

Abstract
Background & Aims
Adherent-invasive Escherichia coli are implicated in inflammatory bowel disease, and mitochondrial dysfunction has been observed in biopsy specimens from patients with inflammatory bowel disease. As a novel aspect of adherent-invasive E coli–epithelial interaction, we hypothesized that E coli (strain LF82) would elicit substantial disruption of epithelial mitochondrial form and function.
Methods
Monolayers of human colon-derived epithelial cell lines were exposed to E coli–LF82 or commensal E coli and RNA sequence analysis, mitochondrial function (adenosine triphosphate synthesis) and dynamics (mitochondrial network imaging, immunoblotting for fission and fusion proteins), and epithelial permeability (transepithelial resistance, flux of fluorescein isothiocyanate–dextran and bacteria) were assessed.
Results
E coli–LF82 significantly affected epithelial expression of ∼8600 genes, many relating to mitochondrial function. E coli–LF82–infected epithelia showed swollen mitochondria, reduced mitochondrial membrane potential and adenosine triphosphate, and fragmentation of the mitochondrial network: events not observed with dead E coli–LF82, medium from bacterial cultures, or control E coli. Treatment with Mitochondrial Division Inhibitor 1 (Mdivi1, inhibits dynamin-related peptide 1, guanosine triphosphatase principally responsible for mitochondrial fission) or P110 (prevents dynamin-related peptide 1 binding to mitochondrial fission 1 protein) partially reduced E coli–LF82–induced mitochondrial fragmentation in the short term. E coli–LF82–infected epithelia showed loss of the long isoform of optic atrophy factor 1, which mediates mitochondrial fusion. Mitochondrial Division Inhibitor 1 reduced the magnitude of E coli–LF82–induced increased transepithelial flux of fluorescein isothiocyanate dextran. By 8 hours after infection, increased cytosolic cytochrome C and DNA fragmentation were apparent without evidence of caspase-3 or apoptosis inducing factor activation.
Conclusions
Epithelial mitochondrial fragmentation caused by E coli–LF82 could be targeted to maintain cellular homeostasis and mitigate infection-induced loss of epithelial barrier function. Data have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession numbers GSE154121 and GSE154122 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154121).
Keywords: Mitochondrial Fission and Fusion, Human Epithelial Cell Lines, Drp1, Epithelial Permeability, Caspase-3, Bacteria
Abbreviations used in this paper: AIEC, adherent invasive Escherichia coli; AIF, apoptosis inducing factor; ATP, adenosine triphosphate; CCCP, carbonyl cyanide m-chlorophenyl hydrazone; CFU, colony forming unit; Drp1, dynamin-related peptide 1; EPEC, enteropathogenic E coli; Fis1, fission protein-1; GTPase, guanosine triphosphatase; IBD, inflammatory bowel disease; IL, interleukin; OPA1, optic atrophy factor 1; PGC-1α, peroxisome proliferator-activated receptor γ coactivator 1α; ROS, reactive oxygen species; siRNA, small interfering RNA; TER, transepithelial resistance
Graphical abstract

Summary.
Disruption of the mitochondrial network is uncovered as a novel aspect of adherent-invasive Escherichia coli (strain LF82: implicated in inflammatory bowel disease) interaction with epithelial cells; the inhibition of which reduced the magnitude of the bacteria-evoked increases in epithelial permeability.
Studies in animal models and analysis of patients with inflammatory bowel disease (IBD) highlight the importance of the gut microbiota in the etiology and pathophysiology of enteric inflammation.1 Tissues from patients with IBD often have increased numbers of Escherichia coli compared with those from healthy individuals,2 and a subgroup therein, the adherent-invasive E coli (AIEC), has emerged as pathobionts.3,4 Attaching via pili and long polar fimbriae,5,6 AIEC can cause reactive oxygen species (ROS) and proinflammatory cytokine production by epithelial cells7,8 and increase epithelial permeability.9 AIECs have enhanced survival within macrophages,10 and can exaggerate the severity of murine colitis.11,12 A comprehensive knowledge of the mechanism(s) by which AIECs participate in IBD is lacking. Intriguingly, the description of the reference strain of AIECs, LF82, showed infected enterocytes with swollen mitochondria that lacked cristae,3 which are cardinal features of dysfunctional mitochondria.
Mitochondrial disease can present with gastrointestinal symptoms,13 and perturbed mitochondrial form and function is an aspect of enteric inflammation.14 Colonic biopsy specimens from patients with IBD can have swollen epithelial mitochondria, reduced adenosine triphosphate (ATP), an altered mitochondrial proteome, and increased sensitivity to uncoupling of oxidative phosphorylation.14, 15, 16, 17 Furthermore, mutation in the IRGM1 gene, which is associated with mitochondria, is a susceptibility trait for IBD,18 as are mutations in the OCTN2 gene, which facilitates carnitine uptake for fatty acid β-oxidation and ATP synthesis by mitochondria.19 Cell culture and murine studies have defined mechanisms whereby dysfunctional mitochondria contribute to enteric inflammatory disease, such as increased epithelial paracellular permeability, internalization of commensal bacteria, and increased ROS production.20,21
Typically depicted as kidney-bean–shaped organelles, mitochondria actually exist in a network that is constantly remodeling by fission and fusion processes dictated in large part by the guanosine triphosphatases (GTPases), dynamin-related protein 1 (Drp1), and mitofusins 1 and 2 and the long isoform of optic atrophy factor 1 (OPA1-L), respectively.22 Fusion facilitates homogeneity of molecules throughout the network, subcellular targeting of ATP, and compensation for mutated mitochondrial DNA. Mitochondrial fission is necessary for cell division, mitophagy, and apoptosis. Excessive fragmentation of the mitochondrial network has been described in neurodegenerative disease, cardiac ischemia–reperfusion injury, kidney disease, and metabolic disease: all of which can have an inflammatory component.23, 24, 25 Systemic delivery of P110, a molecule designed to reduce mitochondrial fission by preventing Drp1–mitochondrial fission 1 protein (Fis1) interaction, reduced the severity of chemical-induced colitis in mice.26
The mitochondrial network can be disrupted significantly in cultured cells infected with microbial pathogens,27 although the functional significance of this is poorly understood. Thus, juxtaposing the concepts of a bacterial etiology for IBD and mitochondrial dysfunction as a contributor to enteric pathophysiology, we sought to determine if AIEC affected epithelial mitochondrial dynamics. E coli–LF82, unlike commensal E coli, caused fragmentation of the epithelial mitochondrial network: at early time points after infection (2–4 h) this was reduced by inhibition of Drp1 activity, while loss of OPA1-L occurred by 16 hours after infection. The reduction in barrier function that accompanied infection was ameliorated in part by Mitochondrial Division Inhibitor 1 (Mdivi1), a pharmacologic that selectively inhibits Drp1 activity. Thus, fragmentation of the mitochondrial network is shown as a novel aspect of E coli–LF82 epithelial interaction with implications for barrier function that could be relevant to AIEC triggering initiation or disease relapse in IBD.
Results
Transcriptomics Indicates Altered Mitochondrial Function in E coli–LF82–Infected Epithelia
T84 epithelia exposed to E coli–LF82 or E coli–F18 showed massive changes in gene transcription after 4 and 16 hours (P < .05) (Figure 1A), with principal component analysis showing clustering of the experimental groups indicative of different transcriptomic programs (Figure 1B). The majority of gene expression changes were small (<2-fold, and statistically significant) and there was considerable overlap in the genes affected by infection; however, gene pathway analysis showed differences in the epithelial response to the 2 strains of E coli that were statistically significant (Figure 1C). Specifically, gene pathways relating to cell viability, cellular stress, cell cycle, antigen handling and response to pathogens, signal transduction, cell-to-cell communication, and mitochondrial function were time-dependently differentially regulated by E coli–LF82 compared with epithelia exposed to E coli–F18 (Figure 1C). E coli–LF82 infection (108 colony-forming unit [CFU]/mL, 4 h) affected 1265 genes and 105 signaling pathways that were not affected by E coli–F18 (Figure 2A and B). With a lower E coli inoculum and longer exposure (104 CFU/mL, 16 h), 964 genes and 69 pathways were uniquely affected by E coli–LF82 (Figure 2C and D) in T84 epithelia. Changes in mitochondrial function were noteworthy (eg, the master transcriptional regulator of mitochondrial biogenesis, peroxisome proliferator-activated receptor γ coactivator 1α [PGC-1α], was reduced approximately 2-fold by exposure to E coli–LF82 [4 h, 108 CFU/mL], but not E coli–F18).
Figure 1.

RNA sequence analysis shows significant differences in the epithelial cell response to E coli strains. T84 epithelia were treated with adherent-invasive E coli–LF82 or commensal E coli–F18. (A) The number of significant gene expression changes. (B) Principle component (PC) analysis shows obvious grouping of the epithelia by treatment, and (C) pathway analysis shows distinct E coli–evoked changes in epithelial gene expression as a function of time and dose of inoculum.
Figure 2.

Distribution of statistically significant changes in (A and C) gene expression and (B and D) gene pathway analysis in T84 epithelia treated with E coli–LF82 or E coli–F18 (dose and time shown above graphs) (red, number of gene expressions increased; blue, number of gene expressions decreased; numbers above bars indicate the total number of genes and pathways altered in the infected epithelia; dots and connections under each bar indicate that the expressions were either unique to that group or shared by the groups).
E coli–LF82 Induces Mitochondrial Dysfunction
Immunostaining showed significant epithelial-associated E coli–LF82 but not E coli–HB101 (Figure 3A). Transmission electron microscopy showed mitochondrial ultrastructural abnormalities in epithelia exposed to E coli–LF82 but not E coli–HB101 (Figure 3B): the mitochondria were electron-lucent, swollen, and lacked cristae. In accordance with these data, E coli–LF82 evoked a loss of mitochondrial membrane potential similar to that evoked by carbonyl cyanide m-chlorophenyl hydrazone (CCCP) (Figure 3C). After 4 hours of infection with E coli–LF82, but not E coli–F18, epithelial ATP levels were reduced by approximately 20% (12–24 epithelia, 6 experiments; data not shown), with greater reductions in ATP occurring 16 hours after exposure to E coli–LF82 (Figure 3D).
Figure 3.

Adherent-invasive E coli induces mitochondrial dysfunction in human colonic epithelium. (A) Immunostaining shows significant association of E coli–LF82 with T84 epithelia in contrast to epithelia treated with the commensal E coli–HB101 (representative images; n = 3 monolayers). (B) Transmission electron microscopy shows swollen and electron-lucent mitochondria in T84 cells infected with E coli–LF82 (n = 4). e, E coli; n, nucleus; pc, paracellular space. Arrows indicate mitochondria. (C) Quantification of tetramethylrhodamine ethyl ester (TMRE) dye average integrated fluorescence intensity, as an indicator of mitochondrial membrane potential, showed a significant reduction by 3.5 and 8.5 hours of exposure to E coli–LF82, 108 and 104 CFU/mL, respectively, but not E coli–HB101 (104 CFU/mL, 8.5 h) (4 μmol/L CCCP was used as a positive control). ∗P < .05 Kruskal–Wallis, followed by the Dunn multiple comparison test compared with control, n = 5–16. (D) T84 cell levels of ATP are reduced after treatment with the pathobiont E coli–LF82 (∗P < .05, t test, n = 8 and 13 epithelial preparations) (data are means ± SEM).
Infection With E coli–LF82 Causes Mitochondrial Fragmentation
Assessment of the T84-epithelial mitochondrial network after exposure to E coli–LF82 (2–4 h 108 CFU/mL or 16 h 104 CFU/mL) showed significant fragmentation that consistently exceeded the negligible or minor changes evoked by either of the commensal E coli strains (Figure 4A–C). Loss of fused mitochondrial networks and increased fragmented networks occurred as early as 2 hours after exposure to E coli–LF82 (108 CFU/mL), with almost complete disruption by 16 hours after treatment with 104 CFU/mL. E coli–LF82 also evoked mitochondrial fragmentation in human colon-derived Caco2 and HT-29 epithelial cell lines (data not shown). In contrast, neither dead E coli–LF82 nor spent medium from E coli (cultured alone or with enterocytes) caused appreciable mitochondrial fragmentation in T84 cells (Figure 4D). Although infection with wild-type E coli–LF82 consistently led to significant fragmentation of the epithelial mitochondrial network, cells infected with E coli–LF82ΔFimH had mitochondrial networks similar to uninfected cells (Figure 4E). Another AIEC, NRG857c, which is less invasive in T84 cells than E coli–LF82, evoked a small increase in mitochondrial fragmentation that was not statistically different from uninfected cells (Figure 5). Growth curves showed negligible differences between the E coli strains and growth was unaffected by any of the drugs used herein. T84 epithelia infected with enteropathogenic E coli (strain E2348; 108 CFU/mL, 4 h) showed a significant increase in the number of cells with a fragmented mitochondrial network (6 epithelial preparations from 2 experiments; data not shown).
Figure 4.

Adherent-invasive E coli–induced mitochondrial fragmentation is dependent on FimH attachment/invasion. (A) Representative confocal images of T84 mitochondrial networks stained with MitoTracker and (B and C) network quantification. ∗P < .05 compared with control, #P < .05 compared with HB101; 2-way analysis of variance (ANOVA) followed by the Tukey multiple comparison test, n = 5–6 epithelial monolayers (20 cells/monolayer) from 3 experiments. (D) Representative images and quantification showing that neither dead E coli–LF82 or spent medium (Med.) from E coli causes mitochondrial fragmentation in T84 epithelia. ∗P < .05 compared with control, 2-way ANOVA followed by the Tukey multiple comparison test, n = 8–9 epithelial preparations (20 cells/monolayer) from 3 experiments. (E) Bacteria lacking the FimH molecule are less invasive than the wild-type E coli–LF82 and fail to significantly induce fragmentation of the T84 epithelial cell mitochondrial network (representative images match the quantification bars). ∗P < .05 compared with control, 2-way ANOVA followed by the Tukey multiple comparison test, n = 6–18 epithelial monolayers from 5 experiments; 4 μmol/L CCCP. Data are means ± SEM. Ctrl, control; wt, wild type.
Figure 5.

Degree of epithelial mitochondrial fragmentation parallels pathobiont invasiveness. T84 epithelia were infected with either the commensal E coli–HB101 or one of the adherent-invasive strains of E coli–LF82 or E coli–NRG857c. (A) Significantly (∗P < .05, 1-way analysis of variance [ANOVA], Tukey multiple comparison test) greater invasion by E coli–LF82, compared to (B) fragmentation of the mitochondrial network 2 hours after infection with E coli-NRG857c resembling that of uninected cells (data are means ± SEM; ∗P < .05 compared with control [Ctrl], 2-way ANOVA, Tukey multiple comparison test in panel B, n = 9 epithelial preparations [20 cells/monolayer] from 3 experiments; control, noninfected epithelia).
Fragmentation of mitochondria can be associated with increased ROS production28 and E coli–LF82 has been shown to induce ROS production in epithelial cells.29 Co-treatment of E coli–LF82–infected epithelia with the antioxidant vitamin C, or the mitochondrial-targeted antioxidant Mito-TEMPO, did not prevent the mitochondrial fragmentation (data not shown) (antioxidant capacity of vitamin C and mito-TEMPO was confirmed by reductions in measurable ROS after treatment with rotenone; data not shown).
Targeting Drp1 Limits E coli–LF82–Induced Epithelial Mitochondrial Fragmentation
Total Drp1 and activated ser616p-Drp1 levels in isolated mitochondria (data not shown) or whole-cell extracts were not altered consistently in T84 epithelia treated with E coli–LF82 (2–8 h 108 CFU/mL; 8–16 h 104 CFU/mL) (Figure 6A). Because of many post-translational modifications in Drp1,22 which were not assayed here, an absence of detectable increases in Drp1 does not negate a role for this GTPase in mitochondrial fragmentation. Subsequently, co-treatment with P110 or Mdivi1 was found to significantly decrease E coli–LF82–induced (108 CFU/mL, 2–4 h) mitochondrial fragmentation (Figure 6B and C). Corroborating these data, epithelia in which Drp1 expression was reduced by small interfering RNA (siRNA) showed less E coli–LF82–induced mitochondrial fission after 2 hours of infection (Figure 6C, middle panel). However, neither P110 nor Mdivi1 prevented the degree of mitochondrial fragmentation observed after 6–8 hours of infection with 108 CFU/mL (Figure 6C, lower panel) or 16 hours of treatment with 104 CFU/mL E coli–LF82 (data not shown) (adding P110 every 2 hours also did not ameliorate the E coli–LF82–evoked mitochondrial fragmentation observed at later time points in the infection).
Figure 6.

Inhibition of Drp1 activity reduces mitochondrial fragmentation induced by adherent-invasive E coli–LF82. (A) Western blot and densitometry analysis showing that neither total nor phosphorylated Drp1 (Ser616 site) protein expression in whole-cell lysates is altered consistently in T84 epithelia infected with E coli–LF82. (B and C) Inhibition of Drp1 activity with P110 (10 μmol/L) or Mdivi1 (5 μmol/L) or siRNA (30 nmol/L) knockdown of Drp1 protein reduces the severity of E coli–LF82–evoked T84 epithelial cell mitochondrial fragmentation assessed at 2 and 4 hours after infection, but not at 6 or 8 hours after exposure to the viable bacteria. Representative images of MitoTracker-stained mitochondrial networks. Middle: ∗P < .05 compared with mock transfection uninfected; #P < .05 compared with mock transfection infected. Bottom: ∗P < .05 compared with control (ctrl), 2-way analysis of variance followed by the Tukey multiple comparison test; n = 4–11 epithelial monolayers (20 cells/monolayer), 2–4 experiments. con.siRNA, control siRNA; Sts, 20 μmol/L staurosporine. Data are means ± SEM.
Infection With E coli–LF82 Results in Loss of OPA1-L
Analysis of protein expression showed a time- and dose-dependent loss of OPA1-L in E coli–LF82–infected epithelia. Infection with the control bacteria, E coli–HB101 and E coli–F18, at high inoculum could reduce OPA1-L levels, but this was not a statistically significant consistent observation (Figure 7). With an inoculum of 106 CFU/mL E coli–LF82, OPA1-L levels began to diminish by 10 hours after infection and were statistically significantly reduced by 16 hours after infection (Figure 7B). The loss of epithelial OPA1-L owing to infection with E coli–LF82 was time- and dose-dependent (Figure 7C).
Figure 7.

Infection with E coli–LF82 results in loss of OPA1-L. (A) Western blots showing the time- and concentration-dependent (CFU inoculum) loss of OPA1-L in E coli–LF82–infected T84 epithelia and (B and C) corresponding densitometry analysis. Data are means ± SEM, 4 μmol/L CCCP for 2 hours. ∗P < .05 compared with control (ctrl), 1-way analysis of variance, the Dunnett multiple comparison test. OPA1-S, Optic Atrophy Factor 1 - Short Form.
Mdivi1 Partially Rescues the Epithelial Barrier Defect Evoked by E coli–LF82
Exposure to E coli–LF82 reduces the barrier function of human colon-derived epithelial cell lines.30 The decrease in transepithelial resistance (TER) that occurred by 8 hours after infection with E coli–LF82 (108 CFU/mL) was not affected by either inhibitor of mitochondrial fragmentation, P110 or Mdivi1, or the pan-caspase inhibitor, Z-VAD (Figure 8A). In contrast, assessment of the transepithelial flux of larger molecules showed that in 4 experiments Mdivi1 significantly prevented the E coli–LF82–evoked flux of 70 kilodalton dextran (Figure 8B), and trended toward a reduced apical-to-basolateral passage of E coli across the epithelium (Figure 8C) (Mdivi1 and P110 alone did not affect epithelial barrier function; data not shown).
Figure 8.
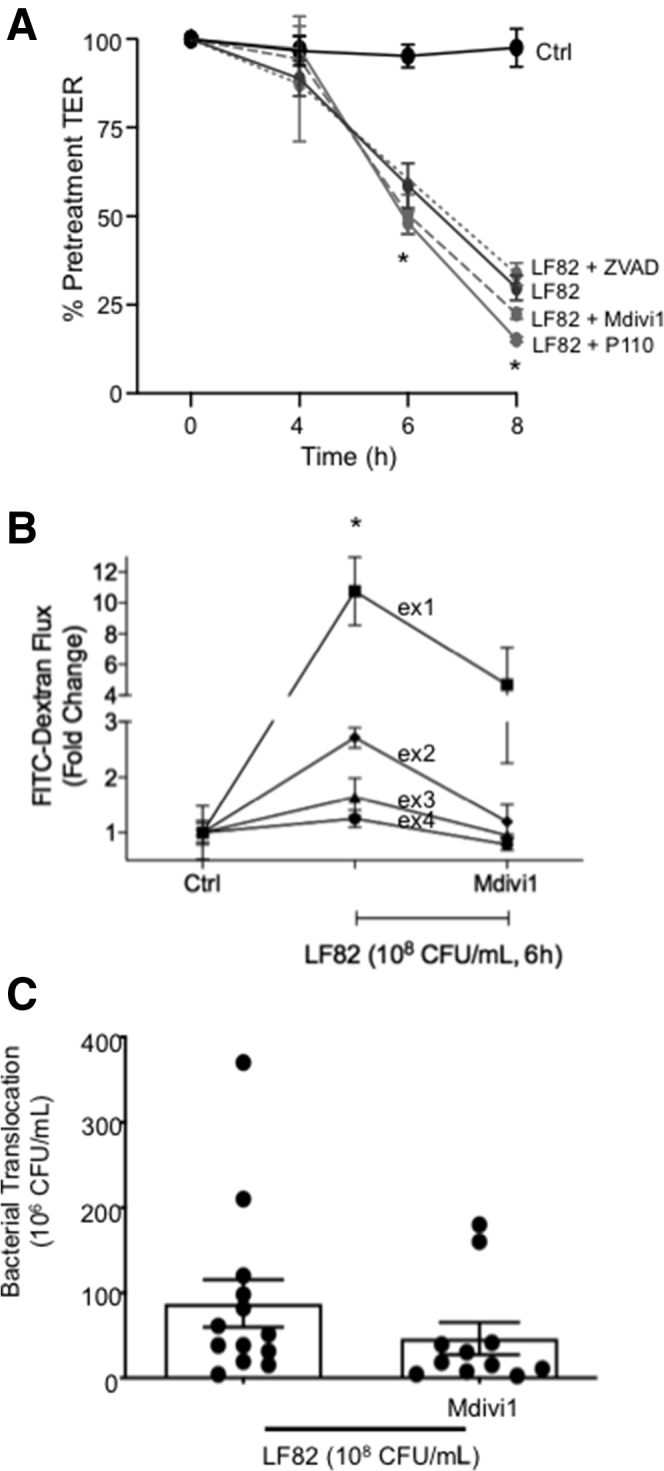
E coli–LF82–induced decreased epithelial barrier function is reduced by co-treatment with an inhibitor of mitochondrial fission. (A) The E coli–induced (108 CFU/mL) decrease in TER across filter-grown T84 monolayers was unaffected by co-treatment with Mdivi1 (5 μmol/L), P110 (10 μmol/L), or the pan-caspase inhibitor Z-VAD (100 μmol/L) (∗P < .05 compared with control [Ctrl], 2-way analysis of variance [ANOVA], Tukey comparison test, n = 12 epithelial preparations from 4 experiments; starting TER = 1000–2000 Ω × cm2). In contrast, (B) barrier assessment by 70 kilodalton dextran–fluorescein isothiocyanate (FITC) flux (∗P < .05 compared with ctrl, 2-way ANOVA, Tukey multiple comparison test, experiment number was considered as a separate variable rather than pooled data owing to large variations in fold change) and (C) transepithelial passage of the E coli were both reduced by Mdivi1 (t test). Data are means ± SEM.
E coli–LF82 Evokes Mitochondrial Release of Cytochrome C
Excessive mitochondrial fragmentation can precede apoptosis. Forty-six percent of cells exposed to staurosporine had cytochrome C in their cytoplasm (Figure 9A and B). With treatment of E coli–LF82 (6 h, 108 CFU/mL), cytoplasmic cytochrome C was not different from control epithelium, whereas by 8 hours after infection approximately 40% of the infected cells were positive for cytosolic cytochrome C (some preparations showed approximately 80% positivity). P110 and Mdivi1 did not prevent the E coli–LF82–evoked mitochondrial release of cytochrome C (Figure 9B), consistent with their inability to block mitochondrial fragmentation at later time points in infection (Figure 9D). Caspase activation can occur after cytochrome C release. T84 epithelia treated with E coli–LF82 (108 CFU/mL) 8–12 hours previously showed no evidence of cleaved caspase-3 in whole-cell protein extracts, and, in accordance with other studies,31 there was a general reduction in total caspase-3 in the treated cells (Figure 9C). Immunostaining did not show a consistent localization of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus in response to E coli–LF82 (108 CFU/mL, 6 h or 8 h), and release of interleukin (IL)18, as a surrogate marker of necroptosis, was not increased after exposure to E coli–LF82 (16 h, 104 CFU/mL; n = 4 epithelial preparations; IL18 <2 pg/mL in control and infected T84 epithelia). However, the nuclei of E coli–LF82–infected T84 cells were pyknotic and gel electrophoresis showed substantial fragmentation of the DNA that was unaffected by co-treatment with either P110 or Mdivi1 (Figure 6D).
Figure 9.

E coli–LF82–infected T84 epithelia show increased cytosolic cytochrome C, absence of caspase-3 activation, and obvious DNA fragmentation. (A) Representative immunostaining of T84 epithelia shows increased cytosolic cytochrome C at 8 hours after infection that was not apparent at 6 hours. (B) Co-treatment with the fission inhibitors, P110 (10 μmol/L) or Mdivi1 (5 μmol/L) did not prevent the mitochondrial release of cytochrome C (∗P < .05 compared with ctrl; 1-way analysis of variance followed by the Dunnett multiple comparison test). Ctrl, control; Sts, staurosporine, 20 μmol/L. (C) Western blot analysis of whole-cell lysates showed a reduction in total caspase-3 protein and an absence of cleaved caspase-3 at 8–12 hours after infection with E coli–LF82 (108 CFU/mL) (Sts was used as a positive control) (blot representative of 4 separate experiments). (D) DNA fragmentation was obvious at 8 and 16 hours after infection. M, Mdivi1; P, P110; n = 3; E, empty lane, no sample added. Data are means ± SEM. bp, basepair; DAPI, 4′,6-diamidino-2-phenylindole.
The ability of P110 and Mdivi1 to suppress E coli–LF82–induced epithelial mitochondrial fragmentation after 2–4 hours of infection but not more than 8 hours infection was intriguing. Killing extracellular bacteria with gentamicin and removal of E coli–LF82 after 4 hours, followed by epithelial assessment 4 hours later, showed significantly less mitochondrial fragmentation and cytoplasmic cytochrome C compared with cells continuously exposed to the bacteria for 8 hours (Figure 10).
Figure 10.
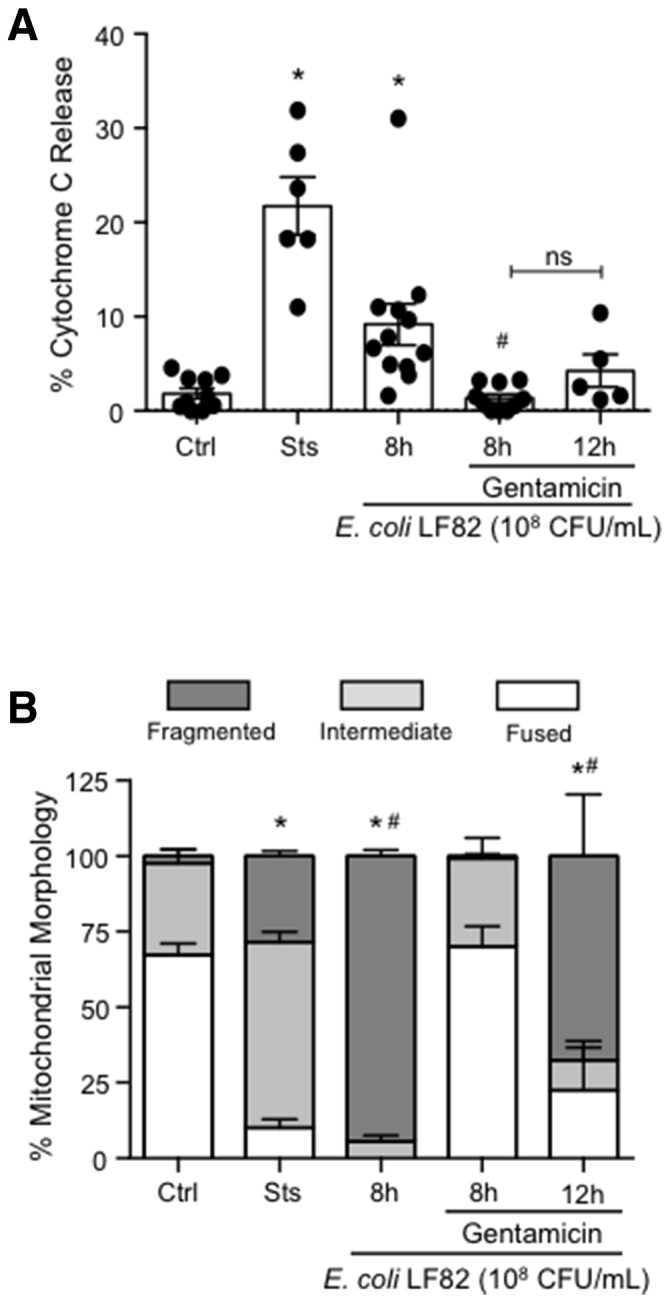
E coli–LF82–evoked release of cytosolic cytochrome C and mitochondrial fragmentation are reduced by gentamicin. T84 epithelia were treated with E coli–LF82 and examined for (A) the presence of cytosolic cytochrome C (∗P < .05 compared with ctrl, #P < .05 compared with 8 hours LF82; 1-way analysis of variance [ANOVA], Tukey multiple comparison test) or (B) pattern of the mitochondrial network 8 hours later. Time-matched epithelia were treated with gentamicin (200 μg/mL, 1 h) 4 hours after the addition of E coli and then examined 4 or 8 hours later (ie, the 8- and 12-hour gentamicin groups) (∗P < .05 compared with ctrl, #P < .05 compared with 8 h LF82 + gentamicin; 2-way ANOVA followed by the Tukey multiple comparison test; n = 5–12 epithelial preparation from 3–4 experiments; Ctrl, control; Sts, 20 μmol/L staurosporine). Data are means ± SEM.
Discussion
The mechanisms responsible for impaired mitochondrial structure and function in biopsy specimens from patients with IBD32 are unknown. The concept of the pathobiont in which environmental conditions promote the emergence of pathogenic traits in a bacterium normally considered a commensal is intriguing.33 By using reductionist models we uncovered that the Crohn’s disease–associated pathobiont, E coli–LF82, can cause massive disruption of the epithelial mitochondrial network, possibly via the sequential activation of Drp1 followed by loss of the profusion GTPase OPA1-L.
Exposure of T84 epithelia to E coli–LF82, E coli–F18, or E coli–HB101 significantly affected gene expression, underscoring the high degree of communication between enterocytes and bacteria. Considerable overlap occurred in the genes affected by exposure to pathobiont or commensal; however, the magnitude and direction of changes often differed, and pathway analysis showed significant differences in the effects of the bacterial strains. Notably, E coli–LF82 affected genes related to mitochondrial form/function and the decrease in PGC-1α (controls mitochondrial biogenesis) messenger RNA distinguished it from the commensal bacteria. Although bacterial-driven perturbation of mitochondrial function is common,34, 35, 36 we are unaware of other reports of microbes affecting PGC-1α expression, with the exception of the hepatitis C virus protein NS5A.37
Epithelia infected with E coli–LF82, but not E coli–HB101, show reduced mitochondrial membrane potential (ΔΨm) that could account for the mitochondrial swelling and loss of cristae, and hence lower levels of ATP. Maintenance of ΔΨm is critical in mitochondrial function, and dissipation of the H+ gradient indicates malfunction that can trigger mitochondrial fission or cell death. Infection with E coli–LF82 evoked a time- and dose-dependent fragmentation of mitochondria in gut-derived epithelia. The bacterial pathogens Listeria monocytogenes, Shigella flexneri, Mycobacterium tuberculosis, and Vibrio cholerae all cause mitochondrial fragmentation,38, 39, 40, 41 and this often was accompanied by reduced ΔΨm.
Listeriolysin O, vacuolating cytotoxin E, early secreted antigen 6 kilodalton, and vacuolating cytotoxin E, have been implicated in mitochondrial fragmentation induced by L monocytogenes, V cholera, M tuberculosis, and Helicobacter pylori, respectively.38,39,42,43 A soluble factor is unlikely to be critical to the effect of E coli–LF82 because medium from bacterial cultures (±T84 cells) did not evoke mitochondrial fragmentation (the possibility of a short-lived mediator contributing to mitochondrial fragmentation cannot be unequivocally ruled out). Elatrech et al29 reported increased ROS production by E coli–LF82–infected T84 epithelia, and ROS can disrupt mitochondrial dynamics.28 Vitamin C and Mito-TEMPO reduced rotenone-induced ROS by T84 cells, yet neither affected E coli–LF82–induced mitochondrial fragmentation. This suggests that ROS are not a major factor contributing to the mitochondrial fragmentation, and could, in fact, be produced as a consequence of the fragmentation and perturbed oxidative phosphorylation.
Lack of an effect of dead E coli–LF82 indicated that viable bacteria attaching to and/or invading the epithelium caused fragmentation of the mitochondrial network. In accordance, epithelia exposed to E coli–LF82 deficient in type 1 pili, which are critical for adhesion and invasion,9 had mitochondrial networks similar to noninfected epithelia. Furthermore, the AIEC strain NRG857c that was less invasive in T84 epithelia caused little mitochondrial fragmentation compared with E coli–LF82. E coli–LF82’s lack of toxins/virulence factors suggests that either attachment to the cell, the process of invasion, and/or the presence of intracellular bacteria triggers mitochondrial fragmentation. Our findings with enteropathogenic E coli (EPEC) suggest that attachment to the epithelial cell membrane could initiate a cascade of events leading to mitochondrial fragmentation. However, injection of EPEC effector molecules may be the critical factor affecting host cell mitochondrial dynamics. The EPEC protein E coli secreted protein F (EspF) contains a mitochondrial localization sequence and was found to lower ΔΨm; however, mitochondrial dynamics were not assessed in that study.36 Consistent with the hypothesis that invasion of E coli–LF82 was critical for mitochondrial fragmentation, it has been suggested that intracellular mechanostimulation resulting from bacteria is sufficient to trigger mitochondrial fission via Drp1.44 This could account for the slight increase in mitochondrial fragmentation in T84 cells exposed to E coli–HB101, some of which are internalized because of epithelial membrane turnover.45
The GTPase Drp1 drives fission by binding to mitochondrial fission factor, mitochondrial dynamics proteins 49 and 51, or Fis1. Pharmacologic inhibition of Drp1 or protein knockdown reduced E coli–LF82–induced mitochondrial fragmentation. Similarly, Mdivi1 has been used to implicate Drp1 in S flexneri–induced41 and H pylori–induced epithelial mitochondrial fragmentation.42 Addition of Mdivi1 or P110 at the start of the E coli–LF82–epithelial co-culture reduced the degree of mitochondrial fragmentation observed 4 hours later, but not at 8–16 hours after infection. These data can be explained by continued replication of the bacteria overwhelming the epithelium or may indicate that mitochondrial fission in the latter stages of infection are Drp1-independent. With respect to the latter, HeLa cells lacking Drp1 showed significant mitochondrial fragmentation when challenged with listeriolysin O.43
OPA1-L facilitates mitochondrial fusion to work in opposition to profission events. E coli–LF82–infected epithelia showed loss of OPA1-L and this lack of a critical profusion molecule would contribute to mitochondrial fragmentation. Similarly, mitochondrial fragmentation in Brucella abortus–infected HeLa cells was accompanied by loss of mitofusin proteins, without evidence of Drp1 involvement.46 Thus, we hypothesize that E coli–LF82–induced fragmentation of epithelial mitochondria occurs via the following sequence: loss of ΔΨm leads to Drp1 activation, driving active mitochondrial fission that subsequently is enhanced by or superseded by reduced capacity to fuse mitochondria resulting from loss of OPA1-L.
Mitochondrial fragmentation may be a common feature of microbial infections,27 whether it benefits the host or microbe is unclear. It was suggested that mitochondrial fragmentation caused by L monocytogenes and S flexneri protected the cell from subsequent infection and promoted cell-to-cell spread of the bacteria, respectively.40,41 Contrarily, hyperfused mitochondrial networks were observed in human umbilical vein endothelial cells (HUVECs) and HeLa cells shortly after infection with Chlamydia trachamatis, and it was speculated that the increase in ATP production favored replication and growth of the bacteria.47
AIEC-infected epithelia show reduced barrier function,30,48 and although the Drp1 inhibitor, Mdivi1, did not affect E coli–LF82–induced reductions in TER (neither did the pan-caspase inhibitor Z-VAD, which blocks caspase-dependent apoptosis), it significantly reduced the concomitant transepithelial passage of fluorescein isothiocyanate dextran (70 kilodalton) and on average the passage of the bacteria was less. The latter are significant because they indicate that a substantial defect in epithelial barrier function evoked by infection with E coli–LF82 can be reduced significantly, at least in the short term, by an inhibitor of mitochondrial fission. Collectively, the data highlight the importance of mitochondrial function (ie, ΔΨm, ATP production, remodeling) in maintaining epithelial barrier function.15,18
E coli–LF82–infected T84 epithelia were characterized by pyknotic nuclei and DNA disintegration indicative of cell death. Lack of IL18 synthesis or receptor-interacting serine/threonine-protein kinase 3 phosphorylation (personal observation) 8–16 hours after infection with E coli–LF82 suggested the enterocytes were not undergoing necroptosis. Reduced ΔΨm can foreshadow release of cytochrome C into the cytoplasm as a precursor to cell death, via caspase-3 activation, as shown in H pylori–induced epithelial apoptosis.42 Increased cytosolic cytochrome C occurred 8 hours after infection with E coli–LF82 (108 CFU/mL). Neither Mdivi1 nor P110 ablated the cytochrome C release, and, furthermore, we found no evidence of caspase-3 activation in E coli–LF82–infected T84 epithelia, which showed a reduction in total caspase-3 protein, recapitulating data from Dunne et al.31 Similarly S flexneri–evoked HeLa cell death after loss of ΔΨm occurred independent of caspase activation.41,49 Apoptosis-inducing factor has been implicated in H pylori– and EPEC-induced epithelial cell death50,51; however, AIF recruitment to the nucleus in E coli–LF82–treated cells was not increased significantly compared with controls. Thus, although E coli–LF82-induced fragmentation of nuclear DNA indicates cell death, no data were obtained to suggest that the reduced epithelial viability was via caspase activation, AIF mobilization, or necroptosis. Although loss of ΔΨm and/or mitochondrial fragmentation can lead to cell death, it remains unclear if these are the underlying reasons for any E coli–LF82–induced decreases in cell viability.
Conclusions
In summary, epithelia exposed to viable E coli–LF82 show loss of mitochondrial function and substantial fragmentation of the mitochondrial network mediated by Drp1 and, potentially, loss of OPA1-L protein. Furthermore, the increased epithelial permeability caused by E coli–LF82 was partially restored by inhibition of mitochondrial fission, showing the importance of normal mitochondrial dynamics in the control of gut barrier function (Figure 11). Using pertinent human-derived epithelial cell lines in conjunction with a Crohn’s disease–relevant pathobiont, the data herein present a novel perspective on AIEC–host interaction, identifying mitochondria as a therapeutic target to alleviate some of the pathophysiology evoked by enteric pathobionts that could be relevant to a cohort of patients with IBD.
Figure 11.
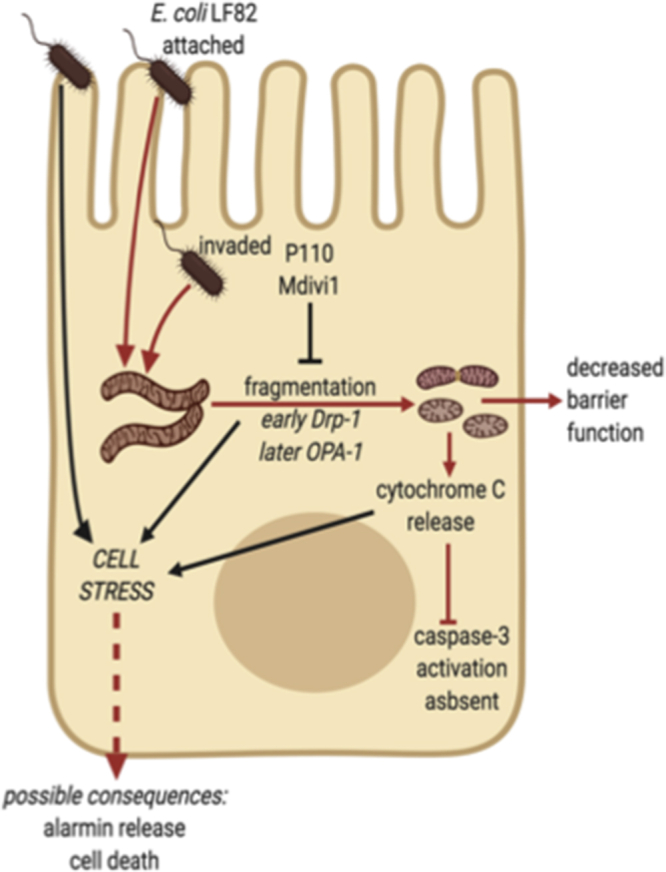
Schematic of model showing how attachment and/or invasion of epithelial cells by adherent-invasive E coli (strain LF82 depicted) can lead to fragmentation of the mitochondrial network via DRP1 activation, and possibly loss of OPA1-L, and mitochondrial disruption as defined by movement of cytochrome C from mitochondria to the cytosol. Infection with E coli–LF82 was not accompanied by evidence of caspase-3 activation. The cell stress experienced results in reduced epithelial function and may have implications for cell activation (ie, alarmin release) and viability (red lines indicate pathway supported by data in the current study).
Methods
Cell Culture
Human colon-derived T84, Caco2, and HT-29 cell lines were maintained as described.52 Briefly, T84 cells (passages 40–120) were maintained in a 1:1 Dulbecco’s modified Eagle medium/F12 Ham medium (Sigma, MO) mixture containing HEPES (2 mmol/L; Sigma), L-glutamine (2.68 mmol/L; Gibco, MA), sodium pyruvate (0.6 mmol/L; Sigma), sodium bicarbonate (0.015%; Gibco), and penicillin-streptomycin (120 U/mL penicillin, 0.12 mg/mL streptomycin; Sigma) and 10% (vol/vol) fetal bovine serum. The HT-29 and Caco2 lines (passages 40–60) were cultured in Dulbecco’s modified Eagle medium and constituents similar to T84 epithelia with the addition of nonessential amino acids (1×; Sigma), and L-glutamine at 2.16 mmol/L, penicillin-streptomycin (108 U/mL penicillin, 0.11 mg/mL streptomycin), and 5% fetal bovine serum.
Commensal E coli (strains HB101, F18; used as controls interchangeably in all experiments to safeguard against any strain-specific effect) and AIEC (strains LF82, LF82ΔFimH, and NRG857c; from Drs P. Sherman [University of Toronto], J.D. Söderholm [Linköping University], and B. Coombes [McMaster University], respectively) and EPEC (strain E2348) (from P. Sherman) were maintained as described.15,30,53 Two infection paradigms were used: short-term, high-dose E coli (4 h, 108 CFU/mL; multiplicity of infection, ∼100) and longer-term, low-dose (16 h, 104 CFU/mL; multiplicity of infection, ∼0.01). Dead E coli–LF82 (108 CFU/mL) were prepared by 2-hour fixation in 2.5% glutaraldehyde (Sigma). Spent medium was prepared by incubating E coli–LF82 (104 CFU/mL) ± T84 cells for 16 hours, followed by centrifugation and 0.2-μm filtration of the supernatant.
The mitochondrial fission protein-1 (Fis1) inhibitor, P110 (designed to block Drp1–Fis1 binding, a gift from Dr D. Mochly-Rosen54) was used as a 30-minute pretreatment at 10 μmol/L (bioactivity of P110 confirmed by its ability to reduce BAPTA [5 μmol/L, 1,2-bis(o-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid] and CCCP [4 μmol/L] induced mitochondrial fragmentation; both Sigma). The selective inhibitor of Drp1 GTPase activity, Mdivi1 (5 μmol/L; Sigma), was applied concomitantly with E coli.55 Staurosporine (20 μmol/L) was used as a positive control in some assays (Sigma). To test a role for ROS in the model, the general antioxidant, vitamin C (0.25 mmol/L), or the mitochondria-targeted antioxidant, Mito-TEMPO (10 μmol/L) (both Sigma), were used as a co-treatment with E coli.21
siRNA Knock-Down of Drp1
Drp1 expression was knocked-down using siRNA (Drp1 siRNA, sc-43732; Santa Cruz Biotechnology, TX; control nontargeted siRNA, sc-37007; Santa Cruz Biotechnology) and Lipofectamine 2000 (ThermoFisher Scientifc).52 Briefly, siRNA and Lipofectamine were prepared in Opti-MEM culture medium (Gibco) and, when combined with cells, would be 2 μL/well Lipofectamine, 30 nmol/L siRNA, 105 T84 cells in 100 μL/well. As described in the manufacturer’s instructions, the siRNA mixture was added to the Lipofectamine mixture, mixed gently, and incubated for 5 minutes at room temperature and the appropriately adjusted concentration of cells was added, mixed gently, and 105 cells/100 μL was seeded into each well of 8-well chamber slides (Lab-Tek; ThermoFisher Scientific). Medium was changed no later than 16 hours after seeding. Cells were stained with MitoTracker dye (see mitochondrial morphology section) 72 hours after transfection, treated with bacteria (2 h, 108 CFU/mL), and imaged live. Efficiency of siRNA knockdown of Drp1 was confirmed by immunoblotting (Figure 12).
Figure 12.

(A) Representative Western blot and (B) accompanying summary densitometry showing the effectiveness of siRNA (30 nmol/L) against Drp1 to reduce Drp1 protein in T84 epithelial cells, control or infected with E coli LF82 (mock represents identically treated epithelia without exposure to siRNA and csiRNA is a control irrelevant siRNA sequence). Data are means ± SEM. Ctrl, control.
RNA Sequence Analysis
RNA sequencing was performed at the Centre for Health Genomics and Informatics at the University of Calgary, where samples were made into libraries using the Illumina TruSeq Stranded messenger RNA LT library preparation kit and were sequenced on a 75-cycle, high-output NextSeq 500 run. Briefly, transcripts were quantified with kallisto 0.43.156 using Homo sapiens GRCh38 (Ensembl release 90) complementary DNA, with sequence bias correction turned on and 50 bootstraps. The tximport package, 1.12.1, was used to import the kallisto results and aggregate transcript abundances to the gene level, and DESeq2 1.24.057 was used for differential expression. Infected epithelia were compared with noninfected control T84 cell monolayers using the Wald test and differentially expressed genes were selected based on a P value cut-off of .05. Enriched gene sets for both KEGG58 and Reactome59 pathways were identified with over-representation tests (P < .05) using clusterProfiler 3.10.060 and ReactomePA 1.26.0,61 respectively. Data were deposited in NCBI's Gene Expression Omnibus and are accessible through GEO series accession numbers: GSE154121 and GSE154122 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE154121).
E coli Localization
T84 cells were seeded on coverslips in 24-well plates and infected with E coli–LF82 or E coli–HB101 (4 h, 108 CFU/mL), fixed with 4% paraformaldehyde for 10 minutes, washed, and permeabilized with 0.1% Triton X-100 in phosphate-buffered saline (Sigma, 2 × 5 min.) and blocked with 10% donkey serum (room temperature, 30 min). After addition of anti–E coli antibody (1:50, 30 min, 37ºC; Abcam, Cambridge, UK), washing (2 × 5 min), donkey anti-goat secondary Alexa Fluor–488 antibody (1:1000, 30 min, room temperature), cells were washed (2 × 5 min), stained with 4′,6-diamidino-2-phenylindole (DAPI, 1:1000, 5 min, room temperature), rinsed, mounted with Dako fluorescent mounting medium (Dako North America, Inc, Agilent Technologies, CA), and viewed on an Olympus (Tokyo, Japan) BX41 wide-field fluorescence microscope.
The gentamycin assay assessed bacterial internalization.15 T84 cells were grown to 50%–70% confluence as determined by phase-contrast microscopy, infected with E coli, a sample of the medium was collected, and then gentamycin (100 μg/mL, 1 h; Sigma) was added to the culture wells. After rinsing, epithelia were lysed with 1% Triton X-100, and the lysate was cultured for 18 hours at 37ºC on blood agar plates: CFUs were counted and data are presented as the percentage of internalization.
Mitochondrial Function
Mitochondrial membrane potential was assessed by seeding T84 epithelia (1 × 105/well) into 96-well plates followed by washing and incubation in tetramethylrhodamine ethyl ester dye (75 nmol/L, 30 min, 37ºC; ThermoFisher). After rinsing, cells were treated with E coli, images were captured on the IncuCyte ZOOM microscope 30 minutes later at 20× magnification (Essen Biosciences, Sartorius, Gottingen, Germany), and analyzed using the IncuCyte ZOOM software. Loss of fluorescence indicates tetramethylrhodamine ethyl ester leakage from the mitochondria resulting from mitochondrial membrane depolarization. The H+ ionophore CCCP (4 μmol/L) was used as a positive control.
Intracellular ATP was assessed using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI) as applied to T84 cells ± E coli–LF82 cultured in 12-well plates and following the manufacturer’s instructions. Samples were collected into white-walled, 96-well plates (Griener Bio-One, KI, Austria), luciferase was measured in a Victor3V 1420 Multilabel Counter (PerkinElmer, Waltham, MA), and data were normalized to protein content in the samples.
Mitochondrial Form and Associated Proteins
T84 epithelia ± E coli were cultured on plastic plates, fixed in gluteraldehyde, processed and sectioned, and the ultrastructure was assessed on a Hitachi (Tokyo, Japan) H-7650 transmission electron microscope.
For mitochondrial network analysis, epithelial cells were seeded on 8-well chamber slides (Lab-Tek, 155409; ThermoFisher Scientific), and stained using the MitoTracker Red CMXRos probe (50 nmol/L, Invitrogen Detection Technologies, Molecular Probes, MA) for 30 minutes at 37ºC, washed, stained with Hoescht dye (1 mg/mL; ThermoFisher), washed, and treated with E coli. Live cell imaging was performed on a Leica (Wetzlar, Germany) DMI6000B Diskovery Flex spinning disk confocal microscope. Cells were chosen randomly based on nuclear staining, fluorescence then was switched to view MitoTracker dye, and mitochondrial morphology was quantified; 20 cells per epithelial preparation were assessed. Mitochondrial networks were classified as fused (interconnecting mitochondrial networks >50% of the cell), fragmented (≥80% spheric mitochondria), or intermediate (combination of fused and fragmented mitochondria, or short tubules not interconnected with each other).62
Whole-cell protein extracts or isolated mitochondria (Mitochondria Isolation Kit for Tissue and Cultured Cells; BioChain, Newark, CA) were subjected to Western blot analysis. Primary antibodies were as follows: OPA1 (1:1000; Abcam), p-Drp1 (1:1000; Cell Signaling, MA), Drp1 (1:2000; Abcam), actin (1:1000; Santa Cruz), cleaved-caspase 3 (1:1000; Cell Signaling), total caspase 3 (1:1000; Cell Signalling), and heat-shock protein-60 (1:1000; Santa Cruz).
Epithelial Viability
Release of cytochrome C from mitochondria into the cytosol and subcellular localization of AIF were determined by fluorescence microscopy in a blinded fashion on paraformaldehyde-fixed epithelial monolayers ± E coli treatment. Antibodies used were rabbit anti–translocase of outer membrane 20 (1:100; Santa Cruz), Alexa Fluor–488 mouse anti–cytochrome C (1:50; BD Pharmingen, CA), and Alexa Fluor–568 goat anti-rabbit (1:1000; ThermoFisher). In other cells, AIF was visualized with a rabbit anti-human antibody (1:100, 4642; Cell Signaling) followed by a Cy3-conjugated anti-rabbit IgG (1:2000, 711-164-152; Jackson Immunoresearch, PA). Images were captured on a Nikon (Tokyo, Japan) A1R laser scanning confocal microscope and image analysis was performed using Ezcolocalisation and ImageJ (National Institutes of Health, Bethesda, MD) plugin (AIF and 4′,6-diamidino-2-phenylindole colocalization compared with Mander’s coefficient of colocalization).63 In addition, DNA was extracted from T84 cells using TRIzol (Invitrogen), 2-μg samples of DNA and 1 kb-plus DNA marker (Invitrogen) were loaded onto a 2% agarose gel with 0.5 μg/mL bromide. After electrophoresis, the gel was examined on a UV transilluminator and images were captured.
Epithelial Barrier Function
TER, flux of fluorescein isothiocyanate dextran (size 70 kilodalton, concentration 200 μg/mL), and transepithelial passage of E coli across filter-grown (3-μm pore) T84 cell monolayers (starting TER, ≥1000 Ω × cm2) were performed as previously described.15
Data Presentation and Analysis
Data are presented as means ± SEM and statistical significance was accepted at P < .05, where n values are the number of epithelial preparations from multiple combined experiments. When comparing 2 groups, the Student t test was performed. When multiple groups were compared, a 1-way analysis of variance followed by either a Dunnett multiple comparison or Tukey multiple comparison post-test was performed. For nonparametric data, a Kruskal–Wallis followed by the Dunn multiple comparison post-test was performed. When analyzing mitochondrial morphology, data are indicated as different if a statistical difference was observed when comparing either the percentage fused or the percentage fragmented morphology between groups. Analyses were conducted using Prism software version 6.0 (GraphPad Software Inc, CA).
Acknowledgments
The authors thank Dr Hena R. Ramay, Bioinformatics Platform of the International Microbiome Centre at the University of Calgary.
CRediT Authorship Contributions
Nicole L. Mancini, PhD (Conceptualization: Equal; Data curation: Lead; Formal analysis: Lead; Methodology: Lead; Project administration: Equal; Writing – original draft: Lead; Writing – review & editing: Equal);
Sruthi Rajeev, BSc (Data curation: Supporting; Writing – review & editing: Supporting);
Timothy S. Jayme, MSc (Data curation: Supporting; Writing – review & editing: Supporting);
Arthur Wang, MD, MSc (Data curation: Supporting; Writing – review & editing: Supporting);
Åsa V. Keita, PhD (Conceptualization: Supporting; Investigation: Supporting; Writing – review & editing: Supporting);
Matthew L. Workentine, PhD (Formal analysis: Supporting; Writing – review & editing: Supporting);
Samira Hamed, BSc (Data curation: Supporting; Writing – review & editing: Supporting);
Johan D. Söderholm, MD PhD (Conceptualization: Supporting; Writing – review & editing: Supporting);
Fernando Lopes, PhD (Data curation: Supporting; Writing – review & editing: Supporting);
Jane Shearer, Phd (Conceptualization: Supporting; Supervision: Supporting; Writing – review & editing: Supporting);
Timothy E. Shutt, PhD (Conceptualization: Supporting; Supervision: Supporting; Writing – review & editing: Supporting);
Derek M. McKay, PhD (Conceptualization: Lead; Funding acquisition: Equal; Project administration: Lead; Supervision: Lead; Writing – original draft: Supporting; Writing – review & editing: Lead).
Footnotes
Conflicts of interest The authors disclose no conflicts.
Funding Supported by grant PJT-153074 from the Canadian Institutes of Health Research (D.M.M.).
References
- 1.Khor B., Gardet A., Xavier R.J. Genetics and pathogenesis of inflammatory bowel disease. Nature. 2011;474:307–317. doi: 10.1038/nature10209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Elliott T.R., Hudspith B.N., Wu G., Cooley M., Parkes G., Quiñones B., Randall L., Mandrell R.E., Fagerquist C.K., Brostoff J., Rayment N.B., Boussioutas A., Petrovska L., Sanderson J.D. Quantification and characterization of mucosa-associated and intracellular Escherichia coli in inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:2326–2338. doi: 10.1097/MIB.0b013e3182a38a92. [DOI] [PubMed] [Google Scholar]
- 3.Darfeuille-Michaud A., Neut C., Barnich N., Lederman E., Di Martino P., Desreumaux P., Gambiez L., Joly B., Cortot A., Colombel J.F. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn's disease. Gastroenterology. 1998;115:1405–1413. doi: 10.1016/s0016-5085(98)70019-8. [DOI] [PubMed] [Google Scholar]
- 4.Chervy M., Barnich N., Denizot J. Adherent-invasive E. coli: update on the lifestyle of a troublemaker in Crohn's disease. Int J Mol Sci. 2020;21:3734. doi: 10.3390/ijms21103734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boudeau J., Barnich N., Darfeuille-Michaud A. Type 1 pili-mediated adherence of Escherichia coli strain LF82 isolated from Crohn's disease is involved in bacterial invasion of intestinal epithelial cells. Mol Microbiol. 2001;39:1272–1284. doi: 10.1111/j.1365-2958.2001.02315.x. [DOI] [PubMed] [Google Scholar]
- 6.Chassaing B., Rolhion N., de Vallee A., Salim S.Y., Prorok-Hamon M., Neut C., Campbell B.J., Söderholm J.D., Hugot J.P., Colombel J.F., Darfeuille-Michaud A. Crohn disease--associated adherent-invasive E. coli bacteria target mouse and human Peyer's patches via long polar fimbriae. J Clin Invest. 2011;121:966–975. doi: 10.1172/JCI44632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jarry A., Cremet L., Caroff N., Bou-Hanna C., Mussini J.M., Reynaud A., Servin A.L., Mosnier J.F., Liévin-Le Moal V., Laboisse C.L. Subversion of human intestinal mucosa innate immunity by a Crohn's disease-associated E. coli. Mucosal Immunol. 2015;8:572–581. doi: 10.1038/mi.2014.89. [DOI] [PubMed] [Google Scholar]
- 8.Palmela C., Chevarin C., Xu Z., Torres J., Sevrin G., Hirten R., Barnich N., Ng S.C., Colombel J.F. Adherent-invasive Escherichia coli in inflammatory bowel disease. Gut. 2018;67:574–587. doi: 10.1136/gutjnl-2017-314903. [DOI] [PubMed] [Google Scholar]
- 9.Keita Å.V., Alkaissi L.Y., Holm E.B., Heil S.D.S., Chassaing B., Darfeuille-Michaud A., McKay D.M., Söderholm J.D. Enhanced E. coli LF82 translocation through the follicle-associated epithelium in Crohn's disease is dependent on long polar fimbriae and CEACAM6 expression, and increases paracellular permeability. J Crohns Colitis. 2020;14:216–229. doi: 10.1093/ecco-jcc/jjz144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vazeille E., Buisson A., Bringer M.A., Goutte M., Ouchchane L., Hugot J.P., de Vallée A., Barnich N., Bommelaer G., Darfeuille-Michaud A. Monocyte-derived macrophages from Crohn's disease patients are impaired in the ability to control intracellular adherent-invasive Escherichia coli and exhibit disordered cytokine secretion profile. J Crohns Colitis. 2015;9:410–420. doi: 10.1093/ecco-jcc/jjv053. [DOI] [PubMed] [Google Scholar]
- 11.Drouet M., Vignal C., Singer E., Djouina M., Dubreuil L., Cortot A., Desreumaux P., Neut C. AIEC colonization and pathogenicity: influence of previous antibiotic treatment and preexisting inflammation. Inflamm Bowel Dis. 2012;18:1923–1931. doi: 10.1002/ibd.22908. [DOI] [PubMed] [Google Scholar]
- 12.Carvalho F.A., Barnich N., Sauvanet P., Darcha C., Gelot A., Darfeuille-Michaud A. Crohn's disease-associated Escherichia coli LF82 aggravates colitis in injured mouse colon via signaling by flagellin. Inflamm Bowel Dis. 2008;14:1051–1060. doi: 10.1002/ibd.20423. [DOI] [PubMed] [Google Scholar]
- 13.Chapman T.P., Hadley G., Fratter C., Cullen S.N., Bax B.E., Bain M.D., Sapsford R.A., Poulton J., Travis S.P. Unexplained gastrointestinal symptoms: think mitochondrial disease. Dig Liver Dis. 2014;46:1–8. doi: 10.1016/j.dld.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 14.Santhanam S., Rajamanickam S., Motamarry A., Ramakrishna B.S., Amirtharaj J.G., Ramachandran A., Pulimood A., Venkatraman A. Mitochondrial electron transport chain complex dysfunction in the colonic mucosa in ulcerative colitis. Inflamm Bowel Dis. 2012;18:2158–2168. doi: 10.1002/ibd.22926. [DOI] [PubMed] [Google Scholar]
- 15.Nazli A., Yang P.C., Jury J., Howe K., Watson J.L., Söderholm J.D., Sherman P.M., Perdue M.H., McKay D.M. Epithelia under metabolic stress perceive commensal bacteria as a threat. Am J Pathol. 2004;164:947–957. doi: 10.1016/S0002-9440(10)63182-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsieh S.Y., Shih T.C., Yeh C.Y., Lin C.J., Chou Y.Y., Lee Y.S. Comparative proteomic studies on the pathogenesis of human ulcerative colitis. Proteomics. 2006;6:5322–5331. doi: 10.1002/pmic.200500541. [DOI] [PubMed] [Google Scholar]
- 17.Söderholm J.D., Olaison G., Peterson K.H., Franzén L.E., Lindmark T., Wirén M., Tagesson C., Sjödahl R. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn's disease. Gut. 2002;50:307–313. doi: 10.1136/gut.50.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho G.T., Aird R.E., Liu B., Boyapati R.K., Kennedy N.A., Dorward D.A., Noble C.L., Shimizu T., Carter R.N., Chew E.T.S., Morton N.M., Rossi A.G., Sartor R.B., Iredale J.P., Satsangi J. MDR1 deficiency impairs mitochondrial homeostasis and promotes intestinal inflammation. Mucosal Immunol. 2018;11:120–130. doi: 10.1038/mi.2017.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russell R.K., Drummond H.E., Nimmo E.R., Anderson N.H., Noble C.L., Wilson D.C., Gillett P.M., McGrogan P., Hassan K., Weaver L.T., Bisset W.M., Mahdi G., Satsangi J. Analysis of the influence of OCTN1/2 variants within the IBD5 locus on disease susceptibility and growth indices in early onset inflammatory bowel disease. Gut. 2006;55:1114–1123. doi: 10.1136/gut.2005.082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Amrouche-Mekkioui I., Djerdjouri B. N-acetylcysteine improves redox status, mitochondrial dysfunction, mucin-depleted crypts and epithelial hyperplasia in dextran sulfate sodium-induced oxidative colitis in mice. Eur J Pharmacol. 2012;691:209–217. doi: 10.1016/j.ejphar.2012.06.014. [DOI] [PubMed] [Google Scholar]
- 21.Wang A., Keita A.V., Phan V., McKay C.M., Schoultz I., Lee J., Murphy M.P., Fernando M., Ronaghan N., Balce D., Yates R., Dicay M., Beck P.L., MacNaughton W.K., Söderholm J.D., McKay D.M. Targeting mitochondria-derived reactive oxygen species to reduce epithelial barrier dysfunction and colitis. Am J Pathol. 2014;184:2516–2527. doi: 10.1016/j.ajpath.2014.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sabouny R.S., Shutt T.E. Reciprocal regulation of mitochondrial fission and fusion. Trends Biomed Sci. 2020;45:564–577. doi: 10.1016/j.tibs.2020.03.009. [DOI] [PubMed] [Google Scholar]
- 23.Archer S.L. Mitochondrial fission and fusion in human diseases. N Engl J Med. 2013;369:2236–2251. doi: 10.1056/NEJMra1215233. [DOI] [PubMed] [Google Scholar]
- 24.del Campo A., Parra V., Vasquez-Trincado C., Gutiérrez T., Morales P.E., López-Crisosto C., Bravo-Sagua R., Navarro-Marquez M.F., Verdejo H.E., Contreras-Ferrat A., Troncoso R., Chiong M., Lavandero S. Mitochondrial fragmentation impairs insulin-dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. Am J Physiol Endocrinol Metab. 2014;306:E1–E13. doi: 10.1152/ajpendo.00146.2013. [DOI] [PubMed] [Google Scholar]
- 25.Ryan J., Dasgupta A., Huston J., Chen K.H., Archer S.L. Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med. 2015;93:229–242. doi: 10.1007/s00109-015-1263-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mancini N.L., Goudie L., Xu W., Sabouny R., Rajeev S., Wang A., Esquerre N., Rajabi A.A., Jayme T.S., van Tilburg Bernardes E., Nasser Y., Ferraz J.G.P., Shutt T.E., Shearer J., McKay D.M. Perturbed mitochondrial dynamics is a novel feature of colitis that can be targeted to lessen disease. Cell Mol Gastroenterol Hepatol. 2020;10:287–307. doi: 10.1016/j.jcmgh.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKay D.M., Mancini N.L., Shearer J., Shutt T.E. Perturbed mitochondrial dynamics, an emerging aspect of epithelial-microbe interactions. Am J Physiol Gastrointest Liver Physiol. 2020;318:G748–G762. doi: 10.1152/ajpgi.00031.2020. [DOI] [PubMed] [Google Scholar]
- 28.Han Y., Kim B., Cho U., Park I.S., Kim S.I., Dhanasekaran D.N., Tsang B.K., Song Y.S. Mitochondrial fission causes cisplatin resistance under hypoxic conditions via ROS in ovarian cancer cells. Oncogene. 2019;38:7089–7105. doi: 10.1038/s41388-019-0949-5. [DOI] [PubMed] [Google Scholar]
- 29.Elatrech I., Marzaioli V., Boukemara H., Bournier O., Neut C., Darfeuille-Michaud A., Luis J., Dubuquoy L., El-Benna J., My-Chan Dang P., Marie J.C. Escherichia coli LF82 differentially regulates ROS production and mucin expression in intestinal epithelial T84 cells: implication of NOX1. Inflamm Bowel Dis. 2015;21:1018–1026. doi: 10.1097/MIB.0000000000000365. [DOI] [PubMed] [Google Scholar]
- 30.Wine E., Ossa J.C., Gray-Owen S.D., Sherman P.M. Adherent-invasive Escherichia coli, strain LF82 disrupts apical junctional complexes in polarized epithelia. BMC Microbiol. 2009;9:180. doi: 10.1186/1471-2180-9-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dunne K.A., Amr A., McIntosh A., Houston S.A., Cervoic V., Goodyear C.S., Roe A.J., Beatson S.A., Milling S.W., Walker D., Wall D.M. Increased S-nitrosylation and proteosomal degradation of caspase-3 during infection contribute to the persistene of adherent invasive Escherichia coli (AIEC) in immune cells. PLoS One. 2013;8 doi: 10.1371/journal.pone.0068386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haberman Y., Karns R., Dexheimer P.J., Schirmer M., Somekh J., Jurickova I., Braun T., Novak E., Bauman L., Collins M.H., Mo A., Rosen M.J., Bonkowski E., Gotman N., Marquis A., Nistel M., Rufo P.A., Baker S.S., Sauer C.G., Markowitz J., Pfefferkorn M.D., Rosh J.R., Boyle B.M., Mack D.R., Baldassano R.N., Shah S., Leleiko N.S., Heyman M.B., Grifiths A.M., Patel A.S., Noe J.D., Aronow B.J., Kugathasan S., Walters T.D., Gibson G., Thomas S.D., Mollen K., Shen-Orr S., Huttenhower C., Xavier R.J., Hyams J.S., Denson L.A. Ulcerative colitis mucosal transcriptomes reveal mitochondriopathy and personalized mechanisms underlying disease severity and treatment response. Nat Commun. 2019;10:38. doi: 10.1038/s41467-018-07841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manfredo Vieira S., Hiltensperger M., Kumar V., Zegarra-Ruiz D., Dehner C., Khan N., Costa F.R.C., Tiniakou E., Greiling T., Ruff W., Barbieri A., Kriegel C., Mehta S.S., Knight J.R., Jain D., Goodman A.L., Kriegel M.A. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science. 2018;359:1156–1161. doi: 10.1126/science.aar7201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He D., Hagen S.J., Pothoulakis C., Chen M., Medina N.D., Warny M., LaMont J.T. Clostridium difficile toxin A causes early damage to mitochondria in cultured cells. Gastroenterology. 2000;119:139–150. doi: 10.1053/gast.2000.8526. [DOI] [PubMed] [Google Scholar]
- 35.Zareie M., Riff J., Donato K., McKay D.M., Perdue M.H., Söderholm J.D., Karmali M., Cohen M.B., Hawkins J., Sherman P.M. Novel effects of the prototype translocating Escherichia coli, strain C25 on intestinal epithelial structure and barrier function. Cell Microbiol. 2005;7:1782–1797. doi: 10.1111/j.1462-5822.2005.00595.x. [DOI] [PubMed] [Google Scholar]
- 36.Nagai T., Abe A., Sasakawa C. Targeting of enteropathogenic Escherichia coli EspF to host mitochondria is essential for bacterial pathogenesis: critical role of the 16th leucine residue in EspF. J Biol Chem. 2005;280:2998–3011. doi: 10.1074/jbc.M411550200. [DOI] [PubMed] [Google Scholar]
- 37.Kuo Y.C., Chen I.Y., Chang S.C., Wu S.C., Hung T.M., Lee P.H., Shimotohno K., Chang M.F. Hepatitis C virus NS5A protein enhances gluconeogenesis through upregulation of Akt-/JNK-PEPCK signalling pathways. Liver Int. 2014;34:1358–1368. doi: 10.1111/liv.12389. [DOI] [PubMed] [Google Scholar]
- 38.Fine-Coulson K., Giguere S., Quinn F.D., Reaves B.J. Infection of A549 human type II epithelial cells with Mycobacterium tuberculosis induces changes in mitochondrial morphology, distribution and mass that are dependent on the early secreted antigen, ESAT-6. Microbes Infect. 2015;17:689–697. doi: 10.1016/j.micinf.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki M., Danilchanka O., Mekalanos J.J. Vibrio cholerae T3SS effector VopE modulates mitochondrial dynamics and innate immune signaling by targeting Miro GTPases. Cell Host Microbe. 2014;16:581–591. doi: 10.1016/j.chom.2014.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stavru F., Bouillaud F., Sartori A., Ricquier D., Cossart P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc Natl Acad Sci U S A. 2011;108:3612–3617. doi: 10.1073/pnas.1100126108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lum M., Morona R. Dynamin-related protein Drp1 and mitochondria are important for Shigella flexneri infection. Int J Med Microbiol. 2014;304:530–541. doi: 10.1016/j.ijmm.2014.03.006. [DOI] [PubMed] [Google Scholar]
- 42.Jain P., Luo Z.Q., Blanke S.R. Helicobacter pylori vacuolating cytotoxin A (VacA) engages the mitochondrial fission machinery to induce host cell death. Proc Natl Acad Sci U S A. 2011;108:16032–16037. doi: 10.1073/pnas.1105175108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stavru F., Palmer A.E., Wang C., Youle R.J., Cossart P. Atypical mitochondrial fission upon bacterial infection. Proc Natl Acad Sci U S A. 2013;110:16003–16008. doi: 10.1073/pnas.1315784110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Helle S.C.J., Feng Q., Aebersold M.J., Hirt L., Grüter R.R., Vahid A., Sirianni A., Mostowy S., Snedeker J.G., Šarić A., Idema T., Zambelli T., Kornmann B. Mechanical force induces mitochondrial fission. Elife. 2017;6 doi: 10.7554/eLife.30292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nazli A., Wang A., Steen O., Prescott D., Lu J., Perdue M.H., Söderholm J.D., Sherman P.M., McKay D.M. Enterocyte cytoskeleton changes are crucial for enhanced translocation of nonpathogenic Escherichia coli across metabolically stressed gut epithelia. Infect Immun. 2006;74:192–201. doi: 10.1128/IAI.74.1.192-201.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lobet E., Willemart K., Ninane N., Demazy C., Sedzicki J., Lelubre C., De Bolle X., Renard P., Raes M., Dehio C., Letesson J.J., Arnould T. Mitochondrial fragmentation affects neither the sensitivity to TNFa-induced apoptosis of Brucella-infected cells nor the intracellular replication of the bacteria. Sci Rep. 2018;8:5173. doi: 10.1038/s41598-018-23483-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chowdhury S.R., Reimer A., Sharan M., Kozjak-Pavlovic V., Eulalio A., Prusty B.K., Fraunholz M., Karunakaran K., Rudel T. Chlamydia preserves the mitochondrial network necessary for replication via microRNA-dependent inhibition of fission. J Cell Biol. 2017;216:1071–1089. doi: 10.1083/jcb.201608063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shawki A., McCole D.F. Mechanisms of intestinal epithelial barrier dysfunction by adherent-invasive Escherichia coli. Cell Mol Gastroenterol Hepatol. 2017;3:41–50. doi: 10.1016/j.jcmgh.2016.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carneiro L.A., Travassos L.H., Soares F., Tattoli I., Magalhaes J.G., Bozza M.T., Plotkowski M.C., Sansonetti P.J., Molkentin J.D., Philpott D.J., Girardin S.E. Shigella induces mitochondrial dysfunction and cell death in nonmyeloid cells. Cell Host Microbe. 2009;5:123–136. doi: 10.1016/j.chom.2008.12.011. [DOI] [PubMed] [Google Scholar]
- 50.Ashktorab H., Dashwood R.H., Dashwod M.M., Zaidi S.I., Hewitt S.M., Green W.R., Lee E.L., Daremipouran M., Nouraie M., Malekzadeh R., Smoot D.T.H. pylori-induced apoptosis in human gastric cancer cells mediated via the release of apoptosis-inducing factor from mitochondria. Helicobacter. 2008;13:506–517. doi: 10.1111/j.1523-5378.2008.00646.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Flynn A.N., Wang A., McKay D.M., Buret A.G. Apoptosis-inducing factor contributes to epithelial cell apoptosis induced by enteropathogenic Escherichia coli. Can J Physiol Pharmacol. 2011;89:143–148. doi: 10.1139/y11-002. [DOI] [PubMed] [Google Scholar]
- 52.Lopes F., Keita A.V., Saxena A., Reyes J.L., Mancini N.L., Al Rajabi A., Wang A., Baggio C.H., Dicay M., van Dalen R., Ahn Y., Carneiro M.B.H., Peters N.C., Rho J.M., MacNaughton W.K., Girardin S.E., Jijon H., Philpott D.J., Söderholm J.D., McKay D.M. ER-stress mobilization of death-associated protein kinase-1-dependent xenophagy counteracts mitochondria stress-induced epithelial barrier dysfunction. J Biol Chem. 2018;293:3073–3087. doi: 10.1074/jbc.RA117.000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nash J.H., Villegas A., Kropinski A.M., Aguilar-Valenzuela R., Konczy P., Mascarenhas M., Ziebell K., Torres A.G., Karmali M.A., Coombes B.K. Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genomics. 2010;11:667. doi: 10.1186/1471-2164-11-667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qi X., Qvit N., Su Y.C., Mochly-Rosen D. A novel Drp1 inhibitor diminishes aberrant mitochondrial fission and neurotoxicity. J Cell Sci. 2013;126:789–802. doi: 10.1242/jcs.114439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bordt E.A., Clerc P., Roelofs B.A. The putative Drp1 inhibitor Mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell. 2017;40:583–594.e6. doi: 10.1016/j.devcel.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bray N.L., Pimentel H., Melsted P., Saladino A.J., Tretter L., Adam-Vizi V., Cherok E., Khalil A., Yadava N., Ge S.X., Francis T.C., Kennedy N.W., Picton L.K., Kumar T., Uppuluri S., Miller A.M., Itoh K., Karbowski M., Sesaki H., Hill R.B., Polster B.M. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 57.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kanehisa M., Furumichi M., Tanabe M., Morishima K., Tanabe M. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:D353–D361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fabregat A., Sidiropoulos K., Garapati P., Sidiropoulos K., Gillespie M., Garapati P., Haw R., Jassal B., Korninger F., May B., Milacic M., Roca C.D., Rothfels K., Sevilla C., Shamovsky V., Shorser S., Varusai T., Viteri G., Weiser J., Wu G., Stein L., Hermjakob H., D'Eustachio P. The reactome pathway Knowledgebase. Nucleic Acids Res. 2016;44:D481–D487. doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu G., Wang L.G., Han Y., Qing-Yu H., et al clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yu G., He Q.Y. ReactomePA: an R/Bioconductor package for reactome pathway analysis and visualization. Mol Biosyst. 2016;12:477–479. doi: 10.1039/c5mb00663e. [DOI] [PubMed] [Google Scholar]
- 62.Chatel-Chaix L., Cortese M., Romero-Brey I., Bender S., Neufeldt C.J., Fischl W., Scaturro P., Schieber N., Schwab Y., Fischer B., Ruggieri A., Bartenschlager R. Dengue virus perturbs mitochondrial morphodynamics to dampen innate immune responses. Cell Host Microbe. 2016;20:342–356. doi: 10.1016/j.chom.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Stauffer W., Sheng H., Lim H.N. EzColocalization: an ImageJ plugin for visualizing and measuring colocalization in cells and organisms. Sci Rep. 2018;8:15764. doi: 10.1038/s41598-018-33592-8. [DOI] [PMC free article] [PubMed] [Google Scholar]