

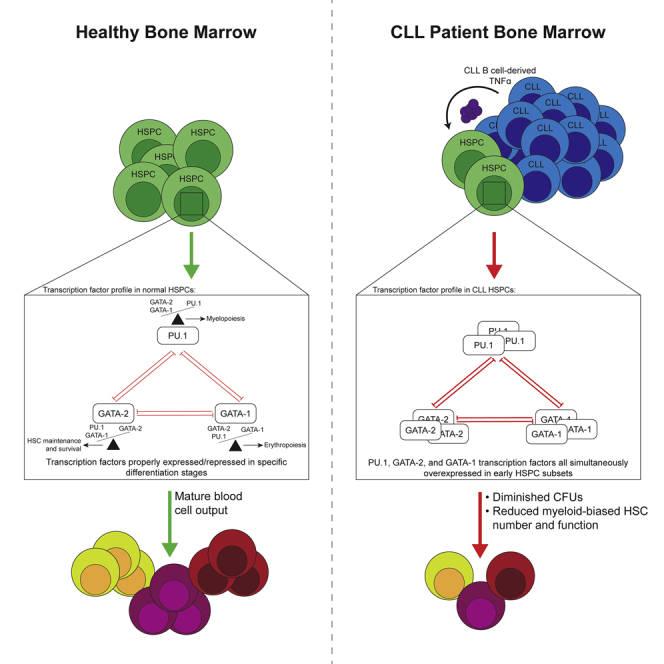
Summary
TNFα is implicated in chronic lymphocytic leukemia (CLL) immunosuppression and disease progression. TNFα is constitutively produced by CLL B cells and is a negative regulator of bone marrow (BM) myelopoiesis. Here, we show that co-culture of CLL B cells with purified normal human hematopoietic stem and progenitor cells (HSPCs) directly altered protein levels of the myeloid and erythroid cell fate determinants PU.1 and GATA-2 at the single-cell level within transitional HSPC subsets, mimicking ex vivo expression patterns. Physical separation of CLL cells from control HSPCs or neutralizing TNFα abrogated upregulation of PU.1, yet restoration of GATA-2 required TNFα neutralization, suggesting both cell contact and soluble-factor-mediated regulation. We further show that CLL patient BM myeloid progenitors are diminished in frequency and function, an effect recapitulated by chronic exposure of control HSPCs to low-dose TNFα. These findings implicate CLL B-cell-derived TNFα in impaired BM myelopoiesis.
Subject areas: Molecular Biology, Immunology, Stem Cells Research
Graphical abstract
Highlights
-
•
CLL patient BM HSPCs exhibit aberrant molecular and functional characteristics
-
•
CLL B-cell-derived TNFα upregulates PU.1 and GATA-2 in BM HSPCs
-
•
The effects of CLL B-cell-derived TNFα are reversible upon TNFα neutralization
-
•
Chronic TNFα exposure in vitro recapitulates ex vivo HSPC functional deficiencies
Molecular Biology; Immunology; Stem Cells Research
Introduction
Chronic lymphocytic leukemia (CLL), a chronic lymphoproliferative neoplasm characterized by accumulation of CD19+CD5+ B cells, is associated with global immunosuppression that clinically manifests as an inability to respond appropriately to immunologic challenges. As CLL B cells are widespread in the host, the immunosuppression may be directly induced by organ infiltrative accumulation of clonal leukemic cells or indirectly via leukemic cell-induced remodeling of tissue microenvironments (Forconi and Moss, 2015; Goldman, 2000; Kipps et al., 2017). In addition to accumulation in blood and secondary lymphoid organs, CLL B cells infiltrate the bone marrow (BM), the primary site of adult hematopoiesis. This latter feature is thus a threat to the normal and critical function of BM.
Steady-state BM hematopoiesis sustains blood cell genesis throughout life (immune cells, erythrocytes, and platelets) (Doulatov et al., 2012) and is a tightly regulated process (Novershtern et al., 2011). Importantly, BM hematopoiesis becomes myeloid-skewed with aging (Pang et al., 2017), a critical consideration as CLL is largely a disease of the aged. Aberrant hematopoiesis and BM dysfunction in CLL is understudied and historically suggested to most likely be an infiltrative remodeling that disrupts normal hematopoietic niches (Lagneaux et al., 1993). Reduced numbers of BM hematopoietic stem cells (HSCs) and their progeny may manifest due to competition with CLL B cells for hematopoietic niches, local increases in CLL-derived factors, or CLL-induced alterations in resident immune cells or components of the BM microenvironment (Fecteau and Kipps, 2012; Sala et al., 1998).
Given the chronic residence of CLL cells in BM, their likely competition with hematopoietic progenitors for essential niches, and the increased potential for leukemic and HSPC interactions, we hypothesized that BM hematopoietic capacity may be compromised in CLL patients. Indeed, our previous report confirmed significantly reduced frequencies of BM HSCs and their progeny in CLL patients compared with age-matched controls (Manso et al., 2019). In addition, we demonstrated hematopoietic functional impairment as evidenced by significantly reduced colony forming unit (CFU) capacity, revealing diminished production of myelo-erythroid progenitors. In this initial work we also showed that select BM hematopoietic progenitor subsets displayed aberrant transcription factor (TF) profiles at the protein level. Importantly, the collective features of impaired BM hematopoiesis in CLL patients were independent of the extent of BM infiltration by CLL B cells (Manso et al., 2019; Sala et al., 1998; Tsopra et al., 2009). Thus, the mechanism(s) responsible for impaired BM hematopoiesis in CLL are likely multifactorial and, at present, largely unexplored.
CLL B cells constitutively produce multiple factors capable of modulating BM hematopoiesis, most prominently tumor necrosis factor alpha (TNFα) (Foa et al., 1990; Michalevicz et al., 1991). TNFα is elevated in patient plasma, and levels associate with disease progression (Bojarska-Junak et al., 2008). The role of TNFα in steady-state hematopoiesis is largely suppressive to maintain BM homeostasis and aberrant exposure can result in BM failure, cytopenias, and anemia, clinical complications often observed in late-stage CLL patients (Broxmeyer et al., 1986; Geissler et al., 1991; Means et al., 1990; Michalevicz et al., 1991; Skobin et al., 2000; Tian et al., 2014; Tsopra et al., 2009). Limited studies have shown that TNFα, in part, can alter TF expression levels in mouse and human hematopoietic progenitors to mediate these modulatory actions (Etzrodt et al., 2019; Grigorakaki et al., 2011). Our previous report was the first to assess this mechanism in the context of CLL where we showed that in vitro exposure of control human BM progenitors (aged-matched to CLL patients) to TNFα upregulated PU.1 and GATA-2 proteins, phenocopying ex vivo findings in CLL patient marrow (Manso et al., 2019). However, questions remain concerning in vivo sources of TNFα in CLL, developmental stage-specific HSPC subset sensitivity to direct CLL B cell exposure or their secreted factors, mechanisms of TNFα modulation of HSPC TF expression, and reversibility of TF alterations upon effective TNFα neutralization. This report details our new findings in relation to these questions at both a global and single-cell level as assayed in primary human BM hematopoietic progenitors from control subjects and CLL patients.
Results
Global and single-cell analysis of control and CLL patient bone marrow progenitors
GATA-2, PU.1, and GATA-1 are cell fate determinants directly modulated by TNFα in BM progenitors (Etzrodt et al., 2019; Grigorakaki et al., 2011). GATA-2 is critical for HSC maintenance and survival and has additional roles in determining erythroid, megakaryocyte, and granulocyte cell fates (Vicente et al., 2012). PU.1 and GATA-1 are master regulators of myeloid and erythroid cell fates, respectively (Burda et al., 2010). Importantly, these factors are cross-antagonistic and the relative abundance of each reinforces bifurcating lineage decisions (Walsh et al., 2002). Given the importance of these TFs in regulation of primitive hematopoiesis, and their modulation by TNFα(Etzrodt et al., 2019; Grigorakaki et al., 2011), we examined global expression levels of these TFs in developmental stage-specific hematopoietic stem and progenitor cell (HSPC) populations: HSC/multipotent (HSC/MPP, Lin−CD34hiCD38−CD45RA−), lymphoid-biased multipotential (LMPP, Lin−CD34hiCD38−CD45RA+), common myeloid/megakaryocyte-erythroid (CMP/MEP, Lin−CD34+CD38+CD45RA−), and granulocyte-monocyte (GMP, Lin−CD34+CD38+CD45RA+) progenitors (Figure S1).
Freshly isolated HSC/MPPs from CLL patient BM exhibited significantly increased protein levels of PU.1 and GATA-2 when evaluated by flow cytometry and compared with age-matched controls (Figures 1A and 1B), consistent with and extending our previous findings that were largely performed in cryopreserved patient samples among simplified progenitor cell fractions (Manso et al., 2019). Here, our refined analysis further revealed that CLL patient-derived LMPPs and CMP/MEPs exhibited significantly higher levels of PU.1 and GATA-2. GATA-2 was also elevated among CLL GMPs. In contrast, an increased frequency of GATA-1+ cells was only observed in HSC/MPPs and CMP/MEPs (Figure S2), and those populations also display elevated levels of GATA-1 protein (among total GATA-1+ cells, Figure 1C). Representative histograms of PU.1, GATA-2, and GATA-1 (shown in red) from control and CLL HSPCs compared with isotype controls (shown in blue) are displayed in Figure 1D.
Figure 1.

Altered expression of PU.1, GATA-2, and GATA-1 in CLL patient bone marrow progenitors
(A–C) Flow cytometry was performed on freshly isolated BM from controls and CLL patients to determine MFI (representative of protein levels) of (A) PU.1, (B) GATA-2, and (C) GATA-1 among HSPC progenitor subsets. Control n = 11 for PU.1, n = 11 for GATA-2, and n = 8 for GATA-1. CLL n = 13 for PU.1, n = 12 for GATA-2, and n = 7 for GATA-1. The displayed number of data points for GATA-1 are reduced due to several individuals containing no detectable GATA-1 protein. We were unable to perform statistical comparisons between control and CLL LMPPs and GMPs for GATA-1 MFI as few cells contained detectable GATA-1 protein: HSC/MPP (Lin−CD34hiCD38−CD45RA−), LMPP (Lin−CD34hiCD38−CD45RA+), CMP/MEP (Lin−CD34+CD38+CD45RA−), and GMP (Lin−CD34+CD38+CD45RA+). Data are presented as the mean ± SEM; individual points represent a unique donor and are representative of 24 individual experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001, and ns by Mann-Whitney U test.
(D) Representative histograms of control and CLL patient BM progenitors. The stained sample is colored red and the isotype is shown in blue (see also Figure S1). (E–H) Combined intranuclear flow cytometry data from control (n = 8) and CLL patient (n = 6) HSPCs was utilized in the cytofkit2 R package to generate UMAP plots. Each point in the UMAP represents a single cell.
(E) Contour plots of the UMAP architecture for all combined samples or separated into control or CLL patients.
(F) Colored overlay of control (teal) and CLL patient (red) HSPCs. The borders of five distinct regions are indicated and individually numbered. (G–H) Expression of PU.1, GATA-2, GATA-1, CD38, and CD45RA was superimposed on the cohort-specific architectures. Note that the data are normalized and expression values are relative to each individual factor.
See also Figures S1–S4.
TF expression levels exist along a dynamic spectrum at the single-cell level, and as a function of cellular differentiation, a feature global analysis of total cell fractions does not wholly represent. Therefore, we employed uniform manifold approximation and projection (UMAP) (Leland McInnes, 2018) dimensionality reduction across the totality of the four HSPC populations from controls and CLL patients to evaluate alterations in TFs and/or phenotypic markers at the single-cell level. We reasoned that, because UMAP normalizes expression of clustering markers across the combination of each input sample, this analysis would allow for the relative relatedness of individual cells to be analyzed. This first-of-a-kind analysis of control and CLL HSPCs enabled us to define global architectures for each cohort and also capture TF alterations at the resolution of single cells (Figures 1E and S3A). As expected, control HSPCs displayed significant heterogeneity in their UMAP occupancy, reflecting a diversity of phenotypes. When overlaid, CLL HSPCs were found, differentially represented, within three distinct control phenotypes (Figure 1F, circled regions 1, 2 and 4), yet largely enrich into region 4. A subset of control HSPCs also uniquely occupied a third region (region 3), which CLL HSPCs were strikingly absent from. Interestingly, region 5 represents a unique population of HSPCs found in CLL BM only.
Using the UMAP architecture, we next visualized the relative expression of PU.1, GATA-2, and GATA-1 (normalized to each specific parameter) across control and CLL HSPCs (Figure 1G). Control HSPCs exhibited considerable PU.1 heterogeneity and moderately uniform, yet variable, GATA-2 and GATA-1 expression. CLL HSPCs exhibited increased expression heterogeneity across all three TFs. When individual regions were evaluated, region 1 displayed variable PU.1 expression and high levels of GATA-2 and GATA-1. CLL HSPCs in region 1 were found to exhibit similar PU.1 and GATA-2 expression patterns as control HSPCs but have a distinct increase in GATA-1 positivity (Figures 1G and S4). Control HSPCs in region 2, a smaller region also occupied by CLL HSPCs, was characterized by low levels of PU.1, intermediate levels of GATA-2, and low levels of GATA-1. CLL HSPCs in region 2 show comparable PU.1 and GATA-2 expression patterns compared with controls, yet there is a small population of PU.1hi cells. CLL HSPCs in this region also display increased GATA-1 expression. The unique control HSPC region 3 is composed of GATA-2hi cells that also express the highest control HSPC levels of PU.1 and GATA-1. Region 4, a CLL-enriched group sharing marginal overlap with controls, displayed variable expression of GATA-2 and low levels of PU.1 and GATA-1 in control HSPCs. In contrast, CLL HSPCs in region 4 exhibit increased yet bimodal expression of PU.1 and GATA-2 and very low levels of GATA-1. Finally, the CLL-enriched region 5 is comprised of cells with low levels of all three TFs.
The cell surface markers CD38 and CD45RA are utilized to distinguish human HSPC subsets (Figures 1H and S4). When assessed by UMAP analysis, significant heterogeneity of CD38 and CD45RA expression was observed in control and CLL HSPCs. Regions 1 and 2 are enriched for cells with higher expression of CD38 and low/intermediate CD45RA. Control HSPCs in region 3 are characterized by intermediate levels of CD38 and increased, although variable, CD45RA expression. Both control and CLL HSPCs exhibit variable CD38 expression in the CLL-enriched region 4. Interestingly, control HSPCs in region 4 show bimodal CD45RA expression, whereas CLL HSPCs are largely CD45RAlow. In contrast, the CLL-specific region 5 displays a unique signature of CD38low levels and uniformly high expression of CD45RA. Together, these data demonstrate that CLL BM HSPCs are distinct from controls and express fundamentally altered TF and cell surface marker profiles when assessed globally and at the single cell level.
CLL cells modulate TF protein levels in specific BM HSPC subsets
CLL B cells constitutively produce soluble and membrane-bound factors that may impact normal hematopoiesis (Burger et al., 2009; Foa et al., 1990; Janel et al., 2014; Kay et al., 2002; Lahat et al., 1991; Lotz et al., 1994; Saulep-Easton et al., 2016; van Attekum et al., 2017). Therefore, we sought to test if CLL cells modulate PU.1, GATA-2, and GATA-1 expression levels in HSPC subsets via cell contact and/or secreted/soluble factors by comparing direct and Transwell co-cultures consisting of primary CLL cells and age-matched control Lin−CD34+ HSPCs. To establish an experimental ratio of CLL B cells:control HSPCs, we assessed the cellular ratio of CD19+CD5− and CD19+CD5+ B cells to Lin−CD34+ HSPCs in control and CLL BM (Figure S5). As expected, the ratio was orders of magnitude higher in CLL patients compared with controls. A 10:1 (CLL cells:HSPCs) ratio was used in all in vitro experiments for two reasons. First, it is reasonably greater than control CD19+CD5+ B cell to HSPC ratios. Second, it allows for evaluation of CLL B-cell-induced alterations in an environment mimicking lower thresholds of leukemic marrow infiltration as would be predicted in early stage disease.
Following 24 h of short-term co-culture allowing direct contact between control HSPCs and primary CLL B cells, the HSC/MPP, LMPP, CMP/MEP, and GMP populations were evaluated for levels of PU.1, GATA-2, and GATA-1 by flow cytometry. We note that the co-culture is serum free, eliminating any contribution of serum-derived factors in TF modulation. Short-term direct exposure to CLL cells significantly increased PU.1 and GATA-2 in HSC/MPPs without affecting levels of GATA-1 compared with HSC/MPPs cultured in media alone (Figure 2A). Similarly, PU.1 was upregulated in the CMP/MEP and GMP (Figures 2C and 2D), but not LMPP, subsets (Figure 2B). In addition to HSC/MPPs, GATA-2 was also increased among CMP/MEPs. Interestingly, GATA-2 displayed a variable response, with some control HSC/MPPs and CMP/MEPs not affected by the co-culture (Figures 2A and 2C). GATA-1 remained unchanged in all populations. These new findings indicate that direct short-term co-culture of CLL B cells with control HSPCs differentially alters TF expression levels across multiple transitional stages of hematopoietic differentiation.
Figure 2.

Expression of PU.1, GATA-2, and GATA-1 are modulated in vitro by direct co-culture with CLL B cells
(A–D′) Freshly isolated control BM CD34+ cells were cultured in media alone, (A–D) directly with peripheral blood CLL cells, or (A′–D′) indirectly by use of a 1.0 μm Transwell insert at a ratio of 10:1 (CLL cells:HSPCs) for 24 h. Flow cytometry was performed to determine the MFI (indicative of relative protein levels) of PU.1, GATA-2, and GATA-1 among (A and A′) Lin−CD34hiCD38−CD45RA− HSC/MPPs, (B and B′) Lin−CD34hiCD38−CD45RA+ LMPPs, (C and C′) Lin−CD34+CD38+CD45RA− CMP/MEPs, and (D and D′) Lin−CD34+CD38+CD45RA+ GMPs. Each point represents a unique pairing of a control HSPC donor and CLL patient, with lines connecting the pairs (n = 11 for direct co-culture and n = 7 for Transwell co-cultures, 11 individual experiments). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001, and ns by paired t test.
(E) Summary table of the in vitro co-culture results. Changes to expression of each TF is indicated by ↑ = increased, ↓ = decreased, and --- = no change.
See also Figures S5 and S8.
Next, we asked whether the TF alterations required direct contact between control HSPCs and CLL B cells or if modulation could be mediated by soluble factors provided by the CLL cells. To test this possibility, we physically separated the CLL B cells from control HSPCs by using Transwell inserts during the short-term co-culture. In contrast to the results obtained in the direct co-cultures, HSPCs separated from CLL cells showed no significant change in PU.1 in any population examined (Figures 2A’–2D′). GATA-2 remained unchanged in HSC/MPPs and LMPPs but significantly increased in CMP/MEPs and GMPs, indicating differential TF sensitivity of individual progenitor stages to CLL-derived soluble factors (Figures 2C′–2D′). HSC/MPPs and CMP/MEPs, but not LMPPs or GMPs, exhibited a decline in GATA-1 levels. Together, these findings suggest differential requirements for direct CLL:HSPC cell contact versus CLL-derived soluble factors in modulation of PU.1, GATA-2, and GATA-1 within developmental stage-specific HSPC subsets (summarized in Figure 2E).
CLL-derived TNFα directly modulates PU.1 and GATA-2 expression
In our previous study we reported that exogenous TNFα rapidly increased expression of PU.1 and GATA-2 in control HSPCs (Manso et al., 2019). We have extended these findings and determined that TNFα exposure reduces levels of GATA-1, consistent with previously published data (Buck et al., 2009; Grigorakaki et al., 2011) (Figure S6A). Further, NF-κB p65 (RelA), a TF at the nexus of inflammatory and TNFα signaling cascades (Liu et al., 2017; Nakagawa et al., 2018), is overexpressed in CLL-derived HSC/MPPs, unchanged in LMPPs and CMP/MEPs, and reduced in GMPs (Figure S7). We also note that the effects of TNFα on human HSPCs may be long-lasting or irreversible (Dybedal et al., 2001). TNFα is constitutively produced by CLL B cells in both membrane-bound and soluble forms (Foa et al., 1990; Michalevicz et al., 1991). Furthermore, TNFRI and TNFRII are expressed on primitive human HSPCs (Wang et al., 2017). To determine if TNFα derived from CLL B cells contributes to the alterations in PU.1, GATA-2, and GATA-1 in control HSPCs, a specific neutralizing antibody against TNFα (anti-TNFα) (Kim et al., 2018; Rothhammer et al., 2018) was added to the short-term direct and Transwell CLL:control HSPC co-cultures. Addition of anti-TNFα to the direct co-cultures significantly reduced levels of PU.1 in HSC/MPP, CMP/MEP, and GMP populations (Figures 3A, 3B, and 3D). GATA-2 was reduced in HSC/MPPs and CMP/MEPs but not GMPs. As PU.1 and GATA-2 were not modulated in LMPPs in the direct co-culture (Figure 2B), they remained unchanged when TNFα was neutralized (Figure 3B). Interestingly, GATA-1 protein levels were increased in HSC/MPPs and LMPPs upon neutralization of TNFα.
Figure 3.

CLL B-cell-derived TNFα modulates transcription factor expression levels in vitro
(A–D′) Freshly isolated control BM CD34+ cells were cultured in the presence or absence of a neutralizing antibody against TNFα (anti-TNFα) either (A–D) directly with peripheral blood CLL cells or (A′–D′) indirectly cultured by use of a 1.0 μm Transwell insert at a ratio of 10:1 (CLL cells:HSPCs) for 24 h. Flow cytometry was performed to determine the MFI (indicative of relative protein levels) of PU.1, GATA-2, and GATA-1 among (A, A′) Lin−CD34hiCD38−CD45RA− HSC/MPPs, (B and B′) Lin−CD34hiCD38−CD45RA+ LMPPs, (C and C′) Lin−CD34+CD38+CD45RA− CMP/MEPs, and (D and D′) Lin−CD34+CD38+CD45RA+ GMPs. Each point represents a unique pairing of a control HSPC donor and CLL patient, with lines connecting the pairs (n = 8 for direct co-culture and n = 7 for Transwell co-cultures, 8 individual experiments). ∗p < 0.05, ∗∗p < 0.01, and ns by paired t test.
(E) Summary table of the in vitro co-culture results. Changes to expression of each TF is indicated by ↑ = increased, ↓ = decreased, and --- = no change.
See also Figures S5, S6, and S8.
In contrast to the direct co-culture results, anti-TNFα did not significantly alter PU.1 levels in HSC/MPPs, LMPPs, or CMP/MEPs in Transwell cultures (Figure 3A’–C′), which was expected, as CLL-derived soluble factors alone did not alter PU.1 expression. However, a reduction in PU.1 was observed in GMPs (Figure 3D’). GATA-2 expression was reduced in HSC/MPPs and CMP/MEPs in Transwell cultures supplemented with anti-TNFα (Figure 3A’ and 3C′). Neutralizing TNFα did not alter GATA-2 levels in LMPPs or GMPs (Figure 3B’ and 3D′). This was expected of LMPPs, as they were refractory to CLL-derived soluble factors in the Transwell culture. The unchanging levels of GATA-2 in GMPs suggest a TNFα-independent effect for GATA-2 modulation in this population. GATA-1 protein remained unchanged upon neutralization of TNFα in all subsets except CMP/MEPs, where it was elevated (Figure 3C’). The effects of anti-TNFα in vitro are summarized in Figure 3E. Importantly, complete normalization of PU.1, GATA-2, and GATA-1 was observed when anti-TNFα was added to recombinant TNFα-exposed control progenitors, confirming effective TNFα neutralization in the co-culture model (Figure S6B).
We next compared media controls with direct CLL B cell:HSPC co-cultures that were subjected to TNFα neutralization to further investigate TNFα-specific modulation (Figure S8A). Compared with media controls, neutralization of TNFα normalized PU.1 in all populations except CMP/MEPs, where PU.1 remained slightly elevated. Anti-TNFα completely restored GATA-2 to control levels in all progenitor subsets except LMPPs, where it was slightly decreased. Notably, although direct co-culture did not change GATA-1 levels, neutralization of TNFα slightly elevated GATA-1 in CMP/MEPs.
When the same comparisons were made in Transwell co-cultures (Figure S8B), no changes were observed in PU.1. However, GATA-2 was slightly decreased in HSC/MPPs and restored in LMPPs, CMP/MEPs, and GMPs. GATA-1 also demonstrated differential responses. GATA-1 was reduced among HSC/MPP and CMP/MEP, but not LMPP and GMP, progenitors. Together, these data demonstrate that CLL B-cell-derived TNFα directly modulates aberrant expression of PU.1 and GATA-2 (and to a lesser extent GATA-1) in BM HSPCs. Further, given that complete TNFα neutralization does not restore every population to media control levels, other CLL-derived factors are likely contributing to TF modulation in BM progenitor cells.
Single-cell analysis of CLL:HSPC co-cultures reveal distinct mechanisms of TF modulation in HSPCs by CLL B cells
To investigate HSPC population changes in our co-culture model at the single-cell level, we performed UMAP analysis collectively of all in vitro experiments (Figures 4 and S3B). The general single-cell distribution of the media controls revealed segregation into three distinct regions that were largely maintained across all experimental conditions. However, unique variations to the overall UMAP architecture were induced by individual experimental manipulations. Next, we assessed the relative expression profiles of PU.1, GATA-2, and GATA-1 across the in vitro UMAP architecture for each experimental condition. Overall, the media controls exhibited high heterogeneity. The in vitro UMAP region 1 exhibited low levels of PU.1 and GATA-1 with intermediate levels of GATA-2. The second region displayed high levels of PU.1 and GATA-2, with lower, yet varying, levels of GATA-1. The third region was comprised of high levels of GATA-2 and GATA-1 with lower levels of PU.1. Addition of recombinant TNFα shifted control progenitors into regions with high levels of PU.1 and GATA-2 with variable, yet declining, GATA-1 expression. When the recombinant TNFα was neutralized, virtually identical UMAP architecture and TF patterning was observed when compared with the media controls.
Figure 4.

Single-cell UMAP analysis of HSPC expression of PU.1, GATA-2, and GATA-1 induced by CLL B cell direct or Transwell co-culture
The relative expression of PU.1, GATA-2, and GATA-1 was overlaid on each individual media (n = 20), TNFα (n = 10), TNFα+anti-TNFα (n = 6), CLL direct co-culture (n = 12), CLL + anti-TNFα direct co-culture (n = 9), CLL Transwell (TW) co-culture (n = 7), and CLL + anti-TNFα TW co-culture (n = 7) in vitro condition UMAP. The expression values and colors of each factor are normalized and relative to themselves. Each point in the UMAP represents a single cell and the total data are comprised of 20 individual experiments.
Direct co-culture with CLL cells shifted control HSPCs to a unique signature of highly variable TF expression (Figure 4). Of note, the majority of cells in this culture condition expressed high levels of PU.1 and GATA-2 while maintaining a similar GATA-1 signature compared with media controls. Interestingly, the signature adopted when TNFα was neutralized in the CLL direct co-culture maintained regions of high PU.1 and GATA-2 expression, yet with a different cellular distribution among the UMAP architecture. The bimodal expression of GATA-1 was maintained. When CLL-derived soluble factors were evaluated in Transwell co-cultures, another unique signature was observed. Compared with media controls, relative levels of PU.1 were increased while GATA-2 and GATA-1 were maintained. Neutralization of TNFα in the Transwell co-cultures largely restored control HSPC TF expression to that of the media control signature, although slight differences were observed, such as an overall reduction in PU.1 levels. Collectively, the UMAP analysis of the in vitro co-culture conditions revealed distinct alterations in the expression of PU.1, GATA-2, and GATA-1 at the single-cell level. Furthermore, direct CLL B cell:HSPC contact versus exposure to CLL soluble factors induced distinct TF signatures within specific HSPC populations and at the single-cell level.
Partial restoration of myelopoiesis upon removal of HSCs from the leukemic bone marrow microenvironment
We previously reported reduced frequencies and functional impairment of BM HPSCs analyzed immediately ex vivo from untreated CLL patients (Manso et al., 2019). We next sought to determine if the cell-intrinsic functional defect documented in the CLL HSPCs analyzed ex vivo was reversible with prolonged removal from the leukemic microenvironment. To make this determination, we utilized the long-term culture initiating cell (LTC-IC) assay (Liu et al., 2013) to test the ability of control and CLL-derived HSCs to initiate and sustain myelopoiesis for 5 weeks across serial input cell concentrations (Figure 5A). As determined by frequencies of wells containing clonal progeny, we observed that CLL myeloid-biased HSCs generated significantly fewer clonal progeny, particularly early in the culture period. This finding is likely a result of maintaining biologic alterations incurred from their in vivo exposure to CLL B cell contact, secreted factors, or other features of a CLL-modified microenvironment. Interestingly, some individual CLL donor HSCs recovered functional capacity and were indistinguishable from controls by weeks 4–5 of culture, although the overall response remained diminished. Notably, this effect was not a function of altered CD34+ isolation purity between controls and CLL patients (Figure 5B). Importantly, when limiting dilution analysis (LDA) was performed (Hu and Smyth, 2009), a near 2-fold decrease in myelopoiesis-potentiating HSC precursor frequency was observed among CLL patients (Figures 5C and 5D).
Figure 5.

CLL HSPCs are functionally compromised, and the dysfunction is recapitulated by exposure to TNFα
(A) CD34+ HSPCs were isolated from control (green) and CLL (blue) BM and plated in the long-term culture initiating cell (LTC-IC) assay under limiting dilution conditions (12 or 36 replicates per dilution). Cultures were evaluated weekly for presence of clonal progeny (as evidenced by the occurrence of at least one colony). Data are presented as the mean ± SD frequency of wells containing clonal progeny. Weeks 1–4 represent n = 4 for both controls and CLL, with week 5 data containing n = 8 controls and n = 9 CLL donors across 18 individual experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗, p < 0.001, and ∗∗∗∗p < 0.0001 by two-way ANOVA.
(B) When available, surplus isolated CD34+ cells (see Transparent methods) were tested by flow cytometry for purity (Control n = 6, CLL n = 4). ns by Mann-Whitney U test. Due to the reduced frequency of CD34+ cells in CLL BM (Manso et al., 2019) and limiting volumes of fresh BM, not every donor was able to be evaluated.
(C) Limiting dilution analysis (LDA) was performed at each time point for control and CLL LTC-IC assays to estimate numbers of responding HSCs among the input CD34+ cells. Circles indicate individual data points, with downward-facing triangles representing a negative (zero) response. Solid lines are the dilution model's fit, with dashed lines indicating the 95% confidence interval. Each time point is represented by a unique color as indicated.
(D) LDA estimates of 1/(stem cell frequency) for control and CLL HSPCs at each time point and fold difference in estimated precursor cell frequency.
(E) LTC-IC assays were performed at limited cell dilutions (12 replicates each) of control BM HSPCs with media alone or supplemented with the indicated concentrations of TNFα. Cells were scored weekly as in (A). Data are presented as the mean ± SD frequency of wells containing clonal progeny at each time point and dilution. n = 4 and is representative of three individual experiments. Statistical comparisons between each dilution at each time point are indicated in the accompanying data tables. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001, and ns by two-way ANOVA adjusted for multiple comparisons (Tukey).
Chronic exposure to TNFα reduces myeloid-biased HSC clonality
Given the impact of TNFα on progenitor TF profiles that regulate myelopoiesis, we next determined the contribution of TNFα to myeloid dysfunction. Confirming previous reports (Broxmeyer et al., 1986; Geissler et al., 1991; Skobin et al., 2000), addition of TNFα to CFU assays significantly reduced colony formation, which was restored when TNFα was neutralized (data not shown). Given these findings, we wished to determine if chronic exposure to TNFα would recapitulate the functional decrease of CLL-derived myeloid HSCs. As it is difficult to experimentally mimic the exact local in vivo TNFα concentrations in CLL patient BM HSPC niches, we performed a dilution series of TNFα in the LTC-IC assay with control BM HSPCs. As previously reported for CD34+CD38- hematopoietic progenitors (Dybedal et al., 2001; Maguer-Satta et al., 2000; Petzer et al., 1996), chronic TNFα exposure of control HSPCs resulted in a dose-dependent inhibition of myelopoiesis (Figure 5E). Interestingly, chronic low-dose exposure effects were more prominent at early culture time points and resolved over time. Importantly, the TNFα-induced reduction in clonal progeny recapitulated the diminished ex vivo CLL functional responses shown in Figure 5A. These data further reinforce a role for TNFα in diminishing myelopoiesis in CLL patient BM.
Discussion
Here, we report a mechanism for diminished BM myelopoiesis in CLL patients driven by leukemic B-cell-derived TNFα. Combining ex vivo and in vitro experimental findings, we show that CLL B cells directly modulate PU.1 and GATA-2 expression in BM HSCs and myelo-erythroid progenitor subsets at the single-cell level. We pursued mechanisms of TF modulation and found that TNFα, produced by the CLL clone, plays an integral role. Importantly, short-term TNFα modulation of PU.1 and GATA-2 is reversible as neutralization of TNFα normalized changes in expression of PU.1 and GATA-2 induced by CLL cells. CLL is a chronic disease and BM infiltration is found even in early stage patients. We determined that removal of HSCs from the leukemic BM microenvironment restored myelopoietic function, although numbers of myeloid-biased HSCs were significantly reduced. Building on this observation, we show that chronic in vitro exposure of control age-matched HSPCs to a range of TNFα concentrations, including low dose and approximating levels reported to be produced by CLL B cells in vitro(Rosati et al., 2005), reduced the numbers and function of myeloid-biased HSCs, largely mimicking CLL patient clinical data. The experimental findings detailed herein provide new insight into the basis of BM hematopoietic dysfunction in CLL patients that likely contribute to impaired immune status.
Our study highlights CLL B-cell-derived TNFα as a major driver of impaired BM myelopoiesis in CLL patients. We focused on TNFα, as it is constitutively produced by CLL B cells (Foa et al., 1990; Michalevicz et al., 1991), has a well-described role in modulating myelopoiesis (Buck et al., 2009; Dybedal et al., 2001; Grigorakaki et al., 2011; Tian et al., 2014), and is implicated in CLL disease progression (Bojarska-Junak et al., 2008). TNFα is produced in two trimeric forms, soluble and membrane-bound, each of which preferentially binds to either TNF receptor 1 or TNF receptor 2 (TNFRI and TNFRII), respectively (Grell et al., 1995). Signaling through both TNF receptors is required for HSC regulation (Grell et al., 1995; Pronk et al., 2011), a critical consideration in Transwell cultures, where membrane-bound TNFα-TNFRII interaction is inhibited, resulting in differential responses compared with direct co-culture. Although we cannot discount the complex combinatorial effects that multiple cell-cell contacts and/or soluble mediators between CLL B cells and HSPCs may have, the reversal of PU.1 and GATA-2 protein elevation by anti-TNFα strongly implicate TNFα in mediating, at least in part, CLL-induced impaired myelopoiesis. Further, when the in vitro cultures and CFU assays were performed with interleukin-10 (IL-10), another cytokine constitutively produced by CLL B cells that is elevated in patient serum (DiLillo et al., 2013; Fayad et al., 2001) and has HSC modulating activity (Kang et al., 2007), no alterations were observed, further demonstrating the specific activity of CLL-derived TNFα (Figure S9). Lastly, the alterations in myelopoiesis we document do not require remodeling of the BM microenvironment, as the changes in expression levels of PU.1 and GATA-2 could be reproduced in short-term in vitro co-cultures comprised of CLL B cells and normal HSPCs that were devoid of stromal and other niche supportive cells.
In CLL patient BM, the hematopoietic dysfunction we show is associated with elevated levels of PU.1, GATA-2, and GATA-1 simultaneously in specific HSPC subsets at the single-cell level. Signaling pathways activated by cytokines influence cell fate trajectories by modulating the activity and/or expression levels of lineage-determining TFs. Here, we focused on TNFα, as levels of this cytokine are elevated in CLL BM (Bojarska-Junak et al., 2008) and CLL-related anemia has been shown to be strongly linked to TNFα(Tsopra et al., 2009). Our in vitro direct CLL B cell:control HSPC co-culture model showed increased PU.1 expression in primitive HSC/MPPs via CLL-derived TNFα. GATA-2 represses the GATA-2/GATA-1 switch critical for erythroid commitment (Bresnick et al., 2012; Moriguchi and Yamamoto, 2014) and was also elevated in progenitors exposed to CLL-derived TNFα. It is well established that overexpression of PU.1 or GATA-2 is disruptive for proper erythroid lineage commitment (Grigorakaki et al., 2011). GATA-2 can also directly antagonize PU.1 (Walsh et al., 2002; Zhang et al., 2000), suppressing myelopoiesis. We show, for the first time, that direct contact between CLL cells and HSPCs is likely required in vivo to increase HSPC expression levels of PU.1 via TNFα, as the PU.1 levels were normalized upon TNFα neutralization. Similarly, we found that CLL B cell direct contact and, to a certain extent, CLL B cell soluble factors (including TNFα), can upregulate GATA-2. Collectively, simultaneously increased levels of PU.1, GATA-2, and GATA-1 may antagonize erythroid and myeloid cell fate programs (Arinobu et al., 2007; Bresnick et al., 2012; Burda et al., 2010; Moriguchi and Yamamoto, 2014; Walsh et al., 2002; Wolff and Humeniuk, 2013), subverting lineage commitment and differentiation, resulting in diminished mature blood cell output from the BM resulting in cytopenias and diminished immune competence.
To further understand the biology of BM hematopoiesis in CLL we also examined ex vivo phenotypic changes among control and CLL HSPCs at the single-cell level using UMAP dimensionality reduction. Distinct global architectures were observed that demonstrated that CLL-derived BM HSPCs are distinct from healthy controls and express fundamentally altered TF and cell surface marker profiles. Intriguingly, the CLL-enriched UMAP region 4 displayed high levels of CD38 with intermediate and variable CD45RA expression, phenotypically overlapping with CMP/MEPs. CLL HSPCs in region 4 also express higher levels of PU.1 and GATA-2 globally and at the single-cell level. We hypothesize that the phenotypic CMP/MEPs may be unable to resolve developmental transitions due to cross-antagonism between TFs. Conversely, the CLL-specific UMAP region 5 displayed a unique expression pattern of low CD38 and high CD45RA, phenotypically representative of LMPPs. Interestingly, this population displays low levels of PU.1, GATA-2, and GATA-1. Given that region 5 is notably enriched in our untreated CLL cohort, and does not express TFs indicative of myeloid or erythroid fates, it is tempting to speculate that this region is enriched for cells that are a CLL stem cell, a CLL-modified LMPP population, and/or a population protected from CLL-induced cellular reductions and is therefore overrepresented.
This study provides, for the first time, a unique and targetable mechanism by which CLL cells negatively regulate BM myelopoiesis. Investigation of mechanisms set a framework for designing interventions that could ameliorate clinically important cytopenias that strongly impact immune competency and clinical outcomes. Importantly, our study challenges the existing paradigm of peripheral immunosuppression as the only mechanism inducing immune cell dysfunction in CLL patients. Together, these new findings explain, in part, the etiology of cytopenias in CLL, particularly of the myelo-erythroid lineage (Manso et al., 2019; Zent et al., 2008). Advances in understanding BM hematopoietic dysfunction in CLL patients offers targetable mechanism(s) for the treatment and reversal of CLL-induced cytopenia and accompanying immunosuppression.
Limitations of the study
A caveat of this study is that it relied on the utilization of freshly prepared, untreated CLL samples and each individual CLL patient sample to be co-cultured with unique control donor HSPCs collected on the same day. This unavoidable constraint imposed by the availability of human experimental research material impaired our ability to further stratify results by clinically relevant CLL patient features (Table S1). We did not measure soluble TNFα levels in the co-cultures. TNFα levels in BM plasma are significantly higher than blood plasma and correlate with disease stage and lymphocytosis (Bojarska-Junak et al., 2008). Regardless, it is very difficult to know precise physiologic levels of secreted or membrane-bound TNFα in the in vivo BM HSPC niche and thus the relevance to in vivo or in vitro levels cannot be determined. Multiple studies have reported that CLL B cells constitutively produce TNFα in vitro in the 20pg/mL range that can be effectively neutralized by an anti-TNF antibody (Aue et al., 2011; Djurdjevic et al., 2009; Rosati et al., 2005). This is important as the levels of TNFα production in vitro are strikingly consistent with reported serum levels of TNFα in patients, which was validated in an independent study with a different cohort of CLL patients (Ferrajoli et al., 2002). An interesting, but potentially relevant, observation by another group was that cellular release of TNFα by primary CLL B cells was higher in early stage (Rai 0–1) compared with advanced stage (Rai II–III) patients, and the majority of CLL B cells used in our in vitro co-cultures were from early stage patients (Foa et al., 1990) (Table S1). Regardless, TNFα inhibits human CFU assays at a range of 1–10ng/mL (Skobin et al., 2000). We show that as little as 0.8μg of anti-TNF antibody mediated complete neutralization of the 25ng/mL exogenous TNFα added to our co-cultures (Figure S6), providing confidence that we are achieving complete TNFα neutralization of either recombinant or CLL-derived TNFα. Importantly, our heterogeneous CLL patient and donor input samples gave a striking uniformity of results, suggesting that the modulation of HSPCs is a shared feature of CLL cells. We also note that TNFα signaling promotes CLL B cell survival and that inhibition of that pathway could result in increased susceptibility to apoptosis (Cordingley et al., 1988; Digel et al., 1989; Durr et al., 2018). However, neutralization of TNFα in our short-term co-culture system did not change the viability of any cell fraction studied (CLL cells or HSPCs).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Kay Medina (medina.kay@mayo.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
This study did not generate or analyze datasets. Further, it did not produce any original code. The code for the R packages used in this manuscript can be found within the references provided.
Methods
All methods can be found in the accompanying Transparent methods supplemental file.
Acknowledgments
We thank Dr. Susan Slager for statistical advice and Susan Schwager for providing CLL patient data. The Mayo Clinic Department of Hematology specimen processing and the Predolin biobanking team provided many of the samples utilized in this study. We also thank Matthew Holets and the Mayo Clinic Charlton Clinical Research and Trials Unit for assisting with donor recruitment and sample collection. This study was supported by Mayo Clinic NIH Grant Relief funding to K.L.M. and NIH T32 funding (NIH T32 AI07425-23) awarded to B.A.M.
Author contributions
B.A.M., K.L.M., and N.E.K. wrote the manuscript. B.A.M., K.A.G., P.K.L., and B.M.W. performed and analyzed all experiments. J.E.K. provided expertise with the R program and assisted with UMAP generation. All authors reviewed and contributed to manuscript revisions.
Declaration of interests
Research funding to S.A.P. has been provided from Pharmacyclics, MorphoSys, Janssen, AstraZeneca, TG Therapeutics, Celgene, AbbVie, and Ascentage Pharma for clinical studies in which S.A.P. is a principal investigator. S.A.P. also participated in Advisory Board meetings of Pharmacyclics, AstraZeneca, Genentech, Gilead, GlaxoSmithKline, Verastem Oncology, and AbbVie (he was not personally compensated for his participation). Research funding to N.E.K. has been provided from Acerta Pharma BV, Celgene, Genentech, Pharmacyclics, and Tolero Pharmaceutical. N.E.K. also participates in the data safety monitoring committees of Agios Pharm, Astra Zeneca, Celgene, Cytomx Therapeutics, Genentech, Infinity Pharm, Morpho-Sys, and Pharmacyclics (he was not personally compensated for his participation). All other authors declare no competing interests.
Published: January 22, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2020.101994.
Supplemental information
References
- Arinobu Y., Mizuno S., Chong Y., Shigematsu H., Iino T., Iwasaki H., Graf T., Mayfield R., Chan S., Kastner P. Reciprocal activation of GATA-1 and PU.1 marks initial specification of hematopoietic stem cells into myeloerythroid and myelolymphoid lineages. Cell Stem Cell. 2007;1:416–427. doi: 10.1016/j.stem.2007.07.004. [DOI] [PubMed] [Google Scholar]
- Aue G., Nelson Lozier J., Tian X., Cullinane A.M., Soto S., Samsel L., McCoy P., Wiestner A. Inflammation, TNFalpha and endothelial dysfunction link lenalidomide to venous thrombosis in chronic lymphocytic leukemia. Am. J. Hematol. 2011;86:835–840. doi: 10.1002/ajh.22114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojarska-Junak A., Hus I., Szczepanek E.W., Dmoszynska A., Rolinski J. Peripheral blood and bone marrow TNF and TNF receptors in early and advanced stages of B-CLL in correlation with ZAP-70 protein and CD38 antigen. Leuk. Res. 2008;32:225–233. doi: 10.1016/j.leukres.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Bresnick E.H., Katsumura K.R., Lee H.Y., Johnson K.D., Perkins A.S. Master regulatory GATA transcription factors: mechanistic principles and emerging links to hematologic malignancies. Nucleic Acids Res. 2012;40:5819–5831. doi: 10.1093/nar/gks281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broxmeyer H.E., Williams D.E., Lu L., Cooper S., Anderson S.L., Beyer G.S., Hoffman R., Rubin B.Y. The suppressive influences of human tumor necrosis factors on bone marrow hematopoietic progenitor cells from normal donors and patients with leukemia: synergism of tumor necrosis factor and interferon-gamma. J. Immunol. 1986;136:4487–4495. [PubMed] [Google Scholar]
- Buck I., Morceau F., Cristofanon S., Reuter S., Dicato M., Diederich M. The inhibitory effect of the proinflammatory cytokine TNFalpha on erythroid differentiation involves erythroid transcription factor modulation. Int. J. Oncol. 2009;34:853–860. doi: 10.3892/ijo_00000212. [DOI] [PubMed] [Google Scholar]
- Burda P., Laslo P., Stopka T. The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis. Leukemia. 2010;24:1249–1257. doi: 10.1038/leu.2010.104. [DOI] [PubMed] [Google Scholar]
- Burger J.A., Quiroga M.P., Hartmann E., Bürkle A., Wierda W.G., Keating M.J., Rosenwald A. High-level expression of the T-cell chemokines CCL3 and CCL4 by chronic lymphocytic leukemia B cells in nurselike cell cocultures and after BCR stimulation. Blood. 2009;113:3050. doi: 10.1182/blood-2008-07-170415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordingley F.T., Hoffbrand A.V., Heslop H.E., Turner M., Bianchi A., Reittie J.E., Vyakarnam A., Meager A., Brenner M.K. Tumour necrosis factor as an autocrine tumour growth factor for chronic B-cell malignancies. Lancet. 1988;331:969–971. doi: 10.1016/s0140-6736(88)91782-5. [DOI] [PubMed] [Google Scholar]
- Digel W., Stefanic M., Schoniger W., Buck C., Raghavachar A., Frickhofen N., Heimpel H., Porzsolt F. Tumor necrosis factor induces proliferation of neoplastic B cells from chronic lymphocytic leukemia. Blood. 1989;73:1242–1246. [PubMed] [Google Scholar]
- DiLillo D.J., Weinberg J.B., Yoshizaki A., Horikawa M., Bryant J.M., Iwata Y., Matsushita T., Matta K.M., Chen Y., Venturi G.M. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia. 2013;27:170–182. doi: 10.1038/leu.2012.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djurdjevic P., Zelen I., Ristic P., Baskic D., Popovic S., Arsenijevic N. Role of decreased production of interleukin-10 and interferon-gamma in spontaneous apoptosis of B-chronic lymphocytic leukemia lymphocytes in vitro. Arch. Med. Res. 2009;40:357–363. doi: 10.1016/j.arcmed.2009.05.007. [DOI] [PubMed] [Google Scholar]
- Doulatov S., Notta F., Laurenti E., Dick J.E. Hematopoiesis: a human perspective. Cell Stem Cell. 2012;10:120–136. doi: 10.1016/j.stem.2012.01.006. [DOI] [PubMed] [Google Scholar]
- Durr C., Hanna B.S., Schulz A., Lucas F., Zucknick M., Benner A., Clear A., Ohl S., Ozturk S., Zenz T. Tumor necrosis factor receptor signaling is a driver of chronic lymphocytic leukemia that can be therapeutically targeted by the flavonoid wogonin. Haematologica. 2018;103:688–697. doi: 10.3324/haematol.2017.177808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dybedal I., Bryder D., Fossum A., Rusten L.S., Jacobsen S.E. Tumor necrosis factor (TNF)-mediated activation of the p55 TNF receptor negatively regulates maintenance of cycling reconstituting human hematopoietic stem cells. Blood. 2001;98:1782–1791. doi: 10.1182/blood.v98.6.1782. [DOI] [PubMed] [Google Scholar]
- Etzrodt M., Ahmed N., Hoppe P.S., Loeffler D., Skylaki S., Hilsenbeck O., Kokkaliaris K.D., Kaltenbach H.M., Stelling J., Nerlov C. Inflammatory signals directly instruct PU.1 in HSCs via TNF. Blood. 2019;133:816–819. doi: 10.1182/blood-2018-02-832998. [DOI] [PubMed] [Google Scholar]
- Fayad L., Keating M.J., Reuben J.M., O'Brien S., Lee B.N., Lerner S., Kurzrock R. Interleukin-6 and interleukin-10 levels in chronic lymphocytic leukemia: correlation with phenotypic characteristics and outcome. Blood. 2001;97:256–263. doi: 10.1182/blood.v97.1.256. [DOI] [PubMed] [Google Scholar]
- Fecteau J.F., Kipps T.J. Structure and function of the hematopoietic cancer niche: focus on chronic lymphocytic leukemia. Front. Biosci. (Schol Ed. 2012;4:61–73. doi: 10.2741/251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrajoli A., Keating M.J., Manshouri T., Giles F.J., Dey A., Estrov Z., Koller C.A., Kurzrock R., Thomas D.A., Faderl S. The clinical significance of tumor necrosis factor-alpha plasma level in patients having chronic lymphocytic leukemia. Blood. 2002;100:1215–1219. [PubMed] [Google Scholar]
- Foa R., Massaia M., Cardona S., Tos A.G., Bianchi A., Attisano C., Guarini A., di Celle P.F., Fierro M.T. Production of tumor necrosis factor-alpha by B-cell chronic lymphocytic leukemia cells: a possible regulatory role of TNF in the progression of the disease. Blood. 1990;76:393–400. [PubMed] [Google Scholar]
- Forconi F., Moss P. Perturbation of the normal immune system in patients with CLL. Blood. 2015;126:573–581. doi: 10.1182/blood-2015-03-567388. [DOI] [PubMed] [Google Scholar]
- Geissler R.G., Ottmann O.G., Eder M., Kojouharoff G., Hoelzer D., Ganser A. Effect of recombinant human transforming growth factor beta and tumor necrosis factor alpha on bone marrow progenitor cells of HIV-infected persons. Ann. Hematol. 1991;62:151–155. doi: 10.1007/BF01703139. [DOI] [PubMed] [Google Scholar]
- Goldman D. Chronic lymphocytic leukemia and its impact on the immune system. Clin. J. Oncol. Nurs. 2000;4:233–234. 236. [PubMed] [Google Scholar]
- Grell M., Douni E., Wajant H., Löhden M., Clauss M., Maxeiner B., Georgopoulos S., Lesslauer W., Kollias G., Pfizenmaier K. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell. 1995;83:793–802. doi: 10.1016/0092-8674(95)90192-2. [DOI] [PubMed] [Google Scholar]
- Grigorakaki C., Morceau F., Chateauvieux S., Dicato M., Diederich M. Tumor necrosis factor alpha-mediated inhibition of erythropoiesis involves GATA-1/GATA-2 balance impairment and PU.1 over-expression. Biochem. Pharmacol. 2011;82:156–166. doi: 10.1016/j.bcp.2011.03.030. [DOI] [PubMed] [Google Scholar]
- Hu Y., Smyth G.K. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods. 2009;347:70–78. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- Janel A., Dubois-Galopin F., Bourgne C., Berger J., Tarte K., Boiret-Dupre N., Boisgard S., Verrelle P., Dechelotte P., Tournilhac O. The chronic lymphocytic leukemia clone disrupts the bone marrow microenvironment. Stem Cells Dev. 2014;23:2972–2982. doi: 10.1089/scd.2014.0229. [DOI] [PubMed] [Google Scholar]
- Kang Y.J., Yang S.J., Park G., Cho B., Min C.K., Kim T.Y., Lee J.S., Oh I.H. A novel function of interleukin-10 promoting self-renewal of hematopoietic stem cells. Stem Cells. 2007;25:1814–1822. doi: 10.1634/stemcells.2007-0002. [DOI] [PubMed] [Google Scholar]
- Kay N.E., Bone N.D., Tschumper R.C., Howell K.H., Geyer S.M., Dewald G.W., Hanson C.A., Jelinek D.F. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia. 2002;16:911. doi: 10.1038/sj.leu.2402467. [DOI] [PubMed] [Google Scholar]
- Kim M., Pyo S., Kang C.H., Lee C.O., Lee H.K., Choi S.U., Park C.H. Folate receptor 1 (FOLR1) targeted chimeric antigen receptor (CAR) T cells for the treatment of gastric cancer. PLoS One. 2018;13:e0198347. doi: 10.1371/journal.pone.0198347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipps T.J., Stevenson F.K., Wu C.J., Croce C.M., Packham G., Wierda W.G., O'Brien S., Gribben J., Rai K. Chronic lymphocytic leukaemia. Nat. Rev. Dis. Primers. 2017;3:17008. doi: 10.1038/nrdp.2017.8. [DOI] [PubMed] [Google Scholar]
- Lagneaux L., Delforge A., Dorval C., Bron D., Stryckmans P. Excessive production of transforming growth factor-beta by bone marrow stromal cells in B-cell chronic lymphocytic leukemia inhibits growth of hematopoietic precursors and interleukin-6 production. Blood. 1993;82:2379–2385. [PubMed] [Google Scholar]
- Lahat N., Aghai E., Maroun B., Kinarty A., Quitt M., Froom P. Increased spontaneous secretion of IL-6 from B cells of patients with B chronic lymphatic leukaemia (B-CLL) and autoimmunity. Clin. Exp. Immunol. 1991;85:302–306. doi: 10.1111/j.1365-2249.1991.tb05723.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leland McInnes J.H. UMAP: uniform manifold approximation and projection for dimension reduction. 2018. https://arxivorg/abs/180203426
- Liu M., Miller C.L., Eaves C.J. Human long-term culture initiating cell assay. Methods Mol. Biol. 2013;946:241–256. doi: 10.1007/978-1-62703-128-8_15. [DOI] [PubMed] [Google Scholar]
- Liu T., Zhang L., Joo D., Sun S.C. NF-kappaB signaling in inflammation. Signal Transduct. Target. Ther. 2017;2:17023. doi: 10.1038/sigtrans.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotz M., Ranheim E., Kipps T.J. Transforming growth factor beta as endogenous growth inhibitor of chronic lymphocytic leukemia B cells. J. Exp. Med. 1994;179:999. doi: 10.1084/jem.179.3.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguer-Satta V., Oostendorp R., Reid D., Eaves C.J. Evidence that ceramide mediates the ability of tumor necrosis factor to modulate primitive human hematopoietic cell fates. Blood. 2000;96:4118–4123. [PubMed] [Google Scholar]
- Manso B.A., Zhang H., Mikkelson M.G., Gwin K.A., Secreto C.R., Ding W., Parikh S.A., Kay N.E., Medina K.L. Bone marrow hematopoietic dysfunction in untreated chronic lymphocytic leukemia patients. Leukemia. 2019;33:638–652. doi: 10.1038/s41375-018-0280-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means R.T., Jr., Dessypris E.N., Krantz S.B. Inhibition of human colony-forming-unit erythroid by tumor necrosis factor requires accessory cells. J. Clin. Invest. 1990;86:538–541. doi: 10.1172/JCI114741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalevicz R., Porat R., Vechoropoulos M., Baron S., Yanoov M., Cycowitz Z., Shibolet S. Restoration of in vitro hematopoiesis in B-chronic lymphocytic leukemia by antibodies to tumor necrosis factor. Leuk. Res. 1991;15:111–120. doi: 10.1016/0145-2126(91)90091-7. [DOI] [PubMed] [Google Scholar]
- Moriguchi T., Yamamoto M. A regulatory network governing Gata1 and Gata2 gene transcription orchestrates erythroid lineage differentiation. Int. J. Hematol. 2014;100:417–424. doi: 10.1007/s12185-014-1568-0. [DOI] [PubMed] [Google Scholar]
- Nakagawa M.M., Chen H., Rathinam C.V. Constitutive activation of NF-kappaB pathway in hematopoietic stem cells causes loss of quiescence and deregulated transcription factor networks. Front. Cell Dev. Biol. 2018;6:143. doi: 10.3389/fcell.2018.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novershtern N., Subramanian A., Lawton L.N., Mak R.H., Haining W.N., McConkey M.E., Habib N., Yosef N., Chang C.Y., Shay T. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144:296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang W.W., Schrier S.L., Weissman I.L. Age-associated changes in human hematopoietic stem cells. Semin. Hematol. 2017;54:39–42. doi: 10.1053/j.seminhematol.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Petzer A.L., Zandstra P.W., Piret J.M., Eaves C.J. Differential cytokine effects on primitive (CD34+CD38-) human hematopoietic cells: novel responses to Flt3-ligand and thrombopoietin. J. Exp. Med. 1996;183:2551–2558. doi: 10.1084/jem.183.6.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pronk C.J., Veiby O.P., Bryder D., Jacobsen S.E. Tumor necrosis factor restricts hematopoietic stem cell activity in mice: involvement of two distinct receptors. J. Exp. Med. 2011;208:1563–1570. doi: 10.1084/jem.20110752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosati E., Sabatini R., Tabilio A., Di Ianni M., Bartoli A., Marconi P. B-chronic lymphocytic leukemia cells exert an in vitro cytotoxicity mediated by tumor necrosis factor alpha. Leuk. Res. 2005;29:829–839. doi: 10.1016/j.leukres.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Rothhammer V., Borucki D.M., Kenison J.E., Hewson P., Wang Z., Bakshi R., Sherr D.H., Quintana F.J. Detection of aryl hydrocarbon receptor agonists in human samples. Sci. Rep. 2018;8:4970. doi: 10.1038/s41598-018-23323-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala R., Mauro F.R., Bellucci R., De Propris M.S., Cordone I., Lisci A., Foa R., de Fabritiis P. Evaluation of marrow and blood haemopoietic progenitors in chronic lymphocytic leukaemia before and after chemotherapy. Eur. J. Haematol. 1998;61:14–20. doi: 10.1111/j.1600-0609.1998.tb01055.x. [DOI] [PubMed] [Google Scholar]
- Saulep-Easton D., Vincent F.B., Quah P.S., Wei A., Ting S.B., Croce C.M., Tam C., Mackay F. The BAFF receptor TACI controls IL-10 production by regulatory B cells and CLL B cells. Leukemia. 2016;30:163–172. doi: 10.1038/leu.2015.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skobin V., Jelkmann W., Morschakova E., Pavlov A.D., Schlenke P. Tumor necrosis factor-alpha and TNF-beta inhibit clonogenicity of mobilized human hematopoietic progenitors. J. Interferon Cytokine Res. 2000;20:507–510. doi: 10.1089/10799900050023924. [DOI] [PubMed] [Google Scholar]
- Tian T., Wang M., Ma D. TNF-alpha, a good or bad factor in hematological diseases? Stem Cell Investig. 2014;1:12. doi: 10.3978/j.issn.2306-9759.2014.04.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsopra O.A., Ziros P.G., Lagadinou E.D., Symeonidis A., Kouraklis-Symeonidis A., Thanopoulou E., Angelopoulou M.K., Vassilakopoulos T.P., Pangalis G.A., Zoumbos N.C. Disease-related anemia in chronic lymphocytic leukemia is not due to intrinsic defects of erythroid precursors: a possible pathogenetic role for tumor necrosis factor-alpha. Acta Haematol. 2009;121:187–195. doi: 10.1159/000220331. [DOI] [PubMed] [Google Scholar]
- van Attekum M.H., Eldering E., Kater A.P. Chronic lymphocytic leukemia cells are active participants in microenvironmental cross-talk. Haematologica. 2017;102:1469–1476. doi: 10.3324/haematol.2016.142679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente C., Conchillo A., Garcia-Sanchez M.A., Odero M.D. The role of the GATA2 transcription factor in normal and malignant hematopoiesis. Crit. Rev. Oncol. Hematol. 2012;82:1–17. doi: 10.1016/j.critrevonc.2011.04.007. [DOI] [PubMed] [Google Scholar]
- Walsh J.C., DeKoter R.P., Lee H.J., Smith E.D., Lancki D.W., Gurish M.F., Friend D.S., Stevens R.L., Anastasi J., Singh H. Cooperative and antagonistic interplay between PU.1 and GATA-2 in the specification of myeloid cell fates. Immunity. 2002;17:665–676. doi: 10.1016/s1074-7613(02)00452-1. [DOI] [PubMed] [Google Scholar]
- Wang W., Fujii H., Kim H.J., Hermans K., Usenko T., Xie S., Luo Z.J., Ma J., Celso C.L., Dick J.E. Enhanced human hematopoietic stem and progenitor cell engraftment by blocking donor T cell-mediated TNFalpha signaling. Sci. Transl. Med. 2017;9:eaag3214. doi: 10.1126/scitranslmed.aag3214. [DOI] [PubMed] [Google Scholar]
- Wolff L., Humeniuk R. Concise review: erythroid versus myeloid lineage commitment: regulating the master regulators. Stem Cells. 2013;31:1237–1244. doi: 10.1002/stem.1379. [DOI] [PubMed] [Google Scholar]
- Zent C.S., Ding W., Schwager S.M., Reinalda M.S., Hoyer J.D., Jelinek D.F., Tschumper R.C., Bowen D.A., Call T.G., Shanafelt T.D. The prognostic significance of cytopenia in chronic lymphocytic leukaemia/small lymphocytic lymphoma. Br. J. Haematol. 2008;141:615–621. doi: 10.1111/j.1365-2141.2008.07086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Zhang X., Iwama A., Yu C., Smith K.A., Mueller B.U., Narravula S., Torbett B.E., Orkin S.H., Tenen D.G. PU.1 inhibits GATA-1 function and erythroid differentiation by blocking GATA-1 DNA binding. Blood. 2000;96:2641–2648. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate or analyze datasets. Further, it did not produce any original code. The code for the R packages used in this manuscript can be found within the references provided.