

Summary:
Obesity is often linked to malignancies including multiple myeloma and the underlying mechanisms remain elusive. Here we showed that acetyl-CoA synthetase 2 (ACSS2) may be an important linker in obesity-related myeloma. ACSS2 is overexpressed in myeloma cells derived from obese patients and contributes to myeloma progression. We identified adipocyte-secreted angiotensin II as a direct cause of adiposity in increased ACSS2 expression. ACSS2 interacts with oncoprotein interferon regulatory factor 4 (IRF4), enhances IRF4 stability and IRF4-mediated gene transcription through activation of acetylation. The importance of ACSS2 overexpression in myeloma is confirmed by the finding that an inhibitor of ACSS2 reduces myeloma growth both in vitro and in a diet-induced obese mouse model. Our findings demonstrate a key impact for obesity-induced ACSS2 on the progression of myeloma. Given the central role of ACSS2 in many tumors, this mechanism could be important to other obesity-related malignancies.
Keywords: Multiple myeloma, obesity, ACSS2, IRF4, lysine acetylation
Graphical Abstract
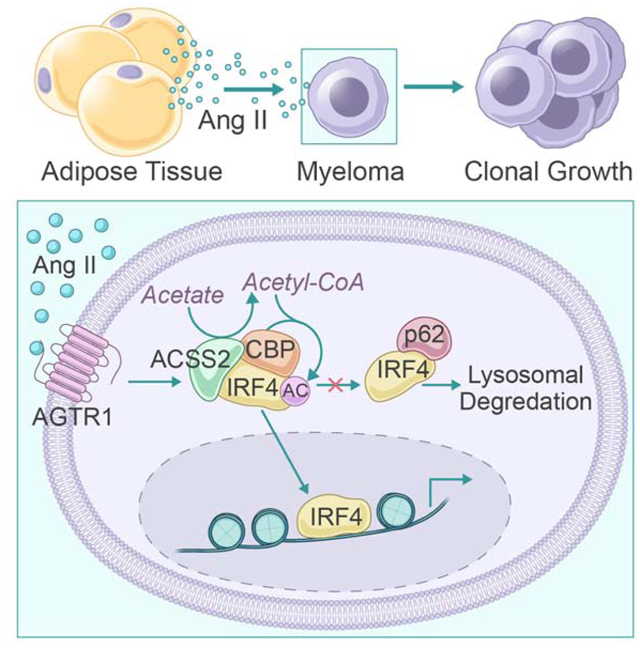
In Brief
Li et al. demonstrate a mechanistic link between obesity and myeloma. Adipocyte-derived angiotensin II stimulates acetyl-CoA synthase 2 (ACSS2) expression in myeloma cells and increased ACSS2 promotes tumorigenesis through the stabilization of interferon regulatory factor 4. Inhibiting the angiotensin II/ACSS2 axis abolishes obesity-associated myeloma progression, suggesting a potential therapeutic target for obesity-associated tumors.
INTRODUCTION
Obesity is characterized by the accumulation of excessive fat mass with a body mass index (BMI) over 30 kg/m2 (Bianchini et al., 2002). The prevalence of obesity is on the rise for the last decades in both United States and worldwide (Lichtman, 2010), and obesity has been shown to cause metabolic, endocrinologic, and immunologic changes that are linked to life-threatening diseases such as diabetes and cancers (Eckel et al., 2005). The aggregate of epidemiologic studies shows obesity to be a major risk factor for multiple tumor types (Lauby-Secretan et al., 2016; Sung et al., 2019). Obesity has been shown to mediate inflammation (Incio et al., 2016; Quail et al., 2017), hormonal alterations (Subbaramaiah et al., 2012), insulin resistance, insulin/insulin-like growth factor 1-mediated PI3K/Akt signaling pathways, hypercholesterolemia (Gallagher and LeRoith, 2015), and gut microbiome dysbiosis (Yoshimoto et al., 2013) which have been identified as mechanistic events in cancer initiation, progression, and response to therapy (Gallagher and LeRoith, 2015; Park et al., 2014). These events implicate the role of obesity in cancer development.
Multiple myeloma, a clonal expansion of malignant plasma cells predominantly accumulated within bone marrow, is the second most common hematologic malignancy in the United States (Kyle and Rajkumar, 2004; Seiden and Anderson, 1994). The American Cancer Society estimated 32,110 newly diagnosed cases and 12,960 myeloma-related deaths in 2019 (Siegel et al., 2019). Even with advanced therapies, the median survival for myeloma patients has stagnated at 5 to 7 years, and a cure remains elusive. The risk of myeloma is higher for people who carry excess weight. High BMI is positively associated with the incidence of myeloma (Lichtman, 2010), poor outcomes (Lauby-Secretan et al., 2016; Sonderman et al., 2016), and the transformation from asymptomatic, benign stage to malignancy (Chang et al., 2017; Landgren et al., 2010). These studies indicate that obesity is etiologically linked to the pathogenesis of myeloma. However, the molecular mechanisms that bridge the two conditions are notably lacking.
In this study, we investigated the role of obesity in metabolic alternation in myeloma patients. By analyzing malignant plasma cells isolated from obese patients, we identified the prominent involvement of the acetyl coenzyme A (acetyl-CoA) metabolic process in obesity-associated myeloma growth. Acetyl-CoA, a key metabolite, plays multiple roles in many cellular processes; it serves as a central metabolic intermediate for pyruvate to enter the tricarboxylic acid cycle, a key precursor for de novo synthesis of fatty acids, and the sole acetyl donor for lysine acetylation (Pietrocola et al., 2015). The production of acetyl-CoA from acetate mostly depends on acetyl-CoA synthetase 2 (ACSS2). Under conditions of metabolic stress, such as hypoxia, glucose deprivation, and low serum, ACSS2 can promote malignant cell growth and survival in some solid tumors through the production of acetyl-CoA, which is further incorporated into metabolic pathways or acts as an epigenetic regulator in the activation of protein acetylation (Comerford et al., 2014; Li et al., 2017; Mashimo et al., 2014; Schug et al., 2015). However, the function and mechanism of ACSS2 in myeloma pathogenesis, especially its role in obesity-induced myeloma progression, are unclear. We found that ACSS2 was highly expressed in malignant plasma cells isolated from patients with myeloma. In obese myeloma patients, the expression of ACSS2 was significantly higher, and the increased ACSS2 level was positively correlated with high BMI values. We also found that ACSS2 expressed by myeloma cells has a function in promoting myelomatous tumorigenesis through enhancing the stability of oncogenic protein interferon regulatory factor 4 (IRF4). Our study thus demonstrates that ACSS2 is a central player in obesity-induced myeloma growth and sheds light on the mechanistic link between obesity and the pathogenesis of myeloma.
RESULTS
Obesity promotes myelomatous tumorigenesis
Epidemiological studies have sent an overwhelmingly consistent message – obesity increases the risk for multiple myeloma no matter which type of risks was measured: relative risk, hazard rate, or odd ratios. Figure S1A is a snapshot of the previous studies from different populations, men, women, or both, and single cohort or pooled data (Birmann et al., 2007; Blair et al., 2005; Brown et al., 2001; Calle et al., 2003; Chiu et al., 2006; Engeland et al., 2007; Hofmann et al., 2013; Khan et al., 2006; Marinac et al., 2018; Pan et al., 2004; Reeves et al., 2007; Samanic et al., 2004; Soderberg et al., 2009; Teras et al., 2014; Yang et al., 2016). Based on the data for the period of 2011 to 2016 from National Program of Cancer Registries and Epidemiology and End Results program, we saw an uptick of the percentage of population-attributable fractions as well as an upward trend in the estimated myeloma cases attributed to obesity (Figure S1B), indicating the increased impact of obesity on myeloma disease.
To determine the influence of host obese conditions in myelomatous tumorigenesis, we used a well-established diet-induced obesity (DIO) C57BL/6J mouse model (Hariri and Thibault, 2010). Fed with a 60% kcal high-fat diet, the DIO mice gained substantial weight after 15 weeks when compared with control mice fed with a 10% kcal normal diet (ND, Figure S1C). To establish myeloma, we injected murine myeloma Vk12598 cells into the femurs of DIO or ND mice. The serum levels of M-proteins, which reflect tumor burden, were significantly higher in DIO mice than in ND mice (Figure S1D). We also found increased numbers of marrow- and spleen-infiltrating CD138+ myeloma cells and enlarged spleens in DIO mice compared with ND mice (Figures S1E and S1F). Using another myeloma DIO mouse model (Lwin et al., 2015), we found that more DIO mice with intravenous injection of 5TGM1 cells developed myeloma than ND mice (Figure S1G). These results indicate increased myeloma incidence and development under obese condition.
Adipocytes are the major cell type in adipose tissue, the mass of which is increased in obese individuals and adipocyte-rich microenvironment can support tumor growth (Caers et al., 2007; Liu et al., 2015). To determine the contribution of adipocytes to myeloma growth, we mixed the luciferase-labeled myeloma ARP-1 cells with purified mature adipocytes, generated from normal bone marrow-derived mesenchymal stem cells (MSCs), and subcutaneously implanted the mixture into NOD-scid IL2Rgnull (NSG) mice (Figure S1H). Implantation of myeloma cells alone in the opposite flank served as control; co-implanting myeloma cells with MSCs served as positive control (Figure S1H). We observed higher levels of bioluminescent activity and tumor weights in the sites implanted with mixtures of adipocytes and myeloma cells than in those with myeloma cells alone (Figures S1H–S1J). We then performed a head-to-head comparison between the adipocytes isolated from ND and DIO mice, and observed higher bioluminescent activity in the flank of mice co-implanted with myeloma cells and adipocytes isolated from DIO mice (Figure S1K). These results indicate the importance of the obese host condition in myelomatous tumorigenesis.
Expression of ACSS2 in malignant plasma cells is elevated in obese myeloma patients and is associated with clinical outcomes
To investigate the potential abnormalities of myeloma cells in the obese setting, we used a microarray to screen the gene expression profile of purified CD138+ malignant plasma cells isolated from the bone marrow aspirates of 9 patients with newly diagnosed myeloma (Supplementary Table S1). Differential gene expression analysis comparing patients with normal weights (BMI < 25 kg/m2) and obese patients (BMI > 30 kg/m2) identified 1903 differentially expressed genes, including 751 downregulated genes and 348 upregulated genes in myeloma cells from obese patients (Figure 1A). Gene ontology analysis further identified highly altered activities in several pathways; the most altered pathways were inflammatory response and metabolic processes (Figure 1B). Because cancer cells live in stressed metabolic microenvironments and metabolic reprogramming is an important hallmark for cancer development, we investigated the metabolic alterations in obese patients with myeloma. Figure 1C depicts a heat map of 127 significantly up- or downregulated metabolic genes, accounting for about 6.7% of total differentially expressed genes. We next analyzed the metabolic alterations between normal-weight and obese patients using Gene Set Enrichment Analysis and found significant differences in acetyl-CoA metabolic process and others such as oxidative phosphorylation. We observed that ACSS1 and ACSS2 were enriched in obese patients, whereas the enrichment of ACLY and PDHA/B was reduced (Figures 1D and 1E). Acetyl-CoA can be produced from several substrates by different enzymes; ATP citrate lyase (ACLY) using tricarboxylic acid cycle-derived citrate as a substrate; pyruvate dehydrogenase (PDH) A and B using glycolysis-derived pyruvate; or mitochondrial ACSS1 and cytosolic ACSS2 converting acetate to acetyl-CoA (Figure 1D). We thus reasoned that under the stress of obesity, myeloma cells may become more dependent on the acetate-derived acetyl-CoA, which is catalyzed by ACSS1 or ACSS2. Analysis of additional 22 patient samples (Supplementary Table S1) demonstrated that both ACSS1 and ACSS2 mRNAs were upregulated in myeloma cells isolated from bone marrow aspirates of newly diagnosed obese myeloma patients as compared with those from normal-weight myeloma patients (Figure 1F). Similar observations were obtained from immunohistochemical staining of marrow-infiltrated malignant plasma cells of obese patients (Figure 1G). More importantly, there was a robust correlation of ACSS1 or ACSS2 mRNA levels in myeloma cells with patients’ BMI values (Figure 1H). Patients with high expression of ACSS2 exhibited poor overall and relapse free survival, while the contribution of ACSS1 in myeloma cells to clinical outcomes was less obvious (Figures 1I–1K). Notably, the patients with BMIs over 30 kg/m2 and high expression of ACSS2 fared the worst (Figure 1I).
Figure 1. Expression of ACSS2 in malignant plasma cells is elevated in obese myeloma patients.
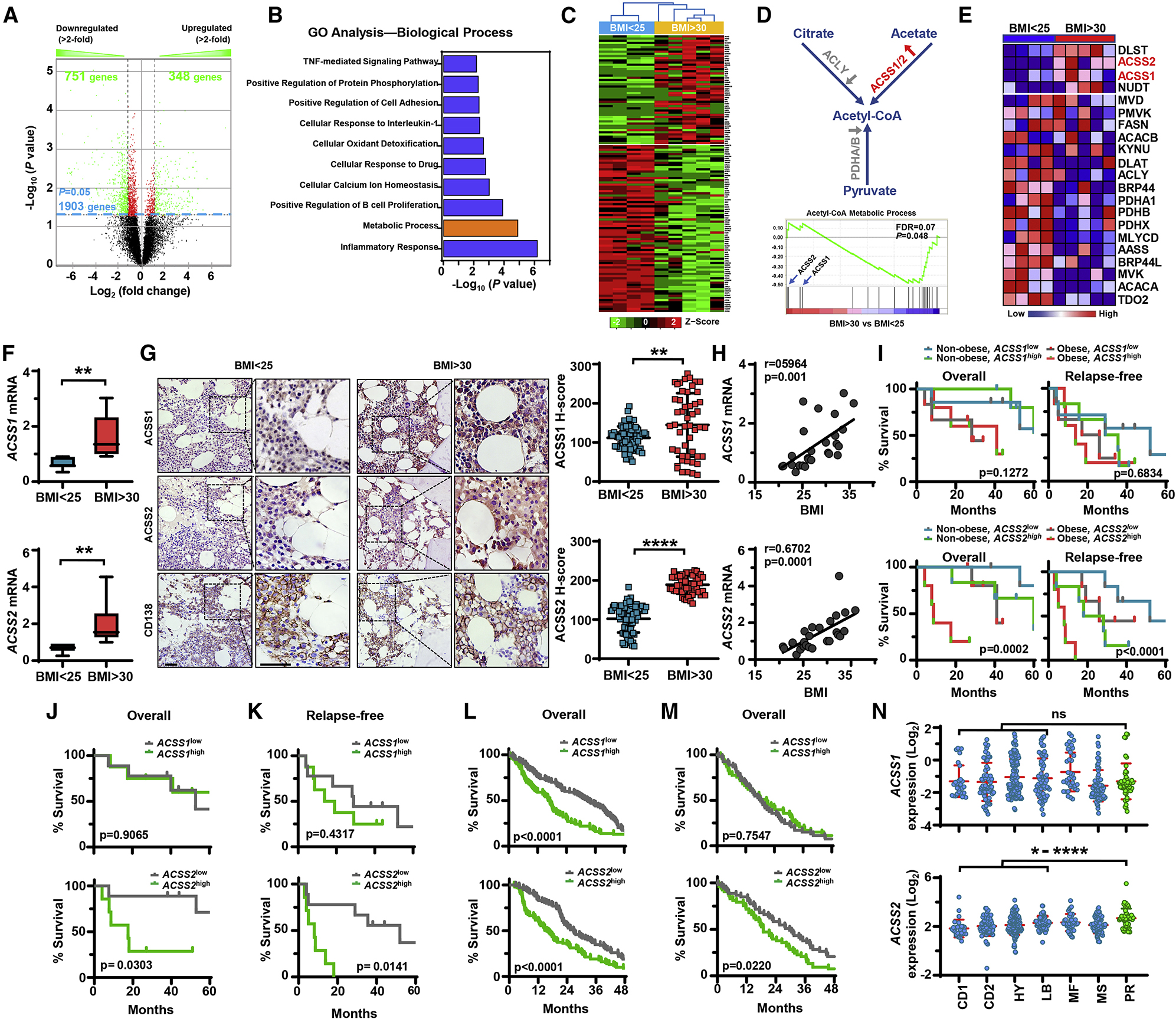
(A-E) RNAs of malignant plasma cells from bone marrow aspirates of patients with newly diagnosed myeloma were subjected to microarray analysis based on patients’ BMI (< 25 kg/m2, n=4; > 30 kg/m2, n=5). (A) Volcano plot shows the differentially up- or down-regulated genes between the two groups. Genes deemed statistically significant are in lime green. (B) Gene ontology analysis shows the biological processes most altered between the compared groups. (C) Heat map of the 127 most significantly up- and down-regulated genes from the metabolic process gene set. (D) Schematic process of acetyl-CoA synthesis (upper) and the GSEA enrichment plot using the “acetyl-CoA metabolic process” gene set in myeloma cells (lower). (E) Heat map of differentially expressed genes in acetyl-CoA synthesis in obese patients when compared with normal-weight patients. (F) ACSS1 or ACSS2 mRNAs in malignant plasma cells isolated from bone marrow aspirates of myeloma patients (n=18). (G) Immunohistochemical staining of patients’ bone marrow biopsies with antibodies against ACSS1, ACSS2, and CD138. Scale bars, 50 μm. Right, summarized data. (H) Correlation coefficient between ACSS1 or ACSS2 mRNAs in malignant plasma cells and patients’ BMIs (n=27). (I) Kaplan–Meier analysis of overall (left) and relapse-free (right) survivals in 4 myeloma patient groups (n=5–7/group). (J-K) Kaplan–Meier analysis of overall (J) and relapse-free (K) survivals in myeloma patients with high (n=9) or low (n=7) ACSS1 or ACSS2 expression. (L-M) Kaplan–Meier analysis of patients’ overall survival in the indicated groups of Zhan et al dataset (n=100/group, L) and MMRF CoMMpass dataset (n = 53/group, M). (N) Relative expression of ACSS1 or ACSS2 in myeloma subgroups (CD1=28, CD2=60, HY=116, LB=55, MF=37, MS=65 and PR=46) characterized by gene expression profiling by Zhan et al, 2006. *P < 0.05; **P < 0.01; ****P < 0.0001. See also Figure S1 and Table S1.
We next analyzed the association of ACSS1 or ACSS2 with myeloma disease using microarray dataset from Oncomine, and we found that patients with high levels of ACSS1 or ACSS2 expression in myeloma cells displayed shorter overall survival than those with low expression (Figure 1L) (Zhan et al., 2006). The association of overall survival with ACSS2 was further confirmed in the latest MMRF CoMMpass RNA-seq database (Figure 1M). Zhan et al. classified multiple myeloma into seven molecular subtypes (CD1 or CD2 of cyclin D translocation; HY: hyperdiploid; LB: low bone disease; MF or MS with activation of MAF, MAFB, or FGRF3/MMSET; PR: proliferation) based on the known genetic lesions (Zhan et al., 2006). Our analysis found that patients in PR subtype, which is a signature of high-risk disease with poor prognosis, had a significantly higher expression of myeloma cell ACSS2 than those in the four subtypes that are associated with favorable prognosis (Figure 1N). However, this pattern was not observed in myeloma cell ACSS1 expression (Figure 1N). Overall, our data strongly suggest that ACSS2 may be a high-risk factor for disease progression.
ACSS2 contributes to obesity-induced myelomatous tumorigenesis
We determined the contribution of ACSS2 to myelomatous tumorigenesis under normal or obese situation. Using lentivirus carrying non-targeted control short-hairpin RNA (shCtrl) or shRNAs for human ACSS2 (shACSS2) gene, we first knocked down ACSS2 expression in myeloma cell lines ARP-1 and MM.1S, both of which highly express ACSS2 (Figures S2A and S2B). We observed a sharp decrease in anchorage-independent growth in shACSS2 myeloma cells compared with shCtrl cells (Figure S2C). On the contrary, knockdown of ACSS1 has little effect on colony formation (Figures S2D and S2E). These results indicate that ACSS2, but not ACSS1, contributes to myelomatous tumorigenesis. The functions of ACSS2 in myeloma cell proliferation and colony formation were further confirmed by knocking out ACSS2 expression in ARP-1, MM.1S, and U266 cells using a CRISPR/Cas9 system (Figures S2F–S2H).
We found that ACSS2 expression was much higher in the marrow-infiltrating malignant plasma cells than in normal plasma cells (Figures S2I and S2J). When myeloma cells were treated with ACSS2 inhibitor (ACSS2i), we observed significantly reduced cellular viability in human myeloma cell lines ARP-1, U266, MM.1S, RPMI8226, and in primary malignant plasma cells isolated from the bone marrow aspirates of myeloma patients (Figure S2K). ACSS2i treatment also impaired the ability of ARP-1 or MM.1S myeloma cells to form colonies and induced myeloma cell apoptosis in a dose-dependent manner in vitro (Figures S2L–S2M). To confirm the specificity of ACSS2i in myeloma, we treated ACSS2-knockout myeloma cells with various concentrations of ACSS2i. We found no or little efficacy in those knockout cells (Figure S2N).
To evaluate the impact of ACSS2 on myeloma growth in vivo, we established a subcutaneous xenograft myeloma mouse model. NSG mice were injected with luciferase-labeled ARP-1 or MM.1S myeloma cells containing shCtrl (left flanks) or shACSS2 (right flanks) (Figure S3A). We observed lower levels of bioluminescent activity and tumor burdens in the flanks implanted with shACSS2 myeloma cells than those implanted with shCtrl myeloma cells (Figures S3A and S3B). Immunohistochemical analysis showed reduced staining of ACSS2 and Ki67 in the tumor tissues from shACSS2 myeloma cell-bearing mice compared with shCtrl cell-bearing mice (Figure S3C). We further confirmed our results with the TetON inducible system both in vitro and in vivo (Figures S3D and S3E). Furthermore, we intravenously injected NSG mice with luciferase-labeled wild-type ARP-1 or MM.1S cells and then treated the mice with ACSS2i at 25 mg/kg, a dose that was effective at reducing myeloma tumor burden but did not cause body weight loss and tissue toxicity in mice (Figures S3F–S3H). Compared with vehicle-treated controls, ACSS2i-treated mice had reduced levels of bioluminescent activity and tumor burden measured by levels of serum M-proteins (Figures S3I and S3J). These findings not only demonstrate the contribution of ACSS2 to myeloma tumorigenesis but also suggest that inhibition of ACSS2 in myeloma can be used as a potential therapeutic strategy.
To determine whether ACSS2 mediates myelomatous tumorigenesis in an obese setting, we examined the efficacy of ACSS2i in the myeloma DIO mouse model (Figure 2A). Figures 2B–2E show the increased tumor burden in DIO mice injected with murine myeloma Vk12598 cells, evidenced by the elevated circulating M-proteins, increased numbers of marrow- and spleen-infiltrating CD138+ myeloma cells, and enlarged spleens, comparing to those in ND mice. After treating DIO or ND mice with 25 mg/kg ACSS2i or with vehicle control, we found that ACSS2i treatment remarkably reduced serum M-protein levels, spleen size, and the number of spleen- and marrow-infiltrating CD138+ cells, and improved mouse survival without affecting mouse body weights comparing with the control (Figures 2B–2F). Using the 5TGM1 mouse model, we observed higher incidence of myeloma established in DIO mice than in ND mice, evidenced by the increased circulating M-proteins and enlarged spleen sizes, while treatment of ACSS2i significantly reduced the rate of myeloma incidence from 60% to 20% (Figure 2G).
Figure 2. Treatment of ACSS2i reduces tumor growth in the myeloma DIO mouse model.
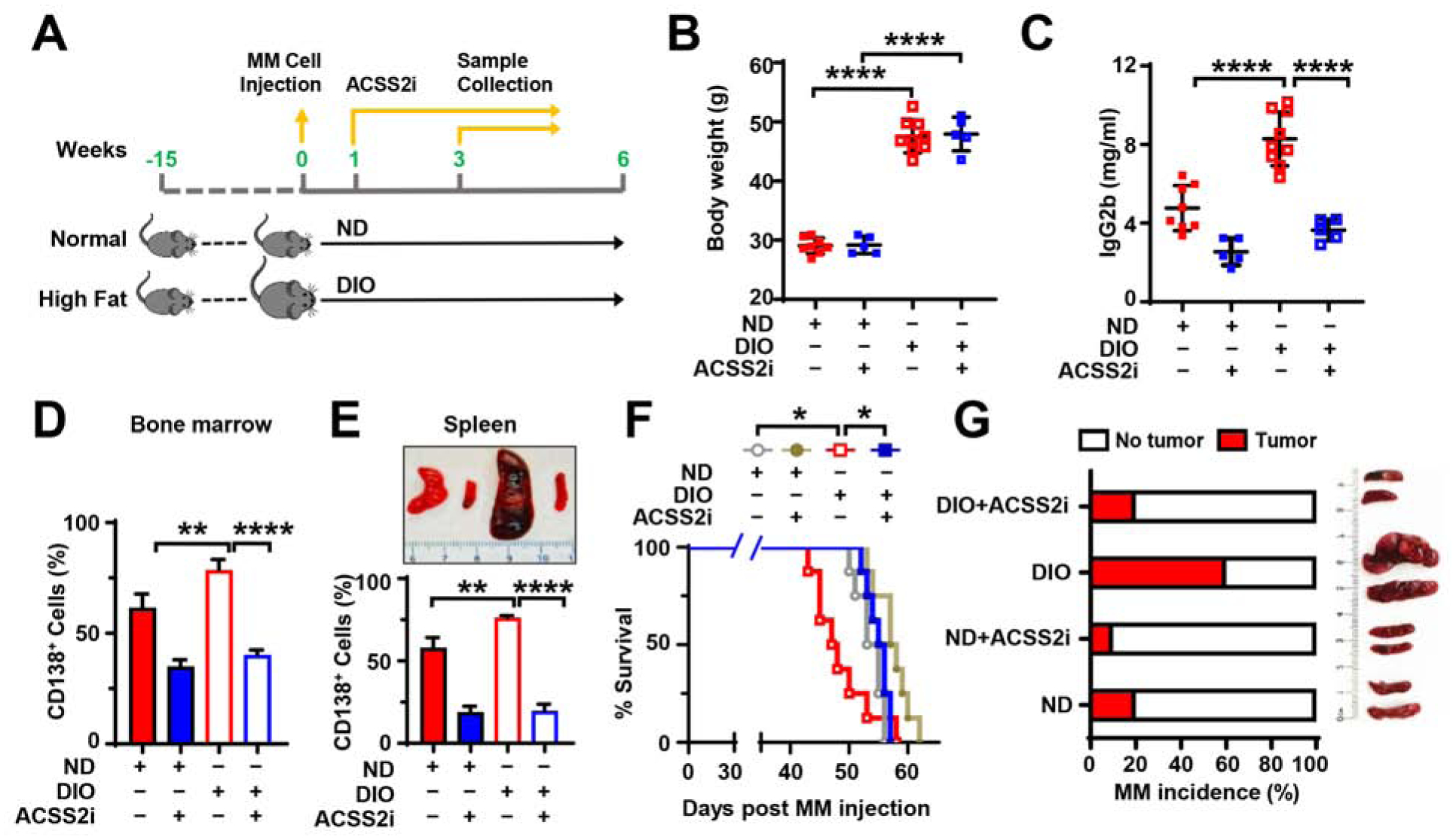
(A-F) ND or DIO C57BL/6J mice intrafemorally injected with Vk12598 cells were treated with ACSS2i (n=5 or 8/group). (A) Schema for the myeloma obesity mouse model. (B) Mouse body weights. (C) Levels of IgG2b in mouse sera. (D) Percentages of marrow-infiltrated CD138+ myeloma cells. (E) Representative images of mouse spleens (upper) and percentages of spleen-infiltrated CD138+ myeloma cells (lower). (F) Kaplan–Meier analysis of overall mouse survival. (G) Incidence rate of myeloma in mice (left) and representative images of mouse spleens (right) 8 weeks after DIO or ND C57BL/6J mice intravenously injected with 5TGM1 cells and treated with ACSS2i (n=10/group). Data shown as mean ± SD (**P < 0.01; ****P < 0.0001). See also Figures S2 and S3.
To examine whether ACSS2 has a function in adipocyte-induced myeloma growth, we subcutaneously implanted the mixture of the luciferase-labeled shCtrl or shACSS2 myeloma ARP-1 cells and purified mature adipocytes into NSG mice. Implantation of myeloma cells alone in the opposite flank served as control. We observed higher levels of bioluminescent activity and tumor weight in the sites implanted with mixtures of adipocytes and myeloma cells than in those with myeloma cells alone (Figures 3A and 3B). These results were further confirmed in mice implanted with adipocytes and sgACSS2 MM.1S myeloma cells as opposed to sgCtrl cells (Figures 3C and 3D). Immunohistochemical staining shows the reduced Ki67 expression in shACSS2 myeloma cells (Figures 3E and 3F). In line with in vivo studies, we found that adding the conditioned medium of adipocytes derived from MSCs (AD-CM) to cultures of ARP-1 or MM.1S myeloma cells significantly increased colony formation, while knockdown of ACSS2 in myeloma cells reduced such effects (Figure 4A). These results suggest that ACSS2 mediates adipocyte-enhanced myelomatous tumorigenesis.
Figure 3. ACSS2 mediates adipocyte-enhanced myeloma growth in vivo.
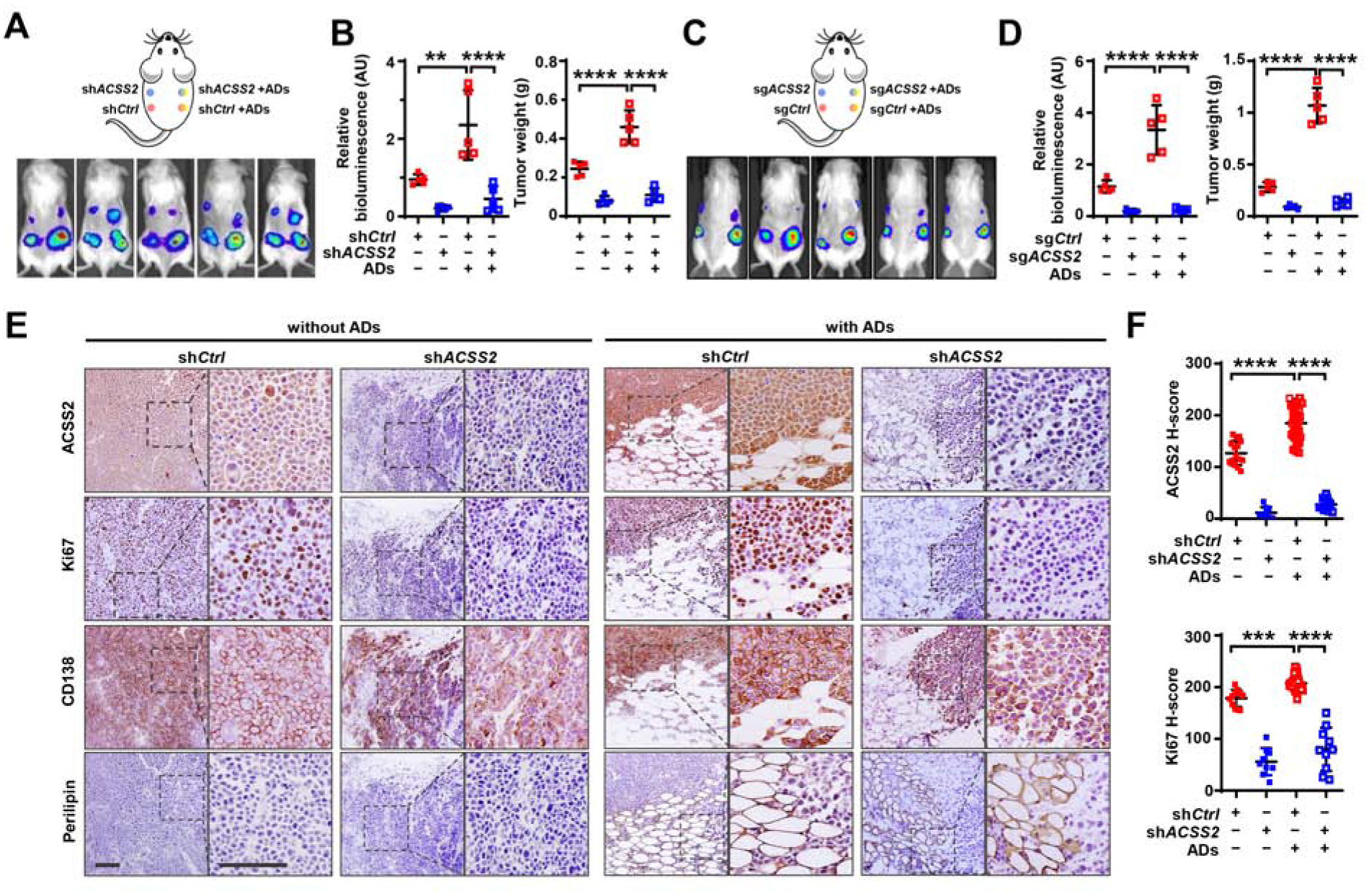
(A-D) Illustrated injection sites (upper), bioluminescent images (lower) (A, C), quantitative bioluminescent signals, and tumor weights (B, D) of NSG mice 4 weeks after subcutaneous injection of luciferase-labeled shCtrl or shACSS2 ARP-1 cells (A-B) or sgCtrl or sgACSS2MM.1S cells (C-D), alone or mixed with human adipocytes. (E-F) Immunohistochemical staining of ACSS2, Ki67, CD138, and perilipin in tumor tissues (E) and H-scores for ACSS2 and Ki67 (F) in shCtrl or shACSS2 ARP-1 cells. Scale bars, 100 μm. Data shown as mean ± SD (n=5 mice/group). **P < 0.01; ***P < 0.001; ****P < 0.0001. See also Figures S2 and S3.
Figure 4. Adipocyte-derived angiotensin II enhances the expression of ACSS2 in myeloma cells.
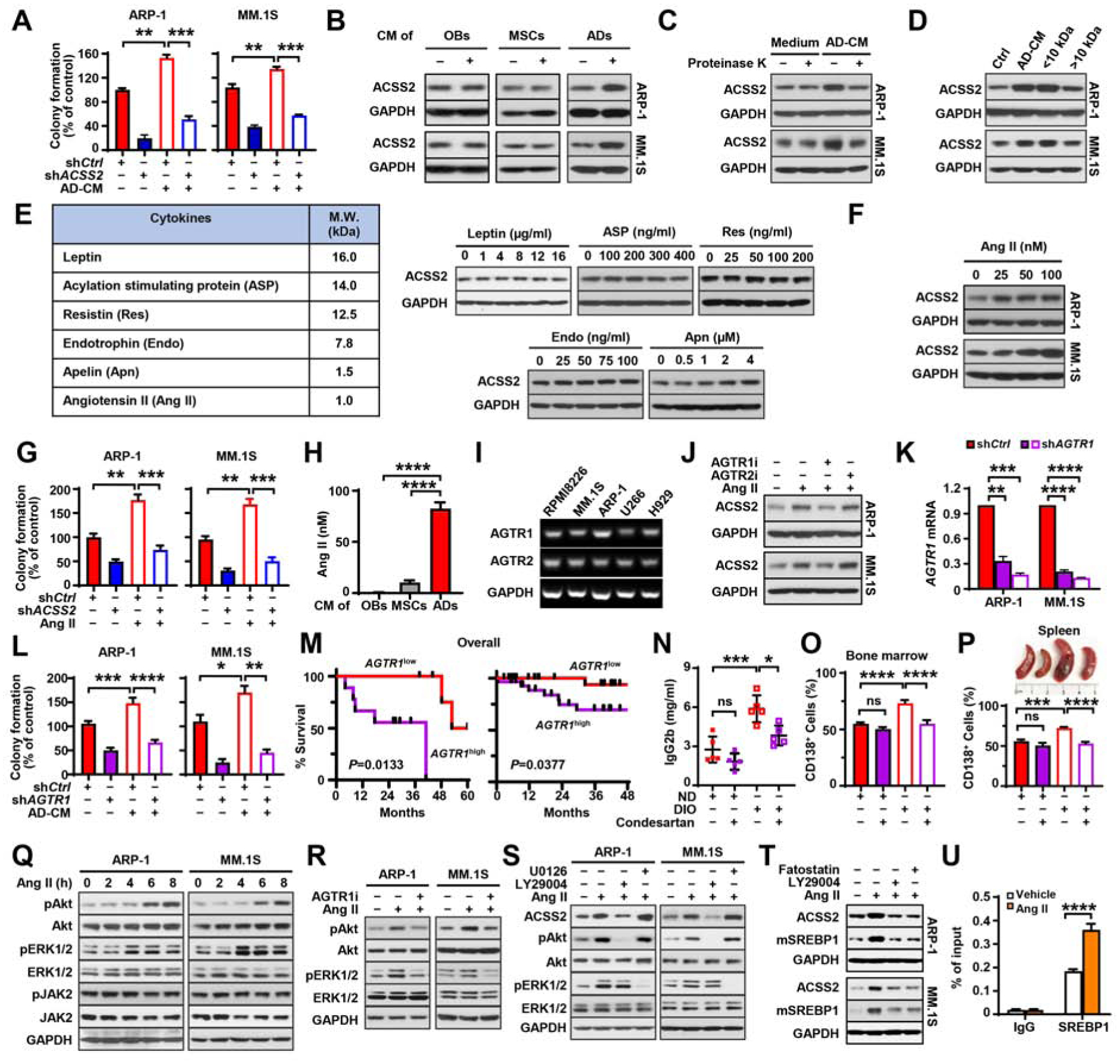
(A) Colony formation in shCtrl or shACSS2 myeloma cells with or without adipocyte condition medium (AD-CM). (B) Expression of ACSS2 in myeloma cells cultured with condition medium of human MSCs, adipocytes, or osteoblasts (OBs). (C-D) ACSS2 protein levels in myeloma cells cultured in AD-CM pretreated with proteinase K-agarose (C) or different molecular weight cutoff fractions (D). (E) List of cytokines secreted by adipocytes with molecular weights around or less than 10 kDa and ACSS2 levels in ARP-1 cells cultured with leptin, ASP, Res, Apn, or Endo. (F) ACSS2 levels in myeloma cells cultured with Ang II. (G) Colony formation of shCtrl or shACCS2-expressing myeloma cells cultured with or without 100 nM Ang II. (H) Concentration of Ang II in the supernatant of adipocytes, OBs, or MSCs after 72 hours. (I) Expression of Ang II receptors AGTR1 and AGTR2 in myeloma cells. (J) ACSS2 expression in myeloma cells cultured in 100 nM Ang II and 10 μM inhibitors against AGTR1 (AGTR1i) or AGTR2 (AGTR2i). (K) AGTR1 mRNAs in shCtrl or shAGTR1 myeloma cells. (L) Colony formation in shCtrl or shAGTR1 myeloma cells cultured with or without AD-CM. (M) Kaplan–Meier analysis of overall survival of myeloma patients with high or low AGTR1 expression (left: our data, high [n=9], low [n=6]; right: MMRF CoMMpass, n=28/group). (N-P) Levels of IgG2b in mouse sera (N), percentages of marrow-infiltrated CD138+ myeloma cells (O), and mouse spleens and percentages of spleen-infiltrated CD138+ myeloma cells (P) in Vk12598-bearing ND or DIO C57BL/6J mice treated with condesartan (n=5/group). (Q) Time course on the levels of non-phosphorylated or phosphorylated Akt, ERK1/2, and JAK2 in myeloma cells treated with 100 nM Ang II. (R-S) Levels of non-phosphorylated or phosphorylated Akt and ERK1/2 in myeloma cells incubated with 100 nM Ang II and 10 μM of AGTR1i (R), LY29004 or U0126 (S). (T) Protein levels of ACSS2 and mature SREBP1 (mSREBP1) in myeloma cells cultured with Ang II (100 nM) and LY29004 (10 μM) or fatostatin (5 μM) for 24 hours. (U) ChIP assay shows the enrichment of SREBP1 at the promoter of ACSS2 gene in ARP-1 cells with or without Ang II (100 nM). Data shown as mean± SD (ns, not significant; *P<0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001). See also Figure S4.
Obesity upregulates the expression of ACSS2 in myeloma cells through adipocyte-secreted angiotensin II
Similar to that seen in obese myeloma patients, we observed an increased ACSS2 expression in myeloma cells co-implanted with adipocytes in vivo (Figures 3E and 3F) or incubated with AD-CM in vitro (Figure 4B). Other stromal cells such as MSCs or osteoblasts had no such effect (Figure 4B). These results indicate that adipocyte-secreted factors mediate obesity-induced ACSS2 upregulation. To pinpoint the relevant adipocyte-derived factor(s), we cultured myeloma cells in AD-CM treated with proteinase K, or in CM fractions with molecular weight cutoff. Few effects on ACSS2 expression were seen in myeloma cells cultured with the proteinase K-treated CM or cultured in a fraction of the CM with molecular weights greater than 10 kDa (Figures 4C and 4D), indicating that the smaller protein components of AD-CM are factor(s) mediating the ACSS2 upregulation. We screened 6 cytokines that are known to be secreted from adipocytes with molecular weights around or less than 10 kDa (Cabia et al., 2016; Johrer et al., 2015; Polak et al., 2017) and found that addition of the recombinant angiotensin II, but not others, to myeloma cell cultures remarkably enhanced ACSS2 expression in a dose-dependent manner (Figures 4E and 4F). Addition of angiotensin II also enhanced colony formation of myeloma cells, while knockdown of ACSS2 reduced such effects (Figure 4G). The secretion of angiotensin II was mainly from adipocytes, while other stromal cells such as MSCs or osteoblasts secreted little (Figure 4H). Adipocytes isolated from DIO mice or human subjects with obesity displayed higher levels of angiotensin II secretion and mRNAs of angiotensin converting enzyme (Ace/ACE) and renin (Ren/REN), two key enzymes responsible for angiotensin II production, than those from ND mice or people with normal weight; culturing myeloma cells with CM of adipocytes isolated from obese subjects had more ACSS2 expression in myeloma cells (Figure S4). These results indicate that under obese condition, the increased angiotensin II secreted from adipocytes upregulates myeloma cell ACSS2 expression and growth.
The two receptors of angiotensin II, angiotensin receptor type (AGTR) 1 and 2, were both expressed in all tested myeloma cells (Figure 4I). Treatment with AGTR1 inhibitor, but not AGTR2 inhibitor, significantly abrogated the effects of angiotensin II on ACSS2 expression (Figure 4J). Not surprisingly, knockdown of the angiotensin II receptor ATGR1 reduced the effects of AD-CM on myeloma cell colony formation (Figures 4K and 4L). High AGTR1 expression was also correlated with worse overall survival in myeloma patients (Figure 4M). While the treatment of the AGTR1 inhibitor showed little effects in myeloma-bearing ND mice, the same treatment significantly abolished obesity-stimulated tumorigenesis in myeloma-bearing DIO mice (Figures 4N–4P), indicating a pivotal contribution of AGTR1 to angiotensin II-induced myeloma tumorigenesis under obese condition.
To investigate the signaling pathways by which angiotensin II upregulates ACSS2 expression in myeloma cells, we examined the PI3K/Akt, ERK1/2, and Jak2 pathways that are known to be activated through AGTR1 (Touyz and Schiffrin, 2000). In myeloma cells, we found that addition of angiotensin II increased the levels of phosphorylated Akt and ERK1/2, but had little or no effects on the levels of phosphorylated Jak2 and the non-phosphorylated kinases (Figure 4Q). Treatment with the AGTR1 inhibitor or the PI3K inhibitor LY294002 significantly abrogated angiotensin II-induced Akt phosphorylation and ACSS2 expression in myeloma cells, while treatment with U0126, an inhibitor of ERK1/2, had no effects on ACSS2 expression (Figures 4R and 4S). Since SREBP-1 is a transcription factor downstream of AGTR1 (Wang et al., 2015), we wondered whether SREBP-1 mediates angiotensin II-induced ACSS2 expression. We found that addition of angiotensin II enhanced the cleavage of SREBP1 to its mature isoform (mSREBP1) and enrichment of SREBP1 at the promoter of ACSS2 gene (Figures 4T and 4U). Treatment with the PI3K inhibitor LY294002 or the SREBP1 inhibitor fatostatin abolished such effects (Figure 4T). These results suggest that angiotensin II binds to its receptor AGTR1, activates the PI3K/Akt/SREBP-1 signaling, thus upregulates myeloma cell ACSS2 expression.
Interaction of ACSS2 with the oncogenic protein IRF4 promotes myelomatous tumorigenesis
To investigate the mechanism underlying ACSS2-induced myeloma growth, we first used mass spectrometry to identify the partners interacting with ACSS2. Myeloma ARP-1 cells were infected with lentivirus carrying FLAG-tagged vector control or human ACSS2 cDNAs (FLAG-ACSS2), and the cell lysates were pulled down by anti-FLAG resin for analysis. In addition to a known ACSS2-associated protein, pyruvate kinase muscle isozyme, we found 4 peptides of interferon regulatory factor 4 (IRF4) in the immunoprecipitates (Figures S5A and S5B), which are among the ones with top coverages. IRF4 plays a critical role in B-lymphocyte development and acts as a transcription factor, regulating the expression of genes that support myeloma growth and survival (Shaffer et al., 2008). To confirm the interaction between ACSS2 and IRF4 proteins, we co-transfected HEK293T cells with FLAG-ACSS2 and HA-tagged IRF4 (HA-IRF4), and immunoprecipitated the cell lysates using anti-FLAG antibody. We detected HA-IRF4 proteins in the immunoprecipitates (Figure S5C). Using anti-FLAG antibody, we also detected the presence of endogenous IRF4 proteins in the immunoprecipitates from myeloma cells infected with lentivirus carrying FLAG-ACSS2 (Figure S5D). To further determine whether the endogenous ACSS2-IRF4 complex exists in myeloma cells, we immunoprecipitated the lysates of ARP-1 and MM.1S cells using an anti-ACSS2 antibody and observed the presence of IRF4 proteins, and vice versa (Figure S5E). Using immunofluorescence-labeled antibodies against ACSS2 or IRF4, we observed a co-localization of the two proteins in the cytoplasm of myeloma cells (Figure S5F). Furthermore, we separated ARP-1 cell lysates into cytoplasmic or nuclear fractions and immunoprecipitated each fraction by the anti-IRF4 antibody. We found that the interaction between ACSS2 and IRF4 overwhelming occurred in cytosol (Figure S5G), which is in line with immunofluorescent staining.
To examine the role of IRF4 in AD-CM or ACSS2-induced myeloma tumorigenesis, we knocked down IRF4 expression in myeloma cells using IRF4 shRNA (shIRF4). We observed that cultures with AD-CM enhanced myeloma cell colony formation, while knockdown of IRF4 reduced such effects (Figure S6A). Similar results were also obtained when we knocked down IRF4 expression in FLAG-ACSS2 expressing myeloma cells (Figure S6B). On the other hand, overexpression of IRF4 in shACSS2-expressing ARP-1 cells overcame shACSS2’s inhibitory effects on colony formation (Figures S6C). We further confirmed our results by overexpressing IRF4 in ACSS2-knockout MM.1S cells in vitro and in vivo (Figures S6D and S6E). These results reveal that IRF4 is a previously unidentified ACSS2-interacting protein, and at least partially mediates ACSS2-induced tumorigenesis in myeloma.
ACSS2 maintains IRF4 protein stability through activation of lysine acetylation in IRF4
We next investigated whether ACSS2 regulates IRF4 expression in myeloma cells. Quantitative PCR analysis showed that addition of AD-CM (Figure S6F, upper panel), knockdown, or inhibition of myeloma cell ACSS2 (Figure 5A) did not affect the levels of IRF4 mRNAs. Western blotting showed the elevation of IRF4 protein levels in myeloma cells cultured with AD-CM (Figure S6F, lower panel), but a drastically reduced level of IRF4 protein in shACSS2-expressing or ACSS2i-treated myeloma cells compared with control cells (Figure 5B). Consequently, the expression of IRF4 target genes was significantly upregulated in myeloma cells cultured with AD-CM, while the levels were reduced upon knockdown or inhibition of ACSS2 (Figures 5C, 5D, and S6G). ChIP assay showed that knockdown of ACSS2 in myeloma cells resulted in decreased enrichment of IRF4 at the promoter of IRF4 target genes (Figure 5E). These results indicate that AD-CM or ACSS2 regulates IRF4 at post-transcriptional levels. When cycloheximide, a eukaryote protein synthesis inhibitor, was added to cultures of myeloma cells, we found that the reduction of IRF4 protein progressed much quicker in ACSS2i-treated cells than in control cells (Figure 5F). This observation led us to hypothesize that ACSS2 stabilizes IRF4 protein by preventing it from degradation. Since proteasome and lysosome are two major organelles responsible for intracellular protein homeostasis, we used MG-132 (a proteasome inhibitor) or leupeptin (a lysosomal-protease inhibitor) in the culture. We observed that when ACSS2 was inhibited, the presence of leupeptin but not MG-132, caused IRF4 protein accumulation in myeloma cells (Figure 5G), suggesting that ACSS2 prevents IRF4 protein degradation through a lysosome pathway. We next asked whether the regulation of IRF4 by ACSS2 is a common mechanism in other tumors, such as melanoma, in which IRF4 is also expressed and may act as a critical factor in melanoma growth (Praetorius et al., 2013; Seberg et al., 2017). We found that either ACSS2i treatment or knockdown of ACSS2 decreased IRF4 protein accumulation and anchorage-independent growth of melanoma A375 cells, while overexpression of IRF4 rescued tumor cell colony formation (Figure S7). These results suggest that there could be a common ACSS2-IRF4 axis responsible for tumorigenesis.
Figure 5. ACSS2 impairs lysosome-mediated IRF4 protein degradation.
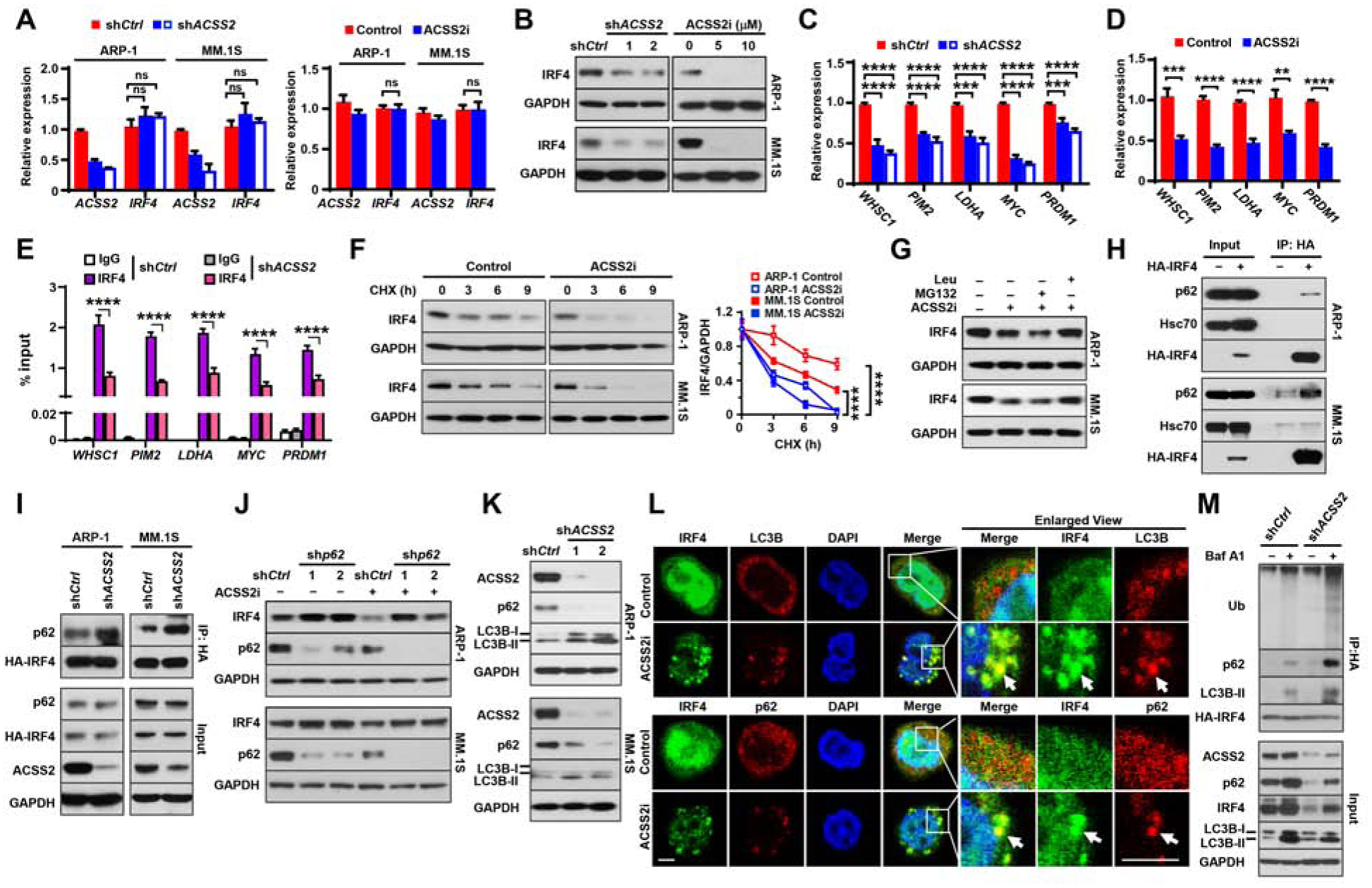
(A-B) IRF4 mRNA (A) and IRF4 protein (B) in shCtrl or shACSS2 myeloma cells, or uninfected cells treated with10 μM ACSS2i for 24 hours. (C-D) Relative expression of IRF4 target genes WHSC1, PIM2, MYC, LDHA, and PRDM1 in shCtrl- and shACSS2-expressing (C) or ACSS2i-treated (D) myeloma cells. (E) ChIP assay shows the enrichment of IRF4 at the promoter of WHSC1, PIM2, MYC, LDHA, or PRDM1 genes. (F) Time course of IRF4 protein in myeloma cells treated with 10 μM ACSS2i and 100 μM cycloheximide (CHX). Right: normalized IRF4 against GAPDH. (G) IRF4 protein levels in myeloma cells treated with 10 μM ACSS2i and 10 μM MG-132 or 10 μM leupeptin for 24 hours. (H) p62, Hsc70, and HA-IRF4 proteins in the immunoprecipitates of lysates from myeloma cells carrying HA-tagged IRF4. (I) Immunoprecipitation (IP) analysis of HA-IRF4 with endogenous p62 protein in HA-tagged IRF4 myeloma cells that were infected with lentivirus carrying shCtrl or shACSS2. Immunoblotting was detected with antibodies against HA or p62. (J) IRF4 and p62 protein levels in shp62-expressing myeloma cells treated with 10 μM ACSS2i for 24 hours. (K) LC3B-I/II, p62, and ACSS2 protein levels in shCtrl or shACSS2 myeloma cells. (L) Immunofluorescent staining of ACSS2i-treated ARP-1 cells with DAPI and antibodies against IRF4, p62, or LC3B. Scale bar, 5 μm. (M) IP of lysates from HA-tagged IRF4 ARP-1 cells that were infected with lentivirus carrying shCtrl or shACSS2 and treated with or without 100 nM bafilomycin A1 for 6 hours. Data shown as mean ± SD (ns, non-significant; **P < 0.01; ***P < 0.001; ****P < 0.0001). See also Figures S5–S7.
Sequestosome 1/ubiquitin-binding protein p62 (p62) and heat shock cognate protein 70 (Hsc70) are two of the key molecules responsible for autophagy-mediated protein degradation (Boya et al., 2013; Wang and Wang, 2015). Co-immunoprecipitation assays showed that p62, but not Hsc70, bound to IRF4 protein (Figure 5H). To examine whether ACSS2 has a function in IRF4-p62 interaction, HA-IRF4 myeloma cells were infected with lentivirus carrying shACSS2, and the cell lysates, pulled down by anti-HA antibody, were subjected to immunoblotting using an anti-p62 antibody. We observed higher levels of p62 in the immunoprecipitates from shACSS2 myeloma cells than in the immunoprecipitates from shCtrl cells (Figure 5I), indicating that ACSS2 knockdown enhanced IRF4-p62 interaction. Furthermore, myeloma cells infected with lentivirus carrying shRNAs against human p62 (shp62) were treated with ACSS2i. ACSS2i treatment induced IRF4 protein degradation, while knockdown of p62 resisted ACSS2i’s effects on IRF4 degradation (Figure 5J). In addition, we found that knockdown or inhibition of ACSS2 increased LC3B-II levels (Figure 5K) and cellular puncta formation (Figure 5L) in myeloma cells, indicating that ACSS2 prevents autophagic activation in myeloma cells. We also observed a reduced p62 accumulation and co-localization of IRF4 protein with either p62 or LC3B-labeled cellular puncta in shACSS2-expressing or ACSS2i-treated myeloma cells, but not in control cells (Figures 5K and 5L). Since p62 binds to the ubiquitinated protein aggregates to form the complex which is recognized by LC3, and subsequently enters autophagosomes (Lamark et al., 2017), we examined ubiquitination levels on IRF4 protein. HA-IRF4-expressing ARP-1 cells were infected with lentivirus carrying shCtrl or shACSS2, treated with the autophagy inhibitor bafilomycin A1, and cell lysates were pulled down by the anti-HA resin. Comparing those in shCtrl cells, bafilomycin A1 treatment induced heavier accumulation of IRF4 polyubiquitination, p62, and LC3B-II proteins in shACSS2 cells (Figure 5M). These results indicate that ACSS2 hinders the binding of IRF4 to p62, thereby reducing p62-mediated IRF4 protein degradation.
Because ACSS2 is involved in the regulation of protein lysine acetylation, we wondered whether ACSS2 stabilizes IRF4 protein in a similar fashion. Indeed, we found reduced levels of acetylated lysine (Ac-K) in the HA-IRF4 immunoprecipitates of ACSS2i-treated myeloma cells (Figure 6A). To identify the acetylated sites in IRF4 protein induced by ACSS2, lysates of ARP-1 cells infected with lentivirus carrying HA-IRF4 and treated with or without ASCC2i were pulled down by an anti-HA antibody, and the bands of HA-IRF4 (Figure 6B) were further analyzed by mass spectrometry. We identified three putative acetylation sites at lysines–K59, K87, and K399–in the band from untreated control cells, but they were absent from ACSS2i-treated cells (Figure 6C). When we mutated each lysine to the acetyl-mimetic amino acid glutamine (Q), we found that mutation of K399Q, but not K59Q or K87Q, resisted ACSS2i-induced IRF4 protein degradation (Figure 6D).
Figure 6. ACSS2 maintains IRF4 protein stability through activation of lysine acetylation.
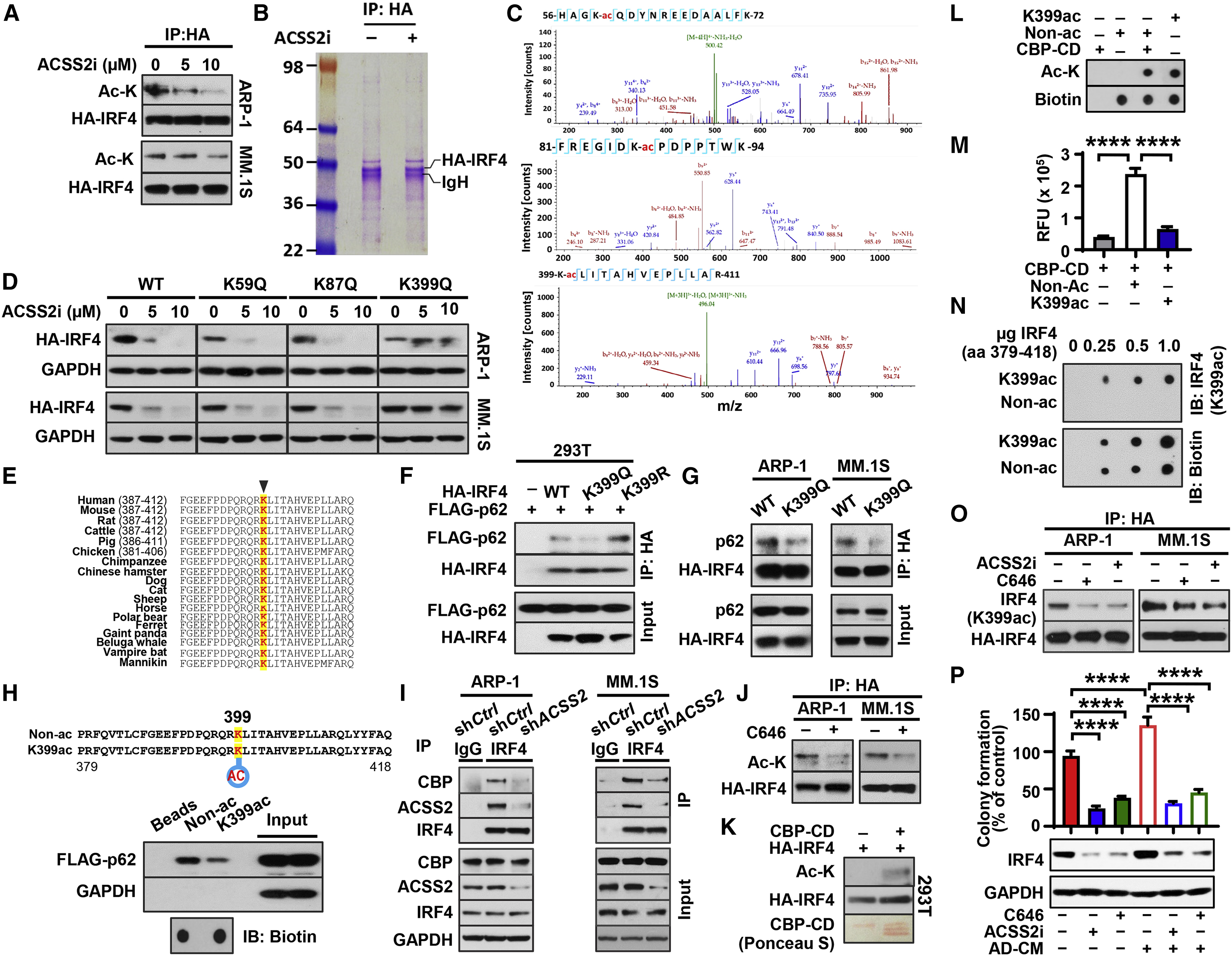
(A-C) Lysine acetylation (Ac-K) and HA-IRF4 protein level (A), Coomassie blue staining of SDS-PAGE gel (B), and mass spectrometry of acetylated lysine (K-ac) at positions 59, 87, and 399 from the cutout HA-IRF4 protein band (C) in HA-IRF4 expressing cell lysates immunoprecipitated with anti-HA resin after 12 hours of ACSS2i treatment. (D) HA-IRF4 protein levels in myeloma cells carrying HA-tagged wild type (WT) or mutation of lysine to glutamine (K to Q) 24 hours after 5 μM ACSS2i treatment (immunoblotting: anti-HA). (E) Conservation of the lysine residue at 399 of IRF4 across different species. (F) IP of p62 and IRF4 with anti-HA resin in lysates of HEK293T cells co-transfected with FLAG-tagged p62 (FLAG-p62) and varied forms of HA-IRF4 (immunoblotting: anti-HA or anti-FLAG). (G) IP of p62 and IRF4 in lysates of myeloma cells carrying WT or K399Q HA-IRF4. (H) Depiction of custom-made biotin-conjugated IRF4 peptides (aa 379 to 418) in acetylated (K399ac) or non-acetylated (Non-ac) form (upper), and their interaction with FLAG-p62 expressed in HEK293T cells (lower). (I) CBP, ACSS2, and IRF4 protein in immunoprecipitates of lysates from shCtrl- or shACSS2-expressing myeloma cells. (J) Immunoprecipitates of lysates from HA-IRF4–expressing myeloma cells treated with 10 μM C646 (immunoblotting: anti-Ac-K or anti-HA). (K) HA-IRF4 and Ac-K levels in the reactive products from in vitro acetylation assays and Ponceau S-stained CBP-CD bands on PVDF membrane. (L-M) Ac-K and biotin spots in dot blotting (L) and the relative fluorescent units measuring acetyltransferase activity (M) when CBP-CD was incubated with custom Non-Ac or K399ac IRF4 peptides. (N) Dot blotting shows the specificity of custom-made antibody (K399ac) for acetylated IRF4 peptide. (O) IP of lysates from HA-IRF4–expressing myeloma cells treated with 10 μM ACSS2i or 10 μM C646 [immunoblotting: anti-IRF4 (K399ac) or anti-HA]. (P) Relative colony formation (upper) and IRF4 protein level (lower) in ARP-1 cells cultured in AD-CM with or without ACSS2i or C646. Data shown as mean ± SD (****P < 0.0001).
K399 is highly conserved among different species (Figure 6E). To examine whether the acetylation of IRF4 at K399 affects its binding to p62, HEK293T cells were co-transfected with FLAG-tagged p62 and various forms of HA-IRF4; wild-type (WT), K399Q, and an acetyl-deficient form with mutation K399 to arginine (K399R). Co-immunoprecipitation assays showed a reduction in binding of p62 to HA-IRF4 K399Q when compared with its binding to WT or K399R of HA-IRF4 (Figure 6F). A similar reduction of endogenous p62 protein was observed in the immunoprecipitates of myeloma cells expressing HA-IRF4 K399Q (Figure 6G). Moreover, we designed a pair of synthetic peptides containing the amino acids 379 to 418 of IRF4 conjugated with biotin; one with an acetylated amino acid at K399 (K399ac), and the other without the acetylation (Non-ac). Using a pulldown assay, we found reduced levels of FLAG-p62 proteins in the immunoprecipitates with K399ac peptides compared with those with control peptides (Figure 6H). These results indicate that the acetylation at K399 of IRF4 protein prevents IRF4-p62 interaction.
We next sought to identify the factor(s) by which ACSS2 activates acetylation in IRF4 protein. The lysine acetyltransferase CREB-binding protein (CBP) is known to bind to ACSS2 (Xu et al., 2014). Using co-immunoprecipitation assays, we observed a complex of CBP/ACSS2/IRF4 in myeloma cells, while knockdown of ACSS2 expression significantly impaired IRF4-CBP interaction (Figure 6I), suggesting a crucial role of ACSS2 in the formation of the CBP/ACSS2/IRF4 complex. Treatment with the inhibitor C646, which impairs CBP enzymatic activity, remarkably downregulated acetylation in HA-IRF4 protein (Figure 6J). In vitro acetylation assays showed that incubation with the catalytic domain of human CBP (CBP-CD) stimulated the acetylation in HA-IRF4 protein and Non-ac IRF4 peptide (Figures 6K–6M). In addition, we generated an antibody specific for IRF4 K399ac (Figure 6N). Using this antibody, we found a decreased level of IRF4 K399ac in myeloma cells treated with ACSS2i or C646 (Figure 6O). These results indicate that CBP mediates ACSS2-induced IRF4 acetylation at K399. Consistent with the biochemical findings, we confirmed that treatment with C646 or ACSS2i abrogated the effect of AD-CM on IRF4 upregulation and anchorage-independent growth in ARP-1 cells (Figure 6P).
The increased IRF4 protein is positively associated with ACSS2 in obese mice and human patients with myeloma
To determine the association between IRF4 and ACSS2 in vivo under obese condition, we collected bone marrow samples from ND or DIO mice. We found increased levels of ACSS2, IRF4 target gene expression, IRF4 protein, but not IRF4 mRNAs, in marrow-infiltrating myeloma cells in DIO mice compared with those in myeloma cells from ND mice (Figures 7A and 7B). Treatment with ACSS2i significantly impaired such effects (Figures 7A and 7B). To assess the clinical relevance of the association between those two molecules, we measured the mRNAs in CD138+ plasma cells isolated from bone marrow aspirates of myeloma patients and examined the protein staining in patients’ bone marrow biopsies. Staining of CD138 showed the presence of marrow-infiltrated myeloma cells. We observed the higher levels of IRF4 protein, but not IRF4 mRNAs, in obese patients than those in patients with normal weight (Figure 7C). While there was little or no correlation at mRNA levels (Figures 7D and 7E), we observed robust positive correlation of IRF4 protein levels with patients’ BMIs (Figure 7D) or myeloma cell ACSS2 expression (Figures 7F and 7G). Both IRF4 and ACSS2 protein were higher in obese patients than those in patients with normal weight (Figures 7F and 7G). A head-to-head comparison between ACSS2 and the target genes of IRF4 also demonstrated similar strong positive correlations (Figure 7H). The results from both mice and human patients affirm that obesity-enhanced ACSS2 promotes myeloma cell IRF4 protein and IRF4-mediated gene transcription.
Figure 7. The levels of IRF4 protein and its target genes are correlated with ACSS2 expression in myeloma-bearing mice and human patients.
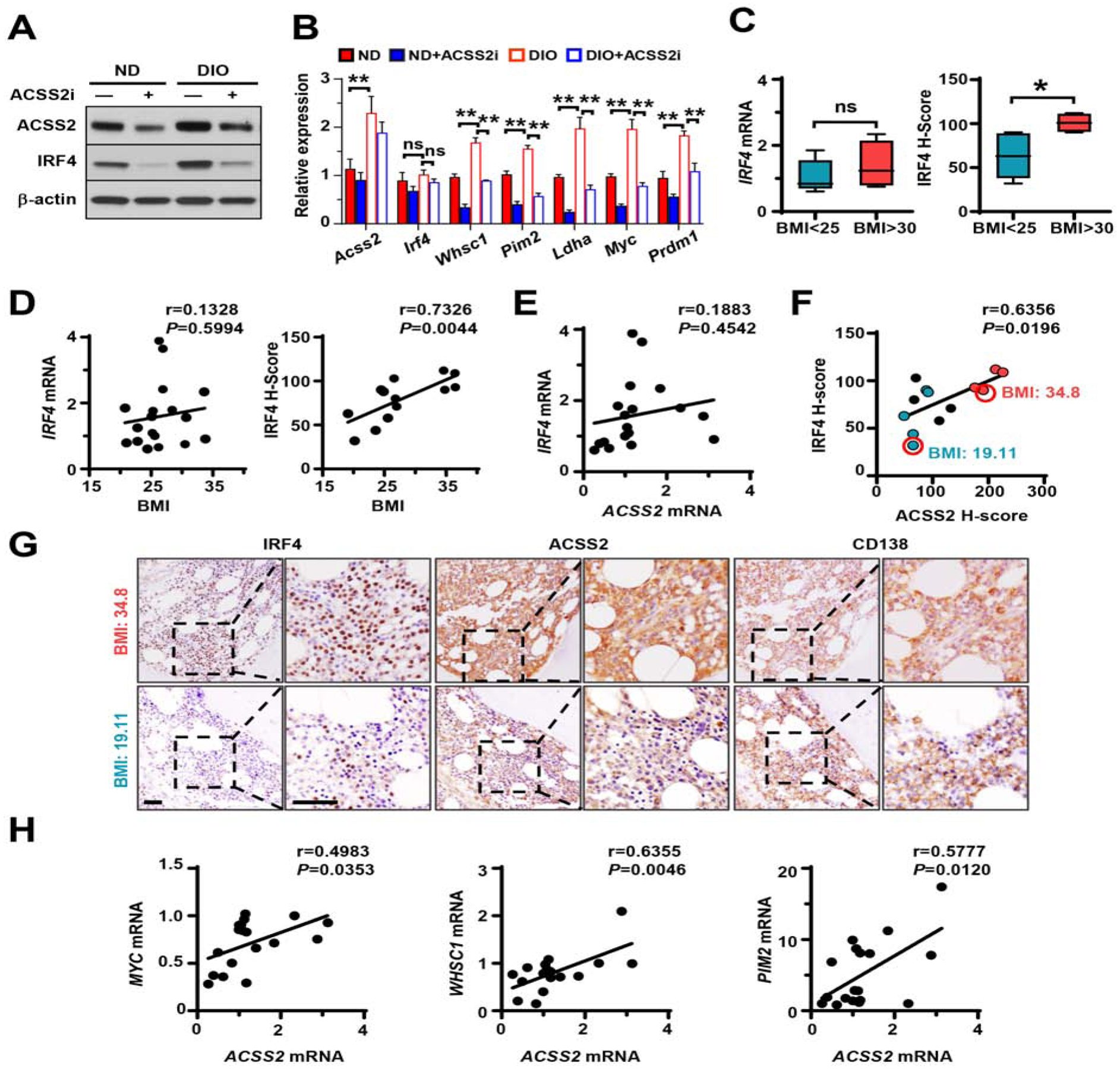
(A-B) ACSS2 and IRF4 protein levels (A) or Acss2, Irf4, Whsc1, Pim2, Ldha, Myc, and Prdm1 mRNAs (B) in myeloma cells isolated from bone marrow of DIO or ND mice that were intrafemorally injected with Vk12598 cells with or without ACSS2i treatment. (C) IRF4 mRNAs in patients’ myeloma cells (n=18) or IRF4 H-scores of patients’ bone marrow biopsies (n=13) immunohistochemically stained with antibodies against IRF4, ACSS2, or CD138. (D-F) Correlation coefficients between the levels of IRF4 mRNAs or IRF4 H-scores in myeloma cells and patients’ BMIs (D), IRF4 and ACSS2 mRNAs in myeloma cells (E), and IRF4 and ACSS2 H-scores in myeloma cells (F). Blue: BMI < 25 kg/m2; black, BMI 25–30 kg/m2; red, BMI > 30 kg/m2. (G) Immunohistochemistry of ACSS2, IRF4, and CD138 in bone marrow of myeloma patients (BMI=19.11 or 34.8). Scale bars, 50 μm. (H) Correlation coefficients between ACSS2 and IRF4 target genes in myeloma cells from patients’ bone marrow (n=18). Data shown as mean ± SD (ns, non-significant; *P < 0.05; **P < 0.01). See also Table S1.
DISCUSSION
Our study has unveiled insights into the molecular mechanisms underlying obesity and myeloma development. In obese patients with myeloma, we found that elevated ACSS2 expression in malignant plasma cells is induced by adipocyte-secreted cytokine angiotensin II. We found that the increased ACSS2 expression promotes myelomatous tumorigenesis through stabilization of IRF4 protein and concomitant upregulation of IRF4 transcriptional activity. We identified a key amino acid as lysine 399 that is central for the regulation of IRF4 by ACSS2. ACSS2 activates lysine acetylation of IRF4 at K399 and disrupts the normal degradation process for IRF4. We found that IRF4 has to bind to p62 first to initiate the lysosomal degradation. Since p62 can only bind to the unacetylated form of IRF4, the ACSS2-mediated acetylation of IRF4 at K399 enables IRF4 protein to escape from p62/lysosome-mediated degradation, resulting in elevated levels of IRF4 protein. The inhibition of ACSS2 further confirmed the importance of the modulation of IRF4 by ACSS2 in myeloma development; ACSS2 inhibition reduced the functions of IRF4 and hindered myeloma growth. Our study thus demonstrates a previously unknown role of ACSS2 in tumorigenesis in the obese setting.
IRF4 belongs to the family of interferon regulatory factors, which are crucial for immune cell development and immune response. IRF4 acts as either an oncogene or a tumor suppressor-like factor in many hematological malignancies. In myeloma, IRF4 controls a wide variety of gene sets that are key for cellular growth, survival, and metabolic processes, many of which are dysregulated (Shaffer et al., 2008). It is believed that loss of IRF4 expression would be a death sentence to myeloma cells regardless of their genetic etiology, making IRF4 an “Achilles’ heel” that may be exploited as a potential therapeutic target. Several approaches have been proven to have anti-myeloma effects through interfering with IRF4 transcription (Conery et al., 2016; Loven et al., 2013). The transcription level of IRF4 can be regulated by its own expression (Shaffer et al., 2008) or other factors, such as IKZF1/3 and NF-κB (Kronke et al., 2014; Xie et al., 2015). Here, we show that ablation of IRF4 protein by ACSS2 inhibition doesn’t change the levels of IRF4 mRNA, suggesting that IRF4 transcription is affected by factors other than the ACSS2/IRF4 protein axis. However, upon the post-translational modification regulated by ACSS2-mediated activation of acetylation, IRF4 becomes resistant to p62-mediated lysosomal degradation and is able to maintain its protein stability. Treatment with ACSS2i significantly reduces IRF4 protein levels in myeloma cells, induces myeloma cell apoptosis, and impairs myeloma growth in vitro and in vivo. Therefore, inhibition of ACSS2 can serve as another potential strategy to target IRF4 for myeloma therapy.
Post-translational modification is an important tactic for the regulation of protein function. Here, we demonstrate that ACSS2-CBP complex–dependent acetylation of IRF4 K399 residue contributes to the stability of IRF4 protein. The highly conserved nature of the K399 residue at IRF4 among different species suggests a common means of regulation of IRF4 protein that is an important event in biologic evolution. In myeloma, K399 of IRF4 is also critical for the interaction between IRF4 and the tumor suppressor PU.1, which can directly repress the transcriptional activity of IRF4 and is usually downregulated (Ueno et al., 2017). Whether acetylation of K399 results in a structural change of IRF4 and impairs their interaction, thus releasing IRF4 from PU.1, is an interesting topic for further study.
The renin-angiotensin system (RAS) regulates blood pressure in cardiovascular system (Billet et al., 2008; Kim and Iwao, 2000). Besides circulating RAS, adipose tissue expresses all the RAS components to form angiotensin II peptide for local function (Yvan-Charvet and Quignard-Boulange, 2011). Adipose tissue-derived angiotensin II is positively correlated with BMI in DIO mice and in people with obesity, and is responsible for the development of obesity-related hypertension (Saiki et al., 2009; Yiannikouris et al., 2012). Our results showed that adipose tissues from obese subjects produce more angiotensin II than those from non-obese subjects, and angiotensin II is one of the major factors mediating adipocyte-induced promotion of anchorage-independent growth of myeloma cells. Thus, our evidence points to the increased release of angiotensin II from expanded adipose tissue mass as a high-risk factor not only for cardiovascular disorders but also for obesity-associated tumors. Mechanistically, angiotensin II exerts its function at least partially through binding to its receptors, AGTR1 and AGTR2, which were expressed in all the myeloma cell lines we tested. In particular, AGTR1 is expressed in endothelial cells and many types of cancer cells (Egami et al., 2003; George et al., 2010). ACE inhibitors and AGTR1 antagonists, which block the production or activity of angiotensin II, are used clinically as potent anti-hypertensive drugs (Azizi and Menard, 2004; Wright et al., 2018). They also offer beneficial effects on impairing tumor progression, vascularization, and metastasis and improve immune-checkpoint blockage therapy (Deshayes and Nahmias, 2005; Kosugi et al., 2007). We demonstrated that AGTR1 mediates angiotensin II-induced upregulation of ACSS2 expression and ACSS2-mediated myeloma cell colony formation, and AGTR1 antagonist candesartan reduces myeloma tumorigenesis in the obese setting, suggesting an important role of angiotensin II/AGTR1 signaling in the pathogenesis of myeloma. Since the levels of angiotensin II are elevated in obese people, blockage of angiotensin II/AGTR1 signaling provides a strong rationale for a potential strategy to augment the efficacy of therapies in patients with myeloma or other obesity-associated cancers.
Collectively, our findings have shed light on a mechanistic insight into the relevance of obesity in myeloma development. Effective blockage of ACSS2 protein could serve as an alternative approach for IRF4-targeted myeloma treatment. Since ACSS2 is highly expressed in a wide range of tumors, our study may elucidate a common pathway of tumor progression in the obese setting. Targeting ACSS2 may have implications as a potential strategy for other obesity-associated tumors.
Limitations of study
Because of the complexity in both obese and tumor microenvironment, stimulation of plasma cell ACSS2 expression by obese adipose tissue-derived angiotensin II is unlikely the only mechanism that dictates obesity-associated tumorigenesis. Our exclusive focus on adipocytes does not tackle the likely contribution of other obesity-associated factors to myeloma progression. In addition to metabolic reprogramming, obesity-induced inflammation may also play an important role in myeloma cell growth and survival. While the co-implantation subcutaneous mouse model highlights the interplay between adipocytes and myeloma cells, the reductionist nature of this approach does not fully recapitulate tumor microenvironment. We also recognize that to fully implement the strategy through inhibition of the angiotensin II-ACSS2 axis, further development and verification is needed in both pre-clinical studies and clinical trials.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Jing Yang (jyang2@houstonmethodist.org)
Materials Availability
Mice used in this study were acquired commercially and available at Jackson Laboratories. Stable cell lines carrying targeted shRNA or with different constructs are available through establishment of Material Transfer Agreement between Houston Methodist Research Institute and request institution.
Data and Code Availability
The mRNA microarray data generated during this study is available at GEO database (GSE132604).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal Models
Male or female NOD-scid IL2Rgnull (NSG) and male C57BL/6J mice, 6 to 8 weeks of age, were purchased from The Jackson Laboratory, maintained, and directly used in American Association of Laboratory Animal Science-accredited facilities. The mice were housed five to a cage in a temperature- and humidity-controlled space (20–25°C, 45–64% humidity) with regulated water and lighting (12-hour light/dark) within the animal facility. All in vivo mouse studies were approved by the Institutional Animal Care and Use Committees of UT MD Anderson Cancer Center and Houston Methodist Research Institute and those protocols were used as guideline to check the mice health status. NSG mice were fed with irradiated rodent diet (Teklad 2920X, Envigo). In the DIO mouse model, the preferential usage of male C57BL/6J mice has been the golden standard due to their ability to gain weight when fed with high fat diet (Hwang et al., 2010). C57BL/6J mice were fed a high-fat diet (60% kcal, Envigo, TD.06414) for 15 weeks to establish DIO model. Control mice were fed with a normal diet (10% kcal, Envigo, TD.120455). Mouse models used in this study were described in the METHOD DETAILS.
Patient Samples
This study was approved by the Institutional Review Boards of UT MD Anderson Cancer Center and Houston Methodist Research Institute. All patient samples were consented by and obtained from the Myeloma Tissue Bank at The University of Texas MD Anderson Cancer Center and Biorepository of Houston Methodist Research Institute. The patient information is shown in Table S1 and the characteristics of the patients among different groups are compatible, where there was no significant difference between our samples from male and female patients. Primary myeloma cells were isolated from bone marrow aspirates of newly diagnosed myeloma patients using human CD138 microbeads (Miltenyi Biotech) according to the manufacturer’s instructions.
Cell Cultures
All cells were maintained at 37°C in a 5% CO2 incubator. The myeloma cell lines ARP-1 and ARK (unknown genders) were kindly provided by the University of Arkansas, murine myeloma cell line Vk12598 (unknown gender) was provided by the Mayo Clinic (Chesi et al., 2012), and 5TGM1 cell line (unknown gender) was provided by the Case Western Reserve University (Vatolin et al., 2016). Other cells, such as MM.1S (female), RPMI8226 (male), and U266 (male), were purchased from the American Type Culture Collection. Myeloma cells were cultured in RPMI1640 medium with 10% fetal bovine serum (FBS); HEK293T cells (female) and A375 cells (female) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS. 5TGM1 cells were cultured in IMDM medium with 10% FBS. In some experiments, cells were treated with various inhibitors. Treatment with vehicle only, such as 1 × PBS or DMSO served as control. Mature adipocytes were either derived from differentiation of human MSCs or isolated from adipose tissues of obese or non-obese mice or human patients (both genders). Detailed methods on generation and isolation of adipocytes and collection of conditioned medium were described in the METHODS DETAILS.
METHOD DETAILS
Culture of adipocytes
Primary mature adipocytes were isolated from visceral adipose tissue of mouse or human subjects as described (Mitry and Hughes, 2011). Briefly, minced visceral adipose tissue were digested with 0.2 % collagenase at 37 °C, centrifuged at 700 rpm for 10 minutes, and filtered through 200 μm membrane to separate from other types of cells. The cells were further washed twice with 1 × PBS. In vitro generation and isolation of human MSCs, osteoblasts, and adipocytes (He et al., 2012; Liu et al., 2019; Liu et al., 2016) were performed as described previously. Briefly, human MSCs from bone marrow aspirates of health donor were characterized by flow cytometry with antibodies against MSC markers (CD44, CD90, and CD166). MSCs were maintained in mesenchymal stem cell medium (ScienCell Research Laboratory, #7501). Mature osteoblasts were generated from MSCs in α-MEM medium with 10% FBS, 100 nM dexamethasone, 10 mM β-glycerophosphate, and 0.05 mM L-ascorbic acid 2-phosphate. MSCs were cultured in DMEM medium with 10% FBS, 1 μM dexamethasone, 0.2 mM indomethacin, insulin (10 μg/ml), and 0.5 mM 3-isobutyl-L-methylxanthine for 2 weeks to differentiate into mature adipocytes. The purity of mature adipocytes was examined by flow cytometry for cells positively expressing BODIPY and not expressing CD44 and CD105 (MSC and fibroblast markers), CD106 (MSC marker), or CD24 and Sca-1 (pre-adipocyte markers); they were also confirmed with Oil Red O staining. Mature adipocytes were maintained in DMEM with 10% FBS. Conditioned medium was collected from supernatants after 3 days of culture. In some experiments, the conditioned medium was further digested by proteinase K or separated into two fractions, > 10 or < 10 kDa, using a Centricon filter unit with a 10-kDa molecular weight cutoff (Millipore). The medium alone was collected as a control.
Proliferation and colony formation assay
To monitor cell proliferation, 1 × 104 cells/well were seeded in triplicate, and cell density was measured every 3 days. For viability assays, cells were plated at 1 × 104 cells/well in triplicate and treated with or without various concentrations of ACSS2i for 48 hours. The dosage of ACSS2i for in vitro studies was based on the previous studies (Marquez et al., 2019; Mews et al., 2017; Mews et al., 2019), and further determined for myeloma cells with dose-response curve (Figure S2K) and off-target analysis (Figure S2N). Assays were performed using a CellTiter-Glo Luminescent Cell Viability Assay Kit (Promega). Anchorage-independent cell growth was evaluated with bilayer soft agar colony formation assays. Briefly, 5×104 cells in 0.4% Noble agar were plated on top of the 0.8% Noble agar bottom layer in a 6-well plate. After 3 weeks, colonies were stained with 1 mg/ml p-iodonitrotetrazolium chloride for visualization and quantitation. Colonies formed in the cells without treatment or in the shCtrl cells were set to 100%. Adipocytes, OBs, or MSCs were cultured for 72 hours and the concentrations of angiotensin II in conditioned medium were measured using an ELISA kit (Sigma Aldrich) according to the manufacturer’s instructions.
Microarray-based gene expression profiling
Microarray analysis was performed as described previously (Ma et al., 2010) to compare the gene expression in primary myeloma cells isolated from bone marrow aspirates of newly diagnosed myeloma patients with obesity vs. those with normal weights. Total RNAs were extracted from primary myeloma cells following standard TRIzol protocol (Life Technologies). After reverse transcription, the Illumina TotalPrep RNA Amplification kit (Ambion) was then used and the samples were processed following manufacture recommended protocol on the Illumina platform. Differential expression was identified by a greater than twofold difference fold change and a P value less than 0.05. Gene ontology (GO) analysis was performed using the enrichment analysis tool of the Gene Ontology Consortium. Heat-map representation of gene expression was generated using MultiExperiment Viewer software. Gene set enrichment analysis (GSEA) was performed using GSEA v2.0.14 software.
Quantitative real-time PCR
Total RNA was isolated using the Purelink RNA Mini Kit (Thermo Fisher Scientific). An aliquot of 0.5 μg of total RNA was subjected to reverse transcription with a SuperScript II RT-PCR kit (Life Technologies). Quantitative PCR was performed using SYBR Green Master Mix (Life Technologies) with the QuantStudio 3 Real-Time PCR System (Life Technologies). The primers used are listed in Table S2.
ORF, shRNA, and CRISPR/Cas9
Human FLAG-tagged ACSS2, HA-tagged IRF4, and FLAG-tagged p62 were subcloned into a pCDH-CMV vector. K59Q, K87Q, K399Q, and K399R mutant forms of IRF4, were mutated from wild type using the QuikChange site-directed mutagenesis kit (Stratagene). shACSS2, shIRF4, shp62, and shAGTR1 were subcloned into a pLKO.1 vector. Control sgRNA (sgCtrl) or ACSS2 gene-targeting sgRNAs were subcloned into the lentiCRISPR v2 vector (Addgene). For transient transfection, cells were transfected with Lipofectamine 3000 reagent (Life Technologies) and assayed 48 hours later. For stable lines, cells were infected with lentiviral particles and selected with 1 μg/mL puromycin. The primer sequences used for shRNA and sgRNA constructs are listed in Table S3 and Table S4, respectively.
Flow cytometry
Cells from murine spleen or bone marrow were stained with anti-CD138 antibody and measured by a BD LSRFortessa flow cytometer (BD Biosciences). The results were analyzed using FlowJo software.
Fluorescent staining
Cells were fixed in 4% paraformaldehyde and permeabilized with 0.3% Triton X-100 in 1 × PBS. After blocking with 2% goat serum, the cells were stained with antibodies against ACSS2 or IRF4 at 4 °C overnight. The cell nuclei were stained with DAPI and mounted with ProLong Gold antifade reagent (Molecular Probes). Immunofluorescent images were acquired with an IX81 confocal microscope system (Olympus America).
Western blot analysis
Cells were harvested and lysed with a 1 × lysis buffer (Cell Signaling Technology). Cell lysates were subjected to SDS-PAGE, transferred to a polyvinylidene difluoride (PVDF) membrane, and immunoblotted with antibodies against ACSS2, IRF4, p62, CBP, Ac-K, biotin, Hsc70, pAkt, Akt, pERK1/2, ERK1/2, pJAK2, JAK2, ubiquitin, LC3B, SREBP1, Lamin A, HA, FLAG, β-actin, and GAPDH. The level of GAPDH or β-actin served as protein loading control. In dot blotting, level of biotin served as control.
Immunoprecipitation (IP) and chromatin IP
Cells were lysed and incubated on ice for 15 min. The total protein lysate (1 mg) was immunoprecipitated with an agarose-immobilized antibody at 4°C overnight. After 6 washes, the beads were spun down and resuspended in 30 μl of 1× SDS buffer. After boiling for 5 min, pulldown samples were run on an SDS-PAGE gel along with a 2% input sample and transferred to a PVDF membrane for immunoblotting. Inputs and immunoprecipitation with IgG served as controls. In some experiment, immunoblotting of immunoprecipitates with anti-HA antibody served as control.
ChIP assay was performed using SimpleChIP Enzymatic Chromatin IP kit (Cell Signaling Technologies) following the manufacturer’s instruction. Cells were fixed in formaldehyde and sonicated to prepare chromatin fragments. Chromatin samples were immunoprecipitated with antibodies against IRF4 and control immunoglobulin G (IgG) at 4°C overnight. Immunoprecipitates and total chromatin inputs were reverse cross-linked; DNA was isolated and analyzed using quantitative PCR with primers targeting the promoter regions of IRF4 target genes (Supplementary Table S5).
In vitro acetylation reaction
Purified HA-IRF4 protein from HA-IRF4–expressing HEK293T cells and the catalytic domain of human CBP (CBP-CD) were prepared for the assay. The mixture of 2 μg of peptides, 0.5 μg of recombinant human CBP-CD, and 4 μg of acetyl-CoA in 1× histone acetyltransferase (HAT) assay buffer was incubated at 30°C with orbital shaking for 4 hours. The reaction products were then separated by SDS-PAGE and immunoblotted with the acetyl-lysine antibody (Wang et al., 2012). An acetyltransferase activity kit (Enzo Life Sciences) was also used to assess the acetylation reaction.
Immunohistochemistry
Formalin-fixed, paraffin-embedded sections of samples from the bone marrow of patients with myeloma singly or in tissue array or from murine subcutaneous tissue were deparaffinized and stained with anti-ACSS2, anti-Ki67, anti-perilipin, anti-IRF4, and anti-CD138 antibodies using an EnVision System (DAKO) following the manufacturer’s instructions and counterstained with hematoxylin. In some experiments, stained slides were scanned with the Vectra 3 automated quantitative pathology imaging system and analyzed by inForm software (Perkin Elmer).
Mass spectrometry
To identify ACSS2-interacting proteins, immunoprecipitates of ARP-1 cells infected with or without FLAG-tagged human ACSS2 were affinity captured by anti-Flag agarose beads and digested by trypsin. The tryptic peptides were then analyzed using a nano–liquid chromatography with tandem mass spectrometry (LC/MS/MS) system (Thermo Fisher Scientific) coupled with an 1100 series high-performance liquid chromatography (HPLC) system (Agilent Technologies). The MS/MS spectra were searched using the SEQUEST software program with the BioWorks Browser (version 3.3.1; Thermo Fisher Scientific) against the National Center for Biotechnology Information database.
To map the acetylation sites in IRF4, HA-IRF4 was immunoprecipitated with anti-HA agarose beads from infected ARP-1 cells treated with or without ACSS2i. The immunoprecipitates were subjected to SDS-PAGE, and the Coomassie blue-stained band corresponding to IRF4 was extracted and subjected to in-gel trypsin digestion. The resulting peptides were analyzed using high-sensitivity LC-MS/MS with an Orbitrap Elite mass spectrometer (Thermo Fisher Scientific). Proteins were identified by searching the fragment spectra against the Swiss-Prot protein database using the Mascot search engine (version 2.3; Matrix Science) via the Proteome Discoverer software program (version 1.4; Thermo Fisher Scientific).
In vivo mouse experiments
To determine the optimal dosage of ACSS2i in vivo, luciferase-expressing ARP-1 cells were intravenously injected into NSG mice (5×105 cells/mouse). After a week, mice were intraperitoneally injected with 0, 12.5, 25 or 50 mg/kg ACSS2i daily. Their body weights and bioluminescent signals were monitored for 4 weeks. The tissues from vital organs were collected and subjected to H&E staining for evaluation. In another intravenous xenograft mouse experiment, 5×105 ARP-1 or 1×106 MM.1S luciferase (luc)-expressing cells were intravenously injected into NSG mice. After a week, mice were treated with intraperitoneally injected 25 mg/kg ACSS2i daily. Treatment with vehicle containing 2% Tween-80 served as control. Tumor burden was monitored weekly by levels of serum M-protein and strength of bioluminescent signal.
For the subcutaneous xenograft model, there are several settings. In Figure S1H, ARP-1 cells (2×105 cells/site) alone or mixed with human MSCs or MSC-derived adipocytes (5×105 cells/site) were injected into opposite flanks of the NSG mice. Tumors were weighed at week 4. Bioluminescent signals shown at week 4. In Figure S4A, shCtrl or shACSS2 myeloma cells were injected into opposite flanks of the mice as diagrammed and the tumors were weighed at week 6. In another set of experiments, there were two implantation spots on each side–shACSS2 and shCtrl ARP-1 cells (2×105 cells/site) alone and each with 5×105 human MSC-derived adipocytes as diagrammed in Figure 3A, and the tumors were weighed at week 4. Bioluminescent signals shown at week 4. At the end point, subcutaneous tissues were extracted, fixed, and paraffin embedded for immunohistochemical staining. Similarly, two spots on each side–sgACSS2 and sgCtrl MM.1S cells (5×105 cells/site) alone and each with 5×105 human MSC-derived adipocytes were implanted as diagrammed in Figure 3C.
In the doxycycline-inducible gene expression model, TetON-shCtrl or TetON-shACSS2 ARP-1 cells were injected into the distal end of the femur in NSG mice. Then the mice were treated with intraperitoneally injected doxycycline (50 mg/kg) daily. Bioluminescent images were taken at 0 week and 3 weeks post-doxycycline treatment. In another set of experiment, luciferase-labeled sgCtrl or sgACSS2 MM.1S cells carrying empty control or HA-IRF4 were intrafemorally injected into NSG mice, and bioluminescent signals were imaged 3 weeks after cell injection.
Using the DIO model (Figure 2A), 6-week-old male C57BL/6J mice were fed a high-fat diet (60% kcal) for 15 weeks, and their weights were monitored. Mice fed a normal diet (10% kcal) served as controls. Vk12598 cells (5×105/mouse) were intrafemorally injected into the mice. Starting 1 week later, mice received daily treatment of intraperitoneally injected ACSS2i (25 mg/kg) or condesartan (10mg/kg) via oral gavage for 5 weeks. Mice receiving equal amount of vehicle control served as control. Tumor burden was evaluated by serum M-protein level. Spleen and bone marrow were extracted, and aliquots of the cells were used to determine the ratios of CD138+ cells by flow cytometry. CD138+ cells were isolated from spleen or bone marrow aspirates and evaluated for protein and mRNA expression. Serum was collected for measuring cytokine levels. To examine the myeloma incidence rate, 5TGM1 cells (1×106 cells/mouse) were intravenously injected into the mice. In one experiment, one week after cell injection, mice were treated with intraperitoneal injection of 25 mg/kg ACSS2i daily. The myeloma occurrences were monitored by the levels of serum M-protein for 8 weeks.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Statistical significance was analyzed using the GraphPad Prism with two tailed unpaired Student t-tests for comparison of two groups (Figures 1F, 1G, 4U, 5A, 5C, 5D, 5E, and 7B), and one-way ANOVA with Newman-Keuls post hoc test (Figures 1N, 4H, 4K, and 6M) or two-way ANOVA with Bonferroni’s post hoc test (Figures 2B–2E, 3B, 3D, 3F, 4A, 4G, 4L, 4N–4P, 5F, 6P, and 7C) for comparison of more than two groups. Pearson’s correlation analysis was used to examine the correlation between two variables and Kaplan-Meier analysis was used in survival analysis. We used the Shapiro-Wilk normality test to confirm that the data met assumptions of the statistical approach. P values less than 0.05 were considered statistically significant. Data shown are mean ± SD that were representative of at least three independent experiments.
Modeling myeloma cases attribute to obesity
Numbers of new myeloma cases in the US by gender group between 2011 and 2016 were obtained from Centers for Disease Control and Prevention’s National Program of Cancer Registries (NPCR) and the National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER) program, which collectively provided complete coverage for the US population between 2011 and 2016 (NPCR/SEER, 2019). Weighted prevalence data of obesities in each gender group was obtained from the 2017 version of the National Health and Nutrition Examination Survey (NCHS, 2018). The relative risk (RR) for obesity classes 1–3 and myeloma were obtained from the study by the International Agency for Research on Cancer (Lauby-Secretan et al., 2016). Statistics analysis followed the established pipeline (Islami et al., 2018). Specifically, cancer cases from the NPCR/SEER were adjusted for delays in reporting to central cancer registries according to North American Association of Central Cancer Registries (Clegg et al., 2002). A simulation method (Greenland, 2004; Islami et al., 2018) was repeated 1000 times to generate RRs, weighted prevalence, and number of cancer cases, allowing for uncertainty in the data. Population attributable fractions (PAF) for obesity (classes 1–3 combined) were calculated, and the number of myeloma cases attributable to each BMI group by gender was calculated by multiplying the number of myeloma cases in each gender by the PAF in that group, and results from obesity class 1–3 were summed to generate the results for combined obesity group. Numbers of attributable myeloma cases are rounded to the nearest 10.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ACSS2 | Cell Signaling Technology | Cat# 3658, RRID: AB_2222710 |
| HA-Tag | Cell Signaling Technology | Cat #3724, RRID: AB_1549585 |
| IRF4 | Cell Signaling Technology | Cat #15106, RRID: AB_2798709 |
| IRF4 | Santa Cruz Biotechnology | Cat #sc-6059, RRID: AB_2127145 |
| IRF4 | Cell Signaling Technology | Cat #62834, RRID: AB_2877647 |
| Histone H3 | Cell Signaling Technology | Cat #4499, RRID: AB_10544537 |
| SQSTM1/p62 | Cell Signaling Technology | Cat #39749, RRID: AB_2799160 |
| SQSTM1/p62 | Cell Signaling Technology | Cat # 88588, RRID: AB_2800125 |
| HSC70 | Cell Signaling Technology | Cat #8444, RRID: AB_10831837 |
| Acetylated-Lysine | Cell Signaling Technology | Cat #9441, RRID: AB_331805 |
| CBP | Cell Signaling Technology | Cat #7389, RRID: AB_2616020 |
| Biotin | Cell Signaling Technology | Cat #5597, RRID: AB_10828011 |
| Perilipin | Cell Signaling Technology | Cat #9349, RRID: AB_10829911 |
| LC3B | Cell Signaling Technology | Cat #83506, RRID: AB_2800018 |
| Akt | Cell Signaling Technology | Cat #9272, RRID: AB_329827 |
| Phospho-Akt (Ser473) | Cell Signaling Technology | Cat #4060, RRID: AB_2315049 |
| p44/42 MAPK (Erk1/2) | Cell Signaling Technology | Cat #4695, RRID: AB_390779 |
| Phosphorylated Erk1/2 | Cell Signaling Technology | Cat #9101, RRID: AB_331646 |
| Jak2 | Cell Signaling Technology | Cat #3230, RRID: AB_2128522 |
| Phospho-Jak2 (Tyr1007) | Cell Signaling Technology | Cat #4406, RRID: AB_10706164 |
| Ubiquitin | Cell Signaling Technology | Cat #3936, RRID: AB_331292 |
| SREBP-1 | Santa Cruz Biotechnology | Cat #sc-13551, RRID: AB_628282 |
| FLAG | Sigma Aldrich | Cat #F1804, RRID: AB_262044 |
| Lamin A | Santa Cruz Biotechnology | Cat #sc-71481, RRID: AB_2136165 |
| GAPDH | Thermo Fisher Scientific | Cat #AM4300, RRID: AB_2536381 |
| Actin | Santa Cruz Biotechnology | Cat #sc-69879, RRID: AB_1119529 |
| Histone H3 (acetyl K27) | Abcam | Cat #ab4729, RRID: AB_2118291 |
| SDC1/Syndecan 1/CD138 | LifeSpan Biosciences | Cat #LS-B9360, RRID: AB_2877650 |
| ACSS2 | LifeSpan Biosciences | Cat #LS‑C334743, RRID: AB_2877651 |
| Ki67 | Thermo Fisher Scientific | Cat #14-5699-82, RRID: AB_2016711 |
| IRF4 (K399ac) | This manuscript, by GeneScript | N/A |
| APC anti-mouse CD138 | Biolegend | Cat #142506, RRID: AB_10962911 |
| Goat anti-Mouse IgG Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat # A-11005, RRID: AB_2534073 |
| Goat anti-Rabbit IgG Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat # A-11008, RRID: AB_143165 |
| Donkey anti-Goat IgG Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat # A-11055, RRID: AB_2534102 |
| Donkey anti-Rabbit IgG Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat # A-21207, RRID: AB_141637 |
| Normal rabbit IgG | Santa Cruz Biotechnology | Cat # sc-2027, RRID: AB_737197 |
| Normal mouse IgG | Santa Cruz Biotechnology | Cat # sc-2025, RRID: AB_737182 |
| Normal goat IgG | Santa Cruz Biotechnology | Cat # sc-2028, RRID: AB_737167 |
| Protein G agarose | Thermo Fisher Scientific | Cat # 22852 |
| anti-HA agarose | Thermo Fisher Scientific | Cat # 26182, RRID: AB_2532162 |
| Neutravidin agarose | Thermo Fisher Scientific | Cat # 29202 |
| Anti-Flag M2 affinity gel | Sigma Aldrich | Cat # A2220, RRID: AB_10063035 |
| Human CD138 microbeads | Miltenyi Biotech | Cat# 130-051-301 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DAPI | Sigma Aldrich | Cat# D9542 |
| Cycloheximide | Cayman Chemical | Cat# 14126 |
| Fatostatin | Cayman Chemical | Cat# 13562 |
| Bafilomycin A1 | Cayman Chemical | Cat# 11038 |
| Leupeptin | Cayman Chemical | Cat# 14026 |
| MG-132 | R&D Systems INC | Cat# 1748/5 |
| Ac-CoA Synthase Inhibitor (ACSS2i) | EMD Millipore | Cat# 533756 |
| C646 | Sigma Aldrich | Cat# SML0002 |
| LY294002 | Cell Signaling Technology | Cat# 9901 |
| U0126 | Cell Signaling Technology | Cat #9903 |
| IRF4 379–418 peptides | This paper, by GeneScript | N/A |
| Recombinant human CBP (catalytic domain) | EMD Millipore | Cat# 03–189 |
| Acylation stimulating protein | R&D Systems | Cat# 9620-C3 |
| Angiotensin II | R&D Systems | Cat# 4474-91-3 |
| Recombinant human resistin | PeproTech | Cat# 450–19 |
| Apelin | Santa Cruz Biotechnology | Cat# sc-351718 |
| Endotrophin | Phoenix Pharmaceuticals | Cat# 062–79 |
| Proteinase K-Agarose | Sigma Aldrich | Cat# P9290 |
| Critical Commercial Assays | ||
| IgG2b Mouse Uncoated ELISA Kit | Thermo Fisher Scientific | Cat# 88-50430-88 |
| NE-PER™ Nuclear and Cytoplasmic Extraction Kit | Thermo Fisher Scientific | Cat# 78833 |
| CellTiter-Glo Luminescent Cell Viability Assay Kit | Promega | Cat# G7570 |
| Purelink RNA Mini Kit | Thermo Fisher Scientific | Cat# 12183025 |
| Human Angiotensin II ELISA Kit | Sigma Aldrich | Cat# RAB0010–1KT |
| SimpleChIP® Plus Enzymatic Chromatin IP Kit | Cell Signaling Technology | Cat# 9004 |
| Acetyltransferase activity kit | Enzo Life Sciences | Cat# ADI-907–026 |
| SignalFire™ Plus ECL Reagent | Cell Signaling Technology | Cat# 12630 |
| Deposited Data | ||
| mRNA array | This manuscript | GSE132604 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratory | Stock No: 000664 |
| NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) | Jackson Laboratory | Stock No: 005557 |
| Oligonucleotides | ||
| Primers for qPCR, see Table S2 | This manuscript | N/A |
| shRNA targeting primers, see Table S3 | This manuscript | N/A |
| sgRNA targeting primers: see Table S4 | This manuscript | N/A |
| Primers for ChIP assay, see Table S5 | This manuscript | N/A |
| Recombinant DNA | ||
| Plasmid: pLKO.1-puro | Addgene | Cat# 8453 |
| Plasmid: lentiCRISPR v2 | Addgene | Cat# 52961 |
| pCDH-CMV-MCS | System Bioscience | Cat# CD500B-1 |
| pLKO.1-shACSS2#1, pLKO.1-shACSS2#2, pLKO.1-shACSS1#1, pLKO.1-shACSS1#2 | This manuscript | N/A |
| pLKO.1-shp62#1, pLKO.1-shp62#2, pLKO.1-shIRF4#1, pLKO.1-shIRF4#2 | This manuscript | N/A |
| lentiCRISPR-sgCtrl, lentiCRISPR-sgACSS2 | This manuscript | N/A |
| pCDH-Flag-ACSS2, pCDH-HA-IRF4, pCDH-Flag-p62 | This manuscript | N/A |
| pCDH-HA-IRF4 (K59Q), pCDH-HA-IRF4 (K87Q), pCDH-HA-IRF4 (K399Q), | This manuscript | N/A |
| Software and Algorithms | ||
| GraphPad Prism 8 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Multiple array viewer (MeV) | Dana-Farber Cancer Institute, | http://mev.tm4.org/#/welcome |
| FlowJo v.10 | FlowJo, LLC | https://www.flowjo.com/solutions/flowjo |
| inForm Advanced Image Analysis Software | Perkin Elmer | |
| Living Image Software | Perkin Elmer | https://www.perkinelmer.com/product/spectrum-200-living-image-v4series-1-128113 |
Significance:
There is a causal role for obesity in the development of many malignancies, but the mechanism for this linkage is unclear. In the setting of multiple myeloma, an obesity-associated malignancy, we identified a unique mechanism, increased angiotensin II production by adipocytes that stimulates ACCS2 overexpression in myeloma cells leading to abnormal proliferation of plasma cells through an IRF4-mediated effect on growth. This mechanism may be applicable to other obesity-related malignancies.
Highlights.
ACSS2 contributes to obesity-induced myelomatous tumorigenesis
Adipocyte-derived angiotensin II enhances ACSS2 expression in myeloma cells
ACSS2-mediated lysine acetylation improves the stability of oncogenic IRF4 protein
Inhibition of the angiotensin II-ACSS2 axis prevents obesity-induced tumor growth
Acknowledgements:
We thank The University of Texas MD Anderson Myeloma Tissue Bank. This research was supported by the National Institutes of Health/National Cancer Institute (R01s: CA190863 and CA193362) and the American Cancer Society (Research Scholar Grant 127337-RSG-15-069-01-TBG). Supported also by the NIH/NCI P30CA016672 for Small Animal Imaging Facility, Research Histology Core Laboratory, and Flow Cytometry and Cellular Imaging Facility. The CoMMpass dataset were generated as part of the Multiple Myeloma Research Foundation Personalized Medicine Initiatives (https://research.themmrf.org and www.themmrf.org)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare no potential conflicts of interest.
REFERENCES
- Azizi M, and Menard J (2004). Combined blockade of the renin-angiotensin system with angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists. Circulation 109, 2492–2499. [DOI] [PubMed] [Google Scholar]
- Bianchini F, Kaaks R, and Vainio H (2002). Overweight, obesity, and cancer risk. Lancet Oncology 3, 565–574. [DOI] [PubMed] [Google Scholar]
- Billet S, Aguilar F, Baudry C, and Clauser E (2008). Role of angiotensin II AT1 receptor activation in cardiovascular diseases. Kidney Int 74, 1379–1384. [DOI] [PubMed] [Google Scholar]
- Birmann BM, Giovannucci E, Rosner B, Anderson KC, and Colditz GA (2007). Body mass index, physical activity, and risk of multiple myeloma. Cancer Epidemiol Biomarkers Prev 16, 1474–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blair CK, Cerhan JR, Folsom AR, and Ross JA (2005). Anthropometric characteristics and risk of multiple myeloma. Epidemiology 16, 691–694. [DOI] [PubMed] [Google Scholar]
- Boya P, Reggiori F, and Codogno P (2013). Emerging regulation and functions of autophagy. Nat Cell Biol 15, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown LM, Gridley G, Pottern LM, Baris D, Swanso CA, Silverman DT, Hayes RB, Greenberg RS, Swanson GM, Schoenberg JB, et al. (2001). Diet and nutrition as risk factors for multiple myeloma among blacks and whites in the United States. Cancer Causes Control 12, 117–125. [DOI] [PubMed] [Google Scholar]
- Cabia B, Andrade S, Carreira MC, Casanueva FF, and Crujeiras AB (2016). A role for novel adipose tissue-secreted factors in obesity-related carcinogenesis. Obes Rev 17, 361–376. [DOI] [PubMed] [Google Scholar]
- Caers J, Deleu S, Belaid Z, De Raeve H, Van Valckenborgh E, De Bruyne E, Defresne MP, Van Riet I, Van Camp B, and Vanderkerken K (2007). Neighboring adipocytes participate in the bone marrow microenvironment of multiple myeloma cells. Leukemia 21, 1580–1584. [DOI] [PubMed] [Google Scholar]
- Calle EE, Rodriguez C, Walker-Thurmond K, and Thun MJ (2003). Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348, 1625–1638. [DOI] [PubMed] [Google Scholar]
- Chang SH, Luo S, Thomas TS, O’Brian KK, Colditz GA, Carlsson NP, and Carson KR (2017). Obesity and the Transformation of Monoclonal Gammopathy of Undetermined Significance to Multiple Myeloma: A Population-Based Cohort Study. J Natl Cancer Inst 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu BC, Gapstur SM, Greenland P, Wang R, and Dyer A (2006). Body mass index, abnormal glucose metabolism, and mortality from hematopoietic cancer. Cancer Epidemiol Biomarkers Prev 15, 2348–2354. [DOI] [PubMed] [Google Scholar]
- Clegg LX, Feuer EJ, Midthune DN, Fay MP, and Hankey BF (2002). Impact of reporting delay and reporting error on cancer incidence rates and trends. J Natl Cancer Inst 94, 1537–1545. [DOI] [PubMed] [Google Scholar]
- Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al. (2014). Acetate dependence of tumors. Cell 159, 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conery AR, Centore RC, Neiss A, Keller PJ, Joshi S, Spillane KL, Sandy P, Hatton C, Pardo E, Zawadzke L, et al. (2016). Bromodomain inhibition of the transcriptional coactivators CBP/EP300 as a therapeutic strategy to target the IRF4 network in multiple myeloma. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshayes F, and Nahmias C (2005). Angiotensin receptors: a new role in cancer? Trends Endocrinol Metab 16, 293–299. [DOI] [PubMed] [Google Scholar]
- Eckel RH, Grundy SM, and Zimmet PZ (2005). The metabolic syndrome. Lancet 365, 1415–1428. [DOI] [PubMed] [Google Scholar]
- Egami K, Murohara T, Shimada T, Sasaki K, Shintani S, Sugaya T, Ishii M, Akagi T, Ikeda H, Matsuishi T, et al. (2003). Role of host angiotensin II type 1 receptor in tumor angiogenesis and growth. J Clin Invest 112, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeland A, Tretli S, Hansen S, and Bjorge T (2007). Height and body mass index and risk of lymphohematopoietic malignancies in two million Norwegian men and women. Am J Epidemiol 165, 44–52. [DOI] [PubMed] [Google Scholar]
- Gallagher EJ, and LeRoith D (2015). Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality. Physiological reviews 95, 727–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George AJ, Thomas WG, and Hannan RD (2010). The renin-angiotensin system and cancer: old dog, new tricks. Nat Rev Cancer 10, 745–759. [DOI] [PubMed] [Google Scholar]
- Greenland S (2004). Interval estimation by simulation as an alternative to and extension of confidence intervals. Int J Epidemiol 33, 1389–1397. [DOI] [PubMed] [Google Scholar]
- Hariri N, and Thibault L (2010). High-fat diet-induced obesity in animal models. Nutr Res Rev 23, 270–299. [DOI] [PubMed] [Google Scholar]
- He J, Liu Z, Zheng Y, Qian J, Li H, Lu Y, Xu J, Hong B, Zhang M, Lin P, et al. (2012). p38 MAPK in myeloma cells regulates osteoclast and osteoblast activity and induces bone destruction. Cancer research 72, 6393–6402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann JN, Moore SC, Lim U, Park Y, Baris D, Hollenbeck AR, Matthews CE, Gibson TM, Hartge P, and Purdue MP (2013). Body mass index and physical activity at different ages and risk of multiple myeloma in the NIH-AARP diet and health study. Am J Epidemiol 177, 776–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang LL, Wang CH, Li TL, Chang SD, Lin LC, Chen CP, Chen CT, Liang KC, Ho IK, Yang WS, et al. (2010). Sex differences in high-fat diet-induced obesity, metabolic alterations and learning, and synaptic plasticity deficits in mice. Obesity (Silver Spring) 18, 463–469. [DOI] [PubMed] [Google Scholar]
- Incio J, Liu H, Suboj P, Chin SM, Chen IX, Pinter M, Ng MR, Nia HT, Grahovac J, Kao S, et al. (2016). Obesity-Induced Inflammation and Desmoplasia Promote Pancreatic Cancer Progression and Resistance to Chemotherapy. Cancer Discovery 6, 852–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, McCullough ML, Patel AV, Ma J, Soerjomataram I, et al. (2018). Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin 68, 31–54. [DOI] [PubMed] [Google Scholar]
- Johrer K, Ploner C, Thangavadivel S, Wuggenig P, and Greil R (2015). Adipocyte-derived players in hematologic tumors: useful novel targets? Expert Opin Biol Ther 15, 61–77. [DOI] [PubMed] [Google Scholar]
- Khan MM, Mori M, Sakauchi F, Matsuo K, Ozasa K, Tamakoshi A, and group JS (2006). Risk factors for multiple myeloma: evidence from the Japan Collaborative Cohort (JACC) study. Asian Pac J Cancer Prev 7, 575–581. [PubMed] [Google Scholar]
- Kim S, and Iwao H (2000). Molecular and cellular mechanisms of angiotensin II-mediated cardiovascular and renal diseases. Pharmacol Rev 52, 11–34. [PubMed] [Google Scholar]
- Kosugi M, Miyajima A, Kikuchi E, Kosaka T, Horiguchi Y, and Murai M (2007). Effect of angiotensin II type 1 receptor antagonist on tumor growth and angiogenesis in a xenograft model of human bladder cancer. Hum Cell 20, 1–9. [DOI] [PubMed] [Google Scholar]
- Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, et al. (2014). Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science 343, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle RA, and Rajkumar SV (2004). Multiple myeloma. N Engl J Med 351, 1860–1873. [DOI] [PubMed] [Google Scholar]
- Lamark T, Svenning S, and Johansen T (2017). Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem 61, 609–624. [DOI] [PubMed] [Google Scholar]
- Landgren O, Rajkumar SV, Pfeiffer RM, Kyle RA, Katzmann JA, Dispenzieri A, Cai Q, Goldin LR, Caporaso NE, Fraumeni JF, et al. (2010). Obesity is associated with an increased risk of monoclonal gammopathy of undetermined significance among black and white women. Blood 116, 1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K, and International Agency for Research on Cancer Handbook Working, G. (2016). Body Fatness and Cancer--Viewpoint of the IARC Working Group. N Engl J Med 375, 794–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Yu W, Qian X, Xia Y, Zheng Y, Lee JH, Li W, Lyu J, Rao G, Zhang X, et al. (2017). Nucleus-Translocated ACSS2 Promotes Gene Transcription for Lysosomal Biogenesis and Autophagy. Mol Cell 66, 684–697 e689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtman MA (2010). Obesity and the risk for a hematological malignancy: leukemia, lymphoma, or myeloma. Oncologist 15, 1083–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, He J, Koh SP, Zhong Y, Liu Z, Wang Z, Zhang Y, Li Z, Tam BT, Lin P, et al. (2019). Reprogrammed marrow adipocytes contribute to myeloma-induced bone disease. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Liu Z, Du J, He J, Lin P, Amini B, Starbuck MW, Novane N, Shah JJ, Davis RE, et al. (2016). Thymidine phosphorylase exerts complex effects on bone resorption and formation in myeloma. Science translational medicine 8, 353ra113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Xu J, He J, Liu H, Lin P, Wan X, Navone NM, Tong Q, Kwak LW, Orlowski RZ, et al. (2015). Mature adipocytes in bone marrow protect myeloma cells against chemotherapy through autophagy activation. Oncotarget 6, 34329–34341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lwin ST, Olechnowicz SW, Fowler JA, and Edwards CM (2015). Diet-induced obesity promotes a myeloma-like condition in vivo. Leukemia 29, 507–510. [DOI] [PubMed] [Google Scholar]
- Ma W, Wang M, Wang ZQ, Sun L, Graber D, Matthews J, Champlin R, Yi Q, Orlowski RZ, Kwak LW, et al. (2010). Effect of long-term storage in TRIzol on microarray-based gene expression profiling. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 19, 2445–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinac CR, Birmann BM, Lee IM, Rosner BA, Townsend MK, Giovannucci E, Rebbeck TR, Buring JE, and Colditz GA (2018). Body mass index throughout adulthood, physical activity, and risk of multiple myeloma: a prospective analysis in three large cohorts. Br J Cancer 118, 1013–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez S, Fernandez JJ, Mancebo C, Herrero-Sanchez C, Alonso S, Sandoval TA, Rodriguez Prados M, Cubillos-Ruiz JR, Montero O, Fernandez N, et al. (2019). Tricarboxylic Acid Cycle Activity and Remodeling of Glycerophosphocholine Lipids Support Cytokine Induction in Response to Fungal Patterns. Cell Rep 27, 525–536 e524. [DOI] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, et al. (2014). Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159, 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P, Donahue G, Drake AM, Luczak V, Abel T, and Berger SL (2017). Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P, Egervari G, Nativio R, Sidoli S, Donahue G, Lombroso SI, Alexander DC, Riesche SL, Heller EA, Nestler EJ, et al. (2019). Alcohol metabolism contributes to brain histone acetylation. Nature 574, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitry RR, and Hughes RD (2011). Human Cell Culture Protocols. (Humana Press; ). [Google Scholar]
- NCHS (2018). National Center for Health Statistics Health, United States, 2017: With special feature on mortality. . (Centers for Disease Control and Prevention’s (CDC) National Center for Health Statistics (NCHS)). [Google Scholar]
- NPCR/SEER (2019). NPCR and SEER Incidence – U.S. Cancer Statistics 2005–2016 Public Use Research Database with Puerto Rico. (US Centers for Disease Control United States Department of Health and Human Services, Centers for Disease Control and Prevention and National Cancer Institute; ). [Google Scholar]
- Pan SY, Johnson KC, Ugnat AM, Wen SW, Mao Y, and Canadian Cancer Registries Epidemiology Research, G. (2004). Association of obesity and cancer risk in Canada. Am J Epidemiol 159, 259–268. [DOI] [PubMed] [Google Scholar]
- Park J, Morley TS, Kim M, Clegg DJ, and Scherer PE (2014). Obesity and cancer--mechanisms underlying tumour progression and recurrence. Nature reviews. Endocrinology 10, 455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, and Kroemer G (2015). Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab 21, 805–821. [DOI] [PubMed] [Google Scholar]
- Polak K, Czyzyk A, Simoncini T, and Meczekalski B (2017). New markers of insulin resistance in polycystic ovary syndrome. J Endocrinol Invest 40, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praetorius C, Grill C, Stacey SN, Metcalf AM, Gorkin DU, Robinson KC, Van Otterloo E, Kim RS, Bergsteinsdottir K, Ogmundsdottir MH, et al. (2013). A polymorphism in IRF4 affects human pigmentation through a tyrosinase-dependent MITF/TFAP2A pathway. Cell 155, 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Olson OC, Bhardwaj P, Walsh LA, Akkari L, Quick ML, Chen IC, Wendel N, Ben-Chetrit N, Walker J, et al. (2017). Obesity alters the lung myeloid cell landscape to enhance breast cancer metastasis through IL5 and GM-CSF. Nature Cell Biology 19, 974–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves GK, Pirie K, Beral V, Green J, Spencer E, Bull D, and Million Women Study C (2007). Cancer incidence and mortality in relation to body mass index in the Million Women Study: cohort study. BMJ 335, 1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiki A, Ohira M, Endo K, Koide N, Oyama T, Murano T, Watanabe H, Miyashita Y, and Shirai K (2009). Circulating angiotensin II is associated with body fat accumulation and insulin resistance in obese subjects with type 2 diabetes mellitus. Metabolism 58, 708–713. [DOI] [PubMed] [Google Scholar]
- Samanic C, Gridley G, Chow WH, Lubin J, Hoover RN, and Fraumeni JF Jr. (2004). Obesity and cancer risk among white and black United States veterans. Cancer Causes Control 15, 35–43. [DOI] [PubMed] [Google Scholar]
- Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, et al. (2015). Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seberg HE, Van Otterloo E, and Cornell RA (2017). Beyond MITF: Multiple transcription factors directly regulate the cellular phenotype in melanocytes and melanoma. Pigment Cell Melanoma Res 30, 454–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiden MV, and Anderson KC (1994). Multiple myeloma. Curr Opin Oncol 6, 41–49. [DOI] [PubMed] [Google Scholar]
- Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, et al. (2008). IRF4 addiction in multiple myeloma. Nature 454, 226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2019). Cancer statistics, 2019. CA Cancer J Clin 69, 7–34. [DOI] [PubMed] [Google Scholar]
- Soderberg KC, Kaprio J, Verkasalo PK, Pukkala E, Koskenvuo M, Lundqvist E, and Feychting M (2009). Overweight, obesity and risk of haematological malignancies: a cohort study of Swedish and Finnish twins. Eur J Cancer 45, 1232–1238. [DOI] [PubMed] [Google Scholar]
- Sonderman JS, Bethea TN, Kitahara CM, Patel AV, Harvey C, Knutsen SF, Park Y, Park SY, Fraser GE, Teras LR, et al. (2016). Multiple Myeloma Mortality in Relation to Obesity Among African Americans. J Natl Cancer Inst 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subbaramaiah K, Morris PG, Zhou XK, Morrow M, Du BH, Giri D, Kopelovich L, Hudis CA, and Dannenberg AJ (2012). Increased Levels of COX-2 and Prostaglandin E-2 Contribute to Elevated Aromatase Expression in Inflamed Breast Tissue of Obese Women. Cancer Discovery 2, 356–365. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Sung H, Siegel RL, Rosenberg PS, and Jemal A (2019). Emerging cancer trends among young adults in the USA: analysis of a population-based cancer registry. Lancet Public Health 4, e137–e147. [DOI] [PubMed] [Google Scholar]
- Teras LR, Kitahara CM, Birmann BM, Hartge PA, Wang SS, Robien K, Patel AV, Adami HO, Weiderpass E, Giles GG, et al. (2014). Body size and multiple myeloma mortality: a pooled analysis of 20 prospective studies. Br J Haematol 166, 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM, and Schiffrin EL (2000). Signal transduction mechanisms mediating the physiological and pathophysiological actions of angiotensin II in vascular smooth muscle cells. Pharmacol Rev 52, 639–672. [PubMed] [Google Scholar]
- Ueno N, Nishimura N, Ueno S, Endo S, Tatetsu H, Hirata S, Hata H, Matsuoka M, Mitsuya H, and Okuno Y (2017). PU.1 acts as tumor suppressor for myeloma cells through direct transcriptional repression of IRF4. Oncogene 36, 4481–4497. [DOI] [PubMed] [Google Scholar]
- Wang C, and Wang X (2015). The interplay between autophagy and the ubiquitin-proteasome system in cardiac proteotoxicity. Biochim Biophys Acta 1852, 188–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TN, Chen X, Li R, Gao B, Mohammed-Ali Z, Lu C, Yum V, Dickhout JG, and Krepinsky JC (2015). SREBP-1 Mediates Angiotensin II-Induced TGF-beta1 Upregulation and Glomerular Fibrosis. J Am Soc Nephrol 26, 1839–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Pan K, Chen Y, Huang C, and Zhang X (2012). The acetylation of transcription factor HBP1 by p300/CBP enhances p16INK4A expression. Nucleic Acids Res 40, 981–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JM, Musini VM, and Gill R (2018). First-line drugs for hypertension. Cochrane Database Syst Rev 4, CD001841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Bi C, Chooi JY, Chan ZL, Mustafa N, and Chng WJ (2015). MMSET regulates expression of IRF4 in t(4;14) myeloma and its silencing potentiates the effect of bortezomib. Leukemia 29, 2347–2354. [DOI] [PubMed] [Google Scholar]
- Xu M, Nagati JS, Xie J, Li J, Walters H, Moon YA, Gerard RD, Huang CL, Comerford SA, Hammer RE, et al. (2014). An acetate switch regulates stress erythropoiesis. Nat Med 20, 1018–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Drake BF, and Colditz GA (2016). Obesity and Other Cancers. 34, 4231–4237. [DOI] [PubMed] [Google Scholar]
- Yiannikouris F, Gupte M, Putnam K, Thatcher S, Charnigo R, Rateri DL, Daugherty A, and Cassis LA (2012). Adipocyte deficiency of angiotensinogen prevents obesity-induced hypertension in male mice. Hypertension 60, 1524–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. (2013). Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 499, 97–101. [DOI] [PubMed] [Google Scholar]
- Yvan-Charvet L, and Quignard-Boulange A (2011). Role of adipose tissue renin-angiotensin system in metabolic and inflammatory diseases associated with obesity. Kidney Int 79, 162–168. [DOI] [PubMed] [Google Scholar]
- Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, et al. (2006). The molecular classification of multiple myeloma. Blood 108, 2020–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mRNA microarray data generated during this study is available at GEO database (GSE132604).