

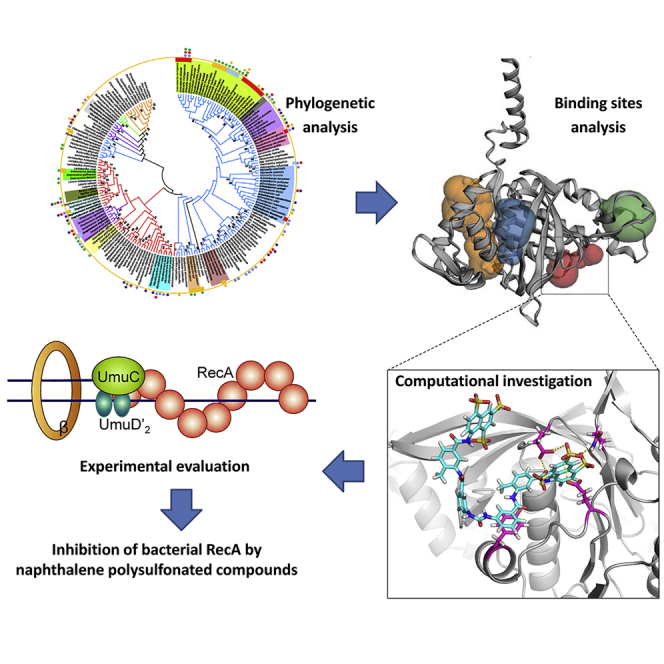
Summary
As a promising target for alternative antimicrobials, bacterial recombinase A (RecA) protein has attracted much attention for its roles in antibiotic-driven SOS response and mutagenesis. Naphthalene polysulfonated compounds (NPS) such as suramin have previously been explored as antibiotic adjuvants targeting RecA, although the underlying structural bases for RecA-ligand interactions remain obscure. Based on our in silico predictions and documented activity of NPS in vitro, we conclude that the analyzed NPS likely interact with Tyr103 (Y103) and other key residues in the ATPase activity center (pocket A). For validation, we generated recombinant RecA proteins (wild-type versus Y103 mutant) to determine the binding affinities for RecA protein interactions with suramin and underexamined NPS in isothermal titration calorimetry. The corresponding dissociation constants (Kd) ranged from 11.5 to 18.8 μM, and Y103 was experimentally shown to be critical to RecA-NPS interactions.
Subject areas: Biochemistry, Microbiology, Molecular Microbiology
Graphical Abstract
Highlights
-
•
RecA is conserved among major bacterial pathogens but deviates from mammalian Rad51
-
•
Naphthalene polysulfonated compounds (NPS) block RecA ATPase and SOS response
-
•
Selected NPS bind recombinant RecA protein with Kd values in micromolar range
-
•
Tyr103 is an amino acid residue critical to RecA-NPS interactions
Biochemistry; Microbiology; Molecular Microbiology
Introduction
Over the past few decades, a draining antimicrobial pipeline and rapid emergence of multidrug-resistant (MDR) organisms have precipitated a global crisis in public health. To galvanize response, the World Health Organization (WHO) published a “global priority pathogens” list for R&D of new antibiotics (2017) (Tacconelli et al., 2018a). Within this list, carbapenem-resistant Gram-negative “superbugs” including MDR Acinetobacter baumannii, Pseudomonas aeruginosa, Escherichia coli, and Klebsiella pneumoniae have already gained notoriety in health care-associated infections (HAIs) (Tacconelli et al., 2018b) by virtue of their extraordinary ability to develop resistance against broad-spectrum antibiotics (Tacconelli et al., 2018b). These nosocomial pathogens cause significant morbidity and mortality among patients with severe injury or compromised immunity. Their infections also greatly increase medical costs and burdens by complicating or foiling fundamental health care practice such as mechanical ventilation, organ transplantation, HIV/AIDS therapy, anti-cancer chemotherapy, and child delivery, among others. There is thus a pressing need for alternative anti-infective solutions less prone to induce selective pressure, in addition to new antibiotics (Brown and Wright, 2016; Seifert et al., 2018).
One alternative anti-infective strategy involves the use of adjuvants and anti-virulence agents, which impose minimized selective pressure in bacterial pathogens compared with conventional antibiotics. The adjuvant approach seeks to exploit novel agents or repurpose existing drugs as antibiotic adjuvants, which themselves do not kill bacteria but enhance the effectiveness of a co-administered antibiotic by blocking antimicrobial resistance (AMR) mechanisms (Wright, 2016; Brown, 2015; Zheng et al., 2016). Examples that have entered into clinical use include a closely allied approach that employs anti-virulence agents to limit a pathogen's infectivity and thus the symptoms caused by its virulence factors (Zheng et al., 2016). Recently, our group has successfully developed small-molecule inhibitors targeting the eukaryotic-like Ser/Thr phosphatase (Stp1) in Staphylococcus aureus (Zheng et al., 2016). Notably, the Stp1 inhibitor aurintricarboxylic acid evidently blunted the expression of staphylococcal virulence in mice, without affecting in vitro growth of S. aureus (Zheng et al., 2015).
The bacterial recombinase A (RecA) protein represents an attractive target for adjuvant development in MDR pathogens, as challenges by exogenous stressors such as UV irradiation, sublethal-dose antibiotics, and host respiratory bursts often converge on the generation of DNA-damaging reactive oxygen/nitrogen species (Hu et al., 2015, 2016; Van Acker and Coenye, 2017). RecA critically regulates an elaborate ATPase-dependent DNA repair process known as SOS response. In the case of unresolved antibiotic stress, RecA hyperactivity could give rise to aberrant expression of inducible error-prone repair systems such as polymerase V (Schlacher et al., 2005, 2006) that promote mutations (Bellio et al., 2017), genomic instability, and ultimately mutagenesis-fueled resistance acquisition (Kohanski et al., 2007). Among naphthalene polysulfonated compounds (NPS), suramin has been found to potently inhibit the ATPase activity of bacterial RecA protein, with IC50 values in the low micromolar range (Nautiyal et al., 2014). Experimentally, suramin displayed efficacy in inhibiting RecA activity and SOS response in Mycobacterium tuberculosis and Mycobacterium smegmatis (Nautiyal et al., 2014). Despite efforts to develop several RecA inhibitors, the binding sites for these inhibitors remain obscure due to the functional versatility and multiple binding positions within RecA. In addition, research on the adjuvant mode of action by NPS has been hampered by a paucity of understanding about the structural details of RecA-suramin interactions.
To provide a theoretical foundation for drug discovery of RecA-targeting adjuvants with novel scaffolds, it is logically desirable to attempt to bridge the abovementioned gap via a combinatorial approach that encompasses molecular docking, dynamics simulation, and experimental validation. The utility of computational approaches to predicting binding sites can be further augmented by a judicious choice of energy-based detection algorithms, 3D structure- or geometry-based algorithms, etc. (Leis et al., 2010). Recently, our group successfully applied these approaches to map out RecA protein-binding determinants and predict binders among curcuminoid compounds (Zhou et al., 2019). In this present study, the phylogenetically relevant features within the RecA protein sequence, probabilistically favorable pocketness in the RecA protein structure, and predicted mechanisms underlying interactions between RecA and NPS of interest were scrutinized. To illustrate, the commercially available suramin was predicted to be a generic RecA inhibitor among untested bacterial pathogens and verified by quantitative real-time PCR (RT-qPCR) in terms of RecA-controlled transcription in E. coli. Additionally, we also performed isothermal titration calorimetry (ITC) to assess binding affinities for interactions between recombinant RecA proteins (wild-type versus mutant) and selected NPS including suramin. The amino acid residue Tyr103 (Y103) in E. coli RecA was critically implicated in RecA-NPS interactions. This work should prove valuable to future studies on the construction and application of structure-based virtual screening protocols that aim to accelerate the discovery of potent small-molecule candidate inhibitors against bacterial RecA, as a potential key regulator of AMR development.
Results and discussion
Phylogenetic analysis
Thus far, no specific study has been devoted to analyzing RecA protein sequences within bacterial species, especially clinically significant pathogens in AMR contexts. As a first step toward evaluating the feasibility and generality of RecA as a drug target, we performed phylogenetic analysis on the evolutionary relationship and sequence features of RecA-like proteins (that is, RecA/Rad51/RadA) among eukaryotic and prokaryotic species, with particular attention to MDR bacterial pathogens high in importance in the WHO “global priority pathogens” list (Tacconelli et al., 2018a). Extensive searches for RecA/Rad51/RadA proteins were performed with sequences extracted from the NCBI database, which were then subjected to multiple phylogenetic analyses. In this study, Rad51 protein sequences from 10 eukaryotic species and RadA protein sequences from four archaebacterial species were included for comparison.
Phylogenetic relationships depicted in Figure 1 were inferred by the neighbor-joining (NJ) method (Saitou and Nei, 1987). A bootstrap consensus tree inferred from 1,000 replicates is taken to represent the evolutionary history of the taxa analyzed (Felsenstein 1985). Branches corresponding to partitions reproduced in less than 50% bootstrap replicates were collapsed. Evolutionary distances were computed by using the Poisson correction method and are denoted in units as number of amino acid substitutions per site. The analysis involved amino acid sequences of 185 RecA and RecA-like proteins (see sequence details in Tables S4, S5, and S6). All positions containing gaps and missing data were eliminated. Evolutionary analyses were conducted with MEGA7 (Kumar et al., 2016). Maximum-likelihood, minimum-evolution, and NJ consensus trees for RecA protein sequences are topologically congruent on most clades (as depicted in the Supplemental information, Figures S3 and S4). Percent bootstrap values above 70 are given. Importantly, we annotated the pathogens' infection sites, based on extensive literature searches (See details in the Tables S2, S3, and S4), with an emphasis on species known to be causative agents of nosocomial infections. Attributes such as Gram status and families are phylogenetically grouped together, whereas other attributes such as infection sites and antibiotic susceptibilities are dispersed. In addition, we analyzed representative antibiotic susceptibility profiles of clinical isolates of major HAI pathogens tested in this study, whose results are presented in two heatmaps (Figures S6 and S7).
Figure 1.

Phylogenetic tree of RecA and RecA-like proteins among bacteria and eukaryotes, with annotations on antibiotic resistance and infection modes for bacterial pathogens
The broad categories of species, relative importance on the WHO “global priority pathogens” list, typical antibiotic resistance, and documented infection sites are indicated with different color branches, bands, closed circles, and dots, respectively. Major bacterial families were indicated as different color shades. Bacteria labeled in bold are known nosocomial pathogens. Detailed information used for annotations is summarized in the Supplemental information (Tables S4, S5, S6, and S9).
Phylogenetically, the evolution of RecA/Rad51 proteins includes both vertical and horizontal gene transfers, whereas the eukaryotic Rad51 proteins are evolutionally close to the archaebacterial RecA-like protein and a branch of Gram-variable bacterial RecA. RecA proteins from several families of pathogenic bacteria (including Enterobacteriaceae, Moraxellaceae, and Pseudomonadaceae) with critical and high WHO priorities were principally clustered into a major branch of Gram-negative bacteria, which is very distant from the eukaryotic Rad51s. RecA-like protein-mediated recombination was reported to be a major reason for the maintenance of large plant chloroplast and mitochondrial genomes, as efficient DNA repair by homologous recombination presumably reduces deleterious effects of mutations.(Xu et al., 2016). On the other hand, the mitochondrial RecA was believed to be lost in the ancestor of animals and fungi (Lin et al., 2006). Phylogenetic relationships of RecA protein between different species indicate that the development of bacterial RecA-selective inhibitor with little or no off-target effects on human Rad51 is feasible. A lack of mitochondrial RecA in human and animals also means that potential adjuvants targeting RecA should be safe for human use.
Structural alignment of RecA-like proteins
To illustrate the structural details of the ATPase core and evaluate the possibility of selective inhibition therein, we performed multiple alignment of amino acid sequences (Figure 2A) and structural alignment of RecA and Rad51 X-ray crystal structures from selected species (Figures 2B, 2C, and S1 in the Supplemental information). In multiple alignment analysis of amino acid sequence, as shown in Figure 2A, Walker A (amino acid residues 66–73), Walker B (amino acid residues 139–144), L1 (amino acid residues 157–164) (Malkov and Camerini-Otero, 1995), and L2 (amino acid residues 195–209) (Malkov and Camerini-Otero, 1995) were marked with red, blue, green, and black boxes, respectively. RecA proteins of 20 representative pathogenetic eubacterial species share a highly conserved core domain (yellow in Figure 2A), in particular, with respect to Walker A and Walker B, which are two highly conserved consensus motifs within the RecA/RAD51 ATPase domain (Walker et al., 1982). Walker A is nestled within the ATPase activity center of RecA (Nahrstedt et al., 2005). Key residues Glu68 and Lys72 in Walker A are pivotal in conferring the RecA its ATP-binding ability and interacting with γ-phosphate of the nucleotide, thus critically regulating ATP hydrolysis (Story and Steitz, 1992). Likewise, Walker B also containing resides within RecA's ATPase activity core and its key residue Asp144 reportedly determines ATPase activity for it involved in ATP-Mg2+ binding (Story and Steitz, 1992). Walker A is relatively conserved from bacteria to eukaryotic species, whereas a considerable variability exists in the Walker B motif of RecA/Rad51 across bacterial species, with a stretch of bulky, hydrophobic amino acids followed by a negatively charged residue as an invariant feature (Koonin, 1993).
Figure 2.

Structural comparisons of RecA and Rad51 crystal structures, and multiple alignment analysis on pivotal regions of amino acid sequences in the RecA/Rad51 superfamily
(A) Multiple alignment of RecA amino acid sequences from 20 bacterial species and Rad51 from 4 eukaryotic species. Domain structures of representative RecA proteins are schematically depicted.
(B) 3D protein structure alignment of E. coli RecA (gray) and M. tuberculosis RecA (cyan).
(C) 3D protein structure alignment of E. coli RecA (gray) and human Rad51 (yellow).
(D) Degree of conservation of sequence homology.
(E) The predicted binding regions are shown as colored spheres: pocket A (red), pocket B (orange), pocket C (blue), pocket D (green).
(F) The ATPase activity center predicted by 3DLigandSite.
Ideally, RecA-specific inhibitors should exhibit adjuvant activity by blocking the ATPase domain of bacterial RecA without inhibiting the eukaryotic RecA homolog Rad51. For eukaryotic RadA and Rad51 and prokaryotic RecA filament, crystal structures are available for the extended form (active state) containing an ATP site at the subunit interface bound with ATP or ATP analogs. Also available are crystal structures corresponding to the compressed form (inactive state) containing an ATP site bound with ADP (Xing and Bell, 2004a, 2004b). The ATPase activity centers within the X-ray structures of E. coli RecA (gray, PDB 3CMT) (Chen et al., 2008) and M. tuberculosis RecA (cyan, PDB 1MO3) (Datta et al., 2003) are almost identical, with all the key residues conserved (Figure 2B). In contrast, due to evident disparity between RecA (gray, PDB 3CMT) and Rad51 (yellow, PDB 5H1B) (Xing and Bell, 2004a, 2004b) in 3D structures, as depicted in Figure 2C, bacterial RecA behaves differently from human Rad51 (Tombline and Fishel, 2002). For instance, unlike bacterial RecA, human Rad51 lacks catalytic efficiency and magnitude of ATP-induced cooperativity (Tombline et al., 2002a). Another difference is that the RecA-ssDNA complex could be stabilized by ATP-γS and destabilized by ADP. Conversely, Rad51-ssDNA complex can be stabilized by ADP or ATP and destabilized by ATP-γS (Tombline et al., 2002b). RecA in the active state binds ssDNA or dsDNA in the presence of ATP (Egelman and Stasiak, 1986). Segment of RecA-DNA-ATP-γS filaments identified by Edward et al. has been suggested to represent two distinct states of RecA (Yu et al., 2001), but later these two states was elaborated to be separable for the slow hydrolysis of ATP-γS, randomly occurring within a filament (VanLoock et al., 2003). Mapping of functional sites on RecA based on degree of conservation of sequence homology as analyzed by ConSurf and the data are depicted with the NGL viewer (Glaser et al., 2003). Yellow parts stand for lack of information (Figure 2D). The locations of ligand-binding sites on RecA are predicted by the prediction server CASTp 3.0 (Dundas et al., 2006; Chen et al., 2018) (Figure 2E). The ATPase activity center (pocket A) is the only binding site predicted by 3DLigandSite (Wass et al., 2010). Figure 2F depicts the view from different directions of the same structure.
Binding mechanisms of naphthalene polysulfonated compounds
Thus far, there have been no attempts to decipher mechanisms underlying the interactions between RecA and NPS via structural or in silico approaches. To unravel details of how NPS interact with RecA, the binding sites on RecA were first computationally predicted by using various algorithms. Compounds that bind RecA nucleoprotein filament in its active conformation can serve as competitive inhibitors of RecA ATPase hydrolysis, whereas compounds that bind the inactive RecA conformation can facilitate the dissociation of DNA from RecA (Wigle et al., 2006). The identification of novel small molecules or peptides targeting RecA could lead to discovery of potential antibiotic adjuvants and a better understanding of the structural determinants for binding between RecA and its conformationally selective ligands (Wigle et al., 2006).
The loop part of RecA in the inactive state is highly flexible in the RecA filament, which has proved difficult to resolve (Yu et al., 2004). Previously, the loop region of much of the DNA-free RecA X-ray crystal structures in the ssDNA binding part has been omitted (Figure S1) (Story and Steitz, 1992; Xing and Bell, 2004a, 2004b; Singleton et al., 2002). In this study, we chose the RecA crystal structure that represents the active state of RecA resolved by Chen et al. (2008) for in silico studies. As noted, Walker A and Walker B are two highly conserved motifs in pocket A. Pocket A is the ATPase center of RecA (Figure 2E), and the underlined residues in Table S8 are the key residues in ATPase function (Story and Steitz, 1992; Chen et al., 2008). Specifically, the highly conserved consensus motifs Walker A (containing Glu68, Ser69, Ser70, Gly71, Lys72, Thr73) and Walker B (containing Asp144) all lie within pocket A. In addition, the catalytic Glu96 (responsible for activating water molecule for nucleophilic attack on γ-phosphate), Gln194 (a key allosteric residue), and Tyr103 (required for stabilization of ATP molecule) are all located within pocket A (Story and Steitz, 1992; VanLoock et al., 2003; Xing and Bell, 2004a, 2004b; Singleton et al., 2002). Pocket C putatively participates in dsDNA binding (Chen et al., 2008; Prabu et al., 2008), whereas pocket D has been found to be an interaction site for ssDNA binding (Shinohara et al., 2015).
Since the discovery of RecA protein, a number of RecA inhibitors (Table 1), including several NPS such as suramin (Figure S2), Congo red, and bis-ANS (4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid), have been reported. Most of them are small molecule inhibitors, whereas several peptide inhibitors have also been proved to be effective RecA inhibitors. However, the structural determinants in RecA that govern its interactions with putative RecA inhibitors have not been examined in detail.
Table 1.
Summary of reported RecA inhibitors
| Compounds | Classification | M .W. (Da) | Target organism | Methods | RecA inhibition (IC50) | Ref. |
|---|---|---|---|---|---|---|
| Congo red (cpd03) | NPS | 1290 | E. coli | Fluorescent ATPase assay | 2 μM | (Wigle and Singleton, 2007) |
| bis-ANS (cpd02) | NPS | 594 | E. coli | Fluorescent ATPase assay | ND | (Wigle and Singleton, 2007) |
| Suramin (cpd01) | NPS | 652 | M. tb | In vitro RecA ATPase; in bacteria | ∼1–2 μM | (Nautiyal et al., 2014; Wigle and Singleton, 2007) |
| Fe/Cu-PcTs (cpd04) | PTA | ∼900 | E. coli | In vitro RecA ATPase; in mice | ND | (Alam et al., 2016) |
| Curcumin | Natural phenols | 368 | E. coli | PCR assay | ND | (Bellio et al., 2014) |
| Sphaerophorins | Lichen metabolites | ∼300 | E. coli | Fluorescent ATPase assay | ≥14.2 μM | (Bellio et al., 2017) |
| 4 × 101 | Peptides | ∼1-2k | E. coli | In vitro RecA ATPase | ND | (Yakimov et al., 2017) |
| INPEP | Peptides | ∼1-2k | E. coli | In vitro RecA ATPase | 3 μM | (Cline et al., 2007) |
| Ara-adenosines | Nucleotide analogs | ∼400 | E. coli | In vitro RecA ATPase | ND | (Wigle et al., 2006) |
| N6-(1-naphthyl)-ADP | Nucleotide analogs | ∼400 | E. coli | PCR assay | ND | (Lee et al., 2005) |
| Zn2+, Cu2+, Hg2+ | Metal ions | 64–200 | E. coli | In vitro RecA ATPase | ND | (Lee and Singleton, 2004) |
NPS, naphthalene polysulfonated compounds; M.W., molecular weight; PTA, phthalocyanine tetrasulfonic acid analogs; ND, not determined; M. tb, Mycobacterium tuberculosis.
To appraise the robustness of currently available docking algorithms for estimating binding free energies that characterize binding between RecA and its ligands, we selected AutoDock and LeDock, and scrutinized their performance with respect to various pockets on RecA. Our results support a good correlation between the experimental inhibitory activities and binding free energies over a training set tested (Figure 3). Several reported RecA inhibitors (cpd01–cpd04 and cpd12) were predicted by the algorithms as the most potent inhibitors (displayed in bold in Table I of Figure 3). In contrast, those compounds determined to lack inhibitory activities against RecA (cpd05–cpd08) have the lowest binding scores. In addition, compounds cpd09–cpd11 were reported to be active in a screening study for RecA targeting inhibitors (Wigle et al., 2009), whereas cpd13 and cpd14 were reported to have less potency than cpd12 in blocking RecA-mediated SOS response (Lee et al., 2005).
Figure 3.

Docking analysis with reported RecA inhibitors or inactive compounds as a training set
Table I depicts the binding free energies estimated by AutoDock Vina (LeDock). Table II depicts the binding free energies between the RecA protein and representative compounds.
To further characterize the binding potency of key molecules, we optimized the complexes of RecA and representative compounds with Amber16 and then carried on a short MD simulation with these complexes; finally, the binding energies were predicted with molecular mechanics/generalized Born surface area (MM-GBSA) calculations. Detailed MM-GBSA results are displayed in Table II of Figure 3. A significant difference was observed between the known RecA inhibitors (cpd01 and cpd02) and known compounds (cpd05 and cpd06) that show no inhibitory activities against RecA. Results for docking and the MM-GBSA calculations are in good agreement, which suggests that these computational approaches could be useful as prediction tools for explaining the ATPase activities of these reported compounds. The MM-GBSA calculation procedure used here is similar to that of our previous work on identification of small-molecule inhibitors against a known protein target (Zhou et al., 2017). The protocol is a multiple step procedure, including format conversion of small molecules by Open Babel, 2,000 steps of energy minimization, 100 ps of molecular dynamics (MD) simulation, and 4,000 steps of energy minimization, and then MM-GBSA method was employed for calculating solvation free energy, using an Amber 16 software package. This procedure is similar to the established binding estimation after refinement (BEAR) protocol. More details about the calculation are listed in the Supplemental information.
In general, binding scores alone are not sufficiently conclusive due to the low accuracy of scoring functions in some docking software. Important aspects of binding free energy such as entropic contributions, long-range electrostatics, and desolvation process upon binding may not be adequately defined in conventional scoring functions built within AutoDock Vina and other algorithms of molecular docking (Boyer and Bryan, 2012; Rastelli et al., 2010). To further delineate details of binding between RecA and NPS, MD simulations were performed to evaluate the stability of RecA-NPS complexes, using suramin, bis-ANS, and Congo red as examples.
Generally, short MD simulations reveal that the predicated binding modes between RecA and suramin and selected NPS are stable within the first 10 ns of MD simulations (Figure 4), although hydrogen bonds could be transiently disrupted and reformed during the simulations. A case in point is that the hydrogen bonds between Tyr103 and Congo red could break and reform, but the overall binding mode remains stable during the process (Figures 4E and 4F).
Figure 4.

MD simulations of the RecA-inhibitor complexes
(A) Key residues (colored in pink: Glu68, Glu96, Tyr103, Gln194) around cpd01 (suramin) (colored in cyan) in the RecA-suramin complex are shown in stick style. Suramin is colored in cyan.
(B) Tracked distance changes in key hydrogen bonds. Tracked distance changes in π-π interactions between Tyr103 and benzene ring of suramin.
(C) Key residues (Tyr103, Lys72) around cpd02 (bis-ANS) in the RecA-bis-ANS complex are shown in stick style. The small-molecule RecA inhibitor bis-ANS is colored in cyan.
(D) Tracked distance changes in hydrogen bonds.
(E) Key residues (Tyr103, Gln194) around cpd03 (Congo red) in the RecA-Congo red complex are shown in stick style. Congo red is colored in cyan.
(F) Tracked distance changes in hydrogen bonds and tracked distance changes in π-π interactions between Tyr103 and naphthalene ring of Congo red.
We next validated the predicted biological effects of RecA inhibition on SOS response in an untested model organism, E. coli, whose RecA protein crystal structure was well resolved and computationally exploited in this study. Ciprofloxacin, a typical quinolone antibiotic, was used in combined treatment with suramin as a commercially available representative NPS. To evaluate the effects of suramin on RecA protein in terms of SOS response, E. coli (strain ATCC 25922) was treated with suramin, ciprofloxacin, or ciprofloxacin plus suramin, according to methods as described previously (Alam et al., 2016). For quantitative assessment by real-time PCR (RT-qPCR) (Figure 5), mRNA levels of four selected SOS response genes including recA (DNA recombination/repair protein), umuD (DNA polymerase V protein), umuC (DNA polymerase V catalytic protein), dinI (DNA damage-inducible protein), and one non-SOS response gene soxR (superoxide response protein) were assayed. In all treatments, soxR expression remained almost unchanged. However, transcriptional expression of SOS response-associated genes was differentially modulated upon treatment of the RecA inhibitor. In the presence of 50 μM suramin alone, transcript levels of recA, umuD, umuC, and dinI moderately decreased. Under antibiotic stress by ciprofloxacin, transcript levels of recA and dinI were elevated, affirming activation of the SOS pathway. Upon addition of 50 μM suramin to ciprofloxacin-challenged cells, up-regulated transcription of recA, umuD, umuC, and dinI was reduced to basal levels, indicating that the interaction of suramin and RecA protein effectively blocked bacterial SOS response, including the transcription of RecA itself.
Figure 5.

Effects of blockage of RecA protein on transcriptional expression of SOS genes in E. coli under antibiotic stress, as assessed by RT-qPCR
(A–F) (A) Schematic representation for polymerase switching that occurs in the transition from a DNA damage checkpoint to translational biosynthesis and replication (Schlacher et al., 2005), (B) recA transcription level, (C) umuC transcription level, (D) umuD transcription level, (E) dinI transcription level, and (F) soxR transcription level. Results are representative of at least three independent experiments. β, beta-sliding clamp. Statistical difference was determined at ∗p < 0.05, and ∗∗p < 0.01.
Prediction of binding free energies in RecA-drug interactions
In conjunction with experimental and theoretical approaches discussed above, in silico prediction of binding free energies for binding between RecA and its ligands is essential for optimizing or discovering RecA inhibitors as potential antibiotic adjuvants. In addition to compounds with experimentally demonstrated RecA inhibitory activities such as cpd01 (suramin), cpd02 (bis-ANS), and cpd03 (Congo red) (Figure 3), we also explored other untested compounds and predicted their RecA binding affinities. For example, sulfanilamide (s01) is one of the first oral antibiotics in chemotherapeutic treatment for bacterial infectious diseases (Al-Omari et al., 2014). Besides sulfanilamide, other antibacterials classified as sulfonamides include sulfapyridine, sulfathiazole, and sulfadiazine (Al-Omari et al., 2014). Chicago sky blue (s05), Evans blue (s06), and direct yellow 50 (s07) are anionic dyes structurally related to suramin. Suramin and these three compounds have been found to be biologically active against infectivity and replication of human T cell lymphtropic virus (Balzarini et al., 1986). Elsewhere, the suramin analogs NF299 (s02), NF301 (s03), and NF307 (s04) reportedly activated the ryanodine receptor in skeletal muscles via a calmodulin binding site (Klinger et al., 1999). Additional NPS have been explored for their antiproliferative and angiostatic activities in mammalian cells. These analogs include NF037 (s08), NF059 (s09), NF061 (s10), NF062 (s11), NF063 (s12), NF064 (s13), NF013 (s14), NF289 (s15), NF031 (s16), NF130 (s17), NF136 (s18), NF291 (s19), NF326 (s20), NF357 (s21), NF036 (s22), NF520 (s23), NF015 (s24), NF033 (s25), and NF066 (s26), as listed in Scheme 1 (Firsching et al., 1995). Another source of reported NPS is the inhibitors and activators of protein tyrosine phosphatases, which include NF504 (s27), NF506 (s28), NF110 (s29), NF250 (s30), NF201 (s31), NF290 (s32), NF336 (s33), NF339 (s34), NF067 (s35), NF069 (s36) in Scheme 1 (Nishimura et al., 2015). Compounds in black boxes in Scheme 1 are NPs that have symmetric urea structures, whereas those in blue boxes are NPs that are asymmetric. Of the 36 structures 11 were denoted in bold in Figure 6, which were predicted to be more potent RecA inhibitors than suramin.
Scheme 1.

Structures of reported naphthalene polysulfonated compounds (NPS) s1–s36 included in this study for prediction of their binding potency with RecA
Figure 6.

Binding energies for interactions between RecA and underexamined NPS, and their trends
(A) Correlation between the binding energies predicted by AutoDock Vina and binding energies by LeDock for NPS.
(B) Correlation between the binding energies predicted by AutoDock Vina and molecular charge of analyzed NPS.
(C) Correlation between the binding energies by AutoDock Vina and molecular weight of analyzed NPS.
(D) Correlation between the binding energies by AutoDock Vina and molecular span of analyzed NPS.
Protein-ligand interactions can be strongly influenced by intrinsic characteristics of small-molecule compounds. Along this line of reasoning, we asked what factors contribute to NPS binding to RecA protein. In our analysis, strong correlations seem to exist between binding affinity and intrinsic molecular properties of NPS, including molecular charge, molecular weight, and molecular span (Table III of Figures 6A–6D). The molecular span was measured with GaussView with the geometry optimized by B3LYP method at the 6-31G(d) level. Robust inter-algorithmic correlations were also observed between binding activity values generated by AutoDock Vina and LeDock (Figure 6A). Grid boxes used for AutoDock Vina and LeDock are identical, and their detailed parameters are given in the Supplemental information. To elaborate, the size of a ligand can be described by the radius of gyration (Rg), which is defined as the root mean squared distance of all elemental scattering volumes from either a given axis or their center of mass weighted by scattering densities (Jacques and Trewhella, 2010). Rg values provide information with respect to the mass distribution within a molecule. Dynamically, proteins can undergo conformational changes upon ligand binding, whereas ligands could also adapt different conformations in the same process. For a target protein, AutoDock Vina was reported to give the best predictions, when the box size is around 2.857Rg (Feinstein and Brylinski, 2015). However, calculations for the box size by this approach are not feasible for RecA, as this protein possesses four possible binding sites with distinct physiological functions.
In general, the structure-activity relationship analysis and docking calculations on these NPS shed light on several trends in RecA-ligand interactions. First, NPS need to be large enough to be stably anchored within the binding pockets in RecA. Second, the negatively charged sulfonate groups formed hydrogen bonds with the conserved key residues (including Glu68, Lys72, Glu96, Asp144, Gln196) in the ATP-binding site of pocket A. Third, a benzene ring, urea group, or other linkers at the center of NPS seems to contribute less to its overall binding affinity and may be a favorable position for further modifications.
Experimental validation of computationally predicted binding affinities for RecA/RecAY103A and NPS analogs
To experimentally verify the strength of binding between RecA and NPS predicted in silico, we ventured to express and purify recombinant wild-type RecA protein, as well as its Tyr103 (Y103) mutant RecAY103A, to test their interactions with NPS in vitro. Subsequently, ITC measurements were performed to determine the dissociation constant (Kd) for interactions between RecA/RecAY103A and suramin, along with three selected NPS for comparison. The binding affinity of RecA and suramin was first assessed in titration, wherein Kd was calculated as 17.6 μM (Figure 7A). We next utilized RecAY103A to appraise the importance of the key residue Tyr103 in RecA binding by potentials inhibitors, including suramin. Consistently, the Kd for binding between RecAY103A and suramin was 40.2 μM, much higher than the case of wild-type RecA (Figure 7B). This confirms Tyr103 as a significant contributor in the interactions between RecA and suramin. In contrast, the binding affinity of RecA and ANS (8-(phenylamino)naphthalene-1-sulfonate), as a negative control, was undetectable (Figure 7C). Furthermore, the binding assay was also carried out with RecA and three other NPS compounds, s01, s05, and s06. As NPS, s05 and s06 were two compounds predicted as potential RecA inhibitors, with binding properties comparable to that of suramin (Table III). In ITC, the binding affinities of s05 and s06 to RecA were experimentally determined to be 18.8 μM and 11.5 μM, respectively (Figures 7D and 7E), which suggests that s05 is similar to suramin, whereas s06 binds more strongly than suramin in terms of binding affinity. On the other hand, our computational prediction suggests that the NPS s01 (sulfanilamide) might not bind RecA (Table III). Its ITC results corroborated this proposition, with no binding affinity detected for s01 and RecA (Figure 7F). Taken as a whole, our experimental results are in good agreement with our in silico predictions, indicating the reliability and utility of our docking approaches to assessing RecA-NPS interactions in general.
Figure 7.

ITC measurements for experimental validation of computationally predicted binding affinities for interactions between RecA/RecAY103A and suramin/NPS
(A–F) (A) RecA and suramin, (B) RecAY103A and suramin, (C) RecA and ANS, (D) RecA and s05, (E) RecA and s06, (F) RecA and s01. RecA: recombinant wild-type RecA protein; RecAY103A: recombinant mutant RecA protein.
Conclusion
AMR among fast evolving MDR pathogens is an urgent, albeit underestimated, global health problem. To cope with an imminent crisis of antibiotic depletion, it is imperative to exploit alternative anti-infective strategies such as antibiotic adjuvants targeting bacterial RecA. Structurally, integration of phylogenetic information and protein crystal structures defining the active and inactive states could yield valuable insights into how bacterial RecA may be pharmacologically targeted. In this respect, we performed detailed phylogenetic analysis on RecA/Rad51/RadA proteins, which confirms that RecA is highly conserved among a large number of clinical bacterial pathogens including M. tuberculosis and E. coli but sequence-wise deviates substantially from mammalian homologs. In detailed crystal structure comparison, the ATP-binding region (pocket A) was demonstrated to be the major binding site. NPS are unlikely to bind RecA in pocket D, precluding interference at the interface between RecA and ssDNA. Molecular modeling and MD simulations consistently demonstrate the possible binding modes of NPS with RecA protein, in which π-π interactions with Tyr103 are anticipated to be critical for sustaining the stability of protein-ligand binding. Additionally, RT-qPCR results also confirmed that suramin as a representative NPS blocked transcription of SOS response genes in E. coli. Extending this knowledge, AutoDock Vina and LeDock were used to screen potential active compounds from untested NPS, based on predicted conformations and binding energies of RecA-NPS complex structures. Again, the ATPase core (pocket A) was predicted to be a primary binding site in RecA. Amid 36 uncharacterized NPS of unreported RecA inhibitory activities, 13 were predicted to be more potent than suramin in inhibiting RecA. Experimentally, suramin and selected NPS with computationally high binding affinities were demonstrated to bind recombinant wild-type RecA protein in ITC with Kd values in the range 11.5–18.8 μM. The amino acid residue Tyr103 was shown to be critical to RecA-NPS interactions, as the recombinant mutant RecAY103A protein displayed a much higher Kd value than that of wild-type RecA. Taken as a whole, our findings here offer structural and computational insights into how RecA protein inhibition can be achieved by known and under-examined NPS. Our work also furnishes an analytical framework for designing or re-engineering compounds with enhanced adjuvant activity against bacterial RecA in the urgent contexts of AMR.
Limitations of the study
Computational investigation on RecA-NPS interactions was largely based on monomeric RecA protein and its ATPase activity center defined by pocket A. In our previous study on curcuminoid compounds as potential RecA inhibitors (Zhou et al., 2019), we discussed the filamentous nature of RecA complexes. However, protein-protein interactions within the RecA hexamer may potentially digress from the focus of our current study such as intra-molecular interactions and their determinants. For biological validation, we endeavored to show only Tyr103 as an amino acid residue crucial to RecA ATPase activity, without elaborating on other amino acid residues co-identified as key determinants. For example, as illustrated in Table S8 and Figure S5, both Tyr103 and Glu96 in RecA are likely to be crucial for ATP binding. NPS such as suramin may interfere with the formation of the RecA hexamer by firmly binding Glu96, the validation of which would require another set of elaborate molecular constructs and protein tools, which is beyond the scope of this study. Similarly, in addition to the ATPase activity center (pocket A), binding events at neighboring pockets such as pocket B and pocket D could modulate RecA protein conformation, with yet unclarified physiological functions. In our previous study (Zhou et al., 2019), structural attributes of the residues in pocket B were analyzed by multiple algorithms. However, none of such residues in pocket B have so far been reported to play significant roles in RecA-related protein-protein interactions.
Resource availability
Lead contact
Further information, requests, and inquiries should be directed to and will be fulfilled by the Lead Contact, Prof. Nai-Kei Wong (wongnksz@163.com).
Materials availability
Compounds were purchased from commercial suppliers including Sigma-Aldrich, Shenzhen Tenglong Logistics Co. or Energy Chemical Co. and were used without further purification unless otherwise stated. This study did not generate new specimens or materials. All images are included in the text and Supplemental information.
Data and code availability
The published article includes all data generated or analyzed during this study.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We thank Prof. Conghui You (Shenzhen University) and Prof. Jiamu Du (Southern University of Science and Technology) for sharing their laboratory facilities. This work was supported by the high-performance computing platform of Peking University. Grants from the Sanming Project of Medicine in Shenzhen (SZSM201812058; SZSM201512005) and Shenzhen Key Medical Discipline Development Fund for J.Y., the Foundation for Basic and Applied Research of Guangdong Province (2019A1515110489) for Z.Z., the High-Level Professional Program of Shenzhen (20191226897H) for Z.Z., the Shenlong High-level talent program (2020011056C) for Z.Z. and the Natural Science Foundation of Jiangsu Province of China (BK20191477) and the Natural Science Foundation of the Higher Education Institutions of Jiangsu Province of China (19KJB150001) for Y.Z. are acknowledged for financial support. We acknowledge Yuan Xue (Kingdee Co.), Chunping Zang (Ruiweisi Co.), and Yanlin Jian (Shenzhen Third People's Hospital) for the insightful discussions. We also acknowledge financial support from grants of the Natural Science Foundation of China (32073002, 21778009, 21977010, 81701818, and 51803006) and the Shenzhen Science and Technology Innovation Committee (JCYJ20170817172023838, JCYJ20180508152213145, and JCYJ20180507181527112).
Author contributions
N.-K.W., J.Y., and Z.L, conceived the project. Z.Z. conducted computational studies. Q.P., M.K., and T.C. performed the phylogenetic analysis. Z.Z., N.-K.W., and T.C. finalized presentation of the figures. X.-M.M., Y.-L.X., and M.-X.Z. handled clinical isolates and performed antimicrobial susceptibility tests (AST) and subsequent analysis. Q.P., R.L., and F.Y. designed the molecular experiments. Q.P., X.M., and Y.X. performed pharmacological studies, RT-qPCR and other molecular experiments. R.L., Q.P., and X.L. performed molecular cloning and protein expression and purification of recombinant RecA. X.L. and R.L. carried out experimental validation of RecA-NPS binding in ITC. Q.P., Y. Zhang, R.L., K.Q., M.L., and N.-K.W. analyzed the resultant data. Z.Z., Q.P., Y.L., F.Y., L.L., Y. Zhang, Z.L., and N.-K.W. drafted the manuscript. N.-K.W., Z.L., Y. Zhou, and J.Y. guided the analysis of this work.
Declaration of interests
The authors declare no competing interests.
Published: January 22, 2021
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101952.
Contributor Information
Rui Liu, Email: liur6@mail.sustech.edu.cn.
Zigang Li, Email: lizg@pkusz.edu.cn.
Nai-Kei Wong, Email: wongnksz@163.com.
Supplemental information
References
- Van Acker H., Coenye T. The role of reactive oxygen species in antibiotic-mediated killing of bacteria. Trends Microbiol. 2017;25:456–466. doi: 10.1016/j.tim.2016.12.008. [DOI] [PubMed] [Google Scholar]
- Al-Omari A., Cameron D.W., Lee C., Corrales-Medina V.F. Oral antibiotic therapy for the treatment of infective endocarditis: a systematic review. BMC. Infect. Dis. 2014;14:1–11. doi: 10.1186/1471-2334-14-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam Md K., Alhhazmi A., DeCoteau John F., Luo Y., Geyer C.R. RecA inhibitors potentiate antibiotic activity and block evolution of antibiotic resistance. Cell Chem. Biol. 2016;23:381–391. doi: 10.1016/j.chembiol.2016.02.010. [DOI] [PubMed] [Google Scholar]
- Balzarini J., Mitsuya H., De Clercq E., Broder S. Comparative inhibitory effects of suramin and other selected compounds on the infectivity and replication of human T-cell lymphotropic virus (HTLV-III)/lymphadenopathy-associated virus (LAV) Int. J. Cancer. 1986;37:451–457. doi: 10.1002/ijc.2910370318. [DOI] [PubMed] [Google Scholar]
- Bellio P., Brisdelli F., Perilli M., Sabatini A., Bottoni C., Segatore B., Setacci D., Amicosante G., Celenza G. Curcumin inhibits the SOS response induced by levofloxacin in Escherichia coli. Phytomedicine. 2014;21:430–434. doi: 10.1016/j.phymed.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Bellio P., Di Pietro L., Mancini A., Piovano M., Nicoletti M., Brisdelli F., Tondi D., Cendron L., Franceschini N., Amicosante G. SOS response in bacteria: inhibitory activity of lichen secondary metabolites against Escherichia coli RecA protein. Phytomedicine. 2017;29:11–18. doi: 10.1016/j.phymed.2017.04.001. [DOI] [PubMed] [Google Scholar]
- Boyer R.D., Bryan R.L. Fast estimation of solvation free energies for diverse chemical species. J. Phys. Chem. B. 2012;116:3772–3779. doi: 10.1021/jp300440d. [DOI] [PubMed] [Google Scholar]
- Brown D. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nat. Rev. Drug Discov. 2015;14:821–832. doi: 10.1038/nrd4675. [DOI] [PubMed] [Google Scholar]
- Brown E.D., Wright G.D. Antibacterial drug discovery in the resistance era. Nature. 2016;529:336–343. doi: 10.1038/nature17042. [DOI] [PubMed] [Google Scholar]
- Chen Z., Yang H., Pavletich N.P. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008;453:489–494. doi: 10.1038/nature06971. [DOI] [PubMed] [Google Scholar]
- Chen C., Tian W., Lei X., Liang J., Zhao J. CASTp 3.0: computed atlas of surface topography of proteins. Nucleic Acids Res. 2018;46:363–367. doi: 10.1093/nar/gky473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cline D.J., Holt S.L., Singleton S.F. Inhibition of Escherichia coli RecA by rationally redesigned N-terminal helix. Org. Biomol. Chem. 2007;5:1525–1528. doi: 10.1039/b703159a. [DOI] [PubMed] [Google Scholar]
- Datta S., Ganesh N., Chandra N.R., Muniyappa K., Vijayan M. Structural studies on MtRecA-nucleotide complexes: insights into DNA and nucleotide binding and the structural signature of NTP recognition. Proteins. 2003;50:474–485. doi: 10.1002/prot.10315. [DOI] [PubMed] [Google Scholar]
- Dundas J., Ouyang Z., Tseng J., Binkowski A., Turpaz Y., Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:116–118. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egelman E.H., Stasiak A. Structure of helical RecA-DNA complexes. Complexes formed in the presence of ATP-gamma-S or ATP. J. Mol. Biol. 1986;191:677–697. doi: 10.1016/0022-2836(86)90453-5. [DOI] [PubMed] [Google Scholar]
- Feinstein W.P., Brylinski M. Calculating an optimal box size for ligand docking and virtual screening against experimental and predicted binding pockets. J. Cheminf. 2015;7:1–10. doi: 10.1186/s13321-015-0067-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39:783–791. doi: 10.1111/j.1558-5646.1985.tb00420.x. [DOI] [PubMed] [Google Scholar]
- Firsching A., Nickel P., Mora P., Allolio B. Antiproliferative and angiostatic activity of suramin analogues. Cancer Res. 1995;55:4957–4961. [PubMed] [Google Scholar]
- Glaser F., Pupko T., Paz I., Bell R., Bechor-Shental D., Martz E., Ben-Tal N. ConSurf: identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics. 2003;19:163–164. doi: 10.1093/bioinformatics/19.1.163. [DOI] [PubMed] [Google Scholar]
- Hu J.J., Wong N.-K., Ye S., Chen X., Lu M.-Y., Zhao A.Q., Guo Y., Ma A.C.-H., Leung A.Y.-H., Shen J., Yang D. Fluorescent probe HKSOX-1 for imaging and detection of endogenous superoxide in live cells and in vivo. J. Am. Chem. Soc. 2015;137:6837–6843. doi: 10.1021/jacs.5b01881. [DOI] [PubMed] [Google Scholar]
- Hu J.J., Wong N.-K., Lu M.-Y., Chen X., Ye S., Zhao A.Q., Gao P., Kao R.Y.-T., Shen J., Yang D. HKOCl-3: a fluorescent hypochlorous acid probe for live-cell and in vivo imaging and quantitative application in flow cytometry and a 96-well microplate assay. Chem. Sci. 2016;7:2094–2099. doi: 10.1039/c5sc03855c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacques D.A., Trewhella J. Small-angle scattering for structural biology-expanding the frontier while avoiding the pitfalls. Protein Sci. 2010;19:642–657. doi: 10.1002/pro.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinger M., Freissmuth M., Nickel P., Stäbler-Schwarzbart M., Kassack M., Suko J., Hohenegger M. Suramin and suramin analogs activate skeletal muscle ryanodine receptor via a calmodulin binding site. Mol. Pharmacol. 1999;55:462–472. [PubMed] [Google Scholar]
- Kohanski M.A., Dwyer D.J., Hayete B., Lawrence C.A., Collins J.J. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797–810. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- Koonin E.V. A common set of conserved motifs in a vast variety of putative nucleic acid-dependent ATPases including MCM proteins involved in the initiation of eukaryotic DNA replication. Nucleic Acids Res. 1993;21:2541–2547. doi: 10.1093/nar/21.11.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.M., Singleton S.F. Inhibition of the Escherichia coli RecA protein: zinc(II), copper(II) and mercury(II) trap RecA as inactive aggregates. J. Inorg. Biochem. 2004;98:1981–1986. doi: 10.1016/j.jinorgbio.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Lee A.M., Ross C.T., Zeng B.-B., Singleton S.F. A molecular target for suppression of the evolution of antibiotic resistance: inhibition of the Escherichia coli RecA protein by N6-(1-Naphthyl)-ADP. J. Med. Chem. 2005;48:5408–5411. doi: 10.1021/jm050113z. [DOI] [PubMed] [Google Scholar]
- Leis S., Schneider S., Zacharias M. In silico prediction of binding sites on proteins. Curr. Med. Chem. 2010;17:1550–1562. doi: 10.2174/092986710790979944. [DOI] [PubMed] [Google Scholar]
- Lin Z., Kong H., Nei M., Ma H. Origins and evolution of the recA/RAD51 gene family: evidence for ancient gene duplication and endosymbiotic gene transfer. Proc. Natl. Acad. Sci. U S A. 2006;103:10328–10333. doi: 10.1073/pnas.0604232103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkov V.A., Camerini-Otero R.D. Photocrosslinks between single-stranded DNA and Escherichia coli RecA protein map to loops L1 (amino acid residues 157-164) and L2 (amino acid residues 195-209) J. Biol. Chem. 1995;270:30230–30233. doi: 10.1074/jbc.270.50.30230. [DOI] [PubMed] [Google Scholar]
- Nahrstedt H., Schröder C., Meinhardt F. Evidence for two RecA genes mediating DNA repair in Bacillus megaterium. Microbiology. 2005;151:775–787. doi: 10.1099/mic.0.27626-0. [DOI] [PubMed] [Google Scholar]
- Nautiyal A., Patil K.N., Muniyappa K. Suramin is a potent and selective inhibitor of Mycobacterium tuberculosis RecA protein and the SOS response: RecA as a potential target for antibacterial drug discovery. J. Antimicrob. Chemother. 2014;69:1834–1843. doi: 10.1093/jac/dku080. [DOI] [PubMed] [Google Scholar]
- Nishimura Y., McLaughlin N.P., Pan J., Goldstein S., Hafenstein S., Shimizu H., Winkler J.D., Bergelson J.M. The suramin derivative NF449 interacts with the 5-fold vertex of the enterovirus A71 capsid to prevent virus attachment to PSGL-1 and heparan sulfate. PLoS Pathog. 2015;11:e1005184. doi: 10.1371/journal.ppat.1005184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabu J.R., Manjunath G.P., Chandra N.R., Muniyappa K., Vijayan M. Functionally important movements in RecA molecules and filaments: studies involving mutation and environmental changes. Acta Crystallogr. D Biol. Crystallogr. 2008;64:1146–1157. doi: 10.1107/S0907444908028448. [DOI] [PubMed] [Google Scholar]
- Rastelli G., Del Rio A., Degliesposti G., Sgobba M. Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 2010;31:797–810. doi: 10.1002/jcc.21372. [DOI] [PubMed] [Google Scholar]
- Saitou N., Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Schlacher K., Leslie K., Wyman C., Woodgate R., Cox M.M., Goodman M.F. DNA polymerase V and RecA protein, a minimal mutasome. Mol. Cell. 2005;17:561–572. doi: 10.1016/j.molcel.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Schlacher K., Pham P., Cox M.M., Goodman M.F. Roles of DNA polymerase V and RecA protein in SOS damage-induced mutation. Chem. Rev. 2006;106:406–419. doi: 10.1021/cr0404951. [DOI] [PubMed] [Google Scholar]
- Seifert H., Blondeau J., Dowzicky M.J. In vitro activity of tigecycline and comparators (2014-2016) among key WHO 'priority pathogens' and longitudinal assessment (2004-2016) of antimicrobial resistance: a report from the T.E.S.T. study. Int. J. Antimicrob. Agents. 2018;52:474–484. doi: 10.1016/j.ijantimicag.2018.07.003. [DOI] [PubMed] [Google Scholar]
- Shinohara T., Ikawa S., Iwasaki W., Hiraki T., Hikima T., Mikawa T., Arai N., Kamiya N., Shibata T. Loop L1 governs the DNA-binding specificity and order for RecA-catalyzed reactions in homologous recombination and DNA repair. Nucleic Acids Res. 2015;43:973–986. doi: 10.1093/nar/gku1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton S.F., Simonette R.A., Sharma N.C., Roca A.I. Intein-mediated affinity-fusion purification of the Escherichia coli RecA protein. Protein Expr. Purif. 2002;26:476–488. doi: 10.1016/s1046-5928(02)00571-5. [DOI] [PubMed] [Google Scholar]
- Story R.M., Steitz T.A. Structure of the recA protein-ADP complex. Nature. 1992;355:374–376. doi: 10.1038/355374a0. [DOI] [PubMed] [Google Scholar]
- Tacconelli E., Carrara E., Savoldi A., Harbarth S., Mendelson M., Monnet D.L., Pulcini C., Kahlmeter G., Kluytmans J., Carmeli Y. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018;18:318–327. doi: 10.1016/S1473-3099(17)30753-3. [DOI] [PubMed] [Google Scholar]
- Tacconelli E., Sifakis F., Harbarth S., Schrijver R., van Mourik M., Voss A., Sharland M., Rajendran N.B., Rodríguez-Baño J., Bielicki J. Surveillance for control of antimicrobial resistance. Lancet Infect. Dis. 2018;18:99–106. doi: 10.1016/S1473-3099(17)30485-1. [DOI] [PubMed] [Google Scholar]
- Tombline G., Fishel R. Biochemical characterization of the human RAD51 protein. I. ATP hydrolysis. J. Biol. Chem. 2002;277:14417–14425. doi: 10.1074/jbc.M109915200. [DOI] [PubMed] [Google Scholar]
- Tombline G., Shim K.-S., Fishel R. Biochemical characterization of the human RAD51 protein. II. Adenosine nucleotide binding and competition. J. Biol. Chem. 2002;277:14426–14433. doi: 10.1074/jbc.M109916200. [DOI] [PubMed] [Google Scholar]
- Tombline G., Heinen C.D., Shim K.-S., Fishel R. Biochemical characterization of the human RAD51 protein. III. Modulation of DNA binding by adenosine nucleotides. J. Biol. Chem. 2002;277:14434–14442. doi: 10.1074/jbc.M109917200. [DOI] [PubMed] [Google Scholar]
- VanLoock M.S., Yu X., Yang S., Lai A.L., Low C., Campbell M.J., Egelman E.H. ATP-Mediated conformational changes in the RecA filament. Structure. 2003;11:187–196. doi: 10.1016/s0969-2126(03)00003-0. [DOI] [PubMed] [Google Scholar]
- Walker J.E., Saraste M., Runswick M.J., Gay N.J. Distantly related sequences in the α- and β-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wass M.N., Kelley L.A., Sternberg M.J.E. 3DLigandSite: predicting ligand-binding sites using similar structures. Nucleic Acids Res. 2010;38:469–473. doi: 10.1093/nar/gkq406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigle T.J., Singleton S.F. Directed molecular screening for RecA ATPase inhibitors. Bioorg. Med. Chem. Lett. 2007;17:3249–3253. doi: 10.1016/j.bmcl.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wigle T.J., Lee A.M., Singleton S.F. Conformationally selective binding of nucleotide analogues to escherichia coli RecA: a ligand-based analysis of the RecA ATP binding site. Biochemistry. 2006;45:4502–4513. doi: 10.1021/bi052298h. [DOI] [PubMed] [Google Scholar]
- Wigle T.J., Sexton J.Z., Gromova A.V., Hadimani M.B., Hughes M.A., Smith G.R., Yeh L.-A., Singleton S.F. Inhibitors of RecA activity discovered by high-throughput screening: cell-permeable small molecules attenuate the SOS response in Escherichia coli. J. Biomol. Screen. 2009;14:1092–1101. doi: 10.1177/1087057109342126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright G.D. Antibiotic adjuvants: rescuing antibiotics from resistance. Trends Microbiol. 2016;24:862–871. doi: 10.1016/j.tim.2016.06.009. [DOI] [PubMed] [Google Scholar]
- Xing X., Bell C.E. Crystal structures of Escherichia coli RecA in a compressed helical filament. J. Mol. Biol. 2004;342:1471–1485. doi: 10.1016/j.jmb.2004.07.091. [DOI] [PubMed] [Google Scholar]
- Xing X., Bell C.E. Crystal structures of Escherichia coli RecA in complex with MgADP and MnAMP−PNP. Biochemistry. 2004;43:16142–16152. doi: 10.1021/bi048165y. [DOI] [PubMed] [Google Scholar]
- Xu J., Zhao L., Xu Y., Zhao W., Sung P., Wang H.-W. Cryo-EM structures of human RAD51 recombinase filaments during catalysis of DNA-strand exchange. Nat. Struct. Mol. Biol. 2016;24:40. doi: 10.1038/nsmb.3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakimov A., Pobegalov G., Bakhlanova I., Khodorkovskii M., Petukhov M., Baitin D. Blocking the RecA activity and SOS-response in bacteria with a short α-helical peptide. Nucleic Acids Res. 2017;45:9788–9796. doi: 10.1093/nar/gkx687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., Jacobs S.A., West S.C., Ogawa T., Egelman E.H. Domain structure and dynamics in the helical filaments formed by RecA and Rad51 on DNA. Proc. Natl. Acad. Sci. U S A. 2001;98:8419. doi: 10.1073/pnas.111005398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X., VanLoock M.S., Yang S., Reese J.T., Egelman E.H. What is the structure of the RecA-DNA filament? Curr. Protein Pept. Sci. 2004;5:73–79. doi: 10.2174/1389203043486883. [DOI] [PubMed] [Google Scholar]
- Zheng W., Liang Y., Zhao H., Zhang J., Li Z. 5,5'-Methylenedisalicylic acid (MDSA) modulates SarA/MgrA phosphorylation by targeting Ser/Thr phosphatase Stp1. ChemBioChem. 2015;16:1035–1040. doi: 10.1002/cbic.201500003. [DOI] [PubMed] [Google Scholar]
- Zheng W., Cai X., Xie M., Liang Y., Wang T., Li Z. Structure-based identification of a potent inhibitor targeting Stp1-mediated virulence regulation in Staphylococcus aureus. Cell Chem. Biol. 2016;23:1002–1013. doi: 10.1016/j.chembiol.2016.06.014. [DOI] [PubMed] [Google Scholar]
- Zhou Z., Yuan Y., Zhou S., Ding K., Zheng F., Zhan C.-G. Selective inhibitors of human mPGES-1 from structure-based computational screening. Bioorg. Med. Chem. Lett. 2017;27:3739–3743. doi: 10.1016/j.bmcl.2017.06.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Yuan J., Pan Q., Mo X.-M., Xie Y.-L., Yin F., Li Z., Wong N.-K. Computational elucidation of the binding mechanisms of curcumin analogues as bacterial RecA inhibitors. RSC Adv. 2019;9:19869–19881. doi: 10.1039/c9ra00064j. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The published article includes all data generated or analyzed during this study.