

Abstract
To identify key gene expression pathways altered with infection of the novel coronavirus SARS-CoV-2, we performed the largest comparative genomic and transcriptomic analysis to date. We compared the novel pandemic coronavirus SARS-CoV-2 with SARS-CoV and MERS-CoV, as well as influenza A strains H1N1, H3N2 and H5N1. Phylogenetic analysis confirms that SARS-CoV-2 is closely related to SARS-CoV at the level of the viral genome. RNAseq analyses demonstrate that human lung epithelial cell responses to SARS-CoV-2 infection are distinct. Extensive Gene Expression Omnibus literature screening and drug predictive analyses show that SARS-CoV-2 infection response pathways are closely related to those of SARS-CoV and respiratory syncytial virus infections. We validated SARS-CoV-2 infection response genes as disease-associated using Kaplan–Meier survival estimates in lung disease patient data. We also analysed COVID-19 patient peripheral blood samples, which identified signalling pathway concordance between the primary lung cell and blood cell infection responses.
Keywords: SARS-CoV-2, SARS-CoV, MERS-CoV, coronaviruses, transcriptomics, RNAseq, COVID-19
Graphical Abstract
Graphical Abstract.
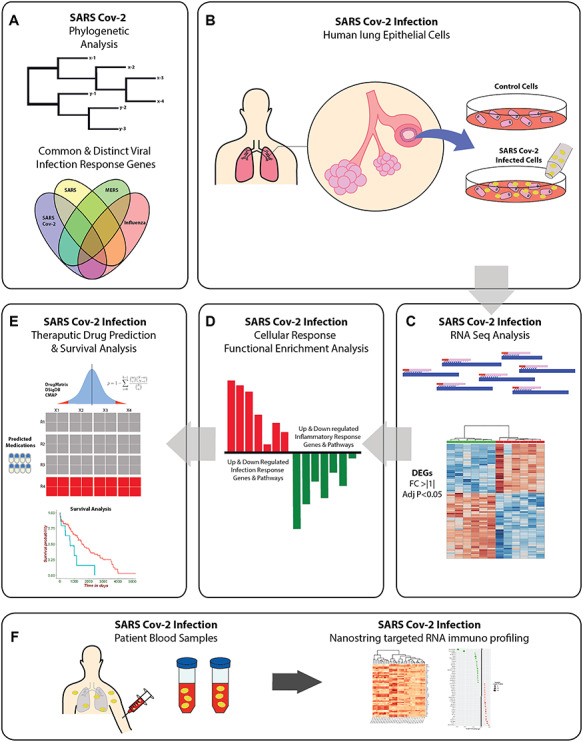
GRAPHIC 1: SARS-CoV-2 graphical abstract depicting the study workflow and analysis pipeline. (A) We first conducted phylogenetic and RNAseq analyses comparing SARS-CoV-2, SARS, MERS, and influenza strains, and the overlapping gene expression effects of infection with these viruses. (B–C) Datasets from a study of transcriptional effects of SARS-CoV-2 infection in human lung epithelial cells (profiled using RNAseq) was analysed to determine differential gene expression. (D) Gene ontology and curated cell signalling pathway databases were used to screen the differentially expressed gene list for functional enrichment. (E) SARS-CoV-2 genes were used for drug prediction and survival analyses in disease datasets, results suggesting pathology associated genes and pathways for subsequent functional analysis. (F) A targeted immune profile in SARS-CoV-2 infected patient blood samples was analysed to assess system wide immune effects of infection.
Introduction
The current global pandemic of the novel coronavirus SARS-CoV-2, causing a severe respiratory disease designated as COVID-19, has as of mid-August 2020 infected >21 million people and caused >750,000 deaths globally [1]. The highly infectious and widespread nature of the disease is attributed in part to its asymptomatic spread, in contrast to earlier pandemic coronaviruses SARS-CoV and MERS-CoV, which were more easily contained as symptom onset co-occurred with infectivity [2].
The development and spread of the novel coronavirus causing COVID-19 has vastly outpaced the rate of vaccine and therapeutic development. Nevertheless, the global research community has rallied; within weeks of the first observations of COVID-19, the virus was isolated and characterised. Central to effective therapy and vaccine development is an improved understanding of the cellular pathway and transcriptional responses to infection in human cells. These are currently very poorly characterised, both in the early stage of infection, as viral load increases before symptom onset; as well as in the later stage of COVID-19 pneumonia, which is associated with a severe cytokine and inflammatory storm in affected patients. It remains a mystery why only a small proportion become severely ill, while a proportion of infected people harbour asymptomatic infections making them difficult to identify [2].
To address these issues and clarify cell signalling pathways affected by SARS-CoV-2 infection, we applied a suite of computational and bioinformatics approaches using SARS-CoV-2 genomic and transcriptomic data. We started by comparing SARS-CoV-2 with SARS-CoV, MERS-CoV and influenza A strains H1N1, H3N2 and H5N1 viral genomes, and assessed the effect of infection on transcriptional responses in human lung epithelial cells [3–5]. This enabled us to characterise a novel set of SARS-CoV-2 acute response genes. On this set of genes, we performed extensive cell signalling pathway and predictive analyses. These assessed gene ontologies and cell signalling cascades associated with SARS-CoV-2 infection in human cells, which enabled us to identify clinically approved drugs [6] that affect aspects of the early cell response to the viral infection, some of which may be of therapeutic relevance. Having identified the distinct transcriptional responses following SARS-CoV-2 infection, we performed bioinformatics validation studies to determine whether these SARS-CoV-2 response genes and pathways are associated with comorbid lung disease, and share concordant signalling pathway responses with the systemic immune response evident in COVID-19 human patient blood samples.
Results
Phylogenetic and RNAseq gene expression analyses define genomic relationships and infection responses between the novel coronavirus SARS-CoV-2, and SARS-CoV, MERS-CoV, and influenza A strains
We first compared the viral phylogenetic tree of known pandemic coronaviruses (SARS-CoV, MERS-CoV and SARS-CoV-2), as well as pandemic influenza A strains (H1N1, H3N2 and H5N1). We found that SARS-CoV-2 is most closely related to SARS-CoV and MERS-CoV, and as expected, these showed significant divergence from the influenza viruses examined (Figure 1A). RNAseq gene expression profiling of human lung epithelial cells infected with SARS-CoV-2, SARS-CoV, MERS-CoV and influenza strains for 24 h revealed a surprisingly sparse overlapping gene expression signature (Figure 1B–C). This is an observation of key importance; despite their close phylogenetic relationship, the novel coronavirus SARS-CoV-2 induces a very different transcriptional response in the host cells compared with SARS-CoV and MERS-CoV, with majority of differentially expressed genes (DEGs) distinct to the SARS-CoV-2 set (Figure 1B). There was no evident relationship between phylogenetic and gene expression relationships, consistent with the notion that COVID-19 is a quite different disease entity (at least in its initial stages of development) to those resulting from the earlier coronaviruses. A similar conclusion was reached when we compared SARS-CoV-2 and influenza-induced gene expression changes in human lung epithelial cells (Figure 1C). For this reason, we then focussed exclusively on the SARS-CoV-2 transcriptome and pathway data to gain insights into early COVID-19 development.
Figure 1.
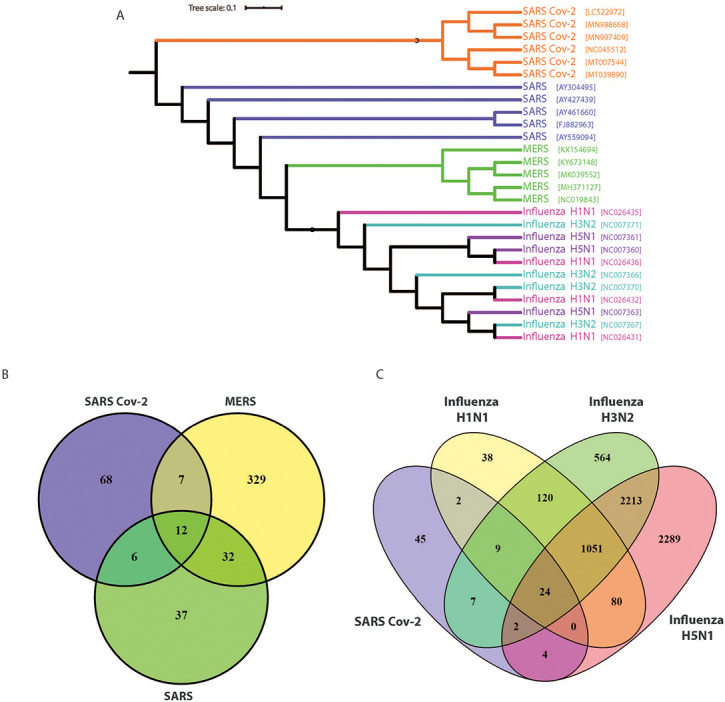
Phylogenetic and RNAseq analyses show the genetic relationship between the novel coronavirus SARS-CoV-2, and SARS, MERS and influenza strains. (A) Phylogenetic viral genome analysis shows SARS-CoV-2 is most closely related to SARS and MERS and is distinct from influenza A strains H1N1, H3N2 and H5N1. (B and C) Comparison of RNAseq analyses of infected primary human lung epithelial cells with these coronaviruses and influenza A strains indicates the common and distinct gene sets with upregulated expression in response to infection.
To understand the magnitude of the unique transcriptional effects of SARS-CoV-2 infection, we stringently assessed DEGs using a cut-off threshold of absolute log2 fold change (LFC) >1, with an adjusted P value <0.05 for initial analyses. Heat map visualisation shows the striking nature of the unique transcriptional signature induced upon SARS-CoV-2 infection (Figure 2A). Of the top 108 SARS-CoV-2 infection response genes, >85% were significantly upregulated compared to MERS-CoV, SARS-CoV and influenza virus infections (Figure 2A). Notably, the top 40 SARS-CoV-2 infection response genes show particularly striking upregulation compared to the other viruses (Figure 2B). Using a more stringent cut-off threshold of absolute log fold change >2, there was evident strong upregulation of a unique suite of inflammatory response genes upon SARS-CoV-2 infection; these included interferon response genes IFI44L, IFI27 and IFI6, interleukins IL6 and Interleukin 8 (IL8), and chemokine and complement gene activation (Figure 2C).
Figure 2.

SARS-CoV-2 infection is associated with upregulation of a unique set of genes not seen with SARS, MERS or influenza A infections in human lung epithelial cells. (A) Heat map depicting the top 108 significant DEGs with infection of the novel coronavirus SARS-CoV-2, compared with top genes related to SARS, MERS and influenza infections. (B) An expanded view of the top 40 genes with increased expression in SARS-CoV-2 infection, indicating a unique expression profile not seen with the other viral infections. (C) Volcano plot highlights the most significant SARS-CoV-2 response genes above a log fold-change of 2 and adjusted P value <0.05. We see that although SARS-CoV-2 is closely related to SARS and MERS by viral phylogeny, the response of cells to infection is significantly different, thus representing another important and novel aspect of this virus.
Collectively, these data further confirm that although the novel coronavirus SARS-CoV-2 genome is closely related to previous pandemic coronaviruses SARS-CoV and MERS-CoV genomes, the transcriptional responses to infection in human lung epithelial cells are markedly different. Similarly, comparing the infection responses of pandemic influenza strains H1N1, H3N2 and H5N1 with SARS-CoV-2 indicates SARS-CoV-2 infection responses in human lung epithelial cells remain unique at the gene expression level.
Gene ontology and cell signalling pathway analysis finds enriched inflammatory and infection responses to SARS-CoV-2 infection
After establishing the unique differential expression profile associated with SARS-CoV-2 infection in human lung epithelial cells, we conducted thorough gene ontology and cell signalling pathway analyses using several curated databases (The Gene Ontology, WikiPathways, BioCarta, Reactome and Panther databases). Assigning significant gene ontologies to SARS-CoV-2 infection found cytokine and interferon cellular responses dominate the infection profile (Figure 3A). Notably, type 1 interferon response and cytokine inflammatory terms were the most enriched, in line with our differential gene expression analysis (Figure 2). Only a small number of viral replication and viral life cycle terms were found significant (Figure 2A and B); this likely reflects the acute nature of the 24 h infection time point used. Cell signalling pathway analyses similarly found striking enrichment of inflammatory interleukin, TNF-alpha and interferon-mediated signalling effects in host cells, distinct from response terms driven by either bacterial or viral infection (Figure 3B).
Figure 3.
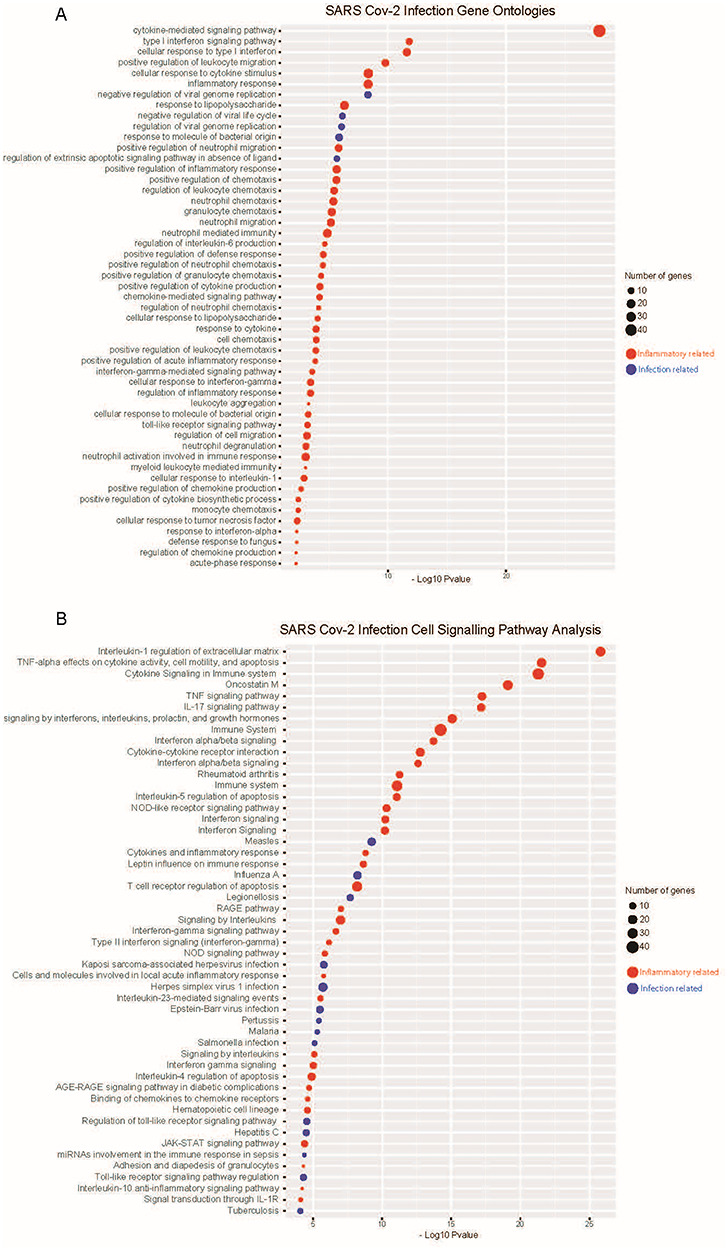
Gene ontology and cell signalling pathway analysis finds enriched inflammatory and infection responses to SARS-CoV-2 infection. (A) Gene ontology analysis finds inflammatory and infection response (bacterial and viral responses pathways combined) significantly enriched with SARS-CoV-2 infection in human lung epithelial cells. (B) Cell signalling pathway analyses similarly find enrichment, predominantly inflammatory related signalling effects seen with SARS-CoV-2 infection. Analyses performed using The Gene Ontology, WikiPathways, BioCarta, Reactome and Panther databases.
Viral infection database screening and drug prediction analyses suggest relevant pathways for subsequent study
To aid in contextualising our findings of a unique SARS-CoV-2 infection gene signature and to provide some external validation, we conducted an extensive Gene Expression Omnibus (GEO) viral literature screen (Figure 4A). Analysing all the publically available viral infection literature in the GEO found infection with the novel coronavirus SARS-CoV-2 matched most closely to studies of SARS-CoV infection, as well as respiration syncytial virus infection between 48 and 72 h (Figure 4A). This is of particular significance, as SARS-CoV, SARS-CoV-2 and respiration syncytial virus all cause significant lower respiratory tract infections in humans.
Figure 4.
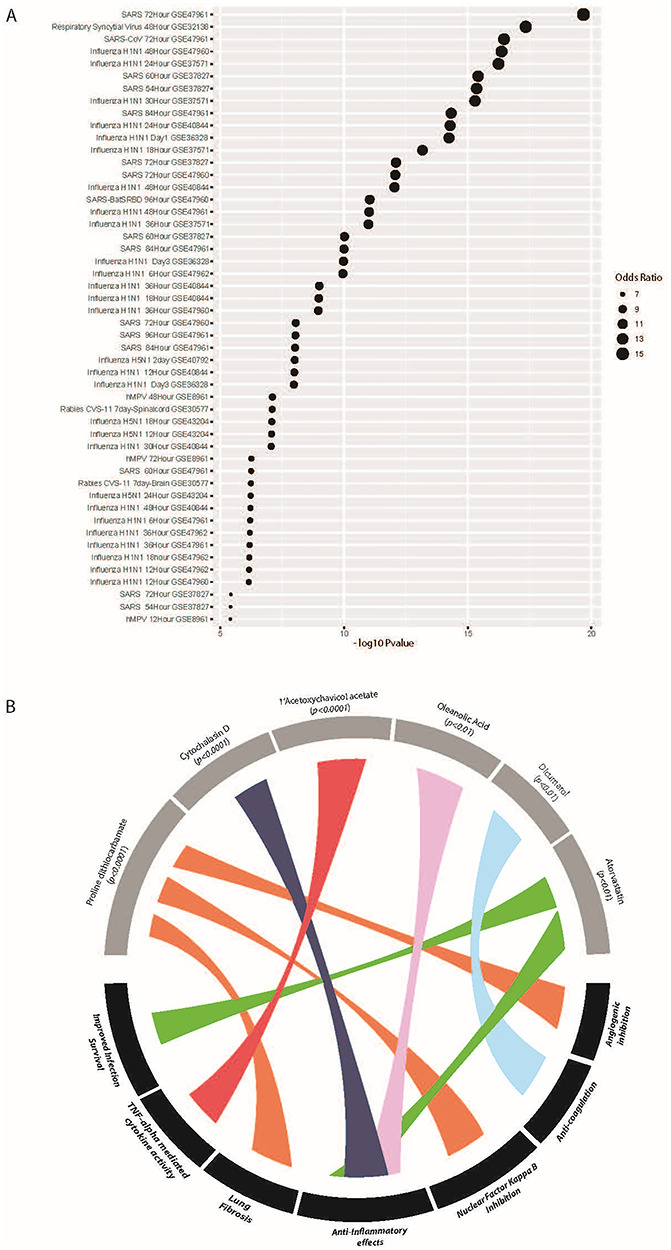
Viral infection literature and drug prediction analysis validates SARS-CoV-2 infection responses and suggests relevant pathways for subsequent study. (A) A validation GEO viral literature screen was conducted of all publically available literature on viral infections. Analysis finds SARS-CoV-2 infection is most closely related to published studies of SARS and respiratory viral infections. (B) Predictive drug screening suggests molecules of interest targeting inflammatory, infection, lung fibrotic and coagulation factors may be of particular relevance to treating early-stage COVID-19 disease.
Next, to understand how the SARS-CoV-2 gene set analysis matched to the actions of previously characterised drugs, we conducted a Drug Signatures Database screen using DSigDB [6]. At an adjusted P value significance cut-off of <0.05, DSigDB screening matched over 600 agents as interacting with SARS-CoV-2 gene signature elements (Supplementary table). While many of these are challenging to interpret or understand in a translational context, they provide insight in terms of broad cell signalling pathways and/or factors that may prove clinically relevant upon further investigation. For example, our analysis predicts several agents with broad anti-inflammatory and TNF-alpha inhibitory effects; including cytochalasin D (P < 0.0001), 1′-acetoxychavicol acetate (P < 0.0001) and statins such as atorvastatin (P < 0.01) (Figure 4B), which are associated with increased survival following infections due to anti-inflammatory effects [7]. Of additional note are the agents proline dithiocarbamate (P < 0.0001), and dicumarol (P < 0.01); inhibitors having anti-angiogenic and anti-coagulation effects [8, 9]. These mechanisms may prove relevant to COVID-19 pneumonia, with reports emerging of significant cardiovascular involvement in COVID-19 [10], as well as asymptomatic cases of acute pulmonary embolism, suggesting significant vascular effects of currently unknown cause [11] (Figure 4B). It can also be noted that our analysis did not predict the broad spectrum anti-inflammatory hydroxychloroquine, or the antiviral remdesivir (agents suggested for the treatment of COVID-19 pneumonia) as significant targets based on this RNAseq analysis of early acute effects of SARS-CoV-2.
Kaplan–Meier survival estimates using lung adenocarcinoma datasets finds a significant relationship between SARS-CoV-2 infection response genes and patient survival
To confirm that there are significant disease associations of the SARS-CoV-2 infection response genes, we performed both univariate and multivariate Cox hazard model analysis and Kaplan–Meier survival estimates of lung cancer (LC) adenocarcinoma patient data (Figure 5 and supplementary table). SARS-CoV-2 infection response genes are able to stratify LC patients, with differential expression of the responding gene associated with significantly reduced survival (Figure 5). Notably, we found that inflammatory and chemokine related genes were particularly associated with poor prognosis in these patients (P < 0.05).
Figure 5.
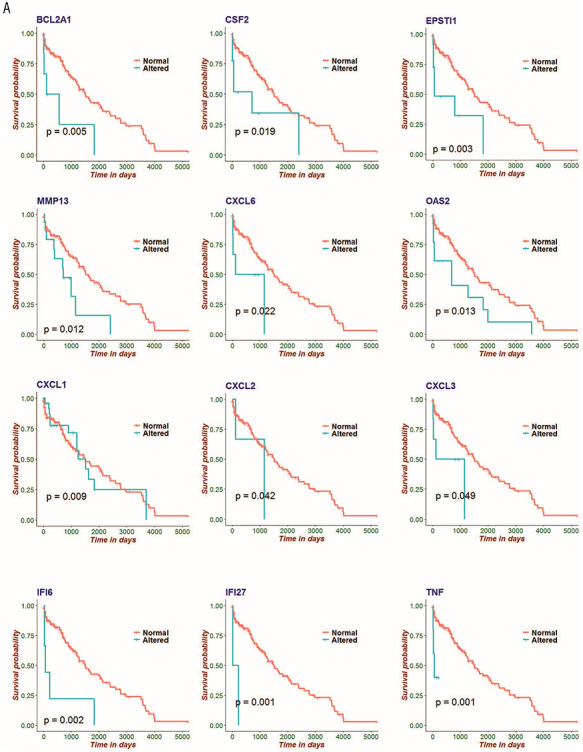
Kaplan–Meier survival estimates using lung adenocarcinoma datasets finds a significant relationship between SARS-CoV-2 infection response genes and patient survival. Kaplan–Meier estimates for the SARS-CoV-2 infection response genes BCL2A1, CSF2, EPSTI1, MMP13, CXCL6, OAS2, CXCL1, CXCL2, CXCL3, IFI6, IFI27 and TNF. These data indicate a significant relationship with increased mortality in LC patients, suggesting these genes as relevant co-morbidity factors in COVID-19 disease.
This analysis provides insight into the acute inflammatory nature of SARS-CoV-2 infection in the lungs and provides data that can be used to examine comorbidity interactions with COVID-19 early-stage disease. This would support the notion of giving particular consideration and critical care to patients with co-morbidities, particularly with lung related diseases such as LC; as survival outcomes in these patients are likely to be significantly impacted by COVID-19 disease.
Targeted immune profiling of SARS-CoV-2 infected patient blood samples reveals systemic immune responses to infection, and perturbed cell signalling pathways similar to that seen in primary lung cell infection
Thus far, we have extensively characterised the acute primary human lung epithelial cell response to SARS-CoV-2 infection. However, in order to understand how this may relate to the system-wide immune effects of SARS-CoV-2 infection, we analysed data from a study of the human immune responses in peripheral blood cells of infected individuals. This study used a targeted immune panel on the NanoString platform (Figure 6). Compared to healthy controls, SARS-CoV-2 infected patient samples show a distinct systemic infection response at the RNA level; forming a discrete group by hierarchical clustering (Figure 6A). A pooled analysis of all patient time-series data found a total of 45 significant DEGs above an absolute log fold-change of 1 (Figure 6B). Notably, compared to our human lung epithelial cell analysis, only three genes (MX1, IRF7, BST2) show concordance between primary lung cell and systemic blood cell responses to SARS-CoV-2 infection in the acute phases (Figure 6C). Nevertheless, cell signalling pathway analyses find the systemic response is characterised by significant perturbation of 140 matched pathways (Supplementary table), of which 32 show concordance with those matched in the primary lung cell infection analysis (Figure 6D).
Figure 6.
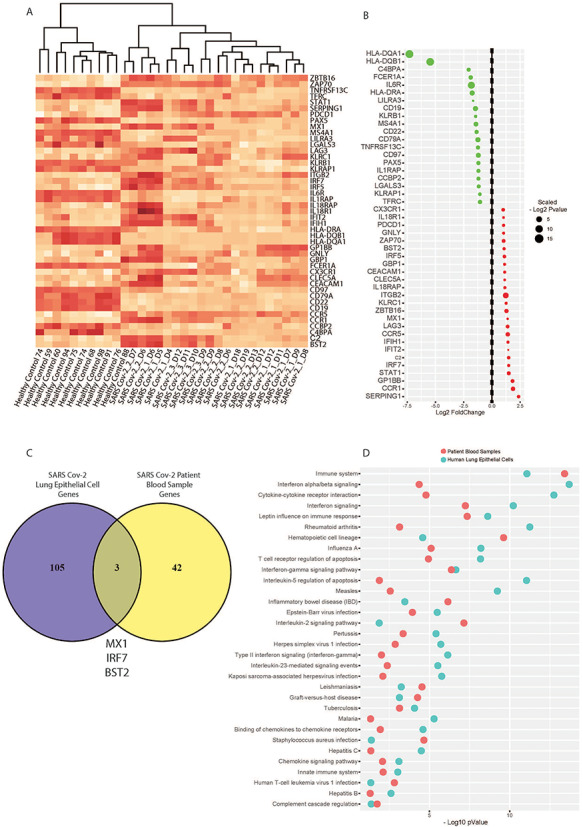
Targeted immune profiling of SARS-CoV-2 infected patient blood samples reveals systemic immune responses to infection, and perturbed cell signalling pathways that overlap with primary lung infection effects. (A) Heat map of targeted immune RNA profile of 32 patients (n = 10 healthy controls, n = 23 SARS-CoV-2 infected patients) using a NanoString-targeted immunology panel shows SARS-CoV-2 infected patients have a distinct systemic infection response. (B) A pooled analysis of time-course data reveals top gene dysregulated in patient blood by SARS-CoV-2 infection. (C) Only three genes are seen to be overlapping between primary lung cell and blood cell infection responses. (D) Nevertheless, we see concordance at the level of perturbed cell signalling pathways related to immune-inflammatory cytokine and interferon responses in the host.
Collectively, these data suggest that while there is a suite of response genes dysregulated in the acute infection phase, the primary infection in lung cells is characterised by a distinct altered gene set compared with systemic immune response genes. Nevertheless, we see common cellular signalling pathways perturbed in both the primary and systemic acute infection responses at the RNA level, and these are dominated by inflammatory-related cytokine and interferon response pathways.
Discussion
In this study, we have performed phylogenetic viral genome analyses and extensive transcriptomic characterisations of the effects of the novel coronavirus SARS-CoV-2, compared with previous human pandemic viruses SARS-CoV, MERS-CoV, and influenza A strains H1N2, H3N2, and H5N1. To our knowledge, this is the largest aggregate comparative genomic and transcriptomic study of the novel coronavirus. We have utilised data from primary human lung epithelial cells, as well as peripheral blood cells from SARS-CoV-2 infected patients to compare the primary acute lung cell responses with changes in the systemic immune response. Most notably, despite their close phylogenetic relationships, SARS-CoV-2 induces a distinct transcriptional response in infected lung cells compared with other pandemic coronaviruses SARS-CoV and MERS-CoV and is also distinct from influenza A strains. We show that inflammatory signalling pathways dominate the acute infection response, and while as expected there was little concordance in the pattern of altered gene expression between infected primary lung cells and circulating COVID-19 patient blood cells, there was concordance seen in a set of perturbed cell signalling pathways. This might be expected if the peripheral blood responses are indirect rather than due to direct blood cell infection, but these observations may prove therapeutically relevant; it suggests that blocking some of these pathways to relieve advanced COVID-19 disease may also reduce some of pathways particularly active in newly infected lung epithelial cells. The pathogenic role of these pathways is not always clear, and while inflammation blockade is desirable for those seriously ill, it may be less so for the early stages of infection. Nevertheless, further analyses using clinical data will be needed to clarify this.
As noted above, the human epithelial lung cell transcriptomic response to SARS-CoV-2 infection after 24 h is surprisingly different to that of other viruses. Our analyses shows that of the top 108 genes differentially expressed upon SARS-CoV-2, MERS-CoV, SARS-CoV and influenza A viral infections, there is little or no concordance in the responses of infected human lung epithelial cells. A notable example is the gene SPRR2E (Small proline-rich protein 2E), which encodes small proteins that form a plasma membrane-associated barrier against extracellular and environmental factors [12, 13]. SPRR2E is upregulated >3-fold upon SARS-CoV-2 infection as shown by our analysis, yet is actually downregulated with SARS-CoV and MERS-CoV infection, and does not significantly change upon influenza infection at all (Figure 2). SPRR2E expression has been associated with pulmonary fibrosis [14, 15]; a clinical presentation significant in COVID-19 pneumonia [16] and is thus a potentially relevant factor important for further investigation. Additional acute inflammatory responses are also noteworthy. IL8 expression is upregulated >2-fold upon SARS-CoV-2 infection in lung cells; however, it is not changed at all with SARS-CoV and the influenza strain infections. IL8 is particularly significant, as it is a chemokine involved in acute inflammatory neutrophil infiltrations and tissue damage responses [17]. Understanding mechanisms involved in the distinct SARS-CoV-2 acute response will further our abilities to develop targeted therapeutics for COVID-19 disease. Indeed, others have also used related transcriptomic study approaches to identified lung epithelium-derived factors in COVID-19 but have focussed on (and confirmed) the interaction with other systems such as blood coagulation, a prominent feature of this disease [18].
Beyond the level of expression of individual genes, we sought to investigate ontologies and pathways affected using unbiased database screening approaches. Gene ontology and cell signalling pathway analyses show a clear acute inflammatory response to SARS-CoV-2 infection in lung cells, dominated by cytokine and interferon signalling (Figure 3A and B). To contextualise this finding with prior published literature on viral infections, we performed an extensive GEO public literature screen of all available viral infection studies (Figure 4A). We found that the SARS-CoV-2 infection transcriptional response in lung cells resembles most that seen in published studies of respiratory viral infections SARS-CoV and respiratory syncytial virus, both of which cause respiratory pneumonias in humans. Our predictive drug target analyses screened hundreds of known agents (most with human safety and therapy approvals already) through the Drug Signatures Database screen using DSigDB. Of particular significance, our analysis matched several broad spectrum anti-inflammatory drugs (Figure 5B). These data are consistent with the notion that cytokine and inflammatory storms are cause of significant morbidity and mortality in COVID-19 disease [19] and that agents aimed at dampening this overactive inflammatory response may prove efficacious. Antimalarial and antiviral drugs have also been examined in COVID-19; notably, Hydroxychloroquine has been subject to much public debate and clinical trials in COVID-19 patients; although its efficacy is unclear [20]. Our analysis did not predict hydroxychloroquine as significant in this case, although this may simply reflect the acute nature of the infection data used in this study rather than the longer term effects of COVID-19.
We gained further insight into the pathogenic nature of genes we identified as altered with SARS-CoV-2 infections by conducting survival analyses using LC patient datasets. We performed this study to identify factors that have a particularly strong effect on mortality (and by implication, severe morbidity) in patients with advanced disease that also affects the lung. This work revealed that many of these SARS-CoV-2 response genes, especially inflammatory-related genes, are indeed associated with significantly higher mortality in such patients (Figure 5). This may have important implications for the treatment of COVID-19 patients that also present with co-morbidities, and particularly so for co-morbid lung conditions that may involve the same inflammatory gene and pathway sets. It also accords with the general observations of the effectiveness of anti-inflammatory drugs in reducing disease severity in COVID-19 and in the known role of inflammatory factors in the progression of this disease [21–23]. This indicates a strategy to identify those patient comorbidities that are at particular risk of having poor COVID-19 outcomes.
Thus far our analyses provide novel comparative insight into SARS-CoV-2 infection in primary human lung cells. While our analysis of SARS-CoV-2 infection represents a model of the acute primary infection responses in the lungs, the nature of the sustained systemic immune responses is also of great therapeutic importance. There are only limited data available on COVID-19 patient peripheral blood analyses, and at the time of writing only two such datasets are publically available [24, 25]. We analysed the unpublished patient data made available in the preprint by Ong et al. [25] and assessed concordant significant genes and pathways between peripheral blood and primary lung cell responses (Figure 6). Our differential gene expression analyses of healthy control and SARS-CoV-2 infected patient peripheral blood cells indicate that the latter cluster separately from healthy controls (Figure 6A). A pooled analysis of the systemic immune patient time-course data finds a suit of affected genes; notably, the human leukocyte antigen genes HLA-DQA1 and DQB1 are significantly downregulated 5–7 fold compared to controls (Figure 6B). Such a striking downregulation is noteworthy; downregulation of the HLAs in circulating immune cells is also characteristic of HIV [26] and influenza [27, 28] infections, as well as seen in a cancer context [17, 29]. This speaks broadly to the ability of the SARS-CoV-2 virus to induce a response in the antigen-presenting machinery required for CD4/8 T-cell recognition of viral peptides. While it remains unclear if the peripheral blood cells themselves become infected with the virus (this may be true only of monocytes although still remains unknown [30]), our analysis provides initial insight into the systemic responses in COVID-19 disease that require further validation.
Given SARS-CoV-2 causing COVID-19 disease is associated with local lung inflammation and damage, and a systemic cytokine and inflammatory storm we investigated whether any of the DEGs and associated signalling pathways showed concordance between lung cell and peripheral blood cell datasets (Figure 6C and D). We observed little overlap in DEGs; indeed only three genes show concordance (MX1, IRF7 and BST2) (Figure 6C). Given the very different nature of the cell types and the infection-reactive nature of the immune responses evident in the blood cells, this is an expected finding. In contrast, however, we see overlap in terms of significant cell signalling pathways matched to the infection datasets (Figure 6D); this suggests that although different genes are responding, the same cell signalling pathways involved in the two different tissues. We also note that the peripheral blood cell study employed a targeted NanoString immune panel and not a complete RNAseq transcriptome analysis in this case. It is thus possible that further work using whole transcriptome RNAseq approaches would detect larger sets of concordant genes and pathways in affected individual patient tissues. Nevertheless, this analysis provides the first comprehensive comparison between human coronaviruses, influenza viruses, as well as data on the immune and inflammatory responses in lung and peripheral blood cells at the level of the transcriptome.
Conclusions
We performed phylogenetic and RNAseq analyses to compare the viral genome, and infection responses between SARS-CoV-2, SARS-CoV, MERS-CoV, and pandemic influenza A strains H1N1, H3N2, and H5N1. We found that SARS-CoV-2 induces a unique transcriptional response in human lung epithelial cells and that this is dominated by inflammatory cytokine and interferon response genes. We provide external validation for these genes and confirmed that although SARS-CoV-2 infection is associated with a unique transcriptional response, it resembles most closely those of SARS-CoV and respiratory syncytial virus infections in the known literature. COVID-19 patient blood sample analyses also indicate concordance with lung cell infection responses at the level of cell signalling pathway perturbations.
These analyses advance our understating of nature of SARS-CoV-2 infection and the cell and immune responses to it and show how the infection might be examined for other diseases and drug interactions. The pathways we identified can indicate a possible role for already approved drugs, although the latter would only be of utility when if there were further clinical data on these drugs in COVID-19. SARS-CoV-2 is only a very recent discovery, so relatively few studies have been performed on its treatment. Given the global importance of this virus, more datasets will become available, enabling further work to better uncover mechanisms of the unique SARS-CoV-2 infection response in humans.
Materials and methods
Genome information and phylogenetic analysis
The complete genomes sequences used in this study were collected from the Virus Pathogen Database and Analysis Resource (ViPR; https://viprbrc.org/). To determine the relationship among all these virus strains, we created a comprehensive phylogram that includes the complete genomes of 27 human beta-coronaviruses: SARS-CoV-2: MN988668, MN997409, LC522972, NC_045512, MT007544 and MT039890; SARS-COV: AY304495, AY427439, AY461660, FJ882963 and AY559094; MERS-COV: NC_019843.3, KX154694, KY673148, MK039552 and MH371127; H1N1: NC_026435, NC_026436, NC_026432 and NC_026431; H3N2: NC_007371, NC_007366, NC_007370 and NC_007367; and H5N1: NC_007363, NC_007361 and NC_007360. The tree was inferred using the FastME program integrated with a ViPR database using genomic data with 1000 bootstrap replications. Then, Newick formatted phylogram was re-designed with the interactive tree of life online tool (https://itol.embl.de/). Furthermore, the genomic distance and G + C difference were calculated through digital DNA–DNA hybridization using Genome-to-Genome Distance Calculator server v2.1 with the default settings. For this analysis, we considered reference genomes of SARS-CoV (Refseq: NC_004718.3) and SARS-CoV-2 (Refseq: NC_045512.2); and formula-2 derived DDH and GC difference scores as per the server’s recommendation.
Data pre-processing and identification of DEGs
We analysed two SARS-CoV-2 related RNAseq transcriptomic datasets. One was an independent biological triplicate of cultured primary human lung epithelial cells that received either infection with SARS-CoV-2 (GSE147507) or a mock-treatment [3]. The second dataset was from a study of immune responses in healthy controls and COVID-19 cases that employed a NanoString Human Immunology Panel to profile collected peripheral blood cells RNA extracted from whole blood samples (E-MTAB-8871). We also analysed data from primary epithelial cells infected with other viruses including SARS-CoV (GSE47963), Mers-CoV (GSE100504) and three different influenza strains (H1N1, H3N2 and H5N1) (GSE89008). The SARS-CoV and Mers-CoV data (and respective controls) were generated from human lung epithelial cells from RNA extracted 24 h post infection. The influenza virus data are human lung epithelial cell transcriptome response to infection with 24 h of H1N1, H3N2, and H5N1 influenza virus infection. We used the DESeq2 R Bioconductor package to analyse all these RNASeq transcriptomic data and the LIMMA (linear models for microarray data) normalization R Bioconductor package was used to analyse gene expression data sets. Note that the dataset we employed was generated within a single experimental study, which formed part of the same data batch; for this reason, no batch effect correction was required in the pre-processing of our data. For each dataset, differential gene expression analysis was run between case and control data, with an adjusted P value <0.05 and the absolute value of LFC ≥1 was regarded as threshold criteria to define significant DEGs of interest. We used the Benjamini–Hochberg method to control the false discovery rate and adjust the P values. DEG lists for each viral infection were then ranked on these criteria and between studies comparisons were made.
Gene ontology and cell signalling pathway analyses
We performed gene set enrichment analysis of Gene Ontology and cell signalling pathways to evaluate the biological relevance and functional pathways of the SARS-CoV-2 associated genes. All functional analyses were performed using the Enrichr (https://amp.pharm.mssm.edu/Enrichr/) software tools [31]. For cell signalling pathway enrichment analyses we employed KEGG [32], WikiPathways [33], BioCarta and Reactome [34] databases. We used GO Biological Process (2018) database for gene ontological analysis [35]. For this work an adjusted P value ≤0.05 was considered as statistically significant for enrichment analysis.
Viral infection database screening
We collected all available virus perturbations datasets in the NCBI GEO database using Enrichr screening. We identified published virus studies that found similar gene expression effects in host cells that we identified in our SARS-CoV-2 infection analyses.
Drug prediction analyses
We have used protein-drug interaction data from the DSigDB, DrugMatrix and CMAP databases to identify potential drugs to be proposed in the SARS-CoV-2. For each gene set of our interest from the selected pathways and GO terms, we calculated the frequency (f) of genes in the study set (s) that interact with a drug, and the frequency (F) of genes in the population set (S) that interact with the same drug. We then performed a test to determine how likely it would be to obtain at least f genes interacting with a drug if s genes would be randomly drawn from the population, given that the frequency F and size S of the population. This can be represented mathematically as follows:
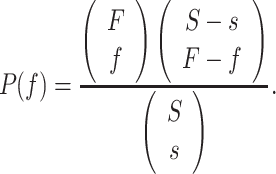 |
Survival prediction analysis
To evaluate how LC patient survival is influenced by expression of candidate genes identified as significant in SARS-CoV-2 infections we collected clinical and RNAseq data for LC from the TCGA genome data analysis centre (http://gdac.broadinstitute.org). The LC clinical dataset (Lung Adenocarcinoma, TCGA, PanCancer Atlas) consists of 566 samples with 81 features, and RNAseq gene expression data included 510 cases [36]. We matched patient ID in both clinical and RNAseq datasets and selected patients with data available for both. Among the clinical variables, we only considered six (ethnicity, anatomical site of cancer, the histological grade of cancer, primary tumour site and neoplasm status with the tumour) to evaluate survival with the significant genes. Hence, we considered z-score values to determine the altered and normal (unaltered) expression of a gene.
To determine the patient survival association of the significant genes identified in SARS-CoV-2 infection, we employed a Cox proportional hazards (PH) model for univariate and multivariate analyses; considering the following equitation:
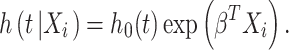 |
Here, h(t|Xi) is the hazard function at time t on a subject i with covariate information as the vector Xi, h0(t), which is the baseline hazard function and is independent of covariate information. Hence, β indicates a corresponding regression coefficients vector to the covariates respectively. We have calculated the hazard ratio (HR) based on the estimated regression coefficients from the fitted Cox PH model to identify whether a specific covariate affects patient survival. The HR for a covariate xr can be expressed as exp (βr). Thus, the HR for any covariate can be measured by applying an exponential function to the respective (βr) coefficient.
To determine survival status we used a non-parametric method to estimate the survival function. Thus, we used the product limit estimator known as the Kaplan–Meier estimator for the survival function which is defined as follows [37]:
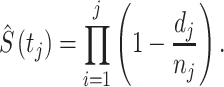 |
Hence 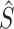 (tj) is the estimated survival function at time tj, dj is the number of events occurred at tj and nj is the number of subjects available at tj. Then two or more groups were compared using a logrank test. We applied logrank tests to detect the most significant genes associated with patient survival time, that is, when comparing groups of patients with altered expression for a particular gene, compared with groups with normal (unaltered) expression, the patient survival of these two groups differ. The null hypothesis for this test can be formulated as follows:
(tj) is the estimated survival function at time tj, dj is the number of events occurred at tj and nj is the number of subjects available at tj. Then two or more groups were compared using a logrank test. We applied logrank tests to detect the most significant genes associated with patient survival time, that is, when comparing groups of patients with altered expression for a particular gene, compared with groups with normal (unaltered) expression, the patient survival of these two groups differ. The null hypothesis for this test can be formulated as follows:
 |
Here H0 is a survival function that is the same for altered and normal genes and HA indicates survival functions that are not the same for these two groups.
Key Points
Phylogenetic analysis of the novel 2019 coronavirus designated as SARS-CoV-2 shows that it is most closely related to SARS and MERS at the level of the viral genome. Comparative RNAseq analyses show SARS-CoV-2 induces a unique transcriptional response in host human cells compared with SARS, MERS, influenza H1N1, H3N2 and H5N1.
The SARS-CoV-2 infection response in human lung epithelial cells is associated with a unique suite of inflammatory gene upregulation, and these genes are enriched in cytokine and interferon-mediated inflammatory processes.
Predictive drug screening based on the unique SARS-CoV-2 signature highlights drugs targeting inflammatory, fibrotic and vascular mechanisms that may prove critical in follow up studies on infection disease mechanisms. And upregulated SARS-CoV-2 genes show significant association with increased mortality in an independent lung disease dataset; providing insight into comorbidity associations in COVID-19.
Analysis of human blood samples from SARS-CoV-2 infected people highlights genes altered in the systemic immune response to infection, and some overlapping pathways and ontologies concordant between the primary lung cell response, and the systemic responses.
Supplementary Material
Mohammad Ali Moni is a Research Fellow and Conjoint Lecturer at the University of New South Wales, Australia. He received his PhD degree in bioinformatics from the University of Cambridge. His research interest encompasses artificial intelligence, machine learning, data science and clinical bioinformatics.
Julian M. W. Quinn received the PhD from the University of Oxford, UK, in 1992, then, moved to Australia for postdoctoral training in bone, joint and cancer biology at the St Vincent’s Institute of Medical Research as a Senior Research Fellow since 2014. His interests are in applications of biostatistics and bioinformatics, and now he works as a Surgical Research Officer at the Surgical Education and Research Training Institute at Royal North Shore Hospital, Sydney Australia.
Nese Sinmaz received her honours degree in cell pathology and immunology from the University of Sydney in 2012, and her PhD in neuroscience and immunology in 2017. She works in drug discovery for immunological disorders in the private sector while maintaining academic research interests in immunology and inflammatory disease.
Matthew A. Summers is a Research Scientist at the Garvan Institute of Medical Research, Australia. His research interests include understanding genetic and molecular mechanisms of rare diseases using cutting-edge single-cell genomic, transcriptomic and bioinformatics methods.
Contributor Information
Mohammad Ali Moni, University of New South Wales, Australia.
Julian M W Quinn, University of Oxford, UK.
Nese Sinmaz, University of Sydney.
Matthew A Summers, Garvan Institute of Medical Research, Australia.
Code and data availability
All differential gene expression, drug predictive, and survival analyses can be freely accessed through the github repository at: https://github.com/m-moni/COVID-19
Funding
The authors have no funding declarations.
References
- 1. Center JHCR. John Hopkins Coronavirus Resource Center.
- 2. Wang Y, et al. Unique epidemiological and clinical features of the emerging 2019 novel coronavirus pneumonia (COVID-19) implicate special control measures. J Med Virol 2020;92(6):568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blanco-Melo D, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020;181(5):1036, e9–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heinz S, et al. Transcription elongation can affect genome 3D structure. Cell 2018;174(6):1522, e22–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heller N. Primary human airway epithelial cell transcriptome response to a wild type Mers-CoV (icMERS-CoV EMC2012). Gene Expression Omnibus, 2017. [Google Scholar]
- 6. Yoo M, et al. DSigDB: drug signatures database for gene set analysis. Bioinformatics 2015;31(18):3069–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Parihar SP, Guler R, Brombacher F. Statins: a viable candidate for host-directed therapy against infectious diseases. Nat Rev Immunol 2019;19(2):104–17. [DOI] [PubMed] [Google Scholar]
- 8. Morais C, et al. Anti-angiogenic actions of pyrrolidine dithiocarbamate, a nuclear factor kappa B inhibitor. Angiogenesis 2009;12(4):365–79. [DOI] [PubMed] [Google Scholar]
- 9. Timson DJ. Dicoumarol: a drug which hits at least two very different targets in vitamin K metabolism. Curr Drug Targets 2017;18(5):500–10. [DOI] [PubMed] [Google Scholar]
- 10. Sarah Zaman AIM, Jennings GLR, Schlaich M, et al. Cardiovascular disease and COVID-19: Australian/New Zealand consensus statement. Med J Aust 2020. [DOI] [PubMed] [Google Scholar]
- 11. Danzi GB, et al. Acute pulmonary embolism and COVID-19 pneumonia: a random association? Eur Heart J 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nozaki I, et al. Small proline-rich proteins 2 are noncoordinately upregulated by IL-6/STAT3 signaling after bile duct ligation. Lab Invest 2005;85(1):109–23. [DOI] [PubMed] [Google Scholar]
- 13. Oany AR, et al. Design of novel viral attachment inhibitors of the spike glycoprotein (S) of severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) through virtual screening and dynamics. Int J Antimicrob Agents 2020;106177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kwapiszewska G, et al. Transcriptome profiling reveals the complexity of pirfenidone effects in idiopathic pulmonary fibrosis. Eur Respir J 2018;52(5). [DOI] [PubMed] [Google Scholar]
- 15. Taz TA, et al. Network-based identification genetic effect of SARS-CoV-2 infections to idiopathic pulmonary fibrosis (IPF) patients. Brief Bioinform 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shi H, et al. Radiological findings from 81 patients with COVID-19 pneumonia in Wuhan, China: a descriptive study. Lancet Infect Dis 2020;20(4):425–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harada A, et al. Essential involvement of interleukin-8 (IL-8) in acute inflammation. J Leukoc Biol 1994;56(5):559–64. [PubMed] [Google Scholar]
- 18. FitzGerald ES, Jamieson AM. Unique transcriptional changes in coagulation cascade genes in SARS-CoV-2-infected lung epithelial cells: a potential factor in COVID-19 coagulopathies. bioRxiv 2020. [Google Scholar]
- 19. Mehta P, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 2020;395(10229):1033–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fabio S Taccone JG, Vincent J-L. Hydroxychloroquine in the management of critically ill patients with COVID-19: the need for an evidence base. Lancet Respir Med 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Conti P, et al. Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by Coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents 2020;34(2):327–31. [DOI] [PubMed] [Google Scholar]
- 22. Villar J, et al. Efficacy of dexamethasone treatment for patients with the acute respiratory distress syndrome caused by COVID-19: study protocol for a randomized controlled superiority trial. Trials 2020;21(1):717–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ahamad MM, et al. A machine learning model to identify early stage symptoms of SARS-Cov-2 infected patients. Expert Syst Appl 2020;160:113661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiong Y, et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerging Microbes & Infections 2020;9(1):761–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eugenia Ziying Ong YFZC, Leong WY, Lee NMY, et al. A dynamic immune response shapes COVID-19 progression. Unpublished - preprint 2020. [DOI] [PMC free article] [PubMed]
- 26. Mwimanzi P, et al. Human leukocyte antigen (HLA) class I down-regulation by human immunodeficiency virus type 1 negative factor (HIV-1 Nef): what might we learn from natural sequence variants? Viruses 2012;4(9):1711–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koutsakos M, et al. Downregulation of MHC class I expression by influenza a and B viruses. Front Immunol 1158;10:2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nain Z, et al. Pathogenetic profiling of COVID-19 and SARS-like viruses. Brief Bioinform 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hicklin DJ, Marincola FM, Ferrone S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol Med Today 1999;5(4):178–86. [DOI] [PubMed] [Google Scholar]
- 30. Jafarzadeh A, et al. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: lessons from SARS and MERS, and potential therapeutic interventions. Life Sci 2020;257:118102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kuleshov MV, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44(W1):W90–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kanehisa M, Goto S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000;28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Slenter DN, et al. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res 2017;46(D1):D661–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Fabregat A, et al. The Reactome pathway knowledgebase. Nucleic Acids Res 2018;46(D1):D649–d655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. The Gene Ontology Resource 20 years and still GOing strong. Nucleic Acids Res 2019;47(D1):D330–d338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hoadley KA, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 Tumors from 33 types of cancer. Cell 2018;173(2):291, e6–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rana HK, et al. Machine learning and bioinformatics models to identify pathways that mediate influences of welding fumes on cancer progression. Sci Rep 2020;10(1):2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All differential gene expression, drug predictive, and survival analyses can be freely accessed through the github repository at: https://github.com/m-moni/COVID-19