

Abstract
Cu(I) active sites in metalloproteins are involved in O2 activation, but their O2 reactivity is difficult to study due to the Cu(I) d10 closed shell which precludes the use of conventional spectroscopic methods. Kβ X-ray emission spectroscopy (XES) is a promising technique for investigating Cu(I) sites as it detects photons emitted by electronic transitions from occupied orbitals. Here, we demonstrate the utility of Kβ XES in probing Cu(I) sites in model complexes and a metalloprotein. Using Cu(I)Cl, emission features from double-ionization (DI) states are identified using varying incident X-ray photon energies, and a reasonable method to correct the data to remove DI contributions is presented. Kβ XES spectra of Cu(I) model complexes, having biologically relevant N/S ligands and different coordination numbers, are compared and analyzed, with the aid of density functional theory (DFT) calculations, to evaluate the sensitivity of the spectral features to the ligand environment. While the low-energy Kβ2,5 emission feature reflects the ionization energy of ligand np valence orbitals, the high-energy Kβ2,5 emission feature corresponds to transitions from molecular orbitals (MOs) having mainly Cu 3d character with the intensities determined by ligand-mediated d–p mixing. A Kβ XES spectrum of the Cu(I) site in preprocessed galactose oxidase (GOpre) supports the 1Tyr/2His structural model that was determined by our previous X-ray absorption spectroscopy and DFT study. The high-energy Kβ2,5 emission feature in the Cu(I)-GOpre data has information about the MO containing mostly Cu character that is the frontier molecular orbital (FMO) for O2 activation, which shows the potential of Kβ XES in probing the Cu(I) FMO associated with small-molecule activation in metalloproteins.
Graphical Abstract
1. INTRODUCTION
Cu active sites in metalloproteins play essential roles in a wide range of biological processes.1,2 One of the most important is O2 activation, which mostly involves Cu(I) active sites. While a variety of spectroscopic methods have been utilized to provide insight into Cu(II) sites, Cu(I) sites are often considered to be “spectroscopically silent” due to their d10 closed-shell nature. X-ray absorption spectroscopy (XAS) has been applied to study Cu(I) sites because the Cu 1s → 4p transition feature is sensitive to the coordination number3 and the extended X-ray absorption fine structure (EXAFS) determines active-site structural parameters.4 With the development of high-resolution spectrometers in the hard X-ray range,5 Kβ X-ray emission spectroscopy (XES) has become a powerful technique for the study of metal sites. This technique is complementary to XAS as it probes electronic transitions from higher-energy occupied orbitals. In this regard, Kβ XES is a promising complementary spectroscopic tool for the study of Cu(I) active sites.
Kβ XES involves the ionization of a metal 1s electron and the detection of the photons emitted from electrons of the occupied orbitals filling the 1s core hole.6–8 Kβ emissions are generally divided into two regions: the Kβ main and the Kβ valence-to-core (VtC) region. The Kβ main features originate from the metal 3p → 1s transition and are dominated by the 3p–3d exchange interaction in the 1s23p53dn final state. Specifically, the antiparallel and parallel exchange interactions between the 3p hole and the unpaired 3d electrons (as in Cu(II) but not Cu(I) sites) lead to the Kβ1,3 and Kβ′ features, respectively.9,10 The splitting of the Kβ1,3 and Kβ′ features becomes larger with increasing 3p–3d exchange interactions, and thus the Kβ main region is sensitive to the metal oxidation and spin state. The electronic transitions from the valence orbitals to the 1s core hole give rise to the Kβ VtC region, which is further divided into the Kβ2,5 and Kβ″ (or “crossover” peak) features. These Kβ VtC features are assigned to transitions from the valence orbitals having mainly ligand np (and metal 3d) and ligand ns characters for the Kβ2,5 and Kβ″ features, respectively. Since the metal np → 1s transition is electric-dipole-allowed, mixing of metal p character into valence orbitals makes a dominant contribution to the Kβ VtC emission intensity. Thus, the Kβ VtC region is very sensitive to the ligand environment that leads to this mixing.
Previous Kβ XES studies using model complexes have provided background knowledge concerning the factors contributing to the spectral features and have demonstrated the ability of this technique to investigate biologically relevant processes such as small-molecule binding and activation and changes in the ligand protonation state.11–25 For the Kβ VtC region, the experimental data were well simulated with ground-state density functional theory (DFT) calculations that can be used as tools for quantitative analysis.12,13,15 Kβ XES has been further applied to metalloproteins. For example, Kβ VtC XES was used to identify bridging oxo groups in the Mn4Ca cluster of photosystem II26 and to identify the central carbon atom in Mo- and V-dependent nitrogenases.27,28 While Kβ XES studies on Cu sites are scarce compared to those on other 3d transition-metal sites, recent studies have applied Kβ XES to investigate Cu sites in various environments including models,29–33 zeolites,34–38 and metalloproteins.39 It is noteworthy that DeBeer, Blackburn, and co-workers demonstrated the first application of Kβ XES to Cu proteins and showed the sensitivity of the Kβ VtC XES spectral features to the ligand environment.39
Kβ XES measurements are conducted using an incident X-ray photon with an energy well above the absorption edge. If the energy of the incident X-ray photon is high enough, then the ionization of a second electron is possible during the 1s core hole creation. This shake-off process can generate additional emission features. Glatzel et al. measured Kβ VtC XES spectra of MnO2 as a function of the excitation energy and assigned emission features that appeared above the Fermi level as the KLβ emissions originating from the double ionization (DI) of 1s and 2p electrons (1s + 2p DI).40 Similar emission features in the high-energy region of Kβ VtC XES data were observed for Cu systems and have been attributed to the same KLβ mechanism.32,39 In the present study, we find that high-energy emission features in Cu(I) Kβ VtC XES data, in fact, are KMβ emissions arising from the 1s + 3d/3p DIs and overlap the Kβ2,5 feature, which contains important information concerning the d10 electronic structure. Thus, to properly utilize Kβ VtC XES spectral features for the study of Cu(I) active sites, these DI emissions must be well understood and properly corrected.
Here, we present a Kβ XES study of Cu(I) model complexes and a Cu(I) metalloprotein. The emission features on the high-energy side of the Kβ VtC region have been investigated using Cu(I)Cl, a model containing Td [Cu(I)Cl4]3− sites with the Cl− ions bridging to adjacent Cu(I) ions. The incident X-ray photon energy has systematically varied to identify and correct the DI emissions, and the corrected Kβ VtC XES data of Cu(I)Cl have been analyzed using DFT calculations. In addition, the Kβ VtC XES spectra of Cu(I) model complexes, having known structures with different coordination numbers and biologically relevant N/S ligands, have been compared and analyzed with DFT calculations. Finally, the Kβ VtC XES spectrum of the Cu(I) site in preprocessed galactose oxidase (GOpre) has been analyzed on the basis of our studies of the structurally defined Cu(I) model complexes and related to our previous XAS and DFT study on this system.41 GO is a secretory fungal enzyme that catalyzes the two-electron oxidation of primary alcohols to aldehydes (Scheme 1A) and can subsequently perform the two-electron oxidation of aldehydes to carboxylic acids at a much slower rate (Scheme 1B).42–46 Each of these catalytic reactions is coupled to the two-electron reduction of O2 to H2O2. The mononuclear Cu active site in GO can provide only one electron, and thus GO generates an additional redox-active cofactor, a Cys–Tyr linked center, at the active site in a cofactor biogenesis reaction (Scheme 1C). Our previous study41 showed that, in the biogenesis, the reaction of the preprocessed Cu(I) site with O2 leads to a triplet Cu(II)-(O2•−) with a π* frontier molecular orbital (FMO) that abstracts a H atom from a nearby Cys residue. This creates the cross-link to a coordinated Tyr residue that enables galactose oxidation in processed enzyme turnover. The present study defines the Cu(I) FMO that enables this O2 activation and demonstrates the considerable potential of Kβ XES in probing the FMO and key bonding interactions in “spectroscopically silent” Cu(I) active sites.
Scheme 1.

2. RESULTS AND ANALYSIS
2.1. Incident X-ray Photon Energy Dependence of Cu(I) Kβ XES Spectra.
Kβ XES data of the Td [Cu(I)Cl4]3− sites in Cu(I)Cl were collected with varying incident X-ray photon energies of 8987, 9000, 9500, 10 000, 10 500, 11 000, 12 000, and 13 000 eV. The Kβ main region (Figure 1A) has an intense peak at 8904 eV and a shoulder at ∼8901.8 eV (indicated by dotted lines). The energy positions of these features are rather invariant with the excitation energy change. These features correspond to the Kβ1 and Kβ3 emissions split by the Cu 3p spin–orbit coupling. Note that this splitting (∼2.2 eV) is close to those reported in the previous studies of Cu sites.39,47,48 For a Cu(I) system having a d10 closed shell, only the Kβ1,3 emission features are expected because there is no exchange interaction between the 3p hole and the paired electrons in the 3d10 subshell. With increasing excitation energy, however, the Kβ main region (Figure 1A) shows spectral changes and gains additional features, one on the low-energy side (∼8895.5 eV; orange arrow) and the other on the high-energy side (∼8909 eV; purple arrow). The inset in Figure 1A presents the maximum-normalized spectra showing the clear change in the spectral shape between excitation energies of 8987 and 10 500 eV. The spectra stop changing at incident X-ray photon energies above 9000 eV. The energy dependence of the additional features on the low- and high-energy sides (orange and purple arrows in Figure 1A) reflects shake-off transitions, with the threshold energy for the DI approximated by a Z + 1 model.49,50 In this Z + 1 model, the increased Zeff due to the ionization of a 1s electron is considered by using a Z + 1 element. For example, the threshold energy for the 1s + 2p DI is estimated from E(Cu KL-edge) ≈ E(Cu K-edge) + E(Zn L-edge), which gives E(1s2p) ≈ 10 008.5 eV (the underlining indicates a vacant orbital) using E(Cu K-edge) = 8979 eV and E(Zn L-edge) = 1029.5 eV (the degeneracy-weighted average of the Zn 2p1/2 and 2p3/2 binding energies).51 For the Cu KM DIs, E(1s3d) and E(1s3p) can be similarly estimated to be 8989.1 and 9068.5 eV, respectively.51 By comparing these estimated threshold energies to the excitation energies used for Cu(I)Cl, the spectrum obtained with 8987 eV (solid black spectrum in Figure 1A) should solely originate from the 1s single ionization (SI). With an excitation energy of 9000 eV (solid red spectrum in Figure 1A) which enables the 1s + 3d DI, the ∼8895.5 eV feature (orange arrow in Figure 1A) has gained intensity while the ∼8909 eV feature (purple arrow in Figure 1A) is still weak. Thus, the onset of these additional features indicates that the ∼8895.5 eV feature arises from the 1s + 3d DI and that the ∼8909 eV feature is likely from the 1s + 3p DI with a minor contribution from the 1s + 3d DI. These assignments are in line with the previous studies of Cu sites.47,48,52,53 The ∼8895.5 eV feature is similar to a Kβ′ feature where, even for d10 Cu(I) complexes, the 3p–3d exchange interaction is present in the 1s + 3d DI state.
Figure 1.

Kβ XES spectra of Cu(I)Cl for (A) Kβ main and (B) Kβ VtC regions obtained with excitation energies of 8987 (solid black), 9000 (solid red), 9500 (solid green), 10 000 (solid blue), 10 500 (dashed black), 11 000 (dashed red), 12 000 (dashed green), and 13 000 eV (dashed blue). The inset in (A) shows the maximum-normalized Kβ main regions obtained with excitation energies of 8987 (solid black) and 10 500 eV (dashed black) for a facile comparison of the spectral shapes. In (B), the high-intensity peaks at 8987 (solid black) and 9000 eV (solid red) are from the elastic scattering of the incident X-ray. Orange and purple arrows indicate the DI emission features.
In Figure 1B, the Kβ VtC region of Cu(I)Cl also exhibits spectral changes with increasing excitation energy, and two additional features appear on the high-energy side (indicated by orange and purple arrows). The Kβ2,5 features at ∼8976 and ∼8972 eV and the Kβ″ feature at ∼8962 eV correspond to the transitions from the valence orbitals and will be analyzed in the following section. While the ∼8980 eV feature (orange arrow in Figure 1B) is not present with an excitation energy of 8987 eV (solid black spectrum in Figure 1B) and becomes reasonably intense with an excitation energy of 9000 eV (solid red spectrum in Figure 1B), the ∼8989 eV feature (purple arrow in Figure 1B) appears with an excitation energy above 9000 eV (e.g., solid blue spectrum in Figure 1B). This indicates that, based on the threshold energies estimated with the Z + 1 model, the ∼8980 and ∼8989 eV features are related to the 1s + 3d DI and the 1s + 3p DI states, respectively.
The Z + 1 approximation can also be used to estimate the emission energies from the DI states. The emission energy from the KM DI (1s3d or 1s3p) is approximated by E(Cu KMβ) ≈ E(Cu Kβ) + [E(Zn M-edge) – E(Cu M-edge)]. E(Cu Kβ) is the Kβ main or VtC peak energy from the 1s SI state, and [E(Zn M-edge) – E(Cu M-edge)] represents the energy shift, which is due to the additional 3d or 3p hole creation (the shake-off process), from the 1s SI emission features. This gives ∼8 and ∼14 eV shifts for the 1s + 3d DI and the 1s + 3p DI, respectively.51,54 Thus, by considering the 1s + 3d DI (∼8 eV shift), the ∼8980 eV DI feature (orange arrow in Figure 1B) correlates to the intense ∼8972 eV SI feature (black spectrum in Figure 1B). The 1s + 3d DI is expected to generate an additional DI emission feature at ∼8984 eV from the ∼8976 eV SI feature, but there is no significant emission feature at ∼8984 eV. This likely reflects the relatively weak nature of the ∼8976 eV SI feature of Cu(I)Cl. We note that, in fact, the ∼8984 eV DI feature is not experimentally observed for any of the Cu(I) model complexes used in this study (section 2.3). The final state for the ∼8984 eV DI feature is 1s23d8 in which both singlet and triplet states are present and the exchange interaction would result in a distribution of the states and thus the intensity of the ∼8984 eV DI feature, which precludes experimental observation. The 1s + 3p DI predicts an ∼14 eV shift, suggesting the correlation of the broad ∼8989 eV DI feature (from fits, the full width at half-maximum is ∼4 to ∼5 eV) with the overall Kβ2,5 SI emission. Therefore, relative to the Kβ2,5 1s SI emission feature, the Z + 1 approximated energy shifts confirm that the ∼8980 and ∼8989 eV features are Kβ2,5 emissions from the 1s + 3d DI and the 1s + 3p DI states, respectively.
Note that these KMβ assignments for Cu(I)Cl are different from the DI emission features in Mn complexes (KLβ) that involve the 1s + 2p DI (with the Kβ main feature as the corresponding 1s SI emission).7,40 For the Cu 1s + 2p DI, the threshold energy and the energy shift from the Kβ 1s SI emission are estimated to be 10 008.5 and ∼90 eV, respectively.51 This indicates that, with the Cu(I) Kβ main 1s SI emission feature at ∼8904 eV, a new DI emission feature at ∼8994 eV is expected with an excitation energy higher than 10 008.5 eV. However, there is no observable feature at ∼8994 eV for any excitation energy (Figure 1B), indicating that the KL DI contribution is substantially weaker than the KM DI contribution. We note that the Kβ VtC XES spectra of Cu(I) Cl were collected up to 9050 eV, but no significant emission feature was observed at emission energies above 8989 eV (Figure S1).
The 1s + 3d DI feature at ∼8980 eV (orange arrow in Figure 1B) appears as a high-energy shoulder on the Kβ2,5 emissions and thus overlaps the ∼8976 eV SI feature, which may lead to a misinterpretation of the Kβ2,5 features. It would be ideal to use an excitation energy lower than the 1s + 3d DI threshold (<8989.1 eV) to collect the Kβ XES data solely originating from the 1s SI state. However, this low excitation energy generally cannot be used, especially for dilute systems (e.g., metalloproteins), because the Compton scattering (which decreases with increasing excitation energy) significantly contributes to the Kβ VtC region and because the data quality becomes poor (low signal-to-noise ratio). Note that the previous Cu Kβ VtC XES studies29–39 used an incident X-ray photon energy higher than 9000 eV and that we used 10 500 eV to collect the Cu(I)-GOpre data presented in section 2.4. The standard correction to remove the 1s + 3d DI contribution from the Kβ VtC XES data obtained with a higher excitation energy is the subtraction of the ∼8980 eV DI feature using fits to the measured spectra. To test this correction, the Kβ VtC XES spectra of Cu(I)Cl were fit with pseudo-Voigt peaks for the spectral features and a spline function modeling the background (high-energy tail of the Kβ1,3 peak) (see the Experimental Section for the fitting process; see Figure S2A for a representative fit). Figure 2A shows the comparison of the Kβ VtC XES spectra of Cu(I)Cl for excitation energies of 8987 (black spectrum; no DI) and 10 500 eV (red spectrum) after the subtraction of the spline background and the DI emission features at ∼8980 and ∼8989 eV (Supporting Information and Figure S2A–C). By increasing the excitation energy from 8987 to 10 500 eV, the contribution from the 1s SI state decreases due to the increased DI probability, and accordingly the intensities of the ∼8972 and ∼8976 eV SI features should decrease. If the subtraction of the ∼8980 eV DI feature using the fit is correct, then the intensity ratio of the ∼8976 to ∼8972 eV features would remain the same for the spectra obtained with excitation energies of 8987 and 10 500 eV. However, Figure 2A shows that the intensity ratio between these two features is different; from the fits, the intensity ratios of the ∼8976 to ∼8972 eV features are 0.51 and 0.75 for excitation energies of 8987 (black spectrum) and 10 500 eV (red spectrum), respectively. This indicates that the simple subtraction of the ∼8980 eV DI feature is not an accurate correction as there remains a contribution from the 1s + 3d DI emission overlapping the ∼8976 eV SI feature for the data obtained with an excitation energy of 10 500 eV. Thus, a further correction is required to account for this residual 1s + 3d DI contribution. The correction factor obtained from the intensity ratios in Figure 2A (i.e., 0.51/0.75 = 0.68) was thus applied to decrease the intensity of the ∼8976 eV feature (Figure S2C,D). This intensity ratio corrected spectrum was scaled to have the same area under the Kβ VtC region as the data obtained with an excitation energy of 8987 eV. The detailed description of this correction method to remove the DI contribution is provided in the Supporting Information. In Figure 2B, the intensity pattern in the corrected spectrum (green) looks very similar to the spectrum solely from the 1s SI state (black; excitation energy of 8987 eV). Note that the corrected intensity ratio is also close to that obtained from the DFT calculations in the next section.
Figure 2.

Kβ VtC XES spectra of Cu(I)Cl. The black spectrum in (A) and (B) is the background-subtracted data for an excitation energy of 8987 eV. The red spectrum in (A) is the data after subtraction of the background and the DI emission features at ∼8980 and ∼8989 eV for an excitation energy of 10 500 eV. The green spectrum in (B) is the data after the intensity ratio correction from the red spectrum in (A).
2.2. DFT Calculations of the Kβ VtC XES Spectrum of Cu(I)Cl.
Cu(I)Cl has the zinc-blende structure with each Cu(I) possessing Td site symmetry and the four Cl− ligands bridging to adjacent Cu(I) ions (Figure S3A).55 The DFT calculations (using BP86) of Cu(I)Cl were first conducted with increasing cluster sizes (from one Td [Cu(I)Cl4]3− cluster up to a large cluster having 113 atoms); these calculated Kβ VtC XES spectra were very similar to one another (Figure S3B). Further DFT calculations using the Td [Cu(I)Cl4]3− cluster were carried out with different functionals: BP86, B3LYP, TPSS, and TPSSh (Figure S3C). While all of the functionals gave qualitatively similar Kβ VtC XES spectra, the BP86 functional best reproduced the energy splittings between the spectral features in the experimental data obtained with an excitation energy of 8987 eV (i.e., no DI) (Table S1). Also, note that the intensity ratio of the ∼8976 to ∼8972 eV features in the DFT/BP86 calculation is 0.48, which is close to the experimental value of 0.51 (Table S1). Thus, further analysis of the Cu(I)Cl data is presented using the DFT calculation for Td [Cu(I)Cl4]3− with the BP86 functional.
Figure 3A compares the calculated spectrum with the experimental spectrum. The schematic molecular orbital (MO) diagram for a Td complex (with σ/π-donor ligands such as Cl−) is presented in Figure 3B. For Td [Cu(I)Cl4]3−, the Cu d orbitals transform as e and t2, and the Cu p orbitals transform as t2. The Cl s orbitals transform as a1 and t2, and the Cl p orbitals transform as a1 and t2 for the σ symmetry set and e, t1, and t2 for the π symmetry set. As the Kβ VtC emission intensity is dominated by the electric dipole p → s transition mechanism, the MOs generating the intense Kβ VtC XES spectral features must have t2 symmetry for Cu p character mixing. In Figure 3A, the experimentally observed features at ∼8962, ∼8972, and ∼8976 eV are well simulated by the DFT calculation, which enables the assignments of these features. The ∼8962 and ∼8972 eV features correspond to the transitions from the MOs having mostly Cl 3s(σ) character (97.8%) and Cl 3p(σ) character (93%), respectively. The transitions from these MOs derive emission intensity from Cu p mixing (i.e., 1.9% and 3.7% Cu p characters for the ∼8962 and ∼8972 eV features, respectively; MOs with t2(σ) symmetry; the contour plots for these MOs are shown in Figure 3A). For the ∼8976 eV feature, two sets of the transitions have been calculated: one set at 8976.4 eV and the other at 8976.2 eV (Figure 3A). The dominant contribution to the ∼8976 eV feature is from the transition at 8976.4 eV, and the MOs for this transition have mainly Cu 3d character (87%) with small Cu p character (1.8%). These MOs correspond to the t2 set of the Cu 3d orbitals (xy, xz, and yz) that gain emission intensity through ligand-mediated Cu d–p mixing (the contour plot for one of these MOs is shown in Figure 3A).39,56 The transition at 8976.2 eV makes a minor contribution to the ∼8976 eV feature; the MOs for this transition correspond to the e set of the Cu 3d orbitals (x2–y2 and z2) (94.3% Cu 3d and 0% Cu p characters). For the e set of the Cu 3d orbitals, Cu p mixing is forbidden by symmetry and thus the Cu 3d → 1s transition from these MOs is calculated to be governed by the electric quadrupole transition mechanism, reflecting their low intensity in Figure 3A.
Figure 3.

(A) Comparison of experimental (black, 8987 eV excitation energy, background subtracted) and DFT/BP86 calculated (blue, using Td [Cu(I)Cl4]3−) Kβ VtC XES spectra of Cu(I)Cl. The calculated spectrum has been scaled to match the ∼8972 eV peak intensity to that in the experimental spectrum. Contour plots of key MOs contributing to calculated transitions are shown (Cu in brown and Cl in green). (B) Schematic MO diagram for a Td complex.
Note that there are two sets of the Cl 3p orbitals (t2(π) and t2(σ)) which have the proper symmetry to mix with the Cu p orbitals (Figure 3B). Both the t2(π) and t2(σ) MOs with dominant Cl 3p character have comparable Cu p character (t2(π), 89.7% Cl 3p and 3.1% Cu p; t2(σ), 93% Cl 3p and 3.7% Cu p), but the ratio of the calculated emission intensity from the t2(π) MOs to that from the t2(σ) MOs is only ∼0.02; note that, in Figure 3A, while the transitions from the t2(σ) MOs (at 8972.4 eV) are shown as the intense stick, the transitions from the t2(π) MOs (at 8972.8 eV) have very low intensity, which is hardly observable on the presented scale. This may be related to the nature of Cu p character (3p vs 4p) mixed into the donor MOs. This is an important issue because, while metal 3p character is borrowed from the Kβ main feature, metal 4p character contributes to the bonding. Through calculations, DeBeer and co-workers suggested that, while most metal p character in the donor MOs is derived from metal 4p mixing, the calculated intensity is more related to metal 3p character as these orbitals are more contracted, giving a large p → 1s transition dipole moment.13,20 For Td [Cu(I)Cl4]3−, the Cl 3p t2(π) orbitals are not directed toward the Cu center, and this limits their mixing with the Cu 3p orbitals, which are radially more contracted than the Cu 4p orbitals.
2.3. Kβ XES Spectra of Cu(I) Model Complexes.
Four Cu(I) model complexes, [Cu(I)-(Me2N-MePY2)]B(C6F5)4 (1),57 [Cu(I)-(BA)]SbF6 (2),58 [Cu(I)-(LASM)]B(C6F5)4 (3),59 and [{Cu(I)-(DMMESE)}2](B(C6F5)4)2 (4),60 having crystal structures61 and more biologically relevant N/S ligations that vary in coordination number have been studied by Kβ XES for calibration and extension to the Cu(I) site in GOpre. Their geometry-optimized structures are given in Figure 4. The calculated and observed structures generally agree within 0.03 Å and 9.8° (see Table S2 for the structural parameters), with 1 being a three-coordinate Cu(I) complex with three N atoms, 2 being a four-coordinate Cu(I) complex with all N atoms, and 3 and 4 being four-coordinate Cu(I) complexes with 3N/1S ligation. Their Kβ XES spectra are shown in Figure 5. These were measured with an excitation energy higher than the 1s + 3p DI threshold, and thus the data contain contributions from both the 1s + 3d and 1s + 3p DIs (arrows in Figure 5). Figure 5A compares the Kβ main regions for complexes 1–4 (also included are Cu(I)Cl with an excitation energy of 10 500 eV and Cu(I)-GOpre). As expected, all of the Cu(I) complexes exhibit very similar Kβ main features as this region is mostly sensitive to the metal oxidation and spin state. Alternatively, the spectral features of complexes 1–4 in the Kβ VtC region vary to a large extent (Figure 5B), indicating that the Kβ VtC region is sensitive to the ligand environment. While the Kβ″ feature at ∼8962 eV is clearly seen for Cu(I)Cl (Figure 1B), this feature is not observed for complexes 1–4 in the 8945–8965 eV region (Figure 5B). The lack of a noticeable Kβ″ feature reflects the fact that the ligand ns → Cu 1s transition is very weak and is overlapped by the strong background from the Kβ main peak.
Figure 4.

DFT-optimized structures of Cu(I) model complexes 1–4 (Cu in brown, N in blue, S in yellow, C in gray, and O in red). H atoms are omitted for clarity. Note that complex 4 is present as a centrosymmetric dimer in the solid state. Structural parameters are given in Table S2.
Figure 5.

Kβ XES spectra of complexes 1 (black), 2 (red), 3 (green), and 4 (blue) for (A) Kβ main and (B) Kβ VtC regions. Kβ main regions of Cu(I)Cl (orange, 10 500 eV excitation energy) and Cu(I)-GOpre (purple) are included in (A) for comparison. Arrows indicate the DI emission features. Insets in (B) show the expanded Kβ VtC XES data for better comparison of the Kβ2,5 and DI emission features.
For comparison to DFT calculations and the analysis of the Kβ VtC regions of complexes 1–4, the background and DI features (modeled with a spline function and pseudo-Voigt peaks, respectively) were subtracted from these data (see Figure S5 for fits). The resultant spectra are presented in Figure 6A–D. In section 2.1, for the Cu(I)Cl data obtained with the 10 500 eV excitation energy, the simple subtraction of the DI features using pseudo-Voigt fits was not sufficient, and the additional intensity ratio correction (using a factor of 0.68) was necessary as the 1s + 3d DI emission at ∼8980 eV largely overlaps the ∼8976 eV SI feature. This suggests that the ∼8976 eV features in Figure 6A–D may still have a residual contribution from the 1s + 3d DI. For Cu(I)Cl, the 1s + 3d DI feature at ∼8980 eV was related to the ∼8972 eV SI feature shifted by ∼8 eV based on the Z + 1 model (see section 2.1 and the orange spectrum in Figure 7). For complexes 1–4, the energy position of the equivalent SI emission on the low-energy side of the Kβ2,5 feature is found to vary (Figure 5B), which results in energy shifts of the corresponding 1s + 3d DI feature. As shown below, the SI emission on the low-energy side of the Kβ2,5 feature corresponds to transitions from the valence MOs having ligand np character whose energy reflects the Zeff of the ligand. For example, in Figure 7, Cl ligands in Cu(I)Cl (orange spectrum) and N ligands in complex 1 (black spectrum) give rise to this SI emission at ∼8972 and ∼8973 eV, respectively, while complex 4 having S (and N) ligation (blue spectrum) generates the equivalent SI emission at the higher energy of ∼8974 eV. Accordingly, the 1s + 3d DI feature is found at ∼8980, ∼8981, and ∼8982 eV for Cu(I)Cl, complex 1, and complex 4, respectively (Figure 7). The 1s + 3d DI emissions of complexes 1 and 4 are ∼5 and ∼6 eV higher in energy than the ∼8976 eV feature, respectively. Thus, these DI emissions in complexes 1 and 4 have less overlap with the ∼8976 eV SI feature, suggesting that the intensity ratio correction factor is larger for complexes 1 and 4 relative to 0.68 obtained for Cu(I)Cl. Accordingly, using the Cu(I)Cl data, for the 1s + 3d DI emission but shifted to different energies (8981–8983 eV), the degree of overlap of this DI emission with the ∼8976 eV SI feature is estimated and proper intensity ratio correction factors are obtained (Table 1) (see the Supporting Information for details in obtaining these factors). Note that the energies of the 1s + 3d DI feature and the corresponding SI feature of complex 3 are very similar to those of complex 4 (green and blue spectra in Figure 5B). For complex 2, the coordinated ligands are all N atoms, which generate the SI emission at ∼8973 eV (as for complex 1), but its 1s + 3d DI feature is observed at ∼8982 eV, which is ∼1 eV higher than that expected from the Z + 1 model (red spectrum in Figure 5B). This may be due to the fact that its ∼8976 eV SI feature is more intense and contributes to the 1s + 3d DI emission intensity envelope. The DI feature associated with the ∼8973 eV SI feature thus shifts the net DI feature to higher energy. The appropriate intensity ratio correction factors were applied to the data of complexes 1–4 based on their 1s + 3d DI emission energies (0.75 for complex 1 and 0.88 for complexes 2–4), and these corrected spectra are shown in Figure 6E–H.
Figure 6.

Kβ VtC XES spectra of complexes 1 (black), 2 (red), 3 (green), and 4 (blue). The ligand names are shown at the top of each column. (A–D, Exp.) Experimental data after subtraction of the background and the DI emission features, (E–H, Corr. Exp.) experimental data after the further intensity ratio correction using correction factors in Table 1, and (I–L, DFT) DFT/BP86 calculated data using the optimized structures shown in Figure 4. Contour plots of selected MOs contributing to calculated transitions are shown at the bottom of each model and are correlated with the dotted lines (see Figures S8–S10 for complete sets of contour plots of MOs associated with intense transitions in complexes 2–4). The molecules are oriented similarly to the structures presented in Figure 4.
Figure 7.

Background-subtracted Kβ VtC XES spectra of Cu(I)Cl (orange, 10 500 eV excitation energy), complex 1 (black), and complex 4 (blue) for an emission energy range of 8970–8985 eV. Arrows, which are drawn with an 8 eV separation for the same colors (considering the Z + 1 model), show correlations for 1s + 3d DI emission features and their corresponding SI emission features.
Table 1.
Intensity Ratio Correction Factorsa
| energy of the 1s + 3d DI emission | correction factor |
|---|---|
| ~8980 eV | 0.68 |
| ~8981 eV | 0.75 |
| ~8982 eV | 0.88 |
| ~8983 eV | 0.94 |
Details of obtaining these factors are described in the Supporting Information.
In Figure 6, although the calculated Kβ″ features in the 8955–8965 eV range are not clear in the experimental data, the Kβ2,5 features are well simulated by DFT calculations (features a–c), which are used for spectral assignments. The Cu(I) center of complex 1 has a geometry between T-shaped and trigonal planar and is coordinated with three N atoms (one amine and two pyridines) (Figure 4 and Table S2). The Kβ VtC region of complex 1 shows two intense features at ∼8976 and ∼8973 eV and a weaker shoulder at ∼8969 eV (features a–c, respectively, of the black spectra in Figure 6). Note that the intensity pattern of these features in the intensity ratio corrected spectrum (Figure 6E) is similar to that in the DFT calculation (Figure 6I). The DFT calculation shows that the MOs contributing to feature a mostly contain Cu 3d character. The major contribution to feature a comes from the MO with an antibonding interaction between the Cu and N(amine) 2p orbitals that is the highest occupied molecular orbital (HOMO). At slightly lower energy, there is the minor contribution to feature a from the MO with an antibonding interaction between the Cu 3dxy and N(pyridine) 2p orbitals. For feature b, most of the intensity comes from the MO having primarily N(pyridine) 2p character which has a bonding interaction with the Cu 3dxy orbital. Feature c corresponds to transitions from the MOs having mostly pyridine character. These key MOs are shown at the bottom of the black spectra in Figure 6. As the transitions are governed by an electric dipole mechanism, the MOs giving the high emission intensity contain Cu p character (∼1–6%). These spectral assignments are similarly relevant to complexes 2–4; features a–c originate from transitions from the MOs having mostly Cu 3d, ligand np, and overall ligand character, respectively. It is noteworthy that the spectral assignments for features a and b in complex 1 parallel those in the DeBeer, Blackburn, and co-workers’ study of three-coordinate Cu(I) sites.39
In proceeding to four-coordinate Cu(I) complexes 2–4, the intensity of feature a at ∼8976 eV becomes comparable to or higher than that of feature b (red, green, and blue spectra in Figure 6). The Cu(I) sites in complexes 2 and 3 have a trigonal pyramidal geometry, and that in complex 4 has a distorted trigonal pyramidal geometry (Figure 4 and Table S2). The DFT calculations show that, for these four-coordinate Cu(I) complexes, the transition from the MO with Cu character (in addition to the MOs with Cu and 3dxy character as in complex 1) gains significant intensity through mixing, which becomes allowed from the site symmetry change. This gives additional intensity to feature a. These MOs having Cu character are the HOMOs for complexes 2–4 and have an antibonding interaction between the Cu and N(amine) 2p orbitals; their contour plots are shown for feature a of complexes 2–4 in Figure 6. We note that, for these four-coordinate Cu(I) complexes 2–4, the relative intensity of feature a to feature b is higher in the DFT calculations (Figure 6J–L) than that in the intensity ratio corrected spectra (Figure 6F–H). While it is possible that the DFT calculations overestimate the feature a intensity for these four-coordinate Cu(I) complexes, it is also possible that the intensity ratio correction factors for the 1s + 3d DI emission at ∼8982 eV (0.88, Table 1) could be slightly low.
For complexes 3 and 4 having S ligation (thioether), feature b appears at ∼8974 eV, an ∼1 eV upshift relative to complexes 1 and 2 (see Figure S11 for an expanded comparison). Also, features a and b are less resolved for complexes 3 and 4 than for complex 2. From the DFT simulations in Figure 6I–L, feature b corresponds to transitions from MOs that mainly have ligand np character, and the energy position of feature b depends on the ionization energy of the ligand np orbitals. The DFT calculations show that, while for complexes 1 and 2 the MOs with N 2p character mostly contribute to feature b, for complexes 3 and 4 the MOs with S 3p character (in addition to N 2p character) contribute to feature b. Thus, the calculated transitions for feature b are primarily at ∼8973 eV for complexes 1 and 2 (Figure 6I,J), but spread over an energy range of ∼8973 to ∼8974 eV for complexes 3 and 4 (Figure 6K,L). This leads to the experimental observation of the ∼1 eV upshift of feature b and the less resolved a and b peaks in the Kβ2,5 region for complexes 3 and 4 having S ligation. Note that while for complexes 1 and 2 the contour plots of the MOs presented for feature b in Figure 6 correspond to the most intense transition in feature b, for complexes 3 and 4 these correspond to the MOs having S 3p character that have a bonding interaction with the Cu 3d orbitals.
2.4. Kβ VtC XES Spectrum of Cu(I)-GOpre.
Because no crystal structure exists for the Cu(I) site in GOpre, we previously used Cu K-edge XAS to obtain insight into its geometric structure.41 The Cu(I) 1s → 4p transition feature and the EXAFS data revealed a three-coordinate Cu(I) center, which is coordinated with three O/N donor atoms at an average distance of 2.01 Å. The possibility of a S–Cu bond was ruled out by the EXAFS analysis, and a bond valence sum analysis showed that the 1O/2N coordination was preferred over the possible 2O/1N coordination. DFT geometry optimizations using both apo GOpre and processed GO indicated that the Cu(I) center in GOpre would be coordinated with Tyr272, His496, and His581 residues (Figure 8) and this optimized structure agreed with the EXAFS data. The optimized structure was used for the further computational study of O2 activation and cofactor biogenesis.41 The present study complements the XAS study with the Kβ VtC XES data of Cu(I)-GOpre to obtain additional insight into its geometric and electronic structure.
Figure 8.

DFT-optimized structure and structural parameters for Cu(I)-GOpre (Cu in brown, N in blue, S in yellow, C in gray, O in red, and H in white).41
An excitation energy of 10 500 eV was used for the Kβ XES measurement of Cu(I)-GOpre to prevent X-ray Raman scattering from the buffer, minimize the Compton scattering background, and achieve a higher signal-to-noise ratio. The background of the Kβ VtC XES spectrum of Cu(I)-GOpre was found to increase toward higher emission energy with a different shape from that typically associated with the tail of the Kβ main peak (Figure S12A). This indicates that, although the excitation energy of 10 500 eV is quite high, the Compton scattering still significantly contributes to the Kβ VtC region for this dilute metalloprotein sample (∼0.6 mM Cu(I)). However, as the Compton scattering affects only the shape of the background without interfering in the Kβ VtC emission features, this was reasonably modeled with a spline function (Figure S12A).
For the Kβ VtC XES spectrum of Cu(I)-GOpre, although its data quality is poorer than that of Cu(I)Cl and complexes 1–4, the spectrum clearly shows the characteristic Kβ2,5 feature (black spectrum in Figure S12A). Thus, for further analysis, the Kβ VtC XES data of Cu(I)-GOpre were processed as described for those of Cu(I)Cl and complexes 1–4 (see the Supporting Information for a detailed description of the data processing). The background and DI emission-subtracted Kβ VtC XES spectrum of Cu(I)-GOpre is presented in Figure 9A (Figure S12A–C). As the 1s + 3d DI emission feature in the Cu(I)-GOpre data was fit with a pseudo-Voigt peak centered on ∼8981 eV (Figure S12B), the a/b intensity ratio was corrected using a factor of 0.75 (Table 1 and Figure S12C,D). This corrected spectrum is shown in Figure 9B. The resultant intensity pattern is similar to that obtained from the DFT simulation (Figure 9C), calculated with the optimized structure shown in Figure 8 and the BP86 functional. Note that this simulation as well as simulations with the different functionals including B3LYP, TPSS, and TPSSh all show qualitatively similar calculated Kβ VtC XES spectra (Figure S13). It should be noted that photodamage from the incident X-ray does not lead to a higher intensity of feature a. Spectral changes from photodamage were carefully monitored and, when present, were found to increase the feature b intensity (Figure S14).
Figure 9.

Kβ VtC XES spectra of Cu(I)-GOpre for (A, black) experimental data after subtraction of the background and the DI emission features, (B, red) experimental data after the further intensity ratio correction, and (C, green) DFT/BP86 calculation using the optimized structure shown in Figure 8.
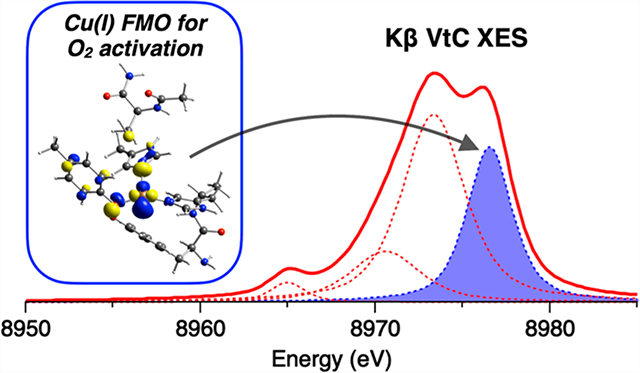
Spectral assignments can be made using the DFT calculation of the optimized structure (1Tyr/2His; Figure 8) shown in Figure 9C. The general spectral assignments are very similar to those made for the Cu(I) model complexes in section 2.3; features a and b correspond to transitions from the MOs having mainly Cu 3d and O/N 2p character, respectively. Specifically, feature a originates mostly from the MOs having antibonding combinations of the Cu and 3dxy orbitals with the O/N 2p orbitals (1 and 2, respectively, in Figures 9C and 10). For feature b, two major transitions are present (3 and 4 in Figure 9C); transition 3 is from the MO having primarily N(His581) 2p character which has a bonding interaction with the Cu orbital (3 in Figure 10), and transition 4 arises from the MO mostly on the Tyr272 and His496 residues (4 in Figure 10). These MOs have ∼1–3% Cu p character giving the higher emission intensity. Note that the HOMO is the MO having mostly Cu character (1 in Figure 10), which is the FMO that plays the key role in O2 binding and activation.
Figure 10.

Contour plots of selected MOs contributing to calculated transitions denoted as 1–4 in Figure 9C. The molecules are oriented similarly to the structure presented in Figure 8.
In our previous study, other possible structures of Cu(I)-GOpre were computationally evaluated.41 These include a two-coordinate structure with Tyr495/His581 ligation and three-coordinate structures with Tyr272/Tyr495/His581 ligands and either Tyr272 or both Tyr272 and Tyr495 deprotonated. Although these structures were found to be at least 5 kcal/mol higher in energy than the 1Tyr/2His structure shown in Figure 8, DFT-simulated Kβ VtC XES spectra have been obtained for these structures to evaluate the sensitivity of the Kβ VtC XES data to structure. Among the DFT calculations from these structures (Figure S15), the 1Tyr/2His structure (Figure S15B, same as Figure 9C) best reproduces the experimental spectrum (Figure S15A), indicating that Kβ VtC XES also supports the 1Tyr/2His model as in our previous XAS and DFT study.41 For the Tyr272/Tyr495/His581 structure with both Tyr’s deprotonated, feature b in Figure S15C mostly reflects O 2p character. Thus, compared to the 1Tyr/2His structure (Figure S15B), feature b appears ∼1 eV lower energy, and the a and b peaks are more separated (Figure S15C), which is not in accord with the experimental spectrum in Figure S15A. The Tyr272/Tyr495/His581 structure with only Tyr272 deprotonated, in fact, has a very long Cu–O(Tyr495) bond (3.127 Å), and thus its DFT-calculated spectrum (Figure S15D) is similar to that from the two-coordinate Tyr495/His581 structure (Figure S15E). For these two calculated spectra (Figure S15D,E), the relative intensity of feature a to feature b is lower than that in the other models, which does not agree with the experimental spectrum (Figure S15A).
In Figure 9B,C, although the DFT-calculated spectrum with the 1Tyr/2His structure is generally similar to the intensity ratio corrected spectrum, the intensity of calculated feature “a” is still low. DeBeer, Blackburn, and co-workers computationally explored the effect of the three-coordinate Cu(I) site geometry on the Kβ VtC XES spectra and found that feature a gains intensity as the geometry is distorted from T-shaped to trigonal planar.39 To explore this, an additional DFT-simulated Kβ VtC XES spectrum has been obtained using a trigonal planar Cu(I) site geometry in GOpre. This trigonal planar Cu(I) geometry was obtained from the Cu(I)-GOpre structure in Figure 8, which was truncated to contain only the first coordination sphere of Cu(I). Note that the DFT-calculated Kβ VtC XES spectrum for the as-truncated Cu(I)-GOpre model shows no significant deviation from that for the full structure (Figure S16). In Figure 11B, relative to the calculated Kβ VtC XES spectrum of the as-truncated Cu(I)-GOpre structure (green spectrum), the calculated spectrum using the trigonal planar Cu(I) geometry (blue spectrum) shows higher intensity in feature a. This intensity change better corresponds to the experimental spectrum in Figure 11A, suggesting that the Cu(I) site in GOpre likely has a more trigonal planar structure than that in Figure 8 where the angle of O–Cu–N(His496) is 149.2° (vs 120° for trigonal planar). However, the HOMO is still found to have mostly Cu character (the contour plots in Figure 11) and thus the FMO for O2 activation is not significantly affected by this distortion.
Figure 11.

Kβ VtC XES spectra of Cu(I)-GOpre for (A, red) experimental data after subtraction of the background along with the DI emission features and the further intensity ratio correction and (B) DFT/BP86 calculations using the as-truncated (green) and the trigonal planar (blue) Cu(I) sites in the GOpre model. Contour plots of the HOMOs contributing to feature a are shown.
3. DISCUSSION
The Kβ XES data of Cu(I)Cl clearly show that the spectra depend on the incident X-ray photon energy (Figure 1). A higher excitation energy increases the probability of the DIs (shake-off transitions), which generate additional features in both the Kβ main and Kβ VtC regions. The energy dependence of the onset of these additional features combined with the Z + 1 model indicates that these additional features arise from the KM DI states. In the Kβ VtC region of Cu(I)Cl, two additional features at ∼8980 and ∼8989 eV are assigned to the 1s + 3d DI and 1s + 3p DI emission features, respectively. While the previous study on MnO2 by Glatzel et al. showed that similar emission features above the Fermi level originate from the 1s + 2p DI (KLβ),40 the present study reveals that the KM DI significantly contributes to the Kβ VtC region of Cu(I) systems. Mukoyama and Taniguchi calculated the probability of the shake-off transitions and found that, for the 3d transition metals going from lower to higher Z, the probability for 1s2p and 1s3p decreases while it remains similar for 1s3d.62 From these calculations, relatively weak KLβ features should also be present in Cu(I) systems, but the KLβ emission for Cu(I)Cl is not experimentally observed as the KMβ emissions dominate the high-energy side of the Kβ VtC region. Furthermore, these calculations indicate that the KM DI contributions should exist in Kβ VtC regions of other 3d transition-metal complexes. However, from the Z + 1 model, the KM DI emissions for other 3d transition-metal complexes are predicted to largely overlap the 1s SI features in their Kβ2,5 regions. Note that Pollock and DeBeer suggested that some features in the Kβ VtC region of MnO are likely from many-electron transitions.13
The Kβ XES data originating solely from the 1s SI state can be obtained with an excitation energy below the 1s + 3d DI threshold (<8989.1 eV), as demonstrated with Cu(I)Cl using an excitation energy of 8987 eV (section 2.1). However, such a low excitation energy generally cannot be used, especially for dilute metalloprotein samples, due to the Compton scattering background and the low signal-to-noise ratio of the data. In Kβ XES data obtained with an excitation energy above the DI threshold, the 1s + 3d DI emission feature appears on the high-energy side of the Kβ2,5 region and overlaps the ∼8976 eV SI feature. This overlap can lead to the incorrect analysis of the Kβ2,5 features, which are particularly important for Cu(I) systems, and thus the data obtained with an excitation energy higher than the DI threshold should be properly corrected. For Cu(I)Cl, it has been found that the subtraction of the 1s + 3d DI emission feature at ∼8980 eV using the pseudo-Voigt fit is not sufficient and an additional intensity ratio correction is required to remove the residual 1s + 3d DI contribution that overlaps the SI emission feature. For Cu(I) model complexes 1–4, the 1s + 3d DI emission appears at a somewhat higher energy (∼8981 or ∼8982 eV), which leads to less overlap with the ∼8976 eV SI feature. Accordingly, reasonable intensity ratio correction factors, depending on the energy of the 1s + 3d DI emission, were obtained (Table 1), and these can be used to obtain Cu(I) Kβ VtC XES data originating solely from the 1s SI state.
The factors contributing to the Kβ VtC XES spectral features for the Cu(I) complexes in the present study are generally in accord with those known from other model studies;11–13 the emission energy reflects the ionization energy of the valence MOs, and the emission intensity is governed by the electric dipole p → s mechanism, which emphasizes the importance of metal p mixing. For Cu(I) complexes, the high-energy Kβ2,5 feature at ∼8976 eV is attributed to the transitions from the MOs having mainly Cu 3d character. It is noteworthy that other 3d transition-metal complexes generally do not show distinct Kβ VtC XES spectral features originating from MOs having mostly metal 3d character, and there are only a few studies that reported a contribution from metal 3d character other than for Cu(I) (e.g., studies of Fe-carbonyl complexes20 and of a Mn model complex activating O223). For most 3d transition-metal complexes (Z < 29), the d band overlaps the ligand valence orbitals and thus is very mixed. For Cu(I), the d10 closed shell leads to an ∼8976 eV d-band feature well separated from the ligand valence orbitals. Also, the 3d10 radial distribution is less contracted, enabling better ligand-mediated d–p mixing for increased intensity.
An analysis of the Kβ XES spectrum of Cu(I)-GOpre supports the three-coordinate Cu(I) site structure (Tyr272/His496/His581, Figure 8) obtained from XAS and DFT.41 The HOMO of Cu(I)-GOpre is found to have mainly Cu character and dominantly contributes to the ∼8976 eV feature (1 in Figures 9C and 10). This HOMO is important since it has a lobe oriented along the open equatorial coordination position and functions as the FMO for O2 activation. We previously investigated the reactivity of the preprocessed Cu(I) active site with O2 through computations,41 finding that an electron is transferred from this Cu orbital into the π* of O2 to form an end-on Cu(II)-(O2•−) species with a triplet ground state (Scheme 2). This originates from two orthogonal MOs, one with an electron having Cu and O2 2p π*σ character and the other with an electron localized on the O2 2p π* orbital oriented perpendicular to the Cu–O2 plane (denoted as π*v in Scheme 2). π*v is a lowest unoccupied orbital that enables H-atom abstraction from a nearby Cys residue. In Figures 9 and 10, Kβ VtC XES of Cu(I)-GOpre shows that the highest-energy spectral feature (1 in Figure 9) originates from the redox-active MO having Cu 3d character. This shows that Kβ XES provides a unique opportunity to probe the Cu(I) FMO for O2 activation that is not accessible with other spectroscopic techniques. In this regard, Kβ XES is expected to give important insight into the reactivity of Cu(I) sites in a range of metalloproteins involved in small-molecule activation.
Scheme 2.

4. EXPERIMENTAL SECTION
4.1. Sample Preparation.
Cu(I)Cl was purchased from Sigma-Aldrich and used without further purification. [Cu(I)-(Me2N-MePY2)] B(C6F5)4 (1, Me2N-MePY2 = N,N-bis(2-((4-dimethylamino)pyridin-2-yl)ethyl)methylamine),57 [Cu(I)-(BA)]SbF6 (2, BA = N-benzyl-6-((bis(pyridin-2-ylmethyl)amino)methyl)pyridin-2-amine),58 [Cu(I)-(LASM)]B(C6F5)4 (3, LASM = 2-(methylthio)-N,N-bis((pyridin-2-yl)methyl)benzenamine),59 and [{Cu(I)-(DMMESE)}2](B(C6F5)4)2 (4, DMMESE = 2-(ethylthio)-N,N-bis((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)ethan-1-amine)60 were prepared as previously described. These solid samples of Cu(I)Cl and complexes 1–4 were stored and prepared in an anaerobic glovebox under a N2 atmosphere. Each solid sample was mixed and finely ground with boron nitride to prevent self-absorption, and this homogeneous mixture was pressed into a 0.5- or 1-mm-thick Al spacer with 38 μm Kapton tape windows. These prepared samples were kept in liquid N2 (LN2) until measurements were made.
Apo GOpre was expressed and purified as previously described.63 Stock solutions (∼20 mg/mL, 50 mM HEPES + 100 mM NaCl, pH 6.5) were frozen and stored at −80 °C until use. An ∼500 μL aliquot was thawed and buffer exchanged into 50 mM phosphate (pH 6.5) by repeatedly concentrating on a centrifuge filter and reconstituting. The apo GOpre solution was transferred to a screw-top conical vial, chilled in an ice bath, and degassed by gently purging the headspace with N2 for 30 min. The solution was transferred to an anaerobic glovebox under a N2 atmosphere and concentrated to ∼210 μL using a centrifuge filter. The protein concentration was determined to be 0.76 mM by UV–vis spectroscopy (ε = 104 900 M−1 cm−1 at 280 nm).43 A 20 mM solution of [Cu(NCMe)4]PF6 was prepared in degassed acetonitrile, and 12.8 μL (0.8 equiv) was added, followed by 20 wt % sucrose. This Cu(I)-GOpre sample was loaded into 2-mm-thick Delrin plastic sample holders wrapped with 38 μm Kapton tape that formed the sample holder windows, and these were immediately frozen and kept in LN2 until measurements. The amount of Cu(II) in the Cu(I)-GOpre sample was <1% as judged by EPR spectroscopy.
4.2. Kβ X-ray Emission Spectroscopy.
The Kβ XES data were collected at the Stanford Synchrotron Radiation Lightsource (SSRL) on 56-pole, 0.9-T wiggler end-station beamline 6–2 under ring conditions of 3 GeV and ∼500 mA. A LN2-cooled Si(111) double-crystal monochromator was used for selection of the excitation energy. The monochromator energy was calibrated with a Cu foil by assigning the first inflection point of the foil’s absorption spectrum to 8978.9 eV.64 For Cu(I)Cl, the incident X-ray photon energy systematically varied from 8987 to 13 000 eV. The presented Kβ XES data of complexes 1, 2, and 4 and Cu(I)-GOpre were collected with an excitation energy of 10 500 eV, and the complex 3 data were measured with an excitation energy of 9300 eV. We note that several incident X-ray photon energies (e.g., 9300, 9700, and 10 500 eV) were used to collect the Kβ VtC XES data of the solid samples of the Cu(I) model complexes and that the excitation energy variation in this energy range (9300–10 500 eV) showed only a very minor difference in the background shape. A multicrystal analyzer spectrometer utilizing seven spherically bent Si(553) crystals arranged in Rowland geometry was employed to select Cu Kβ emission energies, and a Vortex single-element silicon drift detector was used to measure the X-ray emission. Elastic scattering peaks were obtained through the Cu Kβ emission energy range covering both the Kβ main and Kβ VtC regions. These elastic scattering peaks were used for calibrations of the emission energy and intensity. The overall experimental resolution was estimated to be ∼1.5 eV based on the elastic scattering peak measurements. The samples were maintained at a constant temperature of ∼10 K during data collection using a liquid He cryostat. A variable-number Al filter module in the incident X-ray beam path was used when necessary to prevent detector saturation and photodamage. The emitted beam path was enclosed by a He-filled bag to minimize the signal attenuation. Multiple sample spots were used for data collection to minimize photodamage. The Kβ main (8875–8930 eV) and Kβ VtC (8925 to ∼9000 eV) regions were separately measured, and these two regions were merged using a 5 eV overlap (8925–8930 eV). The overall merged Kβ XES spectra were normalized by setting the area under the Kβ main region (8880–8925 eV) to 1000. The data were processed with the Igor Pro program.65
For Cu(I)Cl and complexes 1–4, photodamage was examined by monitoring changes in the spectral shape of three consecutive scans collected at one sample spot. For Cu(I)-GOpre, the comparison of the consecutive scans at one sample spot is difficult due to the significantly low signal-to-noise ratio; thus, the first scan-averaged spectrum is compared with the second scan-averaged spectrum (using many sample spots) to evaluate photodamage. Within the three-scan data acquisition time, for the Kβ main region, photodamage was not observed for any of the Cu(I) complexes. For the Kβ VtC region, while Cu(I)Cl did not show any sign of photodamage, there was a slight decrease in the feature b intensity (at ∼8973 or ∼8974 eV) from photodamage in complexes 1–4. Thus, for Cu(I)Cl and complexes 1–4, only the first scans were used for the final average to minimize the effect of photodamage. Cu(I)-GOpre also showed photodamage that results in an increase in the feature b intensity, as shown in Figure S14.
The Kβ VtC regions were fit using TwinPeaks,66 an in-house-written peak-fitting package for the deconvolution of the spectral contributions to emission spectra. TwinPeaks is written in MATLAB. The package performs nonlinear least-squares fitting with either the trust region reflective67,68 or Levenberg–Marquardt69,70 algorithm. The background contributions to the spectra are fit and subtracted using user-defined spline or polynomial-type functions. Intensities contributing to Kβ VtC peaks are simulated with a pseudo-Voigt profile composed from a linear combination of Gaussian and Lorentzian curves. Initial fits were performed using the trust region reflective method with bounds given to restrict the range of each parameter (peak center, peak width, and peak intensity). The output of trust region reflective fits was then used as the starting point for further fitting performed with the Levenberg–Marquardt algorithm to obtain the final optimized result.
4.3. Computational Methods.
All DFT calculations in this study were performed with the ORCA program, version 3.0.3.71 For Cu(I)Cl, the Kβ VtC XES spectra were calculated with varying cluster sizes of the crystal structure55 (from one Td [Cu(I)Cl4]3− cluster up to a large cluster having 113 atoms) and the different functionals (BP86,72,73 B3LYP,74,75 TPSS,76 and TPSSh77), as described in section 2.2. For complexes 1–4, geometry optimizations were performed from the crystallographic data,61 and these optimized structures were used for the calculations of the Kβ VtC XES spectra (with the BP86 functional for both calculations). Counterions for complexes 1–4 were not included in these calculations. For Cu(I)-GOpre, the Kβ VtC XES spectra were calculated with the various optimized structures shown in ref 41 and the different functionals (BP86, B3LYP, TPSS, and TPSSh), as described in section 2.4. All of the calculations were performed with the CP(PPP) basis set78 on Cu (with a special integration accuracy of 7) and the def2-TZVP basis set79,80 on other atoms. The conductor-like screening model (COSMO)81 with an infinite dielectric constant was utilized for Cu(I)Cl and complexes 1–4, while that with a dielectric constant of 4 was used for Cu(I)-GOpre to account for the protein environment. For complexes 1–4, an infinite dielectric constant was used to compensate for the charge on the complexes because the counterions were not included in the calculations. Note that the COSMO and the chosen dielectric constant hardly affect the calculation of the Kβ VtC XES spectra of complexes 1–4 (Figure S17). The grid4 integration level and tight convergence criteria were used. For close comparison with the experimental data, the DFT/BP86 calculated spectra have been shifted by 230.5, 230, and 230.4 eV for Cu(I)Cl, complexes 1–4, and Cu(I)-GOpre, respectively, and the calculated transitions have been broadened by a Gaussian function with a 2.5 eV line width using the orca_mapspc program (part of the ORCA package). Molecular structures were visualized using the Mercury program.82,83 MO compositions via Mulliken population analysis were calculated with the QMForge program,84 and MOs were visualized with the Lumo program.85
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the National Institutes of Health (DK031450 to E.I.S.; GM28962 to K.D.K.; and GM27659 to D.M.D.) and also by funds provided by the University of Rhode Island. H.L. was supported by an Abbott Laboratories Stanford Graduate Fellowship. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under contract no. DE-AC02–76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by the National Institutes of Health, National Institute of General Medical Sciences (P41GM103393). We thank Uwe Bergmann for helpful discussions on DI emission features.
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c02495.
Additional experimental and calculated Kβ VtC XES data; fits to the Kβ VtC XES spectra; detailed description of the correction method to remove the DI contribution in the Kβ VtC XES spectrum of Cu(I)Cl; crystal structure of Cu(I)Cl; structural parameters for the DFT-optimized and crystal structures of complexes 1–4; details of X-ray crystallography for [Cu(I)-(BA-OMe)] SbF6; detailed description for obtaining the intensity ratio correction factors; complete sets of contour plots of MOs associated with intense transitions in complexes 2–4; description of the data processing of the Kβ VtC XES spectrum of Cu(I)-GOpre; estimation of photodamage in the Cu(I)-GOpre data; and Cartesian coordinates for the optimized structures of complexes 1–4 and truncated Cu(I)-GOpre (PDF)
Accession Codes
CCDC 2008920 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.inorgchem.0c02495
Contributor Information
Hyeongtaek Lim, Department of Chemistry, Stanford University, Stanford, California 94305, United States.
Michael L. Baker, Department of Chemistry, Stanford University, Stanford, California 94305, United States.
Ryan E. Cowley, Department of Chemistry, Stanford University, Stanford, California 94305, United States.
Sunghee Kim, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Mayukh Bhadra, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Maxime A. Siegler, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Thomas Kroll, Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Stanford University, Menlo Park, California 94025, United States.
Dimosthenis Sokaras, Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Stanford University, Menlo Park, California 94025, United States.
Tsu-Chien Weng, Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Stanford University, Menlo Park, California 94025, United States.
Dalia R. Biswas, Department of Chemistry and Biochemistry, Montana State University, Bozeman, Montana 59717, United States
David M. Dooley, Department of Chemistry and Biochemistry, Montana State University, Bozeman, Montana 59717, United States; University of Rhode Island, Kingston, Rhode Island 02881, United States
Kenneth D. Karlin, Department of Chemistry, Johns Hopkins University, Baltimore, Maryland 21218, United States.
Britt Hedman, Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Stanford University, Menlo Park, California 94025, United States.
Keith O. Hodgson, Department of Chemistry and Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Stanford University, Stanford, California 94305, United States
Edward I. Solomon, Department of Chemistry and Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, Stanford University, Stanford, California 94305, United States.
REFERENCES
- (1).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Quist DA; Diaz DE; Liu JJ; Karlin KD Activation of dioxygen by copper metalloproteins and insights from model complexes. JBIC, J. Biol. Inorg. Chem. 2017, 22, 253–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Kau L-S; Spira-Solomon DJ; Penner-Hahn JE; Hodgson KO; Solomon EI X-ray Absorption Edge Determination of the Oxidation State and Coordination Number of Copper. Application to the Type 3 Site in Rhus vernicifera Laccase and Its Reaction with Oxygen. J. Am. Chem. Soc. 1987, 109, 6433–6442. [Google Scholar]
- (4).Penner-Hahn JE Characterization of “spectroscopically quiet” metals in biology. Coord. Chem. Rev. 2005, 249, 161–177. [Google Scholar]
- (5).Sokaras D; Weng T-C; Nordlund D; Alonso-Mori R; Velikov P; Wenger D; Garachtchenko A; George M; Borzenets V; Johnson B; Rabedeau T; Bergmann U A seven-crystal Johann-type hard x-ray spectrometer at the Stanford Synchrotron Radiation Lightsource. Rev. Sci. Instrum. 2013, 84, 053102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).de Groot F High-Resolution X-ray Emission and X-ray Absorption Spectroscopy. Chem. Rev. 2001, 101, 1779–1808. [DOI] [PubMed] [Google Scholar]
- (7).Glatzel P; Bergmann U High resolution 1s core hole X-ray spectroscopy in 3d transition metal complexes—electronic and structural information. Coord. Chem. Rev. 2005, 249, 65–95. [Google Scholar]
- (8).Bergmann U; Glatzel P X-ray emission spectroscopy. Photosynth. Res. 2009, 102, 255–266. [DOI] [PubMed] [Google Scholar]
- (9).In the present study, the Siegbahn nomenclature has been used for Kβ XES. Between the Siegbahn and the IUPAC nomenclatures, Kβ1 corresponds to K-M3; Kβ3, to K-M2; and valence-to-core (VtC), to K-V. Note that, while Kβ2 and Kβ5 technically correspond to K-N2,3 (4p → 1s) and K-M4,5 (3d → 1s), respectively, Kβ2,5 in the present study is used for VtC transitions (as conventionally used for Kβ XES of 3d transition-metal complexes) and thus corresponds to K-V. See ref 10 for a discussion of nomenclature for X-ray spectroscopy.
- (10).Jenkins R; Manne R; Robin R; Senemaud C Nomenclature, symbols, units and their usage in spectrochemical analysis – VIII. Nomenclature system for X-ray spectroscopy (Recommendations 1991). Pure Appl. Chem. 1991, 63, 735–746. [Google Scholar]
- (11).Bauer M HERFD-XAS and valence-to-core-XES: new tools to push the limits in research with hard X-rays? Phys. Chem. Chem. Phys. 2014, 16, 13827–13837. [DOI] [PubMed] [Google Scholar]
- (12).Gallo E; Glatzel P Valence to Core X-ray Emission Spectroscopy. Adv. Mater. 2014, 26, 7730–7746. [DOI] [PubMed] [Google Scholar]
- (13).Pollock CJ; DeBeer S Insights into the Geometric and Electronic Structure of Transition Metal Centers from Valence-to-Core X-ray Emission Spectroscopy. Acc. Chem. Res. 2015, 48, 2967–2975. [DOI] [PubMed] [Google Scholar]
- (14).Bergmann U; Horne CR; Collins TJ; Workman JM; Cramer SP Chemical dependence of interatomic X-ray transition energies and intensities – a study of Mn Kβ″ and Kβ2,5 spectra. Chem. Phys. Lett. 1999, 302, 119–124. [Google Scholar]
- (15).Lee N; Petrenko T; Bergmann U; Neese F; DeBeer S Probing Valence Orbital Composition with Iron Kβ X-ray Emission Spectroscopy. J. Am. Chem. Soc. 2010, 132, 9715–9727. [DOI] [PubMed] [Google Scholar]
- (16).Beckwith MA; Roemelt M; Collomb M-N; DuBoc C; Weng T-C; Bergmann U; Glatzel P; Neese F; DeBeer S Manganese Kβ X-ray Emission Spectroscopy As a Probe of Metal–Ligand Interactions. Inorg. Chem. 2011, 50, 8397–8409. [DOI] [PubMed] [Google Scholar]
- (17).Pollock CJ; DeBeer S Valence-to-Core X-ray Emission Spectroscopy: A Sensitive Probe of the Nature of a Bound Ligand. J. Am. Chem. Soc. 2011, 133, 5594–5601. [DOI] [PubMed] [Google Scholar]
- (18).Pollock CJ; Grubel K; Holland PL; DeBeer S Experimentally Quantifying Small-Molecule Bond Activation Using Valence-to-Core X-ray Emission Spectroscopy. J. Am. Chem. Soc. 2013, 135, 11803–11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Lassalle-Kaiser B; Boron TT III; Krewald V; Kern J; Beckwith MA; Delgado-Jaime MU; Schroeder H; Alonso-Mori R; Nordlund D; Weng T-C; Sokaras D; Neese F; Bergmann U; Yachandra VK; DeBeer S; Pecoraro VL; Yano J Experimental and Computational X-ray Emission Spectroscopy as a Direct Probe of Protonation States in Oxo-Bridged MnIV Dimers Relevant to Redox-Active Metalloproteins. Inorg. Chem. 2013, 52, 12915–12922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Delgado-Jaime MU; DeBeer S; Bauer M Valence-to-Core X-Ray Emission Spectroscopy of Iron–Carbonyl Complexes: Implications for the Examination of Catalytic Intermediates. Chem. - Eur. J. 2013, 19, 15888–15897. [DOI] [PubMed] [Google Scholar]
- (21).Pollock CJ; Delgado-Jaime MU; Atanasov M; Neese F; DeBeer S Kβ Mainline X-ray Emission Spectroscopy as an Experimental Probe of Metal–Ligand Covalency. J. Am. Chem. Soc. 2014, 136, 9453–9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).MacMillan SN; Walroth RC; Perry DM; Morsing TJ; Lancaster KM Ligand-Sensitive But Not Ligand-Diagnostic: Evaluating Cr Valence-to-Core X-ray Emission Spectroscopy as a Probe of Inner-Sphere Coordination. Inorg. Chem. 2015, 54, 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rees JA; Martin-Diaconescu V; Kovacs JA; DeBeer S X-ray Absorption and Emission Study of Dioxygen Activation by a Small-Molecule Manganese Complex. Inorg. Chem. 2015, 54, 6410–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Burkhardt L; Holzwarth M; Plietker B; Bauer M Detection and Characterization of Hydride Ligands in Iron Complexes by High-Resolution Hard X-ray Spectroscopy and Implications for Catalytic Processes. Inorg. Chem. 2017, 56, 13300–13310. [DOI] [PubMed] [Google Scholar]
- (25).Burkhardt L; Mueller C; Groß OA; Sun Y; Sitzmann H; Bauer M The Bonding Situation in the Dinuclear Tetra-Hydrido Complex [{5CpFe}2(μ-H)4] Revisited by Hard X-Ray Spectroscopy. Inorg. Chem. 2019, 58, 6609–6618. [DOI] [PubMed] [Google Scholar]
- (26).Pushkar Y; Long X; Glatzel P; Brudvig GW; Dismukes GC; Collins TJ; Yachandra VK; Yano J; Bergmann U Direct Detection of Oxygen Ligation to the Mn4Ca Cluster of Photosystem II by X-ray Emission Spectroscopy. Angew. Chem., Int. Ed. 2010, 49, 800–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Lancaster KM; Roemelt M; Ettenhuber P; Hu Y; Ribbe MW; Neese F; Bergmann U; DeBeer S X-ray Emission Spectroscopy Evidences a Central Carbon in the Nitrogenase Iron-Molybdenum Cofactor. Science 2011, 334, 974–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rees JA; Bjornsson R; Schlesier J; Sippel D; Einsle O; DeBeer S The Fe–V Cofactor of Vanadium Nitrogenase Contains an Interstitial Carbon Atom. Angew. Chem., Int. Ed. 2015, 54, 13249–13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Mijovilovich A; Hamman S; Thomas F; de Groot FMF; Weckhuysen BM Protonation of the oxygen axial ligand in galactose oxidase model compounds as seen with high resolution X-ray emission experiments and FEFF simulations. Phys. Chem. Chem. Phys. 2011, 13, 5600–5604. [DOI] [PubMed] [Google Scholar]
- (30).Vegelius JR; Kvashnina KO; Klintenberg M; Soroka IL; Butorin SM Cu Kβ2,5 X-ray emission spectroscopy as a tool for characterization of monovalent copper compounds. J. Anal. At. Spectrom. 2012, 27, 1882–1888. [Google Scholar]
- (31).Kumar P; Nagarajan R; Sarangi R Quantitative X-ray absorption and emission spectroscopies: electronic structure elucidation of Cu2S and CuS. J. Mater. Chem. C 2013, 1, 2448–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Müller P; Neuba A; Flörke U; Henkel G; Kühne TD; Bauer M Experimental and Theoretical High Energy Resolution Hard X-ray Absorption and Emission Spectroscopy on Biomimetic Cu2S2 Complexes. J. Phys. Chem. A 2019, 123, 3575–3581. [DOI] [PubMed] [Google Scholar]
- (33).Cutsail GE III; Gagnon NL; Spaeth AD; Tolman WB; DeBeer S Valence-to-Core X-ray Emission Spectroscopy as a Probe of O–O Bond Activation in Cu2O2 Complexes. Angew. Chem., Int. Ed. 2019, 58, 9114–9119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Giordanino F; Borfecchia E; Lomachenko KA; Lazzarini A; Agostini G; Gallo E; Soldatov AV; Beato P; Bordiga S; Lamberti C Interaction of NH3 with Cu-SSZ-13 Catalyst: A Complementary FTIR, XANES, and XES Study. J. Phys. Chem. Lett. 2014, 5, 1552–1559. [DOI] [PubMed] [Google Scholar]
- (35).Borfecchia E; Lomachenko KA; Giordanino F; Falsig H; Beato P; Soldatov AV; Bordiga S; Lamberti C Revisiting the nature of Cu sites in the activated Cu-SSZ-13 catalyst for SCR reaction. Chem. Sci. 2015, 6, 548–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Günter T; Carvalho HWP; Doronkin DE; Sheppard T; Glatzel P; Atkins AJ; Rudolph J; Jacob CR; Casapu M; Grunwaldt J-D Structural snapshots of the SCR reaction mechanism on Cu-SSZ-13. Chem. Commun. 2015, 51, 9227–9230. [DOI] [PubMed] [Google Scholar]
- (37).Lomachenko KA; Borfecchia E; Negri C; Berlier G; Lamberti C; Beato P; Falsig H; Bordiga S The Cu-CHA deNOx Catalyst in Action: Temperature-Dependent NH3-Assisted Selective Catalytic Reduction Monitored by Operando XAS and XES. J. Am. Chem. Soc. 2016, 138, 12025–12028. [DOI] [PubMed] [Google Scholar]
- (38).Zhang R; Li H; McEwen J-S Chemical Sensitivity of Valence-to-Core X-ray Emission Spectroscopy Due to the Ligand and the Oxidation State: A Computational Study on Cu-SSZ-13 with Multiple H2O and NH3 Adsorption. J. Phys. Chem. C 2017, 121, 25759–25767. [Google Scholar]
- (39).Martin-Diaconescu V; Chacón KN; Delgado-Jaime MU; Sokaras D; Weng T-C; DeBeer S; Blackburn NJ Kβ Valence to Core X-ray Emission Studies of Cu(I) Binding Proteins with Mixed Methionine – Histidine Coordination. Relevance to the Reactivity of the M- and H-sites of Peptidylglycine Monooxygenase. Inorg. Chem. 2016, 55, 3431–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Glatzel P; Bergmann U; de Groot FMF; Cramer SP Multiple excitations in the K fluorescence emission of Mn, Fe and Ni compounds. AIP Conf. Proc. 2003, 652, 250–255. [Google Scholar]
- (41).Cowley RE; Cirera J; Qayyum MF; Rokhsana D; Hedman B; Hodgson KO; Dooley DM; Solomon EI Structure of the Reduced Copper Active Site in Preprocessed Galactose Oxidase: Ligand Tuning for One-Electron O2 Activation in Cofactor Biogenesis. J. Am. Chem. Soc. 2016, 138, 13219–13229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Avigad G; Amaral D; Asensio C; Horecker BL The d-Galactose Oxidase of Polyporus circinatus. J. Biol. Chem. 1962, 237, 2736–2743. [PubMed] [Google Scholar]
- (43).Kosman DJ; Ettinger MJ; Weiner RE; Massaro EJ The Molecular Properties of the Copper Enzyme Galactose Oxidase. Arch. Biochem. Biophys. 1974, 165, 456–467. [DOI] [PubMed] [Google Scholar]
- (44).Whittaker JW Free Radical Catalysis by Galactose Oxidase. Chem. Rev. 2003, 103, 2347–2364. [DOI] [PubMed] [Google Scholar]
- (45).Kelleher FM; Bhavanandan VP Re-examination of the Products of the Action of Galactose Oxidase. Evidence for the Conversion of Raffinose to 6″-Carboxyraffinose. J. Biol. Chem. 1986, 261, 11045–11048. [PubMed] [Google Scholar]
- (46).Matsumura S; Kuroda A; Higaki N; Hiruta Y; Yoshikawa S Formation of Uronic Acid by Galactose Oxidase. Chem. Lett. 1988, 17, 1747–1750. [Google Scholar]
- (47).Deutsch M; Hölzer G; Härtwig J; Wolf J; Fritsch M; Förster E Kα and Kβ x-ray emission spectra of copper. Phys. Rev. A: At., Mol., Opt. Phys. 1995, 51, 283–296. [DOI] [PubMed] [Google Scholar]
- (48).Pham TL; Nguyen TV; Lowe JA; Grant IP; Chantler CT Characterization of the copper Kβ x-ray emission profile: an ab initio multi-configuration Dirac–Hartree–Fock approach with Bayesian constraints. J. Phys. B: At., Mol. Opt. Phys. 2016, 49, 035601. [Google Scholar]
- (49).Briand JP; Chevallier P; Tavernier M; Rozet JP Observation of K Hypersatellites and KL Satellites in the X-Ray Spectrum of Doubly K-Ionized Gallium. Phys. Rev. Lett. 1971, 27, 777–779. [Google Scholar]
- (50).Briand JP; Touati A; Frilley M; Chevallier P; Johnson A; Rozet JP; Tavernier M; Shafroth S; Krause MO The structure of Kα hypersatellite spectra of Cu, Ni and Fe as a test of intermediate coupling. J. Phys. B: At. Mol. Phys. 1976, 9, 1055–1064. [Google Scholar]
- (51).Williams GP Electron Binding Energies In X-ray Data Booklet, 3rd ed.; Thompson AC, Ed.; Lawrence Berkeley National Laboratory: Berkeley, CA, 2009; pp 1–1–1–7. [Google Scholar]
- (52).Deutsch M; Förster E; Hölzer G; Härtwig J; Hämäläinen K; Kao CC; Huotari S; Diamant R X-Ray Spectrometry of Copper: New Results on an Old Subject. J. Res. Natl. Inst. Stand. Technol. 2004, 109, 75–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Enkisch H; Sternemann C; Paulus M; Volmer M; Schülke W 3d spectator hole satellites of the Cu Kβ1,3 and Kβ2,5 emission spectrum. Phys. Rev. A: At., Mol., Opt. Phys. 2004, 70, 022508. [Google Scholar]
- (54).Didziulis SV; Cohen SL; Butcher KD; Solomon EI Variable Photon Energy Photoelectron Spectroscopic Studies of Covalent Bonding in 3d10 Transition-Metal Compounds. Inorg. Chem. 1988, 27, 2238–2250. [Google Scholar]
- (55).Wyckoff RWG; Posnjak E The Crystal Structures of the Cuprous Halides. J. Am. Chem. Soc. 1922, 44, 30–36. [Google Scholar]
- (56).DeBeer George S; Brant P; Solomon EI Metal and Ligand K-Edge XAS of Organotitanium Complexes: Metal 4p and 3d Contributions to Pre-edge Intensity and Their Contributions to Bonding. J. Am. Chem. Soc. 2005, 127, 667–674. [DOI] [PubMed] [Google Scholar]
- (57).Zhang CX; Liang H-C; Kim E.-i.; Shearer J; Helton ME; Kim E; Kaderli S; Incarvito CD; Zuberbühler AD; Rheingold AL; Karlin KD Tuning Copper–Dioxygen Reactivity and Exogenous Substrate Oxidations via Alterations in Ligand Electronics. J. Am. Chem. Soc. 2003, 125, 634–635. [DOI] [PubMed] [Google Scholar]
- (58).Kim S; Saracini C; Siegler MA; Drichko N; Karlin KD Coordination Chemistry and Reactivity of a Cupric Hydroperoxide Species Featuring a Proximal H-Bonding Substituent. Inorg. Chem. 2012, 51, 12603–12605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Lee Y; Lee D-H; Park GY; Lucas HR; Narducci Sarjeant AA; Kieber-Emmons MT; Vance MA; Milligan AE; Solomon EI; Karlin KD Sulfur Donor Atom Effects on Copper(I)/O2 Chemistry with Thioanisole Containing Tetradentate N3S Ligand Leading to μ−1,2-Peroxo-Dicopper(II) Species. Inorg. Chem. 2010, 49, 8873–8885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Kim S; Ginsbach JW; Billah AI; Siegler MA; Moore CD; Solomon EI; Karlin KD Tuning of the Copper–Thioether Bond in Tetradentate N3S(thioether) Ligands; O–O Bond Reductive Cleavage via a [CuII2(μ−1,2-peroxo)]2+/[CuIII2(μ-oxo)2]2+ Equilibrium. J. Am. Chem. Soc. 2014, 136, 8063–8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).While the crystal structures of complexes 1, 3, and 4 have been published in refs 57, 59, and 60, respectively, the crystal structure of complex 2 is presented here (Supporting Information). For complex 2, the crystal structure has been obtained with the −OMe group at the para position of the benzyl ring, but this −OMe group is replaced by −H (using the program Avogadro version 1.2.0 (http://avogadro.cc)) for the DFT calculations in the present study.
- (62).Mukoyama T; Taniguchi K Atomic excitation as the result of inner-shell vacancy production. Phys. Rev. A: At., Mol., Opt. Phys. 1987, 36, 693–698. [DOI] [PubMed] [Google Scholar]
- (63).Rogers MS; Hurtado-Guerrero R; Firbank SJ; Halcrow MA; Dooley DM; Phillips SEV; Knowles PF; McPherson MJ Cross-Link Formation of the Cysteine 228–Tyrosine 272 Catalytic Cofactor of Galactose Oxidase Does Not Require Dioxygen. Biochemistry 2008, 47, 10428–10439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Bearden JA; Burr AF Reevaluation of X-Ray Atomic Energy Levels. Rev. Mod. Phys. 1967, 39, 125–142. [Google Scholar]
- (65).Igor Pro, version 8.04; WaveMetrics, Inc.: Lake Oswego, OR, 2019. [Google Scholar]
- (66).Baker ML TwinPeaks; Stanford University: Stanford, CA, 2017. [Google Scholar]
- (67).Coleman TF; Li Y An Interior Trust Region Approach for Nonlinear Minimization Subject to Bounds. SIAM J. Optimiz. 1996, 6, 418–445. [Google Scholar]
- (68).Coleman TF; Li Y On the convergence of interior-reflective Newton methods for nonlinear minimization subject to bounds. Math. Program. 1994, 67, 189–224. [Google Scholar]
- (69).Levenberg K A Method for the Solution of Certain Non-Linear Problems in Least Squares. Q. Appl. Math. 1944, 2, 164–168. [Google Scholar]
- (70).Marquardt DW An Algorithm for Least-Squares Estimation of Nonlinear Parameters. J. Soc. Ind. Appl. Math. 1963, 11, 431–441. [Google Scholar]
- (71).Neese F The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar]
- (72).Becke AD Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- (73).Perdew JP Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B: Condens. Matter Mater. Phys. 1986, 33, 8822–8824. [DOI] [PubMed] [Google Scholar]
- (74).Becke AD Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar]
- (75).Lee C; Yang W; Parr RG Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- (76).Tao J; Perdew JP; Staroverov VN; Scuseria GE Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [DOI] [PubMed] [Google Scholar]
- (77).Staroverov VN; Scuseria GE; Tao J; Perdew JP Comparative assessment of a new nonempirical density functional: Molecules and hydrogen-bonded complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar]
- (78).Neese F Prediction and interpretation of the 57Fe isomer shift in Mössbauer spectra by density functional theory. Inorg. Chim. Acta 2002, 337, 181–192. [Google Scholar]
- (79).Schäfer A; Huber; Ahlrichs R Fully optimized contracted Gaussian basis sets of triple zeta valence quality for atoms Li to Kr. J. Chem. Phys. 1994, 100, 5829–5835. [Google Scholar]
- (80).Weigend F; Ahlrichs R Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- (81).Klamt A; Schüürmann G COSMO: A New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and its Gradient. J. Chem. Soc., Perkin Trans. 2 1993, 799–805. [Google Scholar]
- (82).Macrae CF; Sovago I; Cottrell SJ; Galek PTA; McCabe P; Pidcock E; Platings M; Shields GP; Stevens JS; Towler M; Wood PA Mercury 4.0: from visualization to analysis, design and prediction. J. Appl. Crystallogr. 2020, 53, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Groom CR; Bruno IJ; Lightfoot MP; Ward SC The Cambridge Structural Database. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2016, B72, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Tenderholt AL QMForge: A Program to Analyze Quantum Chemistry Calculations, version 2.4; https://qmforge.net.
- (85).Kieber-Emmons MT LUMO, version 1.0.3; http://www.kieber-emmons.com/Lumo.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.