

SUMMARY
The antigen-presenting molecule MR1 presents microbial metabolites related to vitamin B2 biosynthesis to mucosal-associated invariant T cells (MAIT cells). Although bacteria and fungi drive the MR1 biosynthesis pathway, viruses have not previously been implicated in MR1 expression or its antigen presentation. We demonstrate that several herpesviruses inhibit MR1 cell surface upregulation, including a potent inhibition by herpes simplex virus type 1 (HSV-1). This virus profoundly suppresses MR1 cell surface expression and targets the molecule for proteasomal degradation, whereas ligand-induced cell surface expression of MR1 prior to infection enables MR1 to escape HSV-1-dependent targeting. HSV-1 downregulation of MR1 is dependent on de novo viral gene expression, and we identify the Us3 viral gene product as functioning to target MR1. Furthermore, HSV-1 downregulation of MR1 disrupts MAIT T cell receptor (TCR) activation. Accordingly, virus-mediated targeting of MR1 defines an immunomodulatory strategy that functionally disrupts the MR1-MAIT TCR axis.
Graphical Abstract
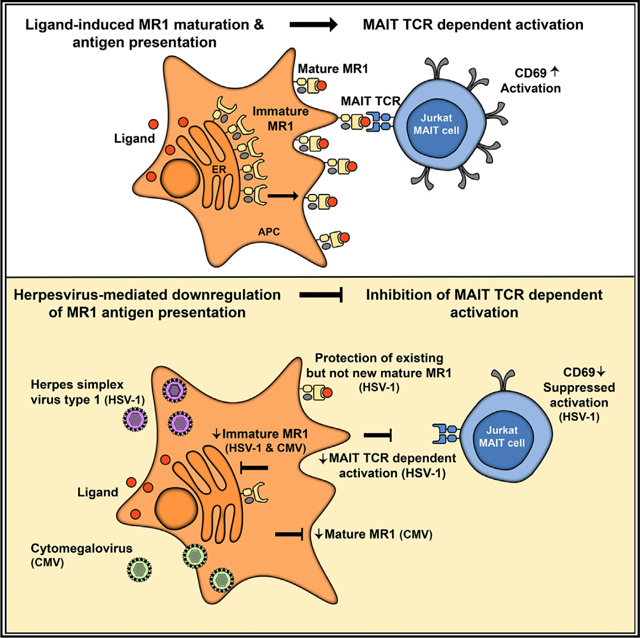
In Brief
The antigen-presenting molecule MR1 presents bacterial and fungal metabolites to MAIT cells. McSharry et al. show that the herpesviruses HSV-1 and CMV disrupt MR1 expression. Downregulation of MR1 by HSV-1 inhibits bacterially driven MAIT TCR-dependent activation. This provides evidence of virus immunomodulatory control of the MR1-restricted immune response.
INTRODUCTION
The major histocompatibility complex (MHC) class I-related gene protein (MR1) is an antigen (Ag) presentation molecule first identified through sequence homology with classical human leukocyte antigen (HLA) molecules (Hashimoto et al., 1995). Like MHC I molecules, the extracellular domain of the MR1 heavy chain has α1, α2, and α3 domains. Unlike MHC I, however, MR1 is monomorphic and is highly conserved across mammalian species (Yamaguchi et al., 1997). A potential biological function for MR1 was initially identified in MR1−/− mice, which lack a subset of T cells known as mucosal associated invariant T cells (MAIT cells) (Treiner et al., 2003). This and subsequent studies have defined MR1 as a restriction element for MAIT cells (Kjer-Nielsen et al., 2012; Treiner et al., 2003).
MAIT cells are primarily a population of αβ T cells, highly abundant in humans, and typically express an invariant T cell receptor (TCR) α chain (TRAV1–2-TRAJ33; Fernandez et al., 2015; Gherardin et al., 2016). MAIT cells are characterized as innate-like cells that have the ability to rapidly respond to a range of bacterial and fungal pathogens (Gold et al., 2010; Howson et al., 2015; Le Bourhis et al., 2013). The mechanism by which MAIT cells can recognize these infections was shown to be via pathogen metabolites derived from a biosynthetic precursor to vitamin B2 (Vit B2), which bind to MR1 to induce trafficking to the cell surface and subsequent TCR-mediated MAIT cell activation (Corbett et al., 2014; Eckle et al., 2014; Kjer-Nielsen et al., 2012; Patel et al., 2013). More recently, it has also been reported that MR1 has the capacity to bind and present a broader repertoire of ligands from both bacterial and non-bacterial sources (Harriff et al., 2018; Keller et al., 2017).
This identification of MR1-bound metabolites has facilitated a characterization of the MR1 processing and presentation pathway. Unlike other antigen presentation molecules, such as MHC I and CD1, that constitutively bind host peptides or lipids, respectively, MR1 primarily resides in an immature state in the endoplasmic reticulum (ER) (McWilliam et al., 2016). Ligand binding to MR1 in the ER triggers a “molecular switch” that drives MR1 to fully mature, associate with β2-microglobulin, and traffic to the cell surface, where it can activate MAIT cells (McWilliam et al., 2016).
Human herpesvirus infections are associated with several significant morbidities. Herpes simplex virus type 1 (HSV-1) is a ubiquitous alpha herpesvirus associated with recurrent orofacial and other infections and remains the most common cause of viral encephalitis (Tyler, 2018). As with all herpesviruses, primary infection is followed by lifelong persistence. It is well established that persistence of herpesvirus infections is facilitated by viral encoded proteins that mediate a multitude of strategies to regulate the host immune response. Definition and elucidation of viral mechanisms of immune control have led to fundamental insights into pathways key to immune effector functions (van de Weijer et al., 2015). Such immunomodulatory activities include the ability of HSV-1 to limit Ag presentation through both MHC I and CD1d to control both classical T cell and NKT cell responses, respectively (Früh et al., 1995; Rao et al., 2011; York et al., 1994; Yuan et al., 2006).
Recently, it has become clear that viral infection can modulate both circulating MAIT cell number and function (Barathan et al., 2016; Cosgrove et al., 2013; Eberhard et al., 2014; Fernandez et al., 2015; Hengst et al., 2016; Hofmann and Thimme, 2016; Paquin-Proulx et al., 2017; Vinton et al., 2016), and MAIT cells can be activated by viral infection; however, this is driven in an MR1-independent fashion by cytokines, including interleukin-12 (IL-12) and IL-18 (Loh et al., 2016; Paquin-Proulx et al., 2018; van Wilgenburg et al., 2016). There have been no reports of a link between viral infection and modulation of MR1. In this study, we have identified multiple herpesviruses that inhibit surface expression of MR1. We focus on that encoded by HSV-1 and identify a viral-encoded immunoevasin that limits MR1 surface expression. This study reports a viral immunomodulatory activity through a pathway previously unidentified as a target of viral infection.
RESULTS
Surface and Cellular MR1 Are Profoundly Downregulated during Herpesvirus Infections
Given the well-defined capacity of a number of viral infections to regulate expression of MHC I and MHC-I-like molecules, the ability of several viruses to modulate MR1 surface expression was tested. Initially, we focused on MR1 regulation during viral infection of fibroblasts. As expected, cell surface expression of MR1 in human fibroblasts (HFFs) in the absence of exogenous ligand was minimal; however, upon addition of the synthetic MR1 ligand, Ac-6-FP, surface MR1 was readily detectable (Figure S1). Infection with the herpesviruses HSV-1, human cytomegalovirus (HCMV), and murine cytomegalovirus (MCMV) prior to ligand addition resulted in a potent inhibition of MR1 surface upregulation in both human and murine fibroblasts, respectively (Figure 1A). In contrast, the DNA virus, adenovirus (Ad) serotype 5, which is well known for its ability to target both MHC and MHC-like molecules (Burgert and Kvist, 1985; Burgert et al., 1987; McSharry et al., 2008), was incapable of inhibiting MR1 surface expression during infection of HFFs (Figure 1A). To extend this initial observation, we confirmed that Ad infection also did not modulate MR1 surface expression in 293 cells, a line in which it has been shown that the Ad-encoded glycoprotein E3/19K can downregulate surface HLA (Figure S2). These data indicate that the ability of HSV-1, HCMV, and MCMV to suppress MR1 was not an inherent response to DNA virus infection.
Figure 1. Viral Infection Inhibits MR1 Protein Expression.
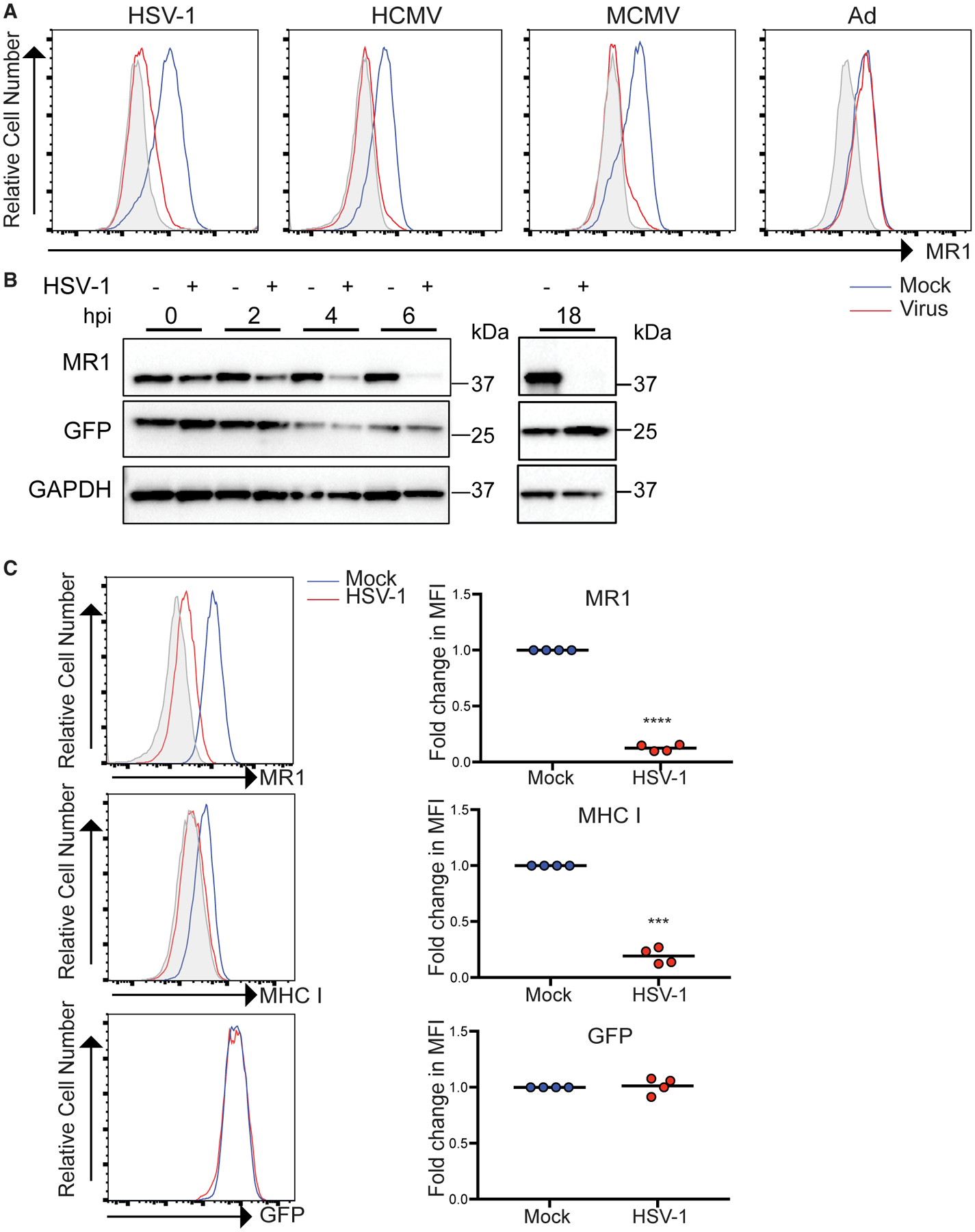
(A) HFFs (HCMV, HSV-1, and Ad) or NIH 3T3 fibroblasts (MCMV) were infected with the indicated viruses. Cells were treated with Ac-6-FP p.i. for 16 h before staining for surface MR1 at the following times p.i. HSV-1 (24 h), HCMV (24 h), Ad (24 h), or MCMV (18 h). Isotype (gray), mock (blue), and virus (red) staining are indicated. Data are representative of at least 3 independent experiments.
(B) Cell lysates from mock or HSV-1-infected ARPE-19 MR1 cells were harvested at 0, 2, 4, 6, and 18 h p.i. before immunoblotting for MR1, GFP, and GAPDH. Data are representative of at least 2 independent experiments.
(C) ARPE-19MR1 were mockor HSV-1 infectedbefore staining for surface MR1,MHC I, or isotype control (gray) at18h p.i.and analysis by flow cytometry. Foldchange relative to mock infected cells is graphed. Statistical significance was calculated by paired Student’s t test; ***p < 0.0005; ****p < 0.0001 (n = 4).
To define the mechanism by which MR1 was targeted during virus infection, we focused on HSV-1. We overexpressed MR1 in an epithelial cell line (ARPE-19) to facilitate western blot analysis of MR1 levels post-infection (p.i.), and co-expressed EGFP from the same promoter via a downstream internal ribosomal entry site (IRES) (McWilliam et al., 2016). A time course analysis (0, 2, 4, and 6 h p.i.) of MR1 protein levels by western blotting in HSV-1-infected cells indicated that MR1 protein was reduced at very early times with a profound loss in MR1 levels obvious by 6 h p.i. and MR1 at near undetectable levels by 18 h p.i. (Figure 1B). At 6 h p.i., there was a significant but limited downregulation (2-fold) of surface levels of MR1 (Figure S3), which by 18 h p.i. showed an almost 10-fold reduction in surface MR1 (Figure 1C), mirroring the loss of total MR1 protein. As previously established (Früh et al., 1995; York et al., 1994), surface MHC I expression was also significantly downregulated by viral infection (Figure 1C). It is important to note that GFP levels, driven from the same expression cassette, were unchanged by HSV-1 infection, as measured by western blotting and flow cytometry at all times assayed (Figures 1B and 1C), suggesting specific targeting of MR1 by HSV-1. Together, this implies that HSV-1 infection can target the MR1 protein to limit surface expression.
Ligand-Induced Surface MR1 Is Protected from HSV-1-Mediated Loss
Current evidence indicates that the functional role for MR1 detected to date is dependent on its ability to present antigen (Kjer-Nielsen et al., 2012). Therefore, we addressed how addition of MR1 ligand affects HSV-1 targeting of MR1. ARPE-19 MR1 cells were either treated pre-infection with the MR1 ligand, Ac-6-FP, or treated at 14 h p.i. before MR1 surface expression was then measured at 18 h p.i. When cells were treated with Ac-6-FP p.i. again there was a potent loss of MR1 surface staining (>40-fold) compared to mock infected cells (Figure 2). Remarkably, when cells were treated with ligand prior to infection, there was no longer a virus-induced suppression of surface MR1 expression, and we even noticed a small but statistically significant increase in MR1 surface expression levels in HSV-1 compared to mock infected cells (Figure 2A). In contrast, MHC I surface expression was strongly inhibited under both conditions (Figure 2), suggesting a fundamental difference in the way in which HSV-1 targets MR1 and MHC I, respectively.
Figure 2. Ligand Binding Blocks HSV-1-Dependent Targeting of MR1.
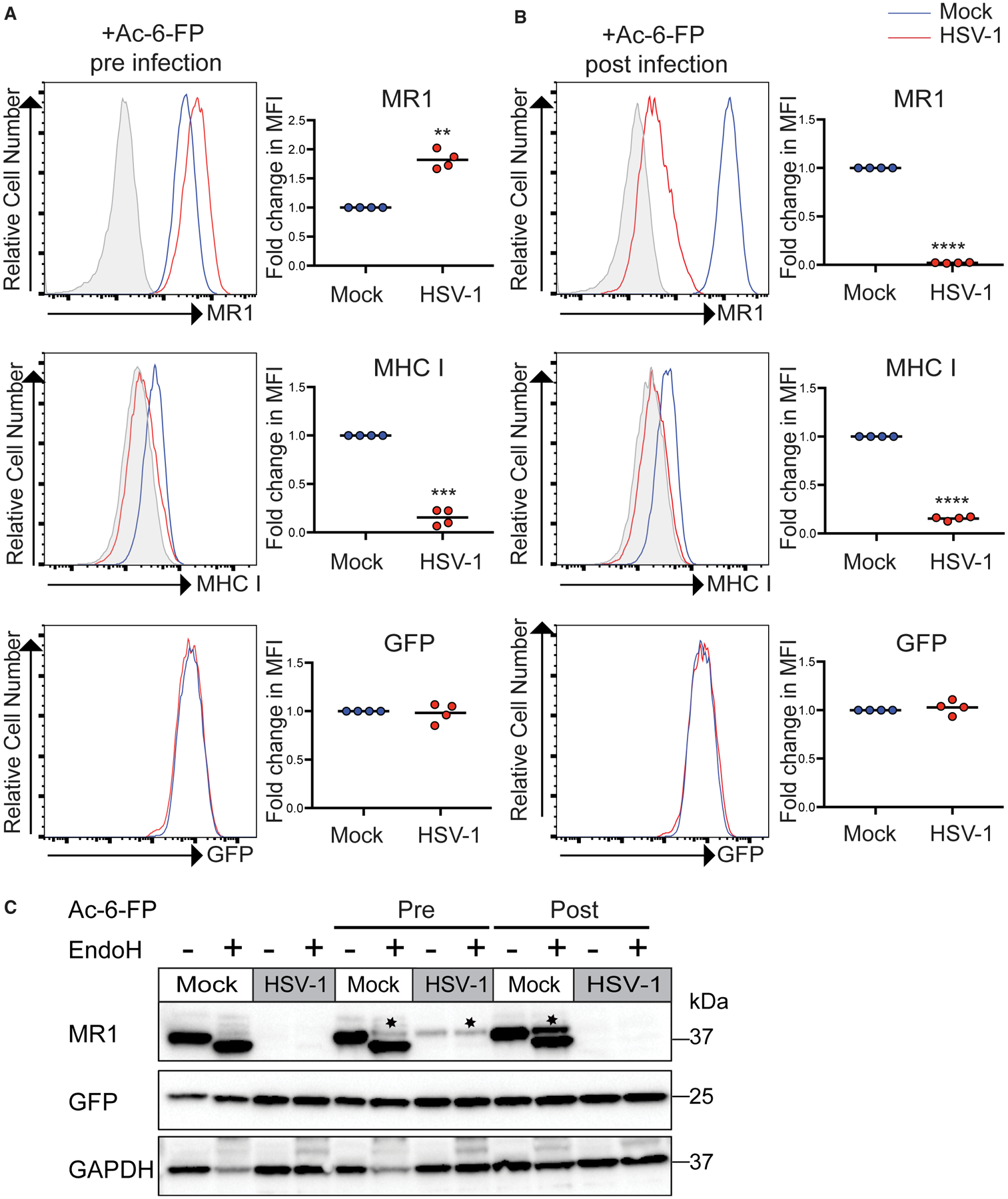
(A and B) ARPE-19 MR1 cells were mock or HSV-1 infected in parallel. Cells were either treated with Ac-6-FP (5 μM; A) for 24 h prior to infection (pre) or (B) at 14 h p.i. (post) before staining for surface MR1, MHC I at 18 h p.i., and analysis by flow cytometry. Fold change relative to mock infected cells is graphed. Statistical significance was calculated by paired Student’s t test; **p < 0.005; ***p < 0.0005; ****p < 0.0001 (n = 4).
(C) ARPE-19 MR1 cells were mock or HSV-1 infected in parallel. Cells were either treated with Ac-6-FP (5 μM) for 24 h prior to infection (pre), at 14 h p.i. (post), or left untreated as indicated. Cell lysates harvested at 18 h p.i. were left undigested or EndoH digested before immunoblotting for MR1, GFP, and GAPDH. Image is representative of two independent experiments. Endo-H-resistant MR1 is denoted with *.
To investigate the mechanism underpinning this regulation of MR1, lysates prepared from mock and HSV-1-infected cells that had been treated with Ac-6-FP pre-infection or p.i. were digested with endoglycosidase H (EndoH) before western blotting as a means to differentiate between the immature MR1 molecules that remain in the ER or the mature MR1 that have been processed through the ER/Golgi apparatus, before western blotting (Figure 2C). As demonstrated previously, Ac-6-FP addition induces the maturation of MR1 (McWilliam et al., 2016) with an EndoH-resistant species detectable in mock infected cells treated with ligand both pre-infection and p.i. In either the cells left untreated or with ligand added p.i., there was an almost complete loss of MR1 protein at 18 h p.i. However, in the cells pre-treated with MR1 ligand, there was a mature EndoH-resistant form of MR1 that was maintained during viral infection, which mirrored the surface MR1 expression observed by flow cytometry. Thus, ligand-bound, mature, surface MR1 appears resistant to HSV-1-dependent MR1 targeting.
This differential targeting of surface MR1, which was dependent on the timing of ligand addition, was also detected in HFFs engineered to overexpress MR1 (Figure S4). Similarly, pre-treatment with Ac-6-FP in parental HFFs protected the endogenously expressed molecule from HSV-1-dependent targeting although MR1 surface expression induced by Ac-6-FP added p.i. was efficiently inhibited by viral infection (Figures 3A and 3B). Interestingly, HCMV infection was capable of potently inhibiting MR1 surface expression whether treated with ligand pre-infection or p.i. in HFFs (Figures 3C and 3D), suggesting distinct herpesviruses do not target MR1 via directly equivalent mechanisms.
Figure 3. Ligand Binding Blocks HSV-1-Mediated, but Not HCMV-Mediated, Inhibition of Endogenous MR1 Surface Expression.
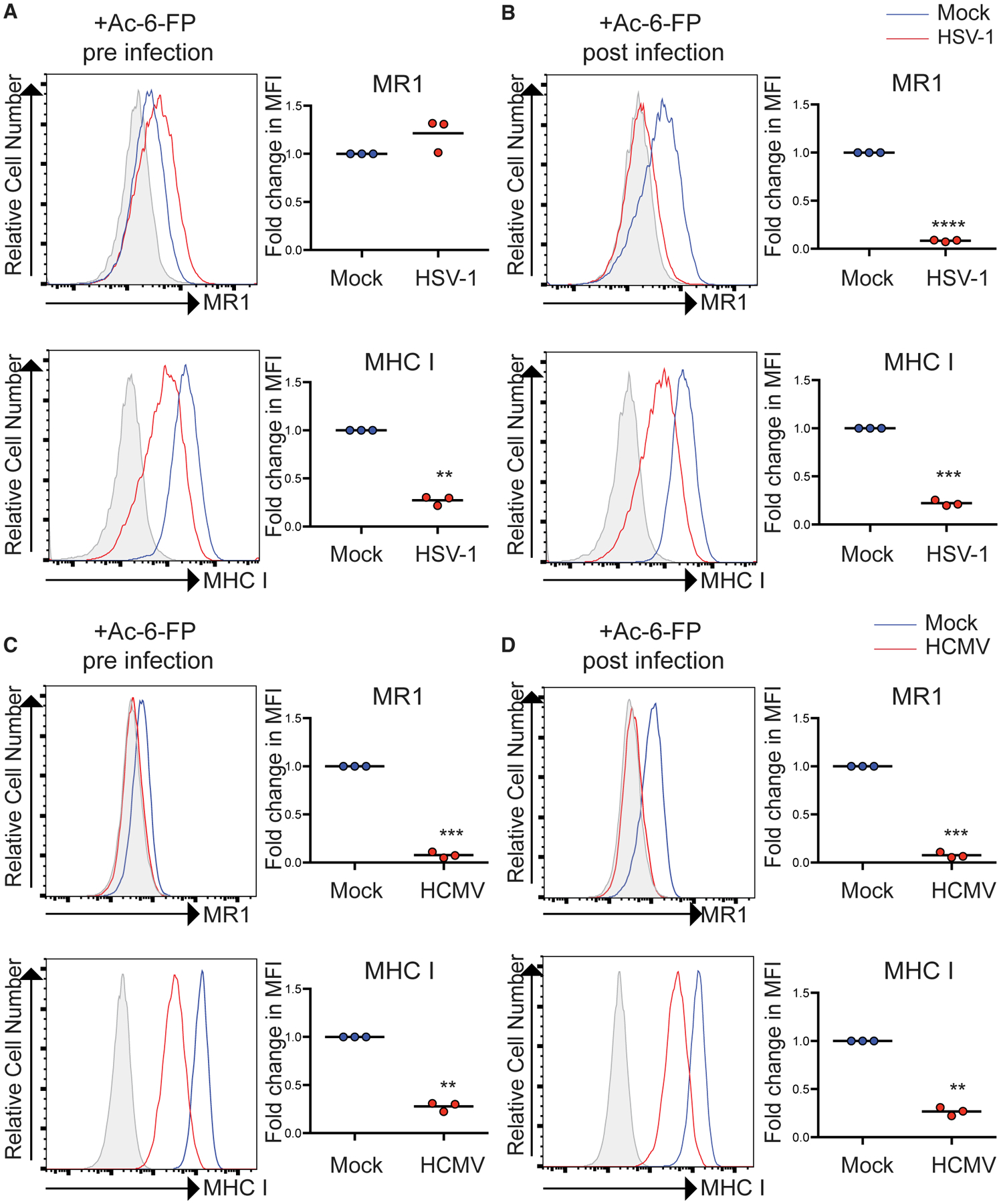
(A and B) HFFs were mock or HSV-1 infected in parallel. Cells were either treated with Ac-6-FP (5 μM) for (A) 24 h prior to infection (pre) or (B) at 14 h p.i. (post) before staining for surface MR1, MHC I at 18 h p.i., and analysis by flow cytometry. Fold change relative to mock infected cells is graphed. Statistical significance was calculated by paired Student’s t test; **p < 0.005; ***p < 0.0005; ****p < 0.0001 (n = 3).
(C and D) HFFs were mock or HCMV infected in parallel. Cells were either treated with Ac-6-FP (5 μM) for (C) 24 h prior to infection (pre) or (D) at 8 h p.i. (post) before staining for surface MR1, MHC I at 24 h p.i., and analysis by flow cytometry. Fold change relative to mock infected cells is graphed. Statistical significance was calculated by paired Student’s t test; **p < 0.005; ***p < 0.0005 (n = 3).
To further investigate HSV-1-dependent targeting of MR1, ARPE-19 cells expressing a C-terminally tagged fusion protein, MR1-GFP, were generated. MR1-GFP-expressing cells demonstrated the same pattern of MR1 surface regulation post-viral expression as untagged MR1, with the timing of ligand addition determining whether surface MR1 expression was protected or inhibited during infection (Figure S5A). In this experimental setup, MR1-GFP fluorescence provided a direct readout for levels of MR1 fusion protein expression that differed from the situation in ARPE-19 MR1 cells, where GFP is expressed via a downstream IRES. MR1-GFP fluorescence levels were found to be reduced in cells treated both pre- and p.i. with Ac-6-FP, and the reduction in MR1-GFP levels was significantly more pronounced when ligand was added p.i. (Figure S5B). This result is consistent with pre-existing, cell-surface-resident, ligand-bound MR1 molecules being resistant to HSV-1-mediated targeting. This was also mirrored by immunoblotting (Figure S5C), in agreement with our observations with untagged MR1.
HSV-1-Mediated Loss of Surface MR1 Inhibits MR1-Mediated Jurkat MAIT Cell Activation
To determine whether the observed viral-dependent targeting of MR1 levels could control MR1-mediated MAIT cell activation, we assayed the ability of viral infection to regulate activation through a MAIT cell TCR. Although Ac-6-FP is an efficient inducer of MR1 surface expression, it does not act as an agonist for MAIT cells. Therefore, we first confirmed that HSV-1 infection was also capable of efficiently inhibiting surface expression driven by the MAIT cell agonist 5-OP-RU (Figure S6A). Ac-6-FP and 5-OP-RU are, for MR1, functionally very similar: they bind to MR1 via a Schiff base (Corbett et al., 2014; Eckle et al., 2014) and upregulate cell surface MR1 to similar levels over the first ~6 h of treatment (Eckle et al., 2014; McWilliam et al., 2016; Mak et al., 2017). However, due to its enhanced stability and relative cost, Ac-6-FP was primarily used for molecular analysis of MR1 regulation during infection with 5-OP-RU used for MAIT functional assays.
Jurkat cells engineered to express a canonical MAIT-cell-specific TCR (Jurkat MAIT) (Reantragoon et al., 2012) were then used to assay MR1 restricted recognition. HFFs were mock or HSV-1 infected before 5-OP-RU (10 μM) or fixed E. coli were added at 14 h p.i. and then, 4 h later, ligand or bacteria were washed from the system and the target cells fixed as described previously (McWilliam et al., 2016). Jurkat MAIT cells were then added to target cells, allowed to incubate overnight before the levels of CD69 expression, as a marker of activation, were measured by flow cytometry. HSV-1 infection alone did not activate Jurkat MAIT cells above mock cells (Figure 4A). Both 5-OP-RU and E. coli-treated HFFs were capable of activating through the MAIT TCR, and this activation was blocked with anti-MR1 antibody (Ab) (Figure S6B). Importantly, Jurkat MAIT cells co-incubated with HSV-1-infected cells treated with E. coli or 5-OP-RU had levels of activation that were significantly less than matched mock infected cells, demonstrating the functional consequence of viral control of MR1 surface expression (Figures 4B and 4C).
Figure 4. HSV-1 Infection Inhibits Ligand and Bacterially Induced MAIT TCR-Driven Activation.
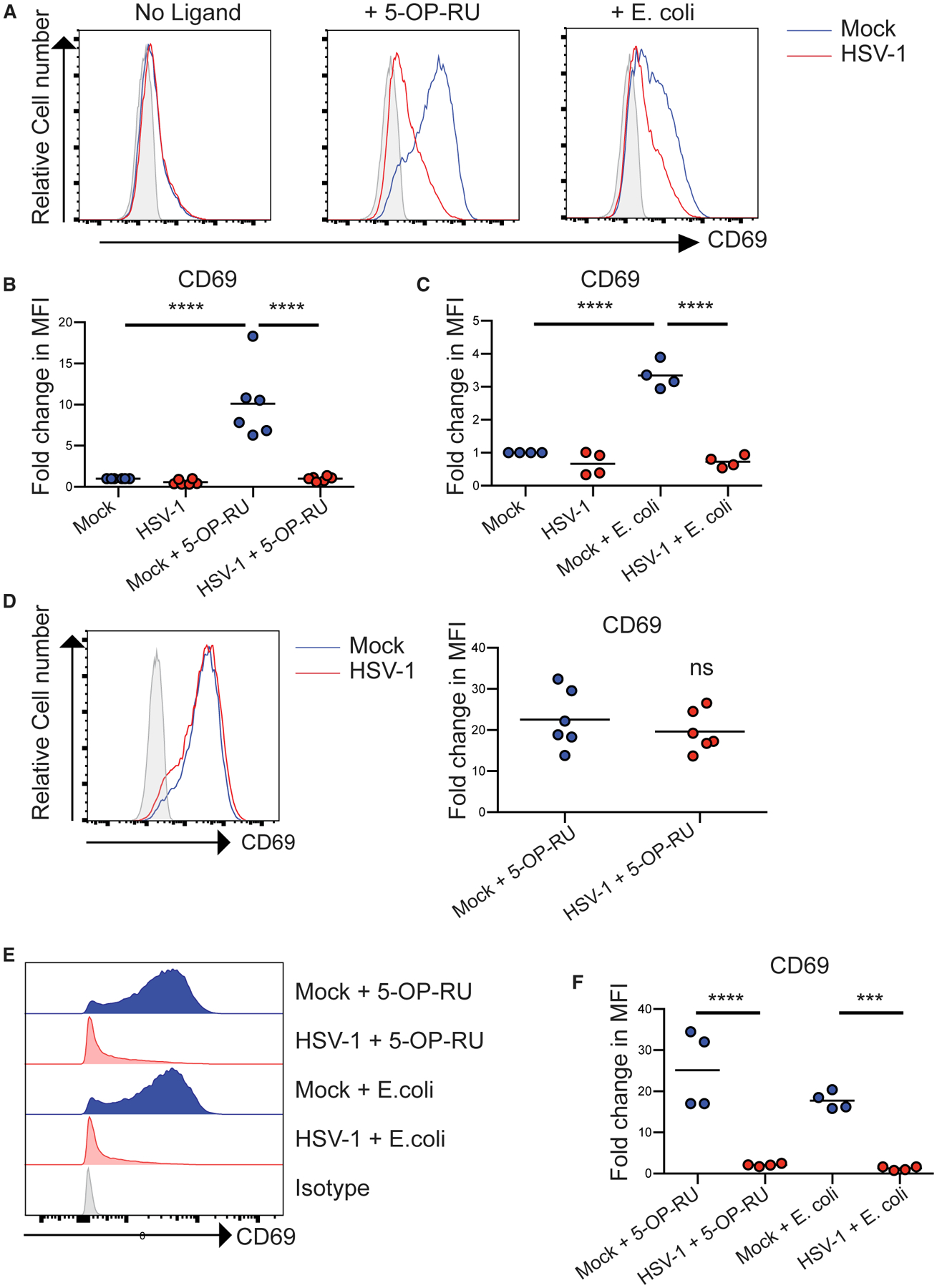
(A) Representative histograms of CD69 expression on Jurkat MAIT (JM) cells incubated with fixed mock or HSV-1-infected HFFs left untreated or treated with 5-OP-RU (10 μM) or E. coli (200 colony-forming units [CFUs]/cell) as indicated.
(B and C) The fold change in CD69 median fluorescence intensity (MFI) compared to mock cells incubated with JM cells for (B) 5-OP-RU, n = 6, and (C) E. coli, n = 4.
(D) Mock or HSV-1-fixed infected fibroblasts in co-culture with JM cells were incubated with 5-OP-RU (10 μM) overnight before staining for surface CD69 expression. The fold change in CD69 MFI compared to mock cells is graphed (n = 6).
(E) Representative histograms of CD69 expression of JM cells incubated with fixed mock or HSV-1-infected ARPE-19 MR1 treated with 5-OP-RU (10 μM) or E. coli (200 CFUs/cell) as indicated.
(F) The fold change in CD69 MFI compared to mock untreated ARPE-19 MR1 co-cultured with JM cells is graphed (n = 4). Statistical significance was calculated by two-way ANOVA; ***p < 0.0005; ****p < 0.0001.
It is known that T cell activation can be inhibited by co-culture with HSV-1-infected cells (Sloan et al., 2003). Thus, we confirmed that Jurkat MAIT cells co-cultured with HSV-1-infected cells were still capable of responding via TCR signaling and were not non-specifically inhibited. 5-OP-RU added directly to the HFFs (mock or HSV-1 infected) and Jurkat MAIT cell co-culture could upregulate CD69 to similar levels (Figure 4D). This is due to the fact that Jurkat MAIT cells are capable of self-presenting antigen through MR1, i.e., 5-OP-RU added directly to Jurkat MAIT cells efficiently upregulates CD69 expression (Figure S6C). HSV-1 infection was also capable of inhibiting both 5-OP-RU and E. coli-induced activation driven by cells overexpressing MR1 (ARPE-19 MR1), where bacterial-induced activation was more pronounced (Figures 4E and 4F).
Proteasomal Inhibition Rescues HSV-1-Mediated Loss of Cellular MR1
As the loss of MR1 protein post-HSV-1 infection is consistent with targeting of this molecule for degradation, we assayed the ability of proteasomal inhibition to recover MR1 levels during infection. Mock or HSV-1-infected ARPE-19 MR1 (Figure 5A) or ARPE-19 MR1-GFP cells (Figure 5B) were treated with the proteasomal inhibitor MG132 immediately post viral adsorption (0 h p.i.) before assaying MR1 levels by western blotting. Although treatment with MG132 led to a reduction in total MR1 levels in mock infected cells, proteasomal inhibition completely blocked the degradation of the remaining MR1 molecules in HSV-1-infected cells, independent of presence/timing of ligand addition. Levels of the viral protein ICP0 were higher in MG132-treated cells, consistent with ICP0 being a known target for proteasomal degradation (Nagel et al., 2011).
Figure 5. HSV-1 Targets the MR1 Protein for Proteasomal Degradation.
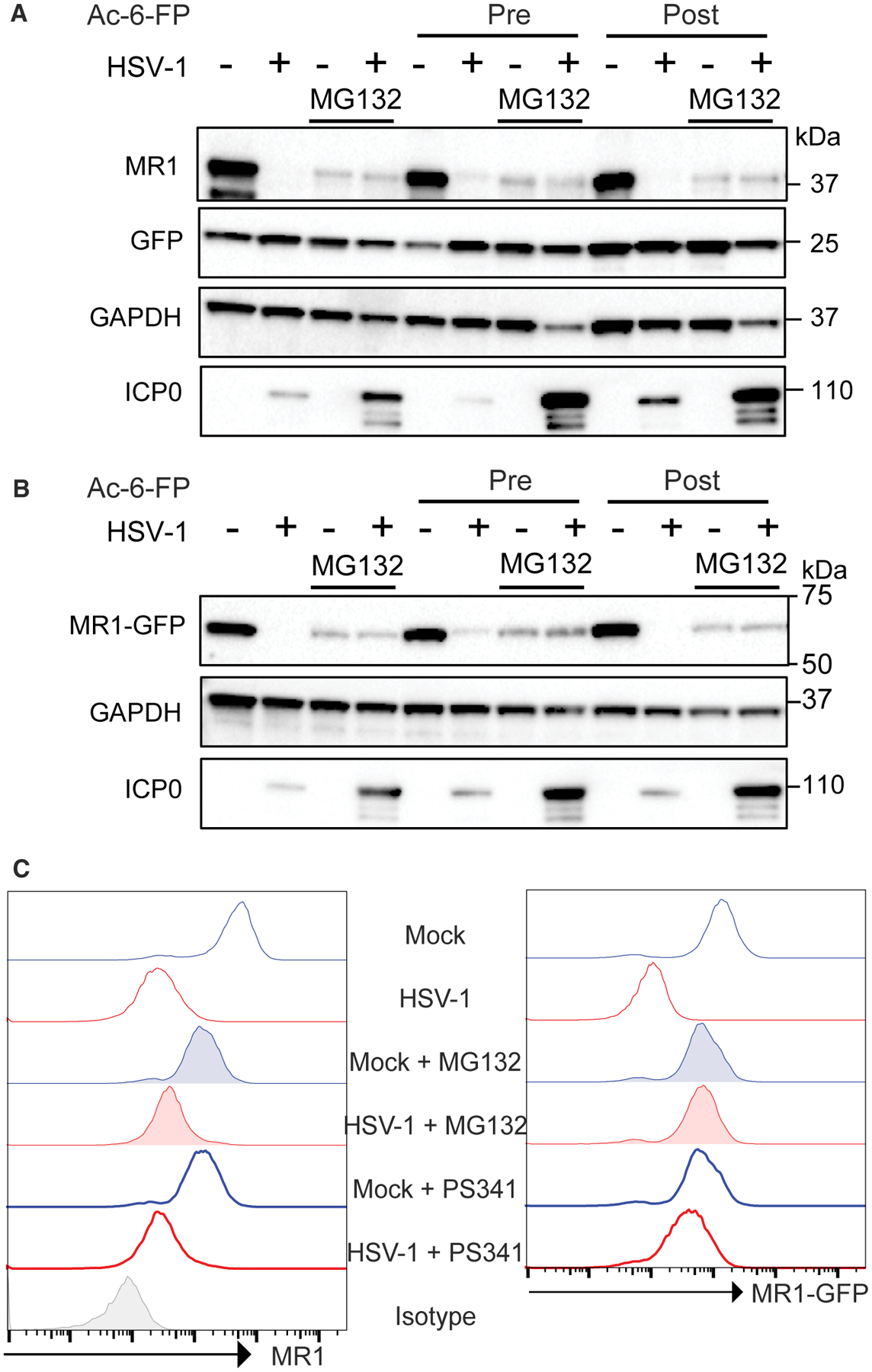
(A and B) ARPE-19 MR1 (A) or ARPE-19 MR1-GFP (B) were mock or HSV-1 infected in parallel and treated with MG132 (5 μM) immediately post-viral adsorption as indicated. Cells were either left untreated or treated with Ac-6-FP (5 μM) for 24 h prior to infection (pre) or at 14 h p.i. (post) before harvesting at 18 h p.i. and immunoblotting for MR1, GFP, GAPDH, or ICP0.
(C) Mock or HSV-1-infected ARPE-19 MR1-GFPs were treated with MG132 (5 μM) or PS-341 (100 nM) immediately post-viral adsorption before addition of Ac-6-FP at 14 h p.i. and staining for surface MR1 at 18 h p.i. Data are representative of 2 independent experiments.
Proteasomal inhibition prior to infection is known to inhibit HSV-1 infection (Delboy et al., 2008). Therefore, we confirmed that treatment with MG132 at 0 h p.i. did not inhibit expression of the HSV-1 late glycoprotein gD (Figure S7), demonstrating that the recovery of MR1 levels post-MG132 treatment in HSV-1 infection was not primarily due to inhibition of viral gene expression. Treatment of mock or HSV-1-infected ARPE-19 MR1-GFP cells with either of the proteasomal inhibitors MG132 or PS-341 could recover MR1-GFP fluorescence (a measure of MR1 fusion protein levels) to that of mock infected cells. This mirrored the immunoblot analysis (Figure 5B), although MR1 surface expression in HSV-1-infected cells was not fully recovered by either inhibitor (Figure 5C). This indicates that MR1 is still retained within the infected cell, even when a major cellular degradation pathway targeting this protein in HSV-1 infection is inhibited, as has been previously described for other MHC-like molecules regulated by herpesvirus infection (Fielding et al., 2014).
Treatment with both MG132 and PS-341 led to a reduction in cellular MR1 levels in mock infected cells (Figures 5A–5C) compared to control-treated cells, suggesting that the proteasome plays an important role in regulation of the homeostatic levels of MR1 in this system. However, whether this is a direct effect on MR1 protein stability or other cellular components that regulate MR1 remains to be elucidated.
HSV-1 Us3 Expression Modulates Surface MR1 Expression
To determine the role of de novo viral gene expression in regulating MR1, the ability of UV-irradiated HSV-1 (UV-HSV-1) to inhibit MR1 surface expression was assayed. ARPE-19 MR1 cells were mock, HSV-1, and UV-HSV-1 infected, with Ac-6-FP added at 14 h p.i. before staining for MR1 at 18 h p.i. Although UV-HSV-1 modestly reduced the surface levels of MR1, viable HSV-1 was far more potent at targeting MR1, suggesting that MR1 targeting is primarily dependent on de novo viral gene expression (Figure 6A). Indeed, the substantial loss of MR1 protein seen at 6 h p.i. with viable virus was not apparent with UV-HSV-1 (Figure 6B).
Figure 6. HSV-1 Gene Expression Is Required to Efficiently Inhibit MR1 Expression.
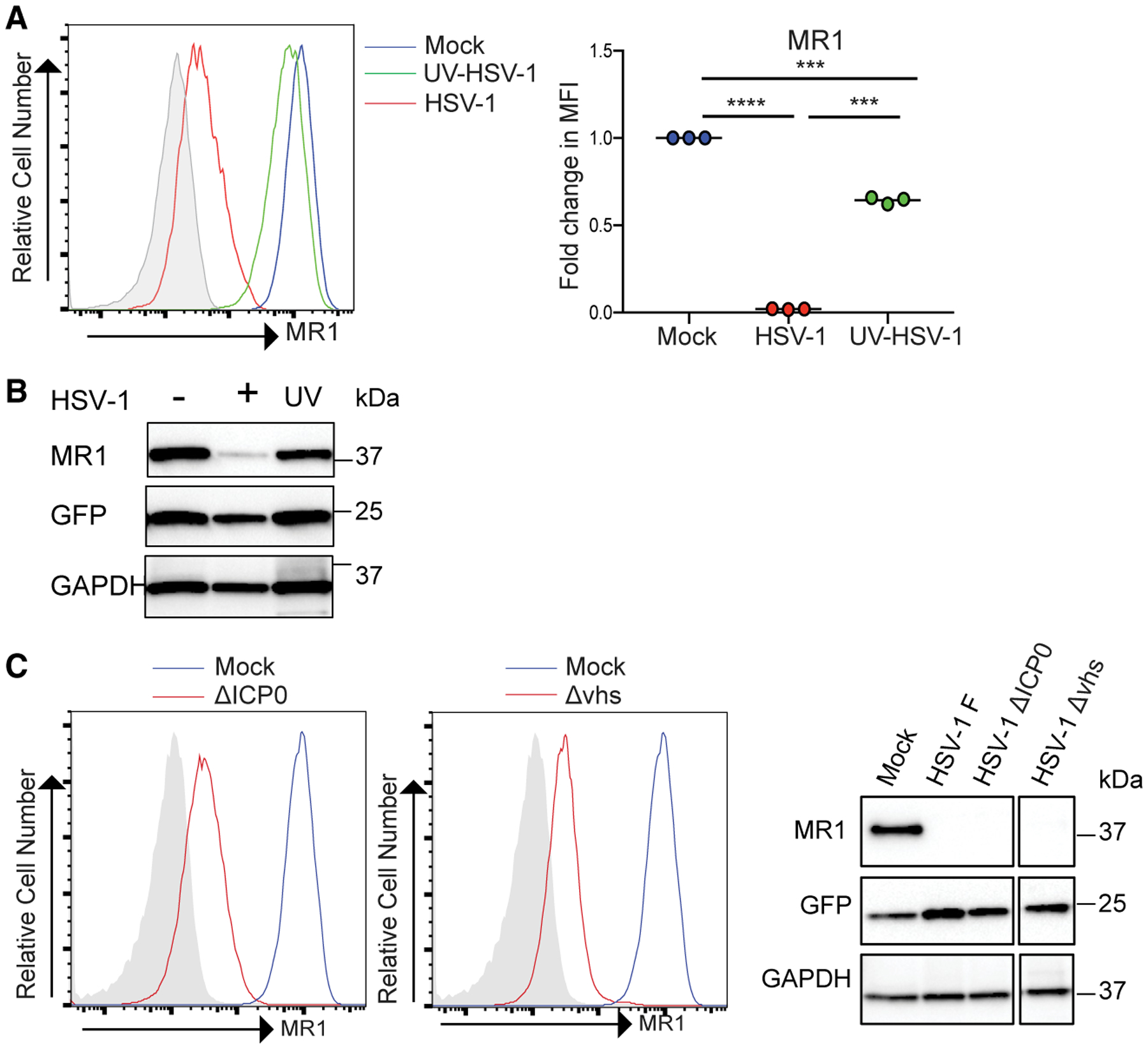
(A) ARPE-19 MR1 were mock, HSV-1, or UV-HSV-1 infected. Cells were treated with Ac-6-FP (5 μM) at 14 h p.i. before staining for surface MR1 at 18 h p.i. and analysis by flow cytometry. Fold change relative to mock infected cells is graphed. Statistical significance was calculated by paired Student’s t test; **p < 0.005; ***p < 0.0005; ****p < 0.0001 (n = 3).
(B) Cell lysates at 6 h p.i. from mock, HSV-1-, or UV-HSV-1-infected ARPE-19 MR1 were immunoblotted for MR1, GFP, or GAPDH.
(C) ARPE-19 MR1 were infected with the indicated viruses and treated with Ac-6-FP at 14 h p.i. before staining for surface MR1 18 h p.i. or immunoblotting for MR1, GFP, or GAPDH.
The ICP0 protein of HSV-1 is known to direct a number of cellular proteins key to viral control for proteasomal degradation (Lanfranca et al., 2014) although the virion host shutoff (vhs) protein is a viral encoded RNase capable of degrading host cell mRNAs to facilitate viral replication (Kwong and Frenkel, 1987). Therefore, we determined whether these proteins could contribute to HSV-1-mediated MR1 regulation using specific viral mutant viruses (Everett et al., 2004; Fenwick and Everett, 1990). We first established that the parental strain of HSV-1 used for the mutants, strain 17, was as effective as the F strain of HSV-1, used in all our previous analyses. Indeed, infection with either the ICP0 and vhs mutants based on the 17 strain both resulted in a loss of MR1 protein levels and surface expression at 18 h p.i. (Figure 6C). Thus, at late time points, neither ICP0 nor vhs are responsible for control of MR1.
We then expanded the study to a third HSV-1 gene. Us3 encodes a serine threonine kinase that is conserved among alphaherpesviruses (McGeoch and Davison, 1986). Us3 has profound impacts on the infected cell while simultaneously promoting the viral replicative cycle (Kato and Kawaguchi, 2018). In particular, the Us3 protein has been shown to have viral immune modulation activities, including inhibition of CD1d and MHC I antigen presentation, resulting in reduced activation of NKT cells and CD8+ T cells, respectively (Imai et al., 2013; Rao et al., 2011; Xiong et al., 2015; Yuan et al., 2006). Thus, the viral protein was expressed via transfection or lentiviral transduction of an expression cassette consisting of Us3 with a downstream T2A ribosome skipping motif and GFP. This enabled the flow-cytometry-based selection of cells expressing Us3. Indeed, GFP-positive, Us3-expressing transfected/transduced cells showed that surface MR1 was profoundly reduced compared to the untransfected GFP-negative cells (Figure 7A). 293T cells transfected with Us3-expressing plasmid and treated with Ac-6-FP ligand 6 h prior to harvesting demonstrated a significant loss of surface MR1 compared to GFP-negative cells. Similarly, transduction of 293T cells with Us3-expressing lentivirus, with or without ligand, also resulted in loss of surface MR1 (Figure S8), although transduced HFFs demonstrated a more modest but still significant downregulation of MR1 (Figure 7B).
Figure 7. HSV-1 Us3 Expression Modulates Surface MR1.
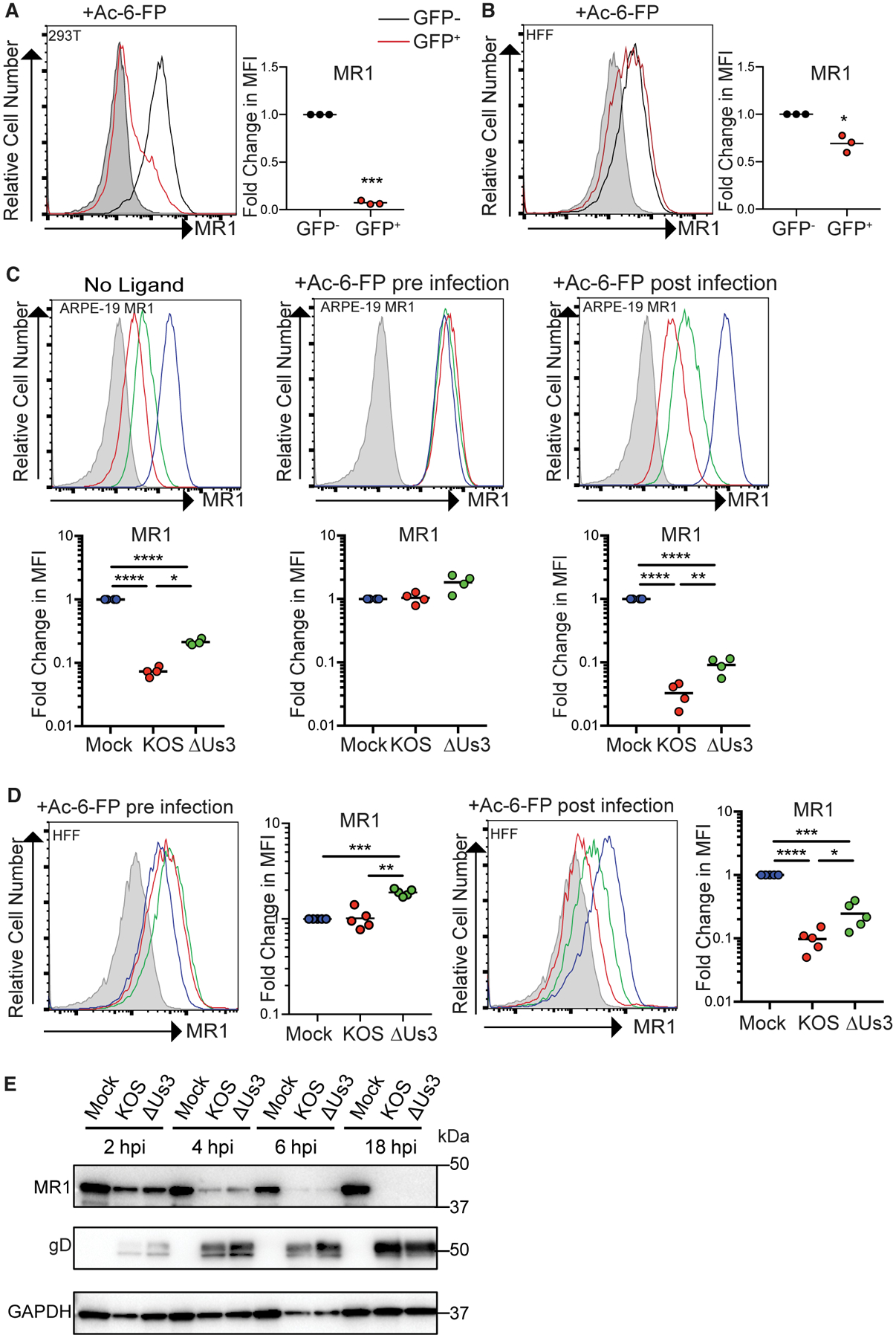
(A) 293T cells transfected with plasmid expressing HSV-1 Us3 (pSY10-Us3).
(B) HFFs transduced with lentivirus expressing HSV-1 Us3 were treated with Ac-6-FP (5 μM) for 6 h prior to staining for surface MR1, analysis by flow cytometry, with gating on GFP expression to identify Us3-expressing GFP+ cells. Fold change relative to GFP− cells is graphed. Statistical significance was calculated by paired Student’s t test (n = 3).
(C and D) ARPE-19 MR1 (C) or HFFs (D) were mock, HSV-1 KOS, or ΔUs3 infected in parallel. Cells were treated with Ac-6-FP (5 μM) at 24 h prior to infection (pre) or at 14 h p.i. (post) before staining for surface MR1 at 18 h p.i. and analysis by flow cytometry. Fold change relative to mock infected cells is graphed. Statistical significance was calculated by ANOVA; *p < 0.05; **p < 0.005; ***p < 0.0005; ****p < 0.0001 (ARPE-19 MR1 n = 4; HFF n = 5).
(E) Cell lysates from mock, HSV-1 KOS-, or ΔUs3-infected ARPE-19 MR1 cells harvested at 2, 4, 6, or 18 h p.i. and immunoblotted for MR1, viral gD, and GAPDH. Data are representative of 2 independent experiments.
We then examined an Us3 deletion mutant HSV-1 to test whether Us3 expression was essential to the modulation of endogenous and overexpressed surface MR1 in the context of productive infection. At 18 h p.i., ARPE-19 MR1 cells infected with the Us3 mutant virus demonstrated a partial rescue of surface MR1 compared to wild-type parental virus, both in the absence of and following p.i. treatment with ligand (Figure 7C). HFFs infected with the Us3 mutant also demonstrated a significant rescue of surface MR1 compared to parental virus (Figure 7D). As expected, pre-treatment of either ARPE-19 MR1 cells or HFFs with ligand prior to infection resulted in MR1 that was resistant to targeting by parental HSV-1, and this remained the case during infection with the Us3 mutant virus (Figures 7C and 7D). To examine whether Us3 is implicated in the loss of the total MR1 pool by HSV-1, parental or mutant-virus-infected ARPE-19 MR1 cells were harvested at 2, 4, 6, and 18 h p.i. and analyzed by immunoblot (Figure 7E), with similar patterns of viral gene expression confirmed using HSV-1 gD expression. Both the wild-type and Us3 mutant demonstrated comparable loss of cellular MR1 across the time course of infection. Collectively, these data suggest that Us3 is responsible in part for loss of surface MR1, but other, as yet unidentified viral product or products are responsible for mediating the loss of total cellular MR1.
DISCUSSION
To date, there has been no infectious agent identified to inhibit MR1 surface expression to modulate immune recognition. Our study provides evidence that multiple herpesvirus infections can inhibit MR1 surface expression, whereas adenovirus infection could not. This indicates that the ability of virus infection to control MR1 surface expression is preserved across multiple herpesviruses, suggesting evolutionary conservation. The failure of adenovirus to mediate this strongly indicates that MR1 regulation is not a global response to viral infection but that it is rather a virus-driven set of specific strategies that herpesviruses have evolved.
Focusing on regulation of MR1 by HSV-1 infection, it is clear that such control was exquisitely sensitive to the timing of ligand addition. Pre-treatment of cells with MR1 ligand induced the protein to mature and become resistant to viral-dependent targeting in both endogenous and overexpressed MR1. It is known that ligand binding to MR1 acts as a driver to initiate the trafficking of molecules to the cell surface, where it can be recognized by MAIT cells. The inability of HSV-1 infection to efficiently target ligand-bound MR1 suggests that such Ag binding could induce a change in the structure/glycosylation of MR1 that allows it to escape viral-mediated regulation. Alternatively, mature MR1 traffics to a cellular compartment not accessible to a HSV-1-encoded immunoevasin(s). It also supports the hypothesis that HSV-1 can only target MR1 in a pre-Golgi compartment and not surface-plasma-membrane-associated MR1. Interestingly, HCMV infection could efficiently target MR1 either pre- or post-treatment with ligand, suggesting that these related viruses may have evolved additional or different mechanisms to target MR1. This reflects the myriad of alternative mechanisms used by individual herpesviruses to control expression of MHC and MHC-like molecules (Schmiedel and Mandelboim, 2017; Schuren et al., 2016). This observation of apparent differential regulation of MR1 expression by individual viruses also suggests that herpesviruses may have evolved specific mechanisms to target MR1 under selective pressure from the MR1:MAIT cell axis in an analogous manner to control exerted by classical MHC-restricted responses.
Using UV-irradiated HSV-1, it was determined that the regulation of MR1 was largely dependent on de novo viral gene expression. However, neither of the viral proteins vhs nor ICP0 was responsible for this phenotype observed. Subsequent analysis identified that expression of the HSV-1 Us3 gene product in isolation was sufficient to downregulate cell surface MR1. Importantly, infection of cells with a HSV-1 Us3 deletion virus demonstrated a partial rescue of cell surface, but not cellular, MR1. These results identify Us3 as a viral mediator of MR1 regulation independent of the capacity of HSV-1 to target MR1 for degradation. This is consistent with our observations that recovery of MR1 protein levels by proteasomal inhibition during HSV-1 infection does not fully restore MR1 cell surface expression. Together, this implicates other, as yet unidentified, viral gene product(s) in this phenotype, which is responsible for mediating the loss of cellular MR1 protein levels detected at early times post-HSV-1 infection.
It is perhaps unsurprising that HSV-1 can apparently encode multiple mechanisms to regulate MR1 expression, given the varied strategies herpesvirus family members use to target the same antigen presentation molecule, e.g., MHC-I or CD1d (van de Weijer et al., 2015). It should also be noted that the major viral gene product (ICP47) identified to regulate MHC-I-dependent antigen presentation in HSV-1 infection targets the transporter associated with antigen processing (TAP) (Früh et al., 1995). However, MR1 processing is TAP independent (Huang et al., 2008; Treiner et al., 2003), suggesting that herpesviruses have evolved independent mechanisms to target these divergent antigen presentation pathways. This is supported by our observation that, although MR1 cell surface expression during HSV-1 infection is differentially regulated dependent on the timing of ligand addition, MHC I surface expression is regulated similarly under both experimental conditions (Figures 2 and 3).
It is becoming increasingly appreciated that interactions between viral, bacterial, fungal, and parasite infections can play key roles in regulating host immunity, as well as in controlling pathogen replication and pathogenicity (Pfeiffer and Virgin, 2016). Infection with HSV-1 predominantly manifests as lesions in the skin, where the resident microbiome contains a vast array of commensal and pathogenic microbiota capable of furnishing MAIT cell antigens (Byrd et al., 2018). Thus, exposure to such potentially activating antigens is likely a common feature of HSV-1 infection. Indeed, HSV-1 infection has been demonstrated to promote both bacterial and fungal infections with intact riboflavin biosynthetic pathways (Plotkin et al., 2016; Wang et al., 2012). In addition, the α-toxin of Staphylococcus aureus can also directly promote HSV-1 infection (Bin et al., 2012), underlining the potential for significant crosstalk between such microbes in the skin where MAIT cells are detected. Thus, herpesvirus-encoded MR1 control may inhibit MR1-restricted responses where Vit B Ag is provided by other infections.
HSV-1 infection could inhibit MR1-restricted MAIT TCR-dependent activation driven either by synthetic ligand or bacteria. Specifically, although HFFs and ARPE-19 cells exposed to 5-OP-RU or E. coli were able to efficiently drive MR1-restricted MAIT TCR-dependent activation, similarly treated HSV-1-infected cells could not, consistent with the potent inhibition of surface MR1 expression observed during infection. This provides evidence of pathogen control of MR1-restricted immune responses.
Initial reports indicated that viral infection (including HSV-1) was not capable of directly activating MAIT cells derived from a transgenic mouse model (Le Bourhis et al., 2010), but more recent studies in humans have demonstrated both in vivo and in vitro that MAIT cells can be activated by viral infections, including influenza, dengue, hepatitis C, and Zika virus (Loh et al., 2016; Paquin-Proulx et al., 2018; van Wilgenburg et al., 2016). Activation was not inhibited by MR1-neutralizing Ab and was primarily dependent on the cytokines IL-12 and IL-18 (Loh et al., 2016; van Wilgenburg et al., 2016). In addition, MAIT cells were activated by influenza infection in a murine model with MR1-deficient mice being significantly more susceptible to severe influenza infection (van Wilgenburg et al., 2018). Several recent reports have also identified dysregulation of MAIT cell number and function in patients as a characteristic of chronic viral infection, in particular, HIV and HCV infection. In HIV/AIDS patients, at both early times p.i. and in chronic viral infection up to 8 years p.i., circulating MAIT cell frequency is reduced with some, but not all, studies detecting a defect in MAIT cell function (Cosgrove et al., 2013; Fernandez et al., 2015; Leeansyah et al., 2013, 2015). MAIT cells are the lymphocyte subset most profoundly affected in chronic HCV infection, with average MAIT cell number reduced by more than 80% compared to healthy individuals (Barathan et al., 2016; Hengst et al., 2016). In addition, MR1-dependent MAIT cell function was reduced from HCV-infected individuals (Hengst et al., 2016). Furthermore, a decrease in MAIT cell number was associated with fatal infections from influenza H7N9 infection (Loh et al., 2016). The factors driving alterations in circulating MAIT cell numbers and function during viral infection, including the potential role of MR1, remain to be fully elucidated.
Although a potential role for MAIT cells during herpesvirus infections in vivo remains to be explored, it is intriguing that a range of primary immunodeficiencies, particularly associated with severe herpesvirus infections, including HSV-1 and HCMV (Latour and Winter, 2018), are characterized by profound reductions in circulating MAIT cell number, including mutations in cytidine triphosphate synthase 1 (Martin et al., 2014), RAS guanyl releasing protein 1 (Winter et al., 2018), CD70 (Izawa et al., 2017), X-linked inhibitor of apoptosis (Gérart et al., 2013), and coronin-1A (Moshous et al., 2013). Circulating MAIT cell number is also reduced in HCMV seropositive compared to seronegative individuals (Patin et al., 2018), suggesting that herpesvirus infections may have the capacity to regulate this specific immune subset. Primary herpesvirus infection is followed by lifelong persistence in a latent state, from which these viruses can reactivate to potentially cause disease. Therefore, it would also be intriguing to investigate whether latently infected cells also retain the capacity to control MR1 surface expression to facilitate evasion of MR1-restricted immune responses throughout the lifespan of the host.
To date, no mammalian and/or virally encoded MR1 ligand has been identified, and studies in the context of viral infection suggest that MAIT cell activity is primarily controlled in an MR1-independent fashion. Therefore, it is not obvious that viruses should encode mechanisms to regulate MR1. However, there are reports indicating that such host-encoded antigens may exist (Lepore et al., 2017; Ling et al., 2016), particularly in the context of MAIT cell development prior to microbial colonization (Koay et al., 2018). In our study, we could not detect activation driven through a MAIT TCR by virus-infected cells alone, consistent with previous reports. However, a more detailed analysis of the potential for viral infection to drive MR1-restricted T cell responses is warranted, given our data indicating that multiple viruses from different species have evolved mechanisms to regulate MR1 surface expression.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Barry Slobedman (barry.slobedman@sydney.edu.au).
There are restrictions to the availability of HFF-1 MR1, ARPE-19 MR1, ARPE-19 MR1-GFP, HSV-1 Strain KOS Us3 mutant, Human adenovirus serotype 5 Ad ΔE3/19K mutant, due to potential requirements for Material Transfer Agreement with the host institution at which these reagents were generated.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells
Human fibroblasts HFF (SCRC-1041), ARPE-19 (CRL-2302), 293T (CRL-3216), 293 (CRL-1573), NIH 3T3 murine fibroblasts (CRL-6442), A549 cells (CCL-185) and Vero cells (CCL-81) (all ATCC), baby kidney hamster cells and U2OS cells (Roger Everett, University of Glasgow) were grown at 37°C and 5% CO2 in Dulbecco’s modified Eagle’s medium (Lonza) supplemented with 10% fetal calf serum (FCS). Jurkat MAIT cells (Reantragoon et al., 2012) were incubated in folate free RPMI1640 (Lonza) supplemented with 10% fetal calf serum (FCS) at 37°C and 5% CO2. HFF and ARPE-19 cell lines expressing MR1 and MR1-GFP under the control of the Murine stem cell virus LTR (plasmids pMIG-MR1 and pMIG-MR1-GFP) were generated as described previously by retroviral transduction (McWilliam et al., 2016) using the parental vector pMIG-II vector, a gift of Dario Vignali, RRID: Addgene_52107 (Holst et al., 2006). The sex of each cell line is listed in the Key Resources Table. Cell lines have not been authenticated by ourselves.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-MR1-biotin (clone 26.5) | Jose Villadangos, The Peter Doherty Institute of Infection and Immunity, The University of Melbourne (McWilliam et al., 2016) | N/A |
| Human recombinant anti-HLA-ABC-PE (clone REA230) | Miltenyi Biotec | Cat# 130-120-055; RRID: AB_2751977 |
| Mouse monoclonal IgG2a-biotin (clone 8A5) isotype | Jose Villadangos, The Peter Doherty Institute of Infection and Immunity, The University of Melbourne | N/A |
| Mouse monoclonal anti-MR1-PE (clone 26.5) | Biolegend | Cat# 361105; RRID: AB_2563042 |
| Mouse anti-IgG2a PE κ isotype (clone G155–178) | BD Biosciences | Cat# 555574; RRID: AB_395953 |
| Mouse monoclonal anti-HLA-ABC-APC (clone G46–2.6) | BD Biosciences | Cat# 555555; RRID: AB_398603 |
| Mouse anti-IgG1 APC κ isotype (clone MOPC-21) | BD Horizon | Cat# 555751; RRID: AB_398613 |
| Mouse monoclonal anti-CD69-BV421 (clone FN50) | BD Biosciences | Cat# 562884; RRID: AB_2687422 |
| Mouse anti-IgG1 BV421 κ isotype | BD Horizon | Cat# 562438; RRID: AB_11207319 |
| Rabbit polyclonal Anti-MR1-CT, generated against the final 15 residues of human MR1 cytosolic tail (PREQNGAIYLPTPDR) | Jose Villadangos, The Peter Doherty Institute of Infection and Immunity, The University of Melbourne (McWilliam et al., 2016) | N/A |
| Mouse monoclonal anti-MR1 | Abcam | Cat# ab55164; RRID: AB_944260 |
| Mouse monoclonal anti-HLA-ABC (clone EMR8–5) | Abcam | Cat# ab70328; RRID: AB_1269092 |
| Mouse monoclonal anti-GFP (B-2) | Santa Cruz Biotechnology | Cat# sc-9996; RRID: AB_627695 |
| Rabbit polyclonal anti-GAPDH (FL-335) | Santa Cruz Biotechnology | Cat# sc-25778; RRID: AB_10167668 |
| Mouse monoclonal anti-HSV-1 ICP0 (clone 11060) | Santa Cruz Biotechnology | Cat# sc-53070; RRID: AB_673704 |
| Mouse monoclonal anti-gD-FITC | Virostat | Cat# 0196 |
| Zombie NIR Fixable Viability Kit | Biolegend | Cat# 423105t |
| Streptavidin-PE | eBioscience | Cat# 12-4317-87 |
| Streptavidin-APC | eBioscience | Cat# 17-4317-82 |
| Bacterial and Virus Strains | ||
| HSV-1 Strain F | Dr Russell Diefenbach (Macquarie University) | GenBank: GU734771 |
| HSV-1 Strain 17 | Prof Roger Everett (University of Glasgow) | GenBank: NC_001806 |
| HSV-1 Strain KOS | Dr P Kinchington, Departments of Ophthalmology, and of Molecular Microbiology and Genetics, University of Pittsburgh | GenBank: JQ780693 |
| HSV-1 Strain 17 ICP0 mutant | Prof Roger Everett (University of Glasgow) (Everett et al., 2004) | N/A |
| HSV-1 Strain 17 vhs mutant | Prof Roger Everett (University of Glasgow) (Fenwick and Everett, 1990) | N/A |
| HSV-1 Strain KOS Us3 mutant | This study | N/A |
| MCMV Murine Cytomegalovirus Smith strain | Prof William Rawlinson (University of New South Wales) (Rawlinson et al., 1996) | GenBank: U68299 |
| HCMV Human cytomegalovirus (Merlin strain) derived from BAC clone pAL1111 | Dr Richard Stanton (Cardiff University) | GenBank: GU179001 |
| Human adenovirus serotype 5 derived from BAC clone pAL908 | (McSharry et al., 2008) | N/A |
| Human adenovirus serotype 5 ΔE3/19K mutant derived from BAC clone pAL918 | (McSharry et al., 2008) | N/A |
| 5-alpha Competent E. coli | NEB | Cat# C2987H |
| E. coli (DH5α) | ThermoFisher Scientific | Cat# 18265017 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dulbecco’s Modified Eagle’s Medium | Lonza | Cat# 12–604F |
| Fetal Calf Serum | Sigma-Aldrich | Cat# 12003C |
| Ac-6-FP Acetyl-6-formylpterin | Schircks | Cat# 11.418 |
| 5-A-RU 5-amino-6-D-ribitylaminouracil | Dr Hamish McWilliam (Corbett et al., 2014) | N/A |
| MG Methylglyoxal | Sigma-Aldrich | Cat# M0252 |
| MG132 | Sigma-Aldrich | Cat# M7449 |
| PS-341 | Selleck Chemicals | Cat# S1013 |
| Fugene HD | Promega | Cat# E2231 |
| Polybrene | Sigma-Aldrich | Cat# TR-1003 |
| folate free RPMI1640 | Lonza | Cat# 12–702F |
| Endo H | NEB | Cat# P0703S |
| BglII | NEB | Cat# R0144 |
| BamHI-HF | NEB | Cat# R3136 |
| SpeI-HF | NEB | Cat# R3133 |
| XbaI | NEB | Cat# R0145 |
| T4 DNA ligase NEB | NEB | Cat3 M0202 |
| Critical Commercial Assays | ||
| GFX PCR DNA and Gel Band Purification Kit | GE Healthcare Life Sciences | Cat# 28903470 |
| NucleoSpin® Gel and PCR Cleanup | Macherey-Nagal | Cat# 740609 |
| Experimental Models: Cell Lines | ||
| Human fibroblasts HFF-1 (male) | ATCC | SCRC-1041; RRID: CVCL_3285 |
| Human ARPE-19 cell line (male) | ATCC | CRL-2302; RRID: CVCL_0145 |
| Human 293T cell line (female) | ATCC | CRL-3216; RRID: CVCL_0063 |
| Human 293 cell line (female) | ATCC | CRL-1573; RRID: CVCL_0045 |
| Green monkey Vero cell line (female) | ATCC | CCL-81; RRID: CVCL_0059 |
| Human U2OS cells (female) | Prof Roger Everett, (University of Glasgow) (Everett et al., 2004) | N/A |
| Murine NIH 3T3 fibroblasts (male) | Prof William Rawlinson (University of New South Wales) | N/A |
| Human Jurkat MAIT (male) | Prof James McCluskey (Reantragoon et al., 2012) | N/A |
| Human A549 (male) | ATCC | Cat# CCL-185; RRID: CVCL_0023 |
| HFF-1 MR1 (male) | This study | N/A |
| ARPE-19 MR1 (male) | This study | N/A |
| ARPE-19 MR1-GFP (male) | This study | N/A |
| Oligonucleotides | ||
| Us3 Forward primer 5′ GTCTACACTAGTATGGCCTGTCGTAAGTTTTGTCG 3′ | This study | N/A |
| Us3 Reverse Primer 5′ GTCTACAGATCTTTTCTGTTGAAACAGCGGCAAAC 3′ | This study | N/A |
| Recombinant DNA | ||
| pSY10-Us3 | This study | N/A |
| pCDH_EF1-MCS-T2A-copGFP vector | Systems Bioscience, USA | Cat3# CD526A-1 |
| pMIG-MR1 | (McWilliam etal., 2016) | N/A |
| pMIG-MR1-GFP | (McWilliam etal., 2016) | N/A |
| Software and Algorithms | ||
| FlowJo software | Treestar Inc. | https://www.flowjo.com/ |
| Paired Student’s t tests or ANOVA analysis performed as indicated using Prism software | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| CLC Main Workbench | QIAGEN | https://digitalinsights.qiagen.com/products-overview/analysis-andvisualization/qiagen-clc-main-workbench/ |
Viruses
HSV-1 strain F as grown and titrated on Vero cells. The ICP0 and vhs mutant viruses based on the HSV-1 strain 17 were grown in Baby kidney hamster cells and titered on U2OS cells (Everett et al., 2004; Fenwick and Everett, 1990). The US3 mutant virus was constructed by inserting the US3 coding sequence in frame and downstream of EGFP in pEGFP-C1 (TaKaRa). The US3 protein was then collapsed between the BglII site (in EGFP) and the unique BamHI site in US3, and the US3 promoter placed upstream of this construct (Figure S9). This was then introduced into HSV-1 strain KOS strain by homologous recombination before mutants were identified by fluorescence and purified by plaque picking. The mutant was confirmed by DNA sequencing and loss of protein kinase function. Human adenovirus type 5 (Ad) and ΔE3/19k mutants derived from BAC clones (pAL908 and pAL918 respectively) were grown and titrated in A549 cells as described previously (McSharry et al., 2008). Murine cytomegalovirus Smith strain (Rawlinson et al., 1996) provided by Prof William Rawlinson (University of New South Wales) was grown and titrated in NIH 3T3 fibroblasts. Human cytomegalovirus (Merlin strain) generated from the BAC clone pAL1111 provided by Dr Richard Stanton (Cardiff University) was grown and titrated in HFFs (Stanton et al., 2010).
Plasmid expression constructs
Primers (Forward: 5′ GTCTACACTAGTATGGCCTGTCGTAAGTTTTGTCG 3′; reverse: 5′ GTCTACAGATCTTTTCTGTTGAAACAGCGGCAAAC 3′; Us3 sequence underlined, restriction sites in bold) were used to amplify the HSV-1 Strain F Us3 sequence excluding the stop codon. PCR products were purified (GFX PCR DNA and Gel Band Purification Kit) and digested (SpeI-HF and BglII, NEB). The plasmid backbone (pCDH_EF1-MCS-T2A-copGFP vector, Systems Bioscience, USA) was digested (XbaI and BamHI-HF, NEB), purified and ligated to the Us3 fragment (T4 DNA ligase NEB) to create the pSY10-Us3 plasmid. This was transformed into NEB 5-alpha E. coli, selected on Luria-Bertani agar with ampicillin (50 μg/ml) and purified (Macherey-Nagal NucleoSpin®). Cells were transfected with pSY10-Us3 using Fugene HD (Promega), and replication-deficient pSY10-Us3 lentivirus were synthesized in 293T cells co-transfected with lentiviral packaging plasmids as previously described (Ashley et al., 2017). Cells were transduced using in the presence of 5 μg/mL Polybrene (Sigma-Aldrich).
METHOD DETAILS
Virus Infections
Cells were infected with HSV-1 at an MOI of either 5 (F and KOS strains) or 3 (17 strain) by incubating for 1 hour with gentle rocking. HCMV and MCMV infections were performed at an MOI of 10. Ad infections were performed at an MOI 30 (HFF) or MOI 10 (293 cells). All cells were incubated at 37°C in 5% CO2.
Flow cytometry analysis
For flow cytometry staining of MR1 in HFFs the mAb 26.5 conjugated to Biotin in combination with Streptavidin conjugated to PE or APC (eBioscience) was utilized. In MR1 overexpressing cells 26.5 directly conjugated to PE (Biolegend) was used. Surface MHC I expression was detected by anti-HLA-A,B,C-PE (Miltenyi Biotec) or anti-HLA-A,B,C-APC (BD Biosciences). HSV-1 gD surface expression was detected using anti-gD-FITC (Virostat). Live cells were identified using Zombie NIR Fixable Viability Kit (Biolegend). Flow cytometry was performed using a LSR Fortessa X-20 (BD Biosciences), with data analyzed using FlowJo software (Treestar Inc.).
MR1 ligands
The MR1 ligands Ac-6-FP (Schircks Laboratories) and 5-OP-RU (synthetic 5-A-RU plus methylglyoxal (Sigma-Aldrich) at equimolar concentrations) were added to the culture medium at the concentrations indicated.
Bacteria
E. coli (DH5a) Life technologies were grown o/n to saturation in Luria-Bertani broth. The following morning the bacteria were pelleted and washed in PBS before fixation in 1% paraformaldehyde for 3 minutes with vortexing for the first 60 s and last 30 s of the fixation (Dias et al., 2017). The cells were washed 4 times with PBS before being added to cells at 200 colony forming units per cell as determined by colony counts.
Jurkat MAIT cell activation assays
5 × 104 (HFFs) or 1 × 105 (ARPE-19 MR1) mock or HSV-1 infected cells in 24-well plates at 14 h p.i. were treated with 5-OP-RU (10 μM) or E. coli for 4 hours before being washed from the system at 18 h p.i. Cells were then fixed with 1% paraformaldehyde and 2% glucose in PBS for 20 min at 4C, before washing 3 times with complete media. Jurkat MAIT cells (3 × 105) were then added in folate free RPMI1640 and allowed to incubate o/n before staining for CD69 (BD Biosciences) expression as a measure of Jurkat MAIT activation. 5-OP-RU (10 μM) was added to HFF/Jurkat MAIT co-cultures or to Jurkat MAIT cells alone and allowed to incubate o/n as indicated.
Immunoblotting
Cells were harvested in cell lysis buffer (50 mM NaCl, 50 mM TRIS pH8, 1% IGEPAL, 1% Triton X-100) supplemented with protease inhibitor cocktail (Sigma) and allowed to incubate on ice for 20 mins. Lysates were then centrifuged (16,000 × g for 20 min at 4°C) and the supernatant collected. Lysates were mock or Endo H (NEB) digested according to the manufacturer’s instructions for 90 mins at 37°C as required. Lysates were denatured by heating at 95°C for 5 mins in reducing sample buffer (Bio-Rad) and resolved by SDS-PAGE on precast polyacrylamide gels (Biorad) before immunoblotting onto PVDF membranes. Membranes were probed with the designated primary antibodies in 3% BSA in PBST, followed by incubation with an appropriate horseradish peroxidase (HRP)-conjugated secondary antibody (all Santa Cruz Biotechnology). The following primary antibodies were utilized: anti-MR1 CT (McWilliam et al., 2016), anti-MR1 and anti-HLA-A, B, C (Abcam), anti-GFP, anti-GAPDH, anti-gD, and anti-ICP0 (all Santa Cruz Biotechnology).
Proteasomal Inhibition
After the 1 h of viral adsorption, cells were treated with either 5 μM MG132 (Sigma-Aldrich), 100 nM PS-341 (Selleck Chemicals) or DMSO control (Sigma) to inhibit proteasomal activity.
QUANTIFICATION AND STATISTICAL ANALYSIS
Paired Student’s t tests or ANOVA analysis were performed as indicated using GraphPad Prism software. Data are presented as dot plots with the mean.
DATA AND CODE AVAILABILITY
This study did not generate any datasets.
Supplementary Material
Highlights.
The herpesviruses HSV-1 and CMV suppress the antigen presentation molecule MR1
HSV-1 targets immature MR1 for degradation although mature MR1 remains protected
The Us3 protein of HSV-1 is partially responsible for suppression of MR1
Suppression of MR1 by HSV-1 inhibits MAIT TCR-dependent activation
ACKNOWLEDGMENTS
We wish to thank Prof. Roger Everett for providing the HSV-1 ICP0 and VHS mutant viruses, Prof. William Rawlinson for the MCMV Smith strain, Dr. Richard Stanton for the bacterial artificial chromosome (BAC)-derived HCMV Merlin strain, and Dr. Russell Diefenbach for HSV-1 strain F. C.S. is supported by an Australian Postgraduate Award. J.R. is supported by an Australian Research Council (ARC) Australian Laureate Fellowship. D.P.F. is supported by a National Health and Medical Research Council of Australia Senior Principal Research Fellowship (1117017) and an ARC Centre of Excellence grant (CE140100011). P.R.K. was supported by awards from the National Institutes of Health USA (R01-EY015291 and R01-AI122640), NEI CORE grant for Vision Research P30-EY08098, and Unrestricted awards from the Research to Prevent Blindness Inc. and The Eye & Ear Foundation of Pittsburgh. H.E.G.M. is supported by an ARC Discovery Early Career Researcher Award (DE170100575). J.A.V. is supported by a National Health and Medical Research Council of Australia Principal Research Fellowship (1154502) and a Program Grant (1113293) and by an ARC Discovery Project Grant (170102471).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.017.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ashley CL, Glass MS, Abendroth A, McSharry BP, and Slobedman B (2017). Nuclear domain 10 components upregulated via interferon during human cytomegalovirus infection potently regulate viral infection. J. Gen. Virol 98, 1795–1805. [DOI] [PubMed] [Google Scholar]
- Barathan M, Mohamed R, Vadivelu J, Chang LY, Saeidi A, Yong YK, Ravishankar Ram M, Gopal K, Velu V, Larsson M, and Shankar EM (2016). Peripheral loss of CD8(+) CD161(++) TCRVα7·2(+) mucosal-associated invariant T cells in chronic hepatitis C virus-infected patients. Eur. J. Clin. Invest 46, 170–180. [DOI] [PubMed] [Google Scholar]
- Bin L, Kim BE, Brauweiler A, Goleva E, Streib J, Ji Y, Schlievert PM, and Leung DYM (2012). Staphylococcus aureus α-toxin modulates skin host response to viral infection. J. Allergy Clin. Immunol 130, 683–691.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgert HG, and Kvist S (1985). An adenovirus type 2 glycoprotein blocks cell surface expression of human histocompatibility class I antigens. Cell 41, 987–997. [DOI] [PubMed] [Google Scholar]
- Burgert HG, Maryanski JL, and Kvist S (1987). “E3/19K” protein of adenovirus type 2 inhibits lysis of cytolytic T lymphocytes by blocking cell-surface expression of histocompatibility class I antigens. Proc. Natl. Acad. Sci. USA 84, 1356–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrd AL, Belkaid Y, and Segre JA (2018). The human skin microbiome. Nat. Rev. Microbiol 16, 143–155. [DOI] [PubMed] [Google Scholar]
- Corbett AJ, Eckle SB, Birkinshaw RW, Liu L, Patel O, Mahony J, Chen Z, Reantragoon R, Meehan B, Cao H, et al. (2014). T-cell activation by transitory neo-antigens derived from distinct microbial pathways. Nature 509, 361–365. [DOI] [PubMed] [Google Scholar]
- Cosgrove C, Ussher JE, Rauch A, Gärtner K, Kurioka A, Hühn MH, Adelmann K, Kang YH, Fergusson JR, Simmonds P, et al. (2013). Early and nonreversible decrease of CD161++ /MAIT cells in HIV infection. Blood 121, 951–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delboy MG, Roller DG, and Nicola AV (2008). Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol 82, 3381–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias J, Leeansyah E, and Sandberg JK (2017). Multiple layers of heterogeneity and subset diversity in human MAIT cell responses to distinct microorganisms and to innate cytokines. Proc. Natl. Acad. Sci. USA 114, E5434–E5443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhard JM, Hartjen P, Kummer S, Schmidt RE, Bockhorn M, Lehmann C, Balagopal A, Hauber J, van Lunzen J, and Schulze zur Wiesch J (2014). CD161+ MAIT cells are severely reduced in peripheral blood and lymph nodes of HIV-infected individuals independently of disease progression. PLoS ONE 9, e111323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckle SB, Birkinshaw RW, Kostenko L, Corbett AJ, McWilliam HE, Reantragoon R, Chen Z, Gherardin NA, Beddoe T, Liu L, et al. (2014). A molecular basis underpinning the T cell receptor heterogeneity of mucosal-associated invariant T cells. J. Exp. Med 211, 1585–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett RD, Boutell C, and Orr A (2004). Phenotype of a herpes simplex virus type 1 mutant that fails to express immediate-early regulatory protein ICP0. J. Virol 78, 1763–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick ML, and Everett RD (1990). Inactivation of the shutoff gene (UL41) of herpes simplex virus types 1 and 2. J. Gen. Virol 71, 2961–2967. [DOI] [PubMed] [Google Scholar]
- Fernandez CS, Amarasena T, Kelleher AD, Rossjohn J, McCluskey J, Godfrey DI, and Kent SJ (2015). MAIT cells are depleted early but retain functional cytokine expression in HIV infection. Immunol. Cell Biol 93, 177–188. [DOI] [PubMed] [Google Scholar]
- Fielding CA, Aicheler R, Stanton RJ, Wang EC, Han S, Seirafian S, Davies J, McSharry BP, Weekes MP, Antrobus PR, et al. (2014). Two novel human cytomegalovirus NK cell evasion functions target MICA for lysosomal degradation. PLoS Pathog. 10, e1004058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Früh K, Ahn K, Djaballah H, Sempé P, van Endert PM, Tampé R, Peterson PA, and Yang Y (1995). A viral inhibitor of peptide transporters for antigen presentation. Nature 375, 415–418. [DOI] [PubMed] [Google Scholar]
- Gérart S, Sibéril S, Martin E, Lenoir C, Aguilar C, Picard C, Lantz O, Fischer A, and Latour S (2013). Human iNKT and MAIT cells exhibit a PLZF-dependent proapoptotic propensity that is counterbalanced by XIAP. Blood 121, 614–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gherardin NA, Keller AN, Woolley RE, Le Nours J, Ritchie DS, Neeson PJ, Birkinshaw RW, Eckle SBG, Waddington JN, Liu L, et al. (2016). Diversity of T cells restricted by the MHC class I-related molecule MR1 facilitates differential antigen recognition. Immunity 44, 32–45. [DOI] [PubMed] [Google Scholar]
- Gold MC, Cerri S, Smyk-Pearson S, Cansler ME, Vogt TM, Delepine J, Winata E, Swarbrick GM, Chua WJ, Yu YY, et al. (2010). Human mucosal associated invariant T cells detect bacterially infected cells. PLoS Biol. 8, e1000407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harriff MJ, McMurtrey C, Froyd CA, Jin H, Cansler M, Null M, Worley A, Meermeier EW, Swarbrick G, Nilsen A, et al. (2018). MR1 displays the microbial metabolome driving selective MR1-restricted T cell receptor usage. Sci. Immunol 3, eaao2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Hirai M, and Kurosawa Y (1995). A gene outside the human MHC related to classical HLA class I genes. Science 269, 693–695. [DOI] [PubMed] [Google Scholar]
- Hengst J, Strunz B, Deterding K, Ljunggren HG, Leeansyah E, Manns MP, Cornberg M, Sandberg JK, Wedemeyer H, and Björkström NK (2016). Nonreversible MAIT cell-dysfunction in chronic hepatitis C virus infection despite successful interferon-free therapy. Eur. J. Immunol 46, 2204–2210. [DOI] [PubMed] [Google Scholar]
- Hofmann M, and Thimme R (2016). MAIT be different-persisting dysfunction after DAA-mediated clearance of chronic hepatitis C virus infection. Eur. J. Immunol 46, 2099–2102. [DOI] [PubMed] [Google Scholar]
- Holst J, Szymczak-Workman AL, Vignali KM, Burton AR, Workman CJ, and Vignali DA (2006). Generation of T-cell receptor retrogenic mice. Nat. Protoc 1, 406–417. [DOI] [PubMed] [Google Scholar]
- Howson LJ, Salio M, and Cerundolo V (2015). MR1-restricted mucosal-associated invariant T cells and their activation during infectious diseases. Front. Immunol 6, 303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Gilfillan S, Kim S, Thompson B, Wang X, Sant AJ, Fremont DH, Lantz O, and Hansen TH (2008). MR1 uses an endocytic pathway to activate mucosal-associated invariant T cells. J. Exp. Med 205, 1201–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai T, Koyanagi N, Ogawa R, Shindo K, Suenaga T, Sato A, Arii J, Kato A, Kiyono H, Arase H, and Kawaguchi Y (2013). Us3 kinase encoded by herpes simplex virus 1 mediates downregulation of cell surface major histocompatibility complex class I and evasion of CD8+ T cells. PLoS ONE 8, e72050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izawa K, Martin E, Soudais C, Bruneau J, Boutboul D, Rodriguez R, Lenoir C, Hislop AD, Besson C, Touzot F, et al. (2017). Inherited CD70 deficiency in humans reveals a critical role for the CD70-CD27 pathway in immunity to Epstein-Barr virus infection. J. Exp. Med 214, 73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, and Kawaguchi Y (2018). Us3 protein kinase encoded by HSV: the precise function and mechanism on viral life cycle. Adv. Exp. Med. Biol 1045, 45–62. [DOI] [PubMed] [Google Scholar]
- Keller AN, Eckle SB, Xu W, Liu L, Hughes VA, Mak JY, Meehan BS, Pediongco T, Birkinshaw RW, Chen Z, et al. (2017). Drugs and drug-like molecules can modulate the function of mucosal-associated invariant T cells. Nat. Immunol 18, 402–411. [DOI] [PubMed] [Google Scholar]
- Kjer-Nielsen L, Patel O, Corbett AJ, Le Nours J, Meehan B, Liu L, Bhati M, Chen Z, Kostenko L, Reantragoon R, et al. (2012). MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 491, 717–723. [DOI] [PubMed] [Google Scholar]
- Koay HF, Godfrey DI, and Pellicci DG (2018). Development of mucosal-associated invariant T cells. Immunol. Cell Biol 96, 598–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong AD, and Frenkel N (1987). Herpes simplex virus-infected cells contain a function(s) that destabilizes both host and viral mRNAs. Proc. Natl. Acad. Sci. USA 84, 1926–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfranca MP, Mostafa HH, and Davido DJ (2014). HSV-1 ICP0: an E3 ubiquitin ligase that counteracts host intrinsic and innate immunity. Cells 3, 438–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latour S, and Winter S (2018). Inherited immunodeficiencies with high pre-disposition to Epstein-Barr virus-driven lymphoproliferative diseases. Front. Immunol 9, 1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bourhis L, Martin E, Péguillet I, Guihot A, Froux N, Coré M, Lévy E, Dusseaux M, Meyssonnier V, Premel V, et al. (2010). Antimicrobial activity of mucosal-associated invariant T cells. Nat. Immunol 11, 701–708. [DOI] [PubMed] [Google Scholar]
- Le Bourhis L, Dusseaux M, Bohineust A, Bessoles S, Martin E, Premel V, Coré M, Sleurs D, Serriari NE, Treiner E, et al. (2013). MAIT cells detect and efficiently lyse bacterially-infected epithelial cells. PLoS Pathog. 9, e1003681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeansyah E, Ganesh A, Quigley MF, Sönnerborg A, Andersson J, Hunt PW, Somsouk M, Deeks SG, Martin JN, Moll M, et al. (2013). Activation, exhaustion, and persistent decline of the antimicrobial MR1-restricted MAIT-cell population in chronic HIV-1 infection. Blood 121, 1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeansyah E, Svärd J, Dias J, Buggert M, Nyström J, Quigley MF, Moll M, Sönnerborg A, Nowak P, and Sandberg JK (2015). Arming of MAIT cell cytolytic antimicrobial activity is induced by IL-7 and defective in HIV-1 infection. PLoS Pathog. 11, e1005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepore M, Kalinichenko A, Calogero S, Kumar P, Paleja B, Schmaler M, Narang V, Zolezzi F, Poidinger M, Mori L, et al. (2017). Functionally diverse human T cells recognize non-microbial antigens presented by MR1. eLife 6, e24476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Lin Y, Zheng W, Hong S, Tang X, Zhao P, Li M, Ni J, Li C, Wang L, and Jiang Y (2016). Circulating and tumor-infiltrating mucosal associated invariant T (MAIT) cells in colorectal cancer patients. Sci. Rep 6, 20358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh L, Wang Z, Sant S, Koutsakos M, Jegaskanda S, Corbett AJ, Liu L, Fairlie DP, Crowe J, Rossjohn J, et al. (2016). Human mucosal-associated invariant T cells contribute to antiviral influenza immunity via IL-18-dependent activation. Proc. Natl. Acad. Sci. USA 113, 10133–10138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mak JY, Xu W, Reid RC, Corbett AJ, Meehan BS, Wang H, Chen Z, Rossjohn J, McCluskey J, Liu L, and Fairlie DP (2017). Stabilizing short-lived Schiff base derivatives of 5-aminouracils that activate mucosal-associated invariant T cells. Nat. Commun 8, 14599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Palmic N, Sanquer S, Lenoir C, Hauck F, Mongellaz C, Fabrega S, Nitschké P, Esposti MD, Schwartzentruber J, et al. (2014). CTP synthase 1 deficiency in humans reveals its central role in lymphocyte proliferation. Nature 510, 288–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeoch DJ, and Davison AJ (1986). Alphaherpesviruses possess a gene homologous to the protein kinase gene family of eukaryotes and retroviruses. Nucleic Acids Res. 14, 1765–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McSharry BP, Burgert HG, Owen DP, Stanton RJ, Prod’homme V, Sester M, Koebernick K, Groh V, Spies T, Cox S, et al. (2008). Adenovirus E3/19K promotes evasion of NK cell recognition by intracellular sequestration of the NKG2D ligands major histocompatibility complex class I chain-related proteins A and B. J. Virol 82, 4585–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam HE, Eckle SB, Theodossis A, Liu L, Chen Z, Wubben JM, Fairlie DP, Strugnell RA, Mintern JD, McCluskey J, et al. (2016). The intracellular pathway for the presentation of vitamin B-related antigens by the antigen-presenting molecule MR1. Nat. Immunol 17, 531–537. [DOI] [PubMed] [Google Scholar]
- Moshous D, Martin E, Carpentier W, Lim A, Callebaut I, Canioni D, Hauck F, Majewski J, Schwartzentruber J, Nitschke P, et al. (2013). Whole-exome sequencing identifies Coronin-1A deficiency in 3 siblings with immunodeficiency and EBV-associated B-cell lymphoproliferation. J. Allergy Clin. Immunol 131, 1594–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel CH, Albrecht N, Milovic-Holm K, Mariyanna L, Keyser B, Abel B, Weseloh B, Hofmann TG, Eibl MM, and Hauber J (2011). Herpes simplex virus immediate-early protein ICP0 is targeted by SIAH-1 for proteasomal degradation. J. Virol 85, 7644–7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquin-Proulx D, Greenspun BC, Costa EA, Segurado AC, Kallas EG, Nixon DF, and Leal FE (2017). MAIT cells are reduced in frequency and functionally impaired in human T lymphotropic virus type 1 infection: potential clinical implications. PLoS ONE 12, e0175345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquin-Proulx D, Avelino-Silva VI, Santos BAN, Silveira Barsotti N, Siroma F, Fernandes Ramos J, Coracini Tonacio A, Song A, Maestri A, Barros Cerqueira N, et al. (2018). MAIT cells are activated in acute Dengue virus infection and after in vitro Zika virus infection. PLoS Negl. Trop. Dis 12, e0006154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel O, Kjer-Nielsen L, Le Nours J, Eckle SB, Birkinshaw R, Beddoe T, Corbett AJ, Liu L, Miles JJ, Meehan B, et al. (2013). Recognition of vitamin B metabolites by mucosal-associated invariant T cells. Nat. Commun 4, 2142. [DOI] [PubMed] [Google Scholar]
- Patin E, Hasan M, Bergstedt J, Rouilly V, Libri V, Urrutia A, Alanio C, Scepanovic P, Hammer C, Jönsson F, et al. ; Milieu Intérieur Consortium (2018). Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat. Immunol 19, 302–314. [DOI] [PubMed] [Google Scholar]
- Pfeiffer JK, and Virgin HW (2016). Viral immunity. Transkingdom control of viral infection and immunity in the mammalian intestine. Science 351, aad5872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin BJ, Sigar IM, Tiwari V, and Halkyard S (2016). Herpes simplex virus (HSV) modulation of Staphylococcus aureus and Candida albicans initiation of HeLa 299 cell-associated biofilm. Curr. Microbiol 72, 529–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao P, Pham HT, Kulkarni A, Yang Y, Liu X, Knipe DM, Cresswell P, and Yuan W (2011). Herpes simplex virus 1 glycoprotein B and US3 collaborate to inhibit CD1d antigen presentation and NKT cell function. J. Virol 85, 8093–8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlinson WD, Farrell HE, and Barrell BG (1996). Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol 70, 8833–8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reantragoon R, Kjer-Nielsen L, Patel O, Chen Z, Illing PT, Bhati M, Kostenko L, Bharadwaj M, Meehan B, Hansen TH, et al. (2012). Structural insight into MR1-mediated recognition of the mucosal associated invariant T cell receptor. J. Exp. Med 209, 761–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmiedel D, and Mandelboim O (2017). Disarming cellular alarm systems-manipulation of stress-induced NKG2D ligands by human herpesviruses. Front. Immunol 8, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuren AB, Costa AI, and Wiertz EJ (2016). Recent advances in viral evasion of the MHC class I processing pathway. Curr. Opin. Immunol 40, 43–50. [DOI] [PubMed] [Google Scholar]
- Sloan DD, Zahariadis G, Posavad CM, Pate NT, Kussick SJ, and Jerome KR (2003). CTL are inactivated by herpes simplex virus-infected cells expressing a viral protein kinase. J. Immunol 171, 6733–6741. [DOI] [PubMed] [Google Scholar]
- Stanton RJ, Baluchova K, Dargan DJ, Cunningham C, Sheehy O, Seirafian S, McSharry BP, Neale ML, Davies JA, Tomasec P, et al. (2010). Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J. Clin. Invest 120, 3191–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiner E, Duban L, Bahram S, Radosavljevic M, Wanner V, Tilloy F, Affaticati P, Gilfillan S, and Lantz O (2003). Selection of evolutionarily conserved mucosal-associated invariant T cells by MR1. Nature 422, 164–169. [DOI] [PubMed] [Google Scholar]
- Tyler KL (2018). Acute viral encephalitis. N. Engl. J. Med 379, 557–566. [DOI] [PubMed] [Google Scholar]
- van de Weijer ML, Luteijn RD, and Wiertz EJ (2015). Viral immune evasion: lessons in MHC class I antigen presentation. Semin. Immunol 27, 125–137. [DOI] [PubMed] [Google Scholar]
- van Wilgenburg B, Scherwitzl I, Hutchinson EC, Leng T, Kurioka A, Kulicke C, de Lara C, Cole S, Vasanawathana S, Limpitikul W, et al. ; STOP-HCV consortium (2016). MAIT cells are activated during human viral infections. Nat. Commun 7, 11653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wilgenburg B, Loh L, Chen Z, Pediongco TJ, Wang H, Shi M, Zhao Z, Koutsakos M, Nüssing S, Sant S, et al. (2018). MAIT cells contribute to protection against lethal influenza infection in vivo. Nat. Commun 9, 4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinton C, Wu F, Rossjohn J, Matsuda K, McCluskey J, Hirsch V, Price DA, and Brenchley JM (2016). Mucosa-associated invariant T cells are systemically depleted in simian immunodeficiency virus-infected rhesus macaques. J. Virol 90, 4520–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhang N, Glorieux S, Holtappels G, Vaneechoutte M, Krysko O, Zhang L, Han D, Nauwynck HJ, and Bachert C (2012). Herpes simplex virus type 1 infection facilitates invasion of Staphylococcus aureus into the nasal mucosa and nasal polyp tissue. PLoS ONE 7, e39875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S, Martin E, Boutboul D, Lenoir C, Boudjemaa S, Petit A, Picard C, Fischer A, Leverger G, and Latour S (2018). Loss of RASGRP1 in humans impairs T-cell expansion leading to Epstein-Barr virus susceptibility. EMBO Mol. Med 10, 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong R, Rao P, Kim S, Li M, Wen X, and Yuan W (2015). Herpes simplex virus 1 US3 phosphorylates cellular KIF3A to downregulate CD1d expression. J. Virol 89, 6646–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Hirai M, Kurosawa Y, and Hashimoto K (1997). A highly conserved major histocompatibility complex class I-related gene in mammals. Biochem. Biophys. Res. Commun 238, 697–702. [DOI] [PubMed] [Google Scholar]
- York IA, Roop C, Andrews DW, Riddell SR, Graham FL, and Johnson DC (1994). A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 77, 525–535. [DOI] [PubMed] [Google Scholar]
- Yuan W, Dasgupta A, and Cresswell P (2006). Herpes simplex virus evades natural killer T cell recognition by suppressing CD1d recycling. Nat. Immunol 7, 835–842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any datasets.