

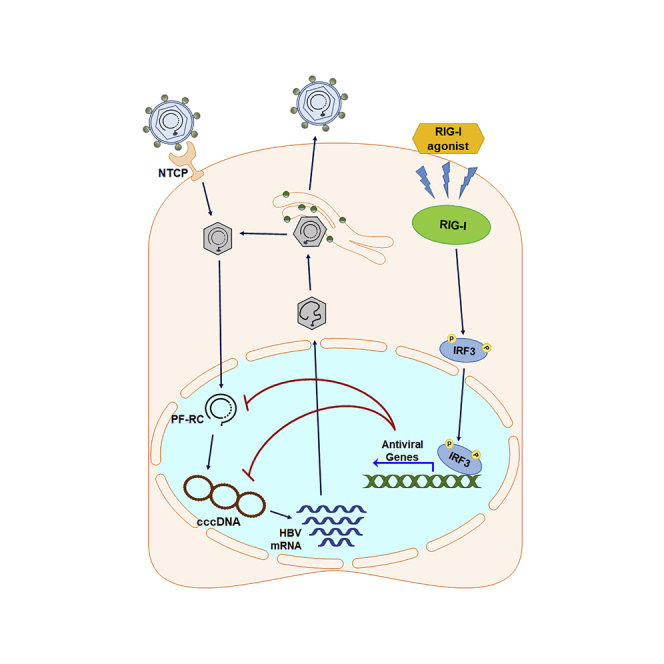
Summary
Hepatitis B virus (HBV) mediates persistent infection, chronic hepatitis, and liver disease. HBV covalently closed circular (ccc)DNA is central to viral persistence such that its elimination is considered the cornerstone for HBV cure. Inefficient detection by pathogen recognition receptors (PRRs) in the infected hepatocyte facilitates HBV persistence via avoidance of innate immune activation and interferon regulatory factor (IRF)3 induction of antiviral gene expression. We evaluated a small molecule compound, F7, and 5′-triphosphate-poly-U/UC pathogen-associated-molecular-pattern (PAMP) RNA agonists of RIG-I, a PRR that signals innate immunity, for ability to suppress cccDNA. F7 and poly-U/UC PAMP treatment of HBV-infected cells induced RIG-I signaling of IRF3 activation to induce antiviral genes for suppression of cccDNA formation and accelerated decay of established cccDNA, and were additive to the actions of entecavir. Our study shows that activation of the RIG-I pathway and IRF3 to induce innate immune actions offers therapeutic benefit toward elimination of cccDNA.
Subject areas: Immunology, Virology
Graphical abstract
Highlights
-
•
Hepatocyte treatment of RIG-I agonists induces IRF3 and antiviral gene expression
-
•
RIG-I agonists direct a block to cccDNA formation in early HBV infection
-
•
Administration of RIG-I agonists imparts cccDNA decay
-
•
Nucleoside analogs with RIG-I agonist is synergistic to block cccDNA formation
Immunology; Virology
Introduction
Hepatitis B virus (HBV) is a global public health problem with more than 250 million people chronically infected worldwide. Chronic HBV infection is a leading cause of liver disease including liver cirrhosis, hepatocellular carcinoma, and liver failure such that HBV infection causes over 700,000 deaths annually (WHO, 2017) (Ott et al., 2012; Jefferies et al., 2018). HBV is a small, hepatotropic DNA virus (Marion and Robinson, 1983) that replicates in part through reverse transcription (Wang and Seeger, 1993). HBV specifically enters hepatocytes through the sodium-taurocholate cotransporting polypeptide (NTCP) receptor to replicate and produce virion (Yan et al., 2012). After entry into the hepatocyte cytosol, the viral nucleocapsid is translocated to the nucleus for disassembly and release of the viral relaxed circular (RC) DNA. In the nucleus, the RC DNA is converted to covalently closed circular (ccc)DNA that is a long-lived viral mini-chromosome serving as the main template for the synthesis of all HBV RNA transcripts including pregenomic (pg) RNA, pre-S, S, and X viral RNAs (Newbold et al., 1995; Shi et al., 2012). After synthesis the mature nucleocapsid-containing RC DNA acquires an envelope via budding at the endoplasmic reticulum (ER) and then produces progeny virion (Wang and Seeger, 1993; Seeger and Mason, 2000). A portion of the pool of mature HBV nucleocapsids is used to facilitate further cccDNA synthesis in a process called the intracellular amplification pathway (Tuttleman et al., 1986; Wu et al., 1990). This process facilitates the pool of cccDNA to be maintained as a steady-state population of 3–50 molecules per cell marking chronic infection (Seeger and Mason, 2000; Beck and Nassal, 2007; Ko et al., 2018). Removal of cccDNA from the liver either through its eradication from infected cells or depletion of infected cells is considered to be essential for HBV cure (Liang et al., 2015; Nassal, 2015; Tang et al., 2017).
Current treatment for chronic hepatitis B infection relies on two main classes of therapy, including (1) nucleot(s)ide analogs (NAs), which inhibit viral reverse transcriptase and DNA polymerase function, and (2) pegylated interferon alpha (peg-IFNα) therapy that induces innate immune defenses for suppression of viral replication and reduced HBV antigen production. Although both treatment modalities can reduce viral burden, IFN-based therapy presents considerable side effects and neither treatment is completely effective (Janssen et al., 2005). Although these therapies can suppress active viral replication, reduce cccDNA levels (Lucifora et al., 2014; Belloni et al., 2012), and can slow disease progression, they do not eliminate the nuclear pool of cccDNA (Wursthorn et al., 2006; Zoulim and Durantel, 2015) and are associated with significant side effects in treated patients (Locarnini et al., 2015; Zoulim and Durantel, 2015; Kwon and Lok, 2011). The persistence of cccDNA is facilitated by long cccDNA half-life, established to be 6–22 weeks in vivo (Huang et al., 2020), in which lifelong treatment with antiviral therapy is required for a majority of patients to continuously suppress viral replication. Problematically, IFN-based therapy for chronic HBV is poorly tolerated and only a low frequency of treated patients show complete loss of HBsAg that defines clinical HBV cure (Dienstag, 2009; Moucari et al., 2009; Perrillo, 2009). Thus new, effective therapeutics that aim to achieve a functional cure by eradicating cccDNA are needed to treat and control chronic HBV infection (Fanning et al., 2019).
Acute viral infection typically triggers intracellular innate immune activation leading to induction of intracellular antiviral defenses. This process serves to control viral replication and spread from the site of infection, and to modulate the adaptive immune response for systemic virus control. Innate immune activation occurs via host cell sensing of viral pathogen-associated molecular patterns (PAMP) embedded in viral replication products, including viral nucleic acid. PAMPs are sensed by cellular pattern recognition receptors (PRRs). PRRs that sense virus infection include Toll-like receptors (TLR); NOD-like receptors (NLRs); intracellular DNA sensors cGAS, STING, IFI16, DAI, and others (Fernandes-Alnemri et al., 2009; Unterholzner et al., 2010); as well as the RIG-I-like receptors (RLRs) including retinoic-acid inducible gene-I (RIG-I) and melanoma differentiation antigen 5 (MDA5) (Pandey et al., 2014; Chow et al., 2018). Each PRR detects specific PAMPs derived from the incoming virus or viral replication products, while certain host cell nucleic acids can also trigger PRR signaling when produced during viral infection (Lee et al., 2019; Pandey et al., 2014). Induction of TLR, RLR, or STING signaling drives the downstream activation of latent transcription factors including interferon regulatory factor (IRF)3 and nuclear factor (NF)-κB to promote the expression of antiviral effector genes and immune regulatory genes including chemokines, IFNs, and other immune regulatory cytokines (Chow et al., 2018). Remarkably, acute HBV infection of primary human hepatocytes (PHHs) neither activates nor inhibits PRR signaling of innate immunity, thus reinforcing the notion that HBV is a “stealth” virus (Mutz et al., 2018) as previously shown in vivo in a nonhuman primate infection model (Wieland et al., 2004). On the other hand, strategies to activate RIG-I signaling have been shown to enhance antiviral defenses against HBV (Wu et al., 2018; Han et al., 2019), suggesting that RLR signaling can direct innate immune programs for control of HBV replication.
Previous studies have shown that RIG-I signaling in response to PAMP RNA can direct innate immune activation and antiviral defenses that suppress replication of hepatitis C virus (HCV) (Saito et al., 2008; Kell et al., 2015; Schnell et al., 2012) that also causes chronic hepatitis (Horner and Gale, 2013). The HCV PAMP is a 100-nt poly uridine/cytosine (poly-U/UC) motif with the HCV genome 3′ nontranslated region (Saito et al., 2008). When introduced into cells as a 5′ppp synthetic RNA the poly-U/UC PAMP specifically activates RIG-I to drive IRF3 activation and antiviral innate immunity that suppresses HCV infection in vitro (Schnell et al., 2012) and activates hepatic innate immunity in vivo (Saito et al., 2008). Moreover, we and others have identified small-molecule benzothiazols (Probst et al., 2017; Pattabhi et al., 2016; Bedard et al., 2012), or 5′ppp RNA ligands that bind and activate RIG-I or induce IRF3 activation and innate immunity to therapeutically suppress RNA virus infection and enhance the immune response (Dassler-Plenker et al., 2019; Hochheiser et al., 2016). Together, these previous studies show that the RIG-I pathway is functional in hepatocytes in which targeted activation of RIG-I confers potent antiviral actions that are fully operational in the liver. However, how the direct targeting and activation of RIG-I and IRF3 impacts HBV is not known.
Various innate immune programs have been shown to impact HBV infection. Beyond IFN, other pro-inflammatory cytokines have suppressive effects on HBV replication (Isorce et al., 2016) (Lanford et al., 2013) and also boost adaptive immunity against chronic HBV infection (Boni et al., 2018). Interferon-gamma and tumor necrosis factor-alpha have been shown to interfere with HBV replication at transcriptional and post-transcriptional steps and to reduce cccDNA stability (Lucifora et al., 2014; Xia et al., 2016). Thus, pharmacological innate immune activation may facilitate intrahepatic antiviral defenses and enhancement of the immune response against chronic HBV.
Here, we evaluated targeted RIG-I activation in the treatment of HBV infection in vitro. RIG-I signaling triggered by poly-U/UC PAMP RNA or small-molecule activator of RIG-I directed robust IRF3 activation and RIG-I-dependent antiviral actions to suppress cccDNA levels. We show that the RIG-I response through IRF3 serves to reduce the half-life (t1/2) of cccDNA to impart cccDNA decay kinetics, and blockage of RC DNA formation and concomitant suppression of HBsAg secretion in HepG2 cells that ectopically express human sodium/taurocholate cotransporting polypeptide (hNTCP) (HepG2-hNTCP), in differentiated HepaRG (dHepaRG) cells and in PHHs. Targeting RIG-I does not promote cell toxicity. Remarkably, when combined with entecavir (ETV) and poly-U/UC PAMP, treatment rapidly depleted established cccDNA pools. Thus targeted RIG-I activation and processes that activate IRF3-directed innate immune activation may offer therapeutic approaches toward HBV cure.
Results
RIG-I and IRF3 agonists trigger innate immune activation in hepatocytes
We previously identified small-molecule agonists of IRF3 that confer innate immune activation leading to induction of IRF3-target genes and antiviral action against a range of RNA viruses (Pattabhi et al., 2016; Probst et al., 2017; Bedard et al., 2012). Based on the published structure (Iadonato et al., 2018), we produced (N-(6-benzamido-1,3-benzothiazol-2-yl)naphthalene-2-carboxamide), referred here as F7, for analyses of anti-HBV activity (Figure 1A). Similarly, we have identified a 100-nt PAMP motif within the HCV genome comprising 5′ppp and the poly-U/UC region of viral RNA that is specifically recognized by RIG-I and confers RIG-I signaling of IRF3 activation leading to antiviral gene expression (Saito et al., 2008; Schnell et al., 2012) (Figure 1A, lower). We evaluated the therapeutic actions of F7 and the poly-U/UC PAMP against HBV infection. We first treated HepG2-hNTCP cells with 10 μM F7 or 200 ng/mL poly-U/UC PAMP formulated in liposome for 24 h. Control cells were treated with DMSO or infected with Sendai virus (SenV; a potent activator of RIG-I-dependent signaling), or transfected with 200 ng/mL X-RNA negative control in liposome, a non-PAMP/non-signaling 5′ppp-containing 100 nt RNA motif from the HCV genome with similar mass to the poly-U/UC PAMP (Saito et al., 2008). Similar to SenV control, both F7 and poly-U/UC PAMP but neither DMSO nor X-RNA treatment specifically induced innate immune activation as marked by IRF3 translocation into the nucleus (Figure 1B). Immunoblot analysis demonstrated that F7 and poly-U/UC PAMP but not X-RNA treatment specifically induced IRF3 phosphorylation and the expression of IFIT1, an IRF3-target gene (Fensterl and Sen, 2011, 2015), in a dose-dependent manner in both HepG2-hNTCP and dHepaRG cells (Figure 1C). We also assessed mRNA expression across a panel of innate immune response genes, including IFNs, ISGs, and direct IRF3-target genes, for response to F7 or poly-U/UC PAMP (Figures 1D and S1). Poly-U/UC PAMP treatment also induced type I and type III IFN and ISG expression, whereas F7 treatment induced only IRF3 direct target gene expression including IFIT1, CXCL10, RSAD2, and IFITM1. This difference is consistent with the signaling properties of each molecule and the nature of IFN expression, as IFN expression relies on activation of both IRF3 and NF-kB, whereas F7 specifically activates IRF3 but not NF-kB (Bedard et al., 2012). In contrast, the poly-U/UC PAMP triggers RIG-I signaling of both transcription factors to induce IRF3-target genes, IFNs, and hence ISGs (Schnell et al., 2012; Saito et al., 2008). Notably, type I IFN, SAMHD1, APOBEC3A, and APOBEC3G, which were induced by poly-U/UC PAMP treatment, have demonstrated antiviral activity against HBV infection (Chen et al., 2014; Bonvin et al., 2006; Lucifora et al., 2014). Taken together, these results show that F7 and poly-U/UC PAMP induce innate immune activation in treated cells that initiates with IRF3 activation and the induction of IRF3-target genes.
Figure 1.

F7 and poly-U/UC PAMP differentially induce innate immune genes
(A) Structure of F7 and sequence poly-U/UC PAMP-RNA.
(B) Immunofluorescence analysis of IRF3 translocation. HepG2-hNTCP cells were cultured in medium containing 2.5% DMSO, infected with Sendai virus (SenV; 10 HAU/ml), or treated with F7 (10 μM), X-RNA (200 ng/mL in liposome), or poly-U/UC PAMP (200 ng/mL in liposome) for 24 h. Cells were fixed with 3% paraformaldehyde, stained by using mouse anti-hNTCP (green) and rabbit anti-IRF3 (red) antibodies, and counterstained with DAPI (blue). Scale bars, 20 μm.
(C) F7 and poly-U/UC induce IRF3 activation and innate immune gene expression. Sendai virus, F7, X-RNA, and poly-U/UC PAMP were administered to HepG2-hNTCP and dHepaRG, as indicated for 24 and 48 h. The cell lysates were resolved by SDS-PAGE and then subjected to immunoblotting. The levels of protein expression by the p-IRF3 (S386 phosphorylation active form of IRF3), IRF3 (total IRF3), and IFIT1 relative to the expression level of tubulin were determined using respective antibodies.
(D) Gene expression analysis. HepG2-hNTCP cells were administered with SenV, F7, X-RNA, and poly-U/UC-RNA as described above for 24 and 72 h; then total cellular RNA was purified from harvested cells. The levels of expression of innate immune genes IFIT1, CXCL10, IFITM1, RSAD2, RIG-I, MDA5, SAMHD1, APOBEC3A, APOBEC3G, IFN-α, IFN-β, and IFN-λ3 were measured by qRT-PCR and normalized to the level of GAPDH expression. Each are shown as the mean fold induction over that achieved with 2.5% DMSO treatment from three independent experiments.
IRF3 activation suppresses HBV cccDNA formation
To determine how IRF3 activation impacts HBV infection and production of cccDNA in infected cells, we treated HepG2-hNTCP cells with F7 or poly-U/UC PAMP (100 ng/mL [ = 2.94 nM] or 200 ng/mL [ = 5.87 nM]) following HBV infection. Cyclosporin A (CsA) treatment was employed as an HBV entry inhibitor antiviral control (Watashi et al., 2014). Cells were harvested 3 days post-infection (dpi), and extracts were prepared using Hirt extraction methods for isolating protein-free DNA, as shown in Figure 2A (Hirt, 1967; Guo et al., 2007). Southern blot analysis revealed that F7 and poly-U/UC PAMP treatment suppressed HBV cccDNA formation compared with the level of expression for the DMSO, or X-RNA treated controls. Additionally, the level of protein free-relaxed circular (PF-RC) DNA, which includes possible precursor forms of cccDNA, was also markedly decreased by treatment with F7 or poly-U/UC PAMP. As both F7 and poly-U/UC PAMP activate IRF3, these results link the IRF3 response with suppression of cccDNA formation in HBV-infected hepatocytes.
Figure 2.

Therapeutic suppression of cccDNA formation
(A) Scheme of infection and treatment (upper) and Southern blot analysis (lower) to measure protein-free DNA including cccDNA. The scheme illustrates the schedule with HBV inoculum (1,000 Geq/cell) and administration of cyclosporin A (CsA), F7, X-RNA (X), and poly-U/UC PAMP. 2.5% DMSO was added to the medium at 1 day post infection (dpi). cccDNA and PF-RC DNA were harvested from HepG2-hNTCP cells using Hirt extraction method and analyzed by Southern blot analysis using an HBV-specific DNA probe. Viral protein-free DNAs (protein-free relaxed circular DNA [PF-RC DNA] and cccDNA) are noted.
(B and C) HepG2-hNTCP (B) or dHepaRG cells (C) were infected with HBV at multiplicity of infection (moi) of 1,000 Geq/cell and administered CsA (10 μM) or F7, in the indicated concentrations. For upper panels of (B and C), DNAs were isolated after Hirt extraction at 3 dpi and subjected to Southern blot analysis using as HBV-DNA probe. For lower panels of (B and C), the inhibitory effects of F7 on cccDNA formation were measured in HepG2-hNTCP and dHepaRG cells, respectively, using qPCR analysis.
(D and E) HepG2-hNTCP (D) or dHepaRG cells (E) were infected with HBV at an moi of 1,000 Geq/cell, and administered CsA (10 μM), X-RNA (X, 100ng/ml), or poly-U/UC PAMP in the indicated concentrations. For upper panels of (D and E), DNAs were analyzed using Southern blot analysis. For lower panels of (D and E), DNAs were analyzed by qPCR. IC90, and IC50 values were calculated based on the decline of the cccDNA relative to DMSO-treated controls. IC90, and IC50 values are the average of three experiments ± 1 SD. Cytotoxicity was determined by measuring cellular ATP content as a measure of cell viability using the CellTiter-Glo reagent. CC50 values shown are the average of three experiments ± 1 SD. Positions of mass markers are indicated on each Southern blot.
To further determine how the activation of IRF3 impacts the HBV replication cycle, we analyzed the expression of HBV DNA, HBV RNA, extracellular HBV DNA, and secreted HBsAg over an infection/treatment time course. As shown in Figure S2A, PF-RC DNA was first detected by 1 dpi. cccDNA synthesis occurred by 2 dpi in control-treated cells, and PF-RC and cccDNA both accumulated over the 20 dpi time course. However, the production of cccDNA was markedly suppressed in cells treated with F7 or poly-U/UC PAMP within 2 dpi. Production of pgRNA across the infection/treatment time course showed that it accumulated from 6 to 20 dpi in the non-treatment control cells but was significantly suppressed in cells treated with F7 or poly-U/UC PAMP (Figure S2B). We also measured the level of capsid-associated intracellular HBV DNA intermediates of reverse transcription produced over the HBV infection/treatment time course. We found that RC DNA, double-stranded linear (DL) DNA, and single-stranded (SS) DNA viral products accumulated from 6 to 20 dpi in control/non-treated cells. However, F7 or poly-U/UC PAMP treatment markedly decreased the levels of these DNA species. Interestingly, the level of incoming viral capsid-associated HBV DNAs (observed from 1–2 dpi) were not affected by treatment, but instead their production was suppressed within 6 dpi (Figure S2C). Suppression of HBV DNA and RNA overall was associated with significant reduction in the level of secreted HBsAg in culture supernatant from cells treated with F7 or poly-U/UC PAMP (Figure S2D). Importantly, both F7 and poly-U/UC PAMP treatment resulted in a block in de novo HBV production revealed by reduction of extracellular HBV DNA (Figure S2E). These results show that F7 and poly-U/UC PAMP treatment impact HBV replication at steps following cccDNA synthesis to impact transcription of viral RNA, reverse transcribed HBV DNAs, and production of progeny virions.
To assess the antiviral activity of F7 and poly-U/UC PAMP against HBV we conducted dose-response analyses. To determine the values of 90% of maximal Inhibitory concentration (IC90) and half-maximal inhibitory concentration (IC50), as well as the cytotoxic concentration, CC50, of F7 and poly-U/UC PAMP, we used two different cell lines, HepG2-hNTCP and dHepaRG cells. Cultures were treated with increasing concentrations of F7 or poly-U/UC followed by HBV infection, then harvested at 3 dpi. Southern blot and qPCR analysis showed that cccDNA level was linearly reduced by increasing concentrations of F7 (Figures 2B and 2C) and poly-U/UC PAMP (Figures 2D and 2E). We defined IC90 and IC50 values for F7 of approximately 17.38 μM and 8.48 μM in HepG2-hNTCP cells, and 12.84 μM and 3.38 μM in differentiated HepaRG cells. The 50% cytotoxic concentration (CC50), as measured by ATP release from treated cells, was over 40 μM (Figures S3A and S3B). We determined the IC90 and IC50 values of poly-U/UC PAMP to be approximately 11.97 and 1.94 nM, respectively, in HepG2-NTCP cells and 3.3 and 1.61 nM in dHepaRG cells, respectively, with CC50 over 23.49 nM (= 800 ng/mL) (Figures S3C and S3D).
Suppression of de novo HBV cccDNA synthesis
To further define inhibitory effect of F7 and poly-U/UC on cccDNA biosynthesis, we conducted time-of-addition experiments in HepG2-hNTCP and dHepaRG cells. For poly-U/UC PAMP, we conducted treatment for 24 h pre-infection (pre-treatment), from 1 to 3 dpi (post-treatment), and from 24 h preinfection onward through 3 dpi (pre/post-treatment) (Figure 3A). For F7, we conducted treatment for 24 h pre-infection (pre-treatment), 24 h initiated at the time of infection (co-treatment), from 1 through 3 dpi (post-treatment), or treatment starting at 24 h before infection and continued through 3dpi (pre/co/post-treatment) (Figure 3D). A co-treatment of CsA with virus inoculation was included as a positive control. Cultures were inoculated with HBV and harvested over the time course for assessment of cccDNA levels using Southern blot and qPCR analyses. Remarkably, we found that the synthesis of cccDNA was uniformly significantly suppressed across the poly-U/UC PAMP regimen, and to a level similar to CsA treatment, in both HepG2-hNTCP and dHepaRG cells (Figures 3B and 3C). By comparison, F7 had little effect on HBV cccDNA level when administered once pre or co-treatment but mediated a significant suppression of cccDNA levels when administered post-infection or from pre-infection throughout the 3-day course (Figures 3D–3F). These results indicate that both poly U/UC and F7 induce innate immune responses that affect cccDNA synthesis post viral entry. However, the innate immune response induced by poly-U/UC shows more sustained effect than F7.
Figure 3.

F7 and poly-U/UC PAMP suppress de novo HBV cccDNA synthesis
(A) Upper: Infection and poly-U/UC PAMP treatment schedule. Cells were treated with 100 ng/mL poly-U/UC PAMP for 24 h before (pre), for 48 h after HBV infection (post), or for 72 h (pre/post).
(B and C) At 3 dpi cells were harvested and Hirt extracts were prepared. (B) HepG2-hNTCP cells and (C) dHepaRG cells were analyzed by Southern blot (upper) and qPCR (lower). For qPCR analysis, values from DMSO-treated control were set to 100%, respectively, and data are shown as mean ± standard deviations (SD) percentage of cccDNA in control samples. ∗∗∗p < 0.005, and ns, non-significant. CSA, Cyclosporin A; X, X-RNA.
(D) HBV infection and F7 treatment schedule. Cells were treated with F7 (10 μM) for 24 h before (pre), 24 h during the time of infection (Co), 48 h after infection (post), or 96 h (pre/co/post).
(E and F) At 3 dpi, the cells were harvested and Hirt extracts prepared. HepG2-hNTCP (E) and dHepaRG cells (F). Upper panels show Southern blot analysis. Lower panels show qPCR analysis and percent of cccDNA remaining in treated cells compared with control cells. Values from DMSO-treated control cultures were set to 100%.
Data was presented as mean ± standard deviations (SD), ∗p < 0.01, ∗∗p < 0.005, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, ns = non-significant. CsA served as a treatment control. For Southern blots the position of mass markers are indicated. CSA, Cyclosporin A; X, X-RNA.
Antiviral actions of IRF3 agonists partition to the nucleus to suppress cccDNA synthesis
To identify the step(s) of HBV cccDNA synthesis impacted by F7 and poly-U/UC PAMP treatment we assessed the level of cccDNA over a 3 dpi time course. Cells were inoculated with HBV for 6 h at 4°C, then the inoculum was removed, and the cells were rinsed and placed in 37 C media to initiate synchronous infection (time 0), at which time the cells were treated with F7 (Figure 4A), or poly-U/UC PAMP (Figures 4C) through 3 dpi. Whole-cell (W), nuclear (N), and cytoplasmic (C) extracts were harvested over the time course, at 3 and 6 h post-infection (hpi), and daily over 1–3 dpi, and PF-RC and cccDNA abundance was analyzed by Southern blot. As shown in Figure 4B, PF-RC DNA was detected in cytoplasmic and whole-cell lysate fractions from 3 hpi and in nuclear fraction by 6 hpi, but accumulation was reduced after 2–3 dpi from F7 treatment. cccDNA was detected by 1 dpi in non-treated cells, but cccDNA accumulation was delayed and reduced in cells treated with F7. For poly-U/UC PAMP treatment we also harvested cells over a similar time course (see Figure 4C). Assessment of HBV DNA in subcellular fractions also showed that PF-RC DNA was present early in non-treatment cells at 3 and 6 hpi in the whole-cell and cytoplasmic extracts with levels accumulating in the nuclear fraction thereafter. Nuclear PF-RC DNA levels were reduced in cells treated with poly-U/UC PAMP from 1–3 dpi concomitantly with reduction of cccDNA in treated cells (Figure 4D). These results indicate that the inhibitory effects of F7 and poly-U/UC on HBV cccDNA biosynthesis occurs in the nucleus at 1–3 dpi during treatment to possibly impact the conversion step of PF-RC DNA to cccDNA early in the HBV replication process.
Figure 4.

Subcellular compartment analysis of antiviral activity
(A and C) Scheme of HBV infection with F7 treatment (A) or poly-U/UC PAMP treatment (C). HepG2-hNTCP cultures were inoculated with HBV at moi 1,000 Geq/cell for 24 h. On treatment day 0 the cultures received F7 (10 μM) or poly-U/UC PAMP (100 ng/mL). Cells were harvested for production of Hirt extracts at each time point shown through 3 days. Parallel cultures were harvested for western blot analysis to monitor Lamin B1 and Calenxin as markers of whole-cell lysate and cytosol, respectively. CSA, cyclosporin A control treatment.
(B and D) DNA was analyzed by Southern blot using HBV-specific DNA probe. Viral protein-free DNAs (PF-RC DNA and cccDNA) are indicated. The position of mass markers is indicated. Lower panels show western blot of Lamin B1 and Clanexin abundance. CSA, cyclosporin A control treatment.
The IRF3 activation restricts the stability of HBV cccDNA alone and in combination with ETV
To determine how the host response to IRF3 activation suppresses HBV cccDNA levels, we evaluated the influence of F7 or poly-U/UC PAMP single treatment and the combination of ETV/F7 or ETV/poly-U/UC PAMP on cccDNA decay kinetics. To assess cccDNA decay we compared F7 and poly-U/UC PAMP treatment of HBV-infected cells with ETV treatment, starting at 3 dpi with treatment maintained through 20 dpi via daily media change with fresh ETV, F7, or poly-U/UC PAMP (Figure 5A). ETV is an NA that prevents the reverse transcription and replication of new synthesized HBV DNAs, thereby blocking the replenishment of cccDNA from intracellular amplification. ETV was applied to HBV-infected cells at a 100-fold IC50 dose thereby allowing for the measurement of cccDNA half-life under conditions in which levels are sustained from early established cccDNA pool but not counterbalanced by the newly produced cccDNA (Langley et al., 2007). Cells were then harvested over a treatment time course, and DNA extracts were analyzed by Southern blot and qPCR assay. As shown in Figure 5B, cccDNA levels modestly increased over the infection time course in non-treatment control cells. Cells from ETV-treated cultures stably maintained cccDNA levels over the time course at levels similar to 3 dpi cultures, demonstrating sustained and stable cccDNA of over 20 dpi in our in vitro culture system, well in line with the reported cccDNA half-life (t1/2) of greater than 40 days (Figure S4, Ko et al., 2018; Huang et al., 2020). However, F7 single treatment stimulated the decay of cccDNA as measured by Southern blot analysis (Figure 5B), and qPCR (Figure 5D). F7 mono-treatment reduced the t1/2 of cccDNA, whereas the combination of ETV with F7 further enhanced their antiviral effects to reduce cccDNA abundance and persistence to barely detectable level by 20 dpi (Figures 5B and 5D). We also found that a mono-treatment with single administration of poly-U/UC PAMP reduced the cccDNA abundance, whereas it continued to accumulate in cells treated with X-RNA control, as analyzed by Southern blot (Figure 5C). The inhibitory effect on cccDNA levels by poly-U/UC PAMP was also confirmed by qPCR assay (Figure 5D). We also found that poly-U/UC PAMP in combination with ETV served to reduce cccDNA abundance and persistence across a treatment time course over ETV or poly-U/UC PAMP alone, with cccDNA being barely detectable after 20 dpi with co-treatment (Figure 5C). Mathematical modeling revealed that combination of F7 or poly-U/UC treatment with ETV reduced the cccDNA t1/2 resulting from monotreatment compared with each respective co-treatment from an average of 8.7 to 6.5 days (F7) and from 7.7 to 6.9 days (poly-U/UC PAMP) (Figures 5E and S5). Neither ETV nor X-RNA treatment of cells had any effect on the production and secretion of HBsAg. However, poly-U/UC PAMP and F7 mono- and combination treatments suppressed HBsAg secretion (Figure 5F). Thus, F7 and poly-U/UC PAMP impart the therapeutic decay of established cccDNA. Moreover, combination treatment of F7 and poly-U/UC PAMP with ETV imparts additive antiviral actions to suppress cccDNA t1/2 and persistence from weeks (Huang et al., 2020; Ko et al., 2018) to less than 7 days.
Figure 5.

F7 and poly-U/UC PAMP treatment directs HBV cccDNA decay
(A) HBV infection and treatment schedule. HepG2-hNTCP cells were infected with HBV at moi of 1,000 Geq/cell, incubated for 24 h, and media was replaced. On day 3 the cells were harvested or treated with DMSO, ETV (500 nM), F7 (10 μM), poly-U/UC (100 ng/mL), or with ETC combination with F7 or poly-U/UC PAMP. Cells were harvested at the indicated points over a 20-day time course.
(B and C) DNA was isolated by Hirt extract and subjected to Southern blot analysis using an HBV-specific DNA probe. Percentage values below each lane indicate the relative amount of cccDNA compared with day 3 control before each treatment. The position of mass markers is indicated.
(D) cccDNA levels were measured by qPCR. Relative cccDNA values to mitochondrial DNA (MT-CO3) are shown. Statistical significance was determined using Student's t test. Data are presented as mean ± standard deviation (SD), ∗p < 0.01, ∗∗p < 0.005, ∗∗∗p < 0.001, ns = non-significant.
(E) Half-life of cccDNA from qPCR analyses was estimated from the simultaneous fitting of three replicates under each treatment strategy. C(t) is the % values of cccDNA at time “t” post-treatment, C(0) is the % values of cccDNA at the start of the treatment.
(F) HBsAg across the time course for each treatment series. Statistical significance was determined using Student's t test. Data are presented as mean ± standard deviations (SD), ∗∗p < 0.005, ns= non-significant.
Suppression of HBV cccDNA by IRF3 activation is RIG-I dependent
To ascertain whether the activation of IRF3 and suppression of cccDNA in HBV-infected cells by F7 or poly-U/UC PAMP treatment was dependent on RIG-I rather than occurring as an off-target effect of IRF3 agonist treatment, we produced HepG2-hNTCP cells expressing non-targeting guide RNA (HepG2-hNTCP-NT), or guide RNA for knockout (KO) of RIG-I expression (HepG2-hNTCP-RKO) or MDA5 (HepG2-hNTCP-MKO) using CRISPR-Cas9 genome editing technology. Immunoblot analysis of the different cell populations shows that HepG2-hNTCP-RKO or HepG2-hNTCP-MKO cells do not express detectable levels of RIG-I or MDA5, respectively (Figure S6). Treatment of cells with F7 (Figure 6A) or poly-U/UC PAMP (Figure 6B) shows that the HepG2-hNTCP-RKO cells do not respond to treatment, whereas HepG2-hNTCP-NT control cells and HepG2-hNTCP-MKO cells fully respond to F7 to accumulate phosphorylated/activated IRF3 concomitant with expression of IFIT1. HepG2-hNTCP-MKO cells serve as an RLR KO control to reveal the specificity of F7 and poly-U/UC PAMP for triggering RIG-I-dependent IRF3 activation (Saito et al., 2008). We next infected each cell population with HBV followed by a single treatment with F7 or poly-U/UC PAMP at 1 dpi. Cells were harvested at 3 dpi for Southern blot analysis of cccDNA levels. Parallel cultures of each cell population were treated with DMSO or single-dose X-RNA (negative control) or with CsA as a treatment control. F7 treatment reduced cccDNA levels in HepG2-hNTCP-NT and HepG2-hNTCP-MKO cells but not in HepG2-hNTCP-RKO cells (Figure 6C). Similarly poly-U/UC PAMP suppression of cccDNA was dependent on RIG-I, as cccDNA was suppressed by poly-U/UC PAMP treatment in HepG2-hNTCP-NT and HepG2-hNTCP-MKO cells but not in the HepG2-hNTCP-RKO cells (Figure 6D). These results demonstrate that the antiviral actions of F7 and poly-U/UC PAMP against HBV specifically signal through RIG-I, defining each as a RIG-I agonist that activates IRF3 to impart suppression of cccDNA.
Figure 6.

F7 and poly-U/UC PAMP specifically signal IRF3 activation through RIG-I to suppress HBV cccDNA
(A) F7 was administered for 24 h to HepG2-hNTCP-NT, RKO, and MKO cells. Cells were harvested and analyzed by immunoblot. The levels of protein expression by p-IRF3 (S386 phosphorylated), IRF3 (total IRF), IFIT1, RIG-I, and MDA5 relative to the expression level of tubulin were determined using respective antibodies.
(B) HepG2-hNTCP-NT, RKO, and MKO cells were treated with 100 ng/mL X-RNA (X) or 100 ng/mL or 200 ng/mL poly-U/UC PAMP (P) for 24 h. Cells were harvested and analyzed by immunoblot as in (A).
(C and D) Cells were infected with HBV at moi of 1,000 Geq/cell. After 24 h cultures were treated with DMSO (-; negative control), CsA (treatment control), (C) 10 μM F7 or (D) X-RNA or poly-U/UC PAMP. Three days later the cells were harvested and Hirt extracts were prepared and subjected to Southern blot analysis using as HBV-DNA probe. The position of mass markers is indicated. Values beneath each lane show percent cccDNA remaining compared with control treatment.
RIG-I signaling of IRF3 activation suppresses HBV infection in primary human hepatocytes
To validate the antiviral actions of RIG-I signaling of IRF3 activation by poly-U/UC PAMP we assessed HBV infection in non-immortalized and terminally differentiated PHHs that retain the expression of hepatocyte marker genes at a level comparable to that of human liver tissue. We treated PHH cultures over a poly-U/UC PAMP dose-response and assessed innate immune activation. Treatment of PHH cultures with 50–200 ng/mL poly-U/UC PAMP induced IRF3 activation marked by accumulation of phosphoserine 386 IRF3 and IFIT1 expression. Treatment with 100 ng/mL X-RNA did not induce innate immune activation of PHH, but cells were fully responsive to acute infection with SenV control (Figure 7A), demonstrating that PHHs harbor an intact RIG-I pathway. Moreover, PHH treatment with poly-U/UC PAMP, but not X-RNA, robustly induced innate immune gene expression (Figure 7B). We then evaluated cccDNA levels in HBV-infected PHH treated with poly-U/UC PAMP. PHH cultures were infected with HBV and subject to a single treatment with poly-U/UC PAMP at 1 dpi. Cells were harvested at 3 dpi and DNA was extracted and subject to Southern blot analysis. As shown in Figure 7C, treatment with poly-U/UC PAMP suppressed cccDNA levels in the infected PHH to levels similar to CsA treatment. Thus, poly-U/UC treatment to trigger RIG-I signaling of IRF3 and innate immune activation directs a response in HBV-infected PHH that suppresses cccDNA. These results demonstrate the efficacy of therapeutic targeting of the RIG-I pathway for viral control in relevant primary cells of HBV infection.
Figure 7.

Suppression of cccDNA in primary human hepatocytes
(A) PHHs were cultured alone or were treated with 100 ng/mL X-RNA (X100), or 50 ng/mL (P50), 100 ng/mL (P100), or 200 ng/mL (P200) poly-U/UC PAMP, or were infected with SenV (control) and harvested 24 and 72 h later. The cell lysates were analyzed by immunoblot for p-IRF3 (S386 phosphorylated), IRF3 (total IRF3), IFIT1, and actin (control) using respective antibodies.
(B) PHH cultures were cultured alone or were treated with 100 ng/mL X-RNA (X100), or 100 ng/mL (P100) or 200 ng/mL (P200) poly-U/UC PAMP, or were infected with SenV (control) and harvested 24 and 72 h later. Cells were harvested and RNA was extracted and analyzed by RT-qPCR to measure the expression levels of the indicated innate immune genes normalized to the level of GAPDH expression. Values are shown on the heatmap as mean fold induction over non-treated cells from three independent experiments.
(C) PHH cultures were inoculated with HBV at moi of 200 Geq/cell. 24 h later the cultures were treated with CsA (treatment control; 10 μM), 100 ng/mL X-RNA (X100), or 100 ng/mL (P100) or 200 ng/mL (P200) poly-U/UC PAMP. Three days later the cells were harvested, DNA isolated by isolated Hirt extraction, and analyzed by Southern blot using an HBV-specific probe. Viral protein-free DNAs (PF-RC DNA and cccDNA) are noted. Values under each lane show the percent remaining cccDNA compared with non-treated control. The positions of mass markers are shown on the left.
Discussion
Persistence of cccDNA in the nucleus of HBV-infected hepatocytes is key to mediating chronic HBV infection, wherein recent analyses indicates that a given pool of cccDNA has a t1/2 in vivo of approximately 5–21 weeks (Huang et al., 2020). Problematically, current therapies for the treatment of chronic HBV infection neither significantly reduce nor eliminate cccDNA (Zoulim and Durantel, 2015; Maynard et al., 2005; Werle-Lapostolle et al., 2004). Members of the current NA class of HBV therapeutics are administered for prolonged, often lifelong periods to keep patients virally suppressed. Moreover, these therapies are leaky in their ability to completely shut down viral replication, such that the nuclear cccDNA pool (Huang et al., 2020) still persists after long-term treatment (Werle-Lapostolle et al., 2004; Zoulim and Locarnini, 2009; Gish et al., 2012), Importantly however, it has been demonstrated that suppression of cccDNA during acute HBV infection can occur through a cytokine-driven non-cytolytic mechanism directed by IFN-α, IFN-γ, or tumor necrosis factor alpha linked with expression of APOBEC3 deaminases (Lucifora et al., 2014; Xia et al., 2016). Pharmacological induction of intrahepatic cytokine responses has been coined as an ideal curative approach to chronic hepatitis B (Chang et al., 2012). While this approach leverages the innate immune and immune modulatory signaling programs driven by specific cytokines to induce the expression of genes whose actions can suppress cccDNA, cytokine therapy for treatment of chronic HBV faces the obstacles of systemic toxicity from the broad off target, nonhepatic actions of cytokine treatment (Kwon and Lok, 2011; Locarnini et al., 2015), underscoring the need for target-directed therapeutic strategies that can also be coupled with current NA therapeutics against HBV.
We demonstrate that targeting RIG-I and the RLR pathway to activate IRF3 via the F7 small-molecule and poly-U/UC PAMP agonists of RIG-I delivered to hepatocytes directs a specific RIG-I-dependent innate immune response through IRF3 that effectively suppresses HBV cccDNA in a cell culture model of HBV infection. Furthermore, mechanistic studies show that the administration of F7 or poly-U/UC PAMP suppresses de novo biosynthesis of cccDNA and enhanced cccDNA degradation rather than inhibiting the production of newly synthesized HBV rcDNA as current NA HBV drugs do. Administration of F7 and poly-U/UC PAMP induces IRF3 activation and expression of IRF3-target genes (Figure 1). Thus, the underlying mechanisms of F7 and poly-U/UC PAMP to suppress cccDNA likely involve the actions of IRF3-target genes to block cccDNA synthesis and impart actions that facilitate the destabilization and degradation of cccDNA to deplete it from the infected cell. We note that F7 treatment of cells does not induce expression of type I or III IFN, owing to the RIG-I activation properties of this class of compounds that do not impart signaling to NF-kB but instead exclusively activate downstream IRF3 (Bedard et al., 2012; Probst et al., 2017). Thus, in the case of F7 the antiviral actions to suppress cccDNA operate independent of IFN actions but through IRF3-responsive genes. Our results show that poly-U/UC PAMP also induces robust IRF3-target gene expression as well as a low level of IFN that can induce ISGs. Among these are the APOBEC3 genes, known antiviral effectors against HBV replication (Turelli et al., 2004; Bonvin et al., 2006). IRF3 activation drives the expression and production of various immune modulatory cytokines and chemokines, including CXCL10, a chemoattractant for T cells (Zhai et al., 2008; Sankar et al., 2006), as well as directs the expression of factors that regulate ubiquitination (Maelfait and Beyaert, 2012) and a variety of cell signaling process (Zhou et al., 2017). Thus we propose that IRF3-target genes include a variety of anti-cccDNA effectors in addition to the known actions of the APOBEC genes. These effector genes then impart pleiotropic actions to (1) suppress cccDNA amplification, (2) destabilize cccDNA and/or enhance cccDNA degradation, (3) impact nuclear translocation or control uncoating/disassembly of the nucleocapsid to release RC DNA for cccDNA formation (Kock et al., 2003; Rabe et al., 2003; Cui et al., 2015), and (4) regulate the extrachromosomal DNA metabolism to impact HBV cccDNA formation (Luo et al., 2020; Blackford and Jackson, 2017). Indeed, we observed a marked reduction in the cccDNA t1/2 when cells were treated with F7 or poly-U/UC PAMP, and this reduction was further enhanced when cells were co-treated with either of these plus ETV. In addition, for poly-U/UC PAMP, the antiviral actions against cccDNA could also include IFN-mediated actions directed by the low-level IFN induction (Lucifora et al., 2014; Phillips et al., 2017; Xu et al., 2018; Robek et al., 2005; Isorce et al., 2016). Interestingly, these actions of IFN and specific ISGs resulting from treatment with poly-U/UC PAMP could have contributed to a suppression of cccDNA resulting in a modestly shorter half-life compared with treatment with F7. How RIG-I/IRF3 signaling activation triggers cccDNA destabilization or degradation remains to be determined. The antiviral action of F7 and poly-U/UC PAMP might also include regulation of extrachromosomal DNA metabolism in addition to function of cytidine deaminases such as APOBEC3 genes. As HBV takes advantage of host DNA repair factors to repair the discontinuity of RC-DNA and convert it into a transcription permissive cccDNA, one or more of these steps of cccDNA production and maintenance could possibly be impacted by RIG-I/IRF3 actions (Long et al., 2017) (Sheraz et al., 2019) (Luo et al., 2020) (Qi et al., 2016; Tang et al., 2019).
Our studies demonstrated that we can specifically target RIG-I and IRF3 to activate the RLR innate immune program for the control of HBV infection through suppression of cccDNA, reducing the t1/2 to days compared with weeks or months in the absence of treatment. Targeting innate immunity and the RLR pathway thus offers an effective strategy toward the design of new antiviral therapies against HBV that can be offered alone or in combination with NA for HBV treatment. Determining the innate immune targets directed by RIG-I and IRF3 that impart depletion of cccDNA will bring insight into the mechanism of action and unique antiviral properties of these unique drug candidates toward an HBV cure.
Limitations of the study
Our data indicate that administration of RIG-I agonists do not confer cytotoxicity using cellular ATP measurement methods. However, the potential cellular toxicity of innate immune modulation is yet to be evaluated in vivo. The biological actions of our RIG-I agonists are currently being examined in animal models.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the corresponding author, Dr. Michael Gale, Jr. (mgale@uw.edu).
Materials availability
The F7 small molecule and poly-U/UC RNA production template are available from the corresponding author upon reasonable request.
Data and code availability
This study did not generate/analyze datasets and code.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We are most grateful to Dr. Ju-Tao Guo (Baruch S. Blumberg Institute), Dr. Aleem Siddiqui (University of California San Diego), and Dr. T. Jake Liang (National Institutes of Health) for HepG2-hNTCP, HepAD38, and HepaRG cell lines.
This study was supported by National Institutes of Health (NIH) grants AI127463 and AI118916, and by NIH contracts HHSN272201300023C, 75N93019C00043, and HHSN272201400055C (M.G.). Portions on this work were also done under the auspices of the U.S. Dept. of Energy under contract 89233218CNA000001 and additionally supported by NIH grants AI078881, AI116868 and OD011095 (A.S.P.).
Author contributions
S.L. and M.G. designed the experiments and interpreted the results. S.L. performed experiments and statistical analysis. A.G. and A.S.P. analyzed the cccDNA half-life using a mathematical model. Y.I. and T.S. performed isolation and culture of primary human hepatocytes. S.L. and M.G. wrote the manuscript with input from all authors.
Declaration of interests
None to report. The authors declare no competing interests.
Published: January 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2020.101969.
Supplemental information
References
- Beck J., Nassal M. Hepatitis B virus replication. World J. Gastroenterol. 2007;13:48–64. doi: 10.3748/wjg.v13.i1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K.M., Wang M.L., Proll S.C., Loo Y.M., Katze M.G., Gale M., Jr., Iadonato S.P. Isoflavone agonists of IRF-3 dependent signaling have antiviral activity against RNA viruses. J. Virol. 2012;86:7334–7344. doi: 10.1128/JVI.06867-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belloni L., Allweiss L., Guerrieri F., Pediconi N., Volz T., Pollicino T., Petersen J., Raimondo G., Dandri M., Levrero M. IFN-alpha inhibits HBV transcription and replication in cell culture and in humanized mice by targeting the epigenetic regulation of the nuclear cccDNA minichromosome. J. Clin. Invest. 2012;122:529–537. doi: 10.1172/JCI58847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackford A.N., Jackson S.P. ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell. 2017;66:801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- Boni C., Vecchi A., Rossi M., Laccabue D., Giuberti T., Alfieri A., Lampertico P., Grossi G., Facchetti F., Brunetto M.R. TLR7 agonist increases responses of hepatitis B virus-specific T cells and natural killer cells in patients with chronic hepatitis B treated with nucleos(T)ide analogues. Gastroenterology. 2018;154:1764–1777.e7. doi: 10.1053/j.gastro.2018.01.030. [DOI] [PubMed] [Google Scholar]
- Bonvin M., Achermann F., Greeve I., Stroka D., Keogh A., Inderbitzin D., Candinas D., Sommer P., Wain-Hobson S., Vartanian J.P., Greeve J. Interferon-inducible expression of APOBEC3 editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology. 2006;43:1364–1374. doi: 10.1002/hep.21187. [DOI] [PubMed] [Google Scholar]
- Chang J., Block T.M., Guo J.T. The innate immune response to hepatitis B virus infection: implications for pathogenesis and therapy. Antivir. Res. 2012;96:405–413. doi: 10.1016/j.antiviral.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Chen Z., Zhu M., Pan X., Zhu Y., Yan H., Jiang T., Shen Y., Dong X., Zheng N., Lu J. Inhibition of Hepatitis B virus replication by SAMHD1. Biochem. Biophys. Res. Commun. 2014;450:1462–1468. doi: 10.1016/j.bbrc.2014.07.023. [DOI] [PubMed] [Google Scholar]
- Chow K.T., Gale M., Jr., Loo Y.M. RIG-I and other RNA sensors in antiviral immunity. Annu. Rev. Immunol. 2018;36:667–694. doi: 10.1146/annurev-immunol-042617-053309. [DOI] [PubMed] [Google Scholar]
- Cui X., Luckenbaugh L., Bruss V., Hu J. Alteration of mature nucleocapsid and enhancement of covalently closed circular DNA formation by hepatitis B virus core mutants defective in complete-virion formation. J. Virol. 2015;89:10064–10072. doi: 10.1128/JVI.01481-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dassler-Plenker J., Paschen A., Putschli B., Rattay S., Schmitz S., Goldeck M., Bartok E., Hartmann G., Coch C. Direct RIG-I activation in human NK cells induces TRAIL-dependent cytotoxicity toward autologous melanoma cells. Int. J. Cancer. 2019;144:1645–1656. doi: 10.1002/ijc.31874. [DOI] [PubMed] [Google Scholar]
- Dienstag J.L. Benefits and risks of nucleoside analog therapy for hepatitis B. Hepatology. 2009;49:S112–S121. doi: 10.1002/hep.22920. [DOI] [PubMed] [Google Scholar]
- Fanning G.C., Zoulim F., Hou J., Bertoletti A. Therapeutic strategies for hepatitis B virus infection: towards a cure. Nat. Rev. Drug Discov. 2019;19:291. doi: 10.1038/s41573-019-0057-9. [DOI] [PubMed] [Google Scholar]
- Fensterl V., Sen G.C. The ISG56/IFIT1 gene family. J. Interferon Cytokine Res. 2011;31:71–78. doi: 10.1089/jir.2010.0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fensterl V., Sen G.C. Interferon-induced Ifit proteins: their role in viral pathogenesis. J. Virol. 2015;89:2462–2468. doi: 10.1128/JVI.02744-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T., Yu J.W., Datta P., Wu J., Alnemri E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gish R., Jia J.D., Locarnini S., Zoulim F. Selection of chronic hepatitis B therapy with high barrier to resistance. Lancet Infect. Dis. 2012;12:341–353. doi: 10.1016/S1473-3099(11)70314-0. [DOI] [PubMed] [Google Scholar]
- Guo H., Jiang D., Zhou T., Cuconati A., Block T.M., Guo J.-T. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: an intermediate of covalently closed circular DNA formation. J. Virol. 2007;81:12472–12484. doi: 10.1128/JVI.01123-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Q., Hou Z., Yin C., Zhang C., Zhang J. 5'-triphosphate siRNA targeting HBx elicits a potent anti-HBV immune response in pAAV-HBV transfected mice. Antiviral Res. 2019;161:36–45. doi: 10.1016/j.antiviral.2018.11.006. [DOI] [PubMed] [Google Scholar]
- Hirt B. Selective extraction of polyoma DNA from infected mouse cell cultures. J. Mol. Biol. 1967;26:365–369. doi: 10.1016/0022-2836(67)90307-5. [DOI] [PubMed] [Google Scholar]
- Hochheiser K., Klein M., Gottschalk C., Hoss F., Scheu S., Coch C., Hartmann G., Kurts C. Cutting edge: the RIG-I ligand 3pRNA potently improves CTL cross-priming and facilitates antiviral vaccination. J. Immunol. 2016;196:2439–2443. doi: 10.4049/jimmunol.1501958. [DOI] [PubMed] [Google Scholar]
- Horner S.M., Gale M., Jr. Regulation of hepatic innate immunity by hepatitis C virus. Nat. Med. 2013;19:879–888. doi: 10.1038/nm.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q., Zhou B., Cai D., Zong Y., Wu Y., Liu S., Mercier A., Guo H., Hou J., Colonno R., Sun J. Rapid turnover of HBV cccDNA indicated by monitoring emergence and reversion of signature-mutation in treated chronic hepatitis B patients. Hepatology. 2020 doi: 10.1002/hep.31240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadonato S.P., Kaiser S., Bedard K.M., Fowler K.W., Madu I. US patent 9,884,876 B2; United States Patent Office: 2018. Anti-viral Compounds, Pharmaceutical Compositions, and Methods of Use Thereof; pp. 23–24. [Google Scholar]
- Isorce N., Testoni B., Locatelli M., Fresquet J., Rivoire M., Luangsay S., Zoulim F., Durantel D. Antiviral activity of various interferons and pro-inflammatory cytokines in non-transformed cultured hepatocytes infected with hepatitis B virus. Antiviral Res. 2016;130:36–45. doi: 10.1016/j.antiviral.2016.03.008. [DOI] [PubMed] [Google Scholar]
- Janssen H.L., Van Zonneveld M., Senturk H., Zeuzem S., Akarca U.S., Cakaloglu Y., Simon C., So T.M., Gerken G., De Man R.A. Pegylated interferon alfa-2b alone or in combination with lamivudine for HBeAg-positive chronic hepatitis B: a randomised trial. Lancet. 2005;365:123–129. doi: 10.1016/S0140-6736(05)17701-0. [DOI] [PubMed] [Google Scholar]
- Jefferies M., Rauff B., Rashid H., Lam T., Rafiq S. Update on global epidemiology of viral hepatitis and preventive strategies. World J. Clin. Cases. 2018;6:589–599. doi: 10.12998/wjcc.v6.i13.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kell A., Stoddard M., Li H., Marcotrigiano J., Shaw G.M., Gale M., Jr. Pathogen-associated molecular pattern recognition of hepatitis C virus transmitted/founder variants by RIG-I is dependent on U-core length. J. Virol. 2015;89:11056–11068. doi: 10.1128/JVI.01964-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko C., Chakraborty A., Chou W.M., Hasreiter J., Wettengel J.M., Stadler D., Bester R., Asen T., Zhang K., Wisskirchen K. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018;69:1231–1241. doi: 10.1016/j.jhep.2018.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kock J., Kann M., Putz G., Blum H.E., Von Weizsacker F. Central role of a serine phosphorylation site within duck hepatitis B virus core protein for capsid trafficking and genome release. J. Biol. Chem. 2003;278:28123–28129. doi: 10.1074/jbc.M300064200. [DOI] [PubMed] [Google Scholar]
- Kwon H., Lok A.S. Hepatitis B therapy. Nat. Rev. Gastroenterol. Hepatol. 2011;8:275–284. doi: 10.1038/nrgastro.2011.33. [DOI] [PubMed] [Google Scholar]
- Lanford R.E., Guerra B., Chavez D., Giavedoni L., Hodara V.L., Brasky K.M., Fosdick A., Frey C.R., Zheng J., Wolfgang G. GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees. Gastroenterology. 2013;144:1508–1517. doi: 10.1053/j.gastro.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley D.R., Walsh A.W., Baldick C.J., Eggers B.J., Rose R.E., Levine S.M., Kapur A.J., Colonno R.J., Tenney D.J. Inhibition of hepatitis B virus polymerase by entecavir. J. Virol. 2007;81:3992–4001. doi: 10.1128/JVI.02395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.H., Chiang C., Gack M.U. Endogenous nucleic acid recognition by RIG-I-like receptors and cGAS. J. Interferon Cytokine Res. 2019;39:450–458. doi: 10.1089/jir.2019.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang T.J., Block T.M., Mcmahon B.J., Ghany M.G., Urban S., Guo J.T., Locarnini S., Zoulim F., Chang K.M., Lok A.S. Present and future therapies of hepatitis B: from discovery to cure. Hepatology. 2015;62:1893–1908. doi: 10.1002/hep.28025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locarnini S., Hatzakis A., Chen D.S., Lok A. Strategies to control hepatitis B: public policy, epidemiology, vaccine and drugs. J. Hepatol. 2015;62:S76–S86. doi: 10.1016/j.jhep.2015.01.018. [DOI] [PubMed] [Google Scholar]
- Long Q., Yan R., Hu J., Cai D., Mitra B., Kim E.S., Marchetti A., Zhang H., Wang S., Liu Y. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog. 2017;13:e1006784. doi: 10.1371/journal.ppat.1006784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucifora J., Xia Y., Reisinger F., Zhang K., Stadler D., Cheng X., Sprinzl M.F., Koppensteiner H., Makowska Z., Volz T. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–1228. doi: 10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J., Luckenbaugh L., Hu H., Yan Z., Gao L., Hu J. Involvement of host ATR-CHK1 pathway in hepatitis B virus covalently closed circular DNA formation. mBio. 2020;11:e03423-19. doi: 10.1128/mBio.03423-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait J., Beyaert R. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol. Mol. Biol. Rev. 2012;76:33–45. doi: 10.1128/MMBR.05012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marion P.L., Robinson W.S. Hepadna viruses: hepatitis B and related viruses. Curr. Top. Microbiol. Immunol. 1983;105:99–121. doi: 10.1007/978-3-642-69159-1_2. [DOI] [PubMed] [Google Scholar]
- Maynard M., Parvaz P., Durantel S., Chevallier M., Chevallier P., Lot M., Trepo C., Zoulim F. Sustained HBs seroconversion during lamivudine and adefovir dipivoxil combination therapy for lamivudine failure. J. Hepatol. 2005;42:279–281. doi: 10.1016/j.jhep.2004.09.017. [DOI] [PubMed] [Google Scholar]
- Moucari R., Korevaar A., Lada O., Martinot-Peignoux M., Boyer N., Mackiewicz V., Dauvergne A., Cardoso A.C., Asselah T., Nicolas-Chanoine M.H. High rates of HBsAg seroconversion in HBeAg-positive chronic hepatitis B patients responding to interferon: a long-term follow-up study. J. Hepatol. 2009;50:1084–1092. doi: 10.1016/j.jhep.2009.01.016. [DOI] [PubMed] [Google Scholar]
- Mutz P., Metz P., Lempp F.A., Bender S., Qu B., Schoneweis K., Seitz S., Tu T., Restuccia A., Frankish J. HBV bypasses the innate immune response and does not protect HCV from antiviral activity of interferon. Gastroenterology. 2018;154:1791–1804.e22. doi: 10.1053/j.gastro.2018.01.044. [DOI] [PubMed] [Google Scholar]
- Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64:1972–1984. doi: 10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- Newbold J.E., Xin H., Tencza M., Sherman G., Dean J., Bowden S., Locarnini S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 1995;69:3350–3357. doi: 10.1128/jvi.69.6.3350-3357.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott J.J., Stevens G.A., Groeger J., Wiersma S.T. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30:2212–2219. doi: 10.1016/j.vaccine.2011.12.116. [DOI] [PubMed] [Google Scholar]
- Pandey S., Kawai T., Akira S. Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 2014;7:a016246. doi: 10.1101/cshperspect.a016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabhi S., Wilkins C.R., Dong R., Knoll M.L., Posakony J., Kaiser S., Mire C.E., Wang M.L., Ireton R.C., Geisbert T.W. Targeting innate immunity for antiviral therapy through small molecule agonists of the RLR pathway. J. Virol. 2016;90:2372–2387. doi: 10.1128/JVI.02202-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrillo R. Benefits and risks of interferon therapy for hepatitis B. Hepatology. 2009;49:S103–S111. doi: 10.1002/hep.22956. [DOI] [PubMed] [Google Scholar]
- Phillips S., Mistry S., Riva A., Cooksley H., Hadzhiolova-Lebeau T., Plavova S., Katzarov K., Simonova M., Zeuzem S., Woffendin C. Peg-interferon lambda treatment induces robust innate and adaptive immunity in chronic hepatitis B patients. Front. Immunol. 2017;8:621. doi: 10.3389/fimmu.2017.00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst P., Grigg J.B., Wang M., Munoz E., Loo Y.M., Ireton R.C., Gale M., Jr., Iadonato S.P., Bedard K.M. A small-molecule IRF3 agonist functions as an influenza vaccine adjuvant by modulating the antiviral immune response. Vaccine. 2017;35:1964–1971. doi: 10.1016/j.vaccine.2017.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y., Gao Z., Xu G., Peng B., Liu C., Yan H., Yao Q., Sun G., Liu Y., Tang D. DNA polymerase kappa is a key cellular factor for the formation of covalently closed circular DNA of hepatitis B virus. PLoS Pathog. 2016;12:e1005893. doi: 10.1371/journal.ppat.1005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe B., Vlachou A., Panté N., Helenius A., Kann M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. U S A. 2003;100:9849–9854. doi: 10.1073/pnas.1730940100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robek M.D., Boyd B.S., Chisari F.V. Lambda interferon inhibits hepatitis B and C virus replication. J. Virol. 2005;79:3851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T., Owen D.M., Jiang F., Marcotrigiano J., Gale M., Jr. Innate immunity induced by composition-dependent RIG-I recognition of hepatitis C virus RNA. Nature. 2008;454:523–527. doi: 10.1038/nature07106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankar S., Chan H., Romanow W.J., Li J., Bates R.J. IKK-i signals through IRF3 and NFkappaB to mediate the production of inflammatory cytokines. Cell. Signal. 2006;18:982–993. doi: 10.1016/j.cellsig.2005.08.006. [DOI] [PubMed] [Google Scholar]
- Schnell G., Loo Y.M., Marcotrigiano J., Gale M., Jr. Uridine composition of the poly-U/UC tract of HCV RNA defines non-self recognition by RIG-I. PLoS Pathog. 2012;8:e1002839. doi: 10.1371/journal.ppat.1002839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger C., Mason W.S. Hepatitis B virus biology. Microbiol. Mol. Biol. Rev. 2000;64:51–68. doi: 10.1128/mmbr.64.1.51-68.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheraz M., Cheng J., Tang L., Chang J., Guo J.T. Cellular DNA topoisomerases are required for the synthesis of hepatitis B virus covalently closed circular DNA. J. Virol. 2019;93:e02230-18. doi: 10.1128/JVI.02230-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L., Li S., Shen F., Li H., Qian S., Lee D.H., Wu J.Z., Yang W. Characterization of nucleosome positioning in hepadnaviral covalently closed circular DNA minichromosomes. J. Virol. 2012;86:10059–10069. doi: 10.1128/JVI.00535-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L., Sheraz M., Mcgrane M., Chang J., Guo J.T. DNA Polymerase alpha is essential for intracellular amplification of hepatitis B virus covalently closed circular DNA. PLoS Pathog. 2019;15:e1007742. doi: 10.1371/journal.ppat.1007742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L., Zhao Q., Wu S., Cheng J., Chang J., Guo J.T. The current status and future directions of hepatitis B antiviral drug discovery. Expert Opin. Drug Discov. 2017;12:5–15. doi: 10.1080/17460441.2017.1255195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli P., Mangeat B., Jost S., Vianin S., Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303:1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- Tuttleman J.S., Pourcel C., Summers J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell. 1986;47:451–460. doi: 10.1016/0092-8674(86)90602-1. [DOI] [PubMed] [Google Scholar]
- Unterholzner L., Keating S.E., Baran M., Horan K.A., Jensen S.B., Sharma S., Sirois C.M., Jin T., Latz E., Xiao T.S. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010;11:997–1004. doi: 10.1038/ni.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G.H., Seeger C. Novel mechanism for reverse transcription in hepatitis B viruses. J. Virol. 1993;67:6507–6512. doi: 10.1128/jvi.67.11.6507-6512.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watashi K., Sluder A., Daito T., Matsunaga S., Ryo A., Nagamori S., Iwamoto M., Nakajima S., Tsukuda S., Borroto-Esoda K. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP) Hepatology. 2014;59:1726–1737. doi: 10.1002/hep.26982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werle-Lapostolle B., Bowden S., Locarnini S., Wursthorn K., Petersen J., Lau G., Trepo C., Marcellin P., Goodman Z., Delaney W.E.T. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology. 2004;126:1750–1758. doi: 10.1053/j.gastro.2004.03.018. [DOI] [PubMed] [Google Scholar]
- Wieland S., Thimme R., Purcell R.H., Chisari F.V. Genomic analysis of the host response to hepatitis B virus infection. Proc. Natl. Acad. Sci. U S A. 2004;101:6669–6674. doi: 10.1073/pnas.0401771101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S., Lin J., Fu Y., Ou Q. RIG-I enhances interferon-alpha response by promoting antiviral protein expression in patients with chronic hepatitis B. Antivir. Ther. 2018;23:575–583. doi: 10.3851/IMP3239. [DOI] [PubMed] [Google Scholar]
- Wu T.T., Coates L., Aldrich C.E., Summers J., Mason W.S. In hepatocytes infected with duck hepatitis B virus, the template for viral RNA synthesis is amplified by an intracellular pathway. Virology. 1990;175:255–261. doi: 10.1016/0042-6822(90)90206-7. [DOI] [PubMed] [Google Scholar]
- Wursthorn K., Lutgehetmann M., Dandri M., Volz T., Buggisch P., Zollner B., Longerich T., Schirmacher P., Metzler F., Zankel M. Peginterferon alpha-2b plus adefovir induce strong cccDNA decline and HBsAg reduction in patients with chronic hepatitis B. Hepatology. 2006;44:675–684. doi: 10.1002/hep.21282. [DOI] [PubMed] [Google Scholar]
- Xia Y., Stadler D., Lucifora J., Reisinger F., Webb D., Hosel M., Michler T., Wisskirchen K., Cheng X., Zhang K. Interferon-gamma and tumor necrosis factor-alpha produced by T cells reduce the HBV persistence form, cccDNA, without cytolysis. Gastroenterology. 2016;150:194–205. doi: 10.1053/j.gastro.2015.09.026. [DOI] [PubMed] [Google Scholar]
- Xu F., Song H., Xiao Q., Li N., Zhang H., Cheng G., Tan G. Type III interferon-induced CBFβ inhibits HBV replication by hijacking HBx. Cell Mol. Immunol. 2018;16:357–366. doi: 10.1038/s41423-018-0006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H., Zhong G., Xu G., He W., Jing Z., Gao Z., Huang Y., Qi Y., Peng B., Wang H. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife. 2012;1:e00049. doi: 10.7554/eLife.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai Y., Shen X.D., Gao F., Zhao A., Freitas M.C., Lassman C., Luster A.D., Busuttil R.W., Kupiec-Weglinski J.W. CXCL10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology. 2008;47:207–214. doi: 10.1002/hep.21986. [DOI] [PubMed] [Google Scholar]
- Zhou Y., He C., Wang L., Ge B. Post-translational regulation of antiviral innate signaling. Eur. J. Immunol. 2017;47:1414–1426. doi: 10.1002/eji.201746959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoulim F., Durantel D. Antiviral therapies and prospects for a cure of chronic hepatitis B. Cold Spring Harb. Perspect. Med. 2015;5:a021501. doi: 10.1101/cshperspect.a021501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoulim F., Locarnini S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology. 2009;137:1593–1608.e1-2. doi: 10.1053/j.gastro.2009.08.063. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze datasets and code.