

Abstract
With the outbreak of the COVID-19 disease, the research community is producing unprecedented efforts dedicated to better understand and mitigate the effects of the pandemic. In this context, we review the data integration efforts required for accessing and searching genome sequences and metadata of SARS-CoV2, the virus responsible for the COVID-19 disease, which have been deposited into the most important repositories of viral sequences. Organizations that were already present in the virus domain are now dedicating special interest to the emergence of COVID-19 pandemics, by emphasizing specific SARS-CoV2 data and services. At the same time, novel organizations and resources were born in this critical period to serve specifically the purposes of COVID-19 mitigation while setting the research ground for contrasting possible future pandemics. Accessibility and integration of viral sequence data, possibly in conjunction with the human host genotype and clinical data, are paramount to better understand the COVID-19 disease and mitigate its effects. Few examples of host-pathogen integrated datasets exist so far, but we expect them to grow together with the knowledge of COVID-19 disease; once such datasets will be available, useful integrative surveillance mechanisms can be put in place by observing how common variants distribute in time and space, relating them to the phenotypic impact evidenced in the literature.
Keywords: epidemic, viral sequences, genomics, metadata, data harmonization, integration and search, COVID-19
Introduction
The outbreak of the COVID-19 disease is presenting novel challenges to the research community, which is rushing toward the delivery of results, pushed by the intent of rapidly mitigating the pandemic effects. During these times, we observe the production of an exorbitant amount of data, often associated with a poor quality of describing information, sometimes generated by insufficiently tested or not peer-reviewed efforts. But we also observe contradictions in published literature, as it is typical of a disease that is still at its infancy, and thus only partially understood.
In this context, the collection of viral genome sequences is of paramount importance, in order to study the origin, wide spreading and evolution of SARS-CoV2 (the virus responsible for the COVID-19 disease) in terms of haplotypes, phylogenetic tree and new variants.
Since the beginning of the pandemic, we have observed an almost exponential growth of the number of deposited sequences within large shared databases, from few hundreds up to thousands; indeed, it is the first time that next-generation sequencing technologies have been used for sequencing a massive amount of viral sequences. As of 8 October 2020, the total number of sequences of SARS-CoV2 available worldwide is about 150 000 (as it can be retrieved by summing up the counts of the main data sources and excluding duplicates based on the comparison of strain names, length and nucleotide percentages). In several cases, also relevant associated data and metadata are provided, although their amount, coverage and harmonization are still limited.
Several institutions provide databases and resources for depositing viral sequences. Some of them, such as NCBI’s GenBank [1], preexist the COVID-19 pandemic, as they host thousands of viral species, including e.g. Ebola, SARS and Dengue, which are also a threat to humanity. Other organizations have produced a new data collection specifically dedicated to the hosting of SARS-CoV2 sequences, such as GISAID [2, 3]—originally created for hosting virus sequences of influenza—which is soon becoming the predominant data source.
Most data sources reviewed in this paper, including GenBank, COG-UK [4] and some new data sources from China, have adopted a fully open-source model of data distribution and sharing. Instead, GISAID is protecting the deposited sequences by controlling users, who must log in from an institutional site and must observe a database access agreement (https://www.gisaid.org/registration/terms-of-use/); probably, such protected use of the deposited data contributes to the success of GISAID in attracting depositors from around the world.
Given that viral sequence data are distributed over many database sources, there is a need for data integration and harmonization, so as to support integrative search systems and analyses; many such search systems have been recently developed, motivated by the COVID-19 pandemic.
In this review paper, we start from describing the database sources hosting viral sequences and related data and metadata, distinguishing between fully open-source and GISAID. We then discuss the data integration issues that are specific to viral sequences, by considering schema integration and value harmonization. Then, we present the various search systems that are available for integrative data access to viral resources. We also briefly discuss how viral genome sequences can be connected to the human phenotype and genotype, so as to build an inclusive, holistic view of how SARS-CoV2 virus sequences can be linked to the COVID-19 disease and to support integrative analyses that can help its mitigation. An active monitoring on variants with their impacts and space-time distribution can provide a first integrative support to viral surveillance, while integrated data about virus sequences, their haplotypes and known epitopes may reveal very useful for supporting vaccine research.
Landscape of data resources for viral sequences
The panorama of relevant initiatives dedicated to data collection, retrieval and analysis of viral sequences is broad. Many resources previously available for viruses have responded to the general call to arms against the COVID-19 pandemic and started collecting data about SARS-CoV2.
According to the WHO’s code of conduct [5], alternative options are available to data providers of virus sequences. The providers who are not concerned about retaining ownership of the data may share it within the many databases that provide full open data access. Among them, GenBank assumes that its submitters have ‘received any necessary informed consent authorizations required prior to submitting sequences’, which includes data redistribution. However, in many cases data providers prefer data sharing options in which they retain some level of data ownership. This attitude has established since the influenza pandemics (around 2006), when the alternative model of GISAID EpiFluTM has emerged as dominant. We next review the sources of virus sequences, starting with the sources that provide full open access, and then presenting GISAID.
Fully open-source resources
Resources coordinated by International Nucleotide Sequence Database (INSDC)
The three main organizations providing open-source viral sequences are NCBI (US), DDBJ (Japan), and EMBL-EBI (Europe); they operate within the broader context of the INSDC (http://www.insdc.org/). INSDC provides what we call a ‘political integration of sequences’ (i.e. three institutions provide agreed submission pipelines, curation process, and points of access to the public, coupled by the use of the same identifiers and a rich interoperability between their portals).
NCBI hosts the two most relevant sequence databases: GenBank [1] contains the annotated collection of publicly available DNA and RNA sequences; RefSeq [6] provides a stable reference for genome annotations, gene identification/characterization and mutation/polymorphism analysis. GenBank is continuously updated thanks to abundant submissions (through https://submit.ncbi.nlm.nih.gov/sarscov2/) from multiple laboratories and data contributors around the world; SARS-CoV2 nucleotide sequences have increased from about 300 around the end of March 2020, to 27 660 by 8 October 2020. EMBL-EBI hosts the European Nucleotide Archive [7], which has a broader scope, accepting submissions of nucleotide sequencing information, including raw sequencing data, sequence assembly information and functional annotations. Several tools are directly provided by the INSDC institutions for supporting the access to their viral resources, such as E-utilities [8], NCBI Virus (https://www.ncbi.nlm.nih.gov/labs/virus) [9] and Pathogens (https://www.ebi.ac.uk/ena/pathogens/).
Coronavirus Disease 2019 Genomics UK Consortium (COG-UK)
The COG-UK [4] is a national-based initiative launched in March 2020 thanks to a big financial support from three institutional partners: UK Research and Innovation, UK Department of Health and Social Care and Wellcome Trust. The primary goal of COG-UK is to sequence about 230 000 SARS-CoV2 patients (with priority to health care workers and other essential workers in the UK) to help the tracking of the virus transmission. They provide data directly on their webpage, open for use, as a single FASTA file on https://www.cogconsortium.uk/data/; this is associated with a csv file for metadata. At the time of writing, the most updated release is dated 3 September 2020, with 48 561 sequences (as declared on the Consortium’s web page).
Chinese sources
Since the early offspring of COVID-19, several resources were made available in China.
The Chinese National Genomic Data Center [10] provides some data resources relevant for COVID-19-related research, including the Genome Warehouse (https://bigd.big.ac.cn/gwh/), which contains genome assemblies with their detailed descriptive information: biological sample, assembly, sequence data and genome annotation.
The National Microbiology Data Center (NMDC, http://nmdc.cn/) provides the ‘Novel Cov National Science and Technology Resource Service System’ to publish authoritative information on resources and data concerning 2019-nCoV to provide support for scientific studies and related prevention/control actions. The resource is provided in Chinese language with only some headers and information translated to English. Its FTP provides a collection of sequences from various coronaviruses, including many from NCBI GenBank, together with a restricted number of NMDC original ones.
The China National GeneBank DataBase [11] (CNGBdb, https://db.cngb.org/) is a platform for sharing biological data and application services to the research community, including internal data resources; it also imports large amounts of external data from INSDC databases.
Around the world there are many other sequence collections not yet included within international repositories, which are hardly accounted; one of them is the CHLA-CPM dataset collected by the Center for Personalized Medicine (CPM, https://www.chla.org/center-personalized-medicine) at the Children’s Hospital, Los Angeles (CHLA), resulting from an initiative launched in March 2020 to test a broad population within the Los Angeles metropolitan area.
GISAID and its resources
During the COVID-19 pandemic, GISAID has proposed again its solution in the form of the new database EpiCoVTM, associated with similar services as the ones provided for influenza. The GISAID restricted open-source model has greatly facilitated the rapid sharing of virus sequence data, but it contemplates constraints on data integration and redistribution, which we later describe in the ‘Discussion’ section. At the time of writing, GISAID has become the most used database for SARS-CoV2 sequence deposition, preferred by the vast majority of data submitters and gathering 140 507 sequences by 8 October 2020.
It is also the case that GISAID formatting/criteria for metadata are generally considered more complete and are thus suggested even outside of the direct submission to GISAID; the SARS-CoV2 sequencing resource guide of the US Centers for Disease Control and Prevention (CDC, https://www.cdc.gov/) reports, in the section regarding recommended formatting/criteria for metadata [12], that the user is invited to submit always using the submission formatting of GISAID EpiCoVTM, ‘which tends to be more comprehensive and structured’. However, in order to check such format, the user is invited to create an account on GISAID, which probably leads to using GISAID directly instead of going back to GenBank.
Some interesting portals are ‘enabled by data from GISAID’, as clearly stated on the top of their pages, with different focuses. NextStrain [13] (https://nextstrain.org/ncov) overviews emergent viral outbreaks based on the visualization of sequence data integrated with geographic information, serology and host species. A similar application for exploring and visualizing genomic analysis has been implemented by Microreact, which has a portal dedicated specifically to COVID-19 (https://microreact.org/project/COVID-19). CoVsurver (https://corona.bii.a-star.edu.sg/), which had a corresponding system for influenza virus called FluSurver (http://flusurver.bii.a-star.edu.sg), enables rapid screening of sequences of coronaviruses for mutations of clinical/epidemiological importance. CoV Genome Tracker [14] (http://cov.genometracker.org/) combines in a dashboard a series of visualizations based on the haplotype network, a map of collection sites and collection dates, and a companion tab with gene-by-gene and codon-by-codon evolutionary rates.
Integration of sources of viral sequences
Next Generation Sequencing is successfully applied to infectious pathogens [15], with many sequencing technology companies developing their assays and workflows for SARS-CoV2 (see Illumina [16] or Nanopore [17]). Some sources provide the corresponding raw data (see European Nucleotide Archive [7] of the INSDC network), but most sources present just the resulting sequences, typically in the form of FASTA, together with some associated metadata. In this review we do not address the topic of sequence pipeline harmonization, as it would require an entire discussion per se (we refer interested readers to forum threads [18] and recent literature contributions [19]). We focus instead on the data integration efforts required for their metadata and value integration.
Metadata integration
Metadata integration is focused on provisioning a global, unified schema for all the data that describe sequences within the various data sources [20]. In the context of viral sequences, as the amount of data is easily manageable, it is common to import data at the integrator site; in this way, data curation/reprocessing can be performed in homogeneous way. In such context, one possible solution is to apply conceptual modeling (i.e. the entity-relationship approach [21]) as a driver of the integration process. The use of conceptual modeling to describe genomics databases dates back to more than 20 years ago [22]. A number of works have targeted integration of human genomic data with a data quality-oriented conceptual modeling approach [23, 24], data warehousing (GEDAW UML Conceptual schema [25]), and metadata-driven search (Genomic Conceptual Model [26], paired with the integration pipeline META-BASE [27]).
In the variety of resources dedicated to viruses [28], very few works relate to conceptual data modeling. Among them, [29] considers host information and normalized geographical location, whereas [30] focuses on influenza A viruses. CoV-GLUE [31] includes a basic conceptual model (http://glue-tools.cvr.gla.ac.uk/images/projectModel.png) for SARS-CoV2. In comparison, the Viral Conceptual Model (VCM, [32]) provides an extensible database and associated query system that works seamlessly with any kind of virus, based on the molecule type, the species and the taxonomic characteristics. VCM has many dimensions and attributes, which are very useful for supporting research queries on virus sequences; it uses the full power of conceptual modeling to structure metadata and to organize data integration and curation.
Value harmonization and ontological efforts
Besides schema unification, data values must be standardized and harmonized in order to fully support integrated query processing. The following value harmonization problems must be solved:
Virus and host species should refer to dedicated controlled vocabularies (the NCBI Taxonomy [33] is widely recognized as the most trusted, even if some concerns apply to the ranking of SARS-CoV2 as a species or just an isolate/group of strains).
Sequence completeness should be calculated using standard algorithms, using the length and the percentage of certain indicative types of basis (e.g. unknown ones = N).
The information on sequencing technology and assembly method should be harmonized, especially the coverage field, which is represented in many ways by each source.
Dates—both collection and submission ones—must be standardized; unfortunately, they often miss the year or the day, and sometimes it is not clear if the submission date refers to transmission of the sequence to the database or to a later article’s publication.
Geographical locations, including continent, country, region and area name, are encoded differently by each source.
In some rare cases, sequences come with gender and age information, hidden in the middle of descriptive fields.
A number of efforts have been directed to the design of ontologies for solving some of these problems:
The Infectious Disease Ontology (IDO) has a focus on the virus aspects; its curators have proposed an extension of the ontology core to include terms relevant for COVID-19 [34].
The Coronavirus Infectious Disease Ontology (CIDO) [35] is a community-based ontology to integrate and share data on coronaviruses, more specifically on COVID-19. Its infrastructure aims to include information about the disease etiology, transmission, epidemiology, pathogenesis, host-coronavirus interactions, diagnosis, prevention and treatment. Currently, CIDO contains more than 4000 terms; in observance of OBO Foundry principles, it aggregates already existing well-established ontologies describing different domains (such as ChEBI [36] for chemical entitites, Human Phenotype Ontology [37] for human host phenotypes, the Disease Ontology [38] for human diseases including COVID-19, the NCBI taxonomy and the IDO itself)—so to not create unnecessary overlaps. New CIDO-specific terms have been developed to meet the special needs arising in the research of COVID-19 and other coronavirus diseases. The work on host-pathogen interactions is described in depth in [39], whereas the inclusion in CIDO of aspects related to drugs and their repurposing is described in [40].
The COVID-19 Disease Map [41] is a visionary project by Elixir Luxembourg, that aims to build a platform for exploration and analyses of molecular processes involved in SARS-CoV2 interactions and immune response.
For the sequence annotation process, there are two kinds of ontologies that are certainly relevant: the Sequence Ontology [42], used by tools such as SnpEff [43] to characterize the different subsequences of the virus, and the Gene Ontology [44], which has dedicated a page to COVID-19 (http://geneontology.org/covid-19.html) that provides an overview of human proteins that are used by SARS-CoV2 to enter human cells, divided by the 29 different virus’ proteins.
Replicated sequences in multiple sources
Record replication is a recurrent problem occurring when integrating different sources; it is solved by ‘Entity Resolution’ tasks i.e. identifying the records that correspond to the same real world entity across and within datasets [45]. This issue arises for SARS-CoV2 sequences, as many laboratories use to submit sequences to multiple sources; in particular, sequences submitted to NCBI GenBank and COG-UK are often also submitted to GISAID.
Such problem is resolved in different manners by the various integrative systems. The main approaches aim to either resolve the redundancy by eliminating from one source records that appear also in another one or by linking records that represent the same sequence, adding to both records an ‘external reference’ pointing to the other source. Along this second solution, a template proposal for data linkage is provided by the CDC [46]: a simple lightweight line list of tab-separated values to hold the name of the sequence, as well as IDs from GISAID and GenBank.
SARS-CoV2 search systems
In this section, we compare the systems that provide search facilities for SARS-CoV2 sequences and related metadata, possibly in addition to those of other viruses. In Table 1 we summarize the content addressed by each system; in the first section, we indicate the target virus species, which either includes the SARS-CoV2 virus only, or also similar viruses (e.g. Coronavirus, other RNA single stranded viruses, other pandemic-related viruses), or an extended set of viruses. In the second section, the table shows which sources are currently integrated by each system. The first five columns refer to portals to resources gathering either NCBI or GISAID data, whereas the following ones refer to integrative systems over multiple sources. These are described in the next two sections.
Table 1.
Top part: characterization of each system based on its focus on general SARS-CoV2 virus only, SARS-CoV2 and similar viruses (e.g. other coronavirus or pandemic-related viruses), or an extended set of viruses. Bottom part: integration of sequences by each portal (columns) from each origin source (rows).
| Portals to NCBI/GISAID resources | Integrative search systems | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| NCBI Virus | COVID-19DP | ViPR | EpiCoVTM | CoV-GLUE | 2019nCoVR | VirusDIP | CARD | CoV-Seq | ViruSurf | ||
| Content | |||||||||||
| SARS-CoV2 specific |
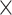
|
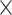
|
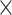
|
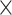
|
|
||||||
| SARS-CoV2 + similar |
|
|
|
||||||||
| Extended virus set |
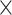
|
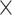
|
|||||||||
| Included sources | |||||||||||
| GenBank |
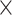
|
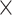
|
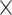
|
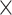
|
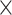
|
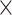
|
|
|
|||
| RefSeq |
|
|
|
|
|
|
|
||||
| GISAID |
|
|
|
|
|
|
|
||||
| COG-UK |
|
||||||||||
| NMDC |
|
|
|
||||||||
| CNGBdb |
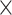
|
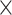
|
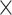
|
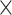
|
|||||||
| Genome Warehouse |
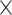
|
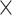
|
|||||||||
| CHLA-CPM |
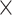
|
||||||||||
Portals to NCBI and GISAID resources
Native portals for accessing NCBI and GISAID resources are hereby described even if they do not provide integrative access to multiple sources, as they are recognized search facilities for SARS-CoV2 sequences collected from laboratories all around the world.
An interesting and rich resource (Virus Variation Resource [9]) is hosted by NCBI, targeting many viruses relevant to emerging outbreaks. At the time of writing, a version for coronaviruses – and SARS-CoV2 in the specific – has not been released yet. Instead, for this virus users are forwarded to the NCBI Virus resource https://www.ncbi.nlm.nih.gov/labs/virus/; this portal provides a search interface to NCBI SARS-CoV2 sequences, with several filter facets and a result table where identifiers are links to NCBI GenBank database pages. It provides a very quick and comprehensive access to SARS-CoV2 data, but is not well aligned with the API provided for external developers, making integration efforts harder, as differences need to be understood and synchronized.
COVID-19 Data Portal (https://www.covid19dataportal.org/) joins the efforts of ELIXIR and EMBL-EBI to provide an integrated view of resources spanning from raw reads/sequences, to expression data, proteins and their structures, drug targets, literature and pointers to related resources. Coupling raw and sequence data in the same portal can be very useful for users who wish to resort to original data and recompute sequences or compare with existing ones. We focus on their contribution to data search, which is given through a data table containing different data types (sequences, raw reads, samples, variants, etc.). The structure of the table changes based on the data types (the metadata provided for nucleotide sequence records are overviewed next).
The Virus Pathogen Database and Analysis Resource (ViPR [47], https://www.viprbrc.org/) is a rich repository of data and analysis tools for multiple virus families, supported by the Bioinformatics Resource Centers program. It provides GenBank strain sequences with UniProt proteins, 3D protein structures and experimentally determined epitopes (immune epitope database, IEDB [48]). For SARS-CoV2 many different views are provided for genome annotation, comparative genomics, ortholog groups, host factor experiments, and phylogenetic tree visualization. It provides the two functions ‘Remove Duplicate Genome Sequences’ and ‘Remove Identical Protein Sequences’ to resolve redundancy, respectively, of nucleotide and amino acid sequences.
GISAID EpiCoVTM portal provides a search interface upon GISAID metadata. Nine filters are available to design the user search, whereas the results table shows 11 metadata attributes. By clicking on single entries, the user accesses a much richer information, consisting of 31 metadata attributes. The browsing power of the interface is limited by the use of few attributes. Although more information is given on single entries, the users are prevented to take advantage of these additional attributes to order results or see distinct values.
CoV-GLUE [31] (http://cov-glue.cvr.gla.ac.uk/) has a database of replacements, insertions and deletions observed in sequences sampled from the pandemic. It also provides a quite sophisticated metadata-based search system to help filtering GISAID sequences with mutations. Although other systems are sequence-based, meaning that the users can select filters to narrow down their search on sequences, CoV-GLUE is variant-based: the user is provided with a list of variants (on amino acids); by selecting given variants, the sequences that present such variants can be accessed.
Integrative search systems
The following systems provide integrative data access from multiple sources, as illustrated in Table 1.
2019nCoVR [49] (https://bigd.big.ac.cn/ncov/) at the Chinese National Genomics Data Center (at the Beijing Institute of Genomics) is a rich data portal with several search facets, tables and visual charts. This resource includes most sources publicly reachable, including GISAID; however, it is unclear if this is compliant with the GISAID data sharing agreement (see the ‘Discussion’ section). 2019nCoVR handles sequence records redundancy by conveniently providing a ‘Related ID’ field that allows to map each sequence from its primary database to others that also contain it.
The Virus Data Integration Platform (VirusDIP [50], https://db.cngb.org/virus/ncov) is a system developed at CNGBdb to help researchers find, retrieve and analyze viruses quickly. It declares itself as a general resource for all kinds of viruses; however, to date includes only coronaviruses sequences.
The COVID-19 Analysis Research Database (CARD [51], https://covid19.cpmbiodev.net/) is a rich and interesting system giving the possibility to rapidly identify SARS-CoV2 genomes using various online tools. However, the data search engine seems to be still under development and to date does not allow to build complex queries combining filters yet.
CoV-Seq [52] (http://covseq.baidu.com/) collects tools to aggregate, analyze, and annotate genomic sequences. It claims to integrate sequences from GISAID, NCBI, EMBL and CNGB. It has to be noted that sequences from NCBI and EMBL are the same ones, as part of the INSDC. The search can only be done on very basic filters directly on the columns of the table, providing poor functionalities. It also provides a basic search system over a few metadata and compute the ‘Identical_Seq’ field, where a sequence is mapped to many identical ones.
ViruSurf [53] (http://gmql.eu/virusurf/), dual of the human genomics search engine GenoSurf [54], is based on a conceptual model [31] that describes sequences and their metadata from their biological, technical, organizational and analytical perspectives. It provides many options for building search queries, by combining—within rich Boolean expressions—metadata attributes about viral sequences and nucleotide and amino acid variants. Full search capabilities are used for open-source databases, whereas search over the GISAID database is suitably restricted to be compliant with the GISAID data sharing agreement. ViruSurf also solves the problem of record redundancy among different databases by using different external references IDs available and by exploiting in-house computations.
Comparison
Table 2 shows a comprehensive view of which metadata information is included by each search system. Attributes are partitioned into macro-areas that reflect the structure of metadata attributes in VCM [32], concerning the ‘biological aspects’ of the virus and of the host organism sample, the ‘technology’ producing the sequence, the ‘sequence’ details and the ‘organization’ producing the sequence. For each of them we provide three kinds of columns: S = search filters (attributes supporting search queries, typically using conjunctive queries), T = columns in result tables (providing direct attribute comparisons), and E = columns of single entries or records (once a record is clicked, search systems enable reading rich metadata but only for each individual sequence entry).
Table 2.
Inspection of metadata fields in different search portals for SARS-CoV2 sequences. 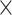 is used when the attribute is present in the search filters (S), in the table of results (T), in single entries (E).
is used when the attribute is present in the search filters (S), in the table of results (T), in single entries (E).
| Attribute description | NCBI Virus | COVID-19DP | ViPR | EpiCoVTM | CoV-GLUE | 2019nCoVR | VirusDIP | CARD | CoV-Seq | ViruSurf | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| S | T | S | T | S | T | S | T | E | S | T | E | S | T | E | S | T | S | T | S | T | ||
| Biology: virus | Accession |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||
| Related ID |
|
|
|
|
||||||||||||||||||
| Strain |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|||||||
| Virus Taxonomy ID |
|
|
|
|||||||||||||||||||
| Virus Species |
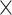
|
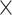
|
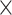
|
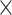
|
|
|
|
|
||||||||||||||
| Virus Genus |
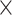
|
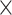
|
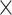
|
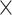
|
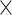
|
|||||||||||||||||
| Virus Subfamily |
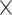
|
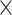
|
||||||||||||||||||||
| Virus Family |
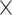
|
|
|
|
||||||||||||||||||
| Lineage |
|
|
|
|
|
|
|
|||||||||||||||
| CoV-GLUE Lineage |
|
|
||||||||||||||||||||
| Pangolin Lineage |
|
|
||||||||||||||||||||
| Total LWR |
|
|||||||||||||||||||||
| MoleculeType |
|
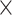
|
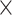
|
|||||||||||||||||||
| SingleStranded |
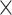
|
|||||||||||||||||||||
| PositiveStranded |
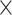
|
|||||||||||||||||||||
| Passage detail |
|
|
||||||||||||||||||||
| Biology: sample | Collection date |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Location |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| Origin lab |
|
|
|
|
|
|
|
|
||||||||||||||
| Host Taxonomy ID |
|
|
||||||||||||||||||||
| Host organism |
|
|
|
|
|
|
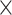
|
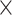
|
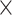
|
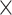
|
|
|
|
|
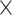
|
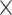
|
||||||
| Host gender |
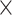
|
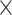
|
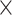
|
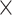
|
||||||||||||||||||
| Host age |
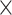
|
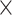
|
|
|
||||||||||||||||||
| Host status |
|
|
||||||||||||||||||||
| Environmental source |
|
|||||||||||||||||||||
| Specimen source |
|
|
|
|
|
|||||||||||||||||
| BiosampleId |
|
|||||||||||||||||||||
| Technology | Sequencing Technology |
|
|
|
|
|||||||||||||||||
| Assembly method |
|
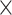
|
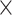
|
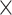
|
||||||||||||||||||
| Coverage |
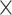
|
|
|
|
||||||||||||||||||
| Quality Assessment |
|
|
|
|
|
|||||||||||||||||
| Sequence Quality |
|
|
||||||||||||||||||||
| SRA Accession |
|
|||||||||||||||||||||
| Sequence | Complete |
|
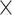
|
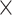
|
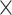
|
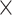
|
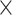
|
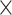
|
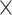
|
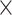
|
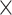
|
|||||||||||
| Length |
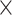
|
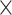
|
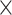
|
|
|
|
|
|
|
|
||||||||||||
| IsReference |
|
|
||||||||||||||||||||
| GC bases% |
|
|
||||||||||||||||||||
| Unknown bases % |
|
|
|
|||||||||||||||||||
| Degegerate bases % |
|
|||||||||||||||||||||
| Organization | Authors |
|
|
|
|
|
|
|
||||||||||||||
| Publications |
|
|
||||||||||||||||||||
| Submission date |
|
|
|
|
|
|
|
|
|
|||||||||||||
| Submission lab |
|
|
|
|
|
|
|
|
|
|
||||||||||||
| Submitter |
|
|
||||||||||||||||||||
| Release date |
|
|
|
|
||||||||||||||||||
| Data source |
|
|
|
|
|
|
|
|
|
|
||||||||||||
| Last Update Time |
|
|||||||||||||||||||||
| Bioproject ID |
|
|
||||||||||||||||||||
The different systems provide the same attribute concepts in very many terminology forms. For example:
‘Virus name’ in EpiCoVTM is called ‘Virus Strain Name’ in 2019nCoVR, just ‘Virus’ in CoV-Seq, ‘Strain’ or ‘Title’ CARD (as they provide two similar search filters), and ‘StrainName’ in ViruSurf.
Geographic information is named ‘Geo Location’ in NCBI Virus results table and ‘Geographic region’ in the search interface; it is given using the pattern Continent/Country/Region/SpecificArea in GISAID EpiCoVTM, whereas the four levels are kept separate in CARD and ViruSurf.
The database source is referred to as ‘Sequence Type’, ‘Data Source’, ‘Data source platform’, ‘Data_source’, ‘Data_Source’ or ‘DatabaseSource’.
It must be mentioned that some systems include additional metadata that we did not added in this comparison, as they were not easily comparable. For example, GISAID has a series of additional location details and sample IDs that can be provided by submitters (but normally they are omitted), whereas NCBI Virus adds info about provirus, lab host and vaccine strains (but these are omitted for SARS-CoV2 related sequences).
Table 3 provides a quick report on which additional data features are provided in each search system, beyond classic metadata; they include haplotypes, philogenetic tree, nucleotide and protein sequences and their pre-calculated variants.
Table 3.
Additional features provided by search systems in addition to standard metadata.
| Data features | Portals to NCBI/GISAID resources | Integrative search systems | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| NCBI Virus | COVID-19DP | ViPR | EpiCoVTM | CoV-GLUE | 2019nCoVR | VirusDIP | CARD | CoV-Seq | ViruSurf | |
| Haplotype network |
|
|||||||||
| Pylogenetic tree |
|
|
|
|
|
|
||||
| Nucl. sequences |
|
|
|
|
||||||
| Aa sequences |
|
|
|
|
||||||
| Prec. nucl. variants |
|
|
|
|
||||||
| Prec. aa variants |
|
|
|
|
|
|||||
Integrating host-pathogen information
Together with virus sequences and their metadata, it is critical to integrate also information about the related host phenotype and genotype; this is key to allow supporting the paramount host-pathogen genotype-phenotype analyses. In this section, we briefly mention how these crucial aspects are being investigated.
The virus genotype—host phenotype connection
At the time of writing, big integration search engines are starting to provide clinical information related to hosts of virus sequences. 2019nCoVR [49] has currently 208 clinical records (https://bigd.big.ac.cn/ncov/clinic) related to specific assemblies (as FASTA files), including information such as the onset date, travel/contact history, clinical symptoms and tests in a semi-structured format (i.e. attribute-value), where values are free-text and not homogeneous w.r.t. any dictionary. GISAID is also progressively adding information regarding the ‘patient status’ (e.g. ‘ intensive care unit, serious’; ‘hospitalized, stable’; ‘released’, ‘discharged’) to its records (in 7862 out of 140 507 on 8 October 2020).
So far this kind of effort has not been systematized. Some early findings connecting virus sequences with the human phenotype have been already published, but these include very small datasets (e.g. [55] with only 5 patients, [56] with 9 patients [57] and [58] with 103 sequenced SARS-CoV2 genomes). We are not aware of big sources comprising linked phenotype data and viral sequences, where the link connects the phenotype of the virus host organism to the viral genome. There is a compelling need for the combination of phenotypes with virus sequences, so to enable more interesting queries e.g. concerning the impact of sequence variants. We are confident that in the near future there will be many more studies like [55, 54, 58]. Along this direction, the efforts mentioned in this last section are providing an initial signal, which should be encouraged. There is need for additional comprehensive studies linking the viral sequences of SARS-CoV2 to the phenotype of patients affected by COVID-19.
The host genotype—host phenotype connection
In addition to investigating the relationship between viral sequences and host conditions, much larger efforts are being conducted for linking the genotype of the human host to the COVID-19 phenotype. In this direction, an important proposal is the COVID-19 Host Genetics Initiative [59] (https://www.covid19hg.org/), aiming at ‘bringing together the human genetics community to generate, share and analyze data to learn the genetic determinants of COVID-19 susceptibility, severity and outcomes.’
To find important genotype-phenotype correlations, well-defined phenotypes need to be ascertained in a quantitative and reproducible way [60]; also the activity of sampling from clearly defined case and control groups is fundamental. For this reason, many efforts in the scientific community have been dedicated to harmonizing clinical records of COVID-19 patients. This topic would require by itself a separate survey; we next illustrate the data dictionary produced by the COVID-19 Host Genetics Initiative, contributed by about 50 active participants and released on 16 April 2020 and updated on 16 August 2020 in the current version (available at http://gmql.eu/phenotype/); genotype data is currently being collected and hosted by EGA [61], the European Genome-phenome Archive of EMBL-EBI.
The dictionary is illustrated by the entity-relationship diagram in Figure 1; phenotype information is collected at admission and during the course of hospitalizations, hosted by a given Hospital. For ease of visualization, attributes are clustered within ‘Attribute Groups,’ describing: Demography&Exposure, RiskFactors, Comorbidities, AdmissionSymptoms, HospitalizationCourse. Attributes within groups can be further clustered within subgroups, denoted by white circles; for instance, Comorbidities include the subgroups ImmuneSystem, Respiratory, GenitoUrinary, CardioVascular, Neurological, Cancer; for brevity, these are not further expanded into specific attributes. Each patient is characterized by multiple Encounters; attribute groups of encounters describe EncounterSymptoms, Treatments and LaboratoryResults. Each patient is connected to human genome information, in particular single nucleotide variations.
Figure 1.
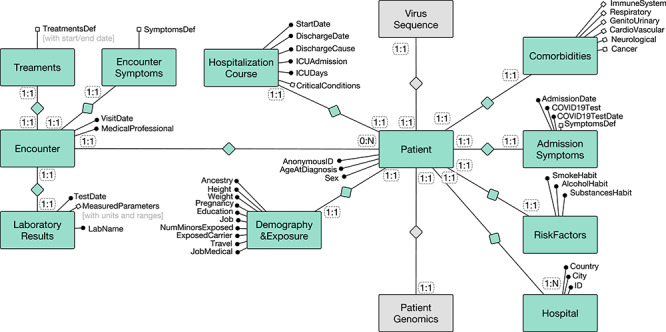
Entity-relationship diagram of the Phenotype Data Dictionary proposed within the COVID-19 Host Genetics Initiative.
Researchers can extract the patient phenotype and differentiate cases and controls in a number of ways. For example, one analysis will discriminate between mild, severe or critical COVID-19 disease severity, based on a set of EncounterSymptomps and HospitalizationCourse conditions; another analysis will distinguish cases and controls based on ‘ Comorbidities’ and AdmissionSymptoms.
Many other efforts to systematize clinical data collection and harmonization are proposed by international organizations (e.g. WHO [62]), national projects (e.g. AllOfUs [63]) or private companies (e.g. 23AndMe https://www.23andme.com/).
Genome Wide Association Studies are being conducted massively at this time [64], whereas some other efforts have attempted to find genetic determinants of COVID-19 severity in specific human genes [65], also in comparisons to SARS-CoV [66].
Note that Figure 1 illustrates the possibility of connecting each patient also to the viral sequence of the SARS-CoV2 virus. In general, there is a strong need to connect human genotype, human phenotype and viral genotype, so as to build a complete and fully encompassing scenario for data analysis.
Active monitoring of SARS-CoV2 variations
SARS-CoV2 sequences can be connected with their genetic variation (defined by coordinates, size and site along the genome). These variations may affect not only the genetic interaction of particular genes, but also proteomic aspects (e.g. change or deletion of relevant amino acids due to missense or frameshift mutations) that, in turn, correspond to phenotypic behaviours (i.e. a particular deletion may attenuate the pathological phenotype [67] or the ability of the virus to replicate itself [68]).
At present there is no consolidated knowledge about such impacts of mutations: neither of their role in increasing/decreasing pathogenicity, nor of their correlation with viral transmission. However, many studies start being published on particular aspects of the virus regarding, for example, sub-proteins of the long ORF1ab polyprotein [69, 70], as well as the widely-studied D614G mutation in the spike protein [71].
Successful integrative reactive systems may, for instance, put in place a daily surveillance of consensus sequences that are published on the sources described earlier. These systems could then extract the sequence variants and compare them with a knowledge base of literature-based well-curated information on the impact of the mutations; by such means, they could connect each change in the virus to the implications on its spread, properly contextualized using statistics on the temporal/spatial coordinates of the collected sample and on the variant’s representativeness in the observed population. This same paradigm could then be used to monitor the spread of the virus also in hosts different from humans [72], as it should not be excluded that the pandemic could turn into a panzootic [73].
Some form of watch of these variations is already put in place by GISAID, but results are not made publicly available; bulletins from the WHO [74] only give a static picture. Online applications—such as the COVID-19 Viral Genome Analysis Pipeline [75], GESS (https://wan-bioinfo.shinyapps.io/GESS/) and coronApp [76]—provide environments to explore mutations of SARS-CoV2; ViruSurf presents—for demonstration purposes—queries that replicate the results of research papers, so that results deposited at given dates can be continuously checked. However, all these systems still require the user to drive the search and lack an automatic monitoring mechanism; this could be used in the future as a means to provide alerts to the general scientific community on the emergence of potentially dangerous mutations in selected regions or in otherwise refined clusters of collected sequences.
On a different—but equivalently important—note, other kinds of auxiliary data could be integrated with sequences. We mention haplotype descriptors like clades and lineages (computed using the GISAID system, the PANGOLIN system [77], or the operational clustering method described in [78]) and epitopes i.e. short amino acid sequences that are recognized by the host immune system antigens—the substance capable of stimulating antibody responses in the organism invaded by the virus. Epitopes, described by their location in the sequence, evidence and type of response, are candidate binding sites for vaccine design. The IEDB [48] is an open resource where we expect epitope sequences will continuously be deposited and curated through literature review. Although, as of October 2020, there are only 365 epitopes specifically deposited for SARS-CoV2, IEDB has several thousand sequences deposited for other species. Of particular importance are those methods capable of transferring consolidated knowledge from related virus species to the one of interest. A significant example is provided by Grifoni et al. [79], a bioinformatics method to infer putative epitopes of SARS-CoV2 from similar viruses, namely SARS-CoV and MERS-CoV.
Discussion
We articulate the discussion along a number of directions: impact of GISAID’s model, lack of metadata quality, (un)willingness of sequence sharing.
GISAID restrictions
Although most of the resources reviewed in this paper, and in particular all the open data sources and search engines, are available through public Web interfaces, the GISAID portal can be accessed just by using a login account; access is granted in response to an application, which must be presented by using institutional emails and requires agreeing to a database access agreement (https://www.gisaid.org/registration/terms-of-use/). Registered users are invited to ‘help GISAID protect the use of their identity and the integrity of its user base’.
Thanks to this controlled policy, GISAID has been able to gather the general appreciation of many scientists, who hesitate to share data within fully open-source repositories. Some concerns are actually legitimate, such as not being properly acknowledged; acknowledgement of data contributors is required to whoever uses specific sequences from the EpiCoVTM database. However, GISAID policies impose limitations to data integrators. Sequences are not communicated to third parties, such as integration systems; interested users can only access and download them one by one from the GISAID portal. As the reference sequence can be reconstructed from the full knowledge of nucleotide variants, these are similarly not revealed.
Metadata quality
Another long-standing problem is the low quality of input metadata. A commentary [80] from the Genomic Standards Consortium board (https://www.gensc.org/) alerts the scientific community of the pressing need for systematizing the metadata submissions and enforcing metadata sharing good practices. This need is even more evident with COVID-19, where the information on geolocalization and collection time of a sample easily becomes a ‘life and death issue’; they claim that the cost of poor descriptions about the pathogen host and collection process could be greater than the poor quality of nucleotide sequence record itself. To this end, EDGE COVID-19 [81] (https://edge-covid19.edgebioinformatics.org/) has made an attempt to facilitate the preparation of genomes of SARS-CoV2 for submission to public databases providing help both with metadata and with processing pipelines.
For what concerns variant information, it is important that they refer to the same reference sequence. We found that in some cases a different sequence was used as reference for SARS-CoV2 with respect to the one of NCBI GenBank, commonly accepted by the research community. If different reference sequences are used and original sequence data are not shared, it becomes very hard to provide significant statistics about variant impact.
The insufficient submission of enriched contextual metadata is generally imputed to the fact that individual researchers receive little recognition for data submission and that probably they prefer withholding information, being concerned that their data could be reused before they finalize their own publications. This is not only a problem of submission practice, but also of data sharing, as discussed next.
(Un)willingness to share sequence data
In times of pandemic, there is—as discussed next—a strong need for data sharing, creating big databases that can support research. Regardless of this necessity, many researchers or research institutions do not join the data sharing efforts. For example, it looks strange to us that searches for SARS-CoV2 sequence data from Italy, witnessing one of the first big outbreaks of COVID-19 in the world, return only a few sequences (60 on GenBank and 737 on GISAID, of which only 456 with patient status information, as of 8 October 2020); similar numbers apply to many other countries.
Successful provisioning of sequences is the result of a number of conditions: having funds for sequencing, high quality technology to retrieve useful results, willingness to join the FAIR science principles [82]. Sampling activity in the hospitals is essential, as well as its timely processing and sequencing pipelines in laboratories; however, nowadays the most critical ‘impasse’ is met at the stage of submitting sequences and associated metadata (if not even clinical information regarding the host), which has become almost a deliberate political act in the current times [83].
We also observed the opposite attitude: consortia such as the COVID-19 Host Genetics Initiative have been assembled around the objective and principles of open data sharing. As another significant case, the E-ellow Submarine [84] interdisciplinary initiative for exploiting data generated during the COVID-19 pandemic is fully committed to open data. Practical ecosystems for supporting open pathogen genomic analysis [85] will become more widespread if proactively encouraged by a strong institutional support. We hope and trust that events such as the COVID-19 pandemic will move scientists toward open data sharing, as a community effort for mitigating the effects of this and future pandemic events.
Key Points
Integration of sources of viral sequences faces traditional problems of metadata integration, value harmonization and replication resolution.
Controlled-access resources for submitting sequences seem to be more popular than fully open-access ones.
Not enough resources have been dedicated to integrating host-pathogen information; the ideal link between the host phenotype with its corresponding genotype and with the infecting virus genotype could greatly improve research outcomes.
Continuous querying systems should be used for monitoring, both in time and space, the presence of potentially harmful sequence variants.
Sharing of sequence data and of clinical aspects is paramount for the development of future pandemics-mitigation integrated approaches; more community and institutional efforts are needed in this direction.
Supplementary Material
Anna Bernasconi is a PhD candidate at Politecnico di Milano. Her research interests include bioinformatics data and metadata integration methodologies to support biological query answering, with focus on open data resources.
Arif Canakoglu is a Postoctoral fellow at Politecnico di Milano. His research interests include data integration, data-driven genomic computing, big data analysis/processing on cloud and artificial intelligence applications of genomic data.
Marco Masseroli is Associate Professor at Politecnico di Milano. He is the author of more than 200 scientific articles, which have appeared in international journals, books and conference proceedings.
Pietro Pinoli is Research Assistant in Computational Biology at Politecnico di Milano. His research activity focuses on the management, modeling and analysis of omics data.
Stefano Ceri is Professor at Politecnico di Milano. He received two advanced ERC Grants, on Search Computing and Data-Driven Genomic Computing. He received the ACM-SIGMOD Innovation Award and is an ACM Fellow.
Contributor Information
Anna Bernasconi, Politecnico di Milano.
Arif Canakoglu, Politecnico di Milano.
Marco Masseroli, Politecnico di Milano.
Pietro Pinoli, Politecnico di Milano.
Stefano Ceri, Politecnico di Milano.
Funding
This research is funded by the ERC (Advanced Grant 693174 GeCo; data-driven Genomic Computing) and by the EIT Digital innovation activity 20663 ‘DATA against COVID-19’.
References
- 1. Sayers EW, Cavanaugh M, Clark K, et al. GenBank. Nucleic Acids Res 2019;47(D1):D94–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shu Y, McCauley J. GISAID: global initiative on sharing all influenza data–from vision to reality. Eurosurveillance 2017;22(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Elbe S, Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Global Challenges 2017;1(1):33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. The COVID-19 Genomics UK (COG-UK) consortium An integrated national scale SARS-CoV-2 genomic surveillance network. The Lancet Microbe 2020;1(3):E99–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. WHO’s Code of Conduct for Open and Timely Sharing of Pathogen Genetic Sequence Data During Outbreaks of Infectious Disease https://www.who.int/blueprint/what/norms-standards/GSDDraftCodeConduct_forpublicconsultation-v1.pdf. (8 October 2020, date last accessed).
- 6. O’Leary NA, Wright MW, Brister JR, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 2015;44(D1):D733–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Amid C, Alako BT, Balavenkataraman Kadhirvelu V, et al. The European nucleotide archive in 2019. Nucleic Acids Res 2020;48(D1):D70–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sayers E. The E-utilities in-depth: parameters, syntax and more. In Entrez Programming Utilities Help [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 29 May 2009. [Updated 24 October 2018]. https://www.ncbi.nlm.nih.gov/books/NBK25499/. [Google Scholar]
- 9. Hatcher EL, Zhdanov SA, Bao Y, et al. Virus variation resource–improved response to emergent viral outbreaks. Nucleic Acids Res 2017;45(D1):D482–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. National Genomics Data Center Members and Partners Database resources of the national genomics data center in 2020. Nucleic Acids Res 2020;48(D1):D24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. CNGBdb: China National GeneBank DataBase doi: 10.25504/FAIRsharing.9btRvC. (8 October 2020, date last accessed). [DOI] [PubMed]
- 12. Recommended Formatting and Criteria for Sample Metadata https://github.com/CDCgov/SARS-CoV-2_Sequencing/. (8 October 2020, date last accessed).
- 13. Hadfield J, Megill C, Bell SM, et al. Nextstrain: real-time tracking of pathogen evolution. Bioinformatics 2018;34(23):4121–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Akther S, Bezrucenkovas E, Sulkow B, et al. CoV genome tracker: tracing genomic footprints of Covid-19 pandemic. bioRxiv 2020. 2020.04.10.036343. doi: 10.1101/2020.04.10.036343. [Google Scholar]
- 15. Gwinn M, MacCannell D, Armstrong GL. Next-generation sequencing of infectious pathogens. JAMA 2019;321(9):893–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. How Next-Generation Sequencing Can Help Identify and Track SARS-CoV-2 https://www.nature.com/articles/d42473-020-00120-0 (8 October 2020, date last accessed).
- 17. Novel Coronavirus (COVID-19) Overview https://nanoporetech.com/covid-19/overview. (8 October 2020, date last accessed).
- 18. De Maio N. Issues with SARS-CoV-2 Sequencing Data. https://virological.org/t/issues-with-sars-cov-2-sequencing-data/473. (8 October 2020, date last accessed).
- 19. Khan KA, Cheung P. Presence of mismatches between diagnostic PCR assays and coronavirus SARS-CoV-2 genome. R Soc Open Sci 2020;7(6):200636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Batini C, Lenzerini M, Navathe SB. A comparative analysis of methodologies for database schema integration. ACM Compu. Surv. (CSUR) 1986;18(4):323–64. [Google Scholar]
- 21. Batini C, Ceri S, Navathe SB. Conceptual Database Design: An Entity-Relationship Approach. Benjamin-Cummings Publishing Co., Inc., 1991. [Google Scholar]
- 22. Paton NW, Khan SA, Hayes A, et al. Conceptual modelling of genomic information. Bioinformatics 2000;16(6):548–57. [DOI] [PubMed] [Google Scholar]
- 23. Román JFR, Pastor Ó, Casamayor JC, et al. Applying conceptual modeling to better understand the human genome. In: International Conference on Conceptual Modeling. Springer, 2016, 404–12. [Google Scholar]
- 24. Palacio AL, López ÓP, Ródenas JCC. A method to identify relevant genome data: conceptual modeling for the medicine of precision. In: International Conference on Conceptual Modeling. Springer, 2018, 597–609. [Google Scholar]
- 25. Guerin É, Marquet G, Burgun A, et al. Integrating and warehousing liver gene expression data and related biomedical resources in GEDAW. In: International Workshop on Data Integration in the Life Sciences. Springer, 2005, 158–74. [Google Scholar]
- 26. Bernasconi A, Ceri S, Campi A, et al. Conceptual modeling for genomics: building an integrated repository of open data. In: Mayr HC, Guizzardi G, Ma H et al. (eds). Conceptual Modeling. Cham: Springer International Publishing, 2017, 325–39. [Google Scholar]
- 27. Bernasconi A, Canakoglu A, Masseroli M, et al. ``META-BASE: a Novel Architecture for Large-Scale Genomic Metadata Integration''in IEEE/ACM Transactions on Computational Biology and Bioinformatics, doi: 10.1109/TCBB.2020.2998954. [DOI] [PubMed] [Google Scholar]
- 28. Sharma D, Priyadarshini P, Vrati S. Unraveling the web of viroinformatics: computational tools and databases in virus research. J Virol 2015;89(3):1489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tahsin T, Weissenbacher D, Jones-Shargani D, et al. Named entity linking of geospatial and host metadata in GenBank for advancing biomedical research. Database 2017;2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lu G, Buyyani K, Goty N, et al. Influenza A virus informatics: genotype-centered database and genotype annotation. In: Second International Multi-Symposiums on Computer and Computational Sciences (IMSCCS 2007). IEEE, 2007, 76–83. [Google Scholar]
- 31. Singer J, Gifford R, Cotten M, et al. CoV-GLUE: a web application for tracking SARS-CoV-2 genomic variation. Preprints 2020;2020060225. doi: 10.20944/preprints202006.0225.v1. [Google Scholar]
- 32. Bernasconi A, Canakoglu A, Pinoli P, et al. Empowering virus sequences research through conceptual modeling. In: Dobbie G., Frank U., Kappel G., Liddle S.W., Mayr H.C. (eds) Conceptual Modeling. ER 2020. Lecture Notes in Computer Science, vol 12400, 2020. Springer, Cham. https://doi.org/10.1007/978-3-030-62522-1_29. [Google Scholar]
- 33. Federhen S. The NCBI taxonomy database. Nucleic Acids Res 2012;40(D1):D136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Babcock S, Cowell LG, Beverley J, et al. The infectious disease ontology in the age of COVID-19. OSF Preprints 2020;az6u5. doi: 10.31219/osf.io/az6u5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He Y, Yu H, Ong E, et al. CIDO, a community-based ontology for coronavirus disease knowledge and data integration, sharing, and analysis. Scientific Data 2020;7(1):181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hastings J, Owen G, Dekker A, et al. ChEBI in 2016: improved services and an expanding collection of metabolites. Nucleic Acids Res 2016;44(D1):D1214–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Köhler S, Carmody L, Vasilevsky N, et al. Expansion of the human phenotype ontology (HPO) knowledge base and resources. Nucleic Acids Res 2019;47(D1):D1018–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schriml LM, Mitraka E, Munro J, et al. Human disease ontology 2018 update: classification, content and workflow expansion. Nucleic Acids Res 2019;47(D1):D955–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yu H, Li L, Huang H, et al. Ontology-based systematic classification and analysis of coronaviruses, hosts, and host-coronavirus interactions towards deep understanding of COVID-19. arXiv 2020;p. 2006.00639 https://arxiv.org/abs/2006.00639. [Google Scholar]
- 40. Liu Y, Chan WK, Wang Z, et al. Ontological and bioinformatic analysis of anti-coronavirus drugs and their implication for drug repurposing against COVID-19. Preprints 2020. 2020030413. doi: 10.20944/preprints202003.0413.v1. [Google Scholar]
- 41. Ostaszewski M, Mazein A, Gillespie ME, et al. COVID-19 disease map, building a computational repository of SARS-CoV-2 virus-host interaction mechanisms. Scientific Data. 2020;7(1):136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eilbeck K, Lewis SE, Mungall CJ, et al. The sequence ontology: a tool for the unification of genome annotations. Genome Biol 2005;6(5):R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012;6(2):80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet 2000;25(1):25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Getoor L, Machanavajjhala A. Entity resolution: theory, practice & open challenges. Proc VLDB Endowm 2012;5(12):2018–9. [Google Scholar]
- 46. Linking Sequence Accessions https://github.com/CDCgov/SARS-CoV-2_Sequencing#linking-sequence-accessions (8 October 2020, date last accessed).
- 47. Pickett BE, Sadat EL, Zhang Y, et al. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Res 2012;40(D1):D593–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vita R, Mahajan S, Overton JA, et al. The immune epitope database (IEDB): 2018 update. Nucleic Acids Res 2019;47(D1):D339–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao WM, Song SH, Chen ML, et al. The 2019 novel coronavirus resource. Yi chuan= Hereditas 2020;42(2):212–21. [DOI] [PubMed] [Google Scholar]
- 50. Wang L, Chen F, Guo X, et al. VirusDIP: virus data integration platform. bioRxiv 2020. 2020.06.08.139451. doi: 10.1101/2020.06.08.139451. [Google Scholar]
- 51. Shen L, Maglinte D, Ostrow D, et al. Children’s hospital Los Angeles COVID-19 analysis research database (CARD)-a resource for rapid SARS-CoV-2 genome identification using interactive online phylogenetic tools. bioRxiv 2020. 2020.05.11.089763. doi: 10.1101/2020.05.11.089763. [Google Scholar]
- 52. Liu B, Liu K, Zhang H, Zhang L, Bian Y, Huang L. CoV-Seq, a New Tool for SARS-CoV-2 Genome Analysis and Visualization: Development and Usability Study. J Med Internet Res 2020;22(10):e22299. doi: https://doi.org/10.2196/22299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Canakoglu A, Pinoli P, Bernasconi A, et al. ViruSurf: an integrated database to investigate viral sequences. Nucleic Acids Res 2020;(D1):gkaa846. doi: 10.1093/nar/gkaa846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Canakoglu A, Bernasconi A, Colombo A, et al. GenoSurf: metadata driven semantic search system for integrated genomic datasets. Database 2019;2019:baz132. https://doi.org/10.1093/database/baz132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lescure FX, Bouadma L, Nguyen D, et al. Clinical and virological data of the first cases of COVID-19 in Europe: a case series. Lancet Infect Dis 2020;20(6):697–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lu R, Zhao X, Li J, et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. The Lancet 2020;395(10224):565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Böhmer MM, Buchholz U, Corman VM, et al. Investigation of a COVID-19 outbreak in Germany resulting from a single travel-associated primary case: a case series. Lancet Infect Dis 2020;20(8):920–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tang X, Wu C, Li X, et al. On the origin and continuing evolution of SARS-CoV-2. Natl Sci Rev 2020;7(6):1012–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. The COVID-19 Host Genetics Initiative The COVID-19 Host Genetics Initiative, a global initiative to elucidate the role of host genetic factors in susceptibility and severity of the SARS-CoV-2 virus pandemic. Eur J Hum Genet 2020;28:715–718. 10.1038/s41431-020-0636-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Murray MF, Kenny EE, Ritchie MD, et al. COVID-19 outcomes and the human genome. Genet Med 2020;22(7):1175–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Flicek P, Birney E. The European Genotype Archive: Background and Implementation [White paper]; 30 March 2007. https://www.ebi.ac.uk/ega/sites/ebi.ac.uk.ega/files/documents/ega_whitepaper.pdf.
- 62. World Health Organization. Revised case report form for Confirmed Novel Coronavirus COVID-19. https://www.who.int/docs/default-source/coronaviruse/2019-covid-crf-v6.pdf (8 October 2020, date last accessed).
- 63. Collins FS, Varmus H. A new initiative on precision medicine. New England journal of medicine 2015;372(9):793–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ellinghaus D, Degenhardt F, Bujanda L, et al. Genomewide association study of severe Covid-19 with respiratory failure. N Eng J Med 2020. doi: 10.1056/NEJMoa2020283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. LoPresti M, Beck DB, Duggal P, et al. The role of host genetic factors in coronavirus susceptibility: review of animal and systematic review of human literature. Am J Hu Genet 2020;107(3): 381–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Zeberg H, Pääbo S. The major genetic risk factor for severe COVID-19 is inherited from Neandertals. Nature 2020. https://doi.org/10.1038/s41586-020-2818-3. [DOI] [PubMed] [Google Scholar]
- 67. Young BE, Fong SW, Chan YH, et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: an observational cohort study. The Lancet 2020;396(10251):603–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lau SY, Wang P, Mok BWY, et al. Attenuated SARS-CoV-2 variants with deletions at the S1/S2 junction. Emerging microbes & infections 2020;9(1):837–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Laha S, Chakraborty J, Das S, et al. Characterizations of SARS-CoV-2 mutational profile, spike protein stability and viral transmission. Infect Genet Evol 2020;85:104445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Toyoshima Y, Nemoto K, Matsumoto S, et al. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J Hum Genet 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Becerra Flores M, Cardozo T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int J Clin Pract 2020;74(8):e13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Decaro N, Lorusso A. Novel human coronavirus (SARS-CoV-2): a lesson from animal coronaviruses. Vet Microbiol 2020;108693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gollakner R, Capua I. Is COVID-19 the first pandemic that evolves into a panzootic? Vet Ital 2020;56(1):11–2. [DOI] [PubMed] [Google Scholar]
- 74. Koyama T, Platt D, Parida L. Variant analysis of SARS-CoV-2 genomes. Bull World Health Organ 2020;98(7):495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Korber B, Fischer WM, Gnanakaran S, et al. Tracking changes in SARS-CoV-2 spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020;182(4):812–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Mercatelli D, Triboli L, Fornasari E, et al. coronapp: a web application to annotate and monitor SARS-CoV-2 mutations. J Med Virol 2020. Accepted Author Manuscript. https://doi.org/10.1002/jmv.26678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rambaut A, Holmes EC, O'Toole Á, et al. A dynamic nomenclature proposal for SARS-CoV-2 to assist genomic epidemiology. Nat Microbiol 2020;5 :1403–7. 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Chiara M, Horner DS, Gissi C, et al. Comparative genomics provides an operational classification system and reveals early emergence and biased spatio-temporal distribution of SARS-CoV-2. bioRxiv 2020. doi: 10.1101/2020.06.26.172924. [Google Scholar]
- 79. Grifoni A, Sidney J, Zhang Y, et al. A sequence homology and Bioinformatic approach can predict candidate targets for immune responses to SARS-CoV-2. Cell Host Microbe 2020;27(4):671–680.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Schriml LM, Chuvochina M, Davies N, et al. COVID-19 pandemic reveals the peril of ignoring metadata standards. Scientific data 2020;7(1):188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Lo CC, Shakya M, Davenport K, et al. EDGE COVID-19: a web platform to generate submission-ready genomes for SARS-CoV-2 sequencing efforts. arXiv. 2020;p. 2006.08058. https://arxiv.org/abs/2006.08058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wilkinson MD, Dumontier M, Aalbersberg IJ, et al. The FAIR guiding principles for scientific data management and stewardship. Scientific data 2016;3:160018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Promoting best practice in nucleotide sequence data sharing. Scientific Data 2020;7, 1:152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. The E-ellow Submarine https://onehealth.ifas.ufl.edu/activities/circular-health-program/eellow-submarine/. (8 October 2020, date last accessed).
- 85. Black A, MacCannell DR, Sibley TR, et al. Ten recommendations for supporting open pathogen genomic analysis in public health. Nat Med 2020;26(6):832–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.