allows for complete customisation of killer cell engagement. IL‐15 can be swapped for another potent cytokine such as IFNγ or for a scFv blocking TGFβ, IDO or PGE2 signalling. Because these molecules can be engineered in an unlimited number of ways, further research will be necessary to determine the optimal combination of modalities that results in superior NK cell anti‐tumor activity.
Antibody‐mediated checkpoint blockade therapies
Immunotherapy for the treatment of cancer is rapidly growing and even becoming standard of care for some tumor types. 100 These initial breakthroughs have focused on ICB, an antibody (Ab)‐mediated therapy where blocking Abs are used to inhibit regulatory surface proteins such PD‐1, programmed death‐ligand 1 (PD‐L1) and cytotoxic T lymphocyte‐associated antigen 4 (CTLA‐4), which suppress T‐cell activation. 101 In addition to these three targets, a number of new surface receptors such as T‐cell immunoglobulin and mucin‐domain‐containing molecule 3 (TIM‐3), T‐cell immunoreceptor with Ig and immunoreceptor tyrosine‐based inhibition motif domains (TIGIT), LAG‐3 and CD96 have been found on multiple immune cell types such T cells, NK cells and dendritic cells and are being recognised for their potential as additional ICB targets 102 , 103 , 104 and has been reviewed previously. 105 These inhibitory pathways are fairly well understood in T cells but further understanding of the underlying biological mechanisms across the immune landscape in the TME will be useful in designing new ICB therapies. One inhibitory pathway that has been successfully targeted in NK cells in a clinical setting is NKG2A. Tumors can evade immune clearance by expression of HLA class I molecule (HLA‐E), which suppresses NK cell activity via ligation of the NK inhibitory receptor NKG2A. Blocking expression of NKG2A has been shown to overcome tumor resistance to NK cells in a preclinical model. 15 Monalizumab, a humanised IgG4 checkpoint inhibitor targeting NKG2A, is entering Phase III clinical trials in combination with cetuximab, a clinically approved anti‐EGFR antibody. The phase II clinical trial results of the combination of monalizumab and cetuximab, demonstrated a 30% objective response rate in immunotherapy‐refractory in patients with head and neck squamous cell carcinoma compared to 12.6% in previous studies using cetuximab alone. 106 Although these results are promising, it may prove to be even more useful to generate NKG2A deficient NK cells for use in adoptive transfer since NK cells are often found in very low numbers in the TME 18 and may not effectively respond to anti‐NKG2A therapy.
NK cell adoptive cancer therapies
The earliest NK cell therapies using adoptively transferred haploidentical NK cells have been successful against certain liquid tumors like acute myeloid leukaemia (AML). 107 However, adoptive cell therapy using T lymphocytes has been under research for over 30 years 108 and has gained massive attraction over the last decade with the use of Chimeric Antigen Receptor (CAR) T‐cell therapy. CAR molecules are designed to direct T cells to recognise a specific tumor antigen. This has proven to be a very successful therapy against liquid tumors such as lymphomas expressing CD19. 109 However, reports have indicated that CD19− lymphoma cells can arise after CAR T mediated tumor epitope editing. 110 Bispecific CAR T cells have been designed to target CD19 and CD20 to prevent antigen escape, 111 yet it remains possible that CD19–CD20– tumor cells could also develop after using this treatment strategy. In addition, life‐threatening complications related to Cytokine Release Syndrome (CRS) have occurred resulting in the death of some patients receiving CAR T therapy. 112 Indeed, nearly all patients experience at least mild CRS during CAR T‐cell therapy infusion, leading many to try and find alternative treatments with reduced potential complications. 112 CAR‐NK cells have shown promise as an alternative therapy demonstrated by anti‐tumor efficacy against a number of preclinical targets without indications of CRS. 113 A recent clinical trial reported a 73% response rate in non‐Hodgkin’s lymphoma or chronic lymphocytic leukaemia patients using HLA‐mismatched anti‐CD19 CAR‐NK cells derived from cord blood progenitors. 114 NK cells were transduced with a retroviral vector expressing genes for an anti‐CD19 CAR, IL‐15, and an inducible caspase 9 safety switch. Importantly, zero patients experienced CRS, neurotoxicity, or graft‐versus‐host disease, and they reported no increase in inflammatory cytokines across 18 patients. 114 It will be important to determine whether these results hold true across a larger number of patients and whether the responses are durable. One concern is that NK cells do not form lifelong memory, like T cells, so relapse may be more likely using CAR‐NK cells. However, multiple infusions of CAR‐NK cells could possibly circumvent this problem. Another concern is that NK cells are more difficult to expand ex vivo and often require the use of feeder cells to initiate robust ex vivo expansion. 115 Similar to CAR T‐cell treatments, the successes of CAR NK cells against liquid tumors have been encouraging, although response rates during adoptive cell therapies against solid tumors have room for much improvement.
NK cell genetic engineering for adoptive cancer therapy
CRISPR Cas9 is a gene editing technology which has revolutionised the field of molecular biology. The recombinant Cas9 nuclease protein is paired with a guide RNA/scaffold, together this is referred to as CRISPR ribonucleoprotein (RNP) which is capable of making a double stranded DNA break which renders a protein non‐functional. 116 NK cells are some of the most difficult immune cells to edit using transfection or other viral methods. However, recent studies have demonstrated high efficiency gene knockout using CRISPR RNPs is possible in mouse and human NK cells. 76 , 77 , 117 CISH has been successfully targeted in both iPSC derived and primary human NK cells using CRISPR RNPs. 76 , 77 CRISPR RNP technology has been useful in adoptive T‐cell therapy, demonstrated by PD‐1−/− CAR T cells 118 and CAR T cells generated using Homology Directed Repair (HDR). 119 In one elegant study, a CAR construct was inserted into the T‐cell receptor (TCR) TRAC locus thus knocking out TCR protein expression. This resulted in superior CAR T cells with uniform CAR expression, reduced tonic signalling, reduced T‐cell exhaustion and improved anti‐tumor efficacy in a humanised murine tumor model. 119 Although high efficiency CRISPR‐HDR has remained difficult in primary NK cells, 120 a recent study has demonstrated a number of optimised genetic engineering approaches in NK‐92 cells. 121 NK‐92 cells were edited to have multiple gene knockouts of inhibitory receptors, knock‐in of a fluorescent protein, and insertion of constitutive promoters to reactivate endogenous CD16 and DNAM‐1 expression. CRISPR‐engineered NK‐92 cells demonstrated strikingly enhanced cytotoxicity and could even mediate ADCC against previously hard to kill cancer cell lines. 121 Employing these methods in primary human NK cells could significantly improve adoptive NK cell therapy (Figure 5). In theory, tumor biopsies could be sampled for expression of various chemokine ligands and a personalised chemokine panel could be generated for each patient. Reactivation of endogenous promoters for those specific chemokine receptors required for recruitment to their TME could be restored, thus improving migration of ex vivo expanded NK cells to the TME. Once these engineered cells have arrived, they could be devoid of intracellular sensors which respond to hypoxia and other immunosuppressive molecules allowing their full cytotoxic capability. Future studies leveraging new advances in CRISPR editing technology will be important for enhancing adoptive NK cell‐based therapies for cancer.
Figure 5.

Genome engineering in NK cells. CRISPR Cas9 RNPs can be used to delete deleterious genes for NK cell function. CRISPR Cas9 RNP‐mediated HDR insertion of exogenous promoter elements can drive expression of activating genes which may be downregulated in the TME or during ex vivo expansion of NK cells.
Conclusions and future perspectives
Current evidence suggests that NK cells face many obstacles to become potently cytotoxic cells in advanced human solid cancers without clinical intervention. Current ICB strategies which target a single molecule such as PD1 are ineffective for a majority of cancer patients, suggesting multifaceted approaches are necessary to bolster the immune response to cancer. Furthermore, T‐cell‐centric immunotherapies and systemic ICB are associated with increased risk of severe immune adverse events, such as CRS. Recent studies have suggested that double gene knockouts are possible to generate with CRISPR RNPs in primary mouse NK and T cells. 117 , 122 It will be important to screen for signs of autoreactivity in these highly engineered NK cells since multiple ‘brakes’ could be removed during this process. However, the repertoire of non‐redundant inhibitory receptors present on primary NK cells may serve to create safer off‐the‐shelf cellular therapies when compared to engineered T cells. Thus, specifically targeting multiple immunosuppressive checkpoints in human NK cells may prove to be the most effective form of anti‐tumor therapy. Targeted multiplexed immunotherapies will one day be possible thanks to these recent advances in genome engineering and continued understanding of the immunosuppressive mechanisms that exist in the solid TME. We envision creating ‘super’ NK cells that are capable of rapidly entering the TME, are resistant to the immunosuppressive environment, retain their enhanced cytotoxicity against solid tumors and are safely designed to prevent CRS. NK cell‐based therapy for solid tumors is poised to break out in the next decade and may offer more robust results than current T‐cell‐based or systemic therapies.
Conflict of Interest
TEO is a scientific advisor for NKMax America Inc.
Author Contribution
Luke Riggan: Conceptualization; Visualization; Writing‐original draft. Siya Shah: Writing‐original draft. Timothy O'Sullivan: Conceptualization; Project administration; Supervision; Writing‐original draft; Writing‐review & editing.
Acknowledgments
We thank Andrew Hildreth of the O’Sullivan lab for helpful discussion. TEO was supported by the NIH (AI145997) and UC CRCC (CRN‐20‐637105)
References
- 1. Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol 2016; 16: 112–123. [DOI] [PubMed] [Google Scholar]
- 2. López‐Soto A, Gonzalez S, Smyth MJ, Galluzzi L. Control of Metastasis by NK Cells. Cancer Cell 2017; 32: 135–154. [DOI] [PubMed] [Google Scholar]
- 3. Dadi S, Chhangawala S, Whitlock BM et al Cancer Immunosurveillance by tissue‐resident innate lymphoid cells and innate‐like T Cells. Cell 2016; 164: 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. O’Sullivan T, Saddawi‐Konefka R, Vermi W et al Cancer immunoediting by the innate immune system in the absence of adaptive immunity. J Exp Med 2012; 209: 1869–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Souza‐Fonseca‐Guimaraes F, Cursons J, Huntington ND. The emergence of natural killer cells as a major target in cancer immunotherapy. Trends Immunol 2019; 40: 142–158. [DOI] [PubMed] [Google Scholar]
- 6. Li B, Jiang Y, Li G, Fisher GA Jr, Li R. Natural killer cell and stroma abundance are independently prognostic and predict gastric cancer chemotherapy benefit. JCI. Insight 2020; 5: e136570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huntington ND, Cursons J, Rautela J. The cancer‐natural killer cell immunity cycle. Nat Rev Cancer 2020; 20: 437–454. [DOI] [PubMed] [Google Scholar]
- 8. Denkert C, von Minckwitz G, Darb‐Esfahani S et al Tumour‐infiltrating lymphocytes and prognosis in different subtypes of breast cancer: a pooled analysis of 3771 patients treated with neoadjuvant therapy. Lancet Oncol 2018; 19: 40–50. [DOI] [PubMed] [Google Scholar]
- 9. Terren I, Orrantia A, Mikelez‐Alonso I, Vitalle J, Zenarruzabeitia O, Borrego F. NK cell‐based immunotherapy in renal cell carcinoma. Cancers (Basel) 2020; 12: 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bottcher JP, Bonavita E, Chakravarty P et al NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune control. Cell 2018; 172: 1022–1037.e1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Barry KC, Hsu J, Broz ML et al A natural killer‐dendritic cell axis defines checkpoint therapy‐responsive tumor microenvironments. Nat Med 2018; 24: 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barrow AD, Martin CJ, Colonna M. The natural cytotoxicity receptors in health and disease. Front Immunol 2019; 10: 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol 2011; 89: 216–224. [DOI] [PubMed] [Google Scholar]
- 14. O'Sullivan T, Dunn GP, Lacoursiere DY, Schreiber RD, Bui JD. Cancer immunoediting of the NK group 2D ligand H60a. J Immunol 2011; 187: 3538–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Invest 2019; 129: 2094–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Freud AG, Mundy‐Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity 2017; 47: 820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duan S, Guo W, Xu Z et al Natural killer group 2D receptor and its ligands in cancer immune escape. Mol Cancer 2019; 18: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bald T, Krummel MF, Smyth MJ, Barry KC. The NK cell‐cancer cycle: advances and new challenges in NK cell‐based immunotherapies. Nat Immunol 2020; 21: 835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deng W, Gowen BG, Zhang L et al Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science 2015; 348: 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Barrow AD, Edeling MA, Trifonov V et al Natural killer cells control tumor growth by sensing a growth factor. Cell 2018; 172: 534–548.e519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zaretsky JM, Garcia‐Diaz A, Shin DS et al Mutations associated with acquired resistance to PD‐1 blockade in melanoma. N Engl J Med 2016; 375: 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chiossone L, Chaix J, Fuseri N, Roth C, Vivier E, Walzer T. Maturation of mouse NK cells is a 4‐stage developmental program. Blood 2009; 113: 5488–5496. [DOI] [PubMed] [Google Scholar]
- 23. Richards JO, Chang X, Blaser BW, Caligiuri MA, Zheng P, Liu Y. Tumor growth impedes natural‐killer‐cell maturation in the bone marrow. Blood 2006; 108: 246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krneta T, Gillgrass A, Chew M, Ashkar AA. The breast tumor microenvironment alters the phenotype and function of natural killer cells. Cell Mol Immunol 2016; 13: 628–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paul S, Kulkarni N, Shilpi Lal G. Intratumoral natural killer cells show reduced effector and cytolytic properties and control the differentiation of effector Th1 cells. Oncoimmunology 2016; 5: e1235106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Swaminathan S, Hansen AS, Heftdal LD et al MYC functions as a switch for natural killer cell‐mediated immune surveillance of lymphoid malignancies. Nat Commun 2020; 11: 2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mattei F, Schiavoni G, Belardelli F, Tough DF. IL‐15 is expressed by dendritic cells in response to type I IFN, double‐stranded RNA, or lipopolysaccharide and promotes dendritic cell activation. J Immunol 2001; 167: 1179–1187. [DOI] [PubMed] [Google Scholar]
- 28. Melsen JE, Lugthart G, Lankester AC, Schilham MW. Human circulating and tissue‐resident CD56bright natural killer cell populations. Front Immunol 2016; 7: 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Crinier A, Milpied P, Escaliere B et al High‐dimensional single‐cell analysis identifies organ‐specific signatures and conserved nk cell subsets in humans and mice. Immunity 2018; 49: 971–986. e975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Carrega P, Morandi B, Costa R et al Natural killer cells infiltrating human nonsmall‐cell lung cancer are enriched in CD56bright CD16– cells and display an impaired capability to kill tumor cells. Cancer 2008; 112: 863–875. [DOI] [PubMed] [Google Scholar]
- 31. Carrega P, Bonaccorsi I, Di Carlo E et al CD56brightperforinlow noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J Immunol 2014; 192: 3805–3815. [DOI] [PubMed] [Google Scholar]
- 32. Levi I, Amsalem H, Nissan A et al Characterization of tumor infiltrating natural killer cell subset. Oncotarget 2015; 6: 13835–13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mamessier E, Sylvain A, Thibult ML et al Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest 2011; 121: 3609–3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lima M, Leander M, Santos M et al Chemokine Receptor Expression on Normal Blood CD56+ NK Cells Elucidates Cell Partners That Comigrate during the Innate and Adaptive Immune Responses and Identifies a Transitional NK‐Cell Population. J Immunol Res 2015; 2015: 839684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Heng TS, Painter MW. Immunological Genome Project C. The Immunological Genome Project: networks of gene expression in immune cells. Nat Immunol 2008; 9: 1091–1094. [DOI] [PubMed] [Google Scholar]
- 36. Berahovich RD, Lai NL, Wei Z, Lanier LL, Schall TJ. Evidence for NK cell subsets based on chemokine receptor expression. J Immunol 2006; 177: 7833–7840. [DOI] [PubMed] [Google Scholar]
- 37. Wendel M, Galani IE, Suri‐Payer E, Cerwenka A. Natural killer cell accumulation in tumors is dependent on IFN‐λ and CXCR3 ligands. Cancer Res 2008; 68: 8437–8445. [DOI] [PubMed] [Google Scholar]
- 38. Marquardt N, Wilk E, Pokoyski C, Schmidt RE, Jacobs R. Murine CXCR3+CD27bright NK cells resemble the human CD56bright NK‐cell population. Eur J Immunol 2010; 40: 1428–1439. [DOI] [PubMed] [Google Scholar]
- 39. O'Sullivan T, Saddawi‐Konefka R, Gross E et al Interleukin‐17D mediates tumor rejection through recruitment of natural killer cells. Cell Rep 2014; 7: 989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen E‐B, Zhou Z‐J, Xiao K et al The miR‐561‐5p/CX3CL1 Signaling Axis Regulates Pulmonary Metastasis in Hepatocellular Carcinoma Involving CX3CR1+ Natural Killer Cells Infiltration. Theranostics 2019; 9: 4779–4794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lavergne E, Combadiere B, Bonduelle O et al Fractalkine mediates natural killer‐dependent antitumor responses in vivo . Cancer Res 2003; 63: 7468–7474. [PubMed] [Google Scholar]
- 42. Regis S, Caliendo F, Dondero A et al TGF‐β1 Downregulates the Expression of CX3CR1 by Inducing miR‐27a‐5p in Primary Human NK Cells. Front Immunol 2017; 8: 868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sechler JM, Barlic J, Grivel JC, Murphy PM. IL‐15 alters expression and function of the chemokine receptor CX3CR1 in human NK cells. Cell Immunol 2004; 230: 99–108. [DOI] [PubMed] [Google Scholar]
- 44. Kremer V, Ligtenberg MA, Zendehdel R et al Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma. J Immunother Cancer 2017; 5: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Ng YY, Tay JCK, Wang S. CXCR1 Expression to Improve Anti‐Cancer Efficacy of Intravenously Injected CAR‐NK Cells in Mice with Peritoneal Xenografts. Mol Ther Oncolytics 2020; 16: 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Melaiu O, Lucarini V, Cifaldi L, Fruci D. Influence of the tumor microenvironment on NK cell function in solid tumors. Front Immunol 2019; 10: 3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Terren I, Orrantia A, Vitalle J, Zenarruzabeitia O, Borrego F. NK cell metabolism and tumor microenvironment. Front Immunol 2019; 10: 2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Krzywinska E, Kantari‐Mimoun C, Kerdiles Y et al Loss of HIF‐1α in natural killer cells inhibits tumour growth by stimulating non‐productive angiogenesis. Nat Commun 2017; 8: 1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chambers AM, Matosevic S. Immunometabolic Dysfunction of Natural Killer Cells Mediated by the Hypoxia‐CD73 Axis in Solid Tumors. Front Mol Biosci 2019; 6: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sarkar S, Germeraad WT, Rouschop KM et al Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL‐2 activation of the NK cells. PLoS One 2013; 8: e64835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Solocinski K, Padget MR, Fabian KP et al Overcoming hypoxia‐induced functional suppression of NK cells. J Immunother Cancer 2020; 8: e000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Velasquez SY, Killian D, Schulte J, Sticht C, Thiel M, Lindner HA. Short Term Hypoxia Synergizes with Interleukin 15 Priming in Driving Glycolytic Gene Transcription and Supports Human Natural Killer Cell Activities. J Biol Chem 2016; 291: 12960–12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zheng X, Qian Y, Fu B et al Mitochondrial fragmentation limits NK cell‐based tumor immunosurveillance. Nat Immunol 2019; 20: 1656–1667. [DOI] [PubMed] [Google Scholar]
- 54. Ni J, Wang X, Stojanovic A et al Single‐Cell RNA Sequencing of Tumor‐Infiltrating NK Cells Reveals that Inhibition of Transcription Factor HIF‐1α Unleashes NK Cell Activity. Immunity 2020; 52: 1075–1087.e1078. [DOI] [PubMed] [Google Scholar]
- 55. Trotta R, Dal Col J, Yu J et al TGF‐β utilizes SMAD3 to inhibit CD16‐mediated IFN‐λ production and antibody‐dependent cellular cytotoxicity in human NK cells. J Immunol 2008; 181: 3784–3792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF‐β downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol 2010; 12: 7–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lazarova M, Steinle A. Impairment of NKG2D‐Mediated Tumor Immunity by TGF‐β. Front Immunol 2019; 10: 2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cong J, Wang X, Zheng X et al Dysfunction of Natural Killer Cells by FBP1‐Induced Inhibition of Glycolysis during Lung Cancer Progression. Cell Metab 2018; 28: 243–255.e245. [DOI] [PubMed] [Google Scholar]
- 59. Holt D, Ma X, Kundu N, Fulton A. Prostaglandin E2 (PGE2) suppresses natural killer cell function primarily through the PGE2 receptor EP4. Cancer Immunol Immunother 2011; 60: 1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ma X, Holt D, Kundu N et al A prostaglandin E (PGE) receptor EP4 antagonist protects natural killer cells from PGE2‐mediated immunosuppression and inhibits breast cancer metastasis. Oncoimmunology 2013; 2: e22647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Labadie BW, Bao R, Luke JJ. Reimagining ido pathway inhibition in cancer immunotherapy via downstream focus on the tryptophan‐kynurenine‐aryl hydrocarbon axis. Clin Cancer Res 2019; 25: 1462–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loftus RM, Assmann N, Kedia‐Mehta N et al Amino acid‐dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun 2018; 9: 2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Neo SY, Yang Y, Record J et al CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J Clin Invest 2020; 130: 1185–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Chambers AM, Wang J, Lupo KB, Yu H, Atallah Lanman NM, Matosevic S. Adenosinergic signaling alters natural killer cell functional responses. Front Immunol 2018; 9: 2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lokshin A, Raskovalova T, Huang X, Zacharia LC, Jackson EK, Gorelik E. Adenosine‐mediated inhibition of the cytotoxic activity and cytokine production by activated natural killer cells. Cancer Res 2006; 66: 7758–7765. [DOI] [PubMed] [Google Scholar]
- 66. Beavis PA, Divisekera U, Paget C et al Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc Natl Acad Sci USA 2013; 110: 14711–14716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Young A, Ngiow SF, Gao Y et al A2AR adenosine signaling suppresses natural killer cell maturation in the tumor microenvironment. Cancer Res 2018; 78: 1003–1016. [DOI] [PubMed] [Google Scholar]
- 68. Potter M, Newport E, Morten KJ. The Warburg effect: 80 years on. Biochem Soc Trans 2016; 44: 1499–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Husain Z, Huang Y, Seth P, Sukhatme VP. Tumor‐derived lactate modifies antitumor immune response: effect on myeloid‐derived suppressor cells and NK cells. J Immunol 2013; 191: 1486–1495. [DOI] [PubMed] [Google Scholar]
- 70. Brand A, Singer K, Koehl GE et al LDHA‐Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab 2016; 24: 657–671. [DOI] [PubMed] [Google Scholar]
- 71. Harmon C, Robinson MW, Hand F et al Lactate‐mediated acidification of tumor microenvironment induces apoptosis of liver‐resident NK cells in colorectal liver metastasis. Cancer Immunol Res 2019; 7: 335–346. [DOI] [PubMed] [Google Scholar]
- 72. Huntington ND, Legrand N, Alves NL et al IL‐15 trans‐presentation promotes human NK cell development and differentiation in vivo . J Exp Med 2009; 206: 25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhou X, Yu J, Cheng X et al The deubiquitinase Otub1 controls the activation of CD8+ T cells and NK cells by regulating IL‐15‐mediated priming. Nat Immunol 2019; 20: 879–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Delconte RB, Kolesnik TB, Dagley LF et al CIS is a potent checkpoint in NK cell‐mediated tumor immunity. Nat Immunol 2016; 17: 816–824. [DOI] [PubMed] [Google Scholar]
- 75. Putz EM, Guillerey C, Kos K et al Targeting cytokine signaling checkpoint CIS activates NK cells to protect from tumor initiation and metastasis. Oncoimmunology 2017; 6: e1267892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Zhu H, Blum RH, Bernareggi D et al Metabolic reprograming via deletion of CISH in Human iPSC‐Derived NK cells promotes In Vivo persistence and enhances anti‐tumor activity. Cell Stem Cell 2020; 27: 224–237.e226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rautela J, Surgenor E, Huntington ND. Drug target validation in primary human natural killer cells using CRISPR RNP. J Leukocyte Biol 2020; 108: 1397–1408. [DOI] [PubMed] [Google Scholar]
- 78. Beurel E, Grieco SF, Jope RS. Glycogen synthase kinase‐3 (GSK3): regulation, actions, and diseases. Pharmacol Ther 2015; 148: 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Aoukaty A, Tan R. Role for glycogen synthase kinase‐3 in NK cell cytotoxicity and X‐linked lymphoproliferative disease. J Immunol 2005; 174: 4551–4558. [DOI] [PubMed] [Google Scholar]
- 80. Parameswaran R, Ramakrishnan P, Moreton SA et al Repression of GSK3 restores NK cell cytotoxicity in AML patients. Nat Commun 2016; 7: 11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Cichocki F, Valamehr B, Bjordahl R et al GSK3 inhibition drives maturation of NK cells and enhances their antitumor activity. Cancer Res 2017; 77: 5664–5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. O'Sullivan TE, Sun JC, Lanier LL. Natural killer cell memory. Immunity 2015; 43: 634–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rudd CE, Chanthong K, Taylor A. Small Molecule Inhibition of GSK‐3 Specifically Inhibits the Transcription of Inhibitory Co‐receptor LAG‐3 for Enhanced Anti‐tumor Immunity. Cell Rep 2020; 30: 2075–2082.e2074. [DOI] [PubMed] [Google Scholar]
- 84. Duan L, Reddi AL, Ghosh A, Dimri M, Band H. The Cbl family and other ubiquitin ligases: destructive forces in control of antigen receptor signaling. Immunity 2004; 21: 7–17. [DOI] [PubMed] [Google Scholar]
- 85. Chirino LM, Kumar S, Okumura M et al TAM receptors attenuate murine NK‐cell responses via E3 ubiquitin ligase Cbl‐b. Eur J Immunol 2020; 50: 48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Paolino M, Choidas A, Wallner S et al The E3 ligase Cbl‐b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014; 507: 508–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kim HS, Das A, Gross CC, Bryceson YT, Long EO. Synergistic signals for natural cytotoxicity are required to overcome inhibition by c‐Cbl ubiquitin ligase. Immunity 2010; 32: 175–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Sim GC, Martin‐Orozco N, Jin L et al IL‐2 therapy promotes suppressive ICOS+ Treg expansion in melanoma patients. J Clin Invest 2014; 124: 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Charych D, Khalili S, Dixit V et al Modeling the receptor pharmacology, pharmacokinetics, and pharmacodynamics of NKTR‐214, a kinetically‐controlled interleukin‐2 (IL2) receptor agonist for cancer immunotherapy. PLoS One 2017; 12: e0179431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Bentebibel SE, Hurwitz ME, Bernatchez C et al A first‐in‐human study and biomarker analysis of NKTR‐214, a Novel IL2Rβλ‐biased cytokine, in patients with advanced or metastatic solid tumors. Cancer Discov 2019; 9: 711–721. [DOI] [PubMed] [Google Scholar]
- 91. Waldmann TA. The biology of interleukin‐2 and interleukin‐15: implications for cancer therapy and vaccine design. Nat Rev Immunol 2006; 6: 595–601. [DOI] [PubMed] [Google Scholar]
- 92. Margolin K, Morishima C, Velcheti V et al Phase I trial of ALT‐803, a novel recombinant IL15 complex, in patients with advanced solid tumors. Clin Cancer Res 2018; 24: 5552–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Wrangle JM, Velcheti V, Patel MR et al ALT‐803, an IL‐15 superagonist, in combination with nivolumab in patients with metastatic non‐small cell lung cancer: a non‐randomised, open‐label, phase 1b trial. Lancet Oncol 2018; 19: 694–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Pinette A, McMichael E, Courtney NB et al An IL‐15‐based superagonist ALT‐803 enhances the NK cell response to cetuximab‐treated squamous cell carcinoma of the head and neck. Cancer Immunol Immunother 2019; 68: 1379–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Romee R, Rosario M, Berrien‐Elliott MM et al Cytokine‐induced memory‐like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med 2016; 8: 357ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Vivier E, Morin P, Brien C, Druker B, Schlossman SF, Anderson P. Tyrosine phosphorylation of the FcλRIII (CD16): zeta complex in human natural killer cells. Induction by antibody‐dependent cytotoxicity but not by natural killing. J Immunol 1991; 146: 206. [PubMed] [Google Scholar]
- 97. Moore GL, Bautista C, Pong E et al A novel bispecific antibody format enables simultaneous bivalent and monovalent co‐engagement of distinct target antigens. MAbs 2011; 3: 546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Felices M, Sarhan D, Brandt L et al CD16‐IL15‐CD33 trispecific killer engager (TriKE) overcomes cancer‐induced immune suppression and induces natural killer cell‐mediated control of MDS and AML via enhanced killing kinetics. Blood 2016; 128: 4291. [Google Scholar]
- 99. Davis ZB, Vallera DA, Miller JS, Felices M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK‐mediated anti‐tumor immunotherapy. Semin Immunol 2017; 31: 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: An overview of FDA‐approved immune checkpoint inhibitors. Int Immunopharmacol 2018; 62: 29–39. [DOI] [PubMed] [Google Scholar]
- 101. Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD‐1 and CTLA‐4. Mol Cancer 2019; 18: 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Sun H, Sun C. The rise of NK cell checkpoints as promising therapeutic targets in cancer immunotherapy. Front Immunol 2019; 10: 2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Peng Q, Qiu X, Zhang Z et al PD‐L1 on dendritic cells attenuates T cell activation and regulates response to immune checkpoint blockade. Nat Commun 2020; 11: 4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Carenza C, Calcaterra F, Oriolo F et al Costimulatory molecules and immune checkpoints are differentially expressed on different subsets of dendritic cells. Front Immunol 2019; 10: 1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Zhang C, Liu Y. Targeting NK cell checkpoint receptors or molecules for cancer immunotherapy. Front Immunol 2020; 11: 1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cohen RB, Bauman JR, Salas S et al Combination of monalizumab and cetuximab in recurrent or metastatic head and neck cancer patients previously treated with platinum‐based chemotherapy and PD‐(L)1 inhibitors. J Clinical Oncol 2020; 38: 6516. [Google Scholar]
- 107. Carlsten M, Jaras M. Natural killer cells in myeloid malignancies: immune surveillance, NK cell dysfunction, and pharmacological opportunities to bolster the endogenous NK Cells. Front Immunol 2019; 10: 2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Rosenberg SA, Packard BS, Aebersold PM et al Use of tumor‐infiltrating lymphocytes and interleukin‐2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med 1988; 319: 1676–1680. [DOI] [PubMed] [Google Scholar]
- 109. Davila ML, Brentjens RJ. CD19‐targeted CAR T cells as novel cancer immunotherapy for relapsed or refractory B‐cell acute lymphoblastic leukemia. Clin Adv Hematol Oncol 2016; 14: 802–808. [PMC free article] [PubMed] [Google Scholar]
- 110. Majzner RG, Mackall CL. Tumor Antigen Escape from CAR T‐cell Therapy. Cancer Discov 2018; 8: 1219. [DOI] [PubMed] [Google Scholar]
- 111. Zah E, Lin M‐Y, Silva‐Benedict A, Jensen MC, Chen YY. T Cells Expressing CD19/CD20 bispecific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol Res 2016; 4: 498–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 2016; 127: 3321–3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Xie G, Dong H, Liang Y, Ham JD, Rizwan R, Chen J. CAR‐NK cells: A promising cellular immunotherapy for cancer. EBioMedicine 2020; 59: 102975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Liu E, Marin D, Banerjee P et al Use of CAR‐Transduced Natural Killer Cells in CD19‐Positive Lymphoid Tumors. N Engl J Med 2020; 382: 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Fujisaki H, Kakuda H, Shimasaki N et al Expansion of highly cytotoxic human natural killer cells for cancer cell therapy. Cancer Res 2009; 69: 4010–4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual‐RNA‐guided DNA endonuclease in adaptive bacterial immunity. Science 2012; 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Riggan L, Hildreth AD, Rolot M et al CRISPR‐Cas9 Ribonucleoprotein‐Mediated Genomic Editing in Mature Primary Innate Immune Cells. Cell Rep 2020; 31: 107651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. McGowan E, Lin Q, Ma G, Yin H, Chen S, Lin Y. PD‐1 disrupted CAR‐T cells in the treatment of solid tumors: Promises and challenges. Biomed Pharmacother 2020; 121: 109625. [DOI] [PubMed] [Google Scholar]
- 119. Eyquem J, Mansilla‐Soto J, Giavridis T et al Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017; 543: 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Nguyen DN, Roth TL, Li PJ et al Polymer‐stabilized Cas9 nanoparticles and modified repair templates increase genome editing efficiency. Nat Biotechnol 2020; 38: 44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Huang RS, Shih HA, Lai MC, Chang YJ, Lin S. Enhanced NK‐92 Cytotoxicity by CRISPR Genome Engineering Using Cas9 Ribonucleoproteins. Front Immunol 2020; 11: 1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9‐mediated gene knockout in primary T cells. J Exp Med 2018; 215: 985–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Castriconi R, Dondero A, Bellora F et al Neuroblastoma‐derived TGF‐β1 modulates the chemokine receptor repertoire of human resting NK cells. J Immunol 2013; 190: 5321–5328. [DOI] [PubMed] [Google Scholar]
- 124. Cheng M, Ma J, Chen Y et al Establishment, characterization, and successful adaptive therapy against human tumors of NKG cell, a new human NK cell line. Cell Transplant 2011; 20: 1731–1746. [DOI] [PubMed] [Google Scholar]
- 125. Li F, Sheng Y, Hou W et al CCL5‐armed oncolytic virus augments CCR5‐engineered NK cell infiltration and antitumor efficiency. J Immunother Cancer 2020; 8: e000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Pachynski RK, Wang P, Salazar N et al Chemerin suppresses breast cancer growth by recruiting immune effector cells into the tumor microenvironment. Front Immunol 2019; 10: 983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Parodi M, Raggi F, Cangelosi D et al Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front Immunol 2018; 9: 2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Pesce S, Moretta L, Moretta A, Marcenaro E. Human NK cell subsets redistribution in pathological conditions: a role for CCR7 receptor. Front Immunol 2016; 7: 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zhang F, Wei H, Wang H et al Involvement of interaction between fractalkine and CX3CR1 in cytotoxicity of NK cells against tumor cells. Oncol Rep 2006; 15: 485–488. [PubMed] [Google Scholar]
- 130. Bernardini G, Sciume G, Santoni A. Differential chemotactic receptor requirements for NK cell subset trafficking into bone marrow. Front Immunol 2013; 4: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Mayol K, Biajoux V, Marvel J, Balabanian K, Walzer T. Sequential desensitization of CXCR4 and S1P5 controls natural killer cell trafficking. Blood 2011; 118: 4863–4871. [DOI] [PubMed] [Google Scholar]
- 132. Robinson LA, Nataraj C, Thomas DW et al The chemokine CX3CL1 regulates NK cell activity in vivo . Cell Immunol 2003; 225: 122–130. [DOI] [PubMed] [Google Scholar]
- 133. Susek KH, Karvouni M, Alici E, Lundqvist A. The Role of CXC Chemokine Receptors 1–4 on Immune Cells in the Tumor Microenvironment. Front Immunol 2018; 9: 2159. [DOI] [PMC free article] [PubMed] [Google Scholar]