

Abstract
Tuberculosis, caused by Mycobacterium tuberculosis (M. tuberculosis), kills 1.6 million people annually. To bridge the gap between structure- and cell-based drug discovery strategies, we are pioneering a computer-aided discovery paradigm that merges structure-based virtual screening with ligand-based, machine learning methods trained with cell-based data. This approach successfully identified N-(3-methoxyphenyl)-7-nitrobenzo[c][1,2,5]oxadiazol-4-amine (JSF-2164) as an inhibitor of purified InhA with whole-cell efficacy versus in vitro cultured M. tuberculosis. When the intrabacterial drug metabolism (IBDM) platform was leveraged, mechanistic studies demonstrated that JSF-2164 underwent a rapid F420H2-dependent biotransformation within M. tuberculosis to afford intrabacterial nitric oxide and two amines, identified as JSF-3616 and JSF-3617. Thus, metabolism of JSF-2164 obscured the InhA inhibition phenotype within cultured M. tuberculosis. This study demonstrates a new docking/Bayesian computational strategy to combine cell- and target-based drug screening and the need to probe intrabacterial metabolism when clarifying the antitubercular mechanism of action.
Keywords: Bayesian, docking, InhA, tuberculosis, intrabacterial, metabolism
Graphical Abstract
A significant subset of the chemical biology field interrogates the fundamental biology of pathogenic bacteria through the discovery and study of small molecule chemical tools. The ramifications from both academic and humanitarian viewpoints are evident given the global health crisis around drug-resistant bacteria and the need for innovative therapies and therapeutic strategies.1 We face a common and yet significant challenge in the identification of small organic molecules with a set of properties consistent with their value as tools2 to probe the essential biology of the bacteria in the hopes of gaining insights into which proteins and/or metabolic pathways are requisite for infection in model systems of varying degrees of relevance to pathogenesis in humans.
We study the causative agent of tuberculosis (TB), Mycobacterium tuberculosis (M. tuberculosis), and utilize it herein as a model system. However, our approach should be applicable to any class of bacteria or infectious agent. Faced with the challenge of identifying highly useful chemical tools, TB researchers have relied on a range of techniques from target-based approaches (both structure-focused and biochemical high-throughput screens) to whole-cell high-throughput phenotypic screening. Target-based methods, presuming they focus on a gene/protein that is both essential3 and vulnerable4 under experimental conditions relevant to human infection, have often struggled to correlate the inhibition of protein function and whole-cell activity against in vitro cultured M. tuberculosis.5 Computational methods such as high-throughput docking, despite some success outside of the TB arena, in general can make educated guesses to increase the probability of finding small molecules that best fit a protein active site and impair its native function.6 However, the correlation between enzyme functional inhibition and whole-cell activity is often lacking.7
To bridge the disconnection between in vitro enzyme inhibition and whole-cell activity, we have sought to utilize computational studies, in conjunction with chemical and biological techniques, to identify small molecule tool compounds for M. tuberculosis. We and others have found that high-throughput docking through a virtual screen (VS) against a single target can identify novel inhibitors of M. tuberculosis enzymes.8 We hypothesized that high-throughput docking would be complemented by our proven machine learning methods9,10 to enhance the probability of finding compounds with bacterial growth inhibitory properties and the lack of mammalian cell cytotoxicity.
Herein, we describe the design of a high-throughput docking/Bayesian methodology, combining target-based and whole-cell-based screening, and its application to the M. tuberculosis enzyme InhA, an essential11,12 and vulnerable4 enoyl acyl carrier protein reductase inhibited by the front-line drug isoniazid (INH).13,14 This platform identified JSF-2164 as an inhibitor of purified InhA with bacterial growth inhibitory activity. Its mechanism within M. tuberculosis was complicated by intrabacterial metabolism, and we found our intrabacterial drug metabolism (IBDM) platform to be useful in detailing this process.
RESULTS
A Novel High-Throughput Docking/Bayesian Approach.
We examined a previous VS conducted by some of us that docked a 5.6 × 106 member composite library versus InhA as part of the GO Fight Against Malaria project (GO FAM).8 This present work leverages a fraction of this library of commercially available drug-like small molecules, comprising a 5.07 × 105 collection of Asinex compounds docked against the Tonge laboratory structure of InhA complexed with a diaryl ether phenol (PDB ID 2X23).15 This structure was chosen because InhA is bound to PT70, a very potent inhibitor (Ki = 7.8 nM) with a long residence time (24 min).15 The Asinex subset was docked into the active site with Auto Dock Vina.16 The average of the estimated free energy of binding values was −6.93 kcal/mol with a standard deviation of 0.81 kcal/mol. Docking filters narrowed down this subset to the top 370 compounds, on the basis of the requirement that the candidate’s predicted binding mode formed base-stacking interactions with the NAD+/NADH cofactor, displayed at least 2 hydrogen bonds with the active site, and had an estimated free energy of binding of ≤−8.0 kcal/mol. These 370 compounds displayed an average estimated free energy of binding of −8.66 kcal/mol with a standard deviation of 0.55 kcal/mol in the docking scores. At this point, current docking strategies typically transition to an experimental stage where the candidate inhibitors are tested in vitro either with or without prior visual inspection of the compounds. Our strategy diverges and utilizes our published TB Bayesian models to create a new workflow (Figure 1) that combines ligand-based machine learning models and structure-based VS data. We proposed that the VS data versus a validated M. tuberculosis drug target such as InhA could be joined with our Bayesian dual-event models to select for novel InhA inhibitors with whole-cell activity and lack of mammalian cell cytotoxicity as assessed with Vero cells. Our published dual-event models consider both whole-cell efficacy versus in vitro cultured M. tuberculosis (under actively growing conditions) and Vero cell cytotoxicity, and they have learned via training sets containing on the order of 103 drug-like small molecules.10 In this case as in our previous work, we define a selectivity index (SI = CC50/MIC, where the MIC is the minimum inhibitory concentration of the molecule that inhibits 90% growth of the bacteria in culture and CC50 is the concentration of the compound that inhibits the growth of 50% of the cultured mammalian cells (Vero cells). Our published models have demonstrated the ability to favorably score novel whole-cell active compounds with an acceptable selectivity index (SI ≥ 10) from screening collections in comparison to inactives.
Figure 1.
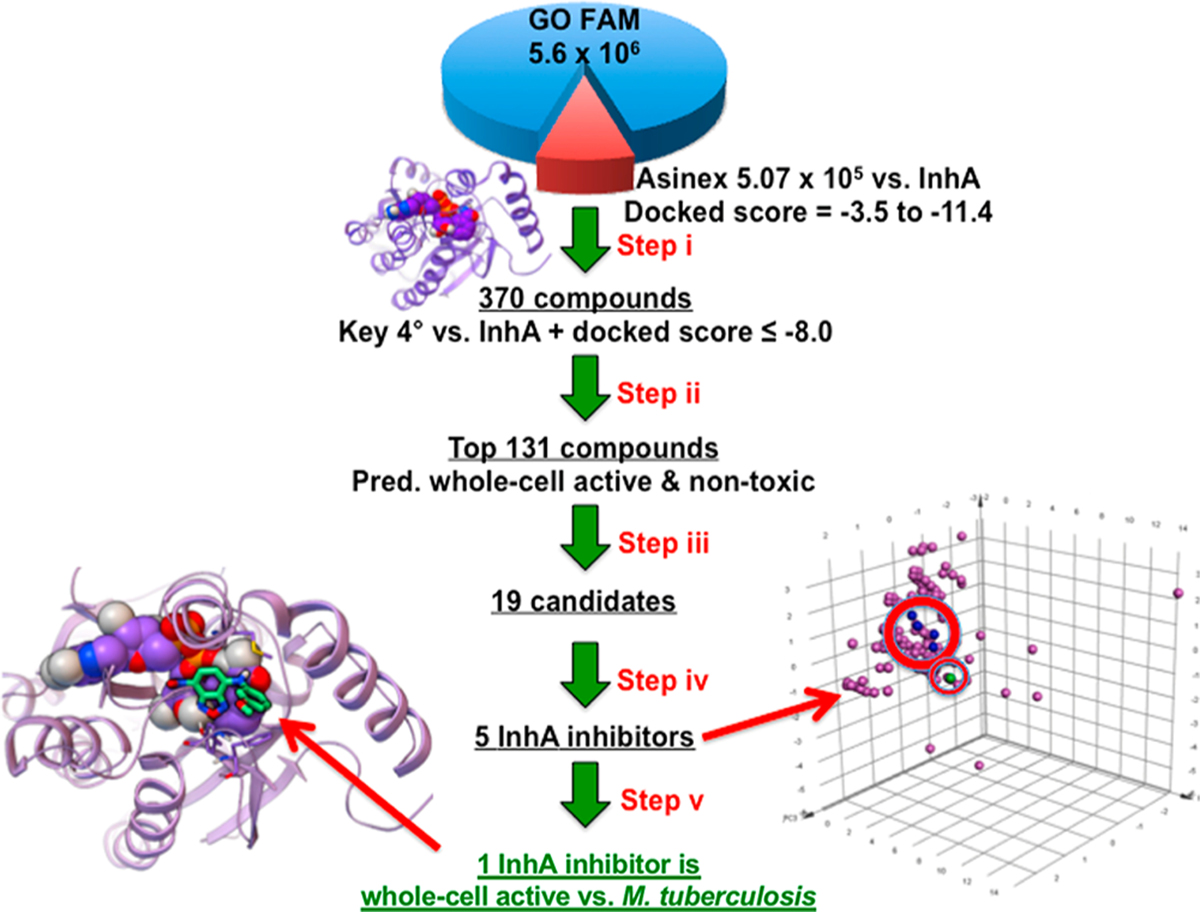
Innovative workflow to advance the discovery of whole-cell active chemical tools against M. tuberculosis that inhibit a drug target and lack significant relative toxicity to mammalian cells. (i) GO Fight Against Malaria experiment 9 docked the Asinex library of 5.07 × 105 models of small molecules against InhA (PDB ID: 2X23). Docking filters narrowed down the results to the top 370 compounds. (ii) These 370 compounds were filtered with two different dual-event Bayesian models to identify 131 compounds predicted to display whole-cell activity against M. tuberculosis and a lack of significant relative toxicity to Vero cells. (iii) The docked binding modes of the top 131 compounds were visually inspected to identify 19 candidate compounds, which were then ordered. (iv) The 19 candidate compounds were each tested at 50 µM to identify five hits with at least 50% inhibition of InhA activity. A principal component analysis is shown on the right that compares the chemical property space of the five hits (four in blue inhibit InhA; one in green is the most active against M. tuberculosis) to 157 known InhA inhibitors (magenta) in the TB Mobile 2 data set. (v) These 5 novel InhA inhibitors were then tested in M. tuberculosis growth inhibition assays to identify one InhA inhibitor that significantly inhibited bacterial growth. Shown on the left with light green carbons is the inhibitor with the best whole-cell activity, an MIC of 8.0 µM.
370 compounds (that passed the initial docking filters) were scored with two of our recently validated Bayesian dual-event models selecting for bacterial growth inhibition with relatively low cytotoxicity in mammalian cells (Table S1). We chose to use the dose−response and cytotoxicity (or TAACF-CB2) model, given its learning based on 1783 compounds screened versus M. tuberculosis and Vero cells by Reynolds and co-workers and its successful identification of four diverse and promising antituberculars from a GlaxoSmithKline antimalarial hit set.10 The second model utilized was our combined TB dose–response and cytotoxicity (or Combined) model that draws on a data set of 5304 small molecules assayed versus M. tuberculosis and Vero cells.9 For the 370 compounds harvested with the initial docking filters, (a) their TAACF-CB2 model scores ranged from −8.61 to 8.38, with 133 compounds predicted as whole-cell active versus M. tuberculosis and relatively nontoxic versus Vero cells having scores ≥0.54, and (b) their Combined model scores ranged from −13.77 to 8.84, with 93 compounds predicted as whole-cell active versus M. tuberculosis and relatively nontoxic versus Vero cells having scores ≥1.17. The top 100 compounds by the TAACF-CB2 model (score ≥1.11) along with the highest scoring 52 from the Combined model afforded a total of 131 compounds for further analysis (given the overlap between the two sets equal to 21 molecules).
Visual inspection of the 131 predicted whole-cell active/noncytotoxic candidate InhA inhibitors was then undertaken. As discussed in our previous publication,8 advantageous attributes included the following: (a) almost all of the heteroatoms were involved in hydrogen bonds, favorable electrostatic interactions,17 or halogen bonds with the target,18,19 (b) the hydrophobic groups packed favorably with hydrophobic side chains of InhA,8,16 and (c) aromatic rings formed T-stacking, base-stacking,20 or π−cation interactions20 with InhA, as measured by the Fox software (available at http://autodock.scripps.edu/resources/raccoon). Base-stacking and T-stacking interactions depend on the distance between the centers of the two aromatic rings and the angle between the planes of each ring, while π−cation interactions can occur when a positively charged group is less than 5−6 Å from the center of an aromatic ring.20 Disadvantageous attributes of a docked pose included displaying (a) one or more large, solvent-exposed hydrophobic groups (such as i-propyl, t-butyl, or benzyl), (b) polar groups packed in hydrophobic pockets (unless they had a complementary group from the target in the vicinity),21 or (c) more than one repulsive electrostatic interaction with charged or polar groups of InhA. This manual inspection afforded 19 candidates for biological testing, which were available and obtained from the commercial source (Asinex; www.asinex.com).
JSF-2164 (BAS01237091) as an InhA Inhibitor with Whole-Cell Activity.
The 19 candidate molecules (Table 1) were assessed for whole-cell growth inhibition of in vitro cultured M. tuberculosis wild type H37Rv strain. Two out of the 19 compounds, BAS01237091 and BAS01237231, exhibited promising whole-cell activity (MIC ≤ 12.5 µM; i.e., better than our goal of 10 µg/mL or ∼33 µM for a 300 g/mol MW compound). INH and triclosan were used as positive controls in all M. tuberculosis growth inhibition assays.22 Interestingly, both compounds (BAS01237091 and BAS01237231) are of the benzofurazan class. The most potent whole-cell inhibitor was BAS01237231, featuring an MIC of 10 µM. From the initial set of 370 candidates passing the docking filters, the worst scoring five molecules (according to their score with the TAACF-CB2 model) were also assayed as a supplementary test of the potential for undesirable “false negative” predictions. All demonstrated MIC values between 125 and 250 µM (Table S2).
Table 1.
In Vitro Efficacy Profiles for Candidate InhA Inhibitors f
| Cmpd Identifiers | Structure | MIC (µM) | Vero Cell Cytotoxicity CC50 (µM) / SIa | InhA Inhibition at 50 µM Cmpd / IC50(µM)b | |
|---|---|---|---|---|---|
| H37Rv | mc24914 | ||||
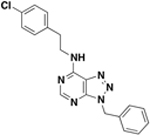
| BAS06264740 ZINC00378804 (resynthesized as JSF-2149) |
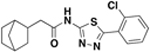 |
>500 | 500 | 9.2 – 18 / <0.018 | 91 ± 2% c / 40 ± 10 |
| BAS00131943 ZINC00805451 |
460 | 250 | 160 – 230 / 0.35 | 65 ± 4 % / 40 ± 5 | |
|
ASN06745 ZINC04411524 |
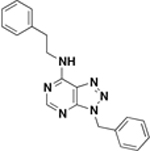 |
>500 | 500 | 4.8 – 9.7 / <0.0096 | 58 ± 5% / 48 ± 9 |
| ASN06744991 ZINC12481353 |
 |
>500 | 500 | <2.2 / <0.0044 | 55 ± 6% / 45 ± 5 |
| BAS01237231 ZINC04894756 (resynthesized as JSF-2164) |
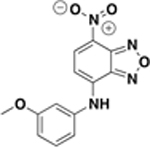 |
10 d | 30 | 44 – 87 / 5.5 | 53 ± 4 % / 51 ± 19 |
| BAS00135708 |  |
125 | n.d. | n.d. | 36 ± 1 %e |
| BAS00704577 |  |
>500 | n.d. | n.d. | 30 ± 5 % |
| BAS00396190 | 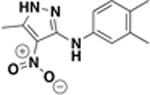 |
>500 | n.d. | n.d. | 27 ± 3% |
| ASN03068448 | 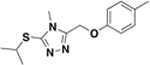 |
250 | n.d. | n.d. | 20 ± 2% |
| BAS01237091 | 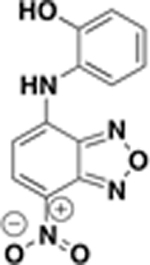 |
12.5 | n.d. | n.d. | 19 ± 1% |
| BAS24802784 | 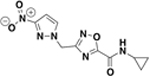 |
>500 | n.d. | n.d. | 4.8 ± 5.6% |
| ASN05988636 | 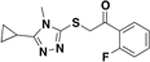 |
500 | n.d. | n.d. | 1.5 ± 4.3% |
| BAS06979351 | 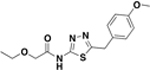 |
250 | n.d. | n.d. | 1.2 ± 6.4% |
| BAS01237237 | 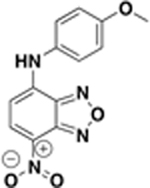 |
500 | n.d. | n.d. | 0.41 ± 11% |
| BAS03794545 | 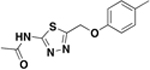 |
500 | n.d. | n.d. | 0 ± 36 %e |
| ASN03986337 | 250 | n.d. | n.d. | 0 ± 2.4% | |
| BAS03069066 | 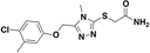 |
250 | n.d. | n.d. | 0 ± 1.4% |
| BAS05024031 | 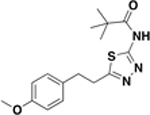 |
200 | n.d. | n.d. | 0 ± 3.7% |
| BAS05667953 | 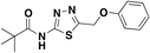 |
200 | n.d. | n.d. | 0 ± 3.1% |
| triclosan | 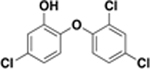 |
62 | 120 | n.d. | 100 ± 6%/0.13 ± 0.02 |
| isoniazid | 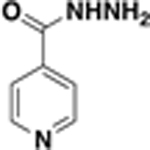 |
0.16 | 2.5 | n.d. | n.d. |
SI = Vero Cell CC50/MIC H37Rv using the more conservative estimate of the CC50 in the reported range.
Values are reported as the mean ± standard error from at least three independent measurements.
Synthesized material demonstrated an InhA inhibition of 84 ± 2%.
While the commercial material demonstrated an MIC of 10 µM and an InhA inhibition of 53 ± 4%, the synthetic material exhibited an MIC value of 8.0 µM versus the H37Rv strain and an InhA inhibition of 44 ± 1%.
Due to compound insolubility at 50 µM, the measurement was made at 25 µM.
n.d. = not determined.
We applied an InhA enzyme assay17 to empirically confirm InhA inhibition efficacy of the 19 predicted hits. Compound inhibition efficacy was calculated by NADH consumption rate when the purified M. tuberculosis InhA was coincubated with compounds and the model substrate 2-dodecenoyl-CoA. Triclosan was included as a positive control in all InhA inhibition assays.22 17/19 candidates were tested at a compound concentration of 50 µM, and 2/19 compounds (thiadiazole BAS03794545 and furanone BAS00135708) were tested at 25 µM due to demonstrated insolubility at 50 µM. 5/19 compounds, including the whole-cell active compound BAS01237231, displayed at least 50% inhibition of InhA at this single concentration (Figure 2A and Table 1). Their respective IC50 values versus InhA ranged from 4.0 to 51 µM (Table 1 and Figure S1). The most efficacious InhA inhibitor was thiadiazole amide BAS06264740 with an IC50 = 4.0 ± 1.0 µM; however, it failed to inhibit bacteria growth (MIC > 500 µM versus the H37Rv strain). The whole-cell active compound BAS01237231 was determined as a moderate inhibitor of purified InhA with an IC50 = 51 ± 19 µM (Table 1 and Figure S1).
Figure 2.
Screening hit JSF-2164 (BAS01237231) is a moderate InhA inhibitor in vitro. (A) InhA inhibition efficacy of the 19 Docking/Bayesian screen hits was assessed by measuring the percent inhibition of purified InhA at 50 µM compound. The dashed line indicates 50% InhA inhibition efficacy. Triclosan was used as a positive control shown in red, and DMSO was used as a negative control. The five screening hits with % InhA inhibition >50% at 50 µM were shown in orange. (B) The InhA enzyme assay was utilized to evaluate the IC50 for JSF-2164 inhibition of purified InhA. Enzyme activity was determined via the consumption rate of NADH read at 340 nm. Enzymatic buffer with or without 0.01 CMC of Tween-20 was shown in red or black, respectively. IC50 values were calculated by nonlinear regression via Prism GraphPad (Version 6.0c). (C−E) Docking evidenced JSF-2164 binding to InhA. In (C), the NAD cofactor was displayed in a ball-and-stick model with magenta carbons, and the residues Tyr158 and Met199 were shown as thin sticks. In (D) and (E), the NAD cofactor was displayed as CPK, and the key residues Phe149, Tyr158, Thr196, Ala198, Met199, and Gln214 were shown as thin sticks with dark purple carbon atoms. The inhibitor that crystallized with InhA in PDB ID: 2×23, PT70, was shown as thick magenta lines in (E) as a reference.
Given their respective InhA inhibition and whole-cell growth inhibition, thiadiazole amide BAS06264740 (renamed JSF-2149) and benzofurazan BAS01237231 (renamed JSF-2164) were selected for further evaluation through synthesis and confirmation of their biological profiles. JSF-2149 was prepared (Figure S2) in three steps from commercially available 2-chlorobenzaldehyde via formation of the thiosemicarbazone 1 that was then cyclized in the presence of ferric chloride to provide 2-aminothiazole 2.23 Acylation of 2 with 2-norbornane acetyl chloride provided JSF-2149. JSF-2164 was prepared (Figure S3) via the reaction of commercial 4-chloro-7-nitrobenzo[c][1,2,5]oxadiazole with 3-methoxyaniline, inspired by previously reported SNAr chemistry.24 The InhA inhibition (84 ± 2% inhibition at 50 µM compound) of synthetic JSF-2149 was similar to that of the commercial material, and the synthetic sample was also inactive versus M. tuberculosis (MIC > 500 µM). The MIC for synthetic JSF-2164 was similar to that of the commercial material (8.0 µM vs 10 µM, respectively) (Table 2) as were their respective percent inhibitions of InhA at 50 µM compound (synthetic vs commercial: 44 ± 1 vs 53 ± 4).
Table 2.
MIC Values for JSF-2164 and Its Two Metabolites versus M. tuberculosis Strains
| Compound | Structure | MIC (µM) | % InhA inhibition at 50 µM with 1% CMC Tween-20 | |||
|---|---|---|---|---|---|---|
| H37Rv | mc24914 | fgd1::tn | ddn::tn | |||
| 1NH | 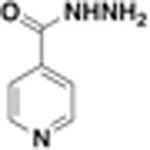 |
0.16 | 2.5 | 0.078 | 0.078 | N.D. |
| JSF-2164 | 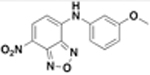 |
8.0 | 25 | >200 | 8.0 | 44 ± 1.0 |
| JSF-3617 | 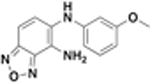 |
250 | 125 | 125 | 250 | 77 ± 0.74 |
| JSF-3616 | 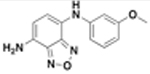 |
32 | 32 | 32 | 32 | 46 ± 2.9 |
IC50 values within the µM range may raise concerns of promiscuous inhibition of purified enzyme InhA due to aggregates formed by compounds in the enzymatic buffer.25 We, therefore, utilized Tween-20 at 1% of its critical micelle concentration (CMC) in the enzymatic buffer as a detergent to mitigate aggregate formation without disrupting enzyme activity (Figure S4). InhA inhibitory potency of the JSF-2149 was evidenced due to unspecific aggregation, judged by an IC50 shift from 4.0 ± 1.0 µM (without Tween-20) to 70 ± 16 µM (with Tween-20) (Figure S5). In contrast, the whole-cell active compound JSF-2164 was a specific InhA inhibitor in vitro given no statistically significant difference between the IC50 with or without Tween-20 (51 ± 19 µM vs 65 ± 16 µM) judged by an unpaired Student’s t test (Figure 2B). Finally, docking results predicted JSF-2164 to form the following quaternary interactions with InhA: base-stacking with the NAD cofactor, two hydrogen bonds with the hydroxyl in the side chain of Tyr158, two hydrogen bonds with the 2′-hydroxyl of the ribose adjacent to the nicotinamide ring of NAD, a hydrogen bond with the 3′-hydroxyl of the ribose adjacent to the nicotinamide ring of NAD, and a hydrogen bond with the amide carbonyl of the nicotinamide ring of NAD (Figure 2C–E).
JSF-2164 Exhibited in Vitro Bacteriostatic Activity Involving More than InhA Inhibition.
Over 7 days, JSF-2164 at 16× MIC demonstrated an ca. 1 log10 reduction in M. tuberculosis colony-forming units/mL (CFU/mL) (Figure 3A). It indicated that JSF-2164 is not formally bactericidal,26 judged by a failure to determine its minimum bactericidal concentration (MBC; minimum compound concentration to afford ≥2 log10 reduction in bacterial CFU/mL) within 4-fold of its MIC range.27 This may be compared to the bactericidal activity of INH as demonstrated by complete sterilization at 4× MIC with a detection limit of 10 CFU/mL.
Figure 3.
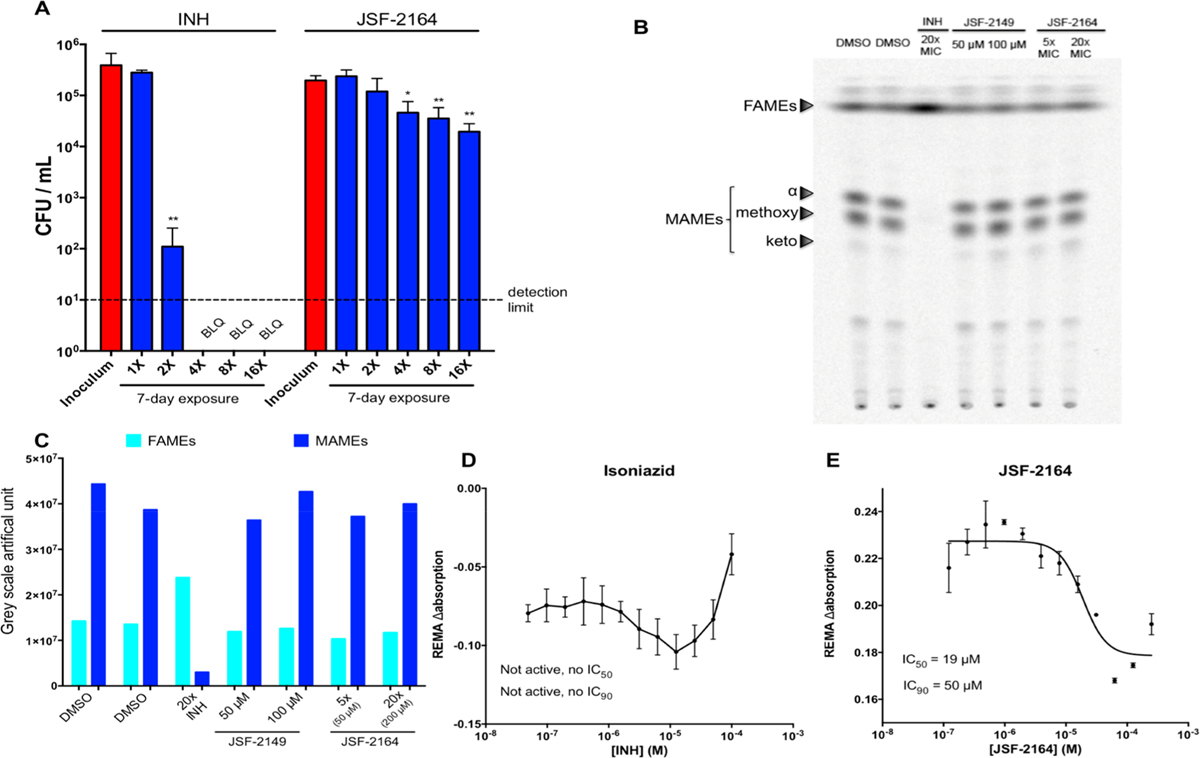
JSF-2164 exhibits an antitubercular profile distinct from INH. (A) 105 CFU/mL M. tuberculosis H37Rv were exposed to INH or JSF-2164 at different folds of the MIC for 7 days, followed by CFU enumeration. The detection limit of 10 CFU/mL is depicted as a dashed line. One-way ANOVA adjusted with Bonferroni’s multiple comparison test was applied to analyze the CFU statistical difference between the inoculation and INH or JSF-2164 treated bacteria. *p < 0.05; **p < 0.01. (B, C) Mid-log phase (OD595 = 0.4) M. tuberculosis H37Rv was pretreated with INH or JSF-2164 for 2 h followed by 14Cacetate labeling for 4 h. Lipids were extracted and esterified to afford the corresponding methyl esters. The mycolic acid methyl esters (MAMEs) and fatty acid methyl esters (FAMEs) were analyzed via thin-layer chromatography (TLC) and developed via a Typhoon phosphorimager. (C) Showed quantification of the gray scale of FAMEs and MAMEs from (B). (D, E) M. tuberculosis streptomycin-sensitive strain ss18b was starved without streptomycin for 2 weeks to maintain OD595 = 0.3, followed by treatment with INH or JSF-2164 for 7 days. The compound activity against ss18b was determined via the resazurin microtiter assay (REMA). IC50 and IC90 were quantified via nonlinear regression with Prism GraphPad (Version 6.0c). Error bars indicated standard errors.
To probe the intrabacterial engagement of InhA by JSF-2164, we assessed the JSF-2164 growth inhibitory effect against the M. tuberculosis inhA overexpression strain mc24914 (Table 2), which harbors an inhA promoter c(−15)t mutation.13 INH demonstrated a 16× MIC shift versus the inhA overexpression strain as compared to H37Rv. JSF-2164, in contrast, exhibited a 3-fold shift in MIC versus the inhA overexpression strain as compared to H37Rv. The effect of JSF-2164 was also assayed on newly synthesized M. tuberculosis short-chain fatty acids and mycolic acids. M. tuberculosis strain H37Rv prelabeled with 14C-acetate was treated with JSF-2164 or INH as a positive control.13 After lipid extraction and esterification to afford the corresponding methyl esters, the samples were analyzed for mycolic acid methyl esters (MAMEs) and their precursors, short-chain fatty acid methyl esters (FAMEs), via thin-layer chromatography (TLC) (Figure 3B,C). INH expectedly13 led to a depletion of MAMEs and an accumulation of FAMEs as a result of InhA inhibition, whereas JSF-2149 served as a negative control given its lack of significant whole-cell activity and failure to inhibit purified InhA in the presence of detergent. JSF-2164 unexpectedly did not exhibit a pattern similar to that for INH. The smaller MIC shift for JSF-2164, as compared to INH, and its lack of a typical InhA (or more generally FAS-II) inhibition profile as judged by TLC, coupled with its IC50 versus purified InhA being ca. 5-fold larger than its MIC against the H37Rv strain, hinted at an intrabacterial process where InhA inhibition is secondary to another mechanism of action. Consistent with this hypothesis was the observation that JSF-2164 exhibited in vitro efficacy versus the M. tuberculosis streptomycin-sensitive (ss18b) strain28 that models aspects of nonreplicating persistence. This was differentiated from the negative control INH, which as the prototypical InhA inhibitor selectively inhibits actively replicating bacteria, and rifampicin as the positive control (Figures 3D,E and S6).
F420H2 Deficiency Linked to JSF-2164 Resistance.
To better comprehend the JSF-2164 mechanism of action within M. tuberculosis, we selected for spontaneous JSF-2164-resistant mutants in the M. tuberculosis strain H37Rv background. JSF-2164 exhibited a relatively high mutation rate (∼10−5), which was similar to the frequency of resistance of pretomanid (10−5 to 10−6),29 a clinical-stage antitubercular requiring activation dependent by the cofactor F420H2.29 We isolated 26 JSF-2164-resistant colonies selected with a JSF-2164 exposure ranging from 8−32× MIC and confirmed their resistance via a liquid culture MIC test. All tested colonies were resistant to JSF-2164: one strain (32 × 4) with a 4-fold MIC shift and the other 23 colonies with ≥16× MIC shift as compared to the wild type strain H37Rv (with an upper limit of 16-fold established by the solubility of JSF-2164 in the assay media) (Figure 4A). 5/24 colonies were cross-resistant to INH, consisting of strains 16 × 6 and 32 × 4 exhibiting 8× MIC shift and strains 32 × 3, 32 × 5, and 32 × 6 showing 4× MIC shift against INH (Figure 4A).
Figure 4.
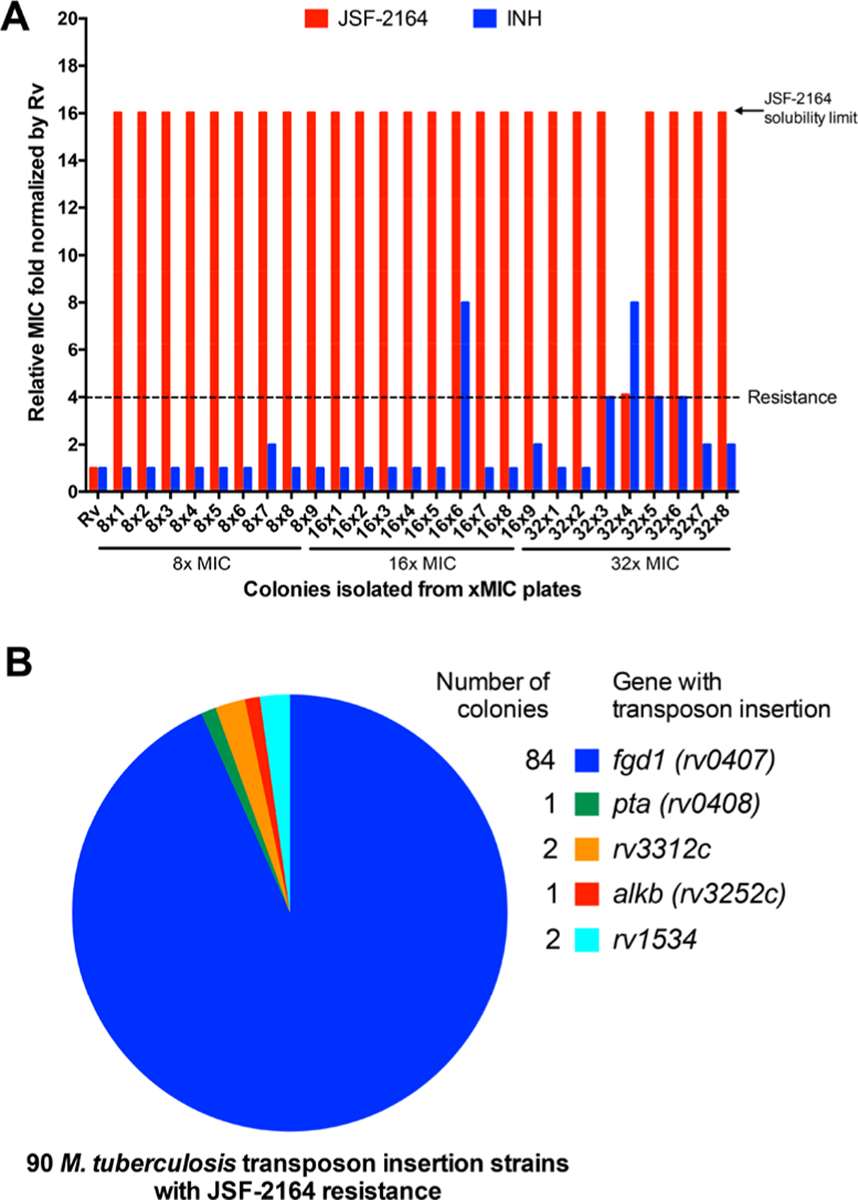
Spontaneous mutagenesis and transposon library selection indicate that mutations in F420H2 biosynthesis result in JSF-2164 resistance. (A) 26 M. tuberculosis colonies spontaneously resistant to JSF-2164 were isolated followed by confirmation of JSF-2164 resistance and evaluation of cross-resistance with INH via the liquid culture MIC test. MIC values for JSF-2164 and INH versus strains were normalized by their corresponding MIC value versus H37Rv, and fold changes were depicted in red (JSF-2164) and in blue (INH). Resistance was determined as a >4-fold MIC shift as compared to H37Rv with an upper limit of 16-fold established by the solubility of JSF-2164 in the assay media. (B) A saturated Himar-1 transposon insertion library with an H37Rv background was selected in the presence of JSF-2164 at 5−30× MIC on 7H10 agar plates supplemented with albumin-dextrose-saline (ADS). 90 viable colonies were isolated, and their resistance to JSF-2164 was verified via liquid culture MIC tests. Transposon insertion sites in the 90 colonies were determined via SacII digestion, ligation, and sequence alignment of the Sanger sequencing fragment with primers annealed to the outward-facing T7 promoter.
Four strains (16 × 2, 16 × 6, 32 × 4, and 32 × 5) were selected for whole-genome sequencing, according to their resistance to JSF-2164 and varying susceptibility to INH (Table S3). Mutations with high-level JSF-2164 resistance in strains 16 × 2, 16 × 6, and 32 × 5 led to single nucleotide polymorphisms (SNPs) in the gene fbiC (F567S) or a frameshift in the gene fgd1 (C insertion in between 411 and 412 resulting in a frameshift at Leu138) (Table S3). We also noted a promoter mutation in the essential30 thymidylate kinase tmk of the sequenced strain 32 × 4. Both enzymes FbiC and Fgd1 comprise the biosynthetic pathway of deazaflavin cofactor F420H2. FbiC is required for biosynthesis of FO, a precursor of the cofactor F420.31 Fgd1, also known as F420-dependent glucose-6-phosphate dehydrogenase, recharges F420H2 through the oxidation of glucose-6-phosphate.29 Loss of function mutations in fgd1 and fbiC led to resistance to pretomanid, a nitro-containing heterocyclic antitubercular requiring F420H2-dependent activation.29 The cross-resistance of pretomanid was indeed observed in these 3 mutant strains judged by the >500× MIC shift as compared to wild type H37Rv (Table S3). A strain fgd1::tn, with fgd1 functionally inactivated by transposon insertion,11,32 led to resistance to both pretomanid (>500× MIC shift) and JSF-2164 (>20× MIC shift) (Table 2). This further confirmed that F420H2 was essential for JSF-2164 activity. Strain 16 × 6, which was resistant to INH, harbored a c(−15)t mutation at the promoter of inhA. This mutation was identical to the one reported in strain mc24914 and other clinical strains,33 which were resistant to INH due to statistically significant inhA overexpression as compared to the H37Rv strain (Figure S7).
The mechanism of action of JSF-2164 was further probed by Himar-1 transposon library screening (Figure 4B).30,34 A saturated transposon insertion library derived from the H37Rv background was exposed to JSF-2164 at concentrations ranging from 5 to 30× MIC for 4 weeks. Select single colonies surviving JSF-2164 pressure were isolated, and the transposon location was determined for each strain.34 A total of 90 colonies (54 colonies from 5× MIC, 8 colonies from 10× MIC, 8 colonies from 20× MIC, and 20 colonies from 30× MIC) exhibited JSF-2164 resistance on agar plates. JSF-2164 resistance for each of the 90 colonies was confirmed by a liquid MIC test and was quantified by a ≥16× MIC shift as compared to wild type H37Rv (again, being limited by the solubility of JSF-2164 in media) for 89/90 strains (Table S4). 89 out of 90 colonies demonstrated significant resistance to pretomanid, quantified by a ≥500× MIC shift (Table S4). All of the colonies were susceptible to INH. Genomic DNA was extracted from each of the colonies followed by SacII digestion, and the Himar-1 transposon sequence was ligated into a plasmid with an oriR6K replication initiation site and a kanamycin selection marker.34 Transposon insertion sites were determined via Sanger sequencing of the plasmids containing the Himar-1 sequence followed by mapping results onto the H37Rv genome. 84/90 colonies harbored a transposon insertion in the open reading frame of fgd1, suggesting the functional inactivation of fgd1 as a leading cause of JSF-2164 resistance. One insertion was observed in the open reading frame of gene pta (rv0408) locating in the same operon as fgd1. This pta disruption may indicate a polar effect of transposon insertion. The other five insertions were harbored in rv3312c (an α/β hydrolase),35 rv1534 (a TetR family transcription regulator),35 and alkB (rv3252c, a transmembrane alkane 1-monooxygenase).35
JSF-2164 Released Intrabacterial NO· Dependent on Cofactor F420H2.
Nitrobenzofurazan family compound NBDHEX (6-(7-nitro,2,1,3-benzoxadiazole-4-ylthio)-hexanol)36 and NBF-SPh37 (7-nitro-4-(phenylthio)-benzofurazan) are reported to release reactive nitrogen species (RNS) and reactive oxygen species (ROS) in cancer cells,37,38 and thus, we hypothesized that the growth inhibitory activity of JSF-2164 may be due to production of intrabacterial nitric oxide (NO·) dependent on the F420H2 cofactor. Although of different chemotypes, the triazine nitrofurans,39 pretomanid,40 and delamanid41 have been found to release NO· within M. tuberculosis through an F420H2-dependent process. We utilized the Griess reagent to quantify the supernatant concentration of nitrite, arising through the intrabacterial extrusion of NO· followed by its oxidation and export from the cell.42 Briefly, the M. tuberculosis strains H37Rv and fgd1::tn were treated with 20 or 100 µM pretomanid or JSF-2164, and for consecutive 5 days, the supernatant nitrite concentration was assessed via Griess reagent. Each compound’s NO· production was quantified via a ratio of nitrite concentration to compound dose. For the H37Rv arm of the experiment, pretomanid treatment led to a burst of NO· production in the initial 2 days, demonstrated by 49% (9.8 µM) and 17% (17 µM) of NO· release for the 20 and 100 µM pretomanid exposures, respectively (Figure 5A). The lower dose of JSF-2164 treatment (20 µM = 2.5× MIC) generated a small yet discernible amount of NO· at 2 days post-treatment of the H37Rv strain, quantified as 0.84 µM, which is slightly above the 0.78 µM limit of detection (Figure 5A). The higher dose of JSF-2164 (100 µM = 12.5× MIC) yielded a larger production of NO·, quantified as 6.7 µM at t = 2 days. F420H2 cofactor deficiency in the fgd1::tn strain abolished NO· production for both the pretomanid and JSF-2164 treatments, indicating that F420H2 was required to produce NO· from JSF-2164 (Figure 5B). Pretomanid has been shown to produce NO· primarily via the catalysis of the nitroreductase Ddn, which utilizes F420H2 as cofactor.40 We excluded Ddn as being responsible for the whole-cell activity of JSF-2164 as a comparison of MIC values of JSF-2164 versus H37Rv and ddn:tn26,40 strains showed no difference (Table 2).
Figure 5.
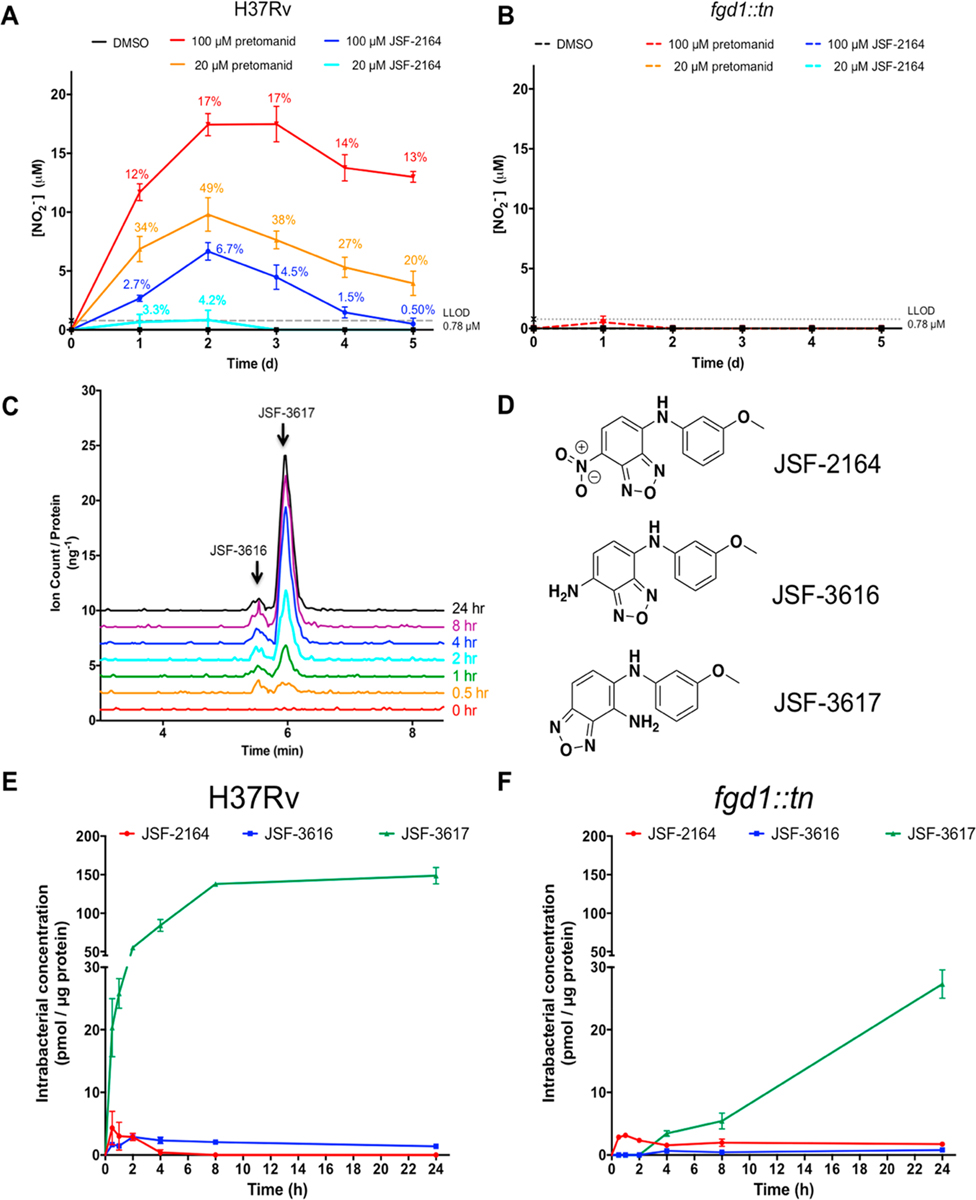
JSF-2164 is metabolized into two amines with NO· release dependent on cofactor F420H2. (A, B) Mid-log phase (OD600 = 0.4) M. tuberculosis H37Rv and fgd1::tn strains were treated with pretomanid or JSF-2019 at 20 or 100 µM. NO· release was measured via Griess reagent in biological triplicates. Percentage numbers indicated a proportion of release NO· from pretomanid or JSF-2019, calculated by a ratio of nitrite concentration versus compound dose. Error bars showed standard errors. (C) Mid-log phase M. tuberculosis H37Rv was treated with 20 µM JSF-2164 for 24 h, and metabolites were extracted at several time points. Two peaks with the same m/z of 257.0 indicated two metabolites JSF-3617 and JSF-3616 with the same molecular weight. (D) The structure of the two amine metabolites JSF-3617 and JSF-3616 were assigned via NMR and high-resolution mass spectrometry experiments with the metabolites isolated from the M. tuberculosis extract. (E, F) Mid-log phase (OD600 = 0.4) M. tuberculosis H37Rv and fgd1::tn strains were treated with 20 µM JSF-2164 in biological triplicates followed by metabolite extraction at various time points ranging from 0 to 24 h. Metabolites were analyzed and quantified via liquid-chromatography mass spectrometry (LC-MS), followed by normalization by protein amount. Error bars indicated standard errors.
JSF-2164 Intrabacterial Metabolism Produces Two Amines. NO· production suggested an intrabacterial metabolism of JSF-2164 contributing to its activity. We applied a liquid chromatography−mass spectrometry (LC-MS) platform for querying intrabacterial drug metabolism (IBDM)39 to probe the fate of JSF-2164 in M. tuberculosis. Briefly, M. tuberculosis H37Rv or fgd1::tn strain was treated with 20 µM JSF-2164 followed by metabolite extraction at various of time points for identification via liquid chromatography−mass spectrometry (LC-MS). After 24 h of treatment of H37Rv with JSF-2164, two peaks (one major and one minor) were observed to gradually accumulate, as detected via LC-MS (250 nm absorption; both peaks were associated with an m/z = 257.0) (Figure 5C). Suggested by a loss of 30 amu as compared to the parent JSF-2164 (m/z = 287.0), we hypothesized that these peaks corresponded to two amines, derived from the reduction of the nitro group. We purified the two metabolites from M. tuberculosis treated with JSF-2164 and assigned their chemical structures (Figure 5D) via NMR and high-resolution mass spectrometry studies. The major metabolite (designated as M1) was determined to be N5-(3-methoxyphenyl)benzo[c][1,2,5]oxadiazole-4,5-diamine, apparently resulting from a shift of the 1,2,5-oxadiazole with a reduction of the nitro group to an amine. The minor metabolite (designated as M2) was identified as N4-(3-methoxyphenyl)-benzo[c][1,2,5]oxadiazole-4,7-diamine, the apparent reduction product of the nitro moiety of JSF-2164. We synthesized authentic M1 and M2 and renamed them as JSF-3617 and JSF-3616, respectively (Figure S3). LC-MS data (Figures S8 and S9) demonstrated the synthetic samples were identical to the metabolites isolated from M. tuberculosis. JSF-3617 was inactive against M. tuberculosis judged by MIC values of 250 and 125 µM against the H37Rv and fgd1::tn strains, respectively (Table 2). JSF-3616 demonstrated modest growth inhibitory efficacy with an MIC of 32 µM versus both the M. tuberculosis and fgd1::tn strains (Table 2). JSF-3616 and JSF-3617 exhibited 46 ± 2.9% and 77 ± 0.73% (Table 2). InhA was inhibited at 50 µM in the presence of 1% CMC Tween-20. We were unable to observe the des-nitro JSF-2164 metabolite via LC-MS comparison to a synthetic standard (JSF-2169; Figure S2).
JSF-2164 underwent a rapid metabolism in H37Rv, demonstrated by intrabacterial accumulation over 2 h and followed by a degradation to a negligible level (lower limit of detection = 7.5 pmol) at t ≥ 4h (Figure 5E). Intrabacterial JSF-3616 and JSF-3617 were detectable after 30 min of treatment (with lower limits of detection of 12 and 94 pmol, respectively). Their concentrations increased over the first ca. 2 and 8 h, respectively, and then leveled off. JSF-3617 was a major intrabacterial metabolite judged by a higher accumulation amount as compared to JSF-3616 and the parent JSF-2164. The lack of cofactor F420H2 in the fgd1::tn strain slowed the JSF-2164 intrabacterial metabolism as compared to the H37Rv strain, judged by an extended intrabacterial half-life of JSF-2164 in fgd1::tn than that in H37Rv. Conversion of JSF-2164 into the metabolites JSF-3616 and JSF-3617 was also slowed as compared to the H37Rv strain. JSF-3616 and JSF-3617 were discernible in fgd1::tn after 4 h of treatment with intrabacterial concentrations significantly lower than concentrations in H37Rv at each same time point, judged by the Student’s t test with p < 0.05. This result indicated an F420-dependent intrabacterial metabolism consuming JSF-2164 to produce two amine metabolites with NO· generation in M. tuberculosis (Figure 6).
Figure 6.
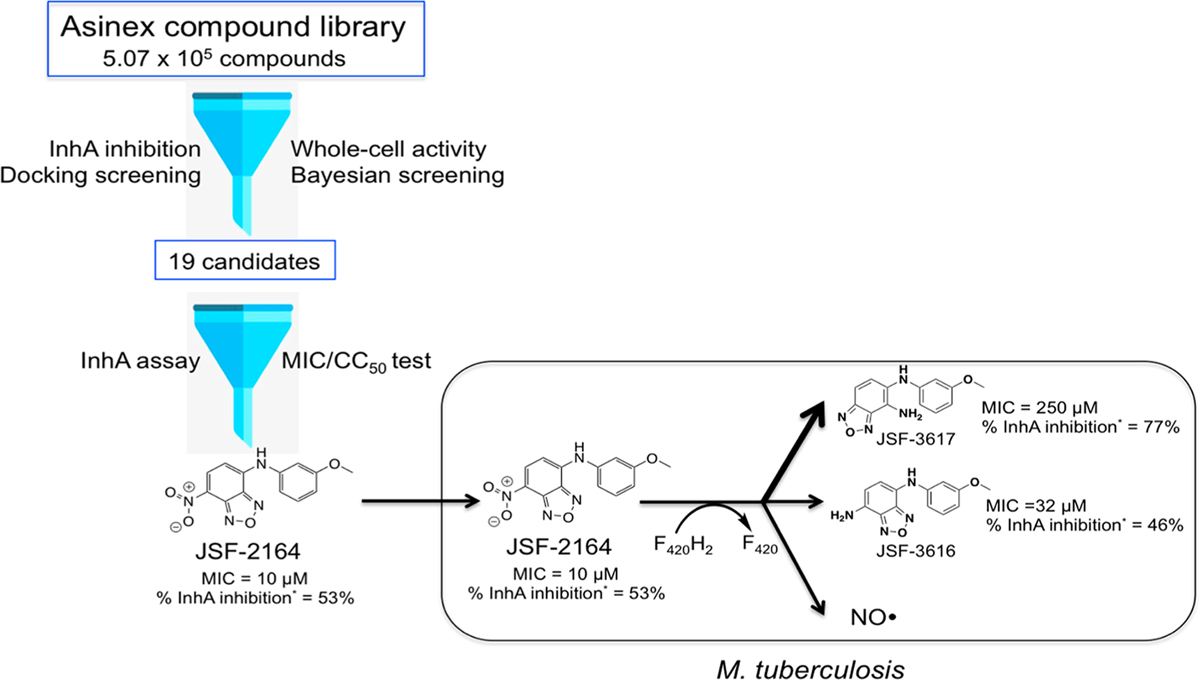
Proposed JSF-2164 intrabacterial metabolism in M. tuberculosis. JSF-2164 was a hit of an in silico Docking/Bayesian screening in search for an InhA inhibitor with antitubercular activity and low cytotoxicity. JSF-2164 suffered an F420H2-dependent metabolism to afford two amine metabolites JSF-3616 and JSF-3617 with NO· production. * InhA inhibition was assessed at 50 µM compound in the presence of 1% CMC Tween-20.
DISCUSSION
Multidrug-resistant TB is a global epidemic crisis that requires new drugs to be added to the current antitubercular arsenal. We hypothesized that an innovative workflow, combining cell-based data via our dual-event Bayesian models and target structural data through high-throughput docking, would offer structurally novel small molecules that inhibit InhA and M. tuberculosis in vitro growth while offering an acceptable lack of Vero cell cytotoxicity. Our initial foray into large-scale virtual screening of commercial chemical space for InhA inhibitors led to two novel fragment-sized enzyme inhibitors of InhA that displayed Ki values of 54 and 59 µM, respectively.8 That previous study used the identical docking program, docking parameters, docking filters, and docking target as the present study, and the same individual (A.L.P.) visually inspected the docking results for both investigations. The most significant difference was the previous docking workflow involved algorithms that knew nothing about the structural and physiochemical properties of small molecules that contribute to antitubercular whole-cell efficacy and lack of relative Vero cell cytotoxicity. While our workflow depended on a Bayesian model component, other machine learning strategies could have been used alternatively. However, we have found that Bayesian models perform at least as well as those derived with Support Vector Machines and Recursive Partitioning.9 Although several previous studies have investigated different ways of combining or comparing docking calculations with Bayesian or SVM models, those hybrid workflows focused on increasing the probability of finding inhibitors of a particular protein target or group of targets (or identifying the target).43–48 They did not involve combining Bayesian models (trained on cell-based data) with docking studies to increase the efficiency of finding whole-cell active, less toxic compounds that could inhibit a particular target within M. tuberculosis. The value of our workflow has been supported by the virtual processing of 5.07 × 105 commercially available small molecules to afford 19 candidates, predicted to exhibit acceptable InhA inhibition (≥50% inhibition at 50 µM compound), an MIC ≤ 10 µg/mL versus M. tuberculosis, and an SI ≥ 10 with respect to Vero cells. One compound, JSF-2164, met the first two criteria and while its Vero cell-based SI of 5.5 (using the most stringent and conservative metrics) did not meet the criterion of 10, it was not prohibitively low (SI ≤ 2) in our opinion. Critically, JSF-2164 represented a novel chemotype for an antitubercular agent inhibiting InhA.
The high-throughput docking/Bayesian workflow was, thus, able to find a small molecule (JSF-2164) with promising InhA inhibition, whole-cell activity, and modest SI versus Vero cells for a virtual screening hit. The hit rate of 2/19 (10.5%) compounds with acceptable antitubercular efficacy is similar to hit rates we have witnessed when implementing Bayesian models in previous ligand-based screens,9,10,49–51 which are more cost- and time-efficient than experimental HTS against M. tuberculosis (hit rates <1%). Additionally, the Bayesian approach removed from consideration whole-cell inactive compounds, as the TAACF-CB2 model’s lowest scoring five candidates all exhibited MIC values between 125 and 250 µM. The two most potent whole-cell actives (JSF-2164 and BAS01237091) were not found when searching the CDD database of antitubercular screening data (www.collaborativedrug.com). This analysis supports the likelihood that optimization efforts with these compounds, or others arising from similar computational screening of different commercial libraries both within and outside of GO FAM, will lead to novel chemical tools. On the topic of chemical similarity, it is also noteworthy that the benzofurazans JSF-2164 and BAS01237091 are dissimilar from the Bayesian training set/s that predicted them to be active as judged by their respective calculated closest distance to the training set (closest distance of identical molecules = 0; JSF-2164: TAACF-CB2 closest distance = 0.55, combined closest distance = 0.54; BAS01237091: combined closest distance = 0.56). It has become clear that our Bayesian models thus far are able to predict actives that are structurally dissimilar to the model’s training set, which supports their general utility when exploring novel areas of chemical space. When scoring a new set of molecules for prospective testing, a Bayesian model gives favorable scores to molecules that contain chemical features recognized among the actives in its training set. Thus, predicted actives may comprise a mixture of training set active molecules’ features that have been “mixed and matched”, as well as additional features that were not part of any training set member (as long as those added features do not have properties that match those of the training set inactives).
A critical assessment of our workflow was tied to the question as to whether JSF-2164 achieved its whole-cell growth inhibition primarily through InhA inhibition. Intrabacterial metabolism is clearly a complicating factor in providing an answer, as this predicted whole-cell active InhA inhibitor has a short lifetime within the bacterium. In addition to producing NO·, the metabolism of JSF-2164 was shown to afford two derivatives (JSF-3617 and JSF-3616), which are less active as growth inhibitors and do not exhibit a loss of efficacy upon inhA overexpression as does JSF-2164 but still maintain modest inhibition of purified InhA. Given the significant consumption of JSF-2164 and production of JSF-3617 within the fgd1::tn strain, we cannot discount the involvement of other genes, despite the results from resistant mutant and transposon insertion experiments, in the production of JSF-2164 metabolites. The potential also exists for metabolites other than those identified to have eluded our LC-MS-based methodology. For the parent molecule and its identified metabolites, some disconnects exist between inhibition of purified InhA and the intracellular hallmarks of InhA inhibition that were assayed. At this level of interrogation, InhA inhibition appears to be a secondary target, but once again, we cannot minimize the role of intrabacterial metabolism in complicating the apparent mechanism of action.
We assert that intrabacterial metabolism must be considered when assessing the connection, or disconnection, between an antitubercular’s interaction with the purified primary protein target and its effect on that protein’s function within the bacterium. Thus, our novel computational workflow may be judged to be successful in that it identified a compound that inhibited purified InhA and achieved significant whole-cell activity. However, neither the docking approach nor the Bayesian modeling was capable of understanding intrabacterial metabolism. It was only comprehended through IBDM studies that were tipped off by the attainment of JSF-2164-resistant mutants that evidenced (1) the engagement of InhA within the cell through the c(−15)t mutation in the inhA promoter region leading to an overexpression of inhA in JSF-2164 spontaneous mutants and (2) the F420H2-based reduction of JSF-2164 to amine metabolites JSF-3617 and JSF-3616 accompanied by the production of NO·. We were unable to observe via LC-MS the presence of the des-nitro JSF-2164 (JSF-2169) that would be formed via loss of NO· in analogy to pretomanid,40 delamanid,41 and JSF-2019.39
Finally, it is significant to note that small molecule nitro-containing heterocycles have been reported as metabolically labile in M. tuberculosis. The recently FDA-approved pretomanid40,52 and EMA-approved delamanid41 are bactericidal in M. tuberculosis due to a release of NO· catalyzed by the nitroreductase Ddn in the presence of the cofactor F420H2. JSF-2164, however, exhibited bacteriostatic activity even with a demonstrated NO· release. We have noted a smaller NO· production amount with JSF-2164 treatment as compared to pretomanid treatment of H37Rv. This quantitative difference in NO· production may play a significant role in determining bactericidal versus bacteriostatic activity. Given the demonstrated need to attain follow-up compounds to pretomanid and delamanid, it is important to better understand nitroheterocycles in terms of their intrabacterial metabolism to afford NO· and other metabolites that have cidal consequences for M. tuberculosis.
CONCLUSIONS
A novel computational workflow combining target-based virtual screening and machine learning from data sets containing whole-cell efficacy and model mammalian cell cytotoxicity has been designed and assessed with regard to M. tuberculosis and InhA. This model successfully identified JSF-2164 from a library of ∼105 compounds as an in vitro InhA inhibitor with whole-cell activity in M. tuberculosis, supporting our goal of further developing and applying this novel hybrid workflow. The F420H2-dependent intrabacterial metabolism, revealed through drug-resistant mutant studies, led to sufficiently rapid biotransformation of JSF-2164 to produce NO· and two amine metabolites that obscured the mechanism of the parent compound. The IBDM platform was crucial to probing this phenomenon. We assert the comprehension of molecule intrabacterial metabolism is critical to investigate the mode of action of antibacterial chemical tools and drug discovery entities.
METHODS
Synthetic Methods.
JSF-2149.
(Synthetic route is depicted in Figure S2.) A mixture of 2-chlorobenzaldehyde (4.5 mL, 40 mmol) and thiosemicarbazide (3.6 g, 40 mmol) in methanol (40 mL) was heated at 70 °C for 3 h. The reaction mixture was cooled in an ice−water bath, and the precipitate formed was filtered to give intermediate 1 (7.1 g, 33 mmol, 83%) as a white powder that was used without characterization. To 1 (4.17 g, 19.5 mmol) in methanol (30 mL) was added an aqueous solution of ferric chloride (9.48 g, 71.0 mmol in 13 mL of H2O). The reaction mixture was heated at 110 °C for 2 h and filtered hot through a plug of activated carbon (4.0 g). To the filtrate, an aqueous solution of citric acid (11.52 g) and sodium citrate (6.42 g) in 40 mL of water was added, and the solution was refluxed for 1 h with stirring. After, the solution was neutralized with 10% aqueous ammonia to pH 4−5 and cooled in an ice−water bath. A precipitate was formed and washed several times with water to give intermediate 2 (2.36 g, 11.5 mmol, 59.0%) as a white powder: 1H NMR (600 MHz, DMSO-d6) δ 8.00 (dd, J = 7.2, 2.1 Hz, 1), 7.65−7.57 (m, 1), 7.51−7.45 (m, 2), 7.44 (s, 2).
To 2-norbornaneacetic acid (0.87 mL, 6.0 mmol) in 30 mL of dichloromethane was added three drops of DMF and oxalyl chloride (0.57 mL, 6.6 mmol). The reaction mixture was stirred at room temperature for 3 h, and then, the solvent was removed to provide a brown liquid. The crude material was diluted in 6.5 mL of dichloromethane to make a theoretically 0.92 M solution of 2-norbornaneacetyl chloride. To intermediate 2 (211 mg, 0.997 mmol) in 5% aqueous NaOH (1.5 mL) was added 2-norbornaneacetyl chloride (1.1 mL, 1.0 mmol, 0.92 M solution in dichloromethane). The reaction mixture was stirred at room temperature for 30 min, diluted in dichloromethane, washed with 1 N aqueous NaOH and saturated aqueous brine solution, and dried over anhydrous sodium sulfate. The mixture was then filtered and concentrated to afford a solid that was crystallized from dichloromethane to give JSF-2149 (210 mg, 0.56 mmol, 56%) as white needles: 1H NMR (500 MHz, DMSO-d6) δ 8.08 (d, J = 7.6 Hz, 1), 7.67 (d, J = 7.8 Hz, 1), 7.58−7.48 (m, 2), 2.48−2.42 (m, 1), 2.35 (dd, J = 14.6, 7.6 Hz, 1), 2.19 (s, 1), 1.97 (s, 1), 1.94−1.86 (m, 1), 1.52−1.39 (m, 3), 1.35 (d, J = 9.7 Hz, 1), 1.20−1.03 (m, 4). Also noted a minor peak at 3.3 (br s, H2O). 13C NMR (126 MHz, DMSO-d6) δ 171.5, 160.4, 158.1, 132.2, 131.5, 131.3, 131.1, 129.5, 128.3, 42.1, 40.9, 38.8, 37.5, 36.6, 35.3, 29.8, 28.7. ESI-HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H19ClN3OS 348.0932; found 348.0943.
JSF-2164.
(Synthetic routes for this compound and its below candidate metabolites are depicted in Figure S3.) To 4-chloro-7-nitrobenzo[c][1,2,5]oxadiazole (0.347 g, 1.70 mmol) in methanol (7 mL) at rt was added 3-methoxyaniline (0.19 mL, 1.7 mmol). The mixture was heated to 60 °C for 3 h, and the solvent was removed under a stream of nitrogen. The resulting solid was chromatographed on silica gel with a 0% to 80% ethyl acetate/hexane gradient. After concentrating fractions, the product was isolated as a brick-red solid (0.307 g, 1.07 mmol, 62.9%): 1H NMR (600 MHz, acetone-d6) δ 9.76 (s, 1), 8.55 (d, J = 8.7 Hz, 1), 7.43 (t, J = 8.1 Hz, 1), 7.16 (ddd, J = 6.0, 3.6, 1.5 Hz, 2), 6.93 (dd, J = 8.0, 4.7 Hz, 2), 3.85 (s, 3). Also noted minor peaks at 6.1 (m), 6.2 (m), 2.8 (s, H2O), 2.8 (s, H2O), 0.75 (s). 13C NMR (126 MHz, DMSO-d6) δ 160.1, 145.0, 144.2, 142.1, 138.9, 137.7, 130.4, 123.2, 115.8, 111.9, 109.5, 102.1, 55.3. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C13H10N4NaO4 309.0600; found 309.0607.
JSF-2169.
A microwave vial was charged with benzo[c]-[1,2,5]oxadiazol-4-amine (0.1174 g, 0.8427 mmol) and cesium carbonate (0.70 g, 2.1 mmol) and flushed with nitrogen. To this mixture was added dioxane (2 mL) and 1-bromo-3-methoxybenzene (0.13 mL, 1.0 mmol), and again, the reaction mixture was flushed with nitrogen. The mixture was subjected to the addition of 2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl (XPhos) (63.5 mg, 0.133 mmol) and tris-(dibenzylideneacetone) dipalladium(0) (25 mg, 0.043 mmol) and was then irradiated in a microwave reactor (120 °C, 30 min). After cooling, the reaction mixture was assayed via LC-MS to demonstrate the presence of product and a small amount of starting amine. The mixture was filtered, concentrated, and chromatographed on silica gel with a 0% to 50% ethyl acetate/hexane gradient. After concentrating fractions, the product was isolated as a red-orange crystalline solid (0.161 g, 0.667 mmol, 79.1%): 1H NMR (600 MHz, acetone-d6) δ 8.20 (s, 1), 7.41 (dd, J = 18.6, 10.0 Hz, 1), 7.29 (t, J = 8.1 Hz, 1), 7.22 (dd, J = 9.0, 0.4 Hz, 1), 7.06−6.96 (m, 3), 6.72−6.62 (m, 1), 3.80 (d, J = 0.9 Hz, 3). Also noted minor peaks at 7.72 (m), 2.85 (s, H2O), 2.83 (s, H2O), 0.80 (s). 13C NMR (126 MHz, DMSO-d6) δ 145.4, 142.0, 134.7, 132.5, 129.9, 112.6, 108.2, 106.3, 103.7, 55.0. LRMS (ES+) m/z: [M + H]+ Calcd for C13H12N3O2 242.1; found 242.0.
JSF-3616.
A solution of JSF-2164 (100 mg, 0.350 mmol) in DCM (12.0 mL) and MeOH (5.00 mL) was subjected to the addition of iron powder (133 mg, 2.38 mmol) and concentrated aqueous HCl (0.750 mL, 7.50 mmol). The reaction mixture was stirred at room temperature overnight, quenched with 1 N aqueous NaHCO3 (15.0 mL) carefully, and extracted with ethyl acetate (2 × 40 mL). The pooled organic extracts were washed with saturated aqueous brine solution (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude material was subjected to flash column chromatography over silica gel, eluted with 0−10% methanol in DCM with 0.1% triethylamine, to afford JSF-3616 as a dark red solid (27.0 mg, 0.105 mmol, 30.0% yield): 1H NMR (500 MHz, CDCl3) δ 7.24 (t, J = 8.1 Hz, 1), 6.93 (d, J = 7.7 Hz, 1), 6.77−6.72 (m, 1), 6.71 (t, J = 2.1 Hz, 1), 6.56 (dd, J = 8.2, 1.7 Hz, 1), 6.34 (d, J = 7.7 Hz, 1), 6.19 (s, 1), 4.27 (br s, 2), 3.82 (s, 3). Also noted minor peaks at 5.3 (s), 5.1 (s), 3.4 (s), 2.4 (t), 2.1 (s), 2.0 (s), 1.2 (s), 0.8 (t). 13C NMR (126 MHz, CDCl3) δ 160.7, 146.5, 145.4, 143.3, 130.2, 127.6, 122.3, 112.9, 110.7, 109.1, 106.8, 104.1, 55.3. LRMS (ES+) m/z: [M + H]+ Calcd for C13H13N4O2 257.1; found 257.0. ESI-HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C13H13N4O2 257.1030; found 257.1026.
JSF-3617.
A solution of 5-chloro-4-nitrobenzo[c][1,2,5]-oxadiazole (100 mg, 0.500 mmol) in DMF (2.0 mL) was subjected to the addition of m-anisidine (56.0 µL, 0.500 mmol) and Cs2CO3 (163 mg, 0.500 mmol). The reaction mixture was stirred at 60 °C overnight, was subjected to the addition of water (20 mL), and was extracted with ethyl acetate (2 × 40 mL). The pooled organic extracts were washed with saturated aqueous brine solution (20 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude material was subjected to flash column chromatography over silica gel, eluted with DCM, to afford N-(3-methoxyphenyl)-4-nitrobenzo[c][1,2,5]-oxadiazol-5-amine as a yellow solid (45.0 mg, 0.161 mmol, 32.2% yield): 1H NMR (500 MHz, DMSO-d6) δ 11.7 (s, 1), 8.13 (d, J = 9.9 Hz, 1), 7.47 (t, J = 8.1 Hz, 1), 7.33 (d, J = 9.9 Hz, 1), 7.09 (s, 1), 7.07−6.99 (m, 2), 3.81 (s, 3). LRMS (ES+) m/z: [M + H]+ Calcd for C13H11N4O4 287.1; found 287.0.
To a solution of N-(3-methoxyphenyl)-4-nitrobenzo[c]-[1,2,5]oxadiazol-5-amine (15.0 mg, 0.0524 mmol) in DCM (1.2 mL) and MeOH (0.50 mL) was added iron powder (32.0 mg, 0.571 mmol) and concentrated aqueous HCl (0.10 mL). The reaction mixture was stirred at room temperature overnight. The reaction mixture was subjected to careful addition of 1 N NaHCO3 (2.0 mL) and was extracted with ethyl acetate (2 × 20 mL). The pooled organic extracts were washed with saturated aqueous brine solution (10 mL), dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude reaction material was subjected to flash column chromatography over silica gel, eluted with 0−10% methanol in DCM with 0.1% triethylamine, to afford JSF-3617 as dark red solid (6.6 mg, 0.026 mmol, 49% yield): 1H NMR (500 MHz, DMSO-d6) δ 7.50 (s, 1), 7.33 (d, J = 9.3 Hz, 1), 7.13 (d, J = 9.3 Hz, 1), 7.07 (t, J = 8.1 Hz, 1), 6.36−6.31 (m, 2), 6.28 (t, J = 2.1 Hz, 1), 5.81 (s, 2), 3.68 (s, 3). 13C NMR (126 MHz, DMSO-d6) δ 160.8, 148.6, 146.7, 145.9, 134.6, 130.3, 126.8, 120.5, 108.1, 104.5, 101.9, 101.3, 55.3. ESI-HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C13H13N4O2 257.1030; found 257.1027.
Structure-Based Virtual Screening.
Docking calculations were performed on the Asinex library of 507 486 compounds against the PT70-bound crystal structure of InhA (PDB ID: 2X23)15 using AutoDock Vina,16 as part of the Global Online Fight Against Malaria project (GO Fight Against Malaria, or GO FAM; http://GOFightAgainstMalaria.scripps.edu).8 The 3-D “mol2” models of the 5.07 × 105 small molecules in the Asinex library were obtained from the ZINC server.53 The methods used to prepare and reformat all of the models of the small molecules used in virtual screens on GO FAM were discussed in our recent paper.8 The pdbqt version of the Asinex library compounds, and of all 5.6 million compounds screened in the GO FAM experiments, is available on request from A.L.P.
These docking calculations focused on the (PDB ID 2X23)15 crystal structure of InhA, because it is bound to PT70, an InhA inhibitor with a Ki = 7.8 nM and a long residence time of 24 min. The MolProbity server54 was used to add the hydrogen atoms to the model of the InhA target. The pdbqt docking format for the target model was then prepared using AutoDockTools 4.2,55 which added the Gasteiger-Marsili charges,56 merged the nonpolar hydrogen atoms onto their respective heavy atoms, and assigned the AutoDock atom types. All crystallographic waters, counterions, and buffer molecules were removed prior to the docking calculations. The “grid box” (the region the small molecules are allowed to explore during the docking calculations) was centered between the N9 atom of adenine and the C1 atom of the adjacent ribose in the NAD cofactor, and it had a size of 30 × 30 × 30 Å3. Since it was more efficient to select a single grid box for all of the different InhA and Pf ENR (Plasmodium falciparum enoyl acyl-carrier protein reductase) targets that were part of the GO FAM experiment, this location and large size of the grid box were chosen. Since a large grid box was involved in these calculations, the “exhaustiveness” setting in AutoDock Vina was increased to 20 (from the default value of 8) to ensure sufficient sampling of the conformations, locations, and orientations of the small molecules during the docking process.
The docking score (estimated free energy of binding) and the number and types of energetically favorable interactions predicted to occur between each small molecule in the Asinex library and InhA were characterized by Python scripts from the Raccoon2 and Fox software, using established protocols.6,57 On the basis of our analysis of the crystal structures of several of the noncovalent, nanomolar inhibitors of InhA,15,22 we initially filtered the docked results to harvest small molecule compounds that were predicted to form similar quaternary interactions (base-stacking with the NAD+/NADH cofactor and at least two hydrogen bonds to the active site of InhA). In addition to these interaction-based docking filters, the top-ranked docking mode of each small molecule also had to display an estimated free energy of binding (FEB) ≤ −8.0 kcal/mol; see Figure 1. This first stage of docking-based filters narrowed down the 5.07 × 106 small molecules in the Asinex library to 370 compounds. These 370 compounds then underwent a second stage of filtering, using the dual-event Bayesian scores.
Filtering the Docked Results with Machine Learning Methods.
Recently, we developed and validated “dual-event” Bayesian models (trained with both M. tuberculosis MIC data and Vero cell cytotoxicity data) that allowed the prediction of the relative likelihood that a compound would display antitubercular activity and lack cytotoxicity to mammalian cells.9,10,49–51 These machine learning models were trained using the following “molecular descriptors” that characterize different aspects of the small molecules: molecular weight, AlogP, FCFP_6 (molecular function class fingerprints of a maximum diameter of 6, which describe topology), number of rotatable bonds, number of aromatic rings, number of all rings, number of hydrogen bond acceptors, number of hydrogen bond donors, and the molecular fractional polar surface area. These descriptors were calculated, and the dual-event Bayesian models were built using Pipeline Pilot and Discovery Studio (BIOVIA Inc., Discovery Studio Modeling Environment, 2013), which were also used to calculate the Bayesian scores on the new, untested compounds from the Asinex library.
Two different dual-event Bayesian models that had performed well in prior validation studies were both utilized to independently filter the docking results to enable a comparison of their hit rates against both InhA and M. tuberculosis: the combined TB dual-event Bayesian model9 and the TAACF-CB2 dual-event Bayesian model.10 The 370 Asinex compounds that passed the docking filters were scored with both the combined TB model and the TAACF-CB2 model to harvest two different sets of compounds that (a) docked well to InhA and (b) had a reasonable likelihood of displaying whole-cell activity against in vitro cultured M. tuberculosis and a relative lack of cytotoxicity to mammalian cells. This docking and Bayesian approach harvested 52 compounds when using the combined TB dual-event Bayesian model, and the top 100 compounds were chosen using the TAACF-CB2 Bayesian model (which included 21 compounds that were also present in the combined TB set). The docked binding modes of these 131 compounds were then visually inspected.
Visual Inspection of the Subset That Passed Both Docking-Based Filters and Dual-Event Bayesian Filters.
Visual inspection is a vague and subjective process, but having experts in macromolecular recognition who also understand the strengths and weaknesses of different modeling approaches view, measure, and judge/prioritize the docked results is an important step. Using expert human knowledge (“in cerebro” quality control) to select which computer-aided (“in silico”) predictions seem best is a strategy supported by previous blind docking challenges, such as SAMPL2 and SAMPL4.58 Our visual inspection process incorporated multiple criteria, in an attempt to decrease the number of false positives that often result from virtual screens. We tended to reject compounds that had (a) one or more large hydrophobic group/s exposed to solvent, (b) more than one polar group packed in hydrophobic pockets (i.e., unless they had a complementary group from the target in the vicinity), (c) docked modes that displayed significantly distorted geometries, or (d) more than one unfavorable electrostatic repulsion interaction with charged or polar groups in the target. We tried to select/accept efficient ligands, according to both their computed ligand efficiencies from AutoDock Vina and the observation that (a) most of their heteroatoms were involved in hydrogen bonds, favorable electrostatic interactions, or halogen bonds with the target, (b) the hydrophobic groups packed well with nonpolar regions of the target, and (c) aromatic rings displayed π−π (base-stacking or T-stacking) or π−cation interactions with the target (as described in the results). When compounds were predicted to display strong interactions with well-conserved features of the target (such as invariant residues, backbone atoms, or the cofactor), they were given extra consideration.
In Vitro Growth Inhibition Assay with M. tuberculosis.
MIC values of the compounds were determined following the microplate-based Alamar Blue assay (MABA) method as previously described.59 50 mM compound stock solutions in DMSO were dissolved in sterile Middlebrook 7H9-OADC broth to afford a 1 mM pretest solution. 100 µL of pretest solution was added into the wells in column 1 of a sterile polystyrene 96-well round-bottom plate (CLS3795, Corning, NY). Wells in columns 2−12 received 50 µL of sterile 7H9-OADC broth. Serial 2-fold dilutions of compounds were performed, and column 12 was used as a drug-free (inoculum-only) control. Final concentrations of compounds were as follows: for INH, 0.012 to 25 µM; for all candidate InhA inhibitors, 0.50 to 500 µM. M. tuberculosis wild type strain H37Rv and inhA overexpression strain mc2491413 were 1:1000 diluted in 7H9+OADC medium at the mid-logarithmic stage of growth (OD595 nm= 0.4). 50 µL of diluted bacteria suspensions was inoculated into each well. Plates were sealed with Parafilm and incubated at 37 °C for 7 days. Twenty microliters of alamarBlue reagent (Invitrogen, Frederick, MD) freshly mixed with 12.5 µL of 20% Tween-80 was added into each well, followed by 24 h of incubation at 37 °C. Absorbance was read at 570 nm, with a reference wavelength of 600 nm, using a microplate reader (ELX808, Biotek Instruments). The MIC end point was defined as the lowest concentration of the test agent that produced at least 90% reduction in absorbance compared with that of the drug-free control.
Determination of InhA Percent Inhibition and IC50.
Enzyme kinetics, following a published procedure,60 were determined in pH 6.8 PIPES buffer supplemented with or without 0.01 CMC Tween-20 (0.6 mg/L) via a Shimadzu UV-2550 UV−visible spectrophotometer. NADH (20 µM), InhA (20 nM; expressed and purified following a literature protocol17), and inhibitor with different concentrations dissolved in DMSO were mixed and incubated for 5 min, followed by addition of trans-2-dodecenoyl-CoA (prepared from trans-2-dodecenoic acid via a mixed anhydride method as described by Parikh et al.61) to initiate the reaction. Assays were followed spectrophotometrically at 25 °C, tracking the consumption of NADH, absorbed at 340 nm, to form NAD+. For the determination of a molecule’s percent inhibition of InhA catalytic activity, the inhibitor concentration was chosen to be 50 µM. For IC50 determination, inhibitor concentrations were varied from 10 nM to 100 µM. Equation 1 was utilized to determine the NAD+ production curve where NNAD+ was the NAD+ concentration, A0 and At were the 340 nm absorbances at time 0 and time t, and CNADH was the initial NADH concentration.
| (1) |
The InhA percent inhibition and IC50 for each inhibitor were calculated with Prism GraphPad version 6.0 and reported as the mean ± standard error. Aggregator formation was experimentally defined as the observation of a significant IC50 shift between assays with and without Tween-20 (p < 0.05).
Minimum Bactericidal Concentration (MBC) Assay.
The bacterial cultures from the 96-well plates utilized for the compound MIC determination instead of undergoing Alamar Blue addition were resuspended, serially diluted with the sterile 7H9 media, and plated on Middlebrook 7H10 plates. Colony-forming units (CFUs) were enumerated following 21 days of incubation at 37 °C. The MBC was reported as the minimum compound concentration at which a 2 log10 reduction in CFUs was observed as compared to the no-compound control.
Cytotoxicity Assay.
The assay was performed on Vero cells as described previously.62
M. tuberculosis Spontaneous Mutagenesis.
Mid-log phase (OD595 = 0.4) M. tuberculosis was harvested and concentrated to ∼109 CFU/mL in fresh 7H9 + ADS followed by CFU enumeration by streaking on the 7H11 agar plates supplemented with OADC. Compound JSF-2164 was supplemented into 7H10 agar plates with 10% (v/v) ADS and 0.5% glycerol at MICs ranging from 4× to 32×. 100 µL of culture (108 CFU) was streaked on JSF-2164 containing agar plates followed by incubation at 37 °C for 4 to 6 weeks. Single colonies were isolated followed by the liquid MIC test for resistance verification. For resistant colonies of interest, genomic DNA was isolated. For resistant colonies of interest, genomic DNA was isolated.63 Genomic libraries were prepared using the Nextera XP Library Prep Kit (Illumina, San Diego, CA) prior to sequencing on the Illumina NextSeq Platform (Illumina, San Diego, CA). Sequence reads were aligned to reference strain H37Rv (AL123456.3) and analyzed for single nucleotide polymorphisms (SNPs) using the SNPTB platform.64 Whole-genome sequencing data for the M. tuberculosis mutants in this paper have been deposited in the NCBI Sequence Read Archive (SRA) with access code PRJNA544744.
Transposon Insertion Locus Determination.
106 CFU of the saturated Himar-1 transposon strain library at the mid-log phase (OD595 = 0.4) were streaked on 7H11 + OADC agar plates supplemented with JSF-2164 concentrations ranging from 1 to 30× MIC. After a 5 week incubation at 37 °C, single colonies were isolated, followed by resistance verification via the liquid MIC test. Genomic DNA from the colonies were extracted, followed by a restrict digestion catalyzed by SacII and a self-ligation mediated by T4 ligase. The digestion and ligation generated plasmids containing the kanamycin resistance cassette and R6Kγ ori site of the MycoMar transposon element plus flanking chromosomal DNA.34 The plasmids were transformed into the E. coli DH5α-pir116 strain (Lucigen, Cat. #60602) containing the pir gene,65 followed by kanamycin selection. Plasmids were isolated from the kanamycin-resistant E. coli followed by Sanger sequencing using the primer annealing to the T7 promoters (primer = 5′-CGCTTCCTCGTGCTTT-ACGGTATCG-3′). The Sanger sequencing results were aligned to the H37Rv genome (NCBI taxid: 83332) to determine the insertion locus.
TLC Assay for Inhibition of M. tuberculosis Fatty Acid Biosynthesis.
For the analysis of mycolic acids and fatty acids,62 5 mL of M. tuberculosis culture (OD595 = 0.4) was pretreated with an antitubercular agent and incubated at 37 °C for 4 h. Radiolabeling of M. tuberculosis lipids with 14C-acetate was carried out via addition of the sodium salt of [1,2-14C] acetic acid (PerkinElmer Inc.) at a final concentration of 2 µCi/mL. Following incubation at 37 °C for another 4 h, the 14C-labeled cells were harvested and washed with PBS. The lipids from the cell pellet were hydrolyzed overnight by treatment with 2 mL of tetra-n-butylammonium hydroxide at 100 °C. Fatty acids were esterified by adding 300 µL of iodomethane in 4 mL of CH2Cl2 and 2 mL of distilled water, followed by mixing at rt for 1 h. After centrifugation, the lower phase was washed with distilled/deionized water and air-dried. The resulting sample was resuspended in 3 mL of diethyl ether, and the organic phase was separated after centrifugation and air-dried. Lipids were resuspended in 200 µL of CH2Cl2. Equal counts of samples (40 000 c.p.m.) were loaded on a silica gel 60 F254 thin-layer chromatography (TLC) plate and resolved using hexane/ethyl acetate (19:1, v/v). The fatty acid methyl esters (FAMEs) and mycolic acid methyl esters (MAMEs) were detected by autoradiography.
Griess Assay for NO· Quantification.
Griess reagent (Invitrogen, G7921) was applied to quantify NO· release from the M. tuberculosis strain in the presence of compound. M. tuberculosis strains were grown to mid-log phase (OD595 = 0.3) followed by compound treatment. At each time point, the supernatant of the culture aliquot was added to a 96-well plate in triplicate (200 µL/well). Twenty microliters of Griess reagent mix was added into each well followed by incubation at rt for 30 min. Results were read at an absorbance of 548 nm, and sample nitrite concentration was converted from the absorbance reading according to a nitrite calibration curve.
Intrabacterial Drug Metabolism (IBDM) Assay.
An M. tuberculosis culture was grown to mid-log phase (OD595= 0.6; ∼2 × 108 CFU/mL) in 7H9 + ADS + tyloxapol for each compound treatment at the desired concentration. After 24 h, the cell pellet was harvested by centrifugation (10 000 rpm for 10 min at 4 °C). The washed pellet was quenched with CH3CN/CH3OH/water (2:2:1) prechilled on dry ice, followed by lysis via bead beating (6.5 m/s, 30 s, 6 times). In between each round of bead beating, the sample was chilled on ice for 2 min to avoid overheating. The pellet metabolite extract in organic solvent was collected via a 0.22 µm filter. After a proper decontamination process, the pellet extract was brought out of the BSL-3 for further analysis.
We typically applied an Agilent 1260 HPLC coupled with an Agilent 6120 quadrupole mass spectrum for metabolite analysis. Verapamil as an internal standard was added into each sample to a final concentration of 1 µg/mL. Metabolites were separated on a Chromolith SpeedRod column with a gradient of H2O and acetonitrile acidified with 0.1% formic acid. The mass resolution ranged from 10 to 2000 with an accuracy of ±0.13 Da within the calibrated mass range in scan mode. After extraction and purification of a certain metabolite from cell lysate, its structure was confirmed by comparison of its 1H NMR spectrum (Bruker Avance 500 MHz spectrometer), LC-MS spectrum (Agilent 6120 single quadrupole LC-MS system), and high-resolution mass spectrum (Agilent 6220 accurate-mass time-of-flight) with those for an authentic/independently synthesized sample. Signal intensity was quantified by a standard curve for an authentic/independently synthesized sample and normalized by sample protein concentration determined via the Pierce BCA protein assay kit.
Supplementary Material
Dose−response curves for InhA inhibition for select compounds; synthetic schemes for JSF-2149 and JSF-2164 and its candidate metabolites; RIF in vitro activity versus the ss18b strain; inhA promoter mutation in 16 × 6 resulting in inhA overexpression; LC-MS data supporting identification of JSF-3616 and JSF-3617 as intrabacterial metabolites of JSF-2164; antitubercular activity of the 5 lowest-scoring compounds among the 370 candidates passing docking filters; profiling of select spontaneous JSF-2164-resistant mutants (PDF)
Table S1: 370 compounds that passed the initial docking filters (XLSX)
Table S4: MIC values for JSF-2164, INH, and pretomanid versus JSF-2164-resistant transposon mutants (XLSX)
ACKNOWLEDGMENTS
This work was supported in part by NIH grants DP2OD008459-01S1 and R44TR000942. Professor Bill Jacobs (Albert Einstein College of Medicine) is acknowledged for the generous gift of the M. tuberculosis strain mc24914. The virtual screening campaigns on GO FAM were supported by the IBM International Foundation and by the volunteers in the public who donated their spare computational power to run the docking calculations. We thank Dassault Systemes for providing S.E. and J.S.F. with Discovery Studio and Pipeline Pilot. We thank Professor Christopher Sassetti for the gift of the saturated Himar-1 transposon insertion library with the background of H37Rv.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.9b00295.
The authors declare no competing financial interest.
REFERENCES
- (1).Fauci AS, and Marston HD (2014) The perpetual challenge of antimicrobial resistance. JAMA 311, 1853–1854. [DOI] [PubMed] [Google Scholar]
- (2).Workman P, and Collins I (2010) Probing the probes: fitness factors for small molecule tools. Chem. Biol 17, 561–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Griffin JE, Gawronski JD, Dejesus MA, Ioerger TR, Akerley BJ, and Sassetti CM (2011) High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog 7, No. e1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Wei JR, Krishnamoorthy V, Murphy K, Kim JH, Schnappinger D, Alber T, Sassetti CM, Rhee KY, and Rubin EJ (2011) Depletion of antibiotic targets has widely varying effects on growth. Proc. Natl. Acad. Sci. U. S. A 108, 4176–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Koul A, Arnoult E, Lounis N, Guillemont J, and Andries K (2011) The challenge of new drug discovery for tuberculosis. Nature 469, 483–490. [DOI] [PubMed] [Google Scholar]
- (6).Cosconati S, Forli S, Perryman AL, Harris R, Goodsell DS, and Olson AJ (2010) Virtual Screening with AutoDock: Theory and Practice. Expert Opin. Drug Discovery 5, 597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lechartier B, Rybniker J, Zumla A, and Cole ST (2014) Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol. Med 6, 158–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Perryman AL, Yu W, Wang X, Ekins S, Forli S, Li SG, Freundlich JS, Tonge PJ, and Olson AJ (2015) A virtual screen discovers novel, fragment-sized inhibitors of Mycobacterium tuberculosis InhA. J. Chem. Inf. Model 55, 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Ekins S, Freundlich JS, and Reynolds RC (2013) Fusing dual-event datasets for Mycobacterium tuberculosis machine learning models and their evaluation. J. Chem. Inf. Model 53, 3054–3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ekins S, Reynolds RC, Kim H, Koo M-S, Ekonomidis M, Talaue M, Paget SD, Woolhiser LK, Lenaerts A, Bunin BA, Connell N, and Freundlich JS (2013) Bayesian models leveraging bioactivity and cytotoxicity information for drug discovery. Chem. Biol 20, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sassetti CM, Boyd DH, and Rubin EJ (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol 48, 77–84. [DOI] [PubMed] [Google Scholar]
- (12).Sassetti CM, Boyd DH, and Rubin EJ (2001) Comprehensive identification of conditionally essential genes in mycobacteria. Proc. Natl. Acad. Sci. U. S. A 98, 12712–12717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Vilcheze C, Wang F, Arai M, Hazbon MH, Colangeli R, Kremer L, Weisbrod TR, Alland D, Sacchettini JC, and Jacobs WR Jr. (2006) Transfer of a point mutation in Mycobacterium tuberculosis inhA resolves the target of isoniazid. Nat. Med 12, 1027–1029. [DOI] [PubMed] [Google Scholar]
- (14).Dessen A, Quemard A, Blanchard JS, Jacobs WR Jr., and Sacchettini JC (1995) Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis. Science 267, 1638–1641. [DOI] [PubMed] [Google Scholar]
- (15).Luckner SR, Liu N, am Ende CW, Tonge PJ, and Kisker C (2010) A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis. J. Biol. Chem 285, 14330–14337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Trott O, and Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Stec J, Vilcheze C, Lun S, Perryman AL, Wang X, Freundlich JS, Bishai W, Jacobs WR Jr., and Kozikowski AP (2014) Biological evaluation of potent triclosan-derived inhibitors of the enoyl-acyl carrier protein reductase InhA in drug-sensitive and drug-resistant strains of Mycobacterium tuberculosis. ChemMedChem 9, 2528–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Donoso-Tauda O, Jaque P, Elguero J, and Alkorta I (2014) Traditional and ion-pair halogen-bonded complexes between chlorine and bromine derivatives and a nitrogen-heterocyclic carbene. J. Phys. Chem. A 118, 9552–9560. [DOI] [PubMed] [Google Scholar]
- (19).Lu Y, Liu Y, Xu Z, Li H, Liu H, and Zhu W (2012) Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug Discovery 7, 375–383. [DOI] [PubMed] [Google Scholar]
- (20).Durrant JD, and McCammon JA (2011) BINANA: a novel algorithm for ligand-binding characterization. J. Mol. Graphics Modell 29, 888–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Deng N, Forli S, He P, Perryman A, Wickstrom L, Vijayan RS, Tiefenbrunn T, Stout D, Gallicchio E, Olson AJ, and Levy RM (2015) Distinguishing Binders from False Positives by Free Energy Calculations: Fragment Screening Against the Flap Site of HIV Protease. J. Phys. Chem. B 119, 976–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Freundlich JS, Wang F, Vilchez̀e C, Gulten G, Langley R, Schiehser GA, Jacobus DP, Jacobs WRJ, and Sacchettini JC (2009) Triclosan Derivatives: Towards Potent Inhibitors of Drug-Sensitive and Drug-Resistant Mycobacterium tuberculosis. ChemMedChem 4, 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Rebolledo CL, Sotelo-Hitschfeld P, Brauchi S, and Olavarria MZ (2013) Design and synthesis of conformationally restricted capsaicin analogues based in the 1, 3, 4-thiadiazole heterocycle reveal a novel family of transient receptor potential vanilloid 1 (TRPV1) antagonists. Eur. J. Med. Chem 66, 193–203. [DOI] [PubMed] [Google Scholar]
- (24).Wang K, Yang L, Zhao C, and Ma H (2013) 4-(8-Quinolyl)amino-7-nitro-2,1,3-benzoxadiazole as a new colorimetric probe for rapid and visual detection of Hg2+. Spectrochim. Acta, Part A 105, 29–33. [DOI] [PubMed] [Google Scholar]
- (25).Shoichet BK (2006) Interpreting steep dose-response curves in early inhibitor discovery. J. Med. Chem 49, 7274–7277. [DOI] [PubMed] [Google Scholar]
- (26).Inoyama D, Paget SD, Russo R, Kandasamy S, Kumar P, Singleton E, Occi J, Tuckman M, Zimmerman MD, Ho HP, Perryman AL, Dartois V, Connell N, and Freundlich JS (2018) Novel Pyrimidines as Antitubercular Agents. Antimicrob. Agents Chemother 62, No. e02063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Xie Z, Siddiqi N, and Rubin EJ (2005) Differential antibiotic susceptibilities of starved Mycobacterium tuberculosis isolates. Antimicrob. Agents Chemother 49, 4778–4780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zhang M, Sala C, Hartkoorn RC, Dhar N, Mendoza-Losana A, and Cole ST (2012) Streptomycin-starved Mycobacterium tuberculosis 18b, a drug discovery tool for latent tuberculosis. Antimicrob. Agents Chemother 56, 5782–5789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Haver HL, Chua A, Ghode P, Lakshminarayana SB, Singhal A, Mathema B, Wintjens R, and Bifani P (2015) Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother 59, 5316–5323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).DeJesus MA, Gerrick ER, Xu W, Park SW, Long JE, Boutte CC, Rubin EJ, Schnappinger D, Ehrt S, Fortune SM, Sassetti CM, and Ioerger TR (2017) Comprehensive Essentiality Analysis of the Mycobacterium tuberculosis Genome via Saturating Transposon Mutagenesis. mBio 8, e02133–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Choi KP, Kendrick N, and Daniels L (2002) Demonstration that fbiC is required by Mycobacterium bovis BCG for coenzyme F(420) and FO biosynthesis. J. Bacteriol 184, 2420–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hazbon MH, Brimacombe M, Bobadilla del Valle M, Cavatore M, Guerrero MI, Varma-Basil M, Billman-Jacobe H, Lavender C, Fyfe J, Garcia-Garcia L, Leon CI, Bose M, Chaves F, Murray M, Eisenach KD, Sifuentes-Osornio J, Cave MD, Ponce de Leon A, and Alland D (2006) Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother 50, 2640–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Muller B, Streicher EM, Hoek KG, Tait M, Trollip A, Bosman ME, Coetzee GJ, Chabula-Nxiweni EM, Hoosain E, Gey van Pittius NC, Victor TC, van Helden PD, and Warren RM (2011) inhA promoter mutations: a gateway to extensively drug-resistant tuberculosis in South Africa? Int. J. Tuberc Lung Dis 15, 344–351. [PubMed] [Google Scholar]
- (34).Siegrist MS, and Rubin EJ (2009) Phage transposon mutagenesis. Methods Mol. Biol 465, 311–323. [DOI] [PubMed] [Google Scholar]
- (35).Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE 3rd, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S, Hamlin N, Holroyd S, Hornsby T, Jagels K, Krogh A, McLean J, Moule S, Murphy L, Oliver K, Osborne J, Quail MA, Rajandream MA, Rogers J, Rutter S, Seeger K, Skelton J, Squares R, Squares S, Sulston JE, Taylor K, Whitehead S, and Barrell BG (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544. [DOI] [PubMed] [Google Scholar]
- (36).Filomeni G, Turella P, Dupuis ML, Forini O, Ciriolo MR, Cianfriglia M, Pezzola S, Federici G, and Caccuri AM (2008) 6-(7-Nitro-2,1,3-benzoxadiazol-4-ylthio)hexanol, a specific glutathione S-transferase inhibitor, overcomes the multidrug resistance (MDR)-associated protein 1-mediated MDR in small cell lung cancer. Mol. Cancer Ther 7, 371–379. [DOI] [PubMed] [Google Scholar]
- (37).Patridge EV, Eriksson ES, Penketh PG, Baumann RP, Zhu R, Shyam K, Eriksson LA, and Sartorelli AC (2012) 7-Nitro-4-(phenylthio)benzofurazan is a potent generator of superoxide and hydrogen peroxide. Arch. Toxicol 86, 1613–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Fershtat LL, and Makhova NN (2017) Molecular Hybridization Tools in the Development of Furoxan-Based NO-Donor Prodrugs. ChemMedChem 12, 622–638. [DOI] [PubMed] [Google Scholar]
- (39).Wang X, Inoyama D, Russo R, Li S-G, Jadhav R, Stratton TP, Mittal N, Bilotta JA, Singleton E, Kim T, Paget SD, Pottorf RS, Ahn Y-A, Davila-Pagan A, Kandasamy S, Grady C, Hussain S, Soteropoulos P, Zimmerman MD, Ho HP, Park S, Dartois V, Ekins S, Connell N, Kumar P, and Freundlich JS Antitubercular Triazines: Optimization and Intrabacterial Metabolism. Cell Chemical Biology, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Singh R, Manjunatha U, Boshoff HI, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, and Barry CE 3rd. (2008) PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 322, 1392–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Matsumoto M, Hashizume H, Tomishige T, Kawasaki M, Tsubouchi H, Sasaki H, Shimokawa Y, and Komatsu M (2006) OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med 3, No. e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Tan MP, Sequeira P, Lin WW, Phong WY, Cliff P, Ng SH, Lee BH, Camacho L, Schnappinger D, Ehrt S, Dick T, Pethe K, and Alonso S (2010) Nitrate respiration protects hypoxic Mycobacterium tuberculosis against acid- and reactive nitrogen species stresses. PLoS One 5, No. e13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Heikamp K, and Bajorath J (2014) Support vector machines for drug discovery. Expert Opin. Drug Discovery 9, 93–104. [DOI] [PubMed] [Google Scholar]
- (44).Chen CY (2013) A novel integrated framework and improved methodology of computer-aided drug design. Curr. Top. Med. Chem 13, 965–988. [DOI] [PubMed] [Google Scholar]
- (45).Di-Wu L, Li LL, Wang WJ, Xie HZ, Yang J, Zhang CH, Huang Q, Zhong L, Feng S, and Yang SY (2012) Identification of CK2 inhibitors with new scaffolds by a hybrid virtual screening approach based on Bayesian model; pharmacophore hypothesis and molecular docking. J. Mol. Graphics Modell 36, 42–47. [DOI] [PubMed] [Google Scholar]
- (46).Klon AE, Glick M, and Davies JW (2004) Application of machine learning to improve the results of high-throughput docking against the HIV-1 protease. J. Chem. Inf. Comput. Sci 44, 2216–2224. [DOI] [PubMed] [Google Scholar]
- (47).Renault N, Laurent X, Farce A, El Bakali J, Mansouri R, Gervois P, Millet R, Desreumaux P, Furman C, and Chavatte P (2013) Virtual screening of CB(2) receptor agonists from bayesian network and high-throughput docking: structural insights into agonist-modulated GPCR features. Chem. Biol. Drug Des 81, 442–454. [DOI] [PubMed] [Google Scholar]
- (48).Ren JX, Li LL, Zheng RL, Xie HZ, Cao ZX, Feng S, Pan YL, Chen X, Wei YQ, and Yang SY (2011) Discovery of novel Pim-1 kinase inhibitors by a hierarchical multistage virtual screening approach based on SVM model, pharmacophore, and molecular docking. J. Chem. Inf. Model 51, 1364–1375. [DOI] [PubMed] [Google Scholar]
- (49).Ekins S, Reynolds RC, Franzblau SG, Wan B, Freundlich JS, and Bunin BA (2013) Enhancing hit identification in Mycobacterium tuberculosis drug discovery using validated dual-event Bayesian models. PLoS One 8, No. e63240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Ekins S, Freundlich JS, and Reynolds RC (2014) Are bigger data sets better for machine learning? Fusing single-point and dual-event dose response data for Mycobacterium tuberculosis. J. Chem. Inf. Model 54, 2157–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Ekins S, Freundlich JS, Hobrath JV, Lucile White E, and Reynolds RC (2014) Combining computational methods for hit to lead optimization in Mycobacterium tuberculosis drug discovery. Pharm. Res 31, 414–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, and Baker WR (2000) A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 405, 962–966. [DOI] [PubMed] [Google Scholar]
- (53).Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, and Coleman RG (2012) ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model 52, 1757–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, and Olson AJ (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Gasteiger J, and Marsili M (1980) Iterative partial equalization of orbital electronegativity - a rapid access to atomic charges. Tetrahedron 36, 3219–3228. [Google Scholar]
- (57).Perryman AL, Santiago DN, Forli S, Santos-Martins D, and Olson AJ (2014) Virtual screening with AutoDock Vina and the common pharmacophore engine of a low diversity library of fragments and hits against the three allosteric sites of HIV integrase: participation in the SAMPL4 protein-ligand binding challenge. J. Comput.-Aided Mol. Des 28, 429–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Voet AR, Kumar A, Berenger F, and Zhang KY (2014) Combining in silico and in cerebro approaches for virtual screening and pose prediction in SAMPL4. J. Comput.-Aided Mol. Des 28, 363–373. [DOI] [PubMed] [Google Scholar]
- (59).Franzblau SG, Witzig RS, McLaughlin JC, Torres P, Madico G, Hernandez A, Degnan MT, Cook MB, Quenzer VK, Ferguson RM, and Gilman RH (1998) Rapid, low-technology MIC determination with clinical Mycobacterium tuberculosis isolates by using the microplate Alamar Blue assay. J. Clin Microbiol 36, 362–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Sivaraman S, Zwahlen J, Bell AF, Hedstrom L, and Tonge PJ (2003) Structure-activity studies of the inhibition of FabI, the enoyl reductase from Escherichia coli, by triclosan: kinetic analysis of mutant FabIs. Biochemistry 42, 4406–4413. [DOI] [PubMed] [Google Scholar]
- (61).Parikh S, Moynihan DP, Xiao G, and Tonge PJ (1999) Roles of tyrosine 158 and lysine 165 in the catalytic mechanism of InhA, the enoyl-ACP reductase from Mycobacterium tuberculosis. Biochemistry 38, 13623–13634. [DOI] [PubMed] [Google Scholar]
- (62).Vilcheze C, Baughn AD, Tufariello J, Leung LW, Kuo M, Basler CF, Alland D, Sacchettini JC, Freundlich JS, and Jacobs WR Jr. (2011) Novel inhibitors of InhA efficiently kill Mycobacterium tuberculosis under aerobic and anaerobic conditions. Antimicrob. Agents Chemother 55, 3889–3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).van Soolingen D, Hermans PW, de Haas PE, Soll DR, and van Embden JD (1991) Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J. Clin Microbiol 29, 2578–2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Gupta A (2017) SNPTB: nucleotide variant identification and annotation in Mycobacterium tuberculosis genomes. bioRxiv, 227066.
- (65).Platt R, Drescher C, Park SK, and Phillips GJ (2000) Genetic system for reversible integration of DNA constructs and lacZ gene fusions into the Escherichia coli chromosome. Plasmid 43, 12–23. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Dose−response curves for InhA inhibition for select compounds; synthetic schemes for JSF-2149 and JSF-2164 and its candidate metabolites; RIF in vitro activity versus the ss18b strain; inhA promoter mutation in 16 × 6 resulting in inhA overexpression; LC-MS data supporting identification of JSF-3616 and JSF-3617 as intrabacterial metabolites of JSF-2164; antitubercular activity of the 5 lowest-scoring compounds among the 370 candidates passing docking filters; profiling of select spontaneous JSF-2164-resistant mutants (PDF)
Table S1: 370 compounds that passed the initial docking filters (XLSX)
Table S4: MIC values for JSF-2164, INH, and pretomanid versus JSF-2164-resistant transposon mutants (XLSX)