

Abstract
The recurrent and recent global outbreak of the severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) has turned into a global concern which has infected more than 42 million people all over the globe, and this number is increasing in hours. Unfortunately, no vaccine or specific treatment is available, which makes it more deadly. A vaccine-informatics approach has shown significant breakthrough in peptide-based epitope mapping and opens the new horizon in vaccine development. In this study, we have identified a total of 15 antigenic peptides [including thymus cells (T-cells) and bone marrow or bursa-derived cells] in the surface glycoprotein (SG) of SARS-CoV-2 which is nontoxic and nonallergenic in nature, nonallergenic, highly antigenic and non-mutated in other SARS-CoV-2 virus strains. The population coverage analysis has found that cluster of differentiation 4 (CD4+) T-cell peptides showed higher cumulative population coverage over cluster of differentiation 8 (CD8+) peptides in the 16 different geographical regions of the world. We identified 12 peptides ((LTDEMIAQY, WTAGAAAYY, WMESEFRVY, IRASANLAA, FGAISSVLN, VKQLSSNFG, FAMQMAYRF, FGAGAALQI, YGFQPTNGVGYQ, LPDPSKPSKR, QTQTNSPRRARS and VITPGTNTSN) that are 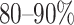 identical with experimentally determined epitopes of SARS-CoV, and this will likely be beneficial for a quick progression of the vaccine design. Moreover, docking analysis suggested that the identified peptides are tightly bound in the groove of human leukocyte antigen molecules which can induce the T-cell response. Overall, this study allows us to determine potent peptide antigen targets in the SG on intuitive grounds, which opens up a new horizon in the coronavirus disease (COVID-19) research. However, this study needs experimental validation by in vitro and in vivo.
identical with experimentally determined epitopes of SARS-CoV, and this will likely be beneficial for a quick progression of the vaccine design. Moreover, docking analysis suggested that the identified peptides are tightly bound in the groove of human leukocyte antigen molecules which can induce the T-cell response. Overall, this study allows us to determine potent peptide antigen targets in the SG on intuitive grounds, which opens up a new horizon in the coronavirus disease (COVID-19) research. However, this study needs experimental validation by in vitro and in vivo.
Keywords: COVID-19, vaccinomics, allergenicity, immunogenicity, epitope prediction, T- and B-cell, molecular docking, population coverage
Introduction
History is witness to the fact that whenever a major disease comes, it brings tremendous destruction with itself, and these destructions are physical and economical. Previously, cholera, smallpox, bubonic plague and influenza are some of the most brutal killers in human history and killed millions of people all over the world. Nowadays, the world is scared of the coronavirus disease 2019 (COVID-19), and its outbreak continues to spread from China to all over the world, and we do not yet know when it will stop. It is a contagious disease caused by a SARS family virus named ‘severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)’, which has not been previously identified in humans. The initial symptoms of COVID-19 are dry cough, throat infection, fever, breath shortness, loss of taste or smell, rashes in skin, conjunctivitis, tiredness, muscles pain and, diarrhea; most of the individuals (about 80%) recover without any special treatment, but older people and a person with preexisting medical conditions or comorbidities (cardiovascular disease, diabetes, lungs disease and cancer) are more likely to develop a serious illness. The incubation period for the infection with SARS-CoV-2 ranges from 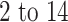 days after its exposure [1]. The transmissibility of the virus is shown by its reproductive number (R0), and it varies from area to area. The World Health Organization (WHO) has estimated R0 between 1.4 and 2.5, but some other studies have estimated it as 2.24–3.58 for COVID-19 [2, 3].
days after its exposure [1]. The transmissibility of the virus is shown by its reproductive number (R0), and it varies from area to area. The World Health Organization (WHO) has estimated R0 between 1.4 and 2.5, but some other studies have estimated it as 2.24–3.58 for COVID-19 [2, 3].
In just 9 months, the COVID-19 that originally emerged from the Wuhan province of China is posing major public health and governance challenges. The cases have now spread in 213 countries and territories around the globe. Till 24 October 2020, more than 42.5 million infected individuals, with over 1 134 940 deaths around the globe, have been reported to WHO (WHO COVID-19 Dashboard), and these numbers are rapidly increasing in hours. At present, unfortunately, no vaccine or specific treatment is available. However, the WHO listed (as per 19 October 2020) more than 200 vaccines in development at various stages (preclinical evaluation: 154, clinical evaluation: 44). The vaccine candidates which are listed in the clinical evaluation stage seven have reached phase III trial, including Ad5-nCoV (CanSino Biologics, China), AZD1222 (The University of Oxford; AstraZeneca; IQVIA; Serum Institute of India), CoronaVac (Sinovac), JNJ-78436735 or Ad26.COV2-S (Johnson & Johnson), mRNA-1273 (Moderna) and unknown vaccine [(no name announced by the Wuhan Institute of Biological Products; China, National Pharmaceutical Group (Sinopharm) and NVX-CoV2373 (Novavax)]. Further, the University of Melbourne and Murdoch Children’s Research Institute, Radboud University Medical Center and the Faustman Lab at the Massachusetts General Hospital’s BCG live-attenuated vaccine are also in the phase II/III combined phase. The AstraZeneca/University of Oxford vaccine candidate (AZD1222) looks the most promising vaccine candidate, which is in the phase II/III combined phase (WHO: Draft landscape of COVID-19 candidate vaccines).
Moreover, there are no chemotherapeutic agents available to curb this menace; however, few agents are being used, including natural compounds [4–6], western medicines [7, 8] and traditional Chinese medicines (TCN) [9, 10], which may have potential efficacy against the SARS-CoV-2. Moreover, other drugs like interferon-α (IFN-α), lopinavir/ritonavir, chloroquine phosphate, ribavirin, favipiravir, disulfiram, arbidol and hydroxychloroquine are recommended as the tentative treatments for COVID-19 [11, 12]. Currently, there is no specific treatment/medicine or vaccine to cure COVID-19, so there is an urgent need to develop new vaccines or drugs against this deadly disease. In this way, the integration of computational techniques provides a novel approach to integrating the vaccine-informatics approach for the development of vaccines. These methods had earlier been used in the development of vaccines against several diseases, including dengue [13], malaria [14], influenza [15], multiple sclerosis [16] and tumor [17]. However, this approach generally works through the identification of major histocompatibility complex (MHC)-1 and II molecules and thymus cells (T-cell) epitopes (CD8+ and CD4+) [18], which particularize the selection of the potential vaccine agents related to the transporter of antigen presentation (TAP) molecules [19, 20].
The beginning of 2020 has seen the emergence of the deadly COVID-19 pandemic, and currently, we are drowning with an enormous amount of articles, with their probable epitope-based peptide vaccine for COVID-19 in which the bone marrow or bursa-derived cells (B-cells) and T-cell epitopes have been analyzed, which have anticipated the possibility of antigenic epitopes which can be used to design a novel vaccine candidate against the SARS-CoV-2 [21–23]. In the current study, we have also predicted epitope-based vaccine candidates against the SARS-CoV-2 using the systematic vaccine-informatics approach. We considered surface glycoprotein (SG) of SARS-CoV-2 due to its specific functions; SARS-CoV-2 uses surface spike protein to mediate entry into the host cells. To fulfill its purpose, the SARS-CoV-2 spike binds to the receptor ‘hACE2’ through its receptor-binding domain (RBD) and is proteolytically triggered by human proteases [24, 25]. Notably, we not only prognosticate the most potential vaccine candidate but have also cross-checked the resemblance, congruity and the compatibility of these selected epitopes with humans to circumvent any possible risk of autoimmunity. Additionally, we had checked the resemblance of our epitopes with those which are already experimentally verified epitopes of different organisms, including SARS-CoV, which not only makes our study more precise and noteworthy but also expands our views for the vaccine-informatics approach in planning for the next global pandemic. Our work can save the time needed to screen a large number of possible epitopes compared with the experimental techniques and also guide the experimental work with high confidence of finding the desired results in vaccine development.
Materials and methods
Sequence retrieval and analysis
The SG sequence (ID: QHO62112.1) was obtained from the NCBI gene bank database (https://www.ncbi.nlm.nih.gov/gene/) in the FASTA format. Additionally, we checked the sequence similarity of peptide sequences with other SG proteins of other SARS-CoV-19 isolates from different geographical regions using the Clustal Omega tool [26] to analyze the variation in epitopes sequences, which can determine whether the epitopes are conserved or have altered peptide ligands.
T-cell peptides (epitopes) prediction
The NETCTL_1.2 server [27] was used to identify the CD8+ T-cell peptides at a set threshold value of 0.95 to sustain the sensitivity and specificity of 0.90 and 0.95, respectively. We used all the expanded 12 MHC class-I supertypes (including A1, A2, A3, A24, A26, B7, B8, B27, B39, B44, B58 and B62) and incorporated the peptide prediction of MHC class-I binding molecules and proteasomal C-terminal cleavage with TAP transport efficiency. These results were accomplished by the weighted TAP transport efficiency matrix. Then MHC class-I binding and proteasomal cleavage efficiency were used to obtain the total scores and translate it into the sensitivity and specificity. We selected peptides as the epitope candidate on the basis of the overall score. Further, we checked the peptides binding to MHC class-I molecules by using the MHC class-I binding predictions tool [28]. The predicted output was given in units of IC50  (IC50 values
(IC50 values 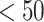
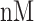 = high affinity, IC50
= high affinity, IC50 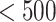
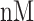 moderate affinity and IC50
moderate affinity and IC50 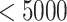
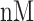 low affinity) [29]. The MHC-NP (Naturally Processed) tool used for assesses the probability of naturally processing and binding of peptides to the given MHC molecules. This tool predicts naturally processed epitopes based on the physiochemical properties and comparison of residual position with experimentally verified epitopes [30]. Further, we identified CD4+ T-cell peptides with IC50 ≤ 100 ((IC50 values
low affinity) [29]. The MHC-NP (Naturally Processed) tool used for assesses the probability of naturally processing and binding of peptides to the given MHC molecules. This tool predicts naturally processed epitopes based on the physiochemical properties and comparison of residual position with experimentally verified epitopes [30]. Further, we identified CD4+ T-cell peptides with IC50 ≤ 100 ((IC50 values 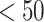
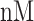 = high affinity, IC50
= high affinity, IC50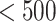
 moderate affinity and IC50
moderate affinity and IC50 
 low affinity) by using MHC class-II binding predictions tool [31].
low affinity) by using MHC class-II binding predictions tool [31].
B-cell peptides (epitopes) prediction
The identification of B-cell peptides (epitopes) in the SG of SARS-CoV-2 was accomplished to find the potent antigen, which gives an assurance of humoral immunity. Here, we used Antibody Epitope Prediction tool [32] to find the B-cell antigenicity with classical propensity scale methods, including Bepipred Linear Epitope Prediction 2.0 [33], Chou & Fasman Beta-Turn Prediction [34], Emini Surface Accessibility Prediction [35], Karplus & Schulz Flexibility Prediction [36], Kolaskar & Tongaonkar Antigenicity [37] and Parker Hydrophilicity Prediction [38]. In this study, we selected that region in the protein sequence, which was cross-referenced, and common findings were considered as the B-cell antigenic region based on the above six classical propensity scale methods.
Epitope conservancy and immunogenicity analysis
The epitope conservation defined the degree of a resemblance betwixt the peptide and query sequences. Hence, we used the Epitope Conservancy Analysis (ECA) tool [39] to find the conservancy of our peptides among the other SARS coronaviruses, bat SARS-like coronaviruses and bat coronaviruses. Additionally, the immunogenicity prediction can reveal the degree of efficiency for individual peptides to produce an immunogenic response. In our study, we used the Class I Immunogenicity [40] tool to predict the immunogenicity of the MHC class-I binding peptides.
Peptide structural modeling and retrieval of human leukocyte antigen molecules
The structure of CD+8 and CD+4 T-cell peptides were modeled by a biased modeling method using an online server, PEP-FOLD 3.5 server, at the RPBS MOBYL portal [41]. The crystal structure of the SARS-CoV-2 spike glycoprotein (6VSB-A) was used as the reference model together with a mask representing the structure fragments for which the local conformation of this structure has been imposed. Additionally, The structure of human leukocyte antigen (HLA) molecules, including HLA-B*53:01 (PDB ID: 1A1M) [42], HLA-B*44:03 (PDB ID: 4JQX) [43] and HLA-DRB1*01:01 (1AQD) [44] were retrieved from the Protein Data Bank (PDB) [45]. The structure of peptides and HLA molecules are depicted in Figure 1.
Figure 1.
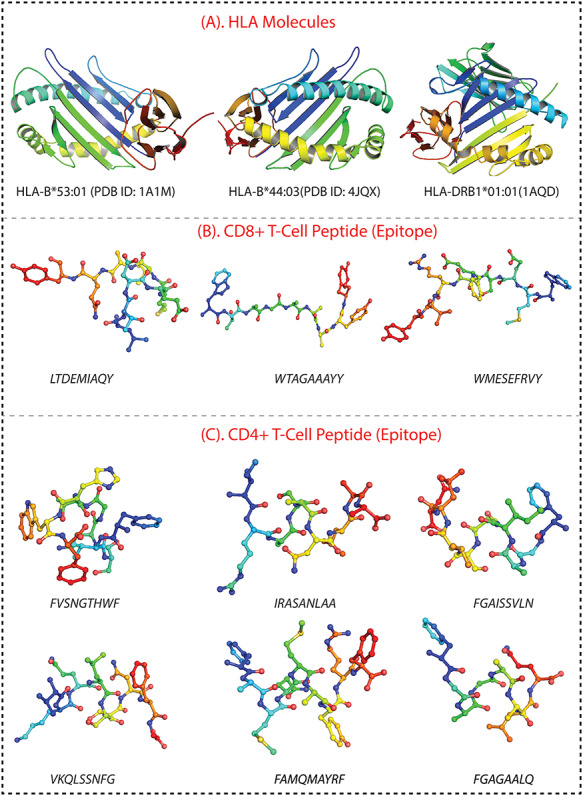
(A) The 3D structure of HLA molecules, including HLA-B*53:01, HLA-B*44:03 and HLA-DRB1*01:01. (B) The peptide structure of three peptides for CD8+ T-cells (LTDEMIAQY, WTAGAAAYY and WMESEFRVY). (C). The structure of six CD4+ T-cells peptides, including FVSNGTHWF, IRASANLAA, FGAISSVLN, VKQLSSNFG, FAMQMAYRF and FGAGAALQ.
Population coverage analysis
The population coverage analysis (PCA) tool [46] gives the idea about the probable response of each peptide in different countries of the world based on peptide–HLA data genotypic frequency. Our CD8+ T-cell (three peptides) and CD4+ T-cell (six peptides) peptides and their respective HLA alleles were used for PCA. We selected 16 geographical areas (115 countries and 21 different ethnicities grouped), which were East Asia, Northeast Asia, South Asia, Southeast Asia, Southwest Asia, Europe, East Africa, West Africa, Central Africa, North Africa, South Africa, West Indies, North America, Central America, South America and Oceania. These 16 geographical regions cover the HLA allele frequencies and associated data for different individual populations from the most popular countries.
Predicted peptides versus epitope database and peptide screening for autoimmune, allergic and toxic response
In this section, we used predicted peptides (including T- and B-cell peptides) to search for homologous epitopes at 80–90% identity in the Epitope database [47, 48]. The current limited accessible data is not sufficient to recognize the antigenic region in the spike proteins of SARS-CoV-2 by human immune responses. Herein, the homologous epitopes (derived from other pathogens) would expedite the evaluation of vaccine candidate immunogenicity, as well as observing the possible outcomes of mutational events and epitope escape as the virus is transmitted through human populations [49]. Based on initially full-length genomic phylogenetic analysis, the precursory studies suggested that SARS-CoV-2 is quite similar to SARS-CoV and the putatively same cell entry mechanism and human cell receptor usage. And due to conspicuous resemblance amid these viruses, the previous study that gave us a basic understanding of protective immune responses against SARS-CoV, which may potentially be leveraged to aid vaccine development against the SARS-CoV-2 [50]. This study also helps to check the peptides identity with human proteome because there is a chance of an autoimmune response due to any kind of molecular mimicry between the predicted peptides (epitopes) and the human proteome. Moreover, we checked the allergic and toxic nature of the predicted peptides using AlgPred [51] and ToxinPred [52] tools, respectively.
Molecular docking studies
In this study, molecular docking studies produced important information regarding the orientation pattern of the peptides in the binding groove of the HLA molecules as well as which residues are actively involved in the binding. In this study, we selected three CD8+ T-cell peptides (LTDEMIAQY, WTAGAAAYY and WMESEFRVY) and five CD4+ T-cell peptides (IRASANLAA, FGAISSVLN, VKQLSSNFG, FAMQMAYRF and FGAGAALQ) for molecular docking against the HLA molecule, including HLA-B*53:01 (PDB ID: 1A1M), HLA-B*44:03 (PDB ID: 4JQX) and HLA-DRB1*01:01 (PDB ID: 1AQD). We used the glide module [53–55] of Schrödinger suite for peptides–HLA molecules docking. All peptides were prepared using the LigPrep module of the Schrodinger suite and docked in the binding site of protein using the SP-peptide mode of Glide. Receptor grid was generated using the receptor grid generation in the Glide application by specifying the binding (active) site residues, which was identified by the SiteMap tool [56]. The docked conformers were evaluated using Glide (G) Score, and the best docked pose with lowest Glide score value was recorded for each peptide.
Results
Sequence retrieval and structure prediction
Viral glycoproteins have a major role in its pathogenesis. The main goal of viral infection is to recognize and bind a receptor on the cell surface of the host. It has been considered that SG plays an important role in immunity and infection, So we have retrieved the envelope SG of SARS-CoV-19 from the NCBI gene bank database (ID: QHO62112.1). Moreover, the sequence similarity of query proteins and peptides was done with the other SG proteins of other SARS-CoV-19 isolates from the various regions of the world (including China, Columbia, Japan, Malaysia, Israel, Iran, India, Sri Lanka, Vietnam, South Korea, Pakistan, United States, Hong Kong, Taiwan, Spain, South Africa, Serbia, Greece, Nederland, France and the Czech Republic), and it was found that all the predicted peptides are conserved in all of the isolates (As per 10 August 2020) (Supplementary table S1 available online at https://academic.oup.com/bib).
Identification of T-cell epitopes from SG protein of SARS-CoV-19
The NETCTL server predicted several peptides in the SARS-CoV-19 SG, but only nine most potent peptides were chosen, which have a high combinatorial score. We considered only those alleles of MHC class-I for which the peptides showed higher binding affinity (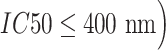 . Proteasomes played a key role in cleaving the peptide bonds and converting the protein into a peptide. The total score of each peptide–HLA interaction was taken into consideration, and a greater score meant greater processing efficiency. The three peptides LTDEMIAQY (P1), WTAGAAAYY (P2) and WMESEFRVY (P3) among the nine were found to bind with most of the MHC class-I molecules, including HLA-B*15:01, HLA-B*53:01, HLA-A*68:02, HLA-B*44:03 and HLA-B*57:01, but peptides P1, P2 and P3 had a maximum probable value of
. Proteasomes played a key role in cleaving the peptide bonds and converting the protein into a peptide. The total score of each peptide–HLA interaction was taken into consideration, and a greater score meant greater processing efficiency. The three peptides LTDEMIAQY (P1), WTAGAAAYY (P2) and WMESEFRVY (P3) among the nine were found to bind with most of the MHC class-I molecules, including HLA-B*15:01, HLA-B*53:01, HLA-A*68:02, HLA-B*44:03 and HLA-B*57:01, but peptides P1, P2 and P3 had a maximum probable value of 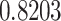 ,
, 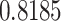 and
and 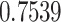 for the HLA-B*53:01, HLA-B*44:03 and HLA-B*44:03, respectively. These peptides (P1, P2 and P3) also have a maximum identity
for the HLA-B*53:01, HLA-B*44:03 and HLA-B*44:03, respectively. These peptides (P1, P2 and P3) also have a maximum identity 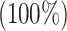 for the conservancy. Moreover, we made the immunogenicity prediction of peptides and got the highest pMHC-I immunogenicity scores of
for the conservancy. Moreover, we made the immunogenicity prediction of peptides and got the highest pMHC-I immunogenicity scores of 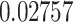 (P1),
(P1), 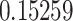 (P2) and
(P2) and 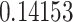 (P3). The details are given in Table 1. Additionally, we identified
(P3). The details are given in Table 1. Additionally, we identified 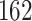 CD4+ T-cell peptides (epitopes) with
CD4+ T-cell peptides (epitopes) with 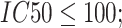 however, only six peptides (FVSNGTHWF, IRASANLAA, FGAISSVLN, VKQLSSNFG, FAMQMAYRF, and FGAGAALQ) were found to interact with most of the HLA-DRB-1 molecules, and the details are given in Table 2.
however, only six peptides (FVSNGTHWF, IRASANLAA, FGAISSVLN, VKQLSSNFG, FAMQMAYRF, and FGAGAALQ) were found to interact with most of the HLA-DRB-1 molecules, and the details are given in Table 2.
Table 1.
The three potential CD8+ T-cell epitopes along with their interacting MHC class-I alleles and NetCTL combine score, epitopes conservancy hits and pMHC-I immunogenicity score
| Peptide | Position | NetCTL combined score | MHC-1 IC50 score < 400 | MHC-NP prediction result | pMHC-I immunogenicity score | Epitope conservancy hit (%) |
|---|---|---|---|---|---|---|
| LTDEMIAQY | 865–873 | A1 = 3.66 A2 = 1.06 |
3.37 (0.2) | 0.9457 HLA-B*57:01 | 0.02757 | 100 |
| 156 (0.74) | 0.8907 HLA-B*57:01 | |||||
| 261 (0.54) | 0.8203 HLA-B*53:01 | |||||
| 330 (0.79) | 0.7935 HLA-B*53:01 | |||||
| 359 (0.81) | 0.7806 HLA-B*53:01 | |||||
| 376 (0.87) | 0.7793 HLA-B*15:01 | |||||
| WTAGAAAYY | 258–266 | A1 = 3.11 A26 = 2.00 |
68.6 (0.18) | 0.9404 HLA-B*44:03 | 0.15259 | 100 |
| 48.4 (0.44) | 0.873 HLA-B*53:01 | |||||
| 132 (0.66) | 0.8287 HLA-B*44:03 | |||||
| 93.7 (0.6) | 0.8185 HLA-B*44:03 | |||||
| 91.8 (0.59) | 0.7519 HLA-B*57:01 | |||||
| 89.9 (0.55) | 0.7412 HLA-A*68:02 | |||||
| WMESEFRVY | 152–160 | A1 = 1.92 B62 = 1.24 |
49.7 (0.29) | 0.8901 HLA-B*57:01 | 0.14153 | 100 |
| 265.7 (0.55) | 0.8378 HLA-B*44:03 | |||||
| 142.2 (0.43) | 0.8341 HLA-B*57:01 | |||||
| 63.8 (0.33) | 0.7539 HLA-B*44:03 | |||||
| 179 (0.47) | 0.734 HLA-B*57:01 |
Table 2.
The selected six most potential CD4+ T-cell epitopes along with their interacting MHC class-II alleles with affinity IC50 < 100
| S. No. | Epitopes | Position | Interacting MHC class-II allele | IC50 values (<100) |
|---|---|---|---|---|
| 1 | FVSNGTHWF | 1095–1103 | HLA-DRB1*01:01 | 21.43 |
| HLA-DRB1*13:02 | 93.35 | |||
| HLA-DRB1*07:01 | 89.95 | |||
| HLA-DRB1*13:02 | 47.25 | |||
| HLA-DRB1*09:01 | 98.31 | |||
| 2 | IRASANLAA | 1018–1026 | HLA-DRB1*01:01 | 19.9 |
| HLA-DRB1*04:01 | 97.67 | |||
| HLA-DRB1*13:02 | 42.12 | |||
| HLA-DRB1*07:01 | 84 | |||
| HLA-DRB1*09:01 | 88.15 | |||
| 3 | FGAISSVLN | 970–978 | HLA-DRB1*01:01 | 20.2 |
| HLA-DRB1*04:01 | 98.03 | |||
| HLA-DRB1*04:05 | 96.39 | |||
| HLA-DRB1*07:01 | 86.61 | |||
| HLA-DRB1*09:01 | 88.46 | |||
| 4 | VKQLSSNFG | 963–971 | HLA-DRB1*01:01 | 22.84 |
| HLA-DRB1*15:01 | 91.6 | |||
| HLA-DRB1*04:01 | 85.68 | |||
| 5 | FAMQMAYRF | 898–906 | HLA-DRB1*01:01 | 12.49 |
| HLA-DRB1*07:01 | 42.09 | |||
| HLA-DRB1*09:01 | 53.55 | |||
| HLA-DRB1*11:01 | 48.76 | |||
| HLA-DRB1*15:01 | 50.31 | |||
| 6 | FGAGAALQI | 888–896 | HLA-DRB1*01:01 | 16.21 |
| HLA-DRB1*09:01 | 24.83 | |||
| HLA-DRB1*07:01 | 48.18 |
Identification of linear B-cell epitopes from SG protein of SARS-CoV-19
The B-cell epitopes comprise of peptides which can easily be used to take the place of antigens for immunizations and antibody production. In this study, we used an amino acid scale-based method in the B-cell antibody epitope prediction tool in which we predict linear epitopes from the protein sequence. We found six B-cell linear epitopes (including LTPGDSSSGWTAG, YQAGSTPCNGV, YGFQPTNGVGYQ, VITPGTNTSN, QTQTNSPRRARS and LPDPSKPSKR) in the surface glycoprotein of SARS-CoV-19, which may be capable of inducing the desired immune response as B-cell epitopes (Figure 2).
Figure 2.
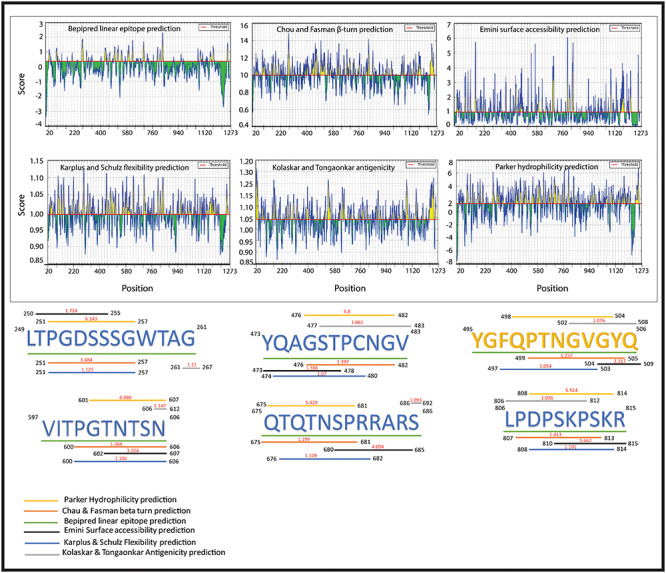
Six Peptides are representing the B-cell epitopes which have highest antigenic propensity and are surrounded by six different colored lines, with each line indicating the different analysis methods (Bepipred linear epitope prediction, Chou & Fasman beta-turn prediction, Emini surface accessibility prediction, Karplus & Schulz flexibility prediction, Kolasker & Tongaonkar antigenicity prediction and Parker hydrophilicity prediction) with the maximum scores.
Population coverage analysis of T-Cell peptides
The population coverage analysis calculates the expected response of the predicted peptides (including B- and T-cells) in populations from different geographical areas. In this study, PCA for 16 different geographical regions was carried out for the predicted peptides by considering all the MHC class-I and II molecules. These results suggested that the expected response of these peptides is varying for populations residing in different geographical regions, as shown in Figure 3. The tool predicted average population coverage of 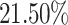 and
and 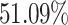 for MHC class-I and II binding peptide fragments, respectively. The PCA of MHC class-II binding peptide fragments of SARS-CoV-19 SG revealed maximum population coverage, for example,
for MHC class-I and II binding peptide fragments, respectively. The PCA of MHC class-II binding peptide fragments of SARS-CoV-19 SG revealed maximum population coverage, for example, 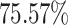 ,
, 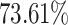 and
and 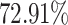 for North America, Europe and East Asia populations (the details are given in Figure 4A and B).
for North America, Europe and East Asia populations (the details are given in Figure 4A and B).
Figure 3.
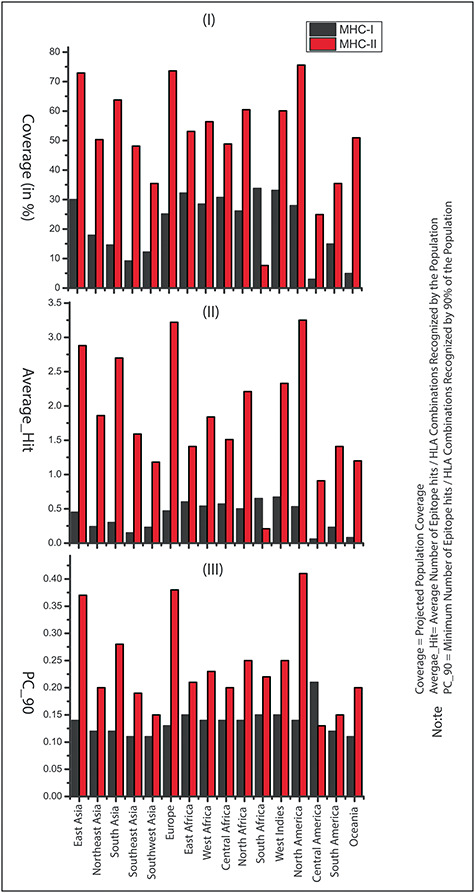
Population coverage of the identified peptides in 16 geographical regions of the world: (A) Population coverage represents the fraction of individuals expected to respond to a given epitope set, (B) average number of epitope hits/HLA combinations recognized by the population and (C) minimum number of epitope hits/HLA combinations recognized by 90% of the population (PC_90).
Figure 4a.
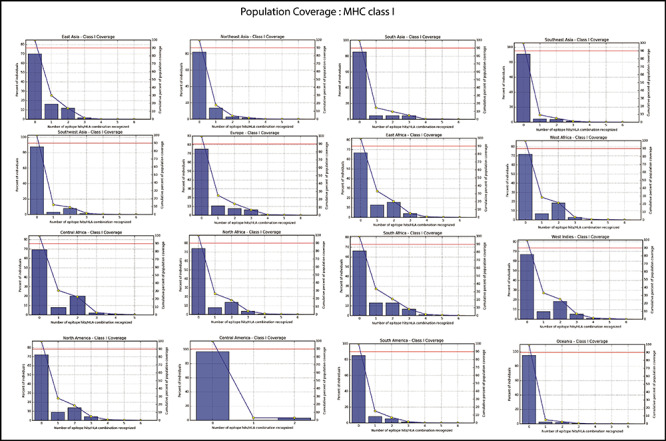
Population coverage of selected peptides binding to the MHC class-I molecules in the 16 geographical regions of the world.
Figure 4b.
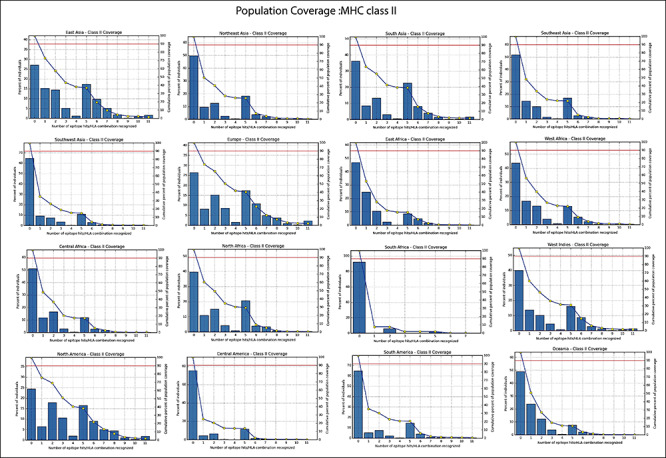
Population coverage of selected peptides binding to the MHC class II molecules in 16 geographical regions of the world.
Autoimmune, allergic and toxic response
In the past, many cases have been reported for the development of autoimmune diseases, like systemic lupus anemia, mysthemia gravis (hepatitis B), multiple sclerosis (swine flu), diabetes mellitus (mumps) and Gullian Barr syndrome (influenza and oral polio vaccine) [57, 58]. To avoid autoimmunity, it becomes important to discard those peptide agents which are similar to the human proteome. So, we have mapped all the predicted peptides sequences against the human and other viruses, including SARS-CoV, which has a maximum sequence identity to SARS-CoV-2 and is the best-characterized coronavirus in respect to epitopic responses. We have identified most of the peptide sequences (including B- and T-cell peptides) that were found to be similar with the experimentally determined epitopes of SARS-CoV virus, except for two peptides (FVSNGTHWF and LTPGDSSSGWTAG) which resemble with Mus musculus and human’s proteome; these peptides were eliminated from further study due to autoimmunity risk. The details of the identified peptides and their resemblance with another organism’s proteome are given in Table 3. Next, we have screened all the peptides to check their allergic and toxic nature, and all these peptides were found to be non-allergen as well as nontoxic in nature (Supplementary table S2 available online at https://academic.oup.com/bib).
Table 3.
List of peptides which are resembled with other organisms’ proteome (homologous epitopes at 80–90% identity), including SARS-CoV, human herpesvirus 4, Leishmania infantum, Bordetella pertussis, Homo sapiens and Mus musculus. The similar fragments of peptides are shown in underline and bold letter along with their source epitopes (epitope ID), antigen and organism
| Peptides (epitopes) | Epitope ID | Antigen | Organism |
|---|---|---|---|
| MHC Class-I | |||
| 1. LTDEMIAQY | |||
| VLPPL LTDEMIAQYT | 1075094 | Spike glycoprotein | SARS-CoV |
| GFIKQYGDCLGDIAARDLICAQKFNGLTVLPPL LTDEMIAQYT | 1074907 | Spike glycoprotein | SARS-COV |
| 2. WTAGAAAYY | |||
| WTAGAAAYY VGY | 1075117 | Spike glycoprotein | SARS-CoV |
| TLLALHRSYLTPGDSSSG WTAGAAAYYVGYLQPRTFLLKYNEN | 1075077 | Spike glycoprotein | SARS-COV |
| ATIASSGIEWT AGAA | 693949 | Major DNA-binding protein | Human herpesvirus 4 |
| EW TAGAARDFLEGVW | 694386 | Major DNA-binding protein | Human herpesvirus 4 |
| SSGIEW TAGAARDFL | 696497 | Major DNA-binding protein | Human herpesvirus 4 |
| 3. WMESEFRVY | |||
| KS WMESEFRVY | 1074961 | Spike glycoprotein | SARS-CoV |
| KVCEFQFCNDPFLGVYYHKNNKS WMESEFRVYSSANNCTFEYV | 1074963 | Spike glycoprotein | SARS-CoV |
| MHC Class-II | |||
| 1. FVSNGTHWF | |||
| RG VSNGTHV | 834339 | Oncoprotein-induced transcript 3 protein | Mus musculus (mouse) |
| REGV FVSNGTHW | 1075025 | Spike glycoprotein | SARS-CoV |
| 2. IRASANLAA | |||
| AE IRASANLA | 999 | Spike glycoprotein | SARS-CoV |
| ASANLA ATK | 4321 | Spike glycoprotein | SARS-CoV |
| QLIRAAEIRASANLAAT | 51379 | Spike glycoprotein | SARS-CoV |
| QQLIRAAE IRASANL | 52057 | Spike glycoprotein | SARS-CoV |
| RASANLAA TKMSECVLG | 53202 | Spike glycoprotein | SARS-CoV |
| QLIRAAE IRASANLAATK | 100428 | Spike glycoprotein | SARS-CoV |
| AE IRASANLAATK | 1074838 | Spike glycoprotein | SARS-CoV |
| 3. FGAISSVLN | |||
| AISSVLN DILSRLDKVE | 2092 | Spike glycoprotein | SARS-CoV |
| KQLSSNF GAISSVLNDI | 33032 | Spike glycoprotein | SARS-CoV |
| SSNF GAISSVLNDIL | 61229 | Spike glycoprotein | SARS-CoV |
| NF GAISSVL | 923559 | Spike glycoprotein | SARS-CoV |
| 4. VKQLSSNFG | |||
| QALNTL VKQLSSNFGAI | 50311 | Spike glycoprotein | SARS-CoV |
| KQLSSNFG AISSVLNDI | 33032 | Spike glycoprotein | |
| LNTL VKQLSSNFGAI | 38353 | Spike glycoprotein | SARS-CoV |
| 5. FAMQMAYRF | |||
| AMQMAYRF | 3176 | Spike glycoprotein | SARS-CoV |
| GAALQIP FAMQMAYR | 18514 | Spike glycoprotein | SARS-CoV |
| GAALQIP FAMQMAYRFN | 18515 | Spike glycoprotein | SARS-CoV |
| P FAMQMAYRFNGIGVTQ | 47479 | Spike glycoprotein | SARS-CoV |
| QIP FAMQMAYRFNGI | 51112 | Spike glycoprotein | SARS-CoV |
| GAALQIP FAMQMAYRF | 100048 | Spike glycoprotein | SARS-CoV |
| LQIP FAMQMAY | 1074986 | Spike glycoprotein | SARS-CoV |
| MIAQYTSALLAGTITSGWTFGAGAALQIP FAMQMAYRFNGIGV | 1074998 | Spike glycoprotein | SARS-CoV |
| 6. FGAGAALQI | |||
| GWT FGAGAALQIPFA | 23293 | Spike glycoprotein | SARS-CoV |
| TAGWT FGAGAALQIPFA | 62872 | Spike glycoprotein | SARS-CoV |
| GWT FGAGAALQIPFA | 23293 | Spike glycoprotein | SARS-CoV |
| GAALQI PFAMQMAYRFN | 18515 | Spike glycoprotein | SARS-CoV |
| GAALQI PFAMQMAYRF | 100048 | Spike glycoprotein | SARS-CoV |
| GAALQI PFAMQMAYR | 18514 | Spike glycoprotein | SARS-CoV |
| SGP GAGAAL | 230418 | Other Leishmania infantum protein | Leishmania infantum |
| B-cell linear epitope | |||
| 1. LTPGDSSSGWTAG | |||
| G GDSSSGPQRLV | 616721 | Transmembrane protein 199 | Homo sapiens (human) |
| GDSSSG PQRLV | 690393 | Transmembrane protein 199 | H. sapiens (human) |
| KG GDSSSGPQRLV | 691072 | Transmembrane protein 200 | H. sapiens (human) |
| GYD GDSSSGSGR | 760719 | Tight junction protein ZO-3 | H. sapiens (human) |
| TLLALHRSY LTPGDSSSGWTAGAAAYYVGYLQPRTFLLKYNEN | 1075077 | Spike glycoprotein | SARS-CoV |
| 2. YGFQPTNGVGYQ | |||
| PTNGVGYQ PYRVVVLSFELLHAPATVCGPKKSTNLVKNKCVNF | 1075016 | Spike glycoprotein | SARS-CoV |
| F QPTNGVGY | 1087346 | Spike glycoprotein | SARS-CoV |
| 3. VITPGTNTSN | |||
| GVS VITPGTNASSEV | 23158 | Spike glycoprotein | SARS-CoV |
| ISPCAFGGVS VITPGTN | 28643 | Spike glycoprotein | SARS-CoV |
| PCSFGGVS VITPGTN | 47041 | Spike glycoprotein | SARS-CoV |
| VITPGTN ASSEVAVLY | 69129 | Spike glycoprotein | SARS-CoV |
| VS VITPGTNASSEVAVL | 71189 | Spike glycoprotein | SARS-CoV |
| 4. QTQTNSPRRARS | |||
| AGCLIGAEHVNNSYECDIPIGAGICASY QTQTNSPRRARSVAS | 1074840 | Spike glycoprotein | SARS-CoV |
| SY QTQTNSPRRARSVA | 1075070 | Spike glycoprotein | SARS-CoV |
| 5. YQAGSTPCNG | |||
| No Hit | No Hit | No Hit | No Hit |
| 6. LPDPSKPSKR | |||
| PSKPSKR SFIEDLLFNKV | 1071808 | Spike glycoprotein | SARS-CoV |
| I LPDPSKPSK | 1074928 | Spike glycoprotein | SARS-CoV |
| KDFGGFNFSQI LPDPSKPSKRSFIEDLLFNKVTLADAGFIKQY | 1074948 | Spike glycoprotein | SARS-CoV |
Peptide–HLA interaction analysis
To ensure the interaction between the CD+8 T-cell peptides (LTDEMIAQY, WTAGAAAYY and WMESEFRVY) and HLA molecules [HLA-B*53:01(1A1M), HLA-B*44:03(4JQX) and HLA-B*44:03(4JQX), respectively], we performed molecular docking analysis and found that the peptide LTDEMIAQY binds with HLA-B*53:01, having a good docking score of 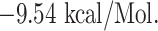 Similarly, WTAGAAAYY and WMESEFRVY bind with HLA-B*44:03, having a binding affinity of
Similarly, WTAGAAAYY and WMESEFRVY bind with HLA-B*44:03, having a binding affinity of 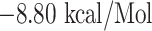 and
and 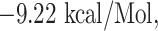 respectively. Moreover, all the CD+4 T-cell peptides were binding into the groove of HLA-DR molecules [HLA-DRB1*01:01(1AQD)] with a good docking score, for example,
respectively. Moreover, all the CD+4 T-cell peptides were binding into the groove of HLA-DR molecules [HLA-DRB1*01:01(1AQD)] with a good docking score, for example, 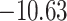
 (with IRASANLAA),
(with IRASANLAA), 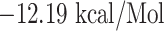 (with FGAISSVLN),
(with FGAISSVLN), 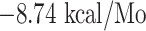 l (with VKQLSSNFG),
l (with VKQLSSNFG), 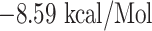 (with FAMQMAYRF), and
(with FAMQMAYRF), and 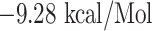 (with FGAGAALQ), and all the interactions are shown in Figures 5 and 6, and the docking details are given in Table 4.
(with FGAGAALQ), and all the interactions are shown in Figures 5 and 6, and the docking details are given in Table 4.
Figure 5.
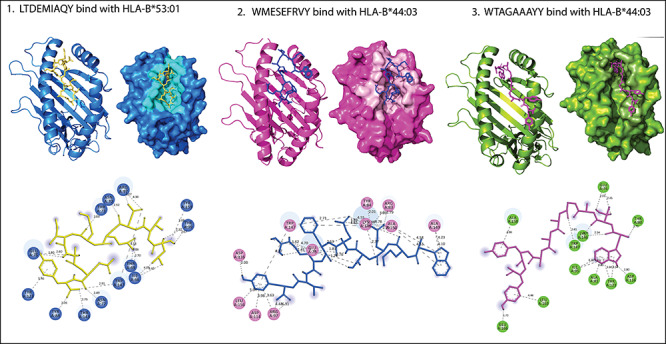
Peptide LTDEMIAQY (yellow) binds in the groove of the HLA-B*53:01. While other two peptides WTAGAAAYY (magenta) and WMESEFRVY (blue) bind in the groove of the HLA-B*44:03. All H-bonds and other type of interactions are represented in dotted lines with bond length (in À).
Figure 6.
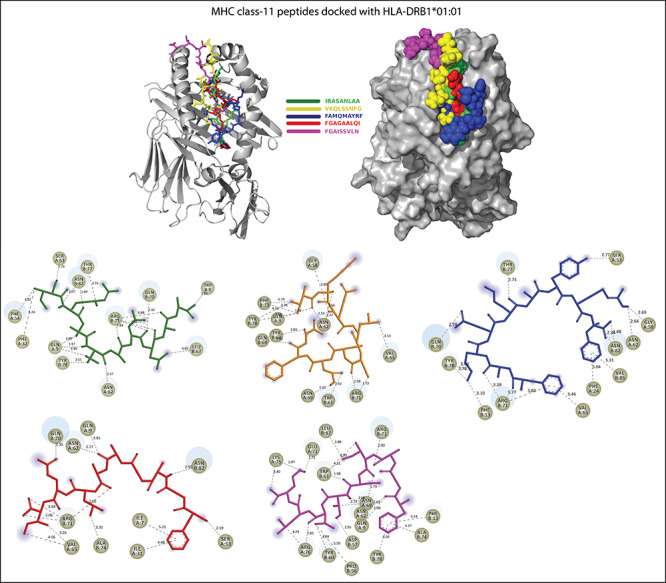
Five peptides including IRASANLAA (green), FGAISSVLN (magenta), VKQLSSNFG (pale yellow), FAMQMAYRF (blue) and FGAGAALQI (red) bind in the groove of HLA-DRB1*01:01. All H-bonds and other type of interactions are represented in dotted lines with bond length (in À).
Table 4.
The details of molecular docking results, including docking score (kcal/Mol), interacting amino acids, and various types of interactions involved in the binding of potential Peptides (epitopes)to HLA molecules.
| Peptides + HLA molecules | Docking score (kcal/Mol) | Interacting residues | Interaction types |
|---|---|---|---|
| CD + 8 T-cell peptides interactions | |||
| LTDEMIAQY + HLA-B*53:01 | −9.54 | LYS-146, TYR-84, ASN-77, GLU-76, THR-73, ASN-70, ARG-97 and TYR-99 | Salt bridges, hydrogen bond, Pi-cation, Pi-anion, Pi-Pi stacked and Akyl |
| WTAGAAAYY + HLA-B*44:03 | −8.80 | GLU-76, THR-80, ALA-81, ILE-95, ASP-116, TYR-123, LYS-146, TRP-147, ALA-158, LEU-163 and GLU-166 | Salt bridges, hydrogen bond, Pi-cation, Pi-anion, Pi-Pi stacked, Amide-Pi stacked and Pi-Akyl |
| WMESEFRVY + HLA-B*44:03 | −9.22 | THR-73 GLU-76 ARG-83, TYR-84, ARG-97, ASP-114, ASP-116, LYS-146 TRP-147, ALA-149, ALA-150 and LEU-156 | Hydrogen bond, Pi-Pi stacked and Pi-Akyl |
| CD + 4 T-cell peptides interactions | |||
| IRASANLAA + HLA-DRB1*01:01 | −10.63 | GLN-09, PHE-32, PHE-54, ASN-62, LEU-67, GLN-70, ARG-71 and TYR-78 | Hydrogen bond, Pi-Sigma, Akyl and Pi-Akyl |
| VKQLSSNFG + LA-DRB1*01:01 | −8.74 | GLN-09, PHE-13, GLY-58, TYR-60, TRP-61, ASN-62, GLN-64, VAL-65, ASN-69, ARG-71 and TYR-78 | Hydrogen bond, Akyl and Pi-Akyl |
| FAMQMAYRF + HLA-DRB1*01:01 | −8.59 | PHE-13, PHE-24, SER-53, GLY-58, ASN-62, VAL-65, GLN-70, ARG-71, THR-77, TRY-78, ASN-82 and VAL-85 | Hydrogen bond, Pi-cation, Pi-sulfur, Pi-Pi T-shaped and Pi-Akyl |
| FGAGAALQ + HLA-DRB1*01:01 | −9.28 | ILE-07, GLN-09, ILE-31, SER-53, ASN-62, VAL-65, GLN-70, ARG-71, ALA-74 and ASN-82 | Salt bridges, hydrogen bond, Akyl and Pi-Akyl |
| FGAISSVLN + HLA-DRB1*01:01 | −12.19 | GLN-09, PHE-13, PRO-56, TYR-60, TRP-61, ASN-62, LEU-67, ASN-69, GLU-71, ALA-74, LYS-75, LYS-75, ATG-76 and TYR-78 | Salt bridges, hydrogen bond, Pi-Pi stacked, Pi-Pi T-shaped and Akyl and Pi-Akyl |
Discussion
The development of vaccines refers to one of the most effective and cost-effective medical and public health achievements of all time. It is a very lengthy, complex and costly process and requires the collaborative involvement of public and private sectors. In each year, vaccination programs save over 3 million lives globally. The peptide, as a choice of vaccine agent, has made the astonishing move toward vaccine design against the viruses, bacteria and cancer. The peptide vaccine is often synthetic and mimics naturally occurring proteins from pathogens, and these peptide-based vaccines have shown promising successful results in the past for diseases like malaria, dengue, multiple sclerosis and influenza. Besides these diseases, the peptide-based vaccines have also been developed against several types of cancer like colorectal cancer, myeloid leukemia and gastric cancer [59–61]. The identification and design of immunogenic peptides (epitopes) are expensive as well as time consuming process. So the vaccine-informatics approach has made it easy to identify potent peptides. In the present study, we have identified CD8+ T-cell (three peptides), CD4+ T-cell (six peptides) and B-cell linear peptides (six peptides) from the SARS-CoV-19 SG; however, we were more emphasized to study T-cell peptides because vaccines against the T-cell epitopes are more promising as they evoke a long-lasting immune response, with antigenic drift, and an antigen can easily escape the memory response of antibody [62, 63].
For the MHC class-I binding peptides, the immune responses for the top five different geographical regions (highest population coverage range 30–34%) were: South Africa: 33.78% (South Africa), West Indies: 33.12% (Cuba, Jamaica, Martinique, Trinidad and Tobago), East Africa: 32.25% (Kenya, Uganda, Zambia and Zimbabwe), Central Africa: 30.70% (Cameroon, Central African Republic, Congo, Equatorial Guinea, Gabon, Rwanda and Sao Tome and Principe) and East Asia: 30% (Japan, South Korea and Mongolia). Similarly, for the MHC class-II binding peptides, the excepted immune response was found to be remarkable (PCA range: 60–76%) for North America (Canada, Mexico and the United States), Europe (Austria, Belarus, Belgium, Bulgaria, Croatia, Czech Republic, Denmark, England, France, Georgia, Germany, Greece, Ireland Northern, Ireland South, Italy, Macedonia, Netherlands, Norway, Poland, Portugal, Romania, Russia, Scotland, Serbia, Slovakia, Spain, Sweden, Switzerland, Turkey, Ukraine, United Kingdom and Wales), East Asia (Japan, South Korea and Mongolia), South Asia (India, Pakistan and Sri Lanka), North Africa (Algeria, Ethiopia, Mali, Morocco, Sudan and Tunisia) and West Indies (Cuba, Jamaica, Martinique, Trinidad and Tobago). Thus, our results suggested that the MHC class-II binding peptides may be potent vaccine agents for different populations of the globe.
We know that vaccination is the utmost prevention of epidemiologic infectious diseases, but it has a low incidence of serious systemic adverse effects (like autoimmune diseases). We have predicted a total of 15 peptides (B- and T-cells),and among those, 12 peptides are very important, including LTDEMIAQY, WTAGAAAYY, WMESEFRVY, IRASANLAA, FGAISSVLN, VKQLSSNFG, FAMQMAYRF, FGAGAALQI, YGFQPTNGVGYQ, LPDPSKPSKR, QTQTNSPRRARS and VITPGTNTSN because their peptide fragments are matching with the experimentally identified epitopes of the glycoprotein of SARS-CoV [64–68]; thus, these peptides are seemingly a more rational set of potential vaccine agents against the SARS-CoV-2.
Moreover, the other two peptides LTPGDSSSGWTAG and FVSNGTHWF, which resemble with the Homo sapiens (human) and Mus musculus proteomes, were eliminated from the study to avoid autoimmunity risk. Besides, we did not find any resemblance of one peptide (YQAGSTPCNG) with any organism’s proteome. Hence, this unique peptide can also be proposed as a candidate for further studies in the area of vaccines development, therapeutic antibodies and diagnostic tools against the SARS-CoV-2.
As we know that HLA alleles are polymorphic in nature; so peptides can interact with them. In our study, MHC class-I binding peptides (8–9 residues long) are bound in the wide groove (~1.25À) of HLA molecules (HLA-B*53:01 and HLA-B*44:03). Among them, the peptide LTDEMIAQY bound with HLA-B*53:01 molecule by. ILE-66, THR-69, ASN-70, THR-73, GLU-76, ASN-77, TYR-84, ARG-97, TYR-99, TYR-123, THR-143, TRP-147, TYR-159 residues and other surrounding residues, which gives it polymorphic nature lie at the interface of this helix and the bottom of the peptide-binding site. In docking with HLA-B*44:03, the peptides WTAGAAAYY (interacted with GLU-76, THR-80, ALA-81, ILE-95, ASP-116, TYR-123, LYS-146, TRP-147, ALA-158, LEU-163 and GLU-166) and WMESEFRVY (interacted with THR-73 GLU-76 ARG-83, TYR-84, ARG-97, ASP-114, ASP-116, LYS-146 TRP-147, ALA-149, ALA-150 and LEU-156) got expanded conformation within the same trench, and both peptides were surrounded by the polymorphic region. Further, we checked the interaction of MHC class-II binding peptides with HLA-DR molecules and noted that all the five peptides are buried by interactions with the most important pocket residues, for example, GLN-09, PHE-13, ASN-62, VAL-65, GLN-70, ARG-71, TYR-78, etc., and are the preferred bind in the groove of HLA-DR molecules. The docking studies suggested that these peptides are preferably bound into the groove of respective alleles and can induce the CD+8 and CD+4 T-cell immunity.
In our study, integrated computational approaches have identified a total of 15 peptides (T- and B-cells) from SARS-CoV-19 SG, of which 12 peptides have resemblance with experimentally identified epitopes of SARS-CoV and other pathogens. There is no vaccine or specific treatment currently available for COVID-19, and vaccine development is a long way from being translated into practical applications. So at this crunch situation, we have suggested that the predicted peptides (epitopes) would be capable to prompt an effective immune response as a peptide vaccine against the SARS-CoV-2. Consequently, these peptides may be used for synthetic vaccine design to combat the emerging COVID-19. However, in vivo and in vitro, experimental validation is needed for their efficient use as a vaccine candidate.
Key Points
As we aware, the COVID-19 infection is the current global health epidemic, with very high infection and spreading rate, which is changing the deadly global figure, hour by hour. Unluckily, no vaccines or specific treatment is available, which makes it more deadly.
This study provides knowledge about the involvement of SARS-CoV-2 spike glycoprotein in the immune pathogenesis of the virus as well as induced the protective immune response.
In this study, we have identified 15 antigenic peptides (epitopes) in SARS-CoV-2 spike glycoprotein. These epitopes are capable of evoking the T-cell immune response via interacting with the MHC class-I and II molecules, and few epitopes are also capable of inducing the B-cell immune response.
Notably, we found 12 peptides (epitopes) that have 80–90% identity with experimentally identified epitopes of SARS-CoV, and this will likely be beneficial for a quick progression of the vaccine design.
All peptides are nontoxic and nonallergenic in nature, highly antigenic and non-mutated in other SARS-CoV-2 virus strains.
Our study provides the knowledge and boosts the ongoing and struggling scientific society for designing an effective vaccine to stop the current global health emergency.
Supplementary Material
Acknowledgements
R.I and A.K are grateful to the Centre for Interdisciplinary Research in Basic Sciences (CIRBSc), Jamia Millia Islamia, for providing the research infrastructure. All the authors are also grateful for the School of Computational and Integrative Sciences (SC&IS), JNU, New Delhi, for providing Glide, Schrödinger software. Nikhat imam is thankful to the Indian Council of Medical Research (ICMR), New Delhi, for award of Senior Research Fellowship (ISRM/11(03)12019). M.F.S. is grateful to Prof. Muratov Zhanybek Kudaibakovich and Dr Syed Ali Abbas from the Osh State University, Kyrgyzstan, for their guidance and support. Aftab Alam acknowledges the Department of Health Research (DHR), New Delhi, India, for the award of ‘Young Scientist’ fellowship (R.12014/06/2019-HR).
Mr Aftab Alam is a Ph.D candidate and working as a ‘Young Scientist’ at the Centre for Interdisciplinary Research in Basic Sciences, Jamia Millia Islamia University, New Delhi 110025, India. He is working on the advance topic of system biology, for example, “Network Medicine or Network Pharmacology” and “Complex Biological Networks”.
Mr Arbaaz Khan is a master student (MSc) at the Department of computer science, Jamia Millia Islamia University, New Delhi, India.
Miss Nikhat Imam is a PhD candidate at the Magadh University (Bihar, India). Currently, she is working as a Senior Research Fellow (SRF) at the Centre for Interdisciplinary Research in Basic Science, Jamia Millia Islamia University, New Delhi, India. She is working on network meta-analysis of endocrine disorders.
Mr Mohd Faizan Siddiqui is a medical student at the International Medical Faculty, Osh State University, Kyrgyz Republic (Kyrgyzstan).
Mr. Mohd Waseem is PhD candidate at at the School of Computational & Integrative Sciences, Jawaharlal Nehru University, New Delhi, India.
Dr. Md. Zubbair Malik is working as ‘Young Scientist’ at the School of Computational & Integrative Sciences, Jawaharlal Nehru University, New Delhi, India. Dr. Zubbair does research in Bioinformatics, computational biology, network biology and computational neuroscience.
Dr Romana Ishrat is an associate professor at the Centre for Interdisciplinary Research in Basic Science, Jamia Millia Islamia University, New Delhi, India. Her research interest is to develop and apply novel statistical/computational methods to solve genetics and genomics problems associated with complex diseases.
Contributor Information
Aftab Alam, Centre for Interdisciplinary Research in Basic Sciences, Jamia Millia Islamia University, New Delhi 110025, India.
Arbaaz Khan, Department of computer science, Jamia Millia Islamia University, New Delhi, India.
Nikhat Imam, Centre for Interdisciplinary Research in Basic Science, Jamia Millia Islamia University, New Delhi, India.
Mohd Faizan Siddiqui, International Medical Faculty, Osh State University, Kyrgyz Republic.
Mohd Waseem, School of Computational & Integrative Sciences, Jawaharlal Nehru University, New Delhi, India.
Md Zubbair Malik, School of Computational & Integrative Sciences, Jawaharlal Nehru University, New Delhi, India.
Romana Ishrat, Centre for Interdisciplinary Research in Basic Science, Jamia Millia Islamia University, New Delhi, India.
Authors’ Contributions
R.I. and A.A. conceived the study design instructed on data analysis. A.K. and A.A. curated data and performed statistical analyses. N.I. and M.F.S. curated data and drew figures. M.W. and M.Z.M. performed molecular docking studies. A.A. improved and revised the manuscript. All the authors read, edited and approved of the manuscript.
Funding
This work did not receive any specific grant from funding agencies in the public, commercial or not-for-profit sectors.
Data Availability Statement
Data is with the authors and will be provided on request through corresponding author.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- 1. Guo Y-R, Cao Q-D, Hong Z-S, et al. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—an update on the status. Mil Med Res 2020;7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zhao S, Lin Q, Ran J, et al. Preliminary estimation of the basic reproduction number of novel coronavirus (2019-nCoV) in China, from 2019 to 2020: a data-driven analysis in the early phase of the outbreak. Int J Infect Dis 2020;92:214–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alam A, Siddiqui MF, Imam N, et al. Covid-19: current knowledge, disease potential, prevention and clinical advances. Turk J Biol Turk Biyol Derg 2020;44:121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Islam MT, Sarkar C, El-Kersh DM, et al. Natural products and their derivatives against coronavirus: a review of the non-clinical and pre-clinical data. Phytother Res 2020;34:2471–92. [DOI] [PubMed] [Google Scholar]
- 5. Chen H, Du Q. Potential Natural Compounds for Preventing SARS-CoV-2 (2019-nCoV) Infection. 2020.
- 6. Moorthy V, Henao Restrepo AM, Preziosi M-P, et al. Data sharing for novel coronavirus (COVID-19). Bull World Health Organ 2020;98:150–0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang K. Is traditional Chinese medicine useful in the treatment of COVID-19? Am J Emerg Med YAJEM-158851; p.1. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Y, Liu X, Guo L, et al. Traditional Chinese herbal medicine for treating novel coronavirus (COVID-19) pneumonia: protocol for a systematic review and meta-analysis. Syst. Dent Rev 2020;9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liu C. Pay attention to situation of SARS-CoV-2 and TCM advantages in treatment of novel coronavirus infection. Chin Herb Med 2020;12:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang Y, Islam MS, Wang J, et al. Traditional Chinese medicine in the treatment of patients infected with 2019-new coronavirus (SARS-CoV-2): a review and perspective. Int J Biol Sci 2020;16:1708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dong L, Hu S, Gao J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov Ther 2020;14:58–60. [DOI] [PubMed] [Google Scholar]
- 12. Alam A, siddiqui MF, Nikhat I, et al. COVID-19: current knowledge, disease potential, prevention and clinical advances. Turk J Biol 2020;44:121–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tambunan n silico analysis of envelope Dengue Virus-2 and envelope Dengue Virus-3 protein as the backbone of Dengue Virus tetravalent vaccine by using homology modeling method. OnLine J Biol Sci 2009;9:6–16. [Google Scholar]
- 14. López JA, Weilenman C, Audran R, et al. A synthetic malaria vaccine elicits a potent CD8(+) and CD4(+) T lymphocyte immune response in humans. Implications for vaccination strategies. Eur J Immunol 2001;31:1989–98. [DOI] [PubMed] [Google Scholar]
- 15. Shahsavandi S, Ebrahimi MM, Sadeghi K, et al. Design of a heterosubtypic epitope-based peptide vaccine fused with hemokinin-1 against influenza viruses. Virol Sin 2015;30:200–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bourdette DN, Edmonds E, Smith C, et al. A highly immunogenic trivalent T cell receptor peptide vaccine for multiple sclerosis. Mult Scler Houndmills Basingstoke Engl 2005;11:552–61. [DOI] [PubMed] [Google Scholar]
- 17. Knutson KL, Schiffman K, Disis ML. Immunization with a HER-2/neu helper peptide vaccine generates HER-2/neu CD8 T-cell immunity in cancer patients. J Clin Invest 2001;107:477–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Petrovsky N, Brusic V. Computational immunology: the coming of age. Immunol Cell Biol 2002;80:248–54. [DOI] [PubMed] [Google Scholar]
- 19. Brusic V, Bajic VB, Petrovsky N. Computational methods for prediction of T-cell epitopes—a framework for modelling, testing, and applications. Methods San Diego Calif 2004;34:436–43. [DOI] [PubMed] [Google Scholar]
- 20. Nielsen M, Lundegaard C, Lund O, et al. The role of the proteasome in generating cytotoxic T-cell epitopes: insights obtained from improved predictions of proteasomal cleavage. Immunogenetics 2005;57:33–41. [DOI] [PubMed] [Google Scholar]
- 21. Bhattacharya M, Sharma AR, Patra P, et al. Development of epitope-based peptide vaccine against novel coronavirus 2019 (SARS-COV-2): immunoinformatics approach. J Med Virol 2020;92:618–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Joshi A, Joshi BC, Mannan MA, et al. Epitope based vaccine prediction for SARS-COV-2 by deploying immuno-informatics approach. Inform Med Unlocked 2020;19:100338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Abdelmageed MI, Abdelmoneim AH, Mustafa MI, et al. Design of a multiepitope-based peptide vaccine against the E protein of human COVID-19: an immunoinformatics approach. Biomed Res Int 2020;2020:2683286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shang J, Wan Y, Luo C, et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A 2020;117:11727–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ou X, Liu Y, Lei X, et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat Commun 2020;11:1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sievers F, Wilm A, Dineen D, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 2011;7:539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Larsen MV, Lundegaard C, Lamberth K, et al. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics 2007;8:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinforma Oxf Engl 2008;24:1397–8. [DOI] [PubMed] [Google Scholar]
- 29. Buchan DWA, Jones DT. The PSIPRED protein analysis workbench: 20 years on. Nucleic Acids Res 2019;47:W402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Giguère S, Drouin A, Lacoste A, et al. MHC-NP: predicting peptides naturally processed by the MHC. J Immunol Methods 2013;400–401:30–6. [DOI] [PubMed] [Google Scholar]
- 31. Wang P, Sidney J, Kim Y, et al. Peptide binding predictions for HLA DR, DP and DQ molecules. BMC Bioinformatics 2010;11:568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chou PY, Fasman GD. Prediction of the secondary structure of proteins from their amino acid sequence. Adv Enzymol Relat Areas Mol Biol 1978;47:45–148. [DOI] [PubMed] [Google Scholar]
- 33. Larsen J, Lund O, Nielsen M. No title found. Immunome Res 2006;2:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chou PY, Fasman GD. Empirical predictions of protein conformation. Annu Rev Biochem 1978;47:251–76. [DOI] [PubMed] [Google Scholar]
- 35. Emini EA, Hughes JV, Perlow DS, et al. Induction of hepatitis a virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol 1985;55:836–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Karplus PA, Schulz GE. Prediction of chain flexibility in proteins: a tool for the selection of peptide antigens. Naturwissenschaften 1985;72:212–3. [Google Scholar]
- 37. Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 1990;276:172–4. [DOI] [PubMed] [Google Scholar]
- 38. Parker JMR, Guo D, Hodges RS. New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and x-ray-derived accessible sites. Biochemistry 1986;25:5425–32. [DOI] [PubMed] [Google Scholar]
- 39. Bui H-H, Sidney J, Li W, et al. Development of an epitope conservancy analysis tool to facilitate the design of epitope-based diagnostics and vaccines. BMC Bioinformatics 2007;8:361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Calis JJA, Maybeno M, Greenbaum JA, et al. Properties of MHC class I presented peptides that enhance immunogenicity. PLoS Comput Biol 2013;9:e1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Thévenet P, Shen Y, Maupetit J, et al. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res 2012;40:W288–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Smith KJ, Reid SW, Harlos K, et al. Bound water structure and polymorphic amino acids act together to allow the binding of different peptides to MHC class I HLA-B53. Immunity 1996;4:215–28. [DOI] [PubMed] [Google Scholar]
- 43. Rist MJ, Theodossis A, Croft NP, et al. HLA peptide length preferences control CD8+ T cell responses. J Immunol 2013;191:561–71. [DOI] [PubMed] [Google Scholar]
- 44. Murthy VL, Stern LJ. The class II MHC protein HLA-DR1 in complex with an endogenous peptide: implications for the structural basis of the specificity of peptide binding. Structure 1997;5:1385–96. [DOI] [PubMed] [Google Scholar]
- 45. Berman HM. The protein data bank. Nucleic Acids Res 2000;28:235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bui H-H, Sidney J, Dinh K, et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinformatics 2006;7:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fleri W, Vaughan K, Salimi N, et al. The immune epitope database: how data are entered and retrieved. J Immunol Res 2017;2017:5974574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dhanda SK, Mahajan S, Paul S, et al. IEDB-AR: immune epitope database-analysis resource in 2019. Nucleic Acids Res 2019;47:W502–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Grifoni A, Sidney J, Zhang Y, et al. A sequence homology and bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe 2020;27:671–680.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ahmed SF, Quadeer AA, McKay MR. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 2020;12:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Saha S, Raghava GPS. AlgPred: prediction of allergenic proteins and mapping of IgE epitopes. Nucleic Acids Res 2006;34:W202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gupta S, Kapoor P, Chaudhary K, et al. In silico approach for predicting toxicity of peptides and proteins. PLoS One 2013;8:e73957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Friesner RA, Murphy RB, Repasky MP, et al. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein−ligand complexes. J Med Chem 2006;49:6177–96. [DOI] [PubMed] [Google Scholar]
- 54. Friesner RA, Banks JL, Murphy RB, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47:1739–49. [DOI] [PubMed] [Google Scholar]
- 55. Halgren TA, Murphy RB, Friesner RA, et al. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem 2004;47:1750–9. [DOI] [PubMed] [Google Scholar]
- 56. Halgren TA. Identifying and characterizing binding sites and assessing druggability. J Chem Inf Model 2009;49:377–89. [DOI] [PubMed] [Google Scholar]
- 57. Cohen AD, Shoenfeld Y. Vaccine-induced autoimmunity. J Autoimmun 1996;9:699–703. [DOI] [PubMed] [Google Scholar]
- 58. Jain S, Baranwal M. Computational analysis in designing T cell epitopes enriched peptides of Ebola glycoprotein exhibiting strong binding interaction with HLA molecules. J Theor Biol 2019;465:34–44. [DOI] [PubMed] [Google Scholar]
- 59. Maslak PG, Dao T, Bernal Y, et al. Phase 2 trial of a multivalent WT1 peptide vaccine (galinpepimut-S) in acute myeloid leukemia. Blood Adv 2018;2:224–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sundar R, Rha SY, Yamaue H, et al. A phase I/Ib study of OTSGC-A24 combined peptide vaccine in advanced gastric cancer. BMC Cancer 2018;18:332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Melief CJM, Burg SH. Immunotherapy of established (pre)malignant disease by synthetic long peptide vaccines. Nat Rev Cancer 2008;8:351–60. [DOI] [PubMed] [Google Scholar]
- 62. Chiou S-S, Fan Y-C, Crill WD, et al. Mutation analysis of the cross-reactive epitopes of Japanese encephalitis virus envelope glycoprotein. J Gen Virol 2012;93:1185–92. [DOI] [PubMed] [Google Scholar]
- 63. Alam A, Ali S, Ahamad S, et al. From ZikV genome to vaccine: in silico approach for the epitope-based peptide vaccine against Zika virus envelope glycoprotein. Immunology 2016;149:386–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. He Y, Zhou Y, Wu H, et al. Identification of immunodominant sites on the spike protein of severe acute respiratory syndrome (SARS) coronavirus: implication for developing SARS diagnostics and vaccines. J Immunol 2004;173:4050–7. [DOI] [PubMed] [Google Scholar]
- 65. Yang J, James E, Roti M, et al. Searching immunodominant epitopes prior to epidemic: HLA class II-restricted SARS-CoV spike protein epitopes in unexposed individuals. Int Immunol 2009;21:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Sidney J, Botten J, Neuman B, Buchmeier M, Sette A. HLA DRB1*01:01 binding capacity of selected SARS-derived peptides IEDB_SUBMISSION 2006;
- 67. Wang B, Chen H, Jiang X, et al. Identification of an HLA-A*0201–restricted CD8+ T-cell epitope SSp-1 of SARS-CoV spike protein. Blood 2004;104:200–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Guo J-P, Petric M, Campbell W, et al. SARS corona virus peptides recognized by antibodies in the sera of convalescent cases. Virology 2004;324:251–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data is with the authors and will be provided on request through corresponding author.