

Abstract
Using gene-regulatory-networks-based approach for single-cell expression profiles can reveal unprecedented details about the effects of external and internal factors. However, noise and batch effect in sparse single-cell expression profiles can hamper correct estimation of dependencies among genes and regulatory changes. Here, we devise a conceptually different method using graphwavelet filters for improving gene network (GWNet)-based analysis of the transcriptome. Our approach improved the performance of several gene network-inference methods. Most Importantly, GWNet improved consistency in the prediction of gene regulatory network using single-cell transcriptome even in the presence of batch effect. The consistency of predicted gene network enabled reliable estimates of changes in the influence of genes not highlighted by differential-expression analysis. Applying GWNet on the single-cell transcriptome profile of lung cells, revealed biologically relevant changes in the influence of pathways and master regulators due to ageing. Surprisingly, the regulatory influence of ageing on pneumocytes type II cells showed noticeable similarity with patterns due to the effect of novel coronavirus infection in human lung.
Keywords: single-cell, COVID, ageing lung, gene network
Introduction
Inferring gene regulatory networks and using them for system-level modelling is being widely used for understanding the regulatory mechanism involved in disease and development. The inter-dependencies among variables in the network is often represented as weighted edges between pairs of nodes, where edge weights could represent regulatory interactions among genes. Gene networks can be used for inferring causal models [1], designing and understanding perturbation experiments, comparative analysis [2] and drug discovery [3]. Due to wide applicability of network inference, many methods have been proposed to estimate inter-dependencies among nodes. Most of the methods are based on pairwise correlation, mutual information or other similarity metrics among gene expression values, provided in a different condition or time point. However, resulting edges are often influenced by indirect dependencies owing to low but effective background similarity in patterns. In many cases, even if there are some true interactions among a pair of nodes, its effect and strength are not estimated properly due to noise, background-pattern similarity and other indirect dependencies. Hence, recent methods have started using alternative approaches to infer more confident interactions. Such alternative approach could be based on partial correlations [4] or Algorithm for the Reconstruction of Accurate Cellular Networks’ (ARACNE’s) method of statistical threshold of mutual information [5].
Single-cell expression profiles often show heterogeneity in expression values even in a homogeneous cell population. Such heterogeneity can be exploited to infer regulatory networks among genes and identify dominant pathways in a cell type. However, due to the sparsity and ambiguity about the distribution of gene expression from single-cell RNA-seq profiles, the optimal measures of gene–gene interaction remain unclear. Hence, recently, Sknnider et al. [6] evaluated 17 measures of association to infer gene co-expression-based network. In their analysis, they found two measures of association, namely phi and rho as having the best performance in predicting co-expression-based gene–gene interaction using scRNA-seq profiles. In another study, Chen et al. [7] performed independent evaluation of a few methods proposed for gene network inference using scRNA-seq profiles such as SCENIC [8], SCODE [9], PIDC [10]. Chen et al. found that for single-cell transcriptome profiles either generated from experiments or simulations, these methods had a poor performance in reconstructing the network. Performance of such methods can be improved if gene expression profiles are denoised. Thus, the major challenge of handling noise and dropout in scRNA-seq profile is an open problem. The noise in single-cell expression profiles could be due to biological and technical reasons. The biological source of noise could include thermal fluctuations and a few stochastic processes involved in transcription and translation such as allele-specific expression [11] and irregular binding of transcription factors (TFs) to DNA, whereas technical noise could be due to amplification bias and stochastic detection due to low amount of RNA. Raser and O’Shea [12] used the term noise in gene expression as measured level of its variation among cells supposed to be identical. Raser and O’Shea categorized potential sources of variation in gene expression in four types: (1) the inherent stochasticity of biochemical processes due to small numbers of molecules, (2) heterogeneity among cells due to cell-cycle progression or a random process such as partitioning of mitochondria, (3) subtle micro-environmental differences within a tissue and (4) genetic mutation. Overall noise in gene expression profiles hinders in achieving reliable inference about the regulation of gene activity in a cell type. Thus, there is demand for pre-processing methods that can handle noise and sparsity in scRNA-seq profiles such that inference of regulation can be reliable.
The predicted gene network can be analyzed further to infer salient regulatory mechanisms in a cell type using methods borrowed from Graph theory. Calculating gene importance in term of centrality, finding communities and modules of genes are common downstream analysis procedures [2]. Just like gene expression profile, inferred gene network could also be used to find differences in two groups of cells(sample) [13] to reveal changes in the regulatory pattern caused due to disease, environmental exposure or ageing. In particular, a comparison of regulatory changes due to ageing has gained attention recently due to a high incidence of metabolic disorder- and infection-based mortality in the older population. Especially in the current situation of pandemics due to novel coronavirus (SARS-CoV-2), when older individuals have a higher risk of mortality, a question is haunting researchers. That question is: Why old lung cells have a higher risk of developing severity due to SARS-CoV-2 infection. However, understanding regulatory changes due to ageing using gene network inference with noisy single-cell scRNA-seq profiles of lung cells is not trivial. Thus there is a need of a noise and batch effect suppression method for investigation of the scRNA-seq profile of ageing lung cells [14] using a network biology approach.
Here we have developed a method to handle noise in gene expression profiles for improving gene network inference. Our method is based on graph wavelet-based filtering of gene expression. Our approach is not meant to overlap or compete with existing network inference methods but its purpose is to improve their performance. Hence, we compared other output of network inference methods with and without graph wavelet-based pre-processing. We have evaluated our approach using several bulk sample and single-cell expression profiles. We further investigated how our denoising approach influences the estimation of graph-theoretic properties of gene network. We also asked a crucial question: how the gene regulatory network differs between young and old individual lung cells. Further, we compared the pattern in changes in the influence of genes due to ageing with differential expression in COVID-infected lung.
Results
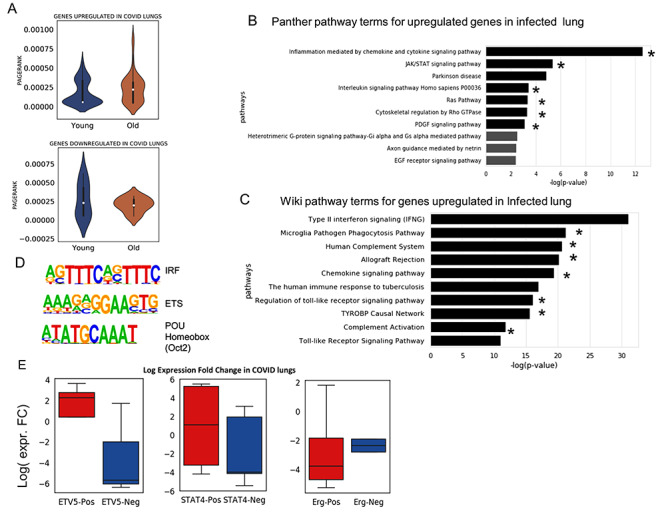
Our method uses a logic that cells (samples), which are similar to each other, would have a more similar expression profile for a gene. Hence, we first make a network such that two cells are connected by an edge if one of them is among the top K-nearest neighbours (KNN) of the other. After building KNN-based network among cells (samples), we use graph wavelet-based approach to filter expression of one gene at a time (see Figure 1). For a gene, we use its expression as a signal on the nodes of the graph of cells. We apply a graph wavelet transform to perform spectral decomposition of graph signal. After graph wavelet transformation, we choose the threshold for wavelet coefficients using SureShrink and BayesShrink or a default percentile value determined after thorough testing on multiple data sets. We use the retained values of the coefficient for inverse graph wavelet transformation to reconstruct a filtered expression matrix of the gene. The filtered gene expression is used for gene network inference and other down-stream process of analysis of regulatory differences. For evaluation purpose, we have calculated inter-dependencies among genes using five different co-expression measurements, namely Pearson and spearman correlations, 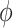 and
and 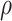 scores and ARACNE.
scores and ARACNE.
Figure 1.
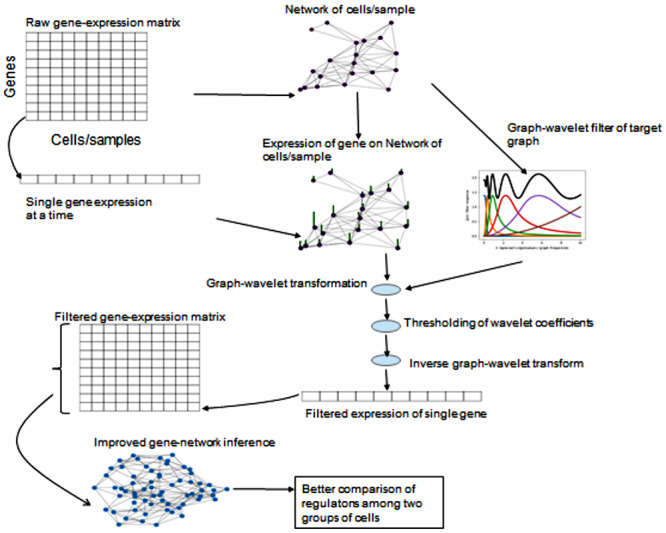
The flowchart of GWNet pipeline. First, a KNN-based network is made between samples/cell. A filter for graph wavelet is learned for the KNN-based network of samples/cells. Gene expression of one gene at a time is filtered using graph wavelet transform. Filtered gene expression data are used for network inference. The inferred network is used to calculate centrality and differential centrality among groups of cells.
Evaluation using bulk expression profiles from DREAM challenge
The biological and technical noise can both exist in a bulk sample expression profile ([12]). In order to test the hypothesis that graph-based denoising could improve gene network inference, we first evaluated the performance of our method on bulk expression data set. We used 4 data sets made available by DREAM5 challenge consortium [15]. Three data sets were based on the original expression profile of bacterium Escherichia coli and the single-celled eukaryotes Saccharomyces cerevisiae and S aureus. While the fourth data set was simulated using in silico network with the help of GeneNetWeaver, which models molecular noise in transcription and translation using chemical Langevin equation [16]. The true positive interactions for all the four data sets are also available. We compared graph Fourier-based low pass filtering with graph wavelet-based denoising using three different approaches to threshold the wavelet coefficients. We achieved 5–25% improvement in score over raw data based on DREAM5 criteria [15] with correlation, ARACNE- and rho-based network prediction. With 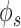 -based gene network prediction, there was an improvement in three of four DREAM5 data sets (Figure 2A). All the 5 network inference methods showed improvement after graph wavelet-based denoising of simulated data (in silico) from DREAM5 consortium (Figure 2A). Moreover, graph wavelet-based filtering had better performance than Chebyshev filter-based low pass filtering in graph Fourier domain. It highlights the fact that even bulk sample data of gene expression can have noise and denoising it with graph wavelet after making KNN-based graph among samples has the potential to improve gene network inference. Moreover, it also highlights another fact, well known in the signal processing field, that wavelet-based filtering is more adaptive than low pass filtering.
-based gene network prediction, there was an improvement in three of four DREAM5 data sets (Figure 2A). All the 5 network inference methods showed improvement after graph wavelet-based denoising of simulated data (in silico) from DREAM5 consortium (Figure 2A). Moreover, graph wavelet-based filtering had better performance than Chebyshev filter-based low pass filtering in graph Fourier domain. It highlights the fact that even bulk sample data of gene expression can have noise and denoising it with graph wavelet after making KNN-based graph among samples has the potential to improve gene network inference. Moreover, it also highlights another fact, well known in the signal processing field, that wavelet-based filtering is more adaptive than low pass filtering.
Figure 2.
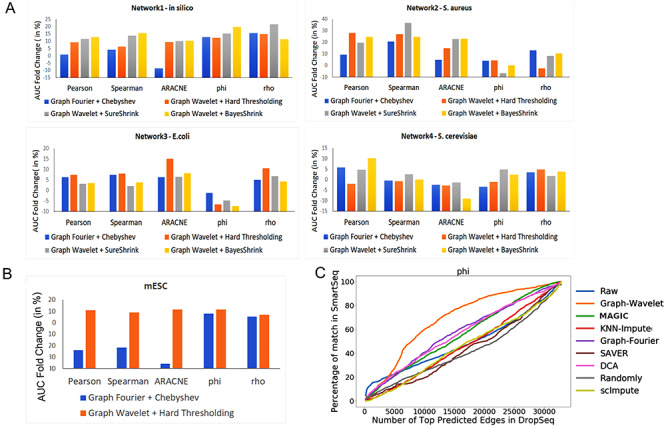
Improvement in gene network inference by graph wavelet-based denoising of gene expression (A) Performance of network inference methods using bulk gene expression data sets of DREAM5 challenge. Three different ways of shrinkage of graph wavelet coefficients were compared with graph-Fourier-based low pass filtering. The Y-axis shows fold change in area under curve (AUC) for receiver operating characteristic curve for overlap of predicted network with golden set of interactions. For hard-threshold, the default value of 70% percentile was used. (B) Performance evaluation using single-cell RNA-seq (scRNA-seq) of mESCs-based network inference after filtering the gene expression. The gold set of interactions was adapted from [19] (C) Comparison of graph-wavelet-based denoising with other related smoothing and imputing methods in terms of consistency in the prediction of the gene interaction network. Here, Phi (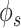 ) score was used to predict network among genes. For results based on other types of scores see Supplementary Figure S1. Predicted networks from two scRNA-seq profile of mESC were compared to check robustness towards the batch effect.
) score was used to predict network among genes. For results based on other types of scores see Supplementary Figure S1. Predicted networks from two scRNA-seq profile of mESC were compared to check robustness towards the batch effect.
Graph wavelet-based denoising of single-cell expression profiles improves gene networks inference
In comparison to bulk samples, there is a higher level of noise and dropout in single-cell expression profiles. Dropouts are caused by non-detection of true expression due to technical issues. Using low pass filtering after graph Fourier transform seems to be an obvious choice as it fills in a background signal at missing values and suppresses high-frequency outlier signal [17]. However, in the absence of information about cell type and cell states, a blind smoothing of a signal may not prove to be fruitful. Hence, we applied graph wavelet-based filtering for processing gene expression data set from the scRNA-seq profile. We first used scRNA-seq data set of mouse embryonic stem cells (mESCs) [18]. In order to evaluate network inference in an unbiased manner, we used gene regulatory interactions compiled by another research group [19]. Our approach of graph wavelet-based pre-processing of mESC scRNA-seq data set improved the performance of gene network inference methods by 8–10% (Figure 2B). However, most often, the gold set of interaction used for evaluation of gene network inference is incomplete, which hinders the true assessment of improvement. Hence, we also used another approach to validate our method. For this purpose, we used a measure of overlap among network inferred from two scRNA-seq data sets of the same cell type but having different technical biases and batch effects. If the inferred networks from both data sets are closer to true gene interaction model, they will show high overlap. For this purpose, we used two scRNA-seq data set of mESC generated using two different protocols (SMARTseq and Drop-seq). For comparison of consistency and performance, we also used a few other imputation and denoising methods proposed to filter and predict the missing expression values in scRNA-seq profiles. We evaluated seven other such methods; graph Fourier-based filtering [17], MAGIC [20], scImpute [21], DCA [22], SAVER [23], Randomly [24], and KNN-impute [25]. Graph wavelet-based denoising provided better improvement in AUC for overlap of predicted network with known interaction than other seven methods meant for imputing and filtering scRNA-seq profiles (Supplementary Figure S1A). Similarly in comparison to graph wavelet-based denoising, the other seven methods did not provided substantial improvement in AUC for overlap among gene network inferred by two data sets of mESC (Figure 2C, Supplementary Figure S1B). However, graph wavelet-based filtering improved the overlap between networks inferred from different batches of scRNA-seq profile of mESC even if they were denoised separately (Figure 2C, Supplementary Figure S1B). With 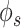 -based edge scores the overlap among predicted gene network increased by 80% due to graph wavelet-based denoising (Figure 2C). The improvement in overlap among networks inferred from two batches hints that graph wavelet denoising is different from imputation methods and has the potential to substantially improve gene network inference using their expression profiles.
-based edge scores the overlap among predicted gene network increased by 80% due to graph wavelet-based denoising (Figure 2C). The improvement in overlap among networks inferred from two batches hints that graph wavelet denoising is different from imputation methods and has the potential to substantially improve gene network inference using their expression profiles.
Improved gene network inference from single-cell profile reveal age-based regulatory differences
Improvement in overlap among inferred gene networks from two expression data sets for a cell type also hints that after denoising predicted networks are closer to true gene interaction profiles. Hence using our denoising approach before estimating the difference in inferred gene networks due to age or external stimuli could reflect true changes in the regulatory pattern. Such a notion inspired us to compare gene networks inferred for young and old pancreatic cells using their scRNA-seq profile filtered by our tool [26]. Martin et al. [26] defined three age groups, namely juvenile (1 month–6 years), young adult (21–22 years) and aged (38–54 years). We applied graph wavelet-based denoising of pancreatic cells from three different groups separately. In other words, we did not mix cells from different age groups while denoising. Graph wavelet-based denoising of a single-cell profile of pancreatic cells caused better performance in terms of overlap with protein-protein interaction (PPI; Figure 3A, Supplementary Figure S2A). Even though like Chen et al. [7], we have used PPI to measure improvement in gene network inference, it may not be reflective of all gene interactions. Hence, we also used the criteria of increase in overlap among predicted networks for same cell types to evaluate our method for scRNA-seq profiles of pancreatic cells. Denoising scRNA-seq profiles also increased overlap between inferred gene network among pancreatic cells of the old and young individuals (Figure 3B, Supplementary Figure S2B). We performed quantile normalization of original and denoised expression matrix taking all three age groups together to bring them on the same scale to calculate the variance of expression across cells of every gene. The old and young pancreatic alpha cells had a higher level of median variance of expression of genes than juvenile. However, after graph wavelet-based denoising, the variance level of genes across all the three age groups became almost equal and had similar median value (Figure 3C). Notice that, it is not trivial to estimate the fraction of variances due to transcriptional or technical noise. Nonetheless, graph wavelet-based denoising seemed to have reduced the noise level in single-cell expression profiles of old and young adults. Differential centrality in the co-expression network has been used to study changes in the influence of genes. However, noise in single-cell expression profiles can cause spurious differences in centrality. Hence we visualized the differential degree of genes in network inferred using young and old cells scRNA-seq profiles. The networks inferred from non-filtered expression had a much higher number of non-zero differential degree values in comparison to the denoised version (Figure 3D, Supplementary Figure S2C). Thus, denoising seems to reduce differences among centrality, which could be due to randomness of noise. Next, we analyzed the properties of genes whose variance dropped most due to graph wavelet-based denoising. Surprisingly, we found that top 500 genes with the highest drop in variance due to denoising in old pancreatic beta cells were significantly associated with diabetes Mellitus and hyperinsulinism, whereas top 500 genes with the highest drop in variance in young pancreatic beta cells had no or insignificant association with diabetes (Figure 3E). A similar trend was observed with pancreatic alpha cells (Supplementary Figure S2D). Such a result hints that ageing causes increase in stochasticity of the expression level of genes associated with pancreas function and denoising could help in properly elucidating their dependencies with other genes.
Figure 3.
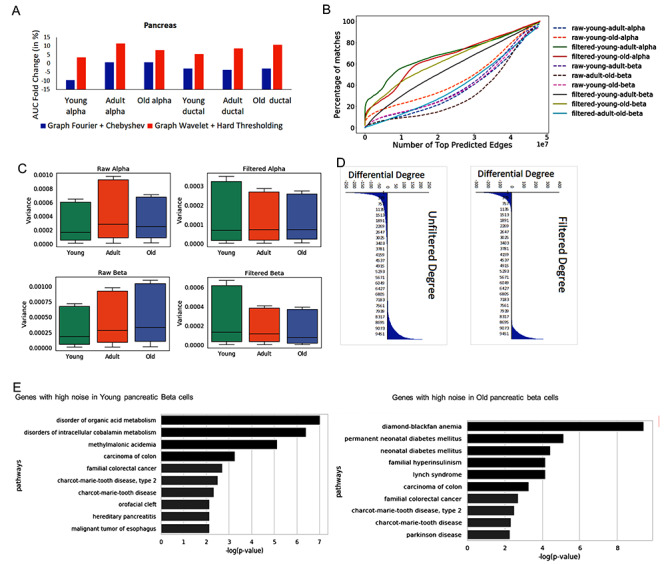
Performance and analysis of noise for single-cell RNA-seq profile of pancreatic cells. (A) Performance based on overlap of predicted network with PPI data set. (B) Evaluation of consistency of predicted network. For comparing two networks, it is important to reduce differences due to noise. Hence, the plot here shows similarity of predicted networks before and after graph wavelet-based denoising. The result shown here are for correlation-based co-expression network, whereas similar results are shown using 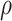 score in Supplementary Figure S2. (C) Variances of expression of genes across single cells before and after denoising (filtering) are shown here. Variances of genes in a cell type were calculated separately for three different stages of ageing (young, adult and old). The variance (estimate of noise) is higher in older alpha and beta cells compared with young. However, after denoising variance of genes in all ageing stage becomes equal (D) Effect of noise in estimated differential centrality is shown is here. The difference in the degree of genes in network estimated for old and young pancreatic beta cells is shown here. The number of non-zero differential-degree estimated using denoised expression is lower than unfiltered expression-based networks.(E) Enriched panther pathway terms for top 500 genes with the highest drop in variance after denoising in old and young pancreatic beta cells.
score in Supplementary Figure S2. (C) Variances of expression of genes across single cells before and after denoising (filtering) are shown here. Variances of genes in a cell type were calculated separately for three different stages of ageing (young, adult and old). The variance (estimate of noise) is higher in older alpha and beta cells compared with young. However, after denoising variance of genes in all ageing stage becomes equal (D) Effect of noise in estimated differential centrality is shown is here. The difference in the degree of genes in network estimated for old and young pancreatic beta cells is shown here. The number of non-zero differential-degree estimated using denoised expression is lower than unfiltered expression-based networks.(E) Enriched panther pathway terms for top 500 genes with the highest drop in variance after denoising in old and young pancreatic beta cells.
Improvement in Gene network inference for studying regulatory differences among young and old lung cells
Studying cell-type-specific changes in regulatory networks due to ageing have the potential to provide better insight about predisposition for disease in the older population. Hence, we inferred gene network for different cell types using scRNA-seq profiles of young and old mouse lung cells published by Kimmel et al. [14]. The lower lung epithelia where a few viruses seem to have the most deteriorating effect consists of multiple types of cells such as bronchial epithelial and alveolar epithelial cells, fibroblast, alveolar macrophages, endothelial and other immune cells. The alveolar epithelial cells also called as pneumocytes are of two major types. The type 1 alveolar (AT1) epithelial cells for major gas exchange surface of lung alveolus have an important role in the permeability barrier function of the alveolar membrane. Type 2 alveolar cells (AT2) are the progenitors of type 1 cells and have the crucial role of surfactant production. AT2 ( or pneumocytes type II) cells are a prime target of many viruses; hence, it is important to understand the regulatory patterns in AT2 cells, especially in the context of ageing.
We applied our method of denoising on scRNA-seq profiles of cells derived from old and young mice lung [14]. Graph wavelet-based denoising lead to an increase in consistency among inferred gene network for young and old mice lung for multiple cell types (Figure 4A). Graph wavelet-based denoising also lead to an increase in consistency in predicted gene network from data sets published by two different groups (Figure 4B). The increase in overlap of gene networks predicted for old and young cells scRNA-seq profile, despite being denoised separately, hints about a higher likelihood of predicting true interactions. Hence, the chances of finding gene network-based differences among old and young cells were less likely to be dominated by noise. We studied ageing-related changes in PageRank centrality of nodes(genes). Since PageRank centrality provides a measure of ‘popularity’ of nodes, studying its change has the potential to highlight the change in the influence of genes. First, we calculated differential PageRank of genes among young and old AT2 cells (Supporting File 1) and performed gene set enrichment analysis using Enrichr [27]. The top 500 genes with higher PageRank in young AT2 cells had enriched terms related to integrin signalling, 5HT2 type receptor-mediated signalling, H1 histamine receptor-mediated signalling pathway, vascular endothelial growth factor (VEGF), cytoskeleton regulation by Rho GTPase and thyrotropin activating receptor signalling (Figure 4C). We ignored oxytocin and thyrotropin-activating hormone receptor-mediated signalling pathways as an artefact as the expression of oxytocin and thyrotropin-releasing hormone (TRH) receptors in AT2 cells was low. Moreover, genes appearing for the terms ‘oxytocin receptor-mediated signalling’ and ‘thyrotropin activating hormone-mediated signalling’ were also present in gene set for 5Ht2 type receptor-mediated signalling pathway. We found literature support for activity in AT2 cells for most of the enriched pathways. However, there were very few studies that showed their differential importance in old and young cells, such as Bayer et al. demonstrated mRNA expression of several 5-HTR including 5-Ht2, 5Ht3 and 5Ht4 in alveolar epithelial cells type II (AT2) cells and their role in calcium ion mobilization. Similarly, Chen et al. [28] showed that histamine 1 receptor antagonist reduced pulmonary surfactant secretion from adult rat alveolar AT2 cells in primary culture. VEGF pathway is active in AT2 cells, and it is known that ageing has an effect on VEGF mediated angiogenesis in lung. Moreover, VEGF-based angiogenesis is known to decline with age [29]. We further performed gene-set enrichment analysis for genes with increased PageRank in older mice AT2 cells. For top 500 genes with higher PageRank in old AT2 cells, the terms that appeared among 10 most enriched in both Kimmel et al. and Angelids et al. data sets were T cell activation, B cell activation, cholesterol biosynthesis and FGF signalling pathway, angiogenesis and cytoskeletal regulation by Rho GTPase (Figure 4D). Thus, there was 60% overlap in results from Kimmel et al. and Angelids et al. data sets in terms of the enrichment of pathway terms for genes with higher PageRank in older AT2 cells (Supplementary Figure S3A, Supporting Files 2 and 3). Overall in our analysis, inflammatory response genes showed higher importance in older AT2 cells. The increase in the importance of cholesterol biosynthesis genes hand in hand with higher inflammatory response points towards the influence of ageing on the quality of pulmonary surfactants released by AT2. Al Saedy et al. [30] recently showed that high level of cholesterol amplifies defects in surface activity caused by oxidation of pulmonary surfactant.
Figure 4.
Improved regulatory inferences with graph wavelet-based pre-processing of the single-cell transcriptome of ageing lung cells (A) Consistency of prediction of the networks using the scRNA-seq profile (Kimmel et al. data set) of young and old lung cells. The coverage of top 10 000 edges in young cells in network inferred for old cells is shown here. For the same type of cells, the predicted networks for old and young cells seem to have higher overlap after graph wavelet-based filtering. The label ‘Raw’ here means that both networks (for old and young) were inferred using unfiltered scRNA-seq profiles. Although the same result from denoised scRNA-seq profile is shown as filtered, networks were inferred using correlation-based co-expression. (B) Plot showing the overlap of networks predicted from two different data sets with their own batch effect. X-axis shows the number of predicted edges in network predicted using Angelidis et al. data set (GEO Id: GSE124872). Y-axis shows the fraction of top 10 000 edges in network estimated using Kimmel et al. data set. (C) A total of 10 most enriched panther pathway terms for top 500 genes with higher PageRank in young AT2 cells compared with old. (D) A total of 10 most enriched panther pathway terms for top 1000 genes with higher PageRank for old AT2 cells compared with young. (E) Scatter plot of differential degree and PageRank (old–young) of TF estimated using networks predicted for old and young AT2 cells from Kimmel et al. data set. Only TFs with a non-zero differential degree are shown.
We also performed Enrichr-based analysis of differentially expressed genes in old AT2 cells (Supporting File 4). For genes upregulated in old AT2 cells compared with young, terms that reappeared were cholesterol biosynthesis, T cell and B cell activation pathways, Angiogenesis and Inflammation mediated by chemokine and cytokine signalling, whereas few terms like RAS pathway, JAK/STAT signalling and cytoskeletal signalling by Rho GTPase did not appear as enriched for genes upregulated in old AT2 cells (Figure 3B, Supporting File 4). However, it has been previously shown that the increase in age changes the balance of pulmonary renin-angiotensin system (RAS), which is correlated with aggravated inflammation and more lung injury [31]. JAK/STAT pathway is known to be involved in the oxidative-stress induced decrease in the expression of surfactant protein genes in AT2 cells [32]. Overall, these results indicate that even though the expression of genes involved in relevant pathways may not show significant differences due to ageing, but their regulatory influence could be changing substantially.
In order to further gain insight, we analyzed the changes in the importance of TFs in ageing AT2 cells. Among top 500 genes with higher PageRank in old AT2 cells, we found several relevant TFs. However, to make a stringent list, we considered only those TFs, which had non-zero value for change in degree among gene network for old and young AT2 cells. Overall, with Kimmel et al. data set, we found 46 TFs with a change in PageRank and degree (Supplementary Table 1) due to ageing for AT2 cells (Figure 4E). The changes in centrality (PageRank and degree) of TFs with ageing was coherent with pathway enrichment results. Such as ETV5, which has higher degree and PageRank in older cells, is known to be stabilized by RAS signalling in AT2 cells [33]. In the absence of Etv5, AT2 cell differentiates to AT1 cells [33]. Another TF Jun (c-jun) having stronger influence in old AT2 cells is known to regulate inflammation lung alveolar cells [34]. We also found Jun to be having co-expression with Jund and Etv5 in old AT2 cell (Supplementary Figure S4). Jund whose influence seems to increase in aged AT2 cells is known to be involved in cytokine-mediated inflammation. Among the TFs Stat 1-4, which are involved in JAK/STAT signalling, Stat4 showed higher degree and PageRank in old AT2. Androgen receptor(AR) also seem to have a higher influence in older AT2 cells (Figure 4E). AR has been shown to be expressed in AT2 cells [35].
We further performed a similar analysis for the scRNA-seq profile of interstitial macrophages(IMs) in lungs and found literature support for the activity of enriched pathways (Supporting File 5), whereas gene set enrichment output for important genes in older IMs had some similarity with results from AT2 cells as both seem to have higher pro-inflammatory response pathway such as T cell activation and JAK/STAT signalling. However, unlike AT2 cells, ageing in IMs seem to cause an increase in glycolysis and pentose phosphate pathway. Higher glycolysis and pentose phosphate pathway activity levels have been previously reported to be involved in the pro-inflammatory response in macrophages by Viola et al. [36]. In our results, RAS pathway was not enriched significantly for genes with a higher importance in older macrophages. Such results show that the pro-inflammatory pathways activated due to ageing could vary among different cell types in lung.
Comparison of the effect of ageing with COVID-19 infection in lung
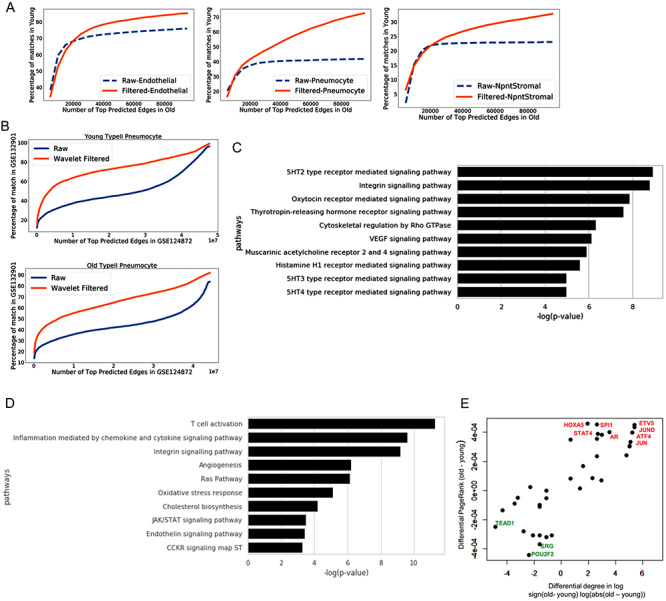
In current pandemic due to SARS-CoV-2, a trend has emerged that older individuals have a higher risk of developing severity and lung fibrosis than the younger population. Since our analysis revealed changes in the influence of genes in lung cells due to ageing, we compared our results with expression profiles of lung infected with SARS-CoV-2 published by Blanco-Melo et al. [37]. Recently, it has been shown that AT2 cells predominantly express ACE2, the host cell surface receptor for SARS-CoV-2 attachment and infection[38]. Thus, COVID infection could have most of the dominant effect on AT2 cells. We found that genes with significant upregulation in SARS-CoV-2-infected lung also had higher PageRank in gene network inferred for older AT2 cells (Figure 5A). We also repeated the process of network inference and calculating differential centrality among old and young using all types of cells in the lung together (Supporting File 6). We performed gene set enrichment for genes upregulated in SARS-CoV-2-infected lung. Majority of the seven panther pathway terms enriched for genes upregulated in SARS-CoV-2-infected lung also had enrichment for genes with higher PageRank in old lung cells (combined). Total six out seven significantly enriched panther pathways for genes upregulated in COVID-19-infected lung were also enriched for genes with higher PageRank in older AT2 cells in either of the two data sets used here (five in Angelids et al.and three in Kimmel et al. data-based results).
Figure 5.
Analysis of gene expression profile in lungs infected with SARS-CoV-2 (COVID). (A) The distribution of PageRank of genes upregulated in COVID-infected lung false detection rate (FDR < 0.05) [37]. The PageRank is shown for network estimated using the scRNA-seq profile of young and old AT2 cells. On similar pattern PageRank is shown for genes downregulated FDR < 0.05 in COVID-infected lung. (B) Top 10 panther pathway enriched for genes upregulated in COVID-infected lung. The terms with * sign also have significant enrichment for genes with higher PageRank in old AT2 cells (C) Top 10 Wiki pathway terms enriched for genes upregulated in COVID-infected lung. The terms with * are also enriched p-value < 0.05 for genes with higher PageRank in old AT2 cells. (D) Top 3 motifs of known TF enriched in promoters of genes upregulated in COVID-infected lung. (E) Fold change of expression in the lung with COVID infection for genes positively and negatively correlated with TFs in old AT2 cells. The results are shown for 2 TFs Etv5 and Stat4, which has higher PageRank in old AT2 cells. As a control, the results are also shown for Erg, which have higher PageRank in young AT2 cells. Most of the genes, which had a positive correlation with Etv5 and Stat4 expression in old murine AT2 cells, were upregulated in COVID-infected lung, whereas for Erg the trend is the opposite. Genes positively correlated with ERG genes in old AT2 had more downregulation than genes with negative correlation. Such results hint that TFs whose influence (PageRank) increase during ageing could be involved activating or poising the genes upregulated in COVID infection.
Among the top 10 enriched Wiki pathway terms for genes upregulated in COVID-infected lung, 7 has significant enrichment for genes with higher PageRank in old AT2 cells (Supporting File 7). However, the term type-II interferon signalling did not have significant enrichment for genes with higher PageRank in old AT2 cells. We further investigated enriched motifs of TFs in promoters of genes upregulated in COVID-infected lungs (Supplementary Methods). For promoters of genes upregulated in COVID-infected lung top two enriched motifs belonged to interferon regulatory factor and erythroblast transformation specific (ETS) family TFs. Notice that, Etv5 belong to sub-family of ETS groups of TFs. Further analysis also revealed that most of the genes whose expression is positively correlated with Etv5 in old AT2 cells are upregulated in COVID-infected lung. In contrast, genes with negative correlation with Etv5 in old AT2 cells were mostly downregulated in COVID-infected lung. A similar trend was found for Stat4 gene. However, for Erg gene with higher PageRank in young AT2 cell, the trend was the opposite. In comparison to genes with negative correlation, positively correlated genes with Erg in old AT2 cell, had more downregulation in COVID-infected lung. Such trend shows that a few TFs like Etv5, Stat4 with higher PageRank in old AT2 cells could be having a role in poising or activation of genes, which gain higher expression level on COVID infection.
Discussion
Inferring regulatory changes in pure primary cells due to ageing and other conditions, using single-cell expression profiles has tremendous potential for various applications. Such applications could be understanding the cause of development of a disorder or revealing signalling pathways and master regulators as potential drug targets. Hence, to support such studies, we developed GWNet to assist biologists in workflow for graph theory-based analysis of single-cell transcriptome. GWNet improves inference of regulatory interaction among genes using graph wavelet-based approach to reduce noise due to technical issues or cellular biochemical stochasticity in gene expression profiles. We demonstrated the improvement in gene network inference using our filtering approach with four benchmark data sets from DREAM5 consortium and several single-cell expression profiles. Using five different ways for inferring network, we showed how our approach for filtering gene expression can help gene network inference methods. Our results of comparison with other imputation, smoothing methods and graph Fourier-based filtering showed that graph wavelet is more adaptive to changes in the expression level of genes with changing neighbourhood of cells. Thus, graph wavelet-based denoising is a conceptually different approach for pre-processing of gene expression profiles. There is a huge body of literature on inferring gene networks from bulk gene expression profile and utilizing it to find differences among two groups of samples. However, applying classical procedures on single-cell transcriptome profiles has not proved to be effective. Our method seems to resolve this issue by increasing consistency and overlap among gene networks inferred using an expression from different sources (batches) for the same cell type even if each data set was filtered independently. Such an increase in overlap among predicted network from independently processed data sets from different sources hints that estimated dependencies among genes reach closer to true values after graph wavelet-based denoising of expression profiles. Having network prediction closer to true values increases the reliability of comparison of a regulatory pattern among two groups of cells. Moreover, recently Chow and Chen [39] have shown that age-associated genes identified using bulk expression profiles of the lung are enriched among those induced or suppressed by SARS-CoV-2 infection. However, they did not perform analysis with systems-level approach. Our analysis highlighted RAS and JAK/STAT pathways to be enriched for genes with stronger influence in old AT2 cells and genes upregulated in COVID-infected lung. RAS/MAPK signalling is considered essential for self-renewal of AT2 cell [33]. Similarly, JAK/STAT pathway is known to be activated in the lung during injury [40] and influence surfactant quality[32]. We have used murine ageing-lung scRNA-seq profiles; however, our analysis provides an important insight that regulatory patterns and master regulators in old AT2 cells are in such a configuration that they could be predisposing it for a higher level of RAS and JAK/STAT signalling. AR, which has been implicated in male pattern baldness and increased risk of males towards COVID infection [41], had higher PageRank and degree in old AT2 cells. However, further investigation is needed to associate AR with severity on COVID infection due to ageing. On the other hand, in young AT2 cells, we find a high influence of genes involved in Histamine H1 receptor-mediated signalling, which is known to regulate allergic reactions in lungs [42]. Another benefit of our approach of analysis is that it can highlight a few specific targets of further study for therapeutics. Such as a kinase that binds and phosphorylates c-Jun called as JNK is being tested in clinical trials for pulmonary fibrosis [43]. Androgen deprivation therapy has shown to provide partial protection against SARS-CoV-2 infection [44]. On the same trend, our analysis hints that Etv5 could also be considered as drug target to reduce the effect of ageing induced RAS pathway activity in the lung.
Methods
We used the term noise in gene expression according to its definition by several researchers such as Raser and O’Shea [12]; as the measured level of variation in gene expression among cells supposed to be identical. Hence, we first made a base graph (networks) where supposedly identical cells are connected by edges. For every gene, we use this base graph and apply graph wavelet transform to get an estimate of variation of its expression in every sample (cells) with respect to other connected samples at different levels of graph-spectral resolution. For this purpose, we first calculated distances among samples (cells). To get a better estimate of distances among samples (cells), one can perform dimension reduction of the expression matrix using tSNE [45] or principal component analysis. We considered every sample (cell) as a node in the graph and connected two nodes with an edge only when one of them was among KNNs of the other. Here we decide the value of K in the range of 10–50 based on the number of samples(cells) in the expression data sets. Thus, we calculated the preliminary adjacency matrix using KNNbased on euclidean distance metric between samples of the expression matrix. We used this adjacency matrix to build a base graph. Thus, each vertex in the base-graph corresponds to each sample and edge weights to the euclidean distance between them.
The weighted graph 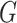 built using KNN-based adjacency matrix comprises of a finite set of vertices
built using KNN-based adjacency matrix comprises of a finite set of vertices 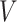 , which corresponds to cells (samples), a set of edges
, which corresponds to cells (samples), a set of edges 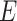 denoting connection between samples (if exist) and a weight function, which gives non-negative weighted connections between cells (samples). This weighted matrix can also be defined as a
denoting connection between samples (if exist) and a weight function, which gives non-negative weighted connections between cells (samples). This weighted matrix can also be defined as a 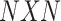 (
(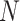 being number of cells) weighted adjacency matrix
being number of cells) weighted adjacency matrix 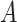 where
where 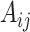 is
is 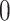 if there is no edge between cells
if there is no edge between cells 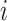 and
and 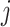 , otherwise
, otherwise 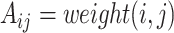 if there exist an edge between
if there exist an edge between 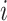 and
and 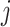 . The degree of a cell in the graph is the sum of weights of edges incident on that cell. Also, diagonal degree matrix D of this graph comprises of degree
. The degree of a cell in the graph is the sum of weights of edges incident on that cell. Also, diagonal degree matrix D of this graph comprises of degree 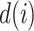 if
if 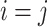 ,
, 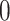 otherwise. A non-normalized graph Laplacian operator
otherwise. A non-normalized graph Laplacian operator 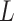 for a graph is defined as
for a graph is defined as 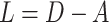 . The normalized form of graph Laplacian operator is defined as:
. The normalized form of graph Laplacian operator is defined as:
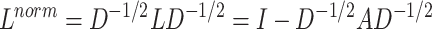 |
Both Laplacian operators produce different eigenvectors [46]. However, we have used a normalized form of Laplacian operator for the graph between cells. The graph Laplacian is further used for graph Fourier transformation of signals on nodes (see Supplementary Methods) ([47] [46]).
For filtering in the Fourier domain, we used Chebyshev filter for gene expression profile. We took the expression of each gene at a time considering it as a signal and projected it onto the raw graph (where each vertex corresponds to each sample) object [17]. We took forward Fourier transform of signal and filtered the signal using Chebyshev filter in the Fourier domain and then inverse transformed the signal to calculate filtered expression. This same procedure was repeated for every gene. This would finally give us filtered gene expression.
Spectral graph wavelet transform
Spectral graph wavelet entails choosing a non-negative real-valued kernel function, which can behave as a bandpass filter and is similar to Fourier transform. The re-scaled kernel function of graph Laplacian gives wavelet operator, which eventually produce graph wavelet coefficients at each scale. However, using continuous functional calculus, one can define a function of self-adjoint operator on the basis of spectral representation of graph. Although for a graph with finite dimensional Laplacian, this can be achieved by eigenvalues and eigenvectors of Laplacian 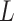 [47]. The wavelet operator is given by
[47]. The wavelet operator is given by 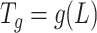 .
. 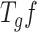 gives wavelet coefficients for a signal
gives wavelet coefficients for a signal 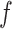 at scale = 1. This operator operates on eigenvectors
at scale = 1. This operator operates on eigenvectors 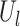 as
as 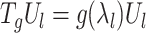 . Hence, for any graph signal, operator
. Hence, for any graph signal, operator 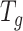 operates on the signal by adjusting each graph Fourier coefficient as
operates on the signal by adjusting each graph Fourier coefficient as
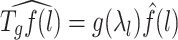 |
and inverse Fourier transform given as
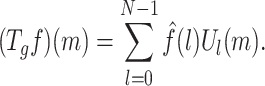 |
The wavelet operator at every scale 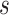 is given as
is given as 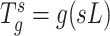 . These wavelet operators are localized to obtain individual wavelets by applying them to
. These wavelet operators are localized to obtain individual wavelets by applying them to 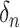 , with
, with 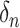 being a signal with
being a signal with 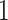 on vertex
on vertex 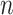 and 0 otherwise [47]. Thus considering coefficients at every scale, the inverse transform can be obtained as
and 0 otherwise [47]. Thus considering coefficients at every scale, the inverse transform can be obtained as
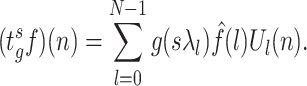 |
Here, in spite of filtering in Fourier domain, we took wavelet coefficients of each gene expression signal at different scales. Thresholding was applied on each scale to filter wavelet coefficients. We applied both hard- and soft-thresholding on wavelet coefficients. For soft-thresholding, we implemented well-known methods Sure Shrink and Bayes Shrink.
Choosing threshold for graph wavelet coefficients
Finding an optimal threshold for wavelet coefficients for denoising linear-signals and images has remained a subject of intensive research. We evaluated both soft- and hard-thresholding approaches and tested an information-theoretic criterion known as the minimum description length (MDL) principle. Using our tool GWNet, user can choose from multiple options of finding threshold such as VisuShrink, SureShrink and MDL. Here, we have used hard-thresholding for most the data sets as proper soft-thresholding of Graph wavelet coefficient is itself a topic of intensive research and may need further fine-tuning. One can also use hard-threshold value based on the best overlap among predicted gene network and PPI. While applying it on multiple data sets, we realized that threshold cut-offs estimated by MDL criteria and best overlap of predicted network with known interaction and PPI were in the range of 60–70 percentile. For comparing predicted network from multiple data sets, we needed uniform percentile cut-off to threshold graph wavelet coefficients. Hence for uniform analysis of several data sets, we have set the default threshold value of 70 percentile. Hence in default mode, wavelet coefficient with absolute value less than 70 percentile was made equal to zero.
Methods used to infer network among genes
GWNet tool is flexible, and any network inferences method can be plugged in it for making regulatory inferences using a graph-theoretic approach. Here, for single-cell RNA-seq data, we have used gene expression values in the form of FPKM (fragments per kilobase of exon model per million reads mapped). We pre-processed single-cell gene expression by quantile normalization and log transformation. To start with, we used spearman and Pearson correlation to achieve a simple estimate of the measure of inter-dependencies among genes. We also used ARACNE to infer network among genes. ARACNE first computes mutual information for each gene-pair. Then it considers all possible triplet of genes and applies the data processing inequality (DPI) to remove indirect interactions. According to DPI, if gene 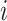 and gene
and gene 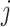 do not interact directly with each other but show dependency via gene
do not interact directly with each other but show dependency via gene 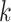 , the following inequality hold
, the following inequality hold
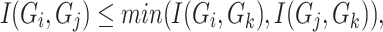 |
where 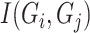 represents mutual information between gene
represents mutual information between gene 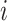 and gene
and gene 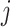 . ARACNE also removes interaction with mutual information less than a particular threshold eps. We have used eps equal to 0.2. Recently Skinnider et al., [6] showed superiority of two measures of proportionality
. ARACNE also removes interaction with mutual information less than a particular threshold eps. We have used eps equal to 0.2. Recently Skinnider et al., [6] showed superiority of two measures of proportionality 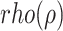 and
and 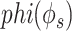 [48] for estimating gene co-expression network using single-cell transcriptome profile. Hence, we also evaluated the benefit of graph wavelet-based denoising of gene expression with measures of proportionality
[48] for estimating gene co-expression network using single-cell transcriptome profile. Hence, we also evaluated the benefit of graph wavelet-based denoising of gene expression with measures of proportionality 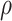 and
and 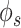 . The measures of proportionality
. The measures of proportionality 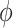 can be defined as
can be defined as
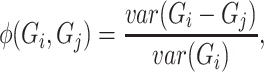 |
where 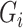 is the vector containing log values of expression of a gene
is the vector containing log values of expression of a gene 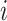 across multiple samples (cells) and var() represents variance function. The symmetric version of
across multiple samples (cells) and var() represents variance function. The symmetric version of 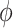 can be written as
can be written as
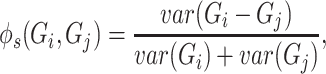 |
whereas rho can be defined as
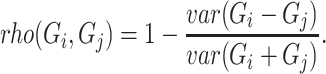 |
To estimate both measures of proportionality, 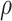 and
and 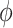 , we used ‘propr’ package2.0 [49].
, we used ‘propr’ package2.0 [49].
Comparison of raw and filtered graph
The networks inferred from filtered and unfiltered gene expression were compared to the ground truth. Ground truth for DREAM5 challenge data set was already available while for single-cell expression; we assembled the ground truth from Human Integrated Protein-Protein Interaction reference database [50]. We considered all edges possible in network, sorted them based on the significance of edge weights. We calculated the area under the Receiver operator curve for both raw and filtered networks by comparing against edges in the ground truth. Receiver operator is a standard performance evaluation metrics from the field of machine learning, which has been used in the DREAM5 evaluation method with some modifications. The modification for Receiver operating curve here is that for X-axis instead of false-positive rate, we used a number of edges sorted according to their weights. For evaluation all possible edges sorted based on their weights in network are taken from the gene network inferred from filtered and raw graphs. We calculated improvement by measuring fold change between raw and filtered scores.
Comparison with other methods
We compared the results of our approach of graph wavelet-based denoising with other methods meant for imputation or reducing noise in scRNA-seq profiles. For comparison, we used graph Fourier-based filtering [17], MAGIC [20], scImpute [21], DCA [22], SAVER [23], Randomly [24], KNN-impute [25]. Brief descriptions and corresponding parameters used for other methods are written in Supplementary Method.
Data Sources
The bulk gene expression data used here evaluation was download from DREAM5 portal (http://dreamchallenges.org/project/dream-5-network-inference-challenge/). The single-cell expression profile of mESC generated using different protocols [18] was downloaded for GEO database (GEO id: GSE75790). Single-cell expression profile of pancreatic cells from individuals with different age groups was downloaded from GEO database (GEO id:GSE81547). The scRNA-seq profile of murine ageing lung published by Kimmel et al. [14] is available with GEO id: GSE132901, while ageing lung scRNA-seq data published by Angelids et al. [51] is available with GEO id: GSE132901.
Availability
The code for graph wavelet-based filtering of gene expression is available at http://reggen.iiitd.edu.in:1207/GraphWavelet/index.html. The codes are present at https://github.com/reggenlab/GWNet/ and supporting files are present at https://github.com/reggenlab/GWNet/tree/master/supporting files.
files.
Key Points
We found that graph wavelet-based denoising of gene expression profiles of bulk samples and single-cells can substantially improve gene regulatory network inference.
More consistent prediction of gene network due to denoising lead to reliable comparison of predicted networks from old and young cells to study the effect of ageing using single-cell transcriptome.
Our analysis revealed biologically relevant changes in regulation due to ageing in lung pneumocyte type II cells, which had similarity with effects of COVID infection in human lung.
Our analysis highlighted influential pathways and master regulators which could be topic of further study for reducing severity due to ageing.
Acknowledgements
We thank Dr Gaurav Ahuja for providing us valuable advice on analysis of single-cell expression profile of ageing cells.
Supplementary Material
Vibhor Kumar is an assistant professor at IIIT Delhi, India. He is also an adjunct scientist at Genome Institute of Singapore. His interest include Genomics and signal processing.
Divyanshu Srivastava completed his thesis on Graph Signal processing for Masters degree at computational biology department in IIIT Delhi, India. He has applied Graph signal processing on protein structures and gene expression data sets.
Shreya Mishra is a PhD student at computational biology department in IIIT Delhi, India. Her interest include data sciences and Genomics.
Contributor Information
Shreya Mishra, IIIT Delhi, India.
Divyanshu Srivastava, IIIT Delhi, India.
Vibhor Kumar, IIIT Delhi, India.
References
- 1. Tu Z, Wang L, Arbeitman MN, et al. An integrative approach for causal gene identification and gene regulatory pathway inference. Bioinformatics 2006; 22:e489–96. [DOI] [PubMed] [Google Scholar]
- 2. Iacono G, Massoni-Badosa R, Heyn H. Single-cell transcriptomics unveils gene regulatory network plasticity. Genome Biol 2019; 20:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pradhan A, Siwo GH, Singh N, et al. Chemogenomic profiling of plasmodium falciparum as a tool to aid antimalarial drug discovery. Sci Rep 2015; 5:15930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Maetschke SR, Madhamshettiwar PB, Davis MJ, et al. Supervised, semi-supervised and unsupervised inference of gene regulatory networks. Brief Bioinform 2014; 15:195–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Margolin AA, Wang K, Lim WK, et al. Reverse engineering cellular networks. Nat Protoc 2006; 1:662–71. [DOI] [PubMed] [Google Scholar]
- 6. Skinnider MA, Squair JW, Foster LJ. Evaluating measures of association for single-cell transcriptomics. Nat Methods 2019; 16:381–6. [DOI] [PubMed] [Google Scholar]
- 7. Chen S, Mar JC. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinformat 2018; 19:232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Aibar S, González-Blas CB, Moerman T, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 2017; 14:1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Matsumoto H, Kiryu H, Furusawa C, et al. SCODE: an efficient regulatory network inference algorithm from single-cell RNA-Seq during differentiation. Bioinformatics (Oxford, England) 2017; 33:2314–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chan TE, Stumpf MPH, Babtie AC. Gene regulatory network inference from single-cell data using multivariate information measures. Cell Systems 2017; 5:251–267.e253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim JK, Kolodziejczyk AA, Ilicic T, et al. Characterizing noise structure in single-cell RNA-seq distinguishes genuine from technical stochastic allelic expression. Nat Commun 2015; 6(1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Raser JM, O’Shea EK. Noise in gene expression: origins, consequences, and control. Science (New York, NY) 2005; 309:2010–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lichtblau Y, Zimmermann K, Haldemann B, et al. Comparative assessment of differential network analysis methods. Brief Bioinform 2017; 18:837–50. [DOI] [PubMed] [Google Scholar]
- 14. Kimmel JC, Penland L, Rubinstein ND, et al. Murine single-cell RNA-seq reveals cell-identity- and tissue-specific trajectories of aging. Genome Res 2019; 29:2088–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marbach D, Costello JC, Küffner R, et al. Wisdom of crowds for robust gene network inference. Nat Methods 2012; 9:796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schaffter T, Marbach D, Floreano D. GeneNetWeaver: in silico benchmark generation and performance profiling of network inference methods. Bioinformatics (Oxford, England) 2011; 27:2263–70. [DOI] [PubMed] [Google Scholar]
- 17. Burkhardt DB, Stanley JS, Perdigoto AL, et al. Enhancing experimental signals in single-cell RNA-sequencing data using graph signal processing. bioRxiv 2019; 10:532846. [Google Scholar]
- 18. Ziegenhain C, Vieth B, Parekh S, et al. Comparative analysis of single-cell RNA sequencing methods. Mol Cell 2017; 65:631–643.e634. [DOI] [PubMed] [Google Scholar]
- 19. Zhou Q, Chipperfield H, Melton DA, et al. A gene regulatory network in mouse embryonic stem cells. Proc Natl Acad Sci 2007; 104:16438–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van Dijk D, Sharma R, Nainys J, et al. Recovering gene interactions from single-cell data using data diffusion. Cell 2018; 174:716–729.e727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li WV, Li JJ. An accurate and robust imputation method scImpute for single-cell RNA-seq data. Nat Commun 2018; 9(1): 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Eraslan G, Simon LM, Mircea M, et al. Single-cell RNA-seq denoising using a deep count autoencoder. Nat Commun 2019; 10(1): 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Huang M, Wang J, Torre E, et al. SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods 2018; 15(7): 539–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aparicio L, Bordyuh M, Blumberg AJ, et al. A random matrix theory approach to denoise single-cell data. Patterns 2020; 4:100035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Troyanskaya O, Cantor M, Sherlock G, et al. Missing value estimation methods for DNA microarrays. Bioinformatics (Oxford, England) 2001; 17:520–5. [DOI] [PubMed] [Google Scholar]
- 26. Enge M, Arda HE, Mignardi M, et al. Single-cell analysis of human pancreas reveals transcriptional signatures of aging and somatic mutation patterns. Cell 2017; 171:321–330.e314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen EY, Tan CM, Kou Y, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform 2013; 14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen M, Brown LA. Histamine stimulation of surfactant secretion from rat type II pneumocytes. Am J Physiol 1990; 258:L195–200. [DOI] [PubMed] [Google Scholar]
- 29. Lecce L, Lam YT, Lindsay LA, et al. Aging impairs VEGF-mediated, androgen-dependent regulation of angiogenesis. Mol Endocrinol 2014; 28:1487–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Al-Saiedy M, Pratt R, Lai P, et al. Dysfunction of pulmonary surfactant mediated by phospholipid oxidation is cholesterol-dependent. Biochimica Et Biophysica Acta General Subjects 2018; 1862:1040–9. [DOI] [PubMed] [Google Scholar]
- 31. Schouten LRA, Helmerhorst HJF, Wagenaar GTM, et al. Age-dependent changes in the pulmonary renin-angiotensin system are associated with severity of lung injury in a model of acute lung injury in rats. Crit Care Med 2016; 44:e1226–35. [DOI] [PubMed] [Google Scholar]
- 32. Park S-K, Dahmer MK, Quasney MW. MAPK and JAK-STAT signaling pathways are involved in the oxidative stress-induced decrease in expression of surfactant protein genes. Cell Physiol Biochem 2012; 30:334–46. [DOI] [PubMed] [Google Scholar]
- 33. Zhang Z, Newton K, Kummerfeld SK, et al. Transcription factor Etv5 is essential for the maintenance of alveolar type II cells. Proc Natl Acad Sci U S A 2017; 114:3903–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Reddy NM, Vegiraju S, Irving A, et al. Targeted deletion of Jun/AP-1 in alveolar epithelial cells causes progressive emphysema and worsens cigarette smoke-induced lung inflammation. Am J Pathol 2012; 180:562–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mikkonen L, Pihlajamaa P, Sahu B, et al. Androgen receptor and androgen-dependent gene expression in lung. Mol Cell Endocrinol 2010; 317:14–24. [DOI] [PubMed] [Google Scholar]
- 36. Viola A, Munari F, Sánchez-Rodríguez R, et al. The metabolic signature of macrophage responses. Front Immunol 2019; 10:1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blanco-Melo D, Nilsson-Payant BE, Liu W-C, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell 2020; 181:1036–1045.e1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qi F, Qian S, Zhang S, et al. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun 2020; 526:135–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chow RD, Chen S. The aging transcriptome and cellular landscape of the human lung in relation to SARS-CoV-2. bioRxiv 2020; 2020.2004.2007.030684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yew-Booth L, Birrell MA, Lau MS, et al. JAK-STAT pathway activation in COPD. Eur Respir J 2015; 46:843–5. [DOI] [PubMed] [Google Scholar]
- 41. Sharifi N, Ryan CJ. Androgen hazards with COVID-19. Endocr Relat Cancer 2020; 27:E1–3. [DOI] [PubMed] [Google Scholar]
- 42. Bryce PJ, Mathias CB, Harrison KL, et al. The H1 histamine receptor regulates allergic lung responses. J Clin Investig 2006; 116:1624–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greenberg S, Horan G, Bennett B, et al. Late breaking abstract - evaluation of the JNK inhibitor, CC-90001, in a phase 1b pulmonary fibrosis trial. Eur Respir J 2017; 50. [Google Scholar]
- 44. Montopoli M, Zumerle S, Vettor R, et al. Androgen-deprivation therapies for prostate cancer and risk of infection by SARS-CoV-2: a population-based study (N = 4532). Ann Oncol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Maaten LVD, Geoffrey H. Visualizing data using t-SNE. J Mach Learn Res 2008; 9:2579–605. [Google Scholar]
- 46. Sandryhaila A, Moura JMF. Discrete signal processing on graphs: frequency analysis. IEEE Trans Signal Proc 2014; 62:3042–54. [Google Scholar]
- 47. Hammond DK, Vandergheynst P, Gribonval R. Wavelets on graphs via spectral graph theory. Appl Comput Harmon Anal 2011; 30:129–50. [Google Scholar]
- 48. Erb I, Notredame C. How should we measure proportionality on relative gene expression data? Theory in Biosciences = Theorie in Den Biowissenschaften 2016; 135:21–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Quinn TP, Richardson MF, Lovell D, et al. Propr: an R-package for identifying proportionally abundant features using compositional data analysis. Sci Rep 2017; 7:16252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Alanis-Lobato G, Andrade-Navarro MA, Schaefer MH. HIPPIE v2.0: enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res 2017; 45:D408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Angelidis I, Simon LM, Fernandez IE, et al. An atlas of the aging lung mapped by single cell transcriptomics and deep tissue proteomics. Nat Commun 2019; 10:963. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.