

Abstract
Functional polymer systems with stimuli responses have attracted great attention over the years due to their diverse range of applications. Such polymers are capable of altering their chemical and/or physical properties, such as chemical structures, chain conformation, solubility, shape, morphologies, and optical properties, in response to single or multiple stimuli. Among various stimuli-responsive polymers, those with aggregation-induced emission (AIE) properties possess the advantages of high sensitivity, fast response, large contrast, excellent photostability, and low background noise. The changes in fluorescence signal can be conveniently detected and monitored using portable instruments. The integration of AIE and stimuli responses into one polymer system provides a feasible and effective strategy for the development of smart polymers with high sensitivity to environmental variations. Here, we review the recent advances in the design, preparation, performance, and applications of functional synthetic polymer systems with AIE and stimuli responses. Various AIE-based polymer systems with responsiveness toward single physical or chemical stimuli as well as multiple stimuli are summarized with specific examples. The current challenges and perspectives on the future development of this research area will also be discussed at the end of this review.
Keywords: Aggregation-induced emission, Stimuli responses, Functional polymers, Fluorescence sensors
Introduction
Stimuli-responsive materials widely exist in living systems and play an important role for the sustainability of life and maintaining biological function. For instance, human bodies can respond to changes in the environment with the assistance of complicated biological processes of proteins and enzymes. Driven by the desire to mimic nature, scientists have developed diverse stimuli-responsive polymer systems [1–4]. These smart polymers are capable of altering their chemical and/or physical properties in response to single or multiple external stimuli, such as mechanical force, temperature, pH, light irradiation, electric/magnetic fields, and specific analytes (gases, ions, bioactive molecules, etc.). The stimuli-induced changes can be chemical structures, chain conformation, solubility, wettability, shape (i.e., contraction or bending), surface charges, morphologies, optical properties, etc. Taking advantage of the stimuli responsiveness, such polymers have been utilized as controlled drug-delivery systems, smart coatings, artificial muscles and actuators, as well as fluorescent sensors [5–7]. Generally speaking, there are at least two parts for a responsive polymer system. One is the stimuli acceptor (input), and the other is the signaling part (output). The stimuli acceptor generally needs to possess high sensitivity, and the signaling part is better to produce readily detectable or visible signals. For instance, mechano-responsive polymer materials that are sensitive enough to spontaneously detect and report the occurrence and propagation of mini-cracks are highly desirable, so that workers can take early actions before small-scale damage develops into macro-damage that compromises the material's performance. Thermo-responsive polymers also require highly sensitive and visualized responses to temperature change. Particularly for those used for drug delivery applications, the direct monitoring of the release process and location of drugs is important for the understanding of the mechanisms of action. However, the responses of many traditional pure polymer systems are often insensitive or hard to detect. Although some detection methods such as UV–vis spectroscopy, electron microscopy, and mechanical performance tests have been employed to evaluate the response signals, these techniques are generally expensive and often require troublesome operation and sample preparation procedures. Therefore, there is still much room for improvement in terms of the signaling part of responsive polymer systems. It is highly demanded to develop functional polymer systems whose responses can be directly and sensitively visualized and monitored in real time, onsite, and in situ using portable instruments.
Stimuli-responsive fluorescent systems generally have the advantages of high sensitivity, fast response, less invasive nature, and large contrast [8–11]. The changes in fluorescence signal can be conveniently detected using portable instruments. In most cases, the fluorescence responses can be directly visualized by the naked eye in real time and in situ. Attracted by these advantages, scientists and researchers have applied fluorescence probes and labels in polymer materials to detect the conformational changes of polymers in response to microenvironment variations via fluorescence signals. For example, the temperature- and viscosity-dependent fluorescence change of molecular rotors can be utilized for the detection of glass transition temperature and polymerization degree [12–15]. The distance-dependent fluorescence of the Förster resonance energy transfer (FRET) process has been introduced as a “spectroscopic ruler” for the measurement of distances in macromolecules [16]. Besides, fluorescence responses have also been applied for the study of complicated protein and DNA folding/unfolding phenomena and processes [17]. However, conventional chromophores often suffer from poor fluorescence efficiency and a photo-bleaching problem in aggregate or solid states due to the aggregation-caused quenching (ACQ) effect, which greatly hinders their use in solid-state responsive systems. The weak fluorescence signal of ACQ dyes was detrimental to both the sensitivity and selectivity of optical indicators, and the stimuli responses were often in a turn-off mode. In the past two decades, materials with aggregation-induced emission (AIE) properties have attracted increasing attention and found significant applications in chemo-/bio-sensing and imaging [18, 19]. AIE luminogens (AIEgens) show highly efficient fluorescence at high concentrations or in aggregate states, which increases the sensitivity of optical indicators in thin films and other solid materials. Moreover, AIE-based sensing systems generally possess excellent photostability and low background noise. The well-accepted working mechanism for AIEgens is the restriction of intramolecular motion (RIM) model [20, 21]. When the intramolecular motion of the rotor-rich AIEgens is restricted, such as at low temperatures or in confined spaces, the fluorescence is turned on or markedly enhanced. Therefore, the fluorescence of AIEgens can exhibit an off–on switching property under various external stimuli, such as organic solvent vapor, pressure, heat, and metal ions [22, 23] The turn-on responses and good photostability can benefit the long-term visualization and monitoring of the response processes. These properties enable AIEgens to serve as excellent signaling components for developing sensitive stimuli-responsive systems. Notably, AIE polymers have many advantages over low-mass AIEgens, including good processability, easy functionalization, complicated structural features, and excellent thermal stability [24, 25]. Hence, by integrating AIE and stimuli responses into one polymer system, we can achieve robustly engineered materials with high sensitivity to environmental variations.
The combinatorial impact of AIE and stimuli-responsive polymers can be independent and synergistic. In some cases, the stimuli-responsive functionalities of polymers and AIE units work independently to perform their own functions as stimuli acceptor and signaling part, respectively. Through elaborative and rational design, the performance of responsive polymer materials and the functionalities of AIE materials can be mutually enhanced and enriched. Actually, the synergistic effect has been observed in most of the AIE-based stimuli-responsive polymer systems. For example, the aggregates of AIEgen-containing polymers often exhibit a super-amplification effect toward explosives or metal ions when they are used as fluorescence chemosensors [24, 26]. This phenomenon has rarely been observed in small molecular AIEgens, indicating that the polymeric structure can enhance the sensing performance of AIEgens. On the other hand, the inherent stimuli responsiveness of AIE components can further enhance the responsive performance and enrich the functionalities of the parent polymer matrix or backbones [27, 28]. In such circumstances, the AIE unit can serve as both signaling component and responsive acceptor. Moreover, AIEgens can also function as additives to improve the inherent physical properties of polymers, such as solubility and shrinking rates [29, 30]. The combination of stimuli-responsive AIEgens and stimuli-responsive polymers makes the development of multi-responsive materials with high sensitivity much easier to realize.
Attracted by the abovementioned advantages, researchers have devoted much effort to the development of functional polymer systems with AIE and stimuli responses, and very impressive progress has been made in recent years. Polymers range from biomacromolecules, synthetic polymers to supramolecular polymer systems. The applications of AIE in stimuli-responsive biomacromolecules and supramolecular polymer systems have already been summarized in several comprehensive review articles [31–37]. Due to the limited space, this review will mainly introduce the recent progress on synthetic polymer systems with AIE and stimuli responses, including the preparation strategies and their responsive behaviors to different stimuli (i.e. physical stimuli, chemical stimuli, and multiple stimuli). The current challenges and perspectives on the future development of this research area will also be discussed. From this review, we hope readers obtain a general grasp on what has been done in this area and meanwhile gain some insights for the future design and development of AIE-based stimuli-responsive polymer systems.
Preparation Strategies
According to the mechanism of AIE, most of the AIE polymer systems have potential for use used as stimuli-responsive materials. Their fluorescence properties are supposed to be sensitive to various environmental changes, such as temperature, pH, light, forces, and solvents. Therefore, the strategies for preparing AIE polymer systems, including both chemical synthesis and physical fabrication, are generally applicable to the fabrication of AIE-based stimuli-responsive polymers.
Chemical Synthesis
Chemical synthesis strategies toward AIE-based stimuli-responsive polymers can be roughly divided into two types. One is covalently linking polymers with AIEgens, and the other is the formation of clusteroluminescent AIE polymers [38]. By chemically incorporating typical AIEgens, such as tetraphenylethene (TPE) and its derivatives, into polymer structures, stimuli-responsive AIE polymers with various compositions and architecture can be obtained. Till now, diverse AIE polymers with linear, graft, hyperbranched, cross-linked, or dendrimeric structures have been constructed by chemical synthesis. Some AIE-active metal–organic frameworks and covalent organic frameworks have also been synthesized by similar strategies [36, 39, 40]. Taking polymers with linear structures as an example, they can be readily synthesized by the homo-polymerization of AIEgen-containing monomers (Fig. 1a). To improve or tune the polymer properties, co-monomers with stimuli-responsive units are often used to copolymerize with the AIEgen-containing monomers (Fig. 1b). Besides the use of stimuli-responsive monomers, the generation of stimuli-responsive chemical bonds in the backbone of AIE polymers is also an effective strategy toward stimuli-responsive fluorescent polymers (Fig. 1c). In some cases, AIEgen core structures can be generated in situ in the polymer main chains through the polymerization of AIE-inactive precursors (Fig. 1d). By attaching AIEgens on a polymerizable monomer, the AIE unit can also be attached on the polymers as side chains (Fig. 1e, f). Another interesting design is using AIEgen-containing initiators to initiate polymerization to produce stimuli-responsive polymers with AIEgens in the terminal or the middle of polymer chains (Fig. 1g, h).
Fig. 1.
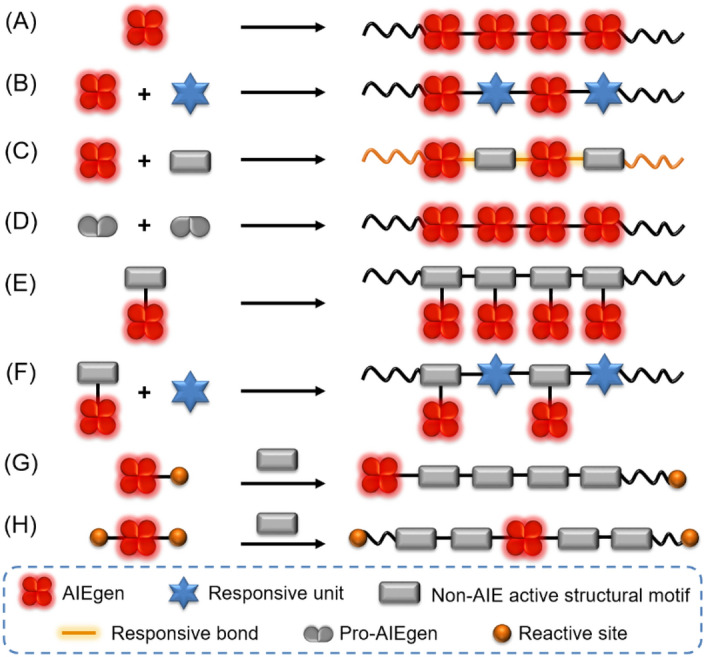
Polymerization strategies towards AIE-based stimuli-responsive polymers
Besides direct polymerizations, AIE-active stimuli-responsive polymers can also be generated through post-modification strategies of polymeric reactants. As schematically illustrated in Fig. 2a, polymer-containing reactive side groups can react with AIEgens to generate AIE polymers with stimuli-responsive chemical bonds. In addition to AIE initiators, AIEgen-containing terminators can also specifically introduce AIE moieties on the terminal of polymers (Fig. 2b). The combined uses of AIE initiator and AIE terminator strategies can benefit the regulation or enhancement of the stimuli-responsive performances (Fig. 2c). Furthermore, it is also possible to achieve AIE diblock copolymers with an AIE unit in the middle of the polymer chains through coupling the reaction of two different polymer block chains with AIEgens (Fig. 2d). The strategies shown in Fig. 2c, d have been rarely reported but are worthy of future exploration.
Fig. 2.

Post-modification strategies towards AIE-based stimuli-responsive polymers
In addition to AIEgen-containing polymers, some non-conjugated polymers have also been reported to show AIE characteristics and stimuli responsiveness. As depicted in Fig. 3, such polymers typically contain multiple electron-rich subgroups, such as N, O, S, and P with lone-pair electrons and/or unsaturated nitrile, carbonyl, aldehyde, carboxylic acid, ester, anhydride, etc. [41]. The uncommon fluorescence of these non-conjugated polymers is often explained by the clusterization-triggered emission (CTE) mechanism [42]. When the electron-rich polymers are molecularly dispersed, such as in dilute solutions, they are weakly or even non-emissive due to the active molecular motion and the lack of efficient electronic conjugation. However, the flexible conformations are rigidified upon aggregation. In aggregate state, the electron-rich subgroups tend to form clusters with different sizes, where their electron clouds are overlapped and shared to extend the electronic conjugation in a through-space way. Clusteroluminescent polymers generally exhibit excitation-dependent emission due to the heterogeneity of emissive clusters [43]. The larger the cluster, the brighter and redder the luminescence will be. According to the CTE mechanism, external stimuli that can affect the clusterization and rigidification processes of clusteroluminescent polymers can readily change their fluorescence. Therefore, clusteroluminescent polymers are promising functional materials with stimuli responsiveness.
Fig. 3.
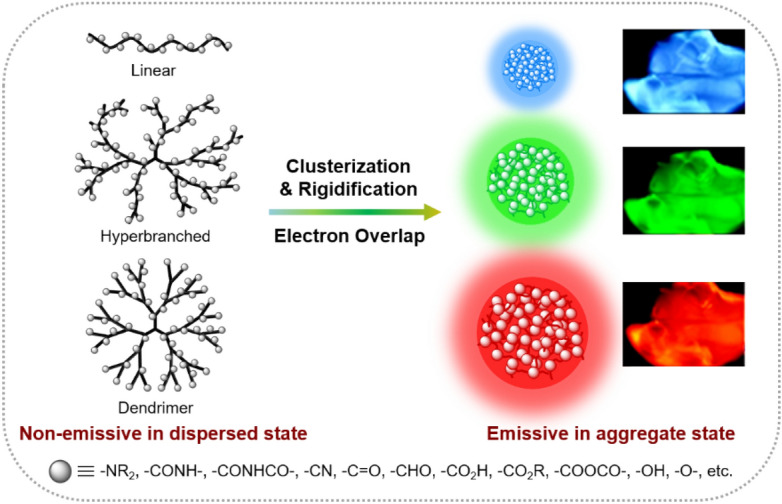
Schematic illustration of the working mechanism for clusteroluminescent AIE polymers. Insets: films of clusteroluminescent polymers under different excitation wavelengths.
Reproduced from [43] with permission from Springer Nature
Physical Fabrication
Chemical strategies normally require the attachment of a specific motif such as AIEgens or stimuli-responsive units to polymer structures via extensive synthetic efforts, which can increase the preparation cost and limit the application scope. By contrast, AIEgens can also be physically combined with polymer materials to construct smart flexible or rigid polymeric optical indicators to external stimuli. The physical blending strategies generally enjoy the advantages of facile fabrication procedures, good economics for raw materials, and wide application range. Taking advantage of the microenvironment-sensitive characteristic of AIEgens and the excellent mechanical processibility and stimuli-responsiveness of the polymer matrix, smart materials with diverse forms can be readily accessed. As shown in Fig. 4, AIEgen-based stimuli-responsive polymer composites can be engineered into films, coatings, fibers, micro/nanoparticles, microcapsules, bulk materials, etc.
Fig. 4.

Fabrication of stimuli-responsive AIEgen-polymer composites by physical blending
The films of AIEgen-embedded stimuli-responsive polymers can be obtained by different processing methods ranging from drop-casting, spin-coating, and electrospinning (Fig. 2a) to dip-coating (Fig. 2b). Among them, drop-casting is one of the most convenient methods. Thin films can be cast into different shapes by simply dropping the blend solution or melt of polymer and AIEgen on a flat substrate or in an open mold, but this method often suffers from the nonuniformity of film products. On the other hand, spin-coating method can produce uniform thin films with the loss of some raw materials during the spinning process. Electro-spinning method is suitable to fabricate stimuli-responsive fibers from the AIEgen/polymer blend solutions or melts. By depositing the nanofibers on a collector, porous polymer films with high surface-to-volume ratio can be obtained. In order to produce high-quality fibers or films, the preparation conditions, such as the concentration and viscosity of solutions or melts, need to be carefully regulated for the electro-spinning method. The dip-coating method is also referred to as the guest-diffusion technique. AIEgen-doped polymer films can be obtained by dipping the polymer sample in AIEgen solutions at various concentrations and elevated temperatures. Early studies also employed melt-processing methods such as the melt-extrusion process to fabricate AIEgen/polymer composites [44].
In addition to films and fibers, the assemblies of AIEgens and polymers, such as nanoparticles and micelles, are another important group of functional stimuli-responsive materials, especially for biological applications. AIE nanoparticles or micelles can be generated through the self-assembly of amphiphilic AIE polymers, but the obtained nanoparticles generally lack proper size control. A more popular and promising approach toward AIEgen/polymer nanoparticles is the physical encapsulation of AIEgens with biocompatible polymer matrices. As illustrated in Fig. 4c, the encapsulation process is induced by transferring the AIEgen/polymer mixture from organic solvents to water. Followed by the sonication treatment and solvent evaporation, functional nanoparticles can be easily formed in the aqueous solutions. This approach possesses the advantages of good control of particle size and excellent colloidal stability as well as the possibility of surface functionalization for targeted delivery [45, 46]. The reproducibility of large-scale production of AIE polymer nanoparticles can be achieved by an automated millifluidic system, which can form uniform AIE dots with a low polydispersity index of less than 0.1 in a high production rate of up to 200 g/day [47]. Microencapsulation technique allows the incorporation of tiny droplets or particles inside small capsules. By embedding the obtained microcapsules in bulk polymer films, stimuli-responsive materials can be facilely achieved based on the stimuli-triggered release of encapsulated core materials (Fig. 2d). Microcapsules loaded with AIEgens can in principle be readily incorporated into any polymer of interest by simple mixing and without the need of chemical modification. This approach is quite simple and versatile in terms of activation modes and responses that can be achieved [48]. In some cases, the chemical synthesis and physical encapsulation methods are combined together to fabricate stimuli-responsive materials. For example, AIEgen-doped hydrogels are often fabricated by mixing AIEgens with monomers and initiators to physically encapsulate AIEgens inside the hydrogels during the polymerization process [29, 49]. Recently, advanced manufacture techniques such as 3D and 4D printing have also been utilized to prepare AIE-based soft actuators [29].
Responses to Physical Stimuli
Most examples of stimuli-responsive AIE polymers only show efficient response to a single stimulus. Materials with sensitive response to multiple stimuli are important but are challenging to develop. For convenience, we will discuss functional AIE polymers with responsiveness from a single stimulus first and then to multiple stimuli. The stimuli are divided into two groups, including physical stimuli and chemical stimuli. In this section, the structure design, responsive performance, and potential applications of functional AIE polymers with responses to a single physical stimulus, such as mechanical force, temperature, light, electricity, γ-rays, and morphology, will be introduced with representative examples.
Force
Mechanochromic luminescent polymer materials whose fluorescence properties can change in response to external mechanical forces can be used as built-in fluorescent force sensors, security inks, damage-reporting coatings, flexible optical devices, etc. The utilization of the AIE effect endows the mechanochromic polymer materials with higher contrast due to their better solid-state luminescence than the conventional ACQ materials. Till now, a variety of AIE-based mechanochromic polymer materials have been reported, including AIE-active linear and cross-linked polymers, clusteroluminescent AIE polymers, blended AIEgen/polymer films, and polymer composites with AIEgen-loaded microcapsules [44].
For example, Chi and coworkers recently reported a linear AIE-active polymer with remarkable mechanochromism in the solid state [28]. As shown in Fig. 5, polymer P1 contains conjugated fluorene and 9,10-distyrylanthracene moieties in the polymer backbone, and TPE units are employed as bulky pendant groups to hinder the π-π stacking of the rigid backbone. The solid powder of P1 exhibits strong yellow fluorescence peaked at 541 nm. Upon grinding with a pestle, the fluorescence obviously changed from yellow to red with a large redshift of 61 nm in the emission maximum (Fig. 5b, c). By heating the ground sample at 200 °C or fuming it with dichloromethane vapor for 30 min, the emission maximum can shift back to 580 nm and 574 nm, respectively. The mechanochromism of P1 was explained by the destruction of its crystalline structure after grinding, which results in the conformational planarization and the increase of molecular conjugation of the polymer backbone. The poor reversibility may result from the slow crystalline rate of the ground sample due to the high molecular weight and the segmental motion of polymers. Besides the AIEgen-bonded polymers, AIE-active polymers based on the CTE mechanism have also been reported to be sensitive to mechanical forces [50]. As depicted in Fig. 5d, the non-conjugated poly-l-proline (PPRO) is weakly emissive under UV illumination, with a fluffy fibrous appearance. However, when PPRO was pressurized and patterned using a traditional Chinese stamp, the wrinkles created by the character on the stamp showed much brighter emission than the surrounding parts. The mechanochromic phenomenon of clusteroluminescent PPRO was attributed to the clustering of the electron-rich amide groups in close distance and the rigidification of molecular conformations after the pressurization process.
Fig. 5.

a Structure of P1. b PL spectra of P1 in different states. c Images of P1 powder in different states without (left) and with (right) the illumination of UV light. Reproduced from [28] with permission from Springer Nature. d Illustration of fluffy poly-l-proline (PPRO) in response to pressure. Reproduced from [50] with permission from the Royal Society of Chemistry
In addition to pure polymer systems, AIEgen/polymer composites are another main group of AIE-based mechano-responsive polymer materials. In earlier studies, such polymer composites were commonly achieved by directly blending mechanochromic AIEgens into various polymer matrices [51]. A more recent approach to realize the mechano-response is dispersing the AIEgen-containing microcapsules into polymeric matrices. Since the first work reported by Moore et al. in 2016 [52], the microencapsulation approach has been demonstrated to be a simple yet powerful strategy to fabricate polymer materials with autonomous detection of damage. As illustrated in Fig. 6a, core–shell microcapsules containing a dilute solution of AIEgen are embedded in a polymeric material. The intact polymer composite is non-emissive under UV light illumination. When the matrix material was subjected to mechanical damage, the microcapsules collapsed to release the AIEgen solution by capillary force. The subsequent solvent evaporation led to the deposition of solid AIEgen at the cracked area to show bright fluorescence. Such self-reporting coatings can provide clear visual indication of surface damage in a single layer, but cannot detect the depth information of crack penetration. In order to autonomously report microcracks and meanwhile differentiate the varying penetration depth, they further developed a self-reporting multilayered polymer system based on a similar strategy [53]. As shown in Fig. 6b, three AIEgens with red, green, and blue emission colors were selected, and their solutions were fabricated into microcapsules. Then, these microcapsules were incorporated into different layers of polymeric coatings. In this way, scratches with various penetration depths can result in the release of different combinations of AIEgens to show a diverse blend fluorescence colors. The microscopic damage depth can thus be visually assessed by correlating the luminescent colors with crack depth. This AIEgen-based detection strategy is applicable to a variety of polymer coatings and can provide high contrast between the damaged and undamaged regions.
Fig. 6.

a Schematic of the autonomous damage-reporting polymer composites using AIEgen-loaded microcapsules. b Schematic of the damage indication in multilayer polymeric materials with varying crack depth using AIEgens.
Reproduced from [52, 53] with permission from the American Chemical Society
In addition to the self-reporting function, the AIE-based microcapsule systems can also be endowed with self-healing properties to achieve dual functions of self-sensing and self-repairing [54, 55]. The damage to the polymer surface or even inside the polymer materials not only can be signaled but can also be instantaneously repaired. This kind of polymeric material can significantly improve the safety and reliability of critical engineering components. Moreover, the life cycle costs resulting from the regular maintenance and inspection of engineering components can be reduced to some extent. Therefore, such materials can have a significant economic and societal impact in terms of structural health monitoring, especially in the aeronautic, automotive, and construction industries.
Temperature
Among all available stimuli, the stimulus of temperature plays a unique role because it can easily be applied and tuned from outside. Polymers with thermo-responsive fluorescence have attracted great attention due to their technological significance and potential applications as fluorescent thermometers, carriers in drug release, intracellular temperature imaging, soft actuators, etc. By chemically or physically incorporating AIEgens into thermo-responsive polymers, the thermal transitions of the polymers can be carefully investigated using the fluorescence technique.
Poly(N-isopropylacrylamide) (PNIPAM) is one of the most investigated synthetic thermo-responsive polymers, exhibiting a characteristic and distinct lower critical solution temperature (LCST) in water. At LCST, a sharp transition from a stable one-phase to an unstable two-phase system will occur. The polymer starts to phase separate from solution to make the solution become turbid at LCST because of the transition from an enthalpy-favored coiled structure to an entropy-favored dense globular structure [56]. By chemically labelling the PNIPAM chain with AIEgens, the temperature-induced microstructure change in PNIPAM chains can directly affect the aggregation behavior of the grafted AIEgens, thus leading to the fluorescence change. In this way, the delicate details of the thermally induced liquid–solid phase transition process can be clearly revealed or monitored according to the change in fluorescence signal. For example, in 2009, Tang et al. prepared a TPE-labelled PNIPAM system through direct polymerizations and utilized its thermo-responsive fluorescence change to probe the thermal transitions of PNIPAM in water [57]. Similarly, Zhu et al. recently reported an AIEgen-grafted copolymer P(NIPAM-co-EM) for constructing a fluorescent thermometer [58]. As shown in Fig. 7a, P(NIPAM-co-EM) is composed of a quinolinemalononitrile-based AIEgen as the signal reporting unit and NIPAM unit as the thermo-responsive unit. The temperature-dependent absorption experiments revealed that the aqueous solution of the copolymer exhibits a specific LCST at about 30 °C. In the temperature region of 30–45 °C, coil–globule transition occurs along with the volume shrinkage, resulting in the formation of compact aggregates. This microstructure change leads to the activation of the RIM process of the AIEgen label to enhance the fluorescence. As depicted in Fig. 7b, c, the fluorescence intensity remained almost unchanged as temperature increased below the LCST value. When heating to 30‒45 °C, a sharp increase in fluorescence intensity for about 3.7-fold was observed. This AIE-active fluorescent thermometer fundamentally solves the serious fluorescence quenching problem of conventional fluorescent thermometers. It is worth mentioning that further heating the polymer mixture to above 45 °C led to fluorescence decrease. Because the phase transition process has already been completed at the LCST, further increasing the temperature may have little effect on the compactness of the polymer aggregates. Instead, the thermally activated molecular motion starts to take effect in the high-temperature region to consume energy of the excited state through the non-radiative pathway and thus result in the continuous decrease in fluorescence intensity. Moreover, the fluorescence signal of P(NIPAM-co-EM) can be switched reversibly. These results demonstrated the great potential of AIEgen-labelled PNIPAM as a nondestructive fluorescent thermometer. By carefully adjusting the hydrophilicity of copolymers, the LCST as well as the detection temperature range of AIEgen-labelled PNIPAM systems can be precisely controlled towards the physiological temperature. In this way, the application of water-soluble thermo-responsive AIE copolymers can be further extended to a series of biological studies such as cell imaging [59].
Fig. 7.

a Chemical structure of the fluorescent thermometer P(NIPAM-co-EM) containing the side-chain grafted AIEgen of quinolinemalononitrile. b Temperature-dependent emission spectra of P(NIPAM-co-EM) (14 mM) in an aqueous solution upon heating. c The relative fluorescent intensity at 607 nm as a function of temperature. Reproduced from [58] with permission from the Royal Society of Chemistry. d Chemical structure of TPE-labeled nylon-6. e Fluorescence spectra of TPE-labeled nylon-6 at different temperatures. f Plots of relative PL intensity and emission wavelength of TPE-labeled nylon-6 versus temperature. g Fluorescence photographs of the TPE-labeled nylon-6 at room temperature and 160 °C. Reproduced from [60] with permission from Wiley-VCH
In addition to PNIPAM and other polyacrylamides, nylon-6 has also been modified by AIEgens to endow it with thermally responsive fluorescence (Fig. 7d) [60]. Different from the PNIPAM systems, the resulting TPE-labeled nylon-6 exhibits remarkable change in both fluorescence intensity and emission wavelength. As shown in Fig. 7e–g, at low temperature, the TPE-labelled nylon-6 showed strong blue fluorescence, with the emission maximum at 485 nm. With the increase of temperature, the photoluminescence (PL) intensity gradually decreased, with little change in emission wavelength. However, a blueshift of the emission began to occur above 120 °C, while little change was seen in the PL intensity. This thermo-responsive fluorescence property arises from the synergistic effect of the chemical structure and conformation of the TPE-labeled nylon-6. PL intensity is closely related to molecular motion. When the temperature was increased from 30 to 120 °C, the intensive hydrogen bonds between nylon chains were gradually destroyed, and the volume between polymer chains expanded. Therefore, the rotation of the phenyl rings of TPE was gradually activated, and the motion rate became faster as the temperature increased, which consequently led to the decreased fluorescence. At 120 °C, the hydrogen bonds might be completely destroyed to cause the free motion to a large extent. Thus, the PL intensity remained almost the same from 120 to 180 °C. Before reaching the melting point, the TPE motion was mainly affected by the segmental motion of nylon-6 chains. However, at a temperature above the melting point, the chain diffusion was completely activated, which further promoted the rotation of all phenyl rings of TPE to completely quench the fluorescence. On the other hand, the emission wavelength was greatly affected by the change in the twisting angle of TPE during the heating process. The thermally induced conformation change of TPE-modified nylon-6 chains resulted in a significant variation in the twisted angle of TPE in the temperature range of 100–140 °C: the larger the twisted angle or the larger the motion amplitude, the bluer the fluorescence due to the decreased conjugation of TPE. In other temperature regions, although the molecular motion was active, the twisted angle of TPE changed little. Therefore, the fluorescence color varied little below 100 °C or above 140 °C.
Yang et al. recently reported a thermo-responsive AIE liquid crystal elastomer (LCE), whose fluorescence intensity was inversely proportional to temperature [61]. As illustrated in Fig. 8A, an AIE-active LCE soft actuator was fabricated through the direct polymerization of a liquid crystal monomer and a TPE-containing cross-linker. The obtained LCE sample exhibited both shape change and fluorescence variation in response to thermal stimuli (Fig. 8B). The PL intensity decreased linearly from 35 to 135 °C, while the longitudinal shape deformations (L/Liso) of the LCE sample presented a jump change around the LC-to-isotropic phase transition temperature (110 °C). This result demonstrated that the fluorescence variation of the AIEgen-functionalized LCE was mainly determined by the temperature effect rather than the macroscopic shape change. Increasing temperature can provide activation energy for the intramolecular motion of TPE, thus leading to the decrease in fluorescence intensity. This work demonstrates the potential applications of AIE-based LCE materials as thermomechanically controllable fluorescent soft actuators.
Fig. 8.

A Chemical composition of an AIE-active LCE film. B Photo images of thermally induced actuation behaviors of one LC elastomer ribbon sample exposed to (a) UV 365 nm radiation and (b) ambient light (scale bar = 5 mm). C Fluorescence spectra of the LC elastomer sample at different temperatures. D Relative fluorescence intensity and the longitudinal shape deformation (L/Liso) of the LC elastomer ribbon versus temperature.
Reproduced from [61] with permission from the American Chemical Society
Taking advantage of the thermo-responsive fluorescence property of AIEgens, in 2017, Tang et al. successfully detected the glass transition temperatures of various polymer systems through the fluorescence-based method by physically doping AIEgens into polymer matrix [62]. More recently, Ji et al. further reported an AIEgen-based fluorescence method to accurately measure the topology freezing transition temperature (Tv) of vitrimers [63]. Vitrimers are reprocessable cross-linked polymers, and their Tv refers to the upper limit temperature for service and lower temperature for recycling. Below Tv, the verified cross-linked network of vitrimers will restrict the intramolecular motion of AIEgens. Therefore, strong fluorescence is expected to be observed in this state (Fig. 9a). However, the intramolecular motion will be significantly activated above Tv due to the network rearrangement of vitrimers, leading to the weakened fluorescence of AIEgens. This design principle works well, as evidenced by the experimental results (Fig. 9b, c). After doping or swelling AIEgens into vitrimers, the fluorescence of the AIEgen-vitrimer composites changed dramatically below and over Tv with different decreasing rate. Thus, Tv can be determined by the turning point of the decreasing rate. This AIEgen-based detection method is straightforward, sensitive, universal, and reliable. Although Tv is measured in a static state in the presented experiments, this method can also be used to monitor the effect of external force on Tv under stress by modifying the sample holder.
Fig. 9.

a Schematic mechanism of detecting the topology freezing transition temperature (Tv) of AIEgen-doped vitrimers. b PL spectra of 1 wt% TPE-doped epoxy vitrimer at a series of rising temperatures. c The variation of PL intensity at 470 nm at different temperatures. All the fluorescence intensities are normalized to 45 °C.
Reproduced from [63] with permission from Springer Nature
Besides the abovementioned examples, a series of other functional polymer systems with thermal responsiveness and AIE characters have also been reported, including AIEgen-modified multiarm star polymers, crystalline polymers, clusteroluminescent AIE polymers, blended AIEgen/polymer films, etc. [64–69] These advancements demonstrate the power of AIE for facilitating the understanding of polymer thermodynamics and advancing the development of functional stimuli-responsive polymers.
Light
Photo-responsive polymers have attracted great attention due to their significance in smart optical and biological applications. The stimulus of light irradiation can be easily and precisely controlled to allow a localized stimulation within a defined area or volume. By changing the irradiation time and light intensity as well as the wavelength range of the light source, the strength of the response can be finely tuned. The most studied photo-responsive molecules include azobenzene, spiropyran, spirooxazine, fulgide, and dithienylethene (DTE) derivatives [70]. Functional photo-responsive polymers are generally constructed by chemically incorporating these photo-responsive units into polymer structures or physically blending the photo-responsive molecules in polymer matrices. The combination of AIE and photo-responsive properties can endow polymers with photo-responsive solid-state fluorescence and promising biomedical applications as photosensitizers.
Taking the DTE-containing photo-responsive polymers as an example, traditional photochromic DTE systems often suffer from the ACQ problem when using fluorescence as the read-out signal, which limits their practical applications in optical systems with high sensitivity and high precision [71]. In 2017, Lin et al. integrated AIEgens into a DTE-based photochromic polymer system to solve the ACQ issue [72]. As shown in Fig. 10a, polymer P-PHT contains DTE, TPE moieties, and a triazole linker in the repeating unit. When the DTE unit was in the open form, the AIE behavior of TPE took effect to enable the solid films of P-PHT to show bright blue fluorescence. Upon UV-irradiation of the P-PHT films, the cyclized DTE completely quenched the solid-state fluorescence of TPE via the non-radiative energy transfer process from TPE (as a fluorophore donor) to the nearby cyclized DTE unit (as an acceptor). In this way, a high fluorescence on/off ratio can be achieved for the photochromic P-PHT solid films. In 2018, Liu et al. reported a physical doping method to generate AIE-DTE-based photo-responsive polymer systems (Fig. 10b) [73]. The intramolecular photochromic property of DTE and the intermolecular AIE behaviors were integrated together using a DTE-bridged bispyridinium salt (DTE-BP). The molecular conformation of the bispyridinium salt located at both sides of the DTE core is sensitive to the restricted environment to endow the open-form DTE-BP with AIE properties. The DTE-BP-embedded polyvinylidene fluoride (PVDF) thin film showed strong green fluorescence. Upon irradiation with 254-nm light for 4 min, the fluorescence of DTE-BP was greatly quenched with a high fluorescence quenching efficiency of up to 91%, which was due to the photocyclization of the DTE core from the emissive open form to non-emissive closed form. In comparison to the photochromic behavior of DTE-BP in glycerin (fluorescence quenching efficiency = 76%), the PVDF matrix greatly accelerated the ring-closing reaction and thus significantly improved the quenching efficiency. Moreover, the quenched fluorescence can be almost completely restored after irradiation with visible light (> 490 nm) for 20 s. Taking advantage of the reversible photoswitchable fluorescence behavior, the DTE-BP-embedded PVDF films can serve as excellent photo-writable and photo-erasable materials with controlled fluorescence. The photo-writing-erasing process could be repeated for many times, indicating the good recyclability of the luminescent film.
Fig. 10.

a Chemical structure of P-PHT and its UV-induced PL spectral changes in solid films before and after UV irradiation. Inset: photo images of P-PHT at 0 min and 40 min UV irradiation. Reproduced from [72] with permission from the Royal Society of Chemistry. b Fluorescent photographs of DTE-BP-embedded PVDF film upon alternating 254-nm UV and visible light irradiations. Reproduced from [73] with permission from the American Chemical Society. c Chemical structure of TPE-PEG and its pattern formation mechanism. d Real-time fluorescent micrographs of pattern evolution assisted by intermittent UV irradiation during the isothermal crystallization of TPE-PEG film at 21 °C from a melt. Scale bars = 100 μm. Reproduced from [74] with permission from the American Chemical Society
Besides the incorporation of photochromic molecules, some AIEgens themselves can show remarkable photo-responsive fluorescence change. For example, TPE, one of the most well-known AIEgens, can undergo photocyclization and photo-oxidation reaction to form 9,10-diphenylphenanthrene (DPP) upon UV light irradiation. TPE shows blue fluorescence with maximum emission at 460 nm, while the fluorescence of cyclized-TPE is largely blueshifted to the invisible region (~ 375 nm). Recently, Tang et al. reported a manipulation strategy on the photocyclization activity of TPE using a crystallizing polymer matrix to generate controlled fluorescent patterns [74]. As illustrated in Fig. 10c, TPE was attached to one end of semicrystalline polyethylene glycol (TPE-PEG). The soft amorphous domain of PEG can facilitate the intramolecular motion of TPE, which is favorable for quenching fluorescence and increasing photoreactivity. By contrast, the intramolecular motion of TPE was restricted in the rigid crystalline phase to allow intense blue fluorescence and low photocyclization activity. Moreover, during the melt-crystallization process, the dynamic movement of PEG segments and chains in the boundary regions across crystalline and amorphous phases further accelerated the intramolecular motion of TPE. Therefore, the photocyclization tendency of TPE in the crystal growth boundary layer was higher than that in the amorphous domain. Taking advantage of this motion-dominated photo-responsive process, desired fluorescent patterns with distinct fluorescence contrast (bright vs. dark) can be readily generated using intermittent UV irradiation during the isothermal crystallization process of TPE-PEG film (Fig. 10d). Based on the photolithography technique, other AIE-active polymers with excellent photosensitivity and good film-forming ability have also been applied for the generation of diverse fluorescent patterns [75–78].
In addition to the photo-induced fluorescence change, AIE-based polyelectrolytes also possess extraordinary potency of reactive oxygen species (ROS) generation under light irradiation, which enables them to serve as excellent photosensitizers for various biomedical applications [79, 80]. For example, Tang et al. recently prepared a series of azonia-containing polyelectrolytes in a one-pot manner using readily accessible nonionic reactants in the presence of cheap ionic species (Fig. 11a) [81]. Among these polymers, P1/2c/3 with a strong donor–acceptor conjugated structure showed significant 1O2 generation ability and excellent photostability. The micelles of P1/2c/3 can efficiently inhibit the Gram-positive S. aureus under white light irradiation with a minimal inhibition concentration of 15 μg/mL. Furthermore, this polyelectrolyte possessed antimicrobial ability against drug-resistant bacteria. As illustrated in Fig. 11b, the high charge density in the polymer main chains endowed P1/2c/3 micelles with high affinity to the negatively charged bacteria membrane, which could physically damage the bacteria membrane to some extent. Upon white light irradiation, P1/2c/3 could efficiently generate ROS. The physical killing together with the photodynamic effect of P1/2c/3 led to strong inhibition against methicillin-resistant Staphylococcus aureus (MRSA), a drug-resistant superbug. The in vivo MRSA-infected mouse burn model further demonstrated that the photodynamic effect of P1/2c/3 could effectively inhibit the infection of MRSA and accelerate the healing process in the burn sites (Fig. 11c).
Fig. 11.

a Synthesis of azonia-containing polyelectrolytes (P1/2/3). b Schematic illustration for the application of the polyelectrolyte as a photosensitizer in superbug killing. c Typical photographs of MRSA-infected burn sites upon different treatments under dark or light irradiation during the therapeutic processes.
Reproduced from [81] with permission from the American Chemical Society
Apart from the abovementioned applications, the photo-responsiveness of AIE-based polymer systems can also find applications in anti-counterfeiting materials, super-resolution fluorescent imaging agents, microlasers, integrated photonics, erasable optical memory, logical gates, light-harvesting films, etc. [71, 82, 83]
Electricity
Electrofluorochromic (EFC) materials with changeable fluorescence in response to the applied potential have attracted great attention due to their potential applications in various optoelectronic devices, including electrofluorochromic devices, organic phototransistor-based memory devices, circularly polarized organic light-emitting diodes (CP-OLED), etc. [84] These practical applications generally require fast response speed, high fluorescence contrast, and stable switching stability for the EFC materials. However, conventional EFC materials often suffer from low fluorescence contrast due to their poor solid-state fluorescence. This problem can be effectively solved by AIE functionality.
Till now, several AIE-based EFC polymer systems have been reported [84–86]. For example, Liou et al. designed and synthesized a series of AIE-active triphenylamine (TPA)-based polyamides for the application of high-performance EFC devices (Fig. 12a) [87]. The solid films of TPA-CN-CH, TPA-CN-TPE, and TPA-OMe-TPE exhibited PL quantum yield of 46%, 16%, and 5%, respectively, due to the AIE characteristic. Moreover, EFC devices with these polyamides as EFC films (Fig. 12b) showed good match between the PL spectra and the absorption pattern in the switched-on state of the devices. These properties are all beneficial for enhancing the fluorescence contrast ratios of the EFC devices. As shown in Fig. 12c, the bright blue fluorescence of the EFC device based on TPA-CN-CH was obviously quenched to a dark state with the applied pulse changing from 0 to 2 V. When the potential was subsequently applied at −2.1 V, the fluorescence of this EFC device was well recovered. A similar phenomenon can also be observed for the EFC devices based on TPA-CN-TPE and TPA-OMe-TPE. The fluorescence quenching process could be attributed to the structural planarization and optical absorption shift of TPA units from the neutral to oxidized states during electrochemical switching. With the introduction of n-heptyl viologen (HV) into the device system as a counter electrochromic (EC) layer for balancing charges, the resulting EFC device based on TPA-CN-CH showed the highest fluorescence contrast ratio (Ioff/Ion) of up to 105 due to its strongest solid-state fluorescence (Fig. 12d). The shortest response time was achieved in the HV-containing TPA-OMe-TPE-based EFC device, whose response time was less than 4.9 s thanks to its conjugated structures and low oxidation potential. These results suggested that the combination of EC and AIE features in one polymer system is a feasible approach for preparing highly efficient EFC devices. However, the advantages of AIE-TPA unit have not been fully utilized in this work.
Fig. 12.

a Chemical structures of three polyamides. b Schematic diagram of electrofluorochromic (EFC) devices based on the polyamides. c Behaviors of EFC devices based on TPA-CN-CH (top), TPA-CN-TPE (middle), and TPA-OMe-TPE (bottom) with a specific applied voltage for oxidation. d Estimation of the fluorescence switching time at different step cycle times of TPA-CN-CH/n-heptyl viologen between 1.4 V and −1.5 V. Reproduced from [87] with permission from the Royal Society of Chemistry. e Chemical structure of TPE-TPA-CH. f Fluorescence spectra of TPE-TPA-CH film at different applied potentials. g Fluorescence switching response under applied potentials between 0.0 and 0.6 V at the cycling time of 20 s for 300 cycles. Reproduced from [86] with permission from the American Chemical Society
Recently, Chen et al. reported an electro- and AIE-active polyamide with a bulky and asymmetric pendant group containing TPE and TPA moieties (Fig. 12e) [86]. The introduction of TPE can improve the fluorescence on/off contrast, while the highly conjugated TPA structure can function as a stable electrochemical modulator to switch the dual emission and color. Moreover, the highly conjugated TPA and bulky pendant substituent can work together to shorten the response time. Such a rational combination of TPE and TPA allowed the resulting polymer (TPE-TPA-CH) to show highly integrated EC and EFC performances, including outstanding emission/color switching behavior (Fig. 12f), high optical contrast of 82 at the duration time of 20 s, and fast response speed (1.8/1.1 s for EC and 0.4/2.9 s for EFC process), as well as excellent long-term switching stability over 300 cycles (Fig. 12g). Therefore, the integration of AIE functionality with a highly conjugated redox unit could be an effective strategy to prepare high-performance emission/color dual-switchable materials in response to the external electric stimulus. In addition, the better device performance of TPE-TPA-CH than those of the polyamides in Fig. 12a also demonstrated that the device structure stability can be improved by attaching electron-donating groups, such as a methoxyl group and TPE, to stabilize the oxidized state of TPA.
Despite these innovations, there is still much room to explore further applications of AIE in EFC materials. With the guidance of the abovementioned strategies, we anticipate that more work could be done to promote the applications of AIE-based EFC materials in sensors, smart windows, and displays.
Gamma Rays
Gamma (γ)-ray radiation is one type of electromagnetic radiation originating from the atomic nucleus and from particle annihilation. γ-rays play an important role in many areas, including astrophysics research, nuclear power industry, sterilization, materials preparation and processing, biomedical applications, etc. For instance, γ-ray-based radiotherapy is an effective therapy for cancer treatment [88]. However, γ-rays can also be very hazardous to human health [89]. For the sake of security, it is of great significance to develop facile and convenient detection approaches for the detection of γ-ray radiation.
Taking advantage of the AIE characteristics and γ-ray-induced polymer degradation, Zhang et al. developed a simple and efficient fluorescence-based detection method for γ-ray radiation [90]. As illustrated in Fig. 13a, b, the negatively charged polyelectrolyte containing sulfone (–SO2–) groups in the main chain is sensitive to γ-rays. Under γ-ray radiation, the C–S bonds in this polyelectrolyte can be cleaved to degrade the polymer into small fragments. When the cationic AIEgen (silole-NI) was mixed with the aqueous solution of polyelectrolyte, bright blue fluorescence could be observed due to the formation of an aggregation complex with the assistance of electrostatic and hydrophobic interactions. However, when the polyelectrolyte was degraded by γ-ray radiation, the interactions between silole-NI and the polymer residues became weak, consequently reducing the coaggregation tendency and releasing the intramolecular motion of the AIEgen to some extent. Therefore, the fluorescence of the mixture was much weaker after exposure to γ-rays. As shown in Fig. 13c, the fluorescence intensity of the ensemble of silole-NI and polyelectrolyte gradually decreased with the increase in the dose of γ-rays. This detection method can be performed at ambient conditions, and the detection limit of γ-ray radiation was as low as 0.13 kGy. The sensitivity can be further improved by optimizing the chemical structures of the AIE probe and the γ-ray-sensitive polymer.
Fig. 13.
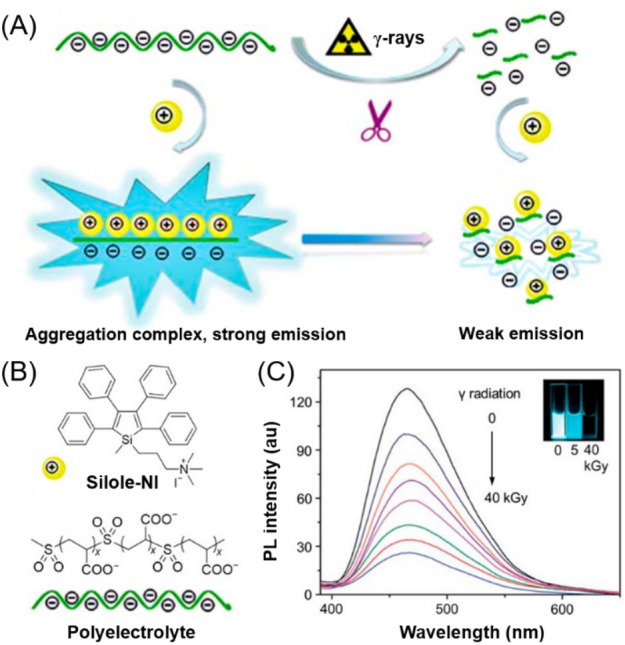
a, b The design rationale and the chemical structures of silole-NI and polyelectrolyte for the fluorescence detection of γ-ray radiation. b Fluorescence response of the aqueous solution of silole-NI and the polyelectrolyte before and after exposure to different doses of γ-ray radiation. Insets: the corresponding fluorescent images.
Reproduced from [90] with permission from the Royal Society of Chemistry
Besides the sulfone groups, diselenide bonds have also been reported to be sensitive to γ-radiation. Under γ-ray radiation, diselenide bonds can be cleaved and oxidized to generate seleninic acid [91]. Seleninic acid was reported to be a chemotherapeutic drug that can upregulate the ROS levels in cancer cells to cause cell apoptosis [92]. Therefore, the diselenide-containing systems are promising for use in the combination applications of radiotherapy and chemotherapy with improved anticancer activity. The incorporation of an AIE unit into the diselenide systems further endows the drugs with bioimaging capability. For example, Xu et al. synthesized an amphiphilic AIEgen with diselenide bonds in the middle of the structure (Fig. 14) [93]. This AIEgen could self-assemble into spherical assemblies, but the assemblies tended to collapse under γ-ray radiation due to the presence of diselenide bonds. This γ-radiation-induced self-assembly change could be monitored in situ through the fluorescence change. The fluorescence intensity of the assemblies dramatically decreased under γ-ray radiation, suggesting the disassembly of the assemblies. The cleaved selenium-containing assemblies showed excellent anticancer activities because of the anticancer efficacy of the generated seleninic acid. This carrier-free assembly system demonstrated the great potential of AIE-based γ-irradiation-responsive materials as nanomedicines for cancer treatment with combination therapy. Although only two examples of hazardous species detection were showcased in this section, countless species can be sensed by virtue of AIE through appropriate structure designs [19].
Fig. 14.

Schematic illustration of selenium-containing AIE assemblies combining cancer radiotherapy with chemotherapy.
Reproduced from [93] with permission from the American Chemical Society
Morphology
The study of polymer morphologies, including the conformations, domain size, shape, composition, texture, as well as phase distribution, is of great significance due to their direct influences on materials properties. Taking advantage of the high sensitivity of AIEgens to subtle microenvironment change, a few AIE-based polymer systems, such as AIEgen/polymer composites and AIEgen-functionalized polymers, have been exploited for the visualization and monitoring of polymer morphologies. Based on the distinct PL behaviors of AIEgens between molecularly dispersed and aggregated states, the conformational change, phase separation structures, self-healing structures, and swelling process of polymers have been clearly detected via fluorescence signals [27, 94–98].
Very recently, Tang et al. reported an AIE-based polymer system that can visualize polymer microstructures based on the distinct emission colors in different phases [99]. As depicted in Fig. 15a, an amphiphilic AIEgen (TPE-EP) with polymorphic characteristics was prepared and doped within the semicrystalline polymer of poly(L-lactide) (PLLA). The fluorescence of TPE-EP aggregates varied from yellow to green depending on the change of polymorphic forms from the metastable crystalline state (Y aggregates) to the thermodynamically stable crystalline state (G aggregates). Through quick and slow evaporation from chloroform solutions, the TPE-EP-loaded amorphous and crystalline PLLA films were produced, respectively. During the film-forming process, the polymorphs of TPE-EP were grown within the polymer network simultaneously. To minimize the influence of TPE-EP on polymer morphology without sacrifice of emission behaviors, 0.1 wt% TPE-EP was used in these films. The amphiphilic TPE-EP molecules were demonstrated to form segregated nanocrystals in the hydrophobic PLLA matrix. In amorphous PLLA, the G aggregates of TPE-EP were stabilized to give a green-emissive film (Fig. 15b) because the loose and mobile network of amorphous PLLA allows the embedded TPE-EP molecules to orient and pack stably. Oppositely, in crystalline PLLA, the Y aggregates were confined between the crystalline lamellae to show yellow emission (Fig. 15c, inset). Under a fluorescence microscope, a number of interlinked spherulites with yellow fluorescence were clearly observed. Therefore, TPE-EP can operate as a good fluorescent marker to distinguish the amorphous and crystalline phases of PLLA by the marked emission color difference. Furthermore, the polymorph selectivity of TPE-EP was also utilized to detect the microphase distribution and composition of polymers. As shown in Fig. 15d, e, the 2D and 3D microscopic images clearly reveal the whole microphase structure of the semicrystalline PLLA films: numerous yellow and circular spherulites randomly distributed within the amorphous green region. These fluorescence imaging results were consistent with those in SEM images. Moreover, a linear relationship between the emission maximum and the crystallinity of the composite film was established, based on which the average degree of polymer crystallinity could be quantitatively measured (Fig. 15f). The high sensitivity of the fluorescence-based imaging technique also enabled the high-contrast visualization of banded spherulites with specific handedness in a straightforward and noninvasive way. As depicted in Fig. 15g and h, the PLLA film prepared from a suitable slow evaporation process showed obvious banded spherulites with alternating bright- and dark-yellow spirals in the anticlockwise (ACW) direction. The AIEgen-embedded polymer films with chiral structured spherulites can efficiently generate circularly polarized light (CPL) upon UV excitation. The film inversion results in the enantiomeric chirality switching of the spiral spherulites to provide an opposite CPL response while the enantiomeric symmetry was kept. These results suggested that the morphology-dependent AIEgen/polymer systems can provide direct and detailed information on polymer microstructures to benefit the understanding of the morphology–property relationship of various polymer materials.
Fig. 15.

a Schematic representation of the preparation of amorphous and crystalline PLLA embedded with TPE-EP and the polymer phase determination. Fluorescence photos of b amorphous and c crystalline polymer films and (insets) their corresponding magnified images. Inset scale bar: 1 cm. d Overview fluorescence micrograph of the outer surface of the PLLA film with a mixture of crystalline and amorphous regions. e Confocal fluorescence 3D images of crystalline spherulites. f Fluorescence images of TPE-EP-embedded PLLA at various degrees of crystallinity. Scale bar: 5 mm. g Representative microscopic fluorescence (2D) and h confocal (3D) representation of the PLLA spherulites with alternative bright and dark yellow spirals in the anticlockwise direction. i Enantiomeric CPL switch upon film inversion.
Reproduced from [99] with permission from the Royal Society of Chemistry
Responses to Chemical Stimuli
Besides the abovementioned physical stimuli, the detection and measurement of chemical stimuli is also of great significance. In this section, we will discuss the representative examples of functional AIE polymer systems with responses to single chemical stimuli, including gas/vapor, pH, ions, explosives, and other chemicals or chemical reactions.
Gas/Vapor
The detection of gas like CO2 and volatile organic chemicals (VOCs) such as NH3 and acid vapor is fundamentally important for a diverse array of real-world applications. In the past years, various AIE-based polymer systems with gas/vapor responsiveness have been reported [100–103]. They are promising for use as selective CO2 detection, monitoring of morphology transformations of nanoassemblies, detection of biogenic amines and seafood spoilage, multi-scale humidity sensing, etc. [104]
For example, Chen and coworkers reported a fluorescent turn-on chemosensor for CO2 gas detection based on AIE (Fig. 16a) [105]. This sensor was constructed by the post-modification of branched polyethyleneimine (PEI) with TPE. Upon purging CO2 into the ethanol solution of TPE-PEI, the transparent solution immediately turned into the turbid precipitation because of the reaction of CO2 with alkylamines moieties of PEI to produce the poorly soluble carbamate salts (TPE-PEI-CO2). This solution-to-precipitation phase transition process induced a turn-on response in fluorescence intensity resulting from the AIE feature of TPE-PEI. This sensing system possessed high selectivity toward CO2 gas as it is highly tolerant to the possibly coexisting CO, water, sulfurous gases, and many common VOC interferents. More recently, Li et al. developed an AIE-active amphiphilic copolymer system, which can undergo reversible transition between polymersomes and micelles upon CO2 activation [106]. As shown in Fig. 16b, amphiphilic block copolymers PEG-b-P(DEAEMA-co-TPEMA) were prepared by the copolymerization of 2-(diethylamino)ethyl methacrylate (DEAEMA) and TPE-pended methacrylate (TPEMA) in the presence of mPEG-CTA. The obtained copolymers can self-assemble into polymersomes with bright fluorescence by the nanoprecipitation method in THF/water or dioxane/water mixtures. Upon bubbling CO2 into the polymersome solution, the DEAEMA segments were protonated and experienced hydrophobic-to-hydrophilic transition, which transformed the polymer vesicles into small spherical micelles. The transmittance of the solution increased and finally reached 97% due to the decrease of the nanoparticle size, whereas the fluorescence intensity slightly decreased due to the higher freedom of intramolecular rotation of TPE moieties in micelles (Fig. 16c). Moreover, this polymersome–micelle transition was demonstrated to be reversible upon the subsequent Ar bubbling. Similar morphology transformation strategy was also reported by Xing et al. They utilized a polymerization-induced self-assembly method to construct AIE-active polymeric nano-objects. With the promotion of CO2, the nano-objects underwent morphology transformation from spheres to a mixture of “jellyfish” and vesicles. This morphology transformation resulted in the variation of AIE behaviors, thus making it a promising CO2-responsive chemosensor with tunable morphologies and sizes [107].
Fig. 16.

a Structure of the sensory polymer TPE-PEI and its chemical reaction with CO2 to form polyammonium carbamates salts TPE-PEI-CO2 at ambient conditions and the schematic representation of its sensing process. Insets: photographs of TPE-PEI in ethanol before and after being purged with CO2. Reproduced from [105] with permission from Elsevier. b Synthesis of the CO2-responsive amphiphilic block copolymer PEG-b-P(DEAEMA-co-TPEMA). c Photograph of the polymer self-assembly solution before and after CO2 treatment and the reversible polymersome–micelle transition with the schematic molecular organization in the vesicle membrane. Reproduced from [106] with permission from Wiley-VCH
Taking advantage of the high sensitivity and excellent photostability of AIE polymers, Tang et al. developed an effective fluorescence-based technique to monitor acid–base homeostasis [108]. As shown in Fig. 17a, TPE-containing polyheterocycles (P1a/2a/3a) were synthesized through a metal-free multicomponent polymerization of terminal diynes, dialdehydes, and ureas. The resulting polymers exhibited typical AIE behaviors, and their thin films showed intense green fluorescence. Interestingly, the protonation of the thin film of P1/2a/3a by HCl vapor caused a marked redshift in the absorption and emission. The fluorescence color changed from chartreuse to dark-red accompanied with a large contrast in emission intensity (Fig. 17b). Similar phenomena were observed on quartz optical fiber coated with P1/2a/3a. When the acid-treated polymer thin film was exposed to ammonia vapor (NH3), the absorption and emission properties were completely recovered. Based on this fast and reversible fluorescence response, the protonated H2P1/2a/3a2+ was developed into a sensitive fluorescence sensor for ammonia with a detection limit of 960 ppb. The acid-treated thin film of P1/2a/3a can serve as a food spoilage sensor based on its sensitive and selective responses toward ammonia and biogenic vapors. As depicted in Fig. 17c, after being sealed in containers with fresh shrimps and scallops for 12 h at room temperature, the H2P1/2a/3a2+ obviously changed from non-emissive orange film to emissive orange-yellow film. The remarkable color change in daylight and turn-on fluorescence response change under UV irradiation made this polymer system a promising chemosensor for the in situ visualization of the food spoilage process. Apart from the abovementioned examples, AIEgen-doped polymer films and fibers with gas/vapor responses have also been constructed through facile physical preparation strategies [23].
Fig. 17.

a Synthetic route to P1a/2a/3a and its protonation and deprotonation. b Absorption and emission spectra of a thin film of P1a/2a/3a (red) after fuming with HCl vapor (black, 10 s) followed by NH3 vapor (blue, 0.08 M, 30 s). Inset: fluorescent microscopy photographs. c Spoilage detection of scallops in sealed packages for 12 h at room temperature using H2P1a/2a/3a2+ thin film.
Reproduced from [108] with permission from Wiley-VCH
pH
The stimulus of pH is a critical factor for cellular functions and other life forms. A subtle variation in environmental pH can have devastating effects on plant and animal lives. Moreover, pH value also plays a crucial role for the function and quality of medicines, foods, drinking water, etc. Therefore, the sensitive detection and monitoring of pH level is very important.
One of the most commonly used strategies for developing multifunctional pH-responsive polymers is the integration of AIEgens and pH-sensitive units in one polymer system. For example, Jin and Ji et al. have designed and synthesized a zwitterionic phosphorylcholine-TPE conjugate for pH-responsive drug delivery and AIE imaging (Fig. 18) [109]. The TPE unit was linked with the polymer backbone via acid-cleavable hydrazone bonds. The obtained AIE-active copolymer can self-assemble into spherical micelles and encapsulate doxorubicin (DOX) through hydrophobic interactions to form the theranostic PC-hyd-TPE-DOX micelles. Under endo/lysosomal acidic conditions, the cleavage of the hydrophobic TPE unit induced the disassembly of micelles followed by the release of DOX. Moreover, the introduction of AIE function enabled this system to show high-quality fluorescence imaging results. Combined with the efficient pH-responsive drug delivery, the multifunctional PC-hyd-TPE-DOX micelles were demonstrated to be a promising nano-platform for a new generation of cancer theranostics.
Fig. 18.

Schematic illustration of phosphorylcholine-TPE conjugate as a cancer theranostic nano-platform for combined AIE imaging and pH-responsive drug delivery.
Reproduced from [109] with permission from the American Chemical Society
The incorporation of pH-sensitive AIEgen into polymers can also achieve pH-responsive functional polymers. As depicted in Fig. 19a, TPE-OX, a pH-sensitive AIEgen, was attached to an amphiphilic copolymer chain with a hydrophilic poly(ethylene glycol) methyl ether methacrylate and hydrophobic polystyrene block [110]. Under acid/base stimuli, TPE-OX switched between the cyan-emissive closed form and the protonated open structure with red emission, thus contributing to the pH-dependent dual-emission of the amphiphilic copolymer. The PL spectra of the self-assembled polymeric micelle of the copolymer showed that its emission intensity at about 489 nm gradually decreased with the decrease of pH value in Tris–HCl buffer solutions (Fig. 19b). A new emission band centered at 640 nm was observed at pH 4.42, and this long-wavelength emission peak became stronger when pH further decreased, resulting in pH-responsive dual emission from cyan to red with a high fluorescence contrast (Fig. 19c). The fluorescence change was attributed to the extended conjugation and the emergence of an intramolecular charge transfer effect when the spiro-ring of the oxazolidine moiety was opened. The cell imaging experiments demonstrated that such polymeric micelles have potential applications in the detection of lysosomal activity and further autophagy in cancer cells based on their pH-dependent reversible AIE fluorescence.
Fig. 19.

a Chemical structures of the TPE-OX-pended copolymer and its acidichromism mechanism. b pH-dependent PL spectra of the polymeric micelles in Tris–HCl buffer solution. c Plot of I489/I638 versus pH.
Reproduced from [110] with permission from the Royal Society of Chemistry
Very recently, Tang et al. utilized the pH-responsive fluorescence of AIEgens and the interactions between AIEgens and hydrogels to fabricate a stimuli-responsive polymer system showing simultaneous changes in fluorescence color, brightness, and shape in response to a single stimulus [29]. As illustrated in Fig. 20a, a simple bilayer hydrogel actuator was designed and prepared with the chemically cross-linked ionomer PAS as the actuator matrix and the pH-responsive tetra-(4-pyridylphenyl)ethylene (TPE-4Py) as the dopant molecule of the active layer. Under acidic stimuli, the protonation of TPE-4Py led to the redshift in fluorescence due to the intramolecular charge transfer. The protonated TPE-4Py acted as the extra cross-linking point because of its electrostatic interactions with the PAS network, causing the shrinkage and the gradual deformation of the active layer. These interactions restricted the intramolecular motion of TPE-4Py to some extent, thus making the redshift emission visible. Further lowering the pH led to the increased solubility of the protonated TPE-4Py, consequently quenching the fluorescence of the active layer due to the enhancement of intramolecular motion (Fig. 20b). Based on this pH-responsive property, a furling flower-shaped bilayer TPE-4Py/PAS hydrogel actuator was further fabricated. As shown in Fig. 20c, when the artificial hydrogel flower was immersed into a pH 3.12 aqueous solution for different immersion time, simultaneous changes in fluorescence brightness, emission color, and shape could be clearly observed. By using the high-tech 3D or 4D printing techniques, pH-responsive hydrogel actuators with diverse shapes can be facilely produced. This example provided an effective strategy for the development of artificial intelligent polymer systems with potential applications in soft robots and smart wearable devices. In addition to the abovementioned examples, functional polymers with AIE and pH responses have also been applied for ratiometric fluorescence sensing of intestinal pH, monitoring of polymer degradation, biomedical probes, etc. [111–114]
Fig. 20.
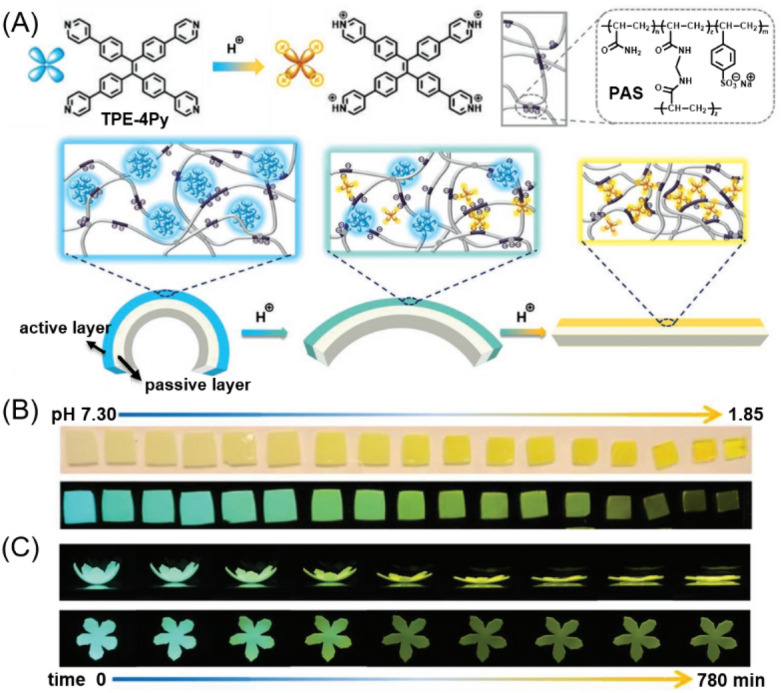
a Chemical structure of the pH-responsive AIEgen TPE-4Py and the chemically cross-linked ionomer PAS and the schematic illustration of design strategy of a TPE-4Py/PAS-based bilayer fluorescent hydrogel actuator. b Photographs of monolayer hydrogel samples after immersion in aqueous solutions with different pH values for 17 h taken under daylight (upper line) and 365-nm UV light illumination (lower line). c Flat view (upper line) and plane view (lower line) showing the simultaneous emission change and complex shape deformation of the pH-responsive hydrogel actuator.
Reproduced from [29] with permission from Wiley-VCH
Ions
Selective fluorescence sensors for ion detection are of great significance to environmental and health issues. In the past decades, diverse AIE-based polymers with sensitive and selective response to Ca2+, Pd2+, Pb2+, Cu2+, Hg2+, Al3+, Ru3+, CN−, etc. have been developed [23, 115]. The main principle of ion detection is based on molecular recognition. Therefore, the main design strategy for ion-responsive fluorescence sensors is the incorporation of AIE-active moieties and ion recognition groups or units into polymers.
For instance, thiourea compounds are well-known ligands for heavy metal ions with strong binding to mercury ion. Recently, Tang and Hu et al. prepared an AIE-active polythiourea through the catalyst-free multicomponent polymerization of sulfur, aliphatic diamines, and TPE-containing diisocyanides at room temperature (Fig. 21a) [116]. The nanoaggregates of the obtained polymer (PTU-TPE) in 50 vol% DMF/water mixtures showed intense fluorescence due to the AIE effect. With the gradual addition of Hg2+ from 0 to 10 μM, the fluorescence of the aqueous suspension of PTU-TPE gradually decreased owing to the formation of polythiourea-Hg2+ complexes. The quenching constant and detection limit reached 224,900 L/mol and 0.1 ppm, respectively, demonstrating the high sensitivity of mercury detection over a large variety of metal cations (Fig. 21b). Apart from ion sensing, this polymer system can also function as the mercury removal adsorbent due to the poor solubility of the polythiourea-Hg2+ complex. After filtration, the removal efficiency reached up to 99.99%, decreasing the amount of Hg2+ to 0.8 μg/L below the standard limit of drinking water (Fig. 21c). Because the concentration of Hg2+ can be correlated with the fluorescence intensity, the nanoaggregates of PTU-TPE can also be used for real-time monitoring of the removal process of mercury contaminants. Different from this turn-off-type fluorescence sensor, Ruan et al. reported an AIE-active thioketal-decorated conjugated polymer for Hg2+ working in a turn-on mode. The fluorescence of the polymer was significantly enhanced upon mixing with a trace amount of Hg2+ due to the Hg2+-promoted deprotection reaction of thioketal. The response of this fluorescence turn-on probe was very fast and efficient together with good selectivity towards Hg2+ because of the specific chemical reaction [117].
Fig. 21.

a Synthesis of the AIE-active polythiourea PTU-TPE by catalyst-free multicomponent polymerization. b Relative intensity (I0/I) at 493 nm of PTU-TPE in a DMF/H2O mixture (v/v, 1/1, 10 μM) in the presence of different metal ions (10 μM) and the corresponding fluorescence photos taken under UV irradiation. I0 = fluorescence intensity in the absence of metal ions. Inset: the proposed mechanism for the fluorescence detection and removal of mercury ion with PTU-TPE. c Schematic diagram of facile mercury removal process.
Reproduced from [116] with permission from the American Chemical Society
Based on the ion-induced conformation change of polymers and AIE, Fumitaka and coworkers developed a solid-state fluorescent sensor for extracellular Ca2+ imaging [118]. As shown in Fig. 22a, this solid-state (gel) sensor consists of a chemically cross-linked polyacrylic acid (PAA) block and TPE pendant. When the TPE pendants were attached to PAA, the obtained linear polymers (PAA-TPE) and chemically cross-linked gel (g-PAA-TPE) became highly fluorescent in the presence of Ca2+ with excellent selectivity over various interference species such as physiological ions, glucose, and amino acids. The PL intensity was intensified with the increase of Ca2+ concentrations. Different from the conventional design strategy of host–guest chemistry using tailored Ca2+-binding sites, the sensing mechanism of this gel sensor relies on polymer chain dynamics triggered by Ca2+. The binding of Ca2+ with PAA chains triggered the folding of non-emissive random coils to form aggregates, thereby activating the AIE effect of TPE pendants to turn on the intense fluorescence (Fig. 22b). The g-PAA-TPE gel can be used in various sizes and shapes. For example, a gel sheet fabricated from g-PAA-TPE can visualize the spatial distribution of Ca2+ concentrations. As depicted in Fig. 22c, a stamp experiment was conducted by attaching the Ca2+-loaded filter papers on the g-PAA-TPE gel. The filter papers were impregnated in aqueous solutions with different concentrations of Ca2+. After removal of the filter papers, the gel exhibited different fluorescence intensity that can be distinguished by the naked eye under UV irradiation. Considering its tailorable size and shapes as well as high sensitivity and selectivity towards Ca2+, this gel sensor can serve as an excellent Ca2+-imaging fluorescence sensor to benefit the understanding of Ca2+-involved biological and environmental events. Later, Shibayama and coworkers further studied the relationship between the mesoscopic structure of the TPE-appended PAA derivatives (PAA-TPE) and its fluorescence behavior, and elucidated the origin of the Ca2+ selectivity on the basis of dynamic and static light-scattering data. By investigating the hydrodynamic radius and molar mass of PAA-TPE in the presence of different metal ions at various concentrations, they concluded that Ca2+ can cause polymer chain folding more effectively than other ions to form aggregates with a much higher inner density, thus leading to more pronounced AIE behaviors and better selectivity in Ca2+ detection [119].
Fig. 22.

a Fluorescence intensities of PAA-TPE (blue bars) and fluorescence quantum yields of g-PAA-TPE (red bars) in the presence of various metal chlorides, glucose (Glc), and glutamine (Gln). Inset: chemical structures of PAA-TPE and g-PAA-TPE. b Schematic illustration of the mechanism of Ca2+ sensing with g-PAA-TPE. c Stamp experiment using filter papers impregnated with CaCl2 aqueous solution with different concentrations. Scale bars: 1.0 cm.
Reproduced from [118] with permission from Springer Nature
Apart from the abovementioned examples, clusteroluminescent AIE polymers with sensitive responses to Fe3+ have also been reported because the paramagnetic nature of Fe3+ can effectively quench the fluorescence of polymer aggregates [120]. However, examples of ion-responsive clusteroluminescent AIE polymers are still very limited. More efforts are needed for the technical investigations of biocompatible AIE polymers for ion detection.
Explosives
Detection of explosives has become an international concern due to the demands of homeland security, global anti-terrorism, landmine detection, forensic research, and environmental pollution assays. AIE-active polymers have been extensively applied for explosive detection, especially for the detection of nitroaromatic-based explosives, such as picric acid (PA), 2,4-dinitrotoluene (DNT), and 2,4,6-trinitrotoluene (TNT) [121–123].
In contrast to small molecular fluorophores, fluorescent conjugated polymers often show an amplification-quenching effect (or so-called “super-amplification effect”) when they are used as fluorescent sensors, resulting from the efficient electronic communication and exciton migration between the conjugated polymer backbones and the quenchers (Fig. 23a) [124]. As reported by Swager et al., one explosive quencher molecule resulted in the fluorescence quenching of the whole polymer chain. This “one-point contact, multi-point response effect” or the “molecular wire effect” could not occur in small molecules but in conjugated polymers owing to their unique long-range conjugated structures [125]. Furthermore, comparing to the linear analogues, hyperbranched polymers often show improved sensing ability toward explosives because their numerous branches provide multiple exciton migration channels and diffusion pathways in the 3D architectures [8]. The enhanced exciton migration as well as the efficient interaction between the explosive analyte and the 3D polymer structure thereby lead to the super-amplification effect of hyperbranched polymers (Fig. 23b) [126].
Fig. 23.
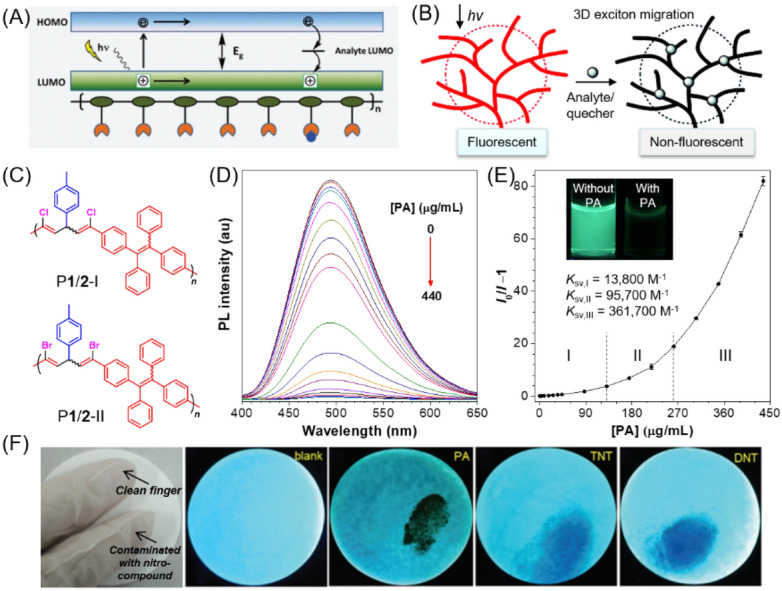
a Schematic diagram illustrating the “the one-point contact, multi-point response effect” of a linear conjugated polymer as an explosive sensor. Reproduced from [124] with permission from the Royal Society of Chemistry. b Schematic illustration of the fluorescence-quenching processes of 3D hyperbranched polymers by the analytes. Reproduced from [126] with permission from Springer Nature. c Structures of P1/2-I and P1/2-II. d PL spectra of P1/2-II in THF/water mixtures with 80% water fraction containing different amounts of picric acid (PA). e Stern–Volmer plots of (I0/I − 1) of P1/2-II versus the PA concentration, where I0 = peak intensity without PA. Inset: fluorescent photographs taken under 365-nm UV illumination [127]. Reproduced from [127] with permission from MDPI. f Detection of nitro-compound contamination by checking the fluorescent images. Reproduced from [128] with permission from the Royal Society of Chemistry
The super-amplification quenching effect was commonly observed in fluorescence sensors based on hyperbranched polymers, but it is worth noting that linear polymers with AIE characteristics have also been reported to show the super-amplification quenching effect in response to explosives. For instance, Tang et al. reported a linear poly(dihalopentadiene) system with AIE activity (P1/2-I and P1/2-II, Fig. 23c) [127]. The strong fluorescence of their nanoaggregates in aqueous media could be sensitively quenched by PA with a low detection limit (Fig. 23d). The Stern–Volmer plot of the AIE polymer bent upward when a large amount of PA was added (Fig. 23e), demonstrating a remarkable super-amplification quenching effect. This phenomenon can be rationalized by the unique working mechanism of AIE. AIE polymers are used in the form of nanoaggregates, which have more cavities to interact with more quenchers and provide additional inter-chain diffusion pathways for exciton migration. In addition to the electronic effect, physical factors also contribute to the super-amplification quenching phenomenon of AIE systems. For example, the detection of PA was generally conducted in aqueous solutions due to the good water solubility of PA. In this circumstance, water molecules could enter into the polymer networks together with PA molecules to result in the swelling of polymer nanoaggregates. The consequent larger free volume between polymer chains facilitated the free motion of AIE units, thus strengthening the fluorescence quenching of polymer nanoaggregates.
Taking advantage of the solid-state sensing of AIE polymers, Xu and coworkers fabricated paper sensors for explosive detection by spraying the nanoparticles of AIE polymers onto filter papers [128]. As depicted in Fig. 23f, the obtained paper sensor with a polymer surface concentration of as low as 1.0 µg cm−2 is effective in detecting various common nitro-aromatic contaminants on fingers, including PA, TNT, DNT, at a scale of less than 1 ng. This type of paper sensor can function as a useful tool for the quick, inexpensive, and highly sensitive detection of nitro-compound-based explosives.
Others
Beside the different chemical stimuli mentioned above, the detection of organic pollutants, redox reaction, and various biomolecules using AIE-based polymer systems have also been widely reported. These systems are too specific and too numerous to be mentioned individually, so they are discussed in just one category with selected examples in this section.
The sensitive detection of organic pollutants in aqueous media is of great significance for the quality evaluation and safety improvement of water resources. Conventional detection methods for organic pollutants often require time-consuming procedures and the use of bulky and expensive devices. Recently, Tang and Liang et al. described a type of sticky nanopad made of crystallizable fluorescent polymers for the facile, rapid, and sensitive detection of toxic organic pollutants in water [129]. As shown in Fig. 24a, b, TPE was chemically linked to one terminal of polyethylene (PE) chains. The sticky nanopads comprised a single layer of the TPE-modified crystalline PE with TPE units located at the surfaces of the nanopads. Through hydrophobic and π–π interactions, the sticky nanopads were able to absorb organic pollutants in water. The absorbed organic pollutants on the surface of the nanopads subsequently quenched the fluorescence of the AIEgens (Fig. 24c), possibly due to the consumption of the excited-state energy of PE-TPE through collision between the excited chromophore with xylene molecules. The detection limit of PE-TPE nanopads to xylene can reach as low as 7 μg/L, and the response was observed in the order of seconds (Fig. 24d). Later, Liang et al. further developed an amphiphilic fluorescent copolymer micelle system with a hydrophobic TPE core, which could absorb the aromatic pollutants and thereby quench the fluorescence. Based on the capture-report strategy, these types of swellable fluorescent polymer micelles can be used for the rapid detection of aromatic pollutants in water in the order of seconds at a concentration of 1 μg/L [130]. Besides the detection of organic pollutants in aqueous media, functional AIE polymers with fast and sensitive fluorescence responses to organophosphorus pesticide, acids, aliphatic amines, etc. have also been reported [131–133].
Fig. 24.

a Synthetic route for polyethylene monoterminated with tetraphenylethene (PE-TPE). b Schematic of sticky PE-TPE nanopads for sensing organic pollutants in water. c PL spectra of PE-TPE nanopads with various amounts of xylene. d Variations in the PL intensity against the xylene concentration. The concentration of PE-TPE was 0.1 mg/mL.
Reproduced from [129] with permission from the Royal Society of Chemistry
Redox-responsive polymer micelles can function as versatile nano-platforms for on-demand drug delivery. The design of redox-responsive polymer materials is often based on the electrochemical processes of redox-sensitive groups or units, such as dithienylethenes, boronate ester, ferrocene, or disulfides, which causes changes in their oxidation states in response to environmental redox variation [134, 135]. However, the in situ evaluation of drug release is challenging. By introducing AIE and FRET effects into the redox-responsive polymer systems, the drug release process from micelles could be well monitored through the variations in fluorescence signal. For example, Zhao and coworkers reported a redox-responsive probe for detection of the intracellular drug release based on the FRET-AIE approach [136]. As illustrated in Fig. 25, poly(ethylene glycol)-polylysine (mPEG-PLys) was selected as the backbone, and the AIE probe (TPE) was chemically linked to the polymer main chain via an amide bond to act as the FRET donor. Meanwhile, curcumin (Cur) was chosen as the model drug and FRET receptor. The ACQ-active Cur was covalently conjugated to the block copolymer backbone via a redox-sensitive disulfide linker. The obtained amphiphilic polymer conjugate could effectively self-assemble into micelles. Upon the supplement of reductants, such as the glutathione (GSH) and tris(2-carboxyethyl)phosphine) (TCEP), the disulfide bond was cleaved to release the Cur drug. This drug release process could turn off the FRET signal and meanwhile turn on the bright AIE fluorescence (Fig. 25). Therefore, both TPE and Cur showed increased fluorescence intensity. This FRET-AIE strategy provided a useful tool for real-time analysis of drug release via stimuli-responsive nano-platforms. However, restricted by the penetration ability of the excitation wavelength, the current system cannot be utilized in situ for in vivo assessment of drug release. Further efforts can be put toward developing redox-responsive polymer systems with NIR absorption and emission properties.
Fig. 25.

Schematic illustration of drug release assessment from redox-responsive micelles via integrating FRET with AIE.
Reproduced from [136] with permission from Wiley-VCH
Functional polymer systems with fluorescence response to biomolecules, such as glucose, enzyme, heparin, adenosine triphosphate (ATP), amino acids, proteins, and lysosomal HClO, are promising candidates for bioprobes [19, 137]. In this aspect, water-soluble AIE polyelectrolytes have been proven to be powerful biosensors for the sensitive and specific detection of various biogenic small molecules and biomacromolecules. For example, Wang et al. reported an AIE-active cationic poly(diketopyrrolopyrrole-co-ethynylfluorene) [138]. This polymer system showed sensitive and selective fluorescence turn-on response to bovine serum albumin (BSA) stimulus based on the BSA-induced deaggregation process of this copolymer. Other biological species bearing negative charge such as alkaline phosphatase, DNA, Con A, lysozyme, ATP, and various anions (Cl−, Br−, I−, NO3−, AcO−, CO32−, NO3−, and PO43−) have no significant effect on the fluorescence, because they failed to induce the deaggregation of the AIE polymer. Compared with conventional ACQ-type turn-off bioprobes, AIE-based bioprobes generally possess turn-on/light-up responses to biomolecules as well as lower background interference and higher resistance to photobleaching. Therefore, biomolecule-responsive AIE polymer probes are promising for use in continuous monitoring of biological processes with high sensitivities and resolution.
Responses to Multiple Stimuli
Intelligent polymer systems that can respond to multiple stimuli are intensively explored in biomimetic design, analytical research, nanotechnology, and biological applications. Compared with the abovementioned single-stimuli-responsive polymer materials, dual- or multi-stimuli-responsive polymers are more intriguing because they can achieve more functions and finer modulations through more parameters. The most commonly used design strategy for developing multi-stimuli-responsive polymers is the combination of two or more stimuli-responsive groups or components into one polymer system [6]. The combinatorial impact of the stimuli-responsive moieties can be independent, serial, or synergistic, depending on the influence of one group’s response on the other’s.
For example, Kim et al. reported a dual-stimuli-responsive polymer material for chemical reservoir coatings [139]. The AIEgen-loaded microcapsules and commercial pH indicator were physically embedded into the polymer matrix for the detection of cracks and pH variation, respectively. These two stimuli-responsive components work independently in this polymer system. The mechanical response has no influence on the capability of pH response because their detection mechanisms are totally different. Zhang and Hadjichristidis chemically combined the pH-responsive building blocks with the thermo-sensitive AIE unit to construct a pH- and thermo-dual-responsive star copolymer system [140]. As depicted in Fig. 26a, the core cross-linked miktoarm star copolymer (PE)n-(PMAA)m-P(TPE-2St) was synthesized, where the hydrophilic poly(methacrylic acid) (PMAA) arm functions as the pH-responsive block, and the hydrophobic polyethylene (PE) arm was introduced to enable the formation of an AIEgen-containing core–shell structure. The PL intensity of the obtained star copolymers showed a negative correlation with temperature (Fig. 26b, c). The decreased fluorescence at high temperatures was attributed to the swelling of the shell, which activates the motion of TPE units in the core to release the excited-state energy in a non-radiative pathway. The pH-responsiveness of the star copolymers was also related to the core–shell structure. As shown in Fig. 26d, e, the PL intensity of (PE)n-(PMAA)m-P(TPE-2St) aggregates gradually increased when pH increased from 1.1 to 5.0, because the PMAA arms were protonated at pH < 4.2, which decreased the solubility of the copolymer to form contracted micelles. In the pH range of 5.0‒10.0, the PL intensity changed little. When pH further increased from 10.0 to 13.3, a dramatically decreased PL intensity was observed due to the higher charge density and better hydrophilicity of PMAA in these conditions. The swelled and extended polymer conformations at high pH provide increased space between the TPE moieties by the separation of solvent molecules, thus leading to the decreased fluorescence. In this example, the microstructure of polymers plays a key role in both the temperature- and pH-responsive properties. Therefore, the stimuli-responsive moieties work synergistically in this dual-stimuli-responsive system.
Fig. 26.

a Synthetic route for the AIE-active core cross-linked multi-miktoarm star copolymers. b PL spectra of (PE)19.4-(PMAA)14.5-P(TPE-2St) in 50/50 vol % THF/n-hexane solution at different temperature. c Plot of PL intensity vs. temperature. d PL spectra of (PE)19.4-(PMAA)14.5-P(TPE-2St) in THF–buffer mixture (10/90 vol %). e Plot of I/I0 vs. pH value; I0 is PL intensity at pH 5.0.
Reproduced from [140] with permission from the American Chemical Society
Similar strategies have also been applied for the development of redox- and pH-dual-responsive polymer systems. For example, Wang et al. developed an AIE-active polymeric micelle system based on AIEgen-pended copolymers with both pH- and redox-sensitive units. This copolymer system can be utilized for ultrasensitive pH and redox triggered drug release and bioimaging [141]. Later, Tang et al. physically combined AIE photosensitizers with stimuli-responsive polymers to form multi-stimuli-responsive nanoparticles for photodynamic therapy [142]. As illustrated in Fig. 27, a far red-emissive AIEgen named MeTTMN with high ROS generation efficiency was used as a photosensitizer. A pH-responsive polymer P-Hyd and a redox-responsive polymer P-SS were designed and synthesized with a hydrazine bond and disulfide bond as the stimuli-responsive sites, respectively. Meanwhile, a nonresponsive polymer P-control with hydrophilic poly(ethylene glycol) and hydrophobic polycaprolactone moieties was also synthesized as a control. All the prepared polymers can spontaneously self-assemble into core–shell-structured nanomicelles in aqueous solution. These nanomicelles with good biocompatibility, excellent stability, and appropriate nanoparticle size can serve as extraordinary carriers for the AIE photosensitizer in high loading efficiency. After efficient uptake of the MeTTMN-loaded nanomicelles by cancer cells, the pH-responsive nanomicelles can release the photosensitizer in a lysosome acid environment due to the cleavage of the hydrazone bond, while the redox-responsive nanomicelles tend to disassemble in cytoplasm of cancer cells to release MeTTMN because of the breakage of the disulfide bond at high concentrations of GSH. The stimuli-responsiveness towards tumor microenvironments can achieve controlled release behaviors. The released MeTTMN exposed more oxygen than in the core of nanomicelles. Therefore, the MeTTMN-loaded stimuli-responsive assemblies exhibited much higher ROS generation efficiency and significantly promoted PDT performance than those of the P-control nanoassemblies. This example revealed that the addition of another stimuli-responsive assembly can greatly improve or enhance the original response performance to a single stimulus. It can be anticipated that when the stimuli-responsive polymers (P-Hyd and P-SS) were used together to form self-assemblies with the AIE photosensitizers, higher delivery efficiency of the photosensitizer could be achieved, leading to more efficient ROS generation and better photodynamic therapeutic effect.
Fig. 27.

Illustration of stimuli-responsive AIE polymer nanoparticles with high ROS generation efficiency and boosted PDT effect.
Reproduced from [142] with permission from Elsevier
Some other design strategies have also been reported occasionally. For instance, Hu, et al. took advantage of the high environmental sensitivity of AIEgen and the inherent stimuli-responsiveness of shape-memory polyurethane (PU) to develop a multi-stimuli-responsive chromic polymer [143]. The TPE units (0.1 wt%) were covalently incorporated into the poly(ε-caprolactone)diol soft segments through the polymerization of TPE-diol with other co-monomers. As shown in Fig. 28, the obtained TPE-containing PU films showed remarkable fluorescence change in response to the stimuli of mechanical force, temperature, and organic solvent. The fluorescence of the shape-memory PU obviously decreased when stretched, which was attributed to the deaggregation of TPE units and the change in the film thickness. Upon heating or solvent treatment, the fluorescence of the PU film was restored together with the almost 100% shape recovery of the film. Actually, the mechano-responsiveness of AIE-based polymer systems was often susceptible to other stimuli, such as solvent vapor and temperature, because the other stimuli can also influence the polymer conformations and the intramolecular motion of AIEgens, thus affecting the fluorescence signals [44].
Fig. 28.

a Schematic illustration of the molecular mechanism during the stretch-recovery process (1), heating–cooling process (2), solvent-dry process (3), and solvent-induced shape recovery process (4) of the TPE-functionalized polyurethane copolymers. b The fluorescent images of the polymer film which was first stretched and then immersed into acetone and then removed from solvent and dried at room temperature.
Reproduced from [143] with permission from Wiley-VCH
Summary and Outlook
Thanks to the enthusiastic endeavors of scientists and researchers, a large variety of functional polymer systems with AIE and stimuli responses have been developed in the past decades. In this review, we summarized the recent advances in this field with selected examples. The combination of AIE property and stimuli-responsive ability in one polymeric system can revitalize the long-history study on stimuli-responsive polymers and advance their applications in different areas. Encouraged by the remarkable progress and fruitful achievements, more explorations and breakthroughs with numerous possibilities can be made in this field. Future directions and challenges might include the following aspects, among others.
Firstly, the fabrication methods can be further advanced. For example, the development of facile and efficient synthetic strategies is fundamentally important to provide powerful tools for the creation of new stimuli-responsive AIE polymers. By choosing suitable chemical methods, different stimuli receptors and signaling units can be readily integrated in one system to enhance materials performance and enrich the functionalities. For physical blending methods, more efforts can be made on the utilization of melt-processing methods such as the melt-extrusion process as well as high-tech fabrication methods such as 3D printing to advance the industrialization of AIE-based stimuli-responsive polymers. In particular, the application of 3D printing in this field might benefit the study of molecular machines. For instance, our group has developed a light-responsive solid-state molecular actuator based on small molecular AIEgen crystals. Remarkable fluorescence change was observed in the moving parts of the crystal actuators due to the ACQ-AIE-transformation under UV irradiation [144]. This proof-of-concept example indicated that the AIE effect may help to visualize the moving positions and mini-motion or monitor the detailed movement processes of molecular machines. By incorporating the AIEgen-based molecular actuators into soft hydrogel matrix, and with the assistance of 3D or even 4D printing techniques, macroscopic actuators with various sizes and shapes as well as amplified responses might be facilely prepared. Secondly, persistent endeavors are demanded for the development of intelligent AIE polymer systems that can show multiple responses to single or multiple stimuli. In other words, the output signals can also be diverse. The appearance, fluorescence color and/or intensity, macroscopic size and/or shape, and other properties and functions of the desired polymers can change simultaneously upon exposure to external stimuli. Such responsive systems are more in line with the responsive behaviors of living systems in nature. The combination of several recognition moieties, stimuli-responsive groups, and functional agents into one AIE polymeric system will surely broaden their applications. Regarding the choice of AIE polymer systems, clusteroluminescent polymers are worthy of more attention and investigations. These kinds of polymers are inherently responsive to external stimuli such as pressure and temperature based on the CTE mechanism. Further efforts are needed to enhance their AIE performance, including the improvement of solid-state fluorescence intensity and the redshift of emission wavelength. Last but not least, the application forms of stimuli-responsive AIE polymers can be further diversified. For instance, the moisture-sensitive and antibacterial AIE polymer fibers can be further made into smart textiles to benefit our daily life.
It is not possible to list all the examples and address all the possibilities in this review due to the limit of space. We hope this review inspires more scientists to investigate AIE-based stimuli-responsive polymer materials and realize their full potential over the conventional ACQ luminescent systems and conventional stimuli-responsive polymers.
Acknowledgements
We acknowledge the funding from the National Natural Science Foundation of China (21905176, 21801169), the Natural Science Foundation for Distinguished Young Scholars of Guangdong Province (2020B1515020011), the Research Grants Council of Hong Kong (16304819, 16305618, and C6009-17G), the Innovation and Technology Commission (ITC-CNERC14SC01), the National Key Research and Development Program of China (2018YFE0190200), and the Science and Technology Plan of Shenzhen (JCYJ20190808142403590, JCYJ20190808153415062).
Compliance with ethical standards
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Dong Wang, Email: wangd@szu.edu.cn.
Ben Zhong Tang, Email: tangbenz@ust.hk.
References
- 1.Wei ML, Gao YF, Li X, Serpe MJ. Polym Chem. 2017;8:127–143. doi: 10.1039/C6PY01585A. [DOI] [Google Scholar]
- 2.Bauri K, Nandi M, De P. Polym Chem. 2018;9:1257–1287. doi: 10.1039/C7PY02014G. [DOI] [Google Scholar]
- 3.Cabane E, Zhang XY, Langowska K, Palivan CG, Meier W (2012) Biointerphases 7 [DOI] [PubMed]
- 4.Vazquez-Gonzalez M, Willner I. Angew Chem Int Ed. 2020;59:15342–15377. doi: 10.1002/anie.201907670. [DOI] [PubMed] [Google Scholar]
- 5.Stuart MAC, Huck WTS, Genzer J, Muller M, Ober C, Stamm M, Sukhorukov GB, Szleifer I, Tsukruk VV, Urban M, Winnik F, Zauscher S, Luzinov I, Minko S. Nat Mater. 2010;9:101–113. doi: 10.1038/nmat2614. [DOI] [PubMed] [Google Scholar]
- 6.Schattling P, Jochum FD, Theato P. Polym Chem. 2014;5:25–36. doi: 10.1039/C3PY00880K. [DOI] [Google Scholar]
- 7.Smith AE, Xu XW, McCormick CL. Prog Polym Sci. 2010;35:45–93. doi: 10.1016/j.progpolymsci.2009.11.005. [DOI] [Google Scholar]
- 8.Wang JA, Mei J, Qin AJ, Sun JZ, Tang BZ. Sci China Chem. 2010;53:2409–2428. doi: 10.1007/s11426-010-4106-9. [DOI] [Google Scholar]
- 9.Li CH, Liu SY. Chem Commun. 2012;48:3262–3278. doi: 10.1039/c2cc17695e. [DOI] [PubMed] [Google Scholar]
- 10.Uchiyama S, Gota C, Tsuji T, Inada N. Chem Commun. 2017;53:10976–10992. doi: 10.1039/C7CC06203F. [DOI] [PubMed] [Google Scholar]
- 11.Hu JM, Zhang GY, Ge ZS, Liu SY. Prog Polym Sci. 2014;39:1096–1143. doi: 10.1016/j.progpolymsci.2013.10.006. [DOI] [Google Scholar]
- 12.Ellison CJ, Kim SD, Hall DB, Torkelson JM. Eur Phys J E. 2002;8:155–166. doi: 10.1140/epje/i2001-10057-y. [DOI] [PubMed] [Google Scholar]
- 13.Kim S, Torkelson JM. Macromolecules. 2011;44:4546–4553. doi: 10.1021/ma200617j. [DOI] [Google Scholar]
- 14.Ellison CJ, Mundra MK, Torkelson JM. Macromolecules. 2005;38:1767–1778. doi: 10.1021/ma047846y. [DOI] [Google Scholar]
- 15.Ellison CJ, Torkelson JM. J Polym Sci Pol Phys. 2002;40:2745–2758. doi: 10.1002/polb.10343. [DOI] [Google Scholar]
- 16.Sobakinskaya E, Busch MSA, Renger T. J Phys Chem B. 2018;122:54–67. doi: 10.1021/acs.jpcb.7b09535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ciruela F. Curr Opin Biotechnol. 2008;19:338–343. doi: 10.1016/j.copbio.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 18.Luo JD, Xie ZL, Lam JWY, Cheng L, Chen HY, Qiu CF, Kwok HS, Zhan XW, Liu YQ, Zhu DB, Tang BZ (2001) Chem Commun 1740–1741. [DOI] [PubMed]
- 19.Mei J, Leung NLC, Kwok RTK, Lam JWY, Tang BZ. Chem Rev. 2015;115:11718–11940. doi: 10.1021/acs.chemrev.5b00263. [DOI] [PubMed] [Google Scholar]
- 20.Mei J, Hong YN, Lam JWY, Qin AJ, Tang YH, Tang BZ. Adv Mater. 2014;26:5429–5479. doi: 10.1002/adma.201401356. [DOI] [PubMed] [Google Scholar]
- 21.Leung NLC, Xie N, Yuan WZ, Liu Y, Wu QY, Peng Q, Miao Q, Lam JWY, Tang BZ. Chem Eur J. 2014;20:15349–15353. doi: 10.1002/chem.201403811. [DOI] [PubMed] [Google Scholar]
- 22.Cai XL, Liu B. Angew Chem Int Ed. 2020;59:9868–9886. doi: 10.1002/anie.202000845. [DOI] [PubMed] [Google Scholar]
- 23.Hu R, Qin AJ, Tang BZ. Prog Polym Sci. 2020;100:101176. doi: 10.1016/j.progpolymsci.2019.101176. [DOI] [Google Scholar]
- 24.Hu R, Leung NLC, Tang BZ. Chem Soc Rev. 2014;43:4494–4562. doi: 10.1039/C4CS00044G. [DOI] [PubMed] [Google Scholar]
- 25.Zhan RY, Pan YT, Manghnani PN, Liu B. Macromol Biosci. 2017;17:1600433. doi: 10.1002/mabi.201600433. [DOI] [PubMed] [Google Scholar]
- 26.Liu JZ, Zhong YC, Lam JWY, Lu P, Hong YN, Yu Y, Yue YN, Faisal M, Sung HHY, Williams ID, Wong KS, Tang BZ. Macromolecules. 2010;43:4921–4936. doi: 10.1021/ma902432m. [DOI] [Google Scholar]
- 27.Han T, Gui C, Lam JWY, Jiang MJ, Xie N, Kwok RTK, Tang BZ. Macromolecules. 2017;50:5807–5815. doi: 10.1021/acs.macromol.7b00973. [DOI] [Google Scholar]
- 28.Chen JR, Zhao J, Xu BJ, Yang ZY, Liu SW, Xu JR, Zhang Y, Wu YC, Lv PY, Chi ZG. Chin J Polym Sci. 2017;35:282–292. doi: 10.1007/s10118-017-1894-9. [DOI] [Google Scholar]
- 29.Li Z, Liu PC, Ji XF, Gong JY, Hu YB, Wu WJ, Wang XN, Peng HQ, Kwok RTK, Lam JWY, Lu J, Tang BZ. Adv Mater. 2020;32:1906493. doi: 10.1002/adma.201906493. [DOI] [PubMed] [Google Scholar]
- 30.Taniguchi R, Yamada T, Sada K, Kokado K. Macromolecules. 2014;47:6382–6388. doi: 10.1021/ma501198d. [DOI] [Google Scholar]
- 31.Wang YJ, Nie JY, Fang W, Yang L, Hu QL, Wang ZK, Sun JZ, Tang B. Chem Rev. 2020;120:4534–4577. doi: 10.1021/acs.chemrev.9b00814. [DOI] [PubMed] [Google Scholar]
- 32.Li B, He T, Shen X, Tang DT, Yin SC. Polym Chem. 2019;10:796–818. doi: 10.1039/C8PY01396A. [DOI] [Google Scholar]
- 33.Li J, Wang JX, Li HX, Song N, Wang D, Tang BZ. Chem Soc Rev. 2020;49:1144–1172. doi: 10.1039/C9CS00495E. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Ji XF, Li ZT, Huang FH. Adv Mater. 2017;29:1606117. doi: 10.1002/adma.201606117. [DOI] [PubMed] [Google Scholar]
- 35.Li YY, Liu SJ, Han T, Zhang HK, Chuah C, Kwok RTK, Lam JWY, Tang BZ. Mater Chem Front. 2019;3:2207–2220. doi: 10.1039/C9QM00404A. [DOI] [Google Scholar]
- 36.Ma L, Feng X, Wang S, Wang B. Mater Chem Front. 2017;1:2474–2486. doi: 10.1039/C7QM00254H. [DOI] [Google Scholar]
- 37.Chen H, Li MH. Chin J Polym Sci. 2019;37:352–371. doi: 10.1007/s10118-019-2204-5. [DOI] [Google Scholar]
- 38.Du LL, Jiang BL, Chen XH, Wang YZ, Zou LM, Liu YL, Gong YY, Wei C, Yuan WZ. Chin J Polym Sci. 2019;37:409–415. doi: 10.1007/s10118-019-2215-2. [DOI] [Google Scholar]
- 39.Dalapati S, Gu C, Jiang DL. Small. 2016;12:6513–6527. doi: 10.1002/smll.201602427. [DOI] [PubMed] [Google Scholar]
- 40.Feng HT, Yuan YX, Xiong JB, Zheng YS, Tang BZ. Chem Soc Rev. 2018;47:7452–7476. doi: 10.1039/C8CS00444G. [DOI] [PubMed] [Google Scholar]
- 41.Yuan WZ, Zhang YM. J Polym Sci Part A: Polym Chem. 2017;55:560–574. doi: 10.1002/pola.28420. [DOI] [Google Scholar]
- 42.Zhang HK, Zhao Z, McGonigal PR, Ye RQ, Liu SJ, Lam JWY, Kwok RTK, Yuan WZ, Xie JP, Rogach AL, Tang BZ. Mater Today. 2020;32:275–292. doi: 10.1016/j.mattod.2019.08.010. [DOI] [Google Scholar]
- 43.Wang RB, Yuan WZ, Zhu XY. Chin J Polym Sci. 2015;33:680–687. doi: 10.1007/s10118-015-1635-x. [DOI] [Google Scholar]
- 44.Han T, Liu LJ, Wang D, Yang JL, Tang BZ. Macromol Rapid Commun. 2020;22:2000311. doi: 10.1002/marc.202000311. [DOI] [PubMed] [Google Scholar]
- 45.Chen SJ, Wang H, Hong YN, Tang BZ. Mater Horizons. 2016;3:283–293. doi: 10.1039/C6MH00060F. [DOI] [Google Scholar]
- 46.Battistelli G, Cantelli A, Guidetti G, Manzi J, Montalti M. Wiley Interdiscip Rev-Nanomed Nanobiotechnol. 2016;8:139–150. doi: 10.1002/wnan.1351. [DOI] [PubMed] [Google Scholar]
- 47.Feng GX, Liu B. Acc Chem Res. 2018;51:1404–1414. doi: 10.1021/acs.accounts.8b00060. [DOI] [PubMed] [Google Scholar]
- 48.Calvino C, Weder C. Small. 2018;14:1802489. doi: 10.1002/smll.201802489. [DOI] [PubMed] [Google Scholar]
- 49.Zheng HY, Li CY, He CC, Dong YQ, Liu QS, Qin PF, Zeng C, Wang HL. J Mater Chem C. 2014;2:5829–5835. doi: 10.1039/C4TC00741G. [DOI] [Google Scholar]
- 50.Ye RQ, Liu YY, Zhang HK, Su HF, Zhang YL, Xu LG, Hu RR, Kwok RTK, Wong KS, Lam JWY, Goddard WA, Tang BZ. Polym Chem. 2017;8:1722–1727. doi: 10.1039/C7PY00154A. [DOI] [Google Scholar]
- 51.Yoon SJ, Chung JW, Gierschner J, Kim KS, Choi MG, Kim D, Park SY. J Am Chem Soc. 2010;132:13675–13683. doi: 10.1021/ja1044665. [DOI] [PubMed] [Google Scholar]
- 52.Robb MJ, Li W, Gergely RCR, Matthews CC, White SR, Sottos NR, Moore JS. ACS Cent Sci. 2016;2:598–603. doi: 10.1021/acscentsci.6b00198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lu XC, Li WL, Sottos NR, Moore JS. ACS Appl Mater Interfaces. 2018;10:40361–40365. doi: 10.1021/acsami.8b16454. [DOI] [PubMed] [Google Scholar]
- 54.Chen SS, Han T, Zhao Y, Luo WJ, Zhang Z, Su HB, Tang BZ, Yang JL. ACS Appl Mater Interfaces. 2020;12:4870–4877. doi: 10.1021/acsami.9b18919. [DOI] [PubMed] [Google Scholar]
- 55.Song YK, Lee TH, Lee KC, Choi MH, Kim JC, Lee SH, Noh SM, Park YI. Appl Surf Sci. 2020;511:145556. doi: 10.1016/j.apsusc.2020.145556. [DOI] [Google Scholar]
- 56.Doberenz F, Zeng K, Willems C, Zhang K, Groth T. J Mater Chem B. 2020;8:607–628. doi: 10.1039/C9TB02052G. [DOI] [PubMed] [Google Scholar]
- 57.Tang L, Jin JK, Qin AJ, Yuan WZ, Mao Y, Mei J, Sun JZ, Tang BZ (2009) Chem Commun 4974–4976. [DOI] [PubMed]
- 58.Yang JF, Gu KZ, Shi CX, Li M, Zhao P, Zhu WH. Mater Chem Front. 2019;3:1503–1509. doi: 10.1039/C9QM00147F. [DOI] [Google Scholar]
- 59.Li TZ, He SC, Qu JN, Wu H, Wu SZ, Zhao ZJ, Qin AJ, Hu RR, Tang B. J Mater Chem C. 2016;4:2964–2970. doi: 10.1039/C5TC03298A. [DOI] [Google Scholar]
- 60.Xu JR, Ji WX, Li C, Lv YF, Qiu ZJ, Gao LC, Chen EQ, Lam JWY, Tang BZ, Jiang L. Adv Opt Mater. 2018;6:1701149. doi: 10.1002/adom.201701149. [DOI] [Google Scholar]
- 61.Liu L, Wang M, Guo LX, Sun Y, Zhang XQ, Lin BP, Yang H. Macromolecules. 2018;51:4516–4524. doi: 10.1021/acs.macromol.8b00677. [DOI] [Google Scholar]
- 62.Qiu ZJ, Chu EKK, Jiang MJ, Gui C, Xie N, Qin W, Alam P, Kwok RTK, Lam JWY, Tang BZ. Macromolecules. 2017;50:7620–7627. doi: 10.1021/acs.macromol.7b01070. [DOI] [Google Scholar]
- 63.Yang Y, Zhang S, Zhang XQ, Gao LC, Wei Y, Ji Y. Nat Commun. 2019;10:3165. doi: 10.1038/s41467-019-11144-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song ZH, Lv XL, Gao LC, Jiang L. J Mater Chem C. 2018;6:171–177. doi: 10.1039/C7TC04771A. [DOI] [Google Scholar]
- 65.Bao SP, Wu QH, Qin W, Yu QL, Wang J, Liang GD, Tang BZ. Polym Chem. 2015;6:3537–3542. doi: 10.1039/C5PY00308C. [DOI] [Google Scholar]
- 66.Echeverri M, Ruiz C, Gamez-Valenzuela S, Alonso-Navarro M, Gutierrez-Puebla E, Serrano JL, Delgado MCR, Gomez-Lor B. ACS Appl Mater Interfaces. 2020;12:10929–10937. doi: 10.1021/acsami.9b21209. [DOI] [PubMed] [Google Scholar]
- 67.Lai CT, Chien RH, Kuo SW, Hong JL. Macromolecules. 2011;44:6546–6556. doi: 10.1021/ma201089j. [DOI] [Google Scholar]
- 68.Saha B, Ruidas B, Mete S, Das Mukhopadhyay C, Bauri K, De P. Chem Sci. 2020;11:141–147. doi: 10.1039/C9SC04338A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang Z, Bilalis P, Zhang HF, Gnanou Y, Hadjichristidis N. Macromolecules. 2017;50:4217–4226. doi: 10.1021/acs.macromol.7b00506. [DOI] [Google Scholar]
- 70.Luo WH, Wang GJ (2020) Adv Opt Mater 2001362.
- 71.Xie NH, Chen Y, Ye H, Li C, Zhu MQ. Front Optoelectron. 2018;11:317–332. doi: 10.1007/s12200-018-0839-4. [DOI] [Google Scholar]
- 72.Singh R, Wu HY, Dwivedi AK, Singh A, Lin CM, Raghunath P, Lin MC, Wu TK, Wei KH, Lin HC. J Mater Chem C. 2017;5:9952–9962. doi: 10.1039/C7TC03071A. [DOI] [Google Scholar]
- 73.Liu GX, Zhang YM, Zhang L, Wang CH, Liu Y. ACS Appl Mater Interfaces. 2018;10:12135–12140. doi: 10.1021/acsami.7b12822. [DOI] [PubMed] [Google Scholar]
- 74.Liu SJ, Cheng YH, Li YY, Chen M, Lam JWY, Tang BZ. ACS Nano. 2020;14:2090–2098. doi: 10.1021/acsnano.9b08761. [DOI] [PubMed] [Google Scholar]
- 75.Han T, Yao ZS, Qiu ZJ, Zhao Z, Wu KY, Wang JG, Poon AW, Lam JWY, Tang BZ. Nat Commun. 2019;10:5483. doi: 10.1038/s41467-019-13308-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zhu JC, Han T, Guo Y, Wang P, Xie HL, Meng ZG, Yu ZQ, Tang BZ. Macromolecules. 2019;52:3668–3679. doi: 10.1021/acs.macromol.9b00221. [DOI] [Google Scholar]
- 77.Han T, Deng HQ, Qiu ZJ, Zhao Z, Zhang HK, Zou H, Leung NLC, Shan GG, Elsegood MRJ, Lam JWY, Tang B. J Am Chem Soc. 2018;140:5588–5598. doi: 10.1021/jacs.8b01991. [DOI] [PubMed] [Google Scholar]
- 78.Chen S, Ma TJ, Bai J, Ma XD, Yin J, Jiang XS. Adv Sci. 2020;7:2002372. doi: 10.1002/advs.202002372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu WB. Chem. 2018;4:1762–1764. doi: 10.1016/j.chempr.2018.07.017. [DOI] [Google Scholar]
- 80.Zhao Z, Zhang HK, Lam JWY, Tang BZ. Angew Chem Int Ed. 2020;59:9888–9907. doi: 10.1002/anie.201916729. [DOI] [PubMed] [Google Scholar]
- 81.Liu XL, Li MG, Han T, Cao B, Qiu ZJ, Li YY, Li QY, Hu YB, Liu ZY, Lam JWY, Hu XL, Tang BZ. J Am Chem Soc. 2019;141:11259–11268. doi: 10.1021/jacs.9b04757. [DOI] [PubMed] [Google Scholar]
- 82.Liu WW, Yu HK, Hu RR, Xu T, Lun YP, Gan JL, Xu SH, Yang ZM, Tang BZ. Small. 2020;16:1907074. doi: 10.1002/smll.201907074. [DOI] [PubMed] [Google Scholar]
- 83.Nakasha K, Fukuhara G. ACS Appl Polym Mater. 2020;2:2303–2310. doi: 10.1021/acsapm.0c00272. [DOI] [Google Scholar]
- 84.Sun NW, Su KX, Zhou ZW, Tian XZ, Wang DM, Vilbrandt N, Fery A, Lissel F, Zhao XG, Chen CH. J Mater Chem C. 2019;7:9308–9315. doi: 10.1039/C9TC02001B. [DOI] [Google Scholar]
- 85.Sun NW, Su KX, Zhou ZW, Yu Y, Tian XZ, Wang DM, Zhao XG, Zhou HW, Chen CH. ACS Appl Mater Interfaces. 2018;10:16105–16112. doi: 10.1021/acsami.8b01624. [DOI] [PubMed] [Google Scholar]
- 86.Sun NW, Su KX, Zhou ZW, Tian XZ, Zhao JH, Chao DM, Wang DM, Lissel F, Zhao XG, Chen CH. Macromolecules. 2019;52:5131–5139. doi: 10.1021/acs.macromol.9b00079. [DOI] [Google Scholar]
- 87.Cheng SW, Han T, Huang TY, Tang BZ, Liou GS. Polym Chem. 2018;9:4364–4373. doi: 10.1039/C8PY00809D. [DOI] [Google Scholar]
- 88.Zhang XD, Chen XK, Jiang YW, Ma NN, Xia LY, Cheng XT, Jia HR, Liu PD, Gu N, Chen Z, Wu FG. ACS Appl Mater Interfaces. 2018;10:10601–10606. doi: 10.1021/acsami.8b00207. [DOI] [PubMed] [Google Scholar]
- 89.Yang CH, Ni X, Mao D, Ren CH, Liu JJ, Gao Y, Ding D, Liu JF. Biomaterials. 2019;188:107–117. doi: 10.1016/j.biomaterials.2018.10.009. [DOI] [PubMed] [Google Scholar]
- 90.Liu ZT, Xue WX, Cai ZX, Zhang GX, Zhang DQ. J Mater Chem. 2011;21:14487–14491. doi: 10.1039/c1jm12400e. [DOI] [Google Scholar]
- 91.Cao W, Zhang XL, Miao XM, Yang ZM, Xu HP. Angew Chem Int Ed. 2013;52:6233–6237. doi: 10.1002/anie.201300662. [DOI] [PubMed] [Google Scholar]
- 92.Ma N, Xu HP, An LP, Li J, Sun ZW, Zhang X. Langmuir. 2011;27:5874–5878. doi: 10.1021/la2009682. [DOI] [PubMed] [Google Scholar]
- 93.Li TY, Pan SJ, Zhuang H, Gao SQ, Xu HP. ACS Appl Bio Mater. 2020;3:1283–1292. doi: 10.1021/acsabm.9b01172. [DOI] [PubMed] [Google Scholar]
- 94.Iasilli G, Battisti A, Tantussi F, Fuso F, Allegrini M, Ruggeri G, Pucci A. Macromol Chem Phys. 2014;215:499–506. doi: 10.1002/macp.201300698. [DOI] [Google Scholar]
- 95.Cheng YH, Liu SJ, Song FY, Khorloo M, Zhang HK, Kwok RTK, Lam JWY, He ZK, Tang BZ. Mater Horiz. 2019;6:405–411. doi: 10.1039/C8MH01092G. [DOI] [Google Scholar]
- 96.Sun JM, Wang JG, Chen M, Pu X, Wang G, Li L, Chen GY, Cai YJ, Gu XG, Tang BZ. Chem Mater. 2019;31:5683–5690. doi: 10.1021/acs.chemmater.9b01611. [DOI] [Google Scholar]
- 97.Wang GJ, Zhang RC, Xu C, Zhou RY, Dong J, Bai HT, Zhan XW. ACS Appl Mater Interfaces. 2014;6:11136–11141. doi: 10.1021/am503171s. [DOI] [PubMed] [Google Scholar]
- 98.Tavakoli J, Gascooke J, Xie N, Tang B, Tang YH. ACS Appl Polym Mater. 2019;1:1390–1398. doi: 10.1021/acsapm.9b00173. [DOI] [Google Scholar]
- 99.Khorloo M, Cheng YH, Zhang HK, Chen M, Sung HHY, Williams ID, Lam JWY, Tang BZ. Chem Sci. 2020;11:997–1005. doi: 10.1039/C9SC04239C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yao W, Tebyetekerwa M, Bian XH, Li WL, Yang SY, Zhu MF, Hu R, Wang ZM, Qin AJ, Tang BZ. J Mater Chem C. 2018;6:12849–12857. doi: 10.1039/C8TC04175J. [DOI] [Google Scholar]
- 101.Jiang XY, Gao HF, Zhang XQ, Pang JH, Li YQ, Li K, Wu YC, Li SZ, Zhu J, Wei Y, Jiang L. Nat Commun. 2018;9:3799. doi: 10.1038/s41467-018-06101-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tao L, Li ML, Yang KP, Guan Y, Wang P, Shen ZH, Xie HL. ACS Appl Mater Interfaces. 2019;11:15051–15059. doi: 10.1021/acsami.9b01476. [DOI] [PubMed] [Google Scholar]
- 103.Huang HC, Qiu ZJ, Han T, Kwok RTK, Lam JWY, Tang BZ. ACS Macro Lett. 2017;6:1352–1356. doi: 10.1021/acsmacrolett.7b00872. [DOI] [PubMed] [Google Scholar]
- 104.Cheng YH, Wang JG, Qiu ZJ, Zheng XY, Leung NLC, Lam JWY, Tang BZ. Adv Mater. 2017;29:1703900. doi: 10.1002/adma.201703900. [DOI] [PubMed] [Google Scholar]
- 105.Lu W, Xiao P, Gu JC, Zhang JW, Huang YJ, Huang Q, Chen T. Sens Actuators B Chem. 2016;228:551–556. doi: 10.1016/j.snb.2016.01.096. [DOI] [Google Scholar]
- 106.Zhang DP, Fan YJ, Chen H, Trepout S, Li MH. Angew Chem Int Ed. 2019;58:10260–10265. doi: 10.1002/anie.201905089. [DOI] [PubMed] [Google Scholar]
- 107.Qiu L, Zhang HR, Wang B, Zhan Y, Xing CF, Pan CY. ACS Appl Mater Interfaces. 2020;12:1348–1358. doi: 10.1021/acsami.9b18792. [DOI] [PubMed] [Google Scholar]
- 108.Hu YB, Han T, Yan N, Liu JK, Liu XL, Wang WX, Lam JWY, Tang BZ. Adv Funct Mater. 2019;29:1902240. doi: 10.1002/adfm.201902240. [DOI] [Google Scholar]
- 109.Chen YJ, Han HJ, Tong HX, Chen TT, Wang HB, Ji J, Jin Q. ACS Appl Mater Interfaces. 2016;8:21185–21192. doi: 10.1021/acsami.6b06071. [DOI] [PubMed] [Google Scholar]
- 110.Qi QK, Li Y, Yan XY, Zhang FL, Jiang S, Su J, Xu B, Fu XQ, Sun LK, Tian WJ. Polym Chem. 2016;7:5273–5280. doi: 10.1039/C6PY01072E. [DOI] [Google Scholar]
- 111.Zhao YM, Zhu W, Ren LX, Zhang K. Polym Chem. 2016;7:5386–5395. doi: 10.1039/C6PY01009A. [DOI] [Google Scholar]
- 112.He N, Chen ZJ, Yuan J, Zhao L, Chen MH, Wang T, Li XH. ACS Appl Mater Interfaces. 2017;9:32534–32544. doi: 10.1021/acsami.7b09519. [DOI] [PubMed] [Google Scholar]
- 113.Ma HH, Zhang AD, Zhang XM, Zhao HT, Cui Z, Fu P, Liu MY, Zhou SZ, Pang XC, Qiao XG (2020) Sensor Actuators B Chem 304
- 114.Huang W, Bender M, Seehafer K, Wacker I, Schroder RR, Bunz UHF. Macromol Rapid Commun. 2019;40:1800774. doi: 10.1002/marc.201800774. [DOI] [PubMed] [Google Scholar]
- 115.Dong WH, Wu HQ, Chen M, Shi Y, Sun JZ, Qin AJ, Tang BZ. Polym Chem. 2016;7:5835–5839. doi: 10.1039/C6PY01202G. [DOI] [Google Scholar]
- 116.Tian T, Hu RR, Tang BZ. J Am Chem Soc. 2018;140:6156–6163. doi: 10.1021/jacs.8b02886. [DOI] [PubMed] [Google Scholar]
- 117.Shan YR, Yao WJ, Liang ZQ, Zhu LH, Yang SB, Ruan ZJ. Dyes Pigm. 2018;156:1–7. doi: 10.1016/j.dyepig.2018.03.060. [DOI] [Google Scholar]
- 118.Ishiwari F, Hasebe H, Matsumura S, Hajjaj F, Horii-Hayashi N, Nishi M, Someya T, Fukushima T. Sci Rep. 2016;6:24275. doi: 10.1038/srep24275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Morishima K, Ishiwari F, Matsumura S, Fukushima T, Shibayama M. Macromolecules. 2017;50:5940–5945. doi: 10.1021/acs.macromol.7b00883. [DOI] [Google Scholar]
- 120.Wang YZ, Bin X, Chen XH, Zheng SY, Zhang YM, Yuan WZ. Macromol Rapid Commun. 2018;39:1800528. doi: 10.1002/marc.201800528. [DOI] [PubMed] [Google Scholar]
- 121.Gao MX, Wu Y, Chen B, He BR, Nie H, Li TY, Wu FP, Zhou WJ, Zhou J, Zhao ZJ. Polym Chem. 2015;6:7641–7645. doi: 10.1039/C5PY01458A. [DOI] [Google Scholar]
- 122.He BZ, Su HF, Bai TW, Wu YW, Li SW, Gao M, Hu RR, Zhao ZJ, Qin AJ, Ling J, Tang BZ. J Am Chem Soc. 2017;139:5437–5443. doi: 10.1021/jacs.7b00929. [DOI] [PubMed] [Google Scholar]
- 123.Chen T, Yin H, Chen ZQ, Zhang GF, Xie NH, Li C, Gong WL, Tang BZ, Zhu MQ. Small. 2016;12:6547–6552. doi: 10.1002/smll.201601410. [DOI] [PubMed] [Google Scholar]
- 124.Zhou H, Chua MH, Tang BZ, Xu JW. Polym Chem. 2019;10:3822–3840. doi: 10.1039/C9PY00322C. [DOI] [Google Scholar]
- 125.Ryu SH, Lee DH, Ko YJ, Lee SM, Kim HJ, Ko KC, Son SU. Macromol Chem Phys. 2019;220:1900157. doi: 10.1002/macp.201900157. [DOI] [Google Scholar]
- 126.Wu YW, Qin AJ, Tang BZ. Chin J Polym Sci. 2017;35:141–154. doi: 10.1007/s10118-017-1882-0. [DOI] [Google Scholar]
- 127.Han T, Zhang Y, He BZ, Lam JWY, Tang BZ. Polymers. 2018;10:821. doi: 10.3390/polym10080821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhou H, Wang XB, Lin TT, Song J, Tang BZ, Xu JW. Polym Chem. 2016;7:6309–6317. doi: 10.1039/C6PY01358A. [DOI] [Google Scholar]
- 129.Liang GD, Ren F, Gao HY, Zhu FM, Wu Q, Tang BZ. J Mater Chem A. 2017;5:2115–2122. doi: 10.1039/C6TA08235A. [DOI] [Google Scholar]
- 130.Zhou YS, Zhang L, Gao HY, Zhu FM, Ge ML, Liang GD. Sens Actuators B Chem. 2019;283:415–425. doi: 10.1016/j.snb.2018.12.018. [DOI] [Google Scholar]
- 131.Qi CX, Zheng C, Hu RR, Tang BZ. ACS Macro Lett. 2019;8:569–575. doi: 10.1021/acsmacrolett.9b00297. [DOI] [PubMed] [Google Scholar]
- 132.Zhou YS, Gao HY, Zhu FM, Ge ML, Liang GD. J Hazard Mater. 2019;368:630–637. doi: 10.1016/j.jhazmat.2019.01.097. [DOI] [PubMed] [Google Scholar]
- 133.Chen JL, Chen XJ, Huang QY, Li WL, Yu QX, Zhu LJ, Zhu TW, Liu SW, Chi ZG. ACS Appl Mater Interfaces. 2019;11:32689–32696. doi: 10.1021/acsami.9b10237. [DOI] [PubMed] [Google Scholar]
- 134.Hirose A, Tanaka K, Yoshii R, Chujo Y. Polym Chem. 2015;6:5590–5595. doi: 10.1039/C5PY00055F. [DOI] [Google Scholar]
- 135.Zhang GY, Liao QB, Liu YF, Wang L, Gou HL, Ke C, Huang X, Xi K, Jia XD. Nanoscale. 2018;10:5503–5514. doi: 10.1039/C7NR09356J. [DOI] [PubMed] [Google Scholar]
- 136.Wang XL, Li JJ, Yan Q, Chen YR, Fan AP, Wang Z, Zhao YJ. Macromol Biosci. 2018;18:1700339. doi: 10.1002/mabi.201700339. [DOI] [PubMed] [Google Scholar]
- 137.Chen C, Ni X, Jia SR, Liang Y, Wu XL, Kong DL, Ding D. Adv Mater. 2019;31:1904914. doi: 10.1002/adma.201904914. [DOI] [PubMed] [Google Scholar]
- 138.Wang LY, Yang LL, Zhu LH, Cao DR, Li L. Sens Actuators B Chem. 2016;231:733–743. doi: 10.1016/j.snb.2016.03.075. [DOI] [Google Scholar]
- 139.Lee TH, Song YK, Park SH, Park YI, Noh SM, Kim JC. Appl Surf Sci. 2018;434:1327–1335. doi: 10.1016/j.apsusc.2017.11.219. [DOI] [Google Scholar]
- 140.Zhang Z, Hadjichristidis N. ACS Macro Lett. 2018;7:886–891. doi: 10.1021/acsmacrolett.8b00329. [DOI] [PubMed] [Google Scholar]
- 141.Zhuang WH, Xu YY, Li GC, Hu J, Ma BX, Yu T, Su X, Wang YB. ACS Appl Mater Interfaces. 2018;10:18489–18498. doi: 10.1021/acsami.8b02890. [DOI] [PubMed] [Google Scholar]
- 142.Li YM, Wu Q, Kang MM, Song N, Wang D, Tang BZ. Biomaterials. 2020;232:119749. doi: 10.1016/j.biomaterials.2019.119749. [DOI] [PubMed] [Google Scholar]
- 143.Wu Y, Hu JL, Huang HH, Li J, Zhu Y, Tang BZ, Han JP, Li LB. J Polym Sci Pol Phys. 2014;52:104–110. doi: 10.1002/polb.23399. [DOI] [Google Scholar]
- 144.Wang HR, Xing H, Gong JY, Zhang HK, Zhang J, Wei PF, Yang GJ, Lam JWY, Lu R, Tang BZ. Mater Horiz. 2020;7:1566–1572. doi: 10.1039/D0MH00447B. [DOI] [Google Scholar]