

Abstract
Objectives
To define key genetic elements, single or in clusters, underlying SARS-CoV-2 (severe acute respiratory syndrome coronavirus-2) evolutionary diversification across continents, and their impact on drug-binding affinity and viral antigenicity.
Methods
A total of 12 150 SARS-CoV-2 sequences (publicly available) from 69 countries were analysed. Mutational clusters were assessed by hierarchical clustering. Structure-based virtual screening (SBVS) was used to select the best inhibitors of 3-chymotrypsin-like protease (3CL-Pr) and RNA-dependent RNA polymerase (RdRp) among the FDA-approved drugs and to evaluate the impact of mutations on binding affinity of these drugs. The impact of mutations on epitope recognition was predicted following Grifoni et al. (Cell Host Microbe 2020;
27
671–80.)
Results
Thirty-five key mutations were identified (prevalence: ≥0.5%), residing in different viral proteins. Sixteen out of 35 formed tight clusters involving multiple SARS-CoV-2 proteins, highlighting intergenic co-evolution. Some clusters (including D614GSpike + P323LRdRp + R203KN + G204RN) occurred in all continents, while others showed a geographically restricted circulation (T1198KPL-Pr + P13LN + A97VRdRp in Asia, L84SORF-8 + S197LN in Europe, Y541CHel + H504CHel + L84SORF-8 in America and Oceania). SBVS identified 20 best RdRp inhibitors and 21 best 3CL-Pr inhibitors belonging to different drug classes. Notably, mutations in RdRp or 3CL-Pr modulate, positively or negatively, the binding affinity of these drugs. Among them, P323LRdRp (prevalence: 61.9%) reduced the binding affinity of specific compounds including remdesivir while it increased the binding affinity of the purine analogues penciclovir and tenofovir, suggesting potential hypersusceptibility. Finally, specific mutations (including Y541CHel + H504CHel) strongly hampered recognition of Class I/II epitopes, while D614GSpike profoundly altered the structural stability of a recently identified B cell epitope target of neutralizing antibodies (amino acids 592–620).
Conclusions
Key genetic elements reflect geographically dependent SARS-CoV-2 genetic adaptation, and may play a potential role in modulating drug susceptibility and hampering viral antigenicity. Thus, a close monitoring of SARS-CoV-2 mutational patterns is crucial to ensure the effectiveness of treatments and vaccines worldwide.
Introduction
The new coronavirus, termed SARS-CoV-2 (severe acute respiratory syndrome coronavirus-2), emerged in China at the end of 2019.1,2 Afterwards, SARS-CoV-2 was declared a pandemic and has been responsible for over 16 million cases with >650 000 deaths (https://www.gisaid.org/, updated 29 July 2020), causing a global health emergency of inconceivable magnitude.2,3
SARS-CoV-2 is an enveloped positive-sense RNA virus characterized by a genome encoding four structural proteins, 16 non-structural proteins (NSPs) and other regulatory proteins. The four structural proteins are: the envelope (E), spike (S), membrane (M) and nucleocapsid (N) protein. The 16 NSPs include the 3-chymotrypsin-like protease (3CL-Pr), the papain-like protease (PL-Pr), the replication complex comprising the RNA-dependent RNA polymerase (RdRp), the helicase (Hel), the 3′,5′-exonuclease (NSP-14) and other NSPs involved in the different steps of viral replication.4 So far, 3CL-Pr and RdRp have been explored as the main drug targets for therapeutic approaches against SARS-CoV-2 infection.5
Preliminary studies suggest that SARS-CoV-2 is evolving during its spread worldwide and its genome is accumulating some new variations with respect to the SARS-CoV-2 strains that originated in China.6,7 Nevertheless, an in-depth definition of mutational profiles underlying SARS-CoV-2 genetic diversification across geographical areas and their functional characterization has not been extensively addressed. Furthermore, given the urgency of the SARS-CoV-2 outbreak, there has been considerable interest in repurposing existing drugs approved for treating other infections or for other medical indications.8 Nevertheless, no information is available on the role of SARS-CoV-2 mutations in affecting, positively or negatively, the binding affinity of these drug candidates. Understanding this issue can provide important information for the development of effective antiviral agents and universal vaccines, as well as for the design of accurate diagnostic assays, thus representing a crucial aspect to consider in ongoing public health measures to contain infection worldwide.
In this light, by analysing one of the largest sets of SARS-CoV-2 sequences, this study aimed to define key genetic elements, single or in clusters, underlying the evolutionary diversification of SARS-CoV-2 across continents, and their impact on protein structural stability by molecular dynamics simulations, on binding affinity of drug candidates by docking analysis and on epitope recognition by in silico prediction models.
Methods
SARS-CoV-2 sequences
A total of 12 150 high-quality and nearly complete SARS-CoV-2 genomic sequences were retrieved from https://www.gisaid.org/ (see Supplementary Information available as Supplementary data at JAC Online). Sequences were obtained from samples collected between 24 December 2019 and 20 April 2020, and cover 69 countries with the following geographic distribution: Europe (N = 6680), America (N = 3274), Oceania (N = 1321), Asia (N = 777) and Africa (N = 98).
The quality filters for sequences inclusion are reported in the Supplementary Material. Sequences were aligned using the NC_045512.2 SARS-Cov-2-Wuhan-Hu-1 isolate as the reference sequence by Bioedit.
Amino acid variability of the different SARS-CoV-2 proteins
The amino acid variability in viral proteins (S, M, N, E, 3CL-Pr, PL-Pr, RdRp, Hel, NSP-14, NSP-7, NSP-8 and ORF-8) was evaluated by estimating the mean evolutionary divergence (ED) compared with the reference NC_045512.2 using the Poisson correction included in the Mega X software.
The Shannon entropy was calculated to measure the extent of amino acid variability at each position of SARS-CoV-2 protein sequences using the formula [Sn = –∑i(pi lnpi)/lnN], where pi was the frequency of each amino acid and N was the total number of sequences analysed.
For each protein, we assessed the number of amino acid positions with a variability ≥0.5%.
Mutational analysis
SARS-CoV-2 mutations were defined according to the sequence of each specific protein using the NC_045512.2 SARS-Cov-2-Wuhan-Hu-1 isolate as the reference sequence. The prevalence of mutations was calculated in the overall population and according to the continent of sequence isolation. Statistically significant differences in the prevalence of mutations between Asia (continent of origin for the epidemic) and the other continents were assessed by Fisher’s exact test and corrected for multiple testing by the Benjamini–Hochberg method (false discovery rate = 0.05).
Covariation analysis
Statistically significant pairs of mutations were investigated by calculating the binomial correlation coefficient (phi) for the simultaneous presence of mutations at two positions in the same isolate, while clusters of mutations were identified by average linkage hierarchical agglomerative clustering described elsewhere9 and in the Supplementary Material.
Phylogenetic analysis
Phylogenetic trees were performed by MEGA X10 using a maximum likelihood tree based on the Jukes–Cantor model,11 and the bootstrap method of 1000 replicates. Phylogenetic trees were rooted and viewed using FigTree v1.4 (http://tree.bio.ed.ac.uk/software/figtree/).
Epitope localization of the identified SARS-CoV-2 mutations and predicted impact on epitope recognition
Mutations were localized in SARS-CoV-2 major histocompatibility complex (MHC) Class I/II T cell epitopes and B cell epitopes defined by Grifoni et al. (2020).12 The impact of each mutation in altering the binding affinity between the epitopes and the MHC molecules was estimated in silico through the Immune Epitope Database and Analysis Resource (IEDB), by following the approach recently used in Grifoni et al. (2020)12 and described in the Supplementary Material.
Structural analysis
Molecular dynamics simulations (described in the Supplementary Material) were performed to assess the impact of mutations on the stability of RdRp, Hel, 3CL-Pr and S proteins, with the available crystallographic models. The molecular recognition studies of 3CL-Pr and RdRp were carried out by structure-based virtual screening techniques using the DrugBank database as library (details in the Supplementary Material).
Results
Differential amino acid variability in SARS-CoV-2 proteins
The amino acid ED from the SARS-CoV-2 reference sequence (NC_045512.2) varied according to the proteins analysed and never exceeded a mean value of 0.0028 amino acid substitutions per site, indicating limited genetic divergence. The highest ED was observed in the N protein and in the ORF-8-encoded regulatory protein (mean ED ± SD: 0.0021 ± 0.0011 and 0.0028 ± 0.0020 amino acid substitutions per site) (Table 1). Notably, in the N protein, the highest ED occurred in the Ser/Arg-rich motif, crucial to mediate the interaction of the nucleocapsid with viral and cellular proteins.13–15
Table 1.
Amino acid evolutionary divergence (ED) and Shannon entropy of SARS-CoV-2 main proteins
| Protein | Amino acid length | Number of sequences | ED (±SD)a | Number of variable amino acid positionsb | Entropy (min–max value)c |
|---|---|---|---|---|---|
| Structural proteins | |||||
| Spike (S) | 1–1273 | 9111 | 0.0006 (±0.0005) | 3 | 0.04–0.67 |
| Membrane (M) | 1–222 | 11 760 | 0.0005 (±0.0003) | 2 | 0.07–0.13 |
| Envelope (E) | 1–75 | 12 003 | 0.0002 (±0.0001) | 0 | 0 |
| Nucleocapsid (N) | 1–419 | 11 380 | 0.0021 (±0.0011) | 7 | 0.04–0.46 |
| Viral enzymes | |||||
| RNA polymerase | 1–932 | 11 185 | 0.0007 (±0.0006) | 4 | 0.03–0.67 |
| NSP-7 | 1–83 | 12 087 | 0.0005 (±0.0004) | 1 | 0.08 |
| NSP-8 | 1–198 | 12 024 | 0.0002 (±0.0000) | 0 | 0 |
| 3′,5′-Exonuclease (NSP-14) | 1–527 | 9526 | 0.0002 (±0.0001) | 2 | 0.05–0.07 |
| Helicase | 1–601 | 11 662 | 0.0007 (±0.0003) | 4 | 0.03–0.28 |
| 3CL-protease | 1–306 | 11 918 | 0.0003 (±0.0001) | 3 | 0.03–0.06 |
| PL-protease | 1–1945 | 9903 | 0.0002 (±0.0000) | 6 | 0.04–0.1 |
| Regulatory protein | |||||
| ORF-8 | 1–121 | 12 023 | 0.0028 (±0.0020) | 3 | 0.07–0.39 |
NS, non-structural; 3CL, 3-chymotrypsin-like; PL, papain-like.
The amino acid ED of each SARS-CoV-2 protein compared with the reference NC_045512.2 sequence was calculated by Mega X software.
A position was defined as variable if amino acid substitutions were detected with a frequency ≥0.5%.
Shannon entropy was used to measure the amino acid variability at each position. The minimum and maximum entropy value is reported.
Despite limited ED, Shannon entropy analysis revealed the presence of key amino acid mutations (with a frequency ≥0.5%) at 35 hot-spot positions (Table 1 and Figure 1). Mutations detected with the highest frequency were D614GSpike (61.2%) and P323LRdRp (61.8%), followed by R203KN and G204RN (17.2% and 17.2%, respectively), L84SORF-8 (13%), P504LHel and Y541CHel (8.1% and 8.2%, respectively) (Figure 1). The remaining mutations occurred with a prevalence <5% (Figure 1). Among them, S193IN, S194LN, S197LN and S202NN (along with R203KN and G204RN) resided in the Ser/Arg-rich motif, corroborating the selective pressure acting on this domain (Figure 1). Furthermore, all the mutations observed in 3CL-Pr and PL-Pr showed a prevalence <5% (Figure 1).
Figure 1.

Mutation prevalence across SARS-CoV-2 proteins. The histogram reports the prevalence of mutations detected with a prevalence ≥0.5% in the Spike protein [number (N) of sequences analysed = 9111)], Membrane protein (N = 11 760), Nucleocapsid protein (N = 11 380) (a), RNA-dependent RNA polymerase (N = 11 185), NSP-7 (N = 12 087), NSP-14 (N = 9526), Helicase (N = 11 662) (b), 3CL-protease (N = 11 918), PL-protease (N = 9903) and ORF-8-encoded protein (N = 12 023) (c). No mutation with a prevalence ≥0.5% was detected in the Envelope protein.
Genetic diversification of SARS-CoV-2 proteins across continents
A dramatic increase (up to 60%) in the prevalence of D614GSpike and P323LRdRp was observed in all continents compared with Asia (adjusted P ≤ 0.01 for all comparisons), indicating that these mutations are emerging as the major circulating viral variants despite a limited initial circulation in Asia (Figure 2).
Figure 2.

Distribution of mutations across continents. The histograms show the mutations with a prevalence ≥1% in Spike, Membrane and Nucleocapsid proteins (a), in RNA-dependent RNA polymerase, NSP-7, NSP-14 and Helicase (b) and in 3CL-protease, PL-protease and ORF-8-encoded protein (c) across continents. Statistically significant differences were calculated by χ2 test comparing each continent with Asia. The Benjamini–Hochberg method was used for correction of multiple comparisons. *P < 0.05, **P < 0.01. G282VPL-Pr, although with an overall prevalence of <1%, was reported due to its peculiar geographic distribution.
Furthermore, specific mutations circulate with higher prevalence in Europe than in other geographical regions. Among them, T175MM, S193IN and K90R3CL-Pr showed a prevalence of 4.8%, 3.5% and 1.8%, respectively, in Europe while being absent or nearly absent in other continents (prevalence <0.9% for T175MM, <0.1% for S193IN and 0.3% for K90R3CL-Pr, adjusted P ≤ 0.05 for all comparisons). A similar scenario was observed for R203KN and G204RN, showing a remarkable increase in their circulation in Europe and Oceania (adjusted P < 0.05) compared with Asia and America (Figure 2). Similarly, G15S3CL-Pr showed a higher prevalence in Europe and Africa compared with the other geographic areas (adjusted P < 0.05) (Figure 2).
Interestingly, the genetic diversification in America mainly involved proteins acting as cofactors of viral polymerase. In particular, P504LHel and Y541CHel circulate predominantly in America (prevalence: 26.5% and 27%, respectively) while being rarely detected in Asia and Europe (prevalence <0.3%) and never in Africa (adjusted P < 0.05 for each comparison) (Figure 2). Similarly, S25LNSP-7 and A320VNSP-14 circulate in America with a prevalence of 5.1% and 4.3%, while their prevalence never exceeded 1.1% and 0.3%, respectively, in other geographic areas.
A peculiar situation was observed in Oceania characterized by an increased circulation of a large variety of specific mutations throughout the different viral proteins, rarely found in Asia. Among them, G282VPL-Pr was the only detected exclusively in Oceania (prevalence: 5.8%) and never in the other continents (Figure 2).
Covariation profiles among SARS-CoV-2 mutations across continents
Statistically significant pairs of mutations
Covariation analysis identified several statistically significant pairs involving mutations localized in different viral proteins, highlighting a process of intergenic co-evolution. In particular, among the above-mentioned 35 SARS-CoV-2 mutations, 16 were tightly associated with each other.
Specific pairs of mutations were detected in all continents, although with a diverse frequency of circulation (Table 2). This is the case of D614GSpik + P323LRdRp (phi from 0.7 to 0.9 in the different continents) predominantly circulating in Europe (71.9%) and Africa (83.3%), followed by America (54.6%) and Oceania (46.6%), and limitedly in Asia (13.6%) (Table 2). Similarly, R203KN + G204RN (phi = 1.0 except for Oceania) circulate in all continents, with the highest frequency in Europe (24.1%), followed by Oceania (13.3%), Africa (11.1%), Asia (6.4%) and America (4.1%) (Table 2).
Table 2.
Significant pairs of mutations across continents
| Mutation | Mutation prevalence, N (%) | Mutation | Mutation prevalence, N (%) | Covariation frequencya, N (%) | Phib | Adjusted P value |
|---|---|---|---|---|---|---|
| Asia | ||||||
|
| ||||||
| P323LRdRp | 108 (18.7) | D614GSpike, | 89 (15.4) | 79 (13.6) | 0.7 | <1.0e-20 |
| R203KN, | 37 (6.4) | 33 (5.7) | 0.5 | 4.5e-18 | ||
| G204RN | 37 (6.4) | 33 (5.7) | 0.5 | 4.5e-18 | ||
| D614GSpike | 89 (15.4) | R203KN, | 37 (6.4) | 37 (6.4) | 0.6 | 3.5e-18 |
| G204RN | 37 (6.4) | 37 (6.4) | 0.6 | 3.5e-18 | ||
| R203KN | 37 (6.4) | G204RN | 37 (6.4) | 37 (6.4) | 1.0 | <1.0e-20 |
| P13LN | 27 (4.6) | A97VRdRp, | 23 (3.9) | 23 (3.9) | 0.9 | <1.0e-20 |
| T1198KPL-Pr | 23 (3.9) | 23 (3.9) | 0.9 | <1.0e-20 | ||
| A97VRdRp | 23 (3.9) | T1198KPL-Pr | 23 (3.9) | 23 (3.9) | 1 | <1.0e-20 |
|
| ||||||
| Europe | ||||||
|
| ||||||
| D614GSpike | 2373 (74.3) | P323LRdRp, | 2318 (72.6) | 2298 (71.9) | 0.9 | <1.0e-20 |
| R203KN, | 771 (24.1) | 771 (24.1) | 0.3 | <1.0e-20 | ||
| G204RN, | 771 (24.1) | 771 (24.1) | 0.3 | <1.0e-20 | ||
| T175MM | 201 (6.2) | 200 (6.3) | 0.1 | 6.4e-13 | ||
| P323LRdRp | 2318 (72.6) | R203KN, | 771 (24.1) | 748 (23.9) | 0.3 | <1.0e-20 |
| G204RN, | 771 (24.1) | 748 (23.9) | 0.3 | <1.0e-20 | ||
| T175MM | 201 (6.2) | 197 (6.3) | 0.1 | 3.5e-11 | ||
| R203KN | 771 (24.1) | G204RN, | 771 (24.1) | 771 (24.1) | 1.0 | <1.0e-20 |
| T175MM | 201 (6.2) | 198 (6.1) | 0.4 | <1.0e-20 | ||
| G204RN | 771 (24.1) | T175MM | 201 (6.2) | 198 (6.1) | 0.4 | <1.0e-20 |
| L84SORF-8 | 107 (3.3) | S197LN | 62 (1.9) | 62 (1.9) | 0.8 | <1.0e-20 |
|
| ||||||
| America | ||||||
|
| ||||||
| D614GSpike | 1554 (60.9) | P323LRdRp, | 1433 (56.1) | 1393 (54.6) | 0.8 | <1.0e-20 |
| R203KN, | 106 (4.1) | 106 (4.2) | 0.2 | 4.0e-18 | ||
| G204RN | 105 (4.1) | 105 (4.1) | 0.2 | 6.8e-18 | ||
| P323LRdRp | 1433 (56.1) | R203KN, | 106 (4.1) | 101 (4.0) | 0.2 | 1.1e-14 |
| G204RN | 105 (4.1) | 100 (3.9) | 0.2 | 1.8e-14 | ||
| L84SORF-8 | 781 (30.6) | P504LHel, | 682 (26.7) | 681 (26.7) | 0.9 | <1.0e-20 |
| Y541CHel, | 694 (27.2) | 694 (27.2) | 0.9 | <1.0e-20 | ||
| P153LPL-Pr, | 42 (1.6) | 39 (1.5) | 1.7 | 5.2e-13 | ||
| V62LORF-8 | 44 (1.6) | 40 (1.6) | 1.7 | 1.3e-12 | ||
| Y541CHel | 694 (27.2) | P504LHel | 682 (26.7) | 681 (26.7) | 0.9 | <1.0e-20 |
| L25SNSP-7 | 134 (5.2) | A320VNSP-14 | 103 (4.0) | 103 (4.0) | 0.9 | <1.0e-20 |
| R203KN | 106 (4.1) | G204RN | 105 (4.1) | 105 (4.1) | 1.0 | <1.0e-20 |
| V62LORF-8 | 44 (1.6) | P153LPL-Pr | 42 (1.6) | 39 (1.5) | 0.9 | <1.0e-20 |
|
| ||||||
| Oceania | ||||||
|
| ||||||
| D614GSpike | 331 (49.4) | P323LRdRp | 314 (46.9) | 312 (46.6) | 0.9 | <1.0e-20 |
| P323LRdRp | 314 (46.9) | R203KN, | 93 (13.9) | 89 (13.3) | 0.4 | 3.7e-20 |
| G204RN | 92 (13.7) | 89 (13.3) | 0.4 | <1.0e-20 | ||
| L84SORF-8 | 148 (22.1) | P13LN, | 69 (10.3) | 46 (6.8) | 0.4 | 2.8e-13 |
| Y541CHel | 57 (8.5) | 56 (8.3) | 0.6 | <1.0e-20 | ||
| L84SORF-8 | 148 (22.1) | P504LHel, | 55 (8.2) | 55 (8.2) | 0.6 | <1.0e-20 |
| S197LN, | 49 (7.3) | 49 (7.3) | 0.5 | <1.0e-20 | ||
| P153LPL-Pr, | 29 (4.3) | 29 (4.3) | 0.4 | 2.9e-16 | ||
| V62LORF-8, | 29 (4.3) | 29 (4.3) | 0.4 | 2.9e-16 | ||
| F233LNSP-14 | 29 (4.3) | 29 (4.3) | 0.4 | 2.9e-16 | ||
| R203KN | 93 (13.9) | G204RN | 92 (13.7) | 89 (13.3) | 0.4 | <1.0e-20 |
| P13LN | 69 (10.3) | S197LN, | 49 (7.3) | 46 (6.8) | 0.8 | <1.0e-20 |
| A97VRdRp, | 23 (3.4) | 23 (3.4) | 0.6 | 1.1e-20 | ||
| T1198KPL-Pr | 22 (3.2) | 22 (3.2) | 0.5 | 1.1e-19 | ||
| Y541CHel | 57 (8.5) | P504LHel | 55 (8.2) | 55 (8.2) | 0.9 | <1.0e-20 |
| P153LPL-Pr | 29 (4.3) | F233LNSP-14, | 29 (4.3) | 29 (4.3) | 1.0 | <1.0e-20 |
| V62LORF-8 | 29 (4.3) | 29 (4.3) | 1.0 | <1.0e-20 | ||
| T1198KPL-Pr | 22 (3.2) | A97VRdRp | 23 (3.4) | 22 (3.2) | 1.0 | <1.0e-20 |
|
| ||||||
| Africa | ||||||
|
| ||||||
| D614GSpike | 30 (8.3) | P323LRdRp | 30 (83.3) | 30 (83.3) | 1.0 | 1.9e-3 |
| R203KN | 4 (11.1) | G204RN | 4 (11.1) | 4 (11.1) | 1.0 | 6.0e-3 |
Covariation analysis was performed using 7024 sequences stratified across continents (Asia, N = 577; Europe, N = 3192; America, N = 2550; Oceania, N = 669; Africa, N = 36). Covariation frequency was calculated as the prevalence of the pairs of mutations in the different continents.
Statistically significant pairs of mutations were assessed by Fisher’s exact test using the Benjamini–Hochberg method for multiple comparison correction [false discovery rate (FDR) = 0.001]. For Africa, FDR = 0.01 was considered due to restricted sample size.
Other specific pairs of mutations showed a preferential circulation in specific geographic areas. In particular, P504LHel + Y541CHel circulate in America and Oceania (phi = 0.9 for both) with a prevalence of 26.7% and 8.6%, respectively (Table 2). Similarly, P13LN + A97VRdRp and P13LN + T1198KPL-Pr were detected in Asia and Oceania with a prevalence ranging from 3.2% to 3.9% and from 3.2% to 7.3%, respectively (phi = 0.9 and 0.6 for P13LN + A97VRdRp and phi = 0.8 and 0.5 for P13LN + T1198KPL-Pr) (Table 2).
Again, a peculiar scenario was observed in Oceania, characterized by the circulation of multiple pairs of mutations occurring solely in this continent: L84SORF-8 + P13LN (phi = 0.4), P153LPL-Pr + F233LNSP-14 (phi = 1.0) and P13LN + S197LN (phi = 0.8).
The 3CL-Pr was the only viral protein whose mutations (G15S, K90R and A266V) were not involved in statistically significant pairs.
Clusters of correlated mutations
By hierarchical clustering analysis, the pair D614GSpike + P323LRdRp was linked to R203KN + G204RN, forming a tight cluster that was detected in all continents, with the highest prevalence in Oceania (13.2%), followed by Europe (6.1%), Asia (5.7%) and America (3.9%) (bootstrap = 1.0 for all continents) (Figure 3). In Europe, this cluster D614GSpike + P323LRdRp + R203KN + G204RN was accompanied by the continent-specific mutation T175MM (bootstrap = 1.0) (Figure 3).
Figure 3.

Dendrogram of correlated mutations. The dendrogram, obtained from average linkage hierarchical agglomerative clustering, shows clusters of mutations localized in different SARS-CoV-2 proteins. The length of branches reflects distances between mutations in the original distance matrix. Bootstrap values, indicating the significance of clusters, are reported in the boxes.
Furthermore, the cluster made up of P504LHel + Y541CHel + L84SORF-8 was detected in America and in Oceania (bootstrap = 1.0 and 0.97, respectively) with a prevalence of 26.7% and 8.2%, respectively.
Finally, two continent-specific clusters were identified: T1198KPL-Pr + P13LN + A97VRdRp in Asia (bootstrap = 1.0) and P153L3CL-Pr + V62LORF-8 + F233LNSP-14 in Oceania (bootstrap = 1.0) (Figure 3). No continent-specific clusters of mutations were found in Africa, presumably due to the limited number of sequences available.
Phylogenetic analysis confirmed the geographically dependent clustering of mutations (Figure S1).
Functional characterization of the identified mutations on SARS-CoV-2 proteins
Predicted impact of the mutations on SARS-CoV-2 recognition by T cell- and B cell-mediated immune response
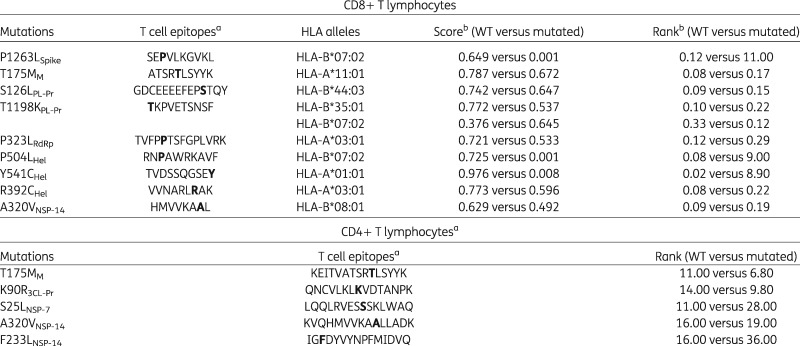
Among the 35 above-mentioned mutations, 16 resided in Class I/II-restricted T cell epitopes (Table S1). Remarkably, by in silico prediction, 12/16 mutations reduced the binding affinity for specific human leukocyte antigens (HLAs) compared with the WT epitope (Table 3). Importantly, a drastic drop in the binding affinity was observed for P1263LSpike (score for HLA-B*07:02 of the WT versus the mutated epitope: 0.649 versus 0.001), P504LHel (score for HLA-B*07:02 of the WT versus the mutated epitope: 0.725 versus 0.001) and Y541CHel (score for HLA-A*01:01 of the WT versus the mutated epitope: 0.976 versus 0.008) (Table 3). This suggests a process of antigenic drift favouring SARS-CoV-2 escape from T cell-mediated immune responses.
Table 3.
Impact of SARS-CoV-2 mutations on binding affinity between epitopes and Class I/II-MHCs
|
Epitopes are based on T cell epitope prediction by Grifoni et al. (2020).12 Letters in bold in the epitopes indicate the mutated amino acid.
The score and the rank were assessed by the Immune Epitope DataBase. A reduced score and an increased rank indicate a decreased binding affinity between the epitope and the related MHC Class I/II allele.
Furthermore, nine mutations were localized in B cell epitopes: two in the S protein, including D614GSpike, and seven in the N protein (Table S1). Notably, six out of seven N mutations were mapped within the same putative B cell epitope spanning positions 177–215.
Impact of mutations on the stability of structural proteins
Structural analysis was focused on mutations with a prevalence >1% localized in 3CL-Pr, RdRp, Hel and S (the only SARS-CoV-2 proteins whose crystallographic models are available).
Most of the analysed mutations determined minimal changes in the stability of the entire proteins compared with the WT (Figure S2a–d). The only exception is represented for K90R3CL-Pr associated with an increased stability of the entire 3CL-Pr compared with the WT (RMSDWT=2.07 Å, RMSDK90R=1.57 Å) (Figure S2d).
Focusing in more detail on the structural localization of mutations, P323LRdRp is located close to the interface with NSP-8. Compared with the WT model, P323LRdR increased the number of total and average hydrogen bonds (HBs) between RdRp and NSP-8, supporting a more stable interaction between these two proteins (WTtot = 241 075 versus P323Ltot = 247 839; WTave = 241 versus P323Lave = 248) (Figure S3a).
Furthermore, as mentioned above, D614GSpike is located in a B cell epitope encompassing residues 592–620. By the root mean square fluctuation analysis, this mutation profoundly decreased the stability of this B cell epitope compared with the WT, suggesting an altered epitope conformation (Figure S3b).
Impact of mutations on viral susceptibility to potential inhibitors of 3CL-Pr and RdRp
Firstly, we applied an in silico drug-repurposing approach to select the best inhibitors of 3CL-Pr and RdRp (the main anticoronavirus pharmacological targets) among the DrugBank compounds. Then, we evaluated if the mutations P323LRdRp, K90R3CL-Pr and G15S3CL-Pr can modulate, positively or negatively, the binding affinity between the enzyme and the inhibitor.
Based on our structure-based virtual screening (SBVS), 20 potential inhibitors of RdRp were identified: five purine analogues (including remdesivir), four cephalosporins, two acetamide derivatives, two flavone compounds, two peptide derivatives, two triazoles, an oxoazepanyl compound, a polyphenol derivative and a pyrimidine analogue (Figure 4). The majority of the identified compounds establish several HBs with specific RdRp residues (including K545, S549, K551, R553, D623 and D761) and with some RNA nucleobases, such as the uracil at position 18, adenine at position 19 and uracil at position 20 (Figure S4 to Figure S23).
Figure 4.



Binding affinity of the different drug candidates for WT and mutated RNA-dependent RNA polymerase (RdRp). This figure reports the absolute best G-score value for each analysed target, expressed as kcal/mol. The molecular recognition studies of RdRp were carried out by structure-based virtual screening techniques using the DrugBank database as library. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
As shown in Figure 5, the best ranked compound cefoperazone in the WT complex was involved in four coordination bonds with Mg2+ cations, two π-cation interactions and two HBs, while in the presence of P323LRdRp the oxoazepanyl derivative RU82209 was well stabilized into the enzyme-binding pocket by means of six coordination bonds between its phosphate groups and the Mg2+ cations.
Figure 5.

3D representation of the best docking pose of (a) cefoperazone (green carbon sticks) and (b) RU82209 (cyan carbon sticks) in the WT RdRp and P323L mutated models, respectively. The enzyme and the RNA are shown as a pale yellow and raspberry cartoon, respectively. The residues involved in crucial contacts with the compounds are reported as pale yellow carbon sticks and the magnesium cations are shown as pink spheres. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC. In the colour version, H bond, stacking and salt bridge interactions are indicated, respectively, as yellow, light blue and violet dashed lines.
Notably, several candidates were better recognized in the P323L complex, as in the case of penciclovir, tenofovir, PF-00610355, zanamivir, diosmin, isavuconazole, resveratrol and, above all, RU82209, associated with the absolute best G-score value (Figure 4).
Conversely, in the presence of P323LRdRp, all cephalosporins and both peptide derivatives showed a decreased binding affinity compared with the WT, as well as rutin and PF-03715455. Interestingly, P323LRdRp decreased the binding affinity of remdesivir compared with the WT (Figure 4).
Regarding 3CL-Pr, SBVS allowed selection of 21 promising inhibitors: four cephalosporins, four peptide derivatives, four purine analogues (including remdesivir), three flavone compounds, three pyrimidine analogues, two triazoles and a benzeneacetamide (Figure 6). The majority of the identified compounds establish several HBs with crucial residues of 3CL-Pr (including N142, G143, C145, E166 and T190), as well as Van der Waals contacts and a pivotal π–π stacking interaction with H41. Both C145 and H41 were involved in the 3CL-Pr catalytic dyad (Figure S24 to Figure S44). Figure 7 reports the best ranked compounds screened against the WT and mutated models.
Figure 6.



Binding affinity of the different drug candidates for WT and mutated 3CL-protease. This figure reports the absolute best G-score value for each analysed target, expressed as kcal/mol. The molecular recognition studies of 3CL-protease were carried out by structure-based virtual screening techniques using the DrugBank database as library. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC.
Figure 7.

3D representation of the best docking pose of (a) sofosbuvir (yellow carbon sticks), (b) N,N-[2,5-O-di-2-fluoro-benzyl-glucaryl]-di-[1-amino-indan-2-ol] (green carbon sticks) and (c) NAD 3-pentanone adduct (cyan carbon sticks) in the WT 3CL-Pr, G15S and K90R mutated models, respectively. The enzyme is shown as a slate cartoon and the residues involved in crucial contacts with the compounds are reported as slate carbon sticks. This figure appears in colour in the online version of JAC and in black and white in the print version of JAC. In the colour version, H bond, stacking and salt bridge interactions are indicated, respectively, as yellow, light blue and violet dashed lines.
Notably, the NAD 3-pentanone adduct, PF-00610355, PF-03715455, reproterol and rutin were found to be better recognized in both K90R3CL-Pr and G15S3CL-Pr compared with the WT, with the NAD 3-pentanone adduct being associated with the absolute best G-score in the presence of K90R3CL-Pr (Figures 6 and 7). This increased theoretical binding affinity could be justified by an additional salt bridge between the ligand pyridine charged nitrogen and 3CL-Pr glutamate at position 166, missing in the WT model. After reproterol recognition, in both mutated complexes, we observed a pivotal π–π stacking interaction with H41 (Figure S35), while in the best poses of both investigational PF compounds, an increased number of HBs was found (Figures S43 and S44). Furthermore, in K90R3CL-Pr, the NAD 3-pentanone adduct engaged seven HBs, a salt bridge contact and a π–π stacking interaction with H41 (Figure 7).
Notably, among the protease inhibitors already used for other viral infections, G15S3CL-Pr determined an increased binding affinity of indinavir (Figure 6). Conversely, both K90R3CL-PR and G15S3CL-Pr strongly reduced the theoretical binding affinity of isavuconazole and sofosbuvir (Figure 6). Furthermore, K90R3CL-Pr determined a remarkable decrease in the binding affinity of adafosbuvir, cefpiramide and ceftibuten, while G15S3CL-Pr affected that of hesperidin, lisinopril and presatovir (Figure 6).
Discussion
Based on one of the largest publicly available datasets of SARS-CoV-2 sequences so far analysed (>12 000), this study identified key mutations, single or in pairs or clusters, underlying geographically dependent viral evolutionary adaptation to human hosts.16,17 Some of these mutations can hamper Class I/II epitope recognition or can profoundly alter the conformation of specific B cell epitopes, suggesting their capability to alter viral antigenicity. Furthermore, to our knowledge, this is the first study highlighting the capability of mutations in RdRp and 3CL-Pr to modulate, either positively or negatively, the binding affinity of specific compounds, suggesting their potential involvement in mechanisms underlying SARS-CoV-2 hypersusceptibility or resistance to drugs.
Among the identified pairs of mutations, D614GSpike + P323LRdRp occurred in all continents, although with a frequency ranging from 13.6% in Asia to 71.9% in Europe and 83.3% in Africa. In particular, in Europe, D614GSpike was first noted in a viral strain isolated from Germany.18 Notably, D614GSpike can introduce a novel protease cleavage site capable of enhancing the fusion between the viral envelope and cell membrane, thus increasing viral infectivity and, in turn, interhuman transmission potential.19–21 Furthermore, D614GSpike lies in a recently identified B cell epitope encompassing amino acids 592–620. Molecular dynamics simulations show that D614GSpike profoundly alters the stability of this epitope, supporting an altered conformation and consequently an impaired recognition by humoral responses. This is in line with a recent finding showing the association of D614GSpike with a reduced antigenicity compared with the SARS-CoV-2 strains isolated in the early epidemic phase.21 On this basis, D614GSpike could also pose concerns for the full effectiveness of vaccine strategies under development and thus deserves further investigation in immunological studies.
P323LRdRp resides in the interface domain (amino acids 250–365) known to connect N- and C-terminal RdRp domains.22 By molecular dynamics simulations, P323LRdRp increased the number of HBs between RdRp and NSP-8, a cofactor which is part of the replication complex along with RdRp, suggesting a stabilized interaction between these two proteins. Furthermore, by in silico prediction, this mutation can reduce the binding affinity for specific HLAs, suggesting that P323LRdRp along with D614GSpike can favour viral evasion from immune responses.
Notably, by SBVS on FDA-approved drugs, P323LRdRp determined a reduced binding affinity for specific compounds including remdesivir. This suggests that P323LRdRp could act as a ‘natural’ drug resistance mutation, hampering the full effectiveness of specific antiviral treatments. This point deserves investigation in ongoing clinical trials.22 At the same time, P323LRdRp determined an increased binding affinity for specific purine analogues, including penciclovir and tenofovir, suggesting a potential viral hypersusceptibility.
Regarding 3CL-Pr, SBVS identified four cephalosporins among the best 3CL-Pr inhibitors, in line with a previous study suggesting the incorporation of a lactam ring in the lead optimization process of SARS-CoV 3CL-Pr inhibitors.23 While K90R3CL-Pr determined a reduced binding affinity for all of them, G15S3CL-Pr was associated with an increased binding affinity for a specific cephalosporin, suggesting differential viral susceptibility to this drug class according to the observed mutational profile.
Among the Pr inhibitors already approved for other viral infections, lopinavir was not included in the list of the best 3CL-Pr inhibitors, due to its low binding affinity towards both WT and mutated 3CL-Pr. This is in line with recent clinical data showing no significant benefits in overall mortality and reduction of SARS-CoV-2 load.24
The overall findings highlight the role of viral genetic variability in modulating, either positively or negatively, drug-binding affinity, a concept that should be taken into account in the current drug-repurposing approach.
The above-mentioned pair D614GSpike + P323LRdRp formed a tight cluster with R203KN + G204RN. This mutational pair resides in the Ser/Arg-rich motif of the N protein, known to be the target of phosphorylation by cellular kinases, to regulate the equilibrium between viral genome replication and morphogenesis25 and to be involved in several intracellular signalling pathways.14,22
Furthermore, R203KN + G204RN along with S193IN, S194LN, S197LN and S202NN reside in a region spanning amino acids 177–215 identified as a B cell epitope.12 Thus, these mutations could alter the antigenicity of the nucleocapsid, potentially affecting its capability to elicit the production of antibodies in infected subjects.26 Similarly, the peculiar enrichment of mutations in the N protein could have important implications in the serological SARS-CoV-2 diagnosis, based on the detection of antibodies against the nucleocapsid, and poses some concerns on the use of this region as a target of molecular diagnostic assays. Indeed, these mutations could contribute to the variations in the sensitivity observed among the assays used for SARS-CoV-2 diagnosis.27 Further comparative studies on the performance of molecular and serological assays for SARS-CoV-2 in the presence of these mutations will be useful to clarify this issue.
Our analysis also showed that the cluster D614GSpike + P323LRdRp + R203KN + G204RN can be linked to continent-specific mutations. Indeed, in Europe, this cluster was accompanied by T175MM. During viral morphogenesis, the M protein interacts with the N and the S protein, acting as a central organizer of viral assembly.25,28 Notably, both R203K and G204R reside in a stretch of amino acids (168–208) that has been proposed to be involved in the interaction with the M protein in SARS-CoV-1.25 Thus, the overall findings suggest that the association of mutations in the above-mentioned proteins can promote the packaging of the encapsidated genome, thus enhancing viral morphogenesis and explaining their emergence in the viral population.
Another widely circulating cluster, characterizing 26.7% and 8.2% of American and Australian viral strains, was made up of H504CHel + Y541CHel + L84SORF-8. Notably, H504CHel and Y541CHel are the only mutations capable of abrogating the binding affinity of CD8+ T cell epitopes for some specific HLAs, supporting that these mutations can act as immune escape mutations, favouring viral evasion from CD8+ T cell responses.
In conclusion, the identification of key geographically dependent genetic elements, single or in pairs or clusters, reflects a geographically dependent viral evolutionary adaptation to human hosts. These mutations may play a potential role in modulating viral susceptibility to drugs either positively or negatively and in favouring vaccine and diagnostic escape events. For this reason, in vitro studies are necessary to further confirm such in silico-based results. In this light, a close and continuous monitoring of SARS-CoV-2 mutational patterns across different geographical areas is crucial to ensure the effectiveness of antiviral treatments and vaccines as well as the accuracy of diagnostic assays worldwide.
Supplementary Material
Acknowledgements
We are grateful to the Johns Hopkins University for collection of SARS-CoV-2 sequences in the GISAID portal and all the laboratories originating and submitting data (Supplementary Material and Supplementary Information). We thank the Aviralia Foundation and the Vironet C Foundation for supporting this study, and Dr Alba Grifoni for her contribution in epitope prediction analysis.
Funding
This study was conducted as part of our routine research work.
Transparency declarations
None to declare.
Supplementary data
Supplementary Information, Supplementary Material, Figures S1 to S44 and Table S1 are available as Supplementary data at JAC Online.
References
- 1. Zhu N, Zhang D, Wang W et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 2020; 382: 727–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Andersen KG, Rambaut A, Lipkin WI et al. The proximal origin of SARS-CoV-2. Nat Med 2020; 26: 2–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Khailany RA, Safdar M, Ozaslan M. Genomic characterization of a novel SARS-CoV-2. Gene Rep 2020; 19: 100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chan JFW, Kok KH, Zhu Z et al. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 2020; 9: 221–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang J, Song W, Huang H et al. Pharmacological therapeutics targeting RNA-dependent RNA polymerase, proteinase and spike protein: from mechanistic studies to clinical trials for COVID-19. 2020; 9: 1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Leary S, Gaudieri S, Chopra A et al. Three adjacent nucleotide changes spanning two residues in SARS-CoV-2 nucleoprotein: possible homologous recombination from the transcription-regulating sequence. bioRxiv 2020; doi: 10.1101/2020.04.10.029454.
- 7. Pachetti M, Marini B, Benedetti F et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J Transl Med 2020; 18: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kiplin Guy R, DiPaola RS, Romanelli F et al. Rapid repurposing of drugs for COVID-19. Science 2020; 368: 829–30. [DOI] [PubMed] [Google Scholar]
- 9. Svicher V, Gori C, Trignetti M et al. The profile of mutational clusters associated with lamivudine resistance can be constrained by HBV genotypes. J Hepatol 2009; 50: 461–70. [DOI] [PubMed] [Google Scholar]
- 10. Kumar S, Stecher G, Li M et al. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 2018; 35: 1547–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jukes TH, Cantor CR. Evolution of protein molecules In: Mammalian Protein Metabolism. Elsevier, 1969; 21–132. [Google Scholar]
- 12. Grifoni A, Sidney J, Zhang Y et al. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe 2020; 27: 671–80.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fung TS, Liu DX. Post-translational modifications of coronavirus proteins: roles and function. Future Virol 2018; 13: 405–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gordon DE, Jang GM, Bouhaddou M et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020; 583: 459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Satarker S, Nampoothiri M. Structural proteins in severe acute respiratory syndrome coronavirus-2. Arch Med Res 2020; 51: 482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Dorp L, Acman M, Richard D et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect Genet Evol 2020; 83: 104351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Laamarti M, Alouane T, Kartti S et al. Large scale genomic analysis of 3067 SARS-CoV-2 genomes reveals a clonal geo-distribution and a rich genetic variations of hotspots mutations. bioRxiv 2020; 3: 2020.05.03.074567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Phan T. Genetic diversity and evolution of SARS-CoV-2. Infect Genet Evol 2020; 81: 104260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bhattacharyya C, Das C, Ghosh A et al. Global spread of SARS-CoV-2 subtype with spike protein mutation D614G is shaped by human genomic variations that regulate expression of TMPRSS2 and MX1 genes. bioRxiv 2020: 2020.05.04.075911.
- 20. Eaaswarkhanth M, Al Madhoun A, Al-Mulla F. Could the D614G substitution in the SARS-CoV-2 spike (S) protein be associated with higher COVID-19 mortality? Int J Infect Dis 2020; 96: 459–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koyama T, Weeraratne D, Snowdon JL et al. Emergence of drift variants that may affect COVID-19 vaccine development and antibody treatment. Pathogens 2020; 9: 324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chand GB, Banjeree A, Azad GK. Identification of novel mutations in RNA-dependent RNA polymerases of SARS-CoV-2 and their implications on its protein structure. PeerJ 2020; 8: e9492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Berry M, Fielding BC, Gamieldien J. Potential broad spectrum inhibitors of the Coronavirus 3CLpro: virtual screening and structure-based drug design study. Viruses 2015; 7: 6642–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cao B, Wang Y, Wen D et al. A trial of lopinavir–ritonavir in adults hospitalized with severe COVID-19. N Engl J Med 2020; 382: 1787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He R, Leeson A, Ballantine M et al. Characterization of protein–protein interactions between the nucleocapsid protein and membrane protein of the SARS coronavirus. Virus Res 2004; 105: 121–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tilocca B, Soggiu A, Sanguinetti M et al. Comparative computational analysis of SARS-CoV-2 nucleocapsid protein epitopes in taxonomically related coronaviruses. Microbes Infect 2020; 22: 188–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Osório NS, Correia-Neves M. Implication of SARS-CoV-2 evolution in the sensitivity of RT-qPCR diagnostic assays. Lancet Infect Dis 2020; S1473–3099(20)30435-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sturman LS, Holmes KV, Behnke J. Isolation of coronavirus envelope glycoproteins and interaction with the viral nucleocapsid. J Virol 1980; 33: 449–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.