

Abstract
The life-threatening coronaviruses MERS-CoV, SARS-CoV-1 and SARS-CoV-2 (SARS-CoV-1/2) have caused and will continue to cause enormous morbidity and mortality to humans. Virus-encoded noncoding RNAs are poorly understood in coronaviruses. Data mining of viral-infection-related RNA-sequencing data has resulted in the identification of 28 754, 720 and 3437 circRNAs encoded by MERS-CoV, SARS-CoV-1 and SARS-CoV-2, respectively. MERS-CoV exhibits much more prominent ability to encode circRNAs in all genomic regions than those of SARS-CoV-1/2. Viral circRNAs typically exhibit low expression levels. Moreover, majority of the viral circRNAs exhibit expressions only in the late stage of viral infection. Analysis of the competitive interactions of viral circRNAs, human miRNAs and mRNAs in MERS-CoV infections reveals that viral circRNAs up-regulated genes related to mRNA splicing and processing in the early stage of viral infection, and regulated genes involved in diverse functions including cancer, metabolism, autophagy, viral infection in the late stage of viral infection. Similar analysis in SARS-CoV-2 infections reveals that its viral circRNAs down-regulated genes associated with metabolic processes of cholesterol, alcohol, fatty acid and up-regulated genes associated with cellular responses to oxidative stress in the late stage of viral infection. A few genes regulated by viral circRNAs from both MERS-CoV and SARS-CoV-2 were enriched in several biological processes such as response to reactive oxygen and centrosome localization. This study provides the first glimpse into viral circRNAs in three deadly coronaviruses and would serve as a valuable resource for further studies of circRNAs in coronaviruses.
Introduction
The ongoing global pandemic COVID-19 has infected 32 730 945 people and caused 991 224 death worldwide as of 27 September 2020 [1]. The agent for COVID-19, SARS-CoV-2, is a coronavirus with a positive and single-stranded RNA genome [2]. Besides the SARS-CoV-2, there have been six human-infecting coronaviruses [2]. Among them, SARS-CoV-1 caused more than 8000 human infections with over 900 deaths in 32 countries in the period of 2002–2004 [3]; MERS-CoV has caused epidemics in more than 20 countries since 2012 [4]. More than 2500 human infections with 866 deaths have been reported as of January 2020 [5]. The high mortality rate of diseases caused by SARS-CoV-1 and MERS-CoV and the ongoing pandemic caused by SARS-CoV-2 suggests that coronaviruses are among the most life-threatening viruses to humans. Unfortunately, no effective vaccines or drugs are currently available for prevention and control of these deadly viruses.
Coronaviruses are the largest kind of RNA viruses with genomes ranging from 26 to 32 kb and with a diameter of 125 nm [6]. Coronaviruses are classified into four genera: Alphacoronavirus, Betacoronavirus, Gammacoronavirus and Deltacoronavirus [7]. The MERS-CoV, SARS-CoV-1 and SARS-CoV-2 (SARS-CoV-1/2) belong to the genus Betacoronavirus [8]. The genome of these three Betacoronaviruses includes 11–14 open reading frames (ORFs) which encode 15–16 nonstructural proteins (nsps), four major structural proteins including spike (S), envelope (E), membrane (M) nucleocapsid protein (N), and 5–8 accessory proteins [9–11]. Besides these canonical ORFs, several virus-encoded noncoding RNAs have been found in infection of coronaviruses [12]. For example, three small RNAs encoded by the SARS-CoV were found to contribute to infection-associated lung pathology [12]. Moreover, a recent work by Kim et al. presented a high-resolution map of the SARS-CoV-2 transcriptome and found many transcripts encoding unknown ORFs [13]. These studies suggest that virus-encoded noncoding RNAs may play important roles in infections of coronaviruses, and the systematic discovery of noncoding RNAs in coronaviruses is of essence due to the complex transcription process.
The circular RNAs (circRNAs) are a kind of covalently closed long noncoding RNAs and have no 3′poly(A) tail and 5′cap [14]. They can be generated by three mechanisms including the complementary sequence-mediated circularization, RNA-binding-protein-driven circularization and lariat-driven circularization [15]. The circRNAs are reported to have diverse functions in mammals and plants [16, 17]. They can function as microRNA (miRNA) sponges, by binding proteins, or by encoding proteins [18–20]. For example, ciRS-7 contains more than 70 binding sites for miR-7 and correlates with the human disease by affecting miR-7 activity [21]. The circRNAs can also function by encoding proteins or interacting with proteins [19, 20]. For example, circFOXO3 binds to the cell cycle proteins cyclin-dependent kinase 2 and cyclin-dependent kinase inhibitor 1, which inhibits cell cycle progression [20]. The circRNAs play essential roles in human diseases and are considered to be ideal biomarkers of these diseases due to their high stability [22]. For example, the up-regulation of hsa_circ_0000064 has been reported in lung cancer tissues and cell lines [22]. Knockdown of hsa_circ_0000064 can attenuate the proliferation, block cell cycle progression, promote cell apoptosis, abate migration and invasion activities of lung cancer cell lines, which indicates this circRNA is a potential prognostic biomarker for lung cancer [22].
In recent years, virus-encoded circRNAs have been identified and characterized in several cancer-associated viruses such as the Epstein–Barr virus [23–25] and the Kaposi’s sarcoma herpesvirus [24, 26]. Several viral circRNAs were associated with the progression of cancers, such as hpv-circE7 [27], ebv-circRPMS1 [28] and ebv-circLMP2A [29]. A recent study by Cai et al. systematically investigated circRNAs in viruses and identified more than 10 000 circRNAs [30]. These circRNAs are encoded in more than 20 viruses including several RNA viruses and RT viruses, which suggest the circRNAs may be a kind of widespread molecules in viruses [30]. However, no circRNAs from coronaviruses have been reported so far. Herein, we identified thousands of circRNAs in MERS-CoV and SARS-CoV-1/2 from the RNA-sequencing data of viral infection. We also characterized the size, expressions, sequence feature, genome location preference and functions of these viral circRNAs. Our systematic characterization of circRNAs in coronaviruses provides a valuable resource that can be used to further explore the mechanisms of action for individual circRNAs in coronaviruses.
Materials and methods
Data collection
The dataset (ID: GSE148729) of RNA sequencing of total RNAs in SARS-CoV-1/2 infection of Calu-3 cells [31], and the dataset (ID: GSE139516) of RNaseR-resistant RNA sequencing in MERS-CoV infection of Calu-3 cells [32], were collected from the Gene Expression Omnibus (GEO: https://www.ncbi.nlm.nih.gov/geo/) [33] and Sequence Read Archive (SRA: https://www.ncbi.nlm.nih.gov/sra/) [34] using the NCBI SRA Toolkit (http://www.ncbi.nlm.nih.gov/books/NBK158900/).
Computational identification of viral circRNAs
The genome annotation of most viruses is not suitable for the detection of circRNAs since most regions of the viral genome encode genes. Therefore, three de novo methods including circRNA_finder [35], find_circ [36] and CIRI2 [37] were used to detect viral circRNAs as described in Cai et al. [30]. More specifically, Fastp [38] was used to trim adaptors and low-quality reads in RNA-seq datasets. The remaining reads were aligned to the human hg38 genome plus the viral reference genome (AY310120.1 for SARS-CoV-1, MN908947.3 for SARS-CoV-2 and NC_019843.3 for MERS-CoV) with different alignment tools. BWA (version 0.7.17, parameter: ‘-T 19’) [39] was used for CIRI2 [37], STAR (version 2.7.1a, parameter: default parameters) [40] for circRNA_finder [35] and Bowtie2 (version 2.1.0, parameter: ‘-p16 --very-sensitive --score-min=C,-15,0’) [41] for find_circ [36]. Viral circRNAs were then identified using three de novo computational tools (CIRI2, find_circ and circRNA_finder) with default parameters. The abundance of viral circRNAs was determined as the average number of reads across the back-splicing junctions in the three de novo methods [42]. For comparison of the abundance of viral circRNAs in MERS-CoV and SARS-CoV-1/2, the TPM (transcripts per million) of each viral circRNAs was calculated by dividing the average number of back-splicing reads by the total number of clean reads in each sample [32, 42].
All the identified viral circRNAs and related information (such as position, strand, abundance and so on) were provided in the VirusCircBase [30] which is available at http://www.computationalbiology.cn/ViruscircBase/home.html.
Genome annotation of coronaviruses
The genome annotations of coronaviruses were adapted from NCBI RefSeq database.
Analysis of sequence conservation of viral circRNAs
Each viral circRNA was queried against the viral circRNAs in other viruses using blastn (version 2.9.0+) [43]. The hits with identity ≥80%, coverage ≥80% and e-value ≤ 1E-5 were considered to be homologous to the query viral circRNA.
Analysis of flanking sequence features of viral circRNAs
For each viral circRNA, the repeat sequences or reverse complementary sequences of 4 bp or longer were searched within the upstream and downstream 10 bp of 5′ and 3′ of the splice site.
Differential expression analysis of host genes and miRNA in viral infection of MERS-CoV and SARS-CoV-1/2
For MERS-CoV, the expression data of human genes and miRNAs measured by FPKM and TPM, respectively, at 6 and 24 hours post infection (hpi) were obtained from the dataset GSE139516 [32] in NCBI GEO database. The differentially expressed (DE) miRNAs and mRNAs at 6 or 24 hpi were obtained by comparing the gene expressions in samples of viral infection and mock infection using the limma package [44] in R. For SARS-CoV-1 and SARS-CoV-2, the expression data of human genes and miRNAs measured by read counts at 4, 12 and 24 hpi were obtained from the dataset GSE148729 [31] in NCBI GEO database. The differentially expressed miRNAs (DEmiRNAs) and mRNAs at 24 hpi were obtained by comparing the gene expressions in samples of viral infection and mock infection using the DESeq2 package [45] in R. The DEmiRNAs and mRNAs at 12 hpi were obtained by comparing the gene expressions in samples of viral infection at 12 hpi and those of mock infection at 4 hpi and 24 hpi, since no mock infections were conducted at 12 hpi. RNAs with at least 2-fold changes and adjusted P-values ≤ 0.05 were considered to be DE.
Prediction of interactions between viral circRNAs and host miRNAs
Previous studies have shown that circRNAs below 200 bp did not exhibit RNase R resistance and were prone to be false positives [46]. Therefore, selection criteria of high-confidence viral circRNAs include those detected by at least two detection tools, with back-splicing junction reads ≥2 and with the length ranging from 200 bp to 2 kbp. This yields 4062 high-confidence viral circRNAs used for interaction and function analysis. The interactions between the high-confidence viral circRNAs and the DEmiRNAs were predicted using TarPmiR (http://hulab.ucf.edu/research/projects/miRNA/TarPmiR/) [47] with default parameters. Further analysis considers only the interactions with definite binding probabilities (P = 1) and binding energies ≤ −20 kcal/mol according to Seclaman’s study [48].
Construction of competing endogenous RNA network of viral circRNAs, human miRNAs and mRNAs
When constructing the competing endogenous RNA (ceRNA) network for a virus, viral circRNAs were connected to the DE human miRNAs if they were predicted to interact with each other; the up-regulated (or down-regulated) miRNAs were connected to down-regulated (or up-regulated) mRNAs if the former targeted the latter based on the database miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/php/index.php) [49]. The circRNA-miRNA-mRNA network was visualized using Cytoscape (version 3.5.1).
Functional enrichment analysis of human genes
The KEGG pathway and GO enrichment analysis was conducted with functions of enrichKEGG and enrichGO in the package clusterProfiler (version 3.12.0) [50] in R (version 3.6.1). All the KEGG pathways and GO terms with q-values less than 0.05 were considered as significant enrichment.
Statistical analysis
All the statistical analyses in this study were conducted in R (version 3.6.1). The Wilcoxon rank-sum test was used to compare the sample means in this study and was conducted with wilcox.test.
Results
Computational identification of circRNAs encoded by MERS-CoV, SARS-CoV-1 and SARS-CoV-2
Three computational methods, i.e. CIRI2, find_circ and circRNA_finder, were used to identify viral circRNAs in Calu-3 cells infected by MERS-CoV, SARS-CoV-1 and SARS-CoV-2. A total of 28 754, 720 and 3437 viral circRNAs were identified for MERS-CoV, SARS-CoV-1 and SARS-CoV-2, respectively (Figure 1). Among them, 4182, 55 and 354 viral circRNAs were detected by at least two methods, and only 319, 3 and 24 viral circRNAs were detected by all three methods in MERS-CoV, SARS-CoV-1 and SARS-CoV-2, respectively.
Figure 1.
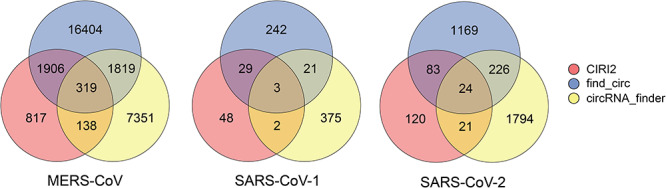
The number of viral circRNAs identified by three computational methods (CIRI2, find_circ and circRNA_finder) in MERS-CoV, SARS-CoV-1 and SARS-CoV-2.
Analysis of the length of circRNAs encoded by three coronaviruses showed that 67–81% of viral circRNAs had lengths ranging from 200 bp to 2 kbp (Figure 2A), which was similar to those observed in other viral circRNAs [30]. The median length of viral circRNAs was 409 bp, 812 bp and 791 bp for MERS-CoV, SARS-CoV-1 and SARS-CoV-2, respectively. The viral circRNAs in MERS-CoV were significantly shorter than those in SARS-CoV-1/2 (P-value < 2.2E-16). Short viral circRNAs with less than 200 bp composed nearly 10% in MERS-CoV but less than 2% in SARS-CoV-1/2, whereas the percentage of long viral circRNA with more than 20 000 bp were less than 5% in all three viruses.
Figure 2.
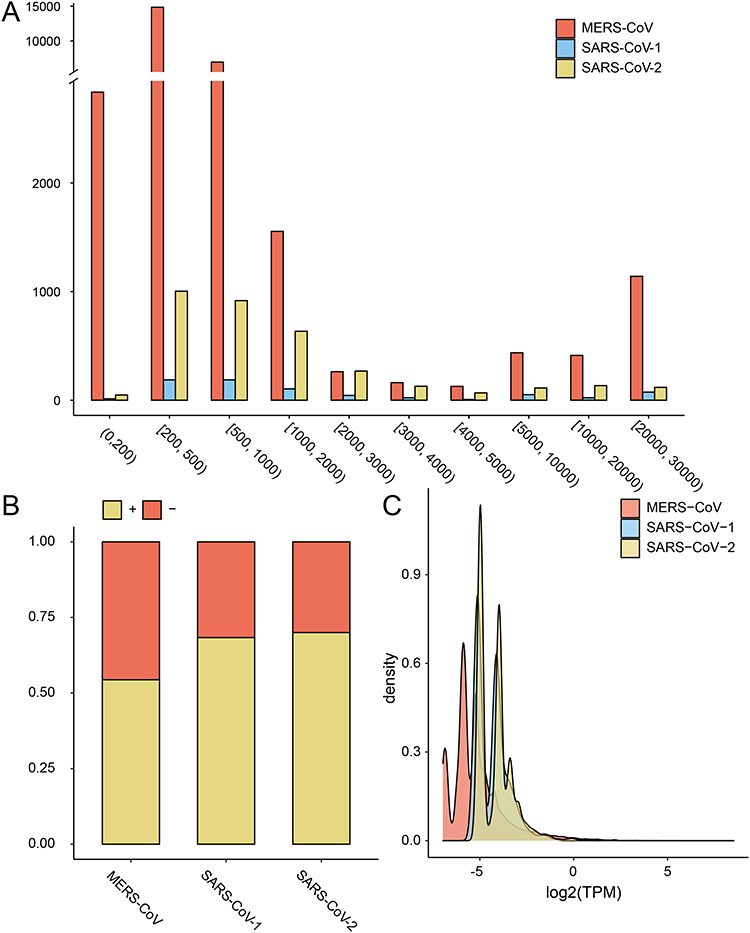
The distribution of length (A), strandness (B) and expression level (C) of viral circRNAs encoded by MERS-CoV and SARS-CoV-1/2. TPM, transcripts per million (Materials and Methods).
Analysis of the strandness of viral circRNAs showed that about half of viral circRNAs were encoded by the positive-strand in the MERS-CoV, whereas nearly 70% of viral circRNAs were encoded by the positive-strand in the SARS-CoV-1 and SARS-CoV-2 (Figure 2B).
The expression level of viral circRNAs showed a bimodal distribution in all three viruses (Figure 2C). Ninety-nine percent of viral circRNAs had less than one TPM. Only a few viral circRNAs had high expression levels. For example, the MERS-CoV_circ_Homo_sapiens_1312 encoded by MERS-CoV had a high expression of 371.7 TPM. The expression level of viral circRNAs in MERS-CoV was significantly lower than those in SARS-CoV-1/2 (P-value < 2.2E-16).
The progressive expression of viral circRNAs in MERS-CoV and SARS-CoV-1/2 were analyzed (Figure 3). MERS-CoV expressed 883 circRNAs in the early stage of infection at 6 hpi, and 28 645 circRNAs in the late stage of infection at 24 hpi. SARS-CoV-1 and SARS-CoV-2 expressed zero and two circRNAs in the early stage (4 hpi), 213 and 1504 circRNAs in the middle stage (12 hpi), and 541 and 2320 circRNAs in the late stage of viral infection (24 hpi), respectively. Five hundred and seven and 1931 viral circRNAs identified in 24 hpi were not identified in 12 hpi for SARS-CoV-1 and SARS-CoV-2, respectively. Comparison of the expression level of common viral circRNAs in different stages of viral infection revealed that MERS-CoV expressed more circRNAs in the late stage than in the early stage for MERS-CoV (P-value < 2.2E-16), whereas SARS-CoV-1/2 expressed similar level of viral circRNAs in both the middle stage and the late stage (Figure S1).
Figure 3.
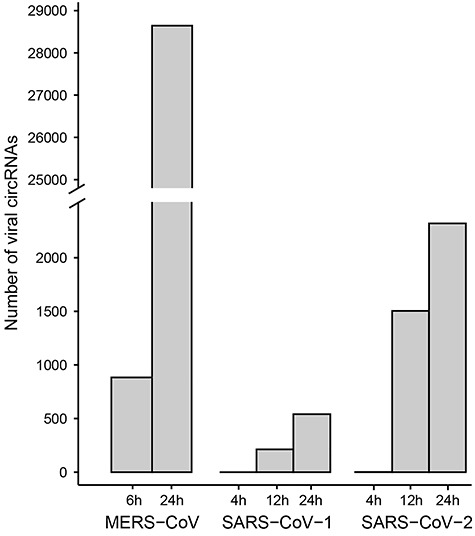
The number of viral circRNAs identified in the progressive infection of MERS-CoV, SARS-CoV-1/2.
Previous studies have reported high variability of circRNAs identified among the biological replicates [51]. We then analyzed the overlapped viral circRNAs identified between the biological replicates of virus infections. As shown in Figure S2, only 3.74% and 19.19% circRNAs were identified in all three biological replicates at 6 hpi and 24 hpi of MERS-CoV, respectively; for SARS-CoV-1 and SARS-CoV-2, 5–13% viral circRNAs were identified among biological replicates of viral infections at different time points. We further analyzed the correlations between the expressions of viral circRNAs in biological replicates. As was shown in Table S1, for all three viruses, median to strong correlations with statistical significance were observed between circRNA expressions in biological replicates.
Location of viral circRNAs along the genome of MERS-CoV and SARS-CoV-1/2
MERS-CoV, SARS-CoV-1 and SARS-CoV-2 share nearly identical genomic structures (Figure 4), but encoded much different numbers of circRNAs (Figure 1). When mapping the viral circRNAs into genomes, we found that the number of viral circRNAs identified per kilobase along the viral genome was the largest in MERS-CoV, next in SARS-CoV-2 and smallest in SARS-CoV-1 (Figure 4). There were no correlations between the number of viral circRNAs identified per kilobase in the three viruses. The ability of encoding circRNAs varied along the viral genomes. The 5′ and 3′ ends of viral genomes encoded less circRNAs than other regions in the viral genomes. In the MERS-CoV genome, the region from 25 kb to 27 kb (marked by a red arrow) encoded much more circRNAs than other regions. In the SARS-CoV-2 genome, the number of viral circRNAs identified per kilobase increased slightly from 5′ to 3′ end (Pearson Correlation Coefficient = 0.72).
Figure 4.
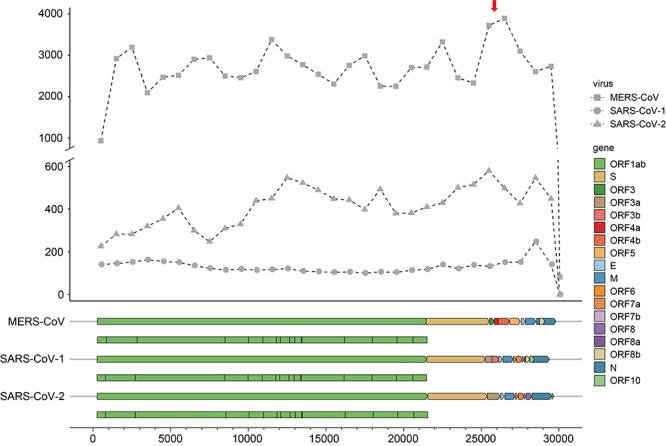
The number of viral circRNAs located within each kilobase along the viral genomes in MERS-CoV (square), SARS-CoV-1 (circle) and SARS-CoV-2 (triangle). (A) circRNA was considered to be located in a kilobase if it overlapped with the region. Genes encoded in each virus were colored according to the figure legend. The nonstructure proteins encoded in ORF1ab was shown below the ORF1ab gene.
Conservation of viral circRNAs in MERS-CoV and SARS-CoV-1/2
The sequence conservation of viral circRNAs in three coronaviruses was analyzed. A total of 456 viral circRNA encoded by SARS-CoV-1 had sequence homology with those in SARS-CoV-2, whereas 1913 viral circRNAs encoded by SARS-CoV-2 had sequence homology with those in SARS-CoV-1. However, none of viral circRNAs encoded by MERS-CoV had homology with those in either SARS-CoV-1 or SARS-CoV-2.
circRNAs in coronaviruses are flanked by short repeat sequences and reverse complementary sequences
To understand the mechanisms of the biogenesis of viral circRNAs, we analyzed the flanking short repeat sequences and reverse complementary sequences of viral circRNAs. For all three viruses, nearly 70% and 80% of viral circRNAs were flanked by reverse complementary sequences and repeat sequences, respectively (Figure S3). More than 90% of viral circRNAs in coronaviruses were flanked by either short repeat sequences or reverse complementary sequences, whereas nearly 60% of viral circRNAs had both. This statistic was comparable to that observed in other viral circRNAs or plant circRNAs [30, 52], suggesting that both flanking short repeat sequences and reverse complementary sequences might be crucial for the biogenesis of circRNAs in coronaviruses.
The competitive interactions of viral circRNAs, human miRNAs and mRNAs
Since circRNAs regulate target genes of miRNAs by acting as miRNA sponges [18], the competitive interactions of viral circRNAs and the DE human miRNAs and mRNAs were analyzed in viral infections of Calu-3 cells to investigate the functions of viral circRNAs. In 6 hpi of MERS-CoV, 238 high-confidence viral circRNAs which were defined as those detected by at least two detection tools, with back-splicing junction reads ≥2 and with the length ranging from 200 bp to 2 kbp (Materials and Methods) were identified (Table 1). Six up-regulated and seven down-regulated human miRNAs were identified, among which two and four were predicted to interact with viral circRNAs; 6214 down-regulated and 2410 up-regulated human mRNAs were identified, among which 93 and 162 were targeted by DEmiRNAs and were therefore regulated indirectly by viral circRNAs. GO enrichment analysis of 162 mRNAs which were up-regulated by viral circRNAs showed that they were enriched in biological processes related to mRNA splicing and processing (Table S2). No GO terms were enriched in the 93 mRNAs which were down-regulated by viral circRNA.
Table 1.
The number of differentially expressed miRNAs and mRNAs, and the high-confidence viral circRNAs interacting with miRNAs in MERS-CoV at 6 and 24 hpi in Calu-3 cells. DEmiRNAs, differentially expressed miRNAs; DEmRNAs, differentially expressed mRNAs
| Time | High-confidence viral circRNAs | Human DEmiRNAs | Human DEmRNAs | |||||
|---|---|---|---|---|---|---|---|---|
| Total | Interact with human DEmiRNAs | Sign. | Total | Interact with viral circRNAs | Sign. | Total | Interact with human DEmiRNAs | |
| 6 hpi | 238 | 59 | Up | 6 | 2 | Down | 6214 | 93 |
| Down | 7 | 4 | Up | 2410 | 162 | |||
| 24 hpi | 3741 | 3311 | Up | 86 | 56 | Down | 7023 | 1165 |
| Down | 41 | 30 | Up | 3963 | 1586 | |||
In 24 hpi of MERS-CoV, 3741 high-confidence viral circRNAs were identified (Table 1); they were predicted to interact with 56 up-regulated and 30 down-regulated human miRNAs, respectively, thus down- and up-regulating 1165 and 1586 human mRNAs indirectly. Analysis of the functions of 1165 mRNAs which were down-regulated by viral circRNAs showed that they were enriched in diverse KEGG pathways associated with cancer, metabolism, bacterial and viral infection, and so on (Figure 5A and Table S3); in the GO enrichment analysis, they were mainly enriched in biological processes of viral infection, protein targeting, in cellular components of cell surface and organelles, and in molecular functions of binding and oxidoreductase (Figure 5A and Table S3). The 1586 mRNAs which were up-regulated by viral circRNAs were also enriched in diverse KEGG pathways associated with cancer, virus infection, protein processing, cellular senescence, autophagy,and so on (Figure 5B and Table S3); in the GO enrichment analysis, they were mainly enriched in biological processes of molecule modification, nucleic acid transport and localization, in cellular components of nucleus, in molecular functions of transcription coactivator, transferase amd phosphatase.
Figure 5.
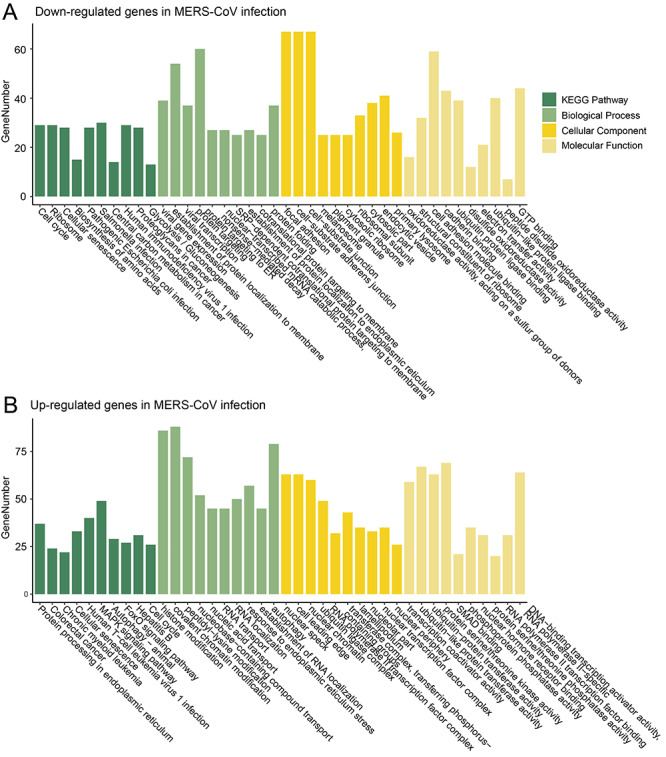
Functional enrichment analysis of up- and down-regulated genes in viral infection of MERS-CoV in Calu-3 cells at 24 hpi.
In 12 and 24 hpi of SARS-CoV-1, two and five human miRNAs were DE, respectively (Table S4). Among them, only one human miRNA was down-regulated and up-regulated by viral circRNAs, respectively. No DE human mRNAs were regulated by human miRNAs and viral circRNAs.
We then analyzed the competitive interactions of viral circRNAs, human miRNAs and mRNAs in 12 and 24 hpi of SARS-CoV-2 in Calu-3 cells. In 12 hpi, only 141 high-confidence viral circRNAs were identified. They were predicted to interact with two up-regulated human miRNAs and thus down-regulated one human mRNA (Table 2). In 24 hpi, 228 viral circRNAs were identified. A ceRNA networks of viral circRNAs, human miRNAs and mRNAs were built in 24 hpi of SARS-CoV-2 (Figure 6) (Materials and Methods). Eighty-six viral circRNAs (yellow circle) were predicted to interact with eight up-regulated miRNAs (red square), whereas the latter targeted 27 down-regulated mRNAs (green triangle). These down-regulated genes were enriched in biological processes of metabolic processes of cholesterol, alcohol, sterol, fatty acid and so on (Figure 7A and Table S5). Twenty-seven viral circRNAs (yellow circle) were predicted to interact with three down-regulated miRNAs (green square), whereas the latter targeted 25 up-regulated mRNAs (red triangle). These up-regulated genes were enriched in KEGG pathway of ‘Longevity regulating pathway’ and in biological processes of cellular responses to oxidative stress (Figure 7B and Table S5).
Table 2.
The number of differentially expressed miRNAs and mRNAs, and the high-confidence viral circRNAs interacting with miRNAs in SARS-CoV-2 at 12 and 24 hpi in Calu-3 cells. DEmiRNAs, differentially expressed miRNAs; DEmRNAs, differentially expressed mRNAs
| Time | High-confidence viral circRNAs | Human DEmiRNAs | Human DEmRNAs | |||||
|---|---|---|---|---|---|---|---|---|
| Total | Interact with human DEmiRNAs | Sign. | Total | Interact with viral circRNAs | Sign. | Total | Interact with human DEmiRNAs | |
| 12 hpi | 141 | 7 | Up | 3 | 2 | Down | 691 | 1 |
| Down | 1 | 0 | Up | 91 | 0 | |||
| 24 hpi | 228 | 96 | Up | 12 | 8 | Down | 1396 | 27 |
| Down | 13 | 3 | Up | 433 | 25 | |||
Figure 6.
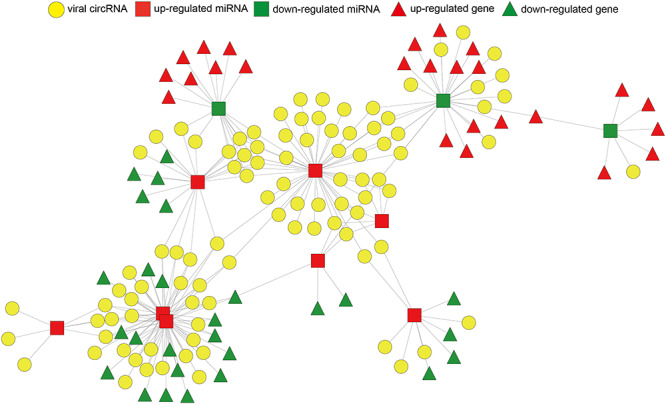
The ceRNA network of viral circRNA, host miRNA and mRNA in SARS-CoV-2. Red represents up-regulation, and green represents down-regulation. Viral circRNAs, host miRNAs and mRNAs in the networks are represented as circles, squares and triangles, respectively.
Figure 7.
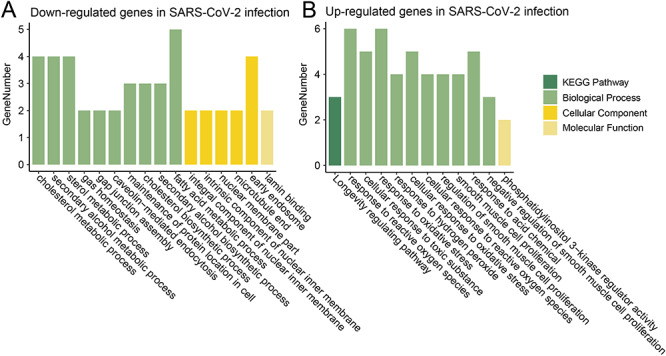
Functional enrichment analysis of up- and down-regulated genes in viral infection of SARS-CoV-2 in Calu-3 cells at 24 hpi.
Comparison of the DEmiRNAs and mRNAs which interacted with viral circRNAs in MERS-CoV and SARS-CoV-1/2 showed that only one miRNA (hsa-miR-4485-3p) was up-regulated by circRNAs of all three viruses in 24 hpi. The miRNA hsa-miR-4485-3p was reported to induce cell cycle arrest and tumor growth inhibition of breast cancer cells through reduction of key cell cycle progression factors including cyclins B1 and D1 [53, 54]. No DE mRNA was regulated by viral circRNAs of all three viruses. Eight mRNAs were up-regulated by viral circRNAs of both MERS-CoV and SARS-CoV-2 in 24 hpi (Table S6), such as SESN2 and SOD2. They were enriched in KEGG pathway of ‘Longevity regulating pathway’ and in biological process of ‘response to reactive oxygen species’ (Table S7). Seven mRNAs were down-regulated by viral circRNAs of both MERS-CoV and SARS-CoV-2 in 24 hpi (Table S6), such as SLC26A2 and SCD. They were enriched in biological process of centrosome localization, bisphosphate metabolic process and so on (Table S7).
Discussion
This work for the first time identified circRNAs encoded by three coronaviruses MERS-CoV, SARS-CoV-1 and SARS-CoV-2. All three coronaviruses belong to Betacoronavirus and have similar genomic structures [55], yet they exhibit largely diverse circRNA-coding ability. MERS-CoV encodes far more circRNA than SARS-CoV-1/2. Even for the SARS-CoV-1 and SARS-CoV-2 which share 80% genome similarity [56], the number of viral circRNAs differs much. We have identified 11 924 viral circRNAs from 23 viral species in a previous study [30]. MERS-CoV, a positive-strand RNA virus with 30 kb genome [55], was predicted to encode more than 20 000 viral circRNAs, which exhibits far greater circRNA-coding ability when compared with several DNA viruses which had genomes larger than 100 kb [30].
Many factors may contribute to the different ability of viruses in encoding circRNAs. Firstly, as was reported in our previous study, the genome size of viruses was observed to have a weak correlation with the coding ability of circRNAs, whereas the genome type (circular or linear genome) contributed little to the coding ability of circRNAs [30]. Secondly, the RNA-binding proteins (RBPs) which may promote the generation of circRNAs were dynamically regulated by viral infections which may result in dynamic expression of viral circRNAs [20]. For example, over 30% of circRNAs are dynamically regulated by the alternative splicing factor, i.e. the RNA binding protein Quaking (QKI), during human epithelial-mesenchymal transition [57]. Thirdly, the expression of circRNAs is tissue- and cell-specific [58] since the expression of RBPs may vary with tissues or cells [20, 59]. Fourthly, the method of library preparation for sequencing has a large influence on the number of circRNAs identified [58]. For example, the RNase R-treated RNA-seq can enrich more circRNAs than the total RNA-seq did [60]. Nevertheless, the key factors that determine the coding ability of circRNAs in viruses await further exploration.
It is worth noting that lots of viral circRNAs identified here are encoded by the negative strand of the coronaviruses which have positive single-stranded RNA genomes. Previous studies have shown that both the full-length negative-stranded genomic RNAs and lots of negative-strand subgenomic RNAs (sgRNAs) are generated during the processes of genome replication and transcription of coronaviruses [13, 61–63]. For example, Kim et al. have shown that numerous discontinuous transcription events led to highly complex transcriptome including lots of negative-strand sgRNAs in the SARS-CoV-2 infection [13]. The circRNAs may be generated from the negative-strand viral sgRNAs through two possible mechanisms: the first is the RNA–RNA interaction of base pairing between the flanking sequence of viral RNAs [15]; the second is the cyclization of viral sgRNAs mediated by the RBPs. The RBPs can promote the interactions between the upstream and downstream of the sgRNAs [57]. More efforts are needed to clarify the detailed mechanism of generating circRNAs in the negative-strand of coronaviruses.
The roles of viral circRNAs in viral infection is poorly understood. Ungerleider et al. investigated the circRNA repertoires in the Lymphocryptovirus and Rhadinovirus genera of Gammaherpesviruses and found that viral circRNAs function in latency and viral replication [64]. The circRNAs may have different functions in the cell and may act as a sustained effector in signaling pathways due to their unique structure and increased stability [64]. Most viral circRNAs identified in this study were expressed in the late stage of infection for coronaviruses. The viral circRNAs in the early stage of MERS-CoV infection were observed to up-regulate genes related to mRNA splicing and processing; whereas in the late stage of viral infection, the viral circRNAs were observed to regulate genes involved in diverse functions including cancer, metabolism, autophagy and viral infection (Figure 5). Interestingly to note, the genes up-regulated by MERS-CoV circRNAs in the late stage of viral infection were enriched in nucleus, whereas those down-regulated by viral circRNAs were enriched in cell surface and organelles (Figure 5). In SARS-CoV-2, the genes down-regulated by viral circRNAs were associated with metabolic processes of cholesterol, alcohol, sterol and fatty acid, whereas those up-regulated genes by viral circRNAs were associated with cellular responses to oxidative stress in the late stage of viral infection (Figure 7). Taken together, viral circRNAs in different viruses may exercise diverse functions during the infection process.
MERS-CoV, SARS-CoV-1 and SARS-CoV-2 are currently the most life-threatening coronaviruses to humans [65]. Despite of their similar genome structures, the pathogenicity varies much, with MERS-CoV the highest, SARS-CoV-1 next and SARS-CoV-2 the lowest [66]. The mechanism behind viral pathogenicity is still poorly understood. MERS-CoV takes DPP4 as the main receptor [67], whereas both SARS-CoV-1 and SARS-CoV-2 take ACE2 as the main receptor [68, 69]. Both DPP4 and ACE2 are expressed in a wide range of human cells [70, 71]. Thus, all three viruses can infect a wide range of human cells, tissues and organs. MERS-CoV can even infect human dendritic cells, macrophages and T cells, which may lead to immune dysregulation [72]. Besides the receptors, the interactions between virus and human proteins (or RNAs) also influence the viral pathogenicity [73]. Previous studies have identified several human or viral RNAs associated with immunity and inflammation on infection of coronaviruses [12, 74]. This study identified a large number of viral circRNAs during the infection process of MERS-CoV, SARS-CoV-1 and SARS-CoV-2. The circRNAs identified herein may regulate human genes with diverse functions and possibly contribute to the viral pathogenicity of coronaviruses, while their mechanism of actions require further exploration.
There are some limitations in this study. Firstly, the viral circRNAs were identified from the RNA-seq dataset of viral infection in vitro. However, viral circRNAs are known to have strong cell and tissue specificity [30]. Therefore, the expression patterns of these viral circRNAs in other types of human cells or tissues in vivo require further investigations. Secondly, the presence of viral circRNAs was identified solely by computational methods in this study, whereas the experimental validation of their presence would require further efforts. Nevertheless, this work for the first time identifies and characterizes a repertoire of circRNAs encoded by MERS-CoV, SARS-CoV-1 and SARS-CoV-2. It would serve as a valuable resource for any further investigations of circRNAs in coronaviruses.
Key Points
We identified thousands of circRNAs encoded by MERS-CoV, SARS-CoV-1 and SARS-CoV-2 for the first time.
Most viral circRNAs were expressed in the late stage of viral infection.
Viral circRNAs had different functions in MERS-CoV and SARS-CoV-2.
Supplementary Material
Zena Cai is a PhD candidate at College of Biology, Hunan University.
Congyu Lu is a PhD candidate at College of Biology, Hunan University.
Jun He is an associate professor in Anhui CDC, China.
Li Liu is a technician in Hunan Yuelu mountain data science and Technology Research Institute Co., Ltd, Changsha, China.
Yuanqiang Zou is a technician in Alibaba Cloud Computing Co., Ltd, Hangzhou, China.
Zheng Zhang is a PhD candidate at College of Biology, Hunan University.
Zhaozhong Zhu is a PhD candidate at College of Biology, Hunan University.
Xingyi Ge is a professor at College of Biology, Hunan University.
Aiping Wu is a professor at Suzhou Institute of Systems Medicine.
Taijiao Jiang is a professor at Suzhou Institute of Systems Medicine.
Heping Zheng is a professor at College of Biology, Hunan University.
Yousong Peng is an associate professor at College of Biology, Hunan University.
Contributor Information
Zena Cai, College of Biology, Hunan University.
Congyu Lu, College of Biology, Hunan University.
Jun He, Anhui CDC, China.
Li Liu, Hunan Yuelu mountain data science and Technology Research Institute Co., Ltd, Changsha, China.
Yuanqiang Zou, Alibaba Cloud Computing Co., Ltd, Hangzhou, China.
Zheng Zhang, College of Biology, Hunan University.
Zhaozhong Zhu, College of Biology, Hunan University.
Xingyi Ge, College of Biology, Hunan University.
Aiping Wu, Suzhou Institute of Systems Medicine.
Taijiao Jiang, Suzhou Institute of Systems Medicine.
Heping Zheng, College of Biology, Hunan University.
Yousong Peng, College of Biology, Hunan University.
Availability of Data
All the RNA-sequencing data used in this study were publicly available at NCBI SRA database. All the identified viral circRNAs were publicly available at the VirusCircBase (http://www.computationalbiology.cn/ViruscircBase/home.html).
Funding
National Key Plan for Scientific Research and Development of China (2016YFD0500300), Hunan Provincial Natural Science Foundation of China (2018JJ3039, 2020JJ3006, 2019JJ20004), the National Natural Science Foundation of China (31671371, 32041001) and the Chinese Academy of Medical Sciences (2016-I2M-1-005).
Conflict of interest
The authors declare that they have no competing interests.
References
- 1. WHO WHO Coronavirus Disease (COVID-19) Dashboard. Available at: https://covid19.who.int/.
- 2. Chen B, Tian EK, He B, et al. Overview of lethal human coronaviruses. Signal Transduct Target Ther 2020;5:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Magro F, Abreu C, Rahier JF. The daily impact of COVID-19 in gastroenterology. United Eur Gastroenterol J 2020;8:520–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Algaissi AA, Alharbi NK, Hassanain M, et al. Preparedness and response to COVID-19 in Saudi Arabia: building on MERS experience. J Infect Public Health 2020;13:834–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. WHO Middle East respiratory syndrome coronavirus (MERS-CoV) – The Kingdom of Saudi Arabia. Available at: https://www.who.int/csr/don/24-february-2020-mers-saudi-arabia/en/.
- 6. Kaul D. An overview of coronaviruses including the SARS-2 coronavirus - molecular biology, epidemiology and clinical implications. Curr Med Res Pract 2020;10:54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Li F. Structure, function, and evolution of coronavirus spike proteins. Annu Rev Virol 2016;3:237–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wu A, Peng Y, Huang B, et al. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe 2020;27:325–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Algaissi A, Agrawal AS, Hashem AM, et al. Quantification of the Middle East respiratory syndrome-coronavirus RNA in tissues by quantitative real-time RT-PCR. Methods Mol Biol 2020;2099:99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Satija N, Lal SK. The molecular biology of SARS coronavirus. Ann N Y Acad Sci 2007;1102:26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Naqvi AAT, Fatima K, Mohammad T, et al. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: structural genomics approach. Biochim Biophys Acta Mol Basis Dis 2020;1866:165878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morales L, Oliveros JC, Fernandez-Delgado R, et al. SARS-CoV-encoded small RNAs contribute to infection-associated lung pathology. Cell Host Microbe 2017;21:344–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kim D, Lee JY, Yang JS, et al. The architecture of SARS-CoV-2 transcriptome. Cell 2020;181:914–921.e910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Quan G, Li J. Circular RNAs: biogenesis, expression and their potential roles in reproduction. J Ovarian Res 2018;11:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lasda E, Parker R. Circular RNAs: diversity of form and function. RNA 2014;20:1829–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li X, Yang L, Chen LL. The biogenesis, functions, and challenges of circular RNAs. Mol Cell 2018;71:428–42. [DOI] [PubMed] [Google Scholar]
- 17. Zhao W, Chu S, Jiao Y. Present scenario of circular RNAs (circRNAs) in plants. Front Plant Sci 2019;10:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma S, Kong S, Wang F, et al. CircRNAs: biogenesis, functions, and role in drug-resistant tumours. Mol Cancer 2020;19:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhong Y, Du Y, Yang X, et al. Circular RNAs function as ceRNAs to regulate and control human cancer progression. Mol Cancer 2018;17:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang A, Zheng H, Wu Z, et al. Circular RNA-protein interactions: functions, mechanisms, and identification. Theranostics 2020;10:3503–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hansen TB, Kjems J, Damgaard CK. Circular RNA and miR-7 in cancer. Cancer Res 2013;73:5609–12. [DOI] [PubMed] [Google Scholar]
- 22. Zhang Z, Yang T, Xiao J. Circular RNAs: promising biomarkers for human diseases. EBioMedicine 2018;34:267–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ungerleider N, Concha M, Lin Z, et al. The Epstein Barr virus circRNAome. PLoS Pathog 2018;14:e1007206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Toptan T, Abere B, Nalesnik MA, et al. Circular DNA tumor viruses make circular RNAs. Proc Natl Acad Sci USA 2018;115:E8737–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang JT, Chen JN, Gong LP, et al. Identification of virus-encoded circular RNA. Virology 2019;529:144–51. [DOI] [PubMed] [Google Scholar]
- 26. Tagawa T, Gao S, Koparde VN, et al. Discovery of Kaposi's sarcoma herpesvirus-encoded circular RNAs and a human antiviral circular RNA. Proc Natl Acad Sci USA 2018;115:12805–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao J, Lee EE, Kim J, et al. Transforming activity of an oncoprotein-encoding circular RNA from human papillomavirus. Nat Commun 2019;10:2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Q, Shuai M, Xia Y. Knockdown of EBV-encoded circRNA circRPMS1 suppresses nasopharyngeal carcinoma cell proliferation and metastasis through sponging multiple miRNAs. Cancer Manag Res 2019;11:8023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gong LP, Chen JN, Dong M, et al. Epstein-Barr virus-derived circular RNA LMP2A induces stemness in EBV-associated gastric cancer. EMBO Rep 2020;21:e49689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cai Z, Fan Y, Zhang Z, et al. VirusCircBase: a database of virus circular RNAs. Brief Bioinform 2020. 10.1093/bib/bbaa052. [DOI] [PubMed] [Google Scholar]
- 31. Emanuel W, Kirstin M, Vedran F, et al. Bulk and single-cell gene expression profiling of SARS-CoV-2 infected human cell lines identifies molecular targets for therapeutic intervention. bioRxiv 2020 10.1101/2020.05.05.079194. [DOI]
- 32. Zhang X, Chu H, Wen L, et al. Competing endogenous RNA network profiling reveals novel host dependency factors required for MERS-CoV propagation. Emerg Microbes Infect 2020;9:733–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Clough E, Barrett T. The gene expression omnibus database. Methods Mol Biol 2016;1418:93–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kodama Y, Shumway M, Leinonen R. The sequence read archive: explosive growth of sequencing data. Nucleic Acids Res 2012;40:D54–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Westholm JO, Miura P, Olson S, et al. Genome-wide analysis of drosophila circular RNAs reveals their structural and sequence properties and age-dependent neural accumulation. Cell Rep 2014;9:1966–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Memczak S, Jens M, Elefsinioti A, et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013;495:333–8. [DOI] [PubMed] [Google Scholar]
- 37. Gao Y, Zhang J, Zhao F. Circular RNA identification based on multiple seed matching. Brief Bioinform 2018;19:803–10. [DOI] [PubMed] [Google Scholar]
- 38. Chen S, Zhou Y, Chen Y, et al. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018;34:i884–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. ArXiv 2013;1303:3997. https://arxiv.org/abs/1303.3997 [Google Scholar]
- 40. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nat Methods 2012;9:357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ruan H, Xiang Y, Ko J, et al. Comprehensive characterization of circular RNAs in ~ 1000 human cancer cell lines. Genome Med 2019;11:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Altschul SF, Gish W, Miller W, et al. Basic local alignment search tool. J Mol Biol 1990;215:403–10. [DOI] [PubMed] [Google Scholar]
- 44. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hansen TB, Venø MT, Damgaard CK, et al. Comparison of circular RNA prediction tools. Nucleic Acids Res 2016;44:e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ding J, Li X, Hu H. TarPmiR: a new approach for microRNA target site prediction. Bioinformatics 2016;32:2768–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Seclaman E, Balacescu L, Balacescu O, et al. MicroRNAs mediate liver transcriptome changes upon soy diet intervention in mice. J Cell Mol Med 2019;23:2263–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chou CH, Shrestha S, Yang CD, et al. miRTarBase update 2018: a resource for experimentally validated microRNA-target interactions. Nucleic Acids Res 2018;46:D296–d302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen TC, Tallo-Parra M, Cao QM, et al. Host-derived circular RNAs display proviral activities in hepatitis C virus-infected cells. PLoS Pathog 2020;16:e1008346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chu Q, Bai P, Zhu X, et al. Characteristics of plant circular RNAs. Brief Bioinform 2018;21:135–143. [DOI] [PubMed] [Google Scholar]
- 53. Sripada L, Singh K, Lipatova AV, et al. Hsa-miR-4485 regulates mitochondrial functions and inhibits the tumorigenicity of breast cancer cells. J Mol Med (Berl) 2017;95:641–51. [DOI] [PubMed] [Google Scholar]
- 54. Fitzpatrick C, Bendek MF, Briones M, et al. Mitochondrial ncRNA targeting induces cell cycle arrest and tumor growth inhibition of MDA-MB-231 breast cancer cells through reduction of key cell cycle progression factors. Cell Death Dis 2019;10:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Souza Silva GA, Silva SP, Costa MAS, et al. SARS-CoV, MERS-CoV and SARS-CoV-2 infections in pregnancy and fetal development. J Gynecol Obstet Hum Reprod 2020;101846 10.1016/j.jogoh.2020.101846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chan KH, Sridhar S, Zhang RR, et al. Factors affecting stability and infectivity of SARS-CoV-2. J Hosp Infect 2020;106:226–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Conn SJ, Pillman KA, Toubia J, et al. The RNA binding protein quaking regulates formation of circRNAs. Cell 2015;160:1125–34. [DOI] [PubMed] [Google Scholar]
- 58. Barrett SP, Salzman J. Circular RNAs: analysis, expression and potential functions. Development 2016;143:1838–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Park JW, Fu S, Huang B, et al. Alternative splicing in mesenchymal stem cell differentiation. Stem Cells 2020;38:1229–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang J, Liu K, Liu Y, et al. Evaluating the bias of circRNA predictions from total RNA-Seq data. Oncotarget 2017;8:110914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wu HY, Brian DA. Subgenomic messenger RNA amplification in coronaviruses. Proc Natl Acad Sci USA 2010;107:12257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sola I, Almazán F, Zúñiga S, et al. Continuous and discontinuous RNA synthesis in coronaviruses. Annu Rev Virol 2015;2:265–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fung TS, Liu DX. Human coronavirus: host-pathogen interaction. Annu Rev Microbiol 2019;73:529–57. [DOI] [PubMed] [Google Scholar]
- 64. Ungerleider NA, Jain V, Wang Y, et al. Comparative analysis of Gammaherpesvirus circular RNA repertoires: conserved and unique viral circular RNAs. J Virol 2019;93:e01952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Dearlove B, Lewitus E, Bai H, et al. A SARS-CoV-2 vaccine candidate would likely match all currently circulating variants. Proc Natl Acad Sci USA 2020;117:23652–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Jafarzadeh A, Chauhan P, Saha B, et al. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: lessons from SARS and MERS, and potential therapeutic interventions. Life Sci 2020;257:118102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Raj VS, Mou H, Smits SL, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013;495:251–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003;426:450–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020;181:271–280.e278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Choi HJ, Kim JY, Lim SC, et al. Dipeptidyl peptidase 4 promotes epithelial cell transformation and breast tumourigenesis via induction of PIN1 gene expression. Br J Pharmacol 2015;172:5096–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Li MY, Li L, Zhang Y, et al. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect Dis Poverty 2020;9:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Wang Y, Sun J, Zhu A, et al. Current understanding of middle east respiratory syndrome coronavirus infection in human and animal models. J Thorac Dis 2018;10:S2260–s2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Li C, Hu J, Hao J, et al. Competitive virus and host RNAs: the interplay of a hidden virus and host interaction. Protein Cell 2014;5:348–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Peng X, Gralinski L, Armour CD, et al. Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. mBio 2010;1:e00206-00210. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the RNA-sequencing data used in this study were publicly available at NCBI SRA database. All the identified viral circRNAs were publicly available at the VirusCircBase (http://www.computationalbiology.cn/ViruscircBase/home.html).