

SUMMARY
Disturbed flow (d-flow) induces atherosclerosis by regulating gene expression in endothelial cells (ECs). For further mechanistic understanding, we carried out a single-cell RNA sequencing (scRNA-seq) and scATAC-seq study using endothelial-enriched single cells from the left- and right carotid artery exposed to d-flow (LCA) and stable-flow (s-flow in RCA) using the mouse partial carotid ligation (PCL) model. We find eight EC clusters along with immune cells, fibroblasts, and smooth muscle cells. Analyses of marker genes, pathways, and pseudotime reveal that ECs are highly heterogeneous and plastic. D-flow induces a dramatic transition of ECs from atheroprotective phenotypes to pro-inflammatory cells, mesenchymal (EndMT) cells, hematopoietic stem cells, endothelial stem/progenitor cells, and an unexpected immune cell-like (EndICLT) phenotypes. While confirming KLF4/KLF2 as an s-flow-sensitive transcription factor binding site, we also find those sensitive to d-flow (RELA, AP1, STAT1, and TEAD1). D-flow reprograms ECs from atheroprotective to proatherogenic phenotypes, including EndMT and potentially EndICLT.
Graphical Abstract
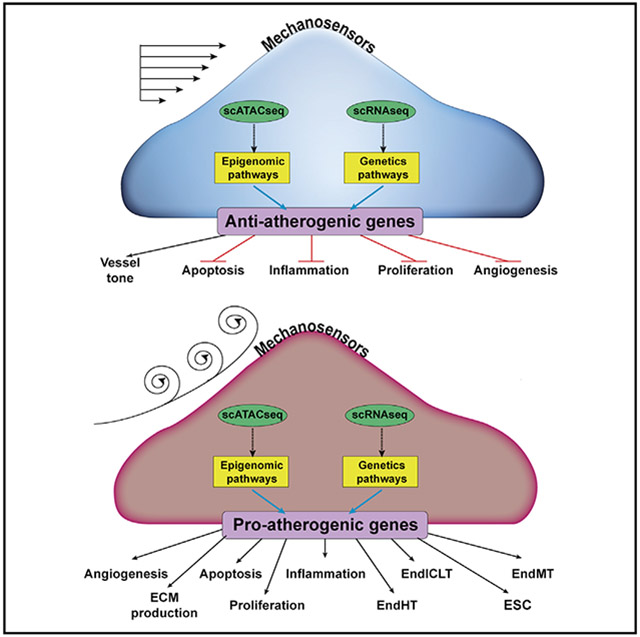
In Brief
To determine the effect of proatherogenic disturbed flow on transcriptomic and epigenomic chromatin accessibility profiles in endothelial cells at single-cell resolution, Andueza et al. perform scRNA-seq and scATAC-seq analyses using mouse carotid arteries following the partial carotid ligation. Disturbed flow reprograms endothelial cells to proatherogenic phenotypes, including EndMT and endothelial-to-immune cell-like transition.
INTRODUCTION
Atherosclerosis is the major underlying cause of myocardial infarction, ischemic stroke, and peripheral arterial disease (PAD), which represent a leading cause of death worldwide (Herrington et al., 2016). Atherosclerosis is a chronic inflammatory disease and occurs preferentially in arterial regions exposed to disturbed blood flow (d-flow), while those exposed to stable flow (s-flow) are protected (Chiu and Chien, 2011; Kwak et al., 2014; Tarbell et al., 2014). Flow is recognized by mechanosensors in endothelial cells (ECs), which in turn activate signaling pathways leading to the regulation of gene expression, endothelial function, and atherogenic pathways (Demos et al., 2020; Simmons et al., 2016). D-flow induces crucial proatherogenic pathways in ECs, including endothelial inflammation and dysfunction, permeability dysfunction, thrombosis, and endothelial-to-mesenchymal transition (EndMT). In contrast, s-flow protects ECs from those proatherogenic pathways (Kumar et al., 2014; Mahmoud et al., 2017; Nigro et al., 2011).
To understand how flow regulates the expression of endothelial genes at the transcript and epigenomic chromatin accessibility levels in vivo, we and others used bulk RNAs or DNAs obtained from the pooled ECs exposed to either s-flow or d-flow using the mouse partial carotid ligation (PCL) model or pig arteries (Dunn et al., 2014; Jiang et al., 2015; Ni et al., 2010; Son et al., 2013). We developed the PCL model and showed that d-flow rapidly induces, whereas s-flow prevents, robust atherosclerosis development within 2 weeks in hypercholesterolemic mice (Kumar et al., 2017; Nam et al., 2010; Son et al., 2013). The PCL model involves the ligation of 3 of 4 distal branches of the left common carotid artery (LCA) to induce d-flow, while using the contralateral right common carotid artery (RCA) that continues to be exposed to s-flow as an internal control. We further developed a lumen flushing method that enabled us to obtain endothelial-enriched RNAs and DNAs from the LCAs and RCAs following the PCL. These pooled bulk RNAs were then analyzed by mRNA microarray, microRNA microarray, and RNA sequencing (RNA-seq) to identify mRNA transcriptomes and microRNAs regulated by flow in ECs. These studies led to the discovery of numerous flow-sensitive genes and microRNAs that have been characterized and studied in detail for their roles in endothelial biology and atherosclerosis. We were also able to obtain bulk DNA samples from the LCAs and RCAs and analyzed by a reduced-representation bisulfite sequencing (RRBS) method (Dunn et al., 2014). This study showed that d-flow and s-flow differentially regulate epigenomic DNA methylation profiles in ECs, identifying many gene loci that were regulated by flow.
While these transcriptome and epigenomic methylome studies using the bulk RNA and DNA samples clearly identified the differential effects of d-flow and s-flow on ECs in vivo, the interpretation of these studies had several limitations. Although our bulk RNA and DNA preparations were highly enriched with ECs from the luminal flushings, it was unavoidable that they contained some other cell types present in the intima, especially in the LCAs following the PCL surgery. For example, we observed an increase in the expression of many genes associated with immune cells and mesenchymal cells in the LCA samples as early as 12 h following PCL; however, we could not ascertain whether those changes were due to non-ECs infiltrating to the intima or were induced by d-flow in ECs.
Recent developments in single-cell RNA-seq (scRNA-seq) and single-cell assay for transposase accessible chromatin sequencing (scATAC-seq) methods has enabled the transcriptomic and epigenomic chromatin accessibility analyses of a large number of cells at single-cell resolution at once. While scRNA-seq study shows an abundance of each gene transcript in individual cells and provides insights into the gene transcript expression profile, scATAC-seq analysis reveals changes in the chromatin accessibility profile, allowing insights into the epigenomic regulation of gene expression genome-wide. Furthermore, scATAC-seq analysis reveals several layers of gene regulation, such as genome-wide identification of cis-regulatory elements, including enhancers, nucleosome positions, and transcription factor (TF) binding sites. Since gene expression is regulated both at the transcriptional and epigenomic levels, the unprecedented power of carrying out concomitant and integrated analyses of scRNA-seq and scATAC-seq data are increasingly evident. These new powerful tools have revealed the heterogeneity of cell types in the liver (Zhao et al., 2020), lungs (Domingo-Gonzalez et al., 2020), heart (Bykov et al., 2020), immune cells (Villani et al., 2017), and ECs (Kalluri et al., 2019; Kalucka et al., 2020) under various physiological and pathological conditions.
Here, we performed a scRNA-seq and a scATAC-seq assay using the endothelial-enriched single cells and nuclei obtained from the mouse carotid arterial lumens following the PCL to determine the differential effect of d-flow and s-flow on genome- and epigenome-wide regulation of gene transcripts and chromatin accessibility profiles. Individual analyses of scRNA-seq and scATAC-seq data as well as an integrated analysis of both datasets revealed that ECs are heterogenous in the carotid artery even under s-flow conditions. D-flow dramatically altered endothelial transcriptome and epigenomic profiles, reprogramming them into inflammatory, mesenchymal, stem/progenitor-like cells, hematopoietic cells, and immune cell-like phenotypes. In addition, we identified a s-flow- and d-flow-dependent enrichment of TF binding sites and cis-regulatory elements.
RESULTS
scRNA-Seq and scATAC-Seq Analyses Using the Mouse PCL Model
To determine the genome-wide effect of s-flow and d-flow on gene transcript expression and chromatin accessibility profiles in ECs at a single-cell resolution in vivo, we carried out a scRNA-seq and a scATAC-seq using the mouse PCL model (Figure S1A). C57BL/6 mice were partially ligated to induce d-flow in the LCA using the contralateral RCA exposed to s-flow as controls. Two days (2D) and 2 weeks (2W) post-partial ligation, RCAs (2D-R and 2W-R) and LCAs (2D-L and 2W-L) were luminally digested with collagenase to obtain single cells and single nuclei, which were used for scRNA-seq and scATAC-seq, respectively.
scRNA-Seq Analysis
Single cells from 10 RCAs and LCAs were prepared, barcoded, and sequenced. The number of single cells used for scRNA-seq was 9,709, pooled from 1,867 (2D-R), 2,119 (2D-L), 1,263 (2W-R), and 4,460 (2W-L) (Table S1). The mean reads per cell ranged from 34,142 to 88,631 and the genes expressed per cell were 2,770–3,537.
scRNA-seq data analysis was performed using Seurat version 3.6 (Stuart et al., 2019). As a quality control, we eliminated cells that expressed gene counts >7,600 or <200, and those expressing >10% mitochondrial unique molecular identifier (UMI) counts to remove doublets and damaged cells during the sample preparation. Following normalization, an unsupervised graph-based clustering partitioned the cells into groups as visualized by the uniform manifold approximation and projection (UMAP) plot (Figure 1A). Our UMAP analysis identified 16 unique cell clusters distributed in a flow- and time-dependent manner (Figures 1A and 1B). Each cell cluster was identified using well-established marker genes (Figure 1C; Cochain et al., 2018; Kalluri et al., 2019; Roberts et al., 2020; Winkels et al., 2018). We found 8 EC clusters (E1–E8), vascular smooth muscle cells (SMCs), fibroblasts (Fibro), 4 monocytes/macrophages (macrophages 1–4), dendritic cells (DCs), and T cells. EC clusters were identified based on the expression of Pecam1, Icam2, Cdh5, and Tie1. The SMC-specific markers were Cnn1, Myl9, and Speg. Likewise, Medag, Dpep1, and Lum were used as markers for Fibro; C1qa, C1qb, C1qc, and C5ar1 for macrophages; Ccr7 and Mmp25 for DCs; and Itk for T cells (Figure 1C).
Figure 1. scRNA-Seq and scATAC-Seq Clustering and Cell Identification.

(A and B) scRNA-seq data plotted on a single UMAP representing 2D-R, 2D-L, 2W-R, and 2W-L. (A) UMAP represents all 4 samples, while (B) represents individual UMAP plots for each condition. Major cell populations include ECs (E1–8), SMCs, Fibro, macrophages, DCs, and T cells.
(C) Dot plot shows the specific marker genes used to classify each cell cluster. The percentage of cells expressing respective transcripts correlate and their expression intensity are depicted by the dot size color intensity, respectively.
(D) Graph shows the cell numbers in each cell cluster for 4 different experimental conditions.
(E and F) scATAC-seq data plotted on a single UMAP representing 2D-R, 2D-L, 2W-R, and 2W-L to identify major cell populations. (E) Single UMAP identifies major cell populations, while (F) represents individual UMAP plots for each condition.
(G) Dot plot shows the specific marker genes used to classify each cell cluster. The percentage of cells with open accessibility and the accessibility scores are depicted by the dot size and color intensity, respectively.
(H) Graph shows the cell numbers in each cell cluster for 4 different experimental conditions.
The unique flow- and time-dependent cell cluster distribution pattern shown in Figure 1B was further quantified in Figure 1D. The E1 cluster consisted of ECs present in all 4 conditions (2D-R, 2D-L, 2W-R, and 2W-L). E1–E4 consisted of ECs exposed to s-flow conditions (2D–R and 2W–R). The majority of E5 and E7 clusters consisted of ECs exposed to acute d-flow (2D-L). Interestingly, E6 and E8 exclusively consisted of ECs exposed to chronic d-flow (2W-L). These results suggest that d-flow induced a phenotypic shift in a time-dependent manner. However, there was no discernable difference between the 2D-R and 2W-R samples in overall cell clusters, suggesting that the cells exposed to s-flow conditions for 2 days or 2 weeks remained largely unchanged. Few SMCs were found in s-flow conditions (2D-R and 2W-R), while SMC numbers increased in acute d-flow conditions (2D-L) to 2.3%, which further increased by the chronic d-flow (2W-L) to 18.2% of the total cells. Fibroblasts were present in all four conditions, with the most in the acute d-flow condition (17% at 2D-L) (Figure 1D).
We found four macrophage clusters, which showed dramatic flow- and time-dependent changes. In the s-flow conditions (2D-R and 2W-R), macrophages were rare. However, acute d-flow increased the number of macrophages to 10.3% (2D-L), which further increased to 28.4% in chronic d-flow (2W-L). This finding is consistent with the known effects of d-flow on immune cell accumulation (Alberts-Grill et al., 2012). Moreover, we found that macrophage 4 was the major population in the acute d-flow (2D-L), whereas macrophage 1 and macrophage 2 were the major populations found in the chronic d-flow (2W-L) group. This suggests flow- and time-dependent phenotypic changes of these macrophage populations. Like macrophages, few DCs and T cells were present in the s-flow conditions, while they increased by chronic d-flow (2W-L) to 4.6% and 3.7%, respectively.
The scRNA-seq analysis performed above was independently validated by using the software package Partek Flow, which produced nearly identical results (Figures S1B-S1F).
scATAC-Seq Analysis
For this study, single nuclei from 12 RCAs and LCAs were prepared, subjected to transposase treatment, barcoded, and sequenced. The number of single nuclei used for sequencing was 18,324, which were pooled from 1,291 (2D-R), 5,351 (2D-L), 5,826 (2W-R), and 5,856 (2W-L), respectively (Table S1). The mean fragments per cell ranged from 19,690 to 24,459 and the total reads ranged from 304 to 372 million reads per group.
The sequence reads from the 4 samples, 2D-R, 2D-L, 2W-R, and 2W-L were analyzed using R software packages. The UMAP analysis partitioned total nuclei populations to 13 cell clusters, which were distributed in flow- and time-dependent manners (Figures 1E and 1F). The scATAC-seq data were converted to a gene-activity index by Signac, and cell identities were determined using the same marker genes described in the scRNA-seq study above. Among the 13 clusters, 8 were endothelial clusters (E1–E8), followed by SMCs, Fibro, macrophages, DCs, T cells, and non-defined (ND) (Figure 1G). Unlike the scRNA-seq result, only one macrophage cluster was identified.
The unique flow-and time-dependent cell cluster distribution pattern shown in Figure 1F was further quantified in Figure 1H. Like the scRNA-seq analysis, E1–E4 clusters consisted of ECs exposed to acute and chronic s-flow conditions (2D-R and 2W-R). Most E5–E7 clusters consisted of ECs exposed to acute d-flow (2D-L). While E6 and E7 were unique to ECs present in the acute d-flow condition (2D-L), E8 cells were exclusively found in the chronic d-flow condition (2W-L). As observed in the scRNA-seq, most SMCs came from the chronic d-flow condition (2W-L), while the fibroblasts were found in all four conditions. The majority of macrophages were found in the chronic d-flow condition (2W-L), followed by the acute d-flow (2D-L), while few were found in the s-flow conditions. Similarly, DCs and T cells were found mainly in chronic d-flow conditions. These results suggest that E2 represents unique s-flow phenotype, E6 and E7 represent the intermediate phenotype in response to acute d-flow, while E8 is a unique phenotype induced by chronic d-flow.
These scRNA-seq and scATAC-seq results showed that d-flow alters genome-wide changes in gene expression and chromatin accessibility, leading to a dramatic shift in the endothelial phenotypes in a time-dependent manner. It also shows heterogeneous endothelial populations even within the carotid artery under the s-flow conditions, and d-flow further increases the heterogeneity in vivo.
Integration of scRNA-Seq and scATAC-Seq
We analyzed the scRNA-seq and scATAC-seq data independently. While scATAC-seq is used to profile epigenomic chromatin accessibility indicating the potential gene activity in each cell, scRNA-seq data show the transcript abundance in each cell, which is a different but complementary aspect of gene regulation. We integrated the scRNA-seq and scATAC-seq datasets to correlate and cross-validate gene expression profiles and the chromatin accessibility landscape in the luminal cells in response to flow using the Harmony algorithm (Korsunsky et al., 2019). The co-embedded UMAP plot showed that the majority of cells analyzed in the scRNA-seq and scATAC-seq datasets overlapped each other, suggesting that the changes in chromatin accessibility and corresponding gene transcript expression in most cell clusters occurred in a concordant manner (Figure 2A). While scRNA-seq identified 4 macrophage clusters (Figure 1A), the scATAC-seq identified 1 macrophage cluster (Figure 1E). However, the integration result showed a near-complete overlap of all macrophage clusters in both datasets, suggesting that all macrophage clusters identified in our study are not clearly distinguishable. The individual UMAP plots for the scATAC-seq and scRNA-seq data are shown with the respective cell cluster identities (Figures 2B and 2C). These results obtained from two independent approaches demonstrate that our two datasets are highly concordant and cross-validate each other. This demonstrates the reproducible and robust effects of d-flow on genome-wide regulation of genes at the transcript and epigenomic chromatin accessibility levels in the aortic luminal cells at a single-cell level in vivo.
Figure 2. Integration of scRNA-Seq and scATAC-Seq Datasets, and Differential Gene Expression and Pathway Analyses.

(A) UMAP representation of the co-embedded scATAC-seq (red) and scRNA-seq (cyan) datasets. Cells from 2 assays were labeled with 2 colors as indicated in the plot.
(B and C) Individual UMAP representation of the co-embedded scATAC-seq and scRNA-seq datasets. Annotations used for the scRNA-seq shown in Figure 1A were used and superimposed onto each cell cluster.
(D) Heatmap represents the top 10 gene transcripts for each cell cluster identified from the scRNA-seq analysis.
(E) Gene Ontology analysis was performed using the top 200 upregulated genes in E8 representing the d-flow phenotype in comparison to E2 representing the s-flow phenotype. The x axis shows the p value.
D-Flow Induces Proatherogenic Pathways in ECs
Having established the high concordance between the scRNA-seq and scATAC-seq data, we then determined the characteristics of each cell cluster by analyzing differential gene transcript expression patterns. For this, we carried out a differentially expressed feature analysis using the scRNA-seq dataset and compared all of the cell clusters with one another. The top 10 upregulated genes from each cell cluster were then used to generate a heatmap plot (Figure 2D). We found that E2 expressed the highest levels of the 2 best-known mechanosensitive genes, Klf2 and Klf4, followed by E3 (Figure S2A). Klf2 and Klf4 expression was much lower in other EC clusters exposed to d-flow conditions (E5–E8). Interestingly, E1 and E4 clusters containing ECs from all 4 flow conditions showed a relatively low Klf2 and Klf4 expression, indicating that this heterogenous EC population is insensitive to flow. These results established E2 as a representative atheroprotective endothelial phenotype under s-flow conditions. In contrast, 3 well-known d-flow-induced proatherogenic genes, Ctgf, Serpine1, and Edn1, were highly expressed in ECs exposed to acute d-flow in E5. The E8 cluster found in the chronic d-flow condition showed a well-known d-flow-induced gene, Thsp1. In addition, E8 cells showed many genes that were also highly expressed in SMCs (Acta2, Tagln), Fibro (Dcn1), and immune cells (Cd74, H2-eb1, H2-aa, H2-ab1), indicating potential for EndMT and acquisition of immune cell-like features by ECs under the chronic d-flow condition. Many of the top genes expressed in the non-endothelial clusters were well-known marker genes used for their identification (Figure 1C). For example, SMCs showed high expression of Acta2, Myh9, Myh11, and Tagln; Fibro expressed Dcn; and macrophages showed high expression of C1qa, C1qb, C1qc, and Ccl4, respectively. Our attempt to compare the characteristics of the 4 macrophage clusters did not reveal whether they represented proinflammatory M1 or reparative M2 phenotypes (Alberts-Grill et al., 2012).
To compare the biological status of the EC clusters exposed to s-flow and chronic d-flow, we selected respective E2’s and E8’s from both scRNA-seq and scATAC-seq datasets, since they were clearly concordant and distinguishable from other endothelial clusters and represented the atheroprotective s-flow and proatherogenic d-flow conditions, respectively. The overexpressed genes found in both datasets in E8 in comparison to E2 were subjected to a Panther Gene Ontology (GO) analysis (Mi et al., 2017). As shown in Figure 2E, ECs exposed to chronic d-flow exhibited the induction of many well-known biological processes associated with proatherogenic pathways (Simmons et al., 2016). These processes include vascular development, inflammation, apoptosis, angiogenesis, EndMT, transforming growth factor-β (TGF-β) pathway, and endothelial permeability (Daniel and van Buul, 2013; DePaola et al., 1999; Fan and Karino, 2010; Jenkins et al., 2013; Johnson and Nerem, 2007; Kutikhin et al., 2018; Rocha et al., 2018; Schnittler, 1998; Seebach et al., 2000; Ueno et al., 2000; Yamamoto et al., 2003). Furthermore, we performed additional GO analyses to determine whether the similar but smaller number of proatherogenic pathways were also induced in other EC clusters (E5, E6, and E7) exposed to acute or chronic d-flow conditions. Interestingly, the E1 cluster, which is found in all 4 conditions independent of time and flow conditions, showed many cellular energetics and mitochondrial pathways indicating high metabolic activities in these cells. The functional significance of these flow-independent EC cohorts is unclear at present. These results suggest that d-flow induces the transition of ECs to proatherogenic phenotypes in a flow- and time-dependent manner by regulating gene transcripts and epigenomic chromatin accessibility profiles.
Pseudotime Trajectory, Chromatin Accessibility, and Gene Expression Analyses Reveal Flow-Dependent Endothelial Reprogramming
The finding that ECs in E8 in response to chronic d-flow expressed the marker genes of mesenchymal and immune cells (Figure 2E) raised the question of whether d-flow induced endothelial reprogramming such as EndMT. To test this hypothesis, we conducted a trajectory analysis with Monocle 2 embedded in Partek Flow software using the scRNA-seq data to track dynamic differentiation status of each cell (Pliner et al., 2018; Qiu et al., 2017). Our analysis clearly identified 3 major groups of differentiated cell types: (1) ECs, (2) immune cells (macrophages, DCs, and T cells), and (3) SMCs and Fibros (Figure 3A). In addition, there were many other cells present along the trajectory paths toward each cell type, indicating their transitional de-differentiation status in response to d-flow. To better understand the trajectory results, we split the analysis by the four experimental conditions (Figure 3B). In the s-flow conditions (2D-R and 2W-R), most cells were well differentiated with few cells in transition. In contrast, d-flow conditions induced many ECs to transitional states, potentially trans-differentiating toward SMCs and Fibro, indicating EndMT. Surprisingly, we also found many ECs transitioning toward immune cells in acute d-flow condition (2D-L), which further increased in the chronic d-flow (2W-L) condition. Further analysis using the E8 cluster revealed that these ECs exposed to chronic d-flow transitioned toward immune cell-like types (EndICLT), but did not achieve fully differentiated immune cell status (Figures 3C and S3).
Figure 3. D-Flow Induced Endothelial-to-Mesenchymal Transition (EndMT) and Endothelial-to-Immune-like Cell Transition (EndICLT) as Shown by Pseudotime Trajectory and Accessibility Profile Analyses.

(A and B) Pseudotime trajectory plot shows differentiated cell types (ECs, SMCs, Fibro, macrophages, DCs, and T cells) at the end of the branches. Dots along the trajectory lines represent the status of the cells transitioning toward differentiated cell types. (A) All cells pooled in 4 different conditions. (B) The cells split according to the time- and flow-dependent conditions.
(C) Pseudotime Trajectory plot for E8.
(D and E) The accessibility profiles of E1 and E2, representing ECs under s-flow and E8 representing ECs under d-flow from scATAC-seq data and violin plots showing the expression of corresponding genes from scRNA-seq data. Genes representing (D) and (E) EndICLT in the EC clusters are shown. Black arrows and * indicate accessibility changes in the promoter regions and 3’UTR, respectively. **p < 0.01 and ***p < 0.001.
To validate the transition of ECs to other cell types, EndMT, and EndICLT, we further examined the chromatin accessibility and gene expression profiles of key marker genes. Figure 3D shows four markers of EndMT, Tagln, Acta2, Cnn1, and Snai1 in EC clusters representing s-flow (E2), acute d-flow (E6), and chronic d-flow (E8) for their chromatin accessibility changes. d-flow increased the chromatin accessibility of these marker genes in the promoter regions (black arrows) or 3′UTR (* in Snai1). Consistent with the accessibility changes, the corresponding gene expression levels were also increased in ECs exposed to d-flow in E8. These results confirm that d-flow induced EndMT as expected (Glaser et al., 2020; Lai et al., 2018; Mahmoud et al., 2017; Moonen et al., 2015; Ten Dijke et al., 2012).
Having validated the EndMT, we further analyzed the gene markers of potential EndICLT using the macrophage markers C1qa, C1qb, C5ar1, and Tnf in EC clusters. As shown in Figure 3D, chronic d-flow increased the accessibility of these genes in their promoter regions (arrows) in E8 compared to the s-flow (E2) and acute d-flow (E6). This result was further confirmed by the increased expression of the corresponding genes in E8 as shown in the violin plot of the scRNA-seq data (Figure 3E). To independently validate whether d-flow induces EndICLT, we tested whether d-flow conditions can induce the transition of cultured ECs toward immune cell-like phenotypes in the absence of any other cell types. To this end, we exposed human aortic ECs (HAECs) to chronic high laminar shear stress (LS, mimicking s-flow conditions) or low oscillating shear (OS, mimicking d-flow conditions) for 7 days in vitro (Ghim et al., 2018). As shown in Figure 4, chronic OS reduced the expression of KLF2 and KLF4, while inducing markers of EndMT (SNAI1 and TAGLN) in comparison to LS. Importantly, chronic OS induced the expression of two macrophage marker genes (C1QC and C5AR1), directly demonstrating that OS can induce EndICLT in vitro. Moreover, we performed an en face co-immunostaining study using the mouse PCL model to determine whether macrophage marker proteins are expressed in CDH5+ ECs in vivo. As shown in Figures 4A-4D, C1QA and LYZ were clearly induced in LCA ECs, but not in the RCAs, providing more strong evidence for flow-induced EndICLT. The fluorescent intensity was measured using ImageJ software (Schneider et al., 2012).
Figure 4. D-Flow Induces Expression of Immune Cell Markers in ECs In Vivo and in Cultured ECs In Vitro.

(A–D) Two weeks following PCL, the LCAs and RCAs were double-immunostained en face using antibodies for C1QA or LYZ along with the CDH5 antibody as the EC marker. Shown are confocal images for CDH5 (red), C1QA or LYZ (white), either individually or merged with nuclear DAPI (blue). The imaging intensity of each was quantified, and the data represent the mean ± SD (n = 8). **p < 0.01 and *** p < 0.001. Scale bars are shown in A and C.
(E and F) HAECs exposed to chronic laminar shear (LS mimicking s-flow) or oscillatory shear (OS, mimicking d-flow) for 1 week were analyzed by qPCR to quantify expression of immune cell markers (C1QC, C5AR1), EndMT markers (SNAI1 and TAGLN), and the flow-sensitive gene markers (KLF2, KLF4). The data represent the mean ± SD (n = 3). *p < 0.05, **p < 0.01, and ***p < 0.001.
The EndMT and EndICLT results suggested that d-flow may induce broad endothelial reprogramming. To test the hypothesis, we examined the expression of marker genes for endothelial-to-hematopoietic transition (EndHT), endothelial stem cell (ESC), endothelial progenitor cell (EPC), and antigen-presenting cells (APCs) transition (Ottersbach, 2019; Santambrogio et al., 2019) using both the scRNA-seq and scATAC-seq datasets. Table S2 shows that d-flow induced expression of those marker genes of EndHT (Sox7, Sox17, Gata2, Kit, Notch1, Eprc, Tie2, Bmp4) and APCs (Cd74, H2-aa, H2-ab1, H2-eb1), as well as previously explained EndMT (Tagln, Cnn1, Acta2, Snai1) and EndICLT (C1qa, C1qb, C5ar1, Tnf, and Lyz2). Furthermore, we found ECs expressing the marker genes of ESCs and EPCs. However, nearly all CD157+ cells were Sca1+ and CD34+, making it difficult to distinguish between ESCs and EPCs. Therefore, we did not attempt to further distinguish ESCs and EPCs in our analysis (Table S2). Interestingly, we found that the expression of some ESC and EPC marker genes was sensitive to blood flow. ECs in E2 in s-flow were Cd157highSca1+Kit−, E5/E6 in acute d-flow showed Cd157lowSca1lowKit−, whereas E8 in chronic d-flow was Cd157highSca1lowKit+. These results clearly demonstrate that d-flow reprograms ECs through EndMT, and potentially via EndICLT and EndHT, by altering the gene expression of key genes at the transcriptional and chromatin accessibility levels.
We next analyzed the proatherogenic pathways activated by d-flow conditions identified from our Gene Ontology results (Figure 2E). As shown in Figure 5, d-flow in E8 altered markers of leukocyte traffic and inflammation (Il1β, Il6, Ccl2, and Ccl3); ECM regulation (Adamts4, Lamb1, Timp3, and Timp4); vasoregulation (Nos3, Ptgs2, and Edn1); and lipid metabolism (Tm6sf2, Lsr, and Mgll), in comparison with s-flow in E2 (Figures 5A-5D). These results show that d-flow activated proatherogenic endothelial responses, while inhibiting antiatherogenic pathways by changing the gene expression at the transcriptional and chromatin accessibility levels.
Figure 5. Chronic d-Flow Reprograms Endothelial Functions.

The accessibility profiles of E1 and E2 (s-flow) and E8 (d-flow) from scATAC-seq data and violin plots showing the expression of corresponding genes from scRNA-seq data are shown. Genes representing (A) leukocyte traffic and inflammation and (B) ECM regulation, (C) vasoregulation, and (D) lipid metabolism in the EC clusters are shown. Black arrows indicate accessibility changes in the promoter region except for Edn1 (*). *p < 0.05 and ***p < 0.001.
Identification of Flow-Dependent TF Binding Motifs In Vivo
We next examined whether flow regulates gene expression through a set of TF binding motifs in response to either s-flow or d-flow. To test this hypothesis, we used the Signac and ChromVar packages to identify flow-sensitive TF binding motifs using our scATAC-seq dataset (Figure 6). The top 20 TF binding motifs were identified in each endothelial cluster (E1–E8) and were plotted as a heatmap (Figure 6A). Figures 6B and 6C show TF binding motifs and the respective cell clusters where they were found. The TF binding motifs for KLF4 were enriched in E2 exposed to s-flow, while NRF1 was enriched in E3 and E4 (exposed to all flow conditions) (Figure 6B). In contrast, TF motifs for TEF, ETV3, RELA, FOS::JUN, TEAD1, and STAT1 were enriched in E5–E8 exposed to d-flow conditions (Figure 6C). Identification of the motifs for some of the well-known flow-sensitive TFs, KLF4 (identical to the KLF2 motif), FOS:JUN (AP1), RELA, and TEAD1 confirms the validity of our analysis (Bondareva et al., 2019; Fraineau et al., 2015; Kempe et al., 2005; Sangwung et al., 2017). To further determine whether any of the newly identified TF binding motifs are also relevant in flow-dependent responses, we examined whether the level of phospo-STAT1 is flow sensitive, as a marker of STAT1 activation. The level of phospho-STAT1 was significantly increased in the CDH5+ LCA ECs compared to the RCA in mice at 2 weeks post-PCL surgery (Figures 6D and 6E). This result demonstrates the potential importance of these TF binding motifs and relevant TFs in the flow-dependent regulation of ECs. We also attempted to look for the shear stress response element (SSRE) (Resnick et al., 1993), but failed due to the lack of a defined list of TFs for the SSRE in our database. Our study validated the role of KLF2/4 as the master TF for s-flow, while identifying both known and new TFs mediating the d-flow effects.
Figure 6. D-Flow Alters Accessibility of Transcription Factors as Determined by Motif Enrichment Analysis and Induces STAT1 Phosphorylation in ECs In Vivo.

(A and B) Heatmap shows the top overrepresented motifs enriched in each endothelial cluster. Fold enrichment of each motif is represented on a color scale, dark blue being the lowest score and yellow being the highest score. UMAP plot and the enriched motif sequences for overrepresented transcription factor binding motifs enriched in 2D-R and 2W-R (s-flow) (B), and 2D-L (acute d-flow) and 2W-L (chronic d-flow) conditions.
(C) The blue dots on the UMAP plot represent the cells in which the motifs are overrepresented.
(D and E) Two weeks following PCL, the LCAs and RCAs were double-immunostained en face using antibodies for CDH5 and phospho-STAT1. Shown are confocal images for CDH5 (red) and phospho-STAT1 (white), either individually or merged with nuclear DAPI (blue). Imaging intensity of each was quantified and the data (E) represents the mean ± SD (n = 4). ***p < 0.001. Scale bars are shown in D.
Accessibility and Co-accessibility Analyses Reveal Flow-Dependent cis-Regulatory Interactions
To identify the flow-dependent regulation of cis-regulatory elements, enhancer regions, and co-accessibility changes, we carried out a Cicero analysis using our scATAC-seq dataset. The prototypical mechanosensitive Klf4 was analyzed, and the results were mapped to the corresponding gene locus using the ENCODE Genome Browser (Figure 7A). For this analysis, we compared E2 (s-flow) and E8 (chronic d-flow) clusters. E2 cells showed a higher co-accessibility between the cis-elements compared to E8, suggesting that d-flow decreased the co-accessibility connections. Next, as additional examples, we showed that the d-flow dramatically reduced the accessibility of the promoter region of Arl4d (Figure 7Bb). Similarly, the d-flow also reduced the co-accessibility and gene expression of Arl4d (Figures 7Bc and 7Bd). In contrast, the d-flow increased the accessibility, co-accessibility, and gene expression of Tgfbi (Figure 7C). These findings suggest that flow regulates the gene expression by not only regulating the TF binding motifs but also altering cis-regulatory elements, and their interactions suggesting both epigenomic and transcriptional mechanisms.
Figure 7. Chronic d-Flow Alters cis-Regulatory Interactions of Flow-Sensitive Genes.

Bioinformatic analysis showing the regulatory elements of (A) Klf4, (B) Arl4d, and (C) Tgfbi genes. Gene structure and location of genes from ENCODE database (a) were mapped with normalized accessibility (b) and co-accessibility score plots (c) of E2 and E8 from our scATAC-seq dataset. Violin plots showing the expression of Klf4 and Arl4d in E1 to E8 from our scRNA-seq data (d). *p < 0.05 and ***p < 0.001.
DISCUSSION
Here, we analyzed scRNA-seq and scATAC-seq datasets and identified that (1) artery ECs are heterogeneous even under s-flow conditions, (2) ECs are highly plastic, (3) d-flow dramatically alters endothelial phenotypes, indicating that ECs transition from the quiescent, anti-inflammatory, and anti-atherogenic phenotype to the pro-inflammatory, pro-atherogenic phenotype, and (4) d-flow induces gene changes, indicating that ECs undergo EndMT, EndHT, and EndICLT, while its effect on ESC and EPC is not clearly defined. In addition to 8 EC clusters, we identified SMCs, Fibro, macrophages, DCs, and T cells in our endothelial-enriched single-cell populations obtained from the luminal flushings of the LCAs and RCAs. The comparison of the prototypical EC profiles (E8 by d-flow versus E2 by s-flow) indicated that the d-flow activated proatherogenic pathways, including inflammation, leukocyte traffic, EndMT, ECM regulation, lipid metabolism, and vasoregulatory gene expression profiles. We also found TF binding motifs enriched by s-flow (KLF4/KLF2) and d-flow (TEF, RELA, ETV3, FOS::JUN, TEAD1, STAT1). Our study also showed that flow regulates chromatin co-accessibility and interactions among cis-regulatory elements. These results demonstrate that d-flow regulates gene expression both at the transcriptional and epigenetic levels, leading to the reprogramming of ECs into a mesenchymal, inflammatory, and immune cell-like phenotype.
Our scRNA-seq and scATAC-seq results are consistent with our previous gene array results using the endothelial-enriched RNA samples in bulk obtained from the same mouse PCL model (Ni et al., 2010). As shown in our bulk RNA data and other reports (Ajami et al., 2017; Jiang et al., 2014; Lee et al., 2017; Ni et al., 2010; Son et al., 2013; Wang et al., 2006, 2016), our single-cell data showed expected changes in many well-known flow-sensitive genes, including Klf2, Klf4, Nos3, Klk10, Angpt2, Ctgf, Bmp4, Timp3, Tgfb1, Thsp1, Col1a1, Tagln, Acta2, Cnn1, and Snai1. The present study also showed that d-flow induced inflammatory and proliferative pathways, consistent with our bulk RNA array result (Ni et al., 2010), further demonstrating the validity of our single-cell results. One of the major limitations of the bulk RNA study was the inability to ascertain the cell types in which the gene changes were occurring. The endothelial-enriched RNA preparation method used for the bulk RNA array study included other cell types such as macrophages and SMCs present in the carotid intima. While our previous bulk RNA results showed that d-flow increased overexpression of many pro-inflammatory genes and immune cell marker genes, it was unclear whether those changes were indeed contributed by ECs in response to d-flow. Current single-cell analysis has addressed this limitation. We also compared our carotid endothelial gene expression data to the previously published endothelial heterogeneity data from murine ECs from various tissue types and arteries (Kalluri et al., 2019; Kalucka et al., 2020). While we found that most of the highly enriched genes (Fbln5, Mecom, Gja4, Azin1, Sox17, Mgp, Nrp1, Kcnj8, Cytl1, Bmx, Eln, Fbln2, Gkn3) found in the arteries were also present in all of our EC clusters, they showed no flow dependence. However, Sema3g was mostly found in our ECs exposed to s-flow (E1 and E2 cluster). Interestingly, the tissue-specific endothelial genes highly enriched in other reported tissue types, including the brain, heart, lungs, and kidney, were expressed at a very low level or not detected in our carotid artery EC clusters. This further highlights the endothelial heterogeneity and the impact of local cues on tissue-specific endothelial gene expression. Our single-cell study clearly demonstrates that d-flow induces the expression of many pro-inflammatory, pro-EndMT, and pro-EndICLT genes, reprogramming ECs from the atheroprotective phenotype under s-flow to the proatherogenic phenotype under d-flow, as discussed further below. Another unexpected finding was that d-flow induced EndHT, although its functional significance in atherosclerosis remains to be studied (Ottersbach, 2019). While we found that some markers of ESCs and EPCs were flow sensitive, we were not able to distinguish between the 2 cell types because nearly all CD157+ ESCs also expressed markers of EPCs (Sca1 and CD34) (Wakabayashi et al., 2018; Xiao et al., 2006). This difficulty in distinguishing ESCs from EPCs is consistent with a recent controversy surrounding the endovascular progenitor cells and ESCs, which could not be distinguished solely based on the expression of the markers CD34, CD45, CD31, and VEGFR2 (Oatley et al., 2020).
The cell-clustering analyses using the scRNA-seq and scATAC-seq data showed that ECs are heterogeneous within the carotid artery under the s-flow condition, which becomes even more diverse in response to d-flow conditions in a time-dependent manner. From our scRNA-seq analysis, we found 4 EC clusters (E1–E4) under s-flow conditions, 2 new populations in response to the acute d-flow (E5 and E7), and 2 additional EC populations (E6 and E8) under chronic d-flow conditions. Likewise, scATAC-seq analysis identified 4 EC clusters (E1–E4) under s-flow conditions, 3 new populations in response to the acute d-flow (E5–E7), and 1 additional EC population (E8) under chronic d-flow conditions. Overall, the comparison of these two datasets and integrated analysis showed that most ECs and other cell populations including macrophages under s-flow and d-flow conditions were highly concordant, cross-validating each other’s data. Our results demonstrate that d-flow induces a dramatic shift in endothelial phenotypes at the genomic and epigenomic levels, suggesting ECs transitioning from the atheroprotective s-flow phenotypes to the proatherogenic d-flow phenotypes.
The differential gene expression analysis of the scRNA-seq data further demonstrated that flow reprograms ECs. We established E2, E5, and E8 as representative endothelial phenotypes for atheroprotective s-flow, acute d-flow, and chronic proatherogenic d-flow conditions, respectively. E2 (s-flow) showed a high expression of Klf2, Klf4, and Klk10, well-known flow-sensitive genes (Ni et al., 2010; Sangwung et al., 2017), while E5 (acute d-flow) showed a high expression of d-flow induced proatherogenic genes (Ctgf, Serpine1, and Edn1) (Brooks et al., 2002; Wang et al., 2016). Interestingly, E8 (chronic d-flow) showed a high expression of not only the flow-dependent genes but also markers of mesenchymal cells, hematopoietic cells, APCs, and immune cells, indicating EndMT, EndHT, and EndICLT (Figure 3; Table S2). While d-flow is widely known to induce EndMT (Mahmoud et al., 2017; Moonen et al., 2015), it was unknown whether d-flow can drive ECs to undergo EndHT, ESC/EPC, and EndICLT status.
To further investigate whether EndICLT occurs, we carried out the pseudotime trajectory analysis and found that chronic d-flow induced ECs to undergo EndMT as expected and also led to a transition toward immune cell-like phenotypes (Figures 3 and S3). The EndICLT hypothesis was further validated by examining the expression of several gene markers of macrophages (C1qa, C1qb, C5ar1, Tnf, and Lyz2) by 2 independent analyses, scRNA-seq and scATAC-seq (Figure 3D; Table S2). However, the most direct and strongest support for the flow-dependent EndICLT is provided by the in vitro result, showing that chronic exposure to OS induced HAECs to express markers of EndICLT (Figures 4E and 4F). Additional support is provided by the en face staining study in the mouse LCAs, demonstrating that chronic d-flow induces the expression of EndICLT marker proteins in vivo (Figures 4A-4D). Taken together, the scRNA-seq and scATAC-seq data analyses, the in vitro shear data, and the in vivo staining results provide strong support for flow-induced EndICLT. However, additional lineage tracing studies would further strengthen the EndICLT hypothesis.
Our finding for EndICLT is supported by previous reports; vascular ECs and lymphatic ECs were shown to serve as APCs expressing major histocompatibility complex class I (MHC class I) and MHC class II (Al-Soudi et al., 2017; Mai et al., 2013). The plasticity of ECs was further demonstrated by the transition of ECs to osteoblasts and hematopoietic cells (Guibentif et al., 2017; Lin et al., 2017). Interestingly, epithelial cell to immune cell-like transition was also reported (Choi et al., 2012). Moreover, recent studies comparing the translatomes and transcriptomes of various tissue-specific ECs demonstrated that they are heterogeneous and plastic, adapting to the tissue-specific environment (Jambusaria et al., 2020; Kalucka et al., 2020). While the functional importance of EndMT in atherosclerosis is well established, the role of flow-dependent EndHT and EndICLT and changes in ESC and EPC status in atherosclerosis will be exciting future research avenues.
Our motif analysis using the scATAC-seq data identified a comprehensive list of flow-sensitive TF binding motifs specific to s-flow and d-flow, respectively. As expected, KLF4 binding motifs were specifically enriched in ECs under s-flow conditions. It is important to note that both KLF4 and KLF2 bind to the same motifs (Nayak et al., 2011). We identified several unreported and expected TF binding motifs enriched under d-flow conditions. RELA, FOS::JUN, and TEAD1 were previously reported as flow-sensitive TF binding motifs, while TEF, ETV3, and STAT1 were reported as new d-flow-sensitive TF-binding motifs (Figure 6). Our results validate the predominant role of KLK2/4-mediated gene regulation under s-flow, whereas d-flow regulates gene expression via several TFs. In addition, we found that d-flow increased the activation of STAT1 (Figures 6D and 6E), identifying it as a new flow-sensitive TF. These results demonstrate that flow regulates access to these TF-binding motifs via epigenomic mechanisms at a genome-wide level.
Our results should be interpreted with some caveats since the single cells and nuclei were obtained from the collagenase digestion, which could have affected the transcriptomic and epigenomic profiles during the incubation period (~45 min). However, the overwhelming list of flow-sensitive marker genes such as Klf2, Klf4, Klk10, and EndMT genes showed the robust expected flow dependency, although we found some exceptions such as Vcam1.
In summary, our scRNA-seq and scATAC-seq studies provide the comprehensive atlas of transcriptomic and epigenomic chromatin accessibility profiles at a single-cell level in response to s-flow and d-flow in vivo. We showed that flow alters the accessibility of TF binding motifs and cis-regulatory elements, thereby regulating gene expression at the genomic and epigenomic levels in ECs. These flow-dependent genomic and epigenomic changes resulted in endothelial heterogeneity and reprogramming, inducing EndMT, EndHT, and EndICLT. These robust and broad flow-dependent changes in ECs, from the quiescent and antiatherogenic phenotype under s-flow conditions to the pro-inflammatory and proatherogenic phenotype under d-flow conditions, would lead to atherosclerosis in the presence of additional risk factors such as hypercholesterolemia. Our single-cell results would serve as valuable resources for researchers in the field, and harnessing the insights provided by our data would lead to a better understanding of the flow-dependent regulation of endothelial biology and pathobiology. These insights will further facilitate the development of therapeutics for atherosclerosis.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hanjoong Jo (hjo@emory.edu).
Materials availability
Further requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hanjoong Jo (hjo@emory.edu). This study did not generate new unique reagents.
Data and code availability
The FastQ files generated from the scRNaseq and scATACseq workflow are available at the NCBI BioProject repository (accession number PRJNA646233). The accession number for the scRNA-seq and scATAC-seq data is https://www.ncbi.nlm.nih.gov/bioproject: PRJNA646233. The code generated during this study are available at GitHub (https://github.com/JoLab-Emory/SingleCell).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All animal procedures used for this study were approved by the Institutional Animal Care and Use Committee at Emory University. To minimize sex-dependent variation and the complication of hypercholesterolemic conditions, we used a total of 44 male C57BL/6 mice, Bar Harbor, 10-week-old) which were fed ad libitum with standard mouse chow diet.
Primary culture cells
Human Aortic Endothelial Cells (HAEC) were purchased from Cell Applications and were used to validate the results obtained from the single-cell study in vitro.
METHOD DETAILS
Mouse partial carotid ligation (PCL) surgery
To induce d-flow, PCL surgery was performed (Nam et al., 2010). Briefly, mice were initially anesthetized with 3.5% isoflurane and subsequently maintained on 1.5% isoflurane during the entire procedure. The LCA bifurcation was exposed by blunt dissection and three of four caudal LCA branches (left external carotid, internal carotid, and occipital arteries) were ligated with 6-0 silk sutures, leaving the superior thyroid artery intact. The contralateral RCA was left untouched and served as an internal control. Following surgery, analgesic buprenorphine (0.1 mg kg−1) was administrated. At 2 days (2D-R and 2D-L) and 2 week post-partial ligation (2W-R and 2W-L), LCAs and RCAs were dissected out and a luminal enzymatic digestion of the carotids was performed to isolate endothelial-enriched single-cell preparations (Figure S1). Single-cell preparations from LCAs and RCAs from 12 mice were pooled to prepare single-nuclei for scATACseq study, while LCAs and RCAs from 10 additional mice were pooled to obtain single cells for scRNaseq study. Both studies were performed back to back.
Single-cells and single-nuclei isolation and library preparation for scRNaseq and scATACseq analyses
After euthanizing the mice using CO2, the LCAs and RCAs were dissected, cleaned, and perfused with normal saline solution before a dissociation buffer was injected. The dissociation buffer was slightly modified from a previously reported recipe (Kalluri et al., 2019)by including 600 U ml−1 Type II Collagenase and 60 U ml−1 DNase I in 0.5% fetal bovine serum (FBS) of 1X phosphate-buffered saline (PBS). Immediately after the introduction of dissociation buffer into the carotid lumens, the ends of the RCAs and LCAs were clamped and dissected out. The explanted carotids were incubated in 35-mm dishes containing HEPES-buffered saline solution (HBSS) at 37 °C. After 45 min, the LCA and RCA lumens were flushed with the dissociation buffer into a 1.5 mL Eppendorf tube. The flushings were washed with ice-cold PBS and centrifuged for 5 min at 500 x g and were further incubated with accutase for 5 min at 37 °C to prepare single-cells.
For scRNaseq, single-cells were resuspended in 1% BSA in PBS, counted and immediately processed at the Emory Integrated Genomics Core (EIGC) using a 10x Genomics Chromium device for single-cell encapsulation, barcoding, and RNA preparation. Subsequently, the cDNA libraries were prepared and sequenced on Illumina NextSeq® to a depth of 15,000 unique molecule identifier (UMI) per cell. UMI counts for each cellular barcode were quantified and used to estimate the number of cells successfully captured and sequenced.
For scATACseq, single-cells were washed with 0.04% BSA and incubated in a lysis buffer containing 10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, 0.1% Tween-20, 0.1% NP40, 0.01% digitonin and 1% BSA in nuclease-free Water. After 5 min, the lysates were washed with wash buffer (10 mM Tris-HCl, 10 mM NaCl, 3 mM MgCl2, and 0.1% Tween-20) and the nuclei were resuspended in a Nuclei Buffer® (10x Genomics), and were immediately processed at the EIGC for scATAC sequencing.
The nuclei (~7,000 each) from 2D (R and L) and 2W (R and L) were individually incubated with ATAC Buffer and ATAC Enzyme (Tn5 transposase, 10x Genomics) to form a transposition mix for 60 min at 37°C following manufacturer’s protocol. A mild detergent condition was chosen to keep the nuclei intact during tagmentation. Then a master mix composed of Barcoding Reagent, Reducing Agent B and Barcoding Enzyme was added. The resulting mixtures were loaded onto a Chromium Chip and single-nuclei gel emulsions with barcoding were prepared following the manufacturer’s instructions.
The Cell Ranger Single-Cell Software suite was then used for demultiplexing, barcode processing, alignment, and initial clustering of the raw scATACseq and scRNaseq profiles.
scRNaseq data analysis
We analyzed the scRNaseq data with Seurat R package. Briefly, the scRNaseq data files were processed with Cell Ranger Software where the sequencing reads were aligned to mouse reference genome using STAR aligner. A read was considered exonic, if at least 50% of it mapped to an exon, intronic (if it was non-exonic and intersected an intron), or intergenic otherwise. For reads that aligned to a single exonic locus but also aligned to 1 or more non-exonic loci, the exonic locus was prioritized and the read was considered to be confidently mapped to the exonic locus. Cell Ranger also aligned exonic reads to annotated transcripts. An annotated transcript that aligned to the same strand was considered to be confidently mapped to the transcriptome. These confidently mapped reads were used for unique molecular identifier (UMI) counting and subsequent analysis to generate h5 files. The h5 file of each sample was then processed with Seurat R package for further analysis. First, a quality check was performed to remove low-quality cells and doublets by selecting the cells containing unique feature counts of over 200 or less than 7,600, and cells with lower than 10% of mitochondrial counts. All datasets were merged, normalized, scaled, clustered, and visualized by UMAP.
As an independent validation, the scRNaseq analysis was also carried using Partek Flow® analysis software following the same filtering criteria. Both Partek Flow and Seurat analyses provided consistent and nearly identical results.
Single-cell ATACseq data analysis
The sequencing data for the scATAC study were processed by Cell Ranger (CR) Software. Briefly, to avoid barcode misidentification, each 16-nucleotide barcode was compared against a ‘whitelist’ of correct barcode sequences, and the frequency of each whitelist barcode is counted. The barcodes with less than 2 nucleotide mismatches were corrected and included for further analysis. Prior to the alignment of the sequences to a mouse reference sequence (mm10), the cutadapt tool was used to trim the barcodes using default parameters. Reads with less than 25 bp were not included in further analysis. Fragments were then identified as reading pairs if their MAPping Quality (MAPQ) scores were larger than 30 on both reads, not mitochondrial, and not chimerically mapped. Once the fragments were filtered, fragments.tsv.gz file was created marking the start and end of the fragment after adjusting the 5′ ends of the read-pair to account for transposition. The file was position-sorted and ran through the SAMtools tabix command with default parameters. Peak calling was also performed with Cell Ranger using the sites as determined by the ends of the fragments in the position-sorted fragments.tsv.gz file, which counts the number of transposition events at each base-pair along the genome. A signal threshold was set up based on an odds-ratio of 1/5 that determines whether a region is a peak signal or noise. Consequently, not all cut sites were within a peak region. Peaks within 500 bp of each other were merged to produce a position-sorted BED file of peaks. Finally, the Peak-Barcode Matrix was produced which consisted of the counts of fragment ends within each peak region for each barcode. This raw Peak-Barcode Matrix captures the enrichment of open chromatin accessibility per barcode.
The h5 files generated in Cell Ranger were subsequently analyzed using Signac which is an extension of Seurat R package for the analysis, interpretation, and exploration of single-cell chromatin datasets. First, for each sample dataset, (2D-R, 2D-L, 2W-R and 2W-L), a Seurat object was created, which contained the Peak Barcode-Matrix and the fragments file. Once the data objects were generated, the enrichment of Tn5 integration events at transcriptional start sites (TSS) was calculated for each sample. As a quality control, the cells with TSS enrichment score >1 and cells with at least 500 peaks were retained and used for further analysis.
The filtered datasets were then merged and the frequency-inverse document frequency (TF-IDF) normalization was performed followed by feature selection and dimensional reduction which returned a low dimensional representation of the object (analogous to the principal component analysis used in scRNaseq). Next, a graph-based clustering and non-linear dimension reduction were performed for UMAP visualization.
The analysis of differentially accessible regions between different cell clusters was performed with Signac, which follows a logistic regression and takes into account the differential sequencing depth (Stuart et al., 2019). Furthermore, chromatin accessibility was studied by Monocle 2 and Cicero R packages through the pseudotime trajectory analysis from scATACseq data (https://cole-trapnell-lab.github.io/cicero-release/docs_m3/#single-cell-accessibility-trajectories). In addition, cis-regulatory networks information obtained using Cicero was applied to our scATACseq dataset following this vignette https://cole-trapnell-lab.github.io/cicero-release/docs/#constructing-cis-regulatory-networks (Pliner et al., 2018).
Moreover, motif analysis was performed with Signac by two complementary methods (https://satijalab.org/signac/articles/motif_vignette.html). The first one performed a hypergeometric analysis to test the probability of observing the motif at the given frequency by chance comparing with a background set of peaks matched for GC content. The second one identified a differential motif activity analysis using ChromVar. In both cases, default parameters were used.
Integration of scRNaseq and scATACseq datasets
Once the scRNaseq and scATACseq datasets were individually analyzed using the same Seurat v 3.6 package, they were further integrated using the Harmony algorithm as described by Raychaudhuri laboratory (Korsunsky et al., 2019).
Gene ontology analysis and functional pathway analysis
Gene ontology analysis was performed using PANTHER (Protein Analysis Through Evolutionary Relationships). Moreover, the term used in these analyses was GO molecular function. For functional pathway analysis, expression data from both scRNaseq and scATACseq analyses were used to generate differentially expressed genes or accessible genes, respectively, among cell clusters identified by the UMAP analyses. A p value of < 0.05 and a false discovery rate (FDR) of < 0.05 served as cutoffs for significance between the groups. Significantly upregulated genes between a group and all remaining groups were selected as the exclusively regulated genes per group. These genes were used for further pathway analysis using Panther (Mi et al., 2017). Significantly regulated functional clusters or single pathways were further grouped by the indicated functional classes and compared by the enrichment score. Significantly enriched (p < 0.05) canonical pathways were selected as representative pathways for each cluster. Additionally, gene ontology and functional pathway analysis were further validated by using Partek Flow® software using internal modules following the above-mentioned parameters.
En face immunofluorescence staining of mouse carotid arteries
Two weeks following the PCL surgery of C57BL/6 mice, LCAs and RCAs were dissected out and used for en face double-immunostaining using CDH5 antibody (an EC marker) with antibodies for C1QA, LYZ (immune cell markers), or phospho-STAT1 (p-STAT1) as we described (Kheirolomoom et al., 2015). AlexaFluor555- or Alexafluor647-conjugated secondary antibodies were used, as well as DAPI to stain the nuclei. Samples were imaged using a Zeiss LSM 510 META confocal microscope (Carl Zeiss). Image analysis was performed using ImageJ software to quantitate average fluorescence.
Chronic exposure of HAECs to shear stress in vitro
HAECs were exposed to chronic laminar shear stress (LS, mimicking s-flow) or oscillatory shear stress (OS, mimicking d-flow) for 7 days as described in Ghim et al. (2018) with a slight modification. Briefly, we replaced the polydimethylsiloxane (PDMS) and stainless-steel ring, which were used previously to segment the inner circle and the outer ring areas, with a tape (Frogtape) with a hydrophobic surface. The wells of a 6-well plate were segmented into two regions of high LS or low OS, by taping either the center (10.5 mm radius) or outer-ring (10.5 to 17.4 mm, inner and outer radius, respectively) using a Frogtape, which prevented cell-growth on its hydrophobic surface. The segmented wells were sterilized with 100% ethanol, air-dried under the UV light for 1h, and coated with 0.1% gelatin on non-taped regions. HAECs (300,000 cells per well) were seeded and grown to confluency in complete media (MCDB 131, 10% FBS, 1% Pen-Strep, 1% L-glutamine, 1% ECGS, 15 ng/mL IGF-1, 1 μg/mL hydrocortisone, 50 μg/mL ascorbic acid, 5 ng/mL VEGF, 5 ng/mL EGF, 5 ng/mL FGF). Once confluent, the cells were placed on an orbital shaker (5mm orbital radius at 150 RPM) in a CO2 incubator for 7 days to expose HAECs to LS (time-averaged wall shear stress of 0.5 to 0.7 Pa) or OS (0.3 Pa), while changing the media every two days. After 7 days, cells were lysed to collect total RNAs. RT-qPCR was performed to determine the effect of chronic LS and OS on the expression of two key shear sensitive genes (KLF2 and KLF4), Mϕ marker genes (C1QC and C5AR1), and EndMT marker genes (SNAI1 and TAGLN). 18S was used as an internal control for quantification of qPCR. See Table S3 for primer sequences. Fold changes were calculated using the 2^-ΔΔCt method as we described (Son et al., 2013).
QUANTIFICATION AND STATISTICAL ANALYSES
The statistical analysis for scRNaseq differential gene expression analysis was performed using Partek® Genomics Suite, GSA (Gene Specific Analysis). This algorithm is a statistical modeling approach used to test for differential expression of genes or transcripts in Partek® Flow®. GSA is capable of considering the following response distributions: Normal, Lognormal, Lognormal with shrinkage, Negative Binomial, Poisson, and ANOVA. Moreover, it also performs multivariate analysis to see which factors are influencing that gene. (*p < 0.05; **p < 0.01; ***p < 0.001).
All validation experiments were performed from at least three independent biological experiments and quantified blindly. Sample sizes, statistical tests and p values are indicated in the text, figures, and figure legends. All the quantitative data are presented in mean ± SD. Statistical analyses were performed using GraphPad Prism software. Initially, the datasets were analyzed for normality using the Shapiro-Wilk test (p < 0.05) and equal variance using the F test (p > 0.05). Data that followed a normal distribution and possessed equal variance were analyzed using 2-tailed Student’s t test or or Mann-Whitney U test (*, p < 0.05; **, p < 0.01; ***, p < 0.001).
Image analysis/Immunostaining quantification
En face In vivo and HAECs In vitro fluorescent quantifications and analyses were performed using ImageJ software to quantitate average fluorescence.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified Rat Anti-Mouse CD144 | BD Biosciences | RRID: AB_395707 |
| Anti-C1qa antibody | LifeSpan Biosciences | LS-C806881-100 |
| Lysozyme Recombinant Rabbit Monoclonal Antibody (ST50-02) | Thermo Fisher Scientific | RRID: AB_2809443 |
| Phospho-Stat1 (Tyr701) (58D6) | Cell-Signaling | RRID: AB_561284 |
| Alexa Fluor® 555 Donkey Anti-Rabbit IgG H&L | Abcam | ab150074; RRID: AB_2636997 |
| Alexa Fluor® 647 Goat anti Rat IgG (H+L) | Thermo Fisher Scientific | ab150075; RRID: AB_141778 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Isoflurane | Patterson vet | 789 313 89 |
| Buprenorphine | Med-Vet International | RXBUPRENOR5-V |
| Collagenase II | MP Biomedicals | 02100502.5 |
| Digitonin | Sigma-Aldrich | D141-100MG |
| QIAzol Lysis Reagent | QIAGEN | 79306 |
| PerfeCTa SYBR Green FastMix ROX | Quantabio | 95073-05K |
| MCDB 131 1X media | Corning | 15-100-CV |
| Fetal Bovine Serum - Premium Select | R&D systems | S11550 |
| Trypsin/EDTA 0.05% | VWR Scientific | 25-052-CI |
| Dnase1 | New England Biolabs Inc | M0303S |
| Penicillin-Streptomycin (10,000 U/mL) | Thermo Fisher Scientific | 15140122 |
| L-Glutamine (200 mM) | Thermo Fisher Scientific | 25030-081 |
| Recombinant Human IGF-I/IGF-1 Protein, CF | R&D Systems | 291-G1-200 |
| Human Recombinant EGF | STEMCELL Technologies | 78006.2 |
| Human Recombinant VEGF | BioAbChem Inc. | 44-VEGF C |
| FGF 2 Human | ProSpec | CYT-218 |
| L-Ascorbic Acid Bioxtra Crystalline | Sigma-Aldrich | A5960-25G |
| Hydrocortisone,bioreagent, suitable for cell culture | Sigma-Aldrich | H0888-5G |
| 6 Well Non-treated Plate, Sterile | CellTreat | 229506 |
| Hoechst 33342, Trihydrochloride, Trihydrate | Thermo Fisher Scientific | H3570 |
| Critical Commercial Assays | ||
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | 4368813 |
| Chromium Next GEM Single Cell ATAC Reagent Kits v1.1 | 10X Genomics | 1000175 |
| Chromium Next GEM Single Cell 3′ Reagent Kits v3.1 | 10X Genomics | 1000121 |
| Deposited Data | ||
| scRNaseq FastQ files | This study | https://www.ncbi.nlm.nih.gov/bioproject Accession # PRJNA646233 |
| scATACseq FastQ files | This study | https://www.ncbi.nlm.nih.gov/bioproject Accession # PRJNA646233 |
| Human Aortic Endothelial Cells (HAEC) | Lonza | CC-2535 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J mice (10-wk-old) | Jackson Laboratory | Stock No: 000664 |
| Oligonucleotides | ||
| qPCR primers, (see Table S3) | This paper | N/A |
| Software and Algorithms | ||
| R version 3.6.2 | R Foundation | https://www.r-project.org |
| Cell Ranger 3.1.0 | 10X Genomics | https://support.10xgenomics.com/ single-cell-gene-exp |
| Seurat 3.1.3 | Stuart et al., 2019 | https://github.com/satijalab/seurat |
| Signac 0.2.5 | Stuart et al., 2019 | https://github.com/timoast/signac |
| Monocle 2.8.0 | Qiu et al., 2017 | https://github.com/cole-trapnell-lab/monocle-release |
| Cicero | Pliner et al., 2018 | https://cole-trapnell-lab.github.io/cicero-release/ |
| Ggplot2 v3.2.1 | Hadley Wickham | https://cran.r-project.org |
| Harmony | Korsunsky et al., 2019 | https://github.com/immunogenomics/harmony |
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov |
| Other | ||
| Mouse reference genome NCBI build GRCm38 (mm10) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/grc/mouse |
| StepOnePlus Real-Time PCR System | Thermo Fisher Scientific | 4376600 |
| PSU-10i orbital platform shaker | VWR | 89194-490 |
| Delicate Surface Painter’s Tape | Frogtape | 280222 |
Highlights.
scRNA-seq and scATAC-seq of mouse carotid arteries after PCL surgery are presented
Carotid artery endothelial cells are heterogeneous and plastic in response to flow
Disturbed flow reprograms ECs to proatherogenic EndMT and EndICLT phenotypes
Disturbed flow regulates access to known and unreported TF binding sites
ACKNOWLEDGMENTS
This work was supported by funding from National Institutes of Health grants HL119798, HL095070, and HL139757 to H.J. H.J. was also supported by the Wallace H. Coulter Distinguished Faculty Chair Professorship. This study was supported in part by the Emory Integrated Genomics Core (EIGC), which is subsidized by the Emory University School of Medicine and is one of the Emory Integrated Core Facilities. Additional support was provided by the Georgia Clinical and Translational Science Alliance of the National Institutes of Health under award no. UL1TR002378. The content is solely the responsibility of the authors and does not necessarily reflect the official views of the National Institutes of Health.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108491.
DECLARATION OF INTERESTS
H.J. is the founder of FloKines Pharma.
REFERENCES
- Ajami NE, Gupta S, Maurya MR, Nguyen P, Li JY, Shyy JY, Chen Z, Chien S, and Subramaniam S (2017). Systems biology analysis of longitudinal functional response of endothelial cells to shear stress. Proc. Natl. Acad. Sci. USA 114, 10990–10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Soudi A, Kaaij MH, and Tas SW (2017). Endothelial cells: from innocent bystanders to active participants in immune responses. Autoimmun. Rev 16, 951–962. [DOI] [PubMed] [Google Scholar]
- Alberts-Grill N, Rezvan A, Son DJ, Qiu H, Kim CW, Kemp ML, Weyand CM, and Jo H (2012). Dynamic immune cell accumulation during flow-induced atherogenesis in mouse carotid artery: an expanded flow cytometry method. Arterioscler. Thromb. Vasc. Biol 32, 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondareva O, Tsaryk R, Bojovic V, Odenthal-Schnittler M, Siekmann AF, and Schnittler HJ (2019). Identification of atheroprone shear stress responsive regulatory elements in endothelial cells. Cardiovasc. Res 115, 1487–1499. [DOI] [PubMed] [Google Scholar]
- Brooks AR, Lelkes PI, and Rubanyi GM (2002). Gene expression profiling of human aortic endothelial cells exposed to disturbed flow and steady laminar flow. Physiol. Genomics 9, 27–41. [DOI] [PubMed] [Google Scholar]
- Bykov Y, Kim SH, and Zamarin D (2020). Preparation of single cells from tumors for single-cell RNA sequencing. Methods Enzymol. 632, 295–308. [DOI] [PubMed] [Google Scholar]
- Chiu JJ, and Chien S (2011). Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol. Rev 91, 327–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Gout PW, Collins CC, and Wang Y (2012). Epithelial immune cell-like transition (EIT): a proposed transdifferentiation process underlying immune-suppressive activity of epithelial cancers. Differentiation 83, 293–298. [DOI] [PubMed] [Google Scholar]
- Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, Wolf D, Saliba AE, and Zernecke A (2018). Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res 122, 1661–1674. [DOI] [PubMed] [Google Scholar]
- Daniel AE, and van Buul JD (2013). Endothelial junction regulation: a pre-requisite for leukocytes crossing the vessel wall. J. Innate Immun 5, 324–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demos C, Williams D, and Jo H (2020). Disturbed Flow Induces Atherosclerosis by Annexin A2-Mediated Integrin Activation. Circ. Res 127, 1091–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaola N, Davies PF, Pritchard WF Jr., Florez L, Harbeck N, and Polacek DC (1999). Spatial and temporal regulation of gap junction connexin43 in vascular endothelial cells exposed to controlled disturbed flows in vitro. Proc. Natl. Acad. Sci. USA 96, 3154–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domingo-Gonzalez R, Zanini F, Che X, Liu M, Jones RC, Swift MA, Quake SR, Cornfield DN, and Alvira CM (2020). Diverse homeostatic and immunomodulatory roles of immune cells in the developing mouse lung at single cell resolution. eLife 9, e56890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn J, Qiu H, Kim S, Jjingo D, Hoffman R, Kim CW, Jang I, Son DJ, Kim D, Pan C, et al. (2014). Flow-dependent epigenetic DNA methylation regulates endothelial gene expression and atherosclerosis. J. Clin. Invest 124, 3187–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan L, and Karino T (2010). Effect of a disturbed flow on proliferation of the cells of a hybrid vascular graft. Biorheology 47, 31–38. [DOI] [PubMed] [Google Scholar]
- Fraineau S, Palii CG, Allan DS, and Brand M (2015). Epigenetic regulation of endothelial-cell-mediated vascular repair. FEBS J. 282, 1605–1629. [DOI] [PubMed] [Google Scholar]
- Ghim M, Pang KT, Arshad M, Wang X, and Weinberg PD (2018). A novel method for segmenting growth of cells in sheared endothelial culture reveals the secretion of an anti-inflammatory mediator. J. Biol. Eng 12, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser SF, Heumüller AW, Tombor L, Hofmann P, Muhly-Reinholz M, Fischer A, Günther S, Kokot KE, Hassel D, Kumar S, et al. (2020). The histone demethylase JMJD2B regulates endothelial-to-mesenchymal transition. Proc. Natl. Acad. Sci. USA 117, 4180–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guibentif C, Rönn RE, Böiers C, Lang S, Saxena S, Soneji S, Enver T, Karlsson G, and Woods NB (2017). Single-Cell Analysis Identifies Distinct Stages of Human Endothelial-to-Hematopoietic Transition. Cell Rep. 19, 10–19. [DOI] [PubMed] [Google Scholar]
- Herrington W, Lacey B, Sherliker P, Armitage J, and Lewington S (2016). Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res 118, 535–546. [DOI] [PubMed] [Google Scholar]
- Jambusaria A, Hong Z, Zhang L, Srivastava S, Jana A, Toth PT, Dai Y, Malik AB, and Rehman J (2020). Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. eLife 9, e51413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins NT, Padilla J, Boyle LJ, Credeur DP, Laughlin MH, and Fadel PJ (2013). Disturbed blood flow acutely induces activation and apoptosis of the human vascular endothelium. Hypertension 61, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YZ, Jiménez JM, Ou K, McCormick ME, Zhang LD, and Davies PF (2014). Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ. Res 115, 32–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YZ, Manduchi E, Jiménez JM, and Davies PF (2015). Endothelial epigenetics in biomechanical stress: disturbed flow-mediated epigenomic plasticity in vivo and in vitro. Arterioscler. Thromb. Vasc. Biol 35, 1317–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TL, and Nerem RM (2007). Endothelial connexin 37, connexin 40, and connexin 43 respond uniquely to substrate and shear stress. Endothelium 14, 215–226. [DOI] [PubMed] [Google Scholar]
- Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, Ellinor PT, Regev A, Kathiresan S, and Gupta RM (2019). Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation 140, 147–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalucka J, de Rooij LPMH, Goveia J, Rohlenova K, Dumas SJ, Meta E, Conchinha NV, Taverna F, Teuwen LA, Veys K, et al. (2020). Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 180, 764–779.e20. [DOI] [PubMed] [Google Scholar]
- Kempe S, Kestler H, Lasar A, and Wirth T (2005). NF-kappaB controls the global pro-inflammatory response in endothelial cells: evidence for the regulation of a pro-atherogenic program. Nucleic Acids Res. 33, 5308–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirolomoom A, Kim CW, Seo JW, Kumar S, Son DJ, Gagnon MK, Ingham ES, Ferrara KW, and Jo H (2015). Multifunctional Nanoparticles Facilitate Molecular Targeting and miRNA Delivery to Inhibit Atherosclerosis in ApoE(−/−) Mice. ACS Nano 9, 8885–8897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh PR, and Raychaudhuri S (2019). Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Kim CW, Simmons RD, and Jo H (2014). Role of flow-sensitive microRNAs in endothelial dysfunction and atherosclerosis: mechanosensitive athero-miRs. Arterioscler. Thromb. Vasc. Biol 34, 2206–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Kang DW, Rezvan A, and Jo H (2017). Accelerated atherosclerosis development in C57Bl6 mice by overexpressing AAV-mediated PCSK9 and partial carotid ligation. Lab. Invest 97, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutikhin AG, Sinitsky MY, Yuzhalin AE, and Velikanova EA (2018). Shear stress: an essential driver of endothelial progenitor cells. J. Mol. Cell. Cardiol 118, 46–69. [DOI] [PubMed] [Google Scholar]
- Kwak BR, Bäck M, Bochaton-Piallat ML, Caligiuri G, Daemen MJ, Davies PF, Hoefer IE, Holvoet P, Jo H, Krams R, et al. (2014). Biomechanical factors in atherosclerosis: mechanisms and clinical implications. Eur. Heart J 35, 3013–3020, 3020a–3020d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai B, Li Z, He M, Wang Y, Chen L, Zhang J, Yang Y, and Shyy JY (2018). Atheroprone flow enhances the endothelial-to-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol 315, H1293–H1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Lin TE, Lee CI, Zhou J, Huang YH, Lee PL, Shih YT, Chien S, and Chiu JJ (2017). MicroRNA-10a is crucial for endothelial response to different flow patterns via interaction of retinoid acid receptors and histone deacetylases. Proc. Natl. Acad. Sci. USA 114, 2072–2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SC, Lee YC, Yu G, Cheng CJ, Zhou X, Chu K, Murshed M, Le NT, Baseler L, Abe JI, et al. (2017). Endothelial-to-Osteoblast Conversion Generates Osteoblastic Metastasis of Prostate Cancer. Dev. Cell 41, 467–480.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud MM, Serbanovic-Canic J, Feng S, Souilhol C, Xing R, Hsiao S, Mammoto A, Chen J, Ariaans M, Francis SE, et al. (2017). Shear stress induces endothelial-to-mesenchymal transition via the transcription factor Snail. Sci. Rep 7, 3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai J, Virtue A, Shen J, Wang H, and Yang XF (2013). An evolving new paradigm: endothelial cells–conditional innate immune cells. J. Hematol. Oncol 6, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, and Thomas PD (2017). PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45 (D1), D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moonen JR, Lee ES, Schmidt M, Maleszewska M, Koerts JA, Brouwer LA, van Kooten TG, van Luyn MJ, Zeebregts CJ, Krenning G, and Harmsen MC (2015). Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc. Res 108, 377–386. [DOI] [PubMed] [Google Scholar]
- Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison DG, Giddens DP, and Jo H (2010). A model of disturbed flow-induced atherosclerosis in mouse carotid artery by partial ligation and a simple method of RNA isolation from carotid endothelium. J. Vis. Exp (40), 1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak L, Lin Z, and Jain MK (2011). “Go with the flow”: how Krüppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid. Redox Signal 15, 1449–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni CW, Qiu H, Rezvan A, Kwon K, Nam D, Son DJ, Visvader JE, and Jo H (2010). Discovery of novel mechanosensitive genes in vivo using mouse carotid artery endothelium exposed to disturbed flow. Blood 116, e66–e73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro P, Abe J, and Berk BC (2011). Flow shear stress and atherosclerosis: a matter of site specificity. Antioxid. Redox Signal 15, 1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oatley M, Bölükbasi ÖV, Svensson V, Shvartsman M, Ganter K, Zirngibl K, Pavlovich PV, Milchevskaya V, Foteva V, Natarajan KN, et al. (2020). Single-cell transcriptomics identifies CD44 as a marker and regulator of endothelial to haematopoietic transition. Nat. Commun 11, 586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ottersbach K (2019). Endothelial-to-haematopoietic transition: an update on the process of making blood. Biochem. Soc. Trans 47, 591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliner HA, Packer JS, McFaline-Figueroa JL, Cusanovich DA, Daza RM, Aghamirzaie D, Srivatsan S, Qiu X, Jackson D, Minkina A, et al. (2018). Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data. Mol. Cell 71, 858–871.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nature Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnick N, Collins T, Atkinson W, Bonthron DT, Dewey CF Jr., and Gimbrone MA Jr. (1993). Platelet-derived growth factor B chain promoter contains a cis-acting fluid shear-stress-responsive element. Proc. Natl. Acad. Sci. USA 90, 4591–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts ME, Barvalia M, Silva JAFD, Cederberg RA, Chu W, Wong A, Tai DC, Chen S, Matos I, Priatel JJ, et al. (2020). Deep Phenotyping by Mass Cytometry and Single-Cell RNA-Sequencing Reveals LYN-Regulated Signaling Profiles Underlying Monocyte Subset Heterogeneity and Lifespan. Circ. Res 126, e61–e79. [DOI] [PubMed] [Google Scholar]
- Rocha HNM, Garcia VP, Batista GMS, Silva GM, Mattos JD, Campos MO, Nóbrega ACL, Fernandes IA, and Rocha NG (2018). Disturbed blood flow induces endothelial apoptosis without mobilizing repair mechanisms in hypertension. Life Sci. 209, 103–110. [DOI] [PubMed] [Google Scholar]
- Sangwung P, Zhou G, Nayak L, Chan ER, Kumar S, Kang DW, Zhang R, Liao X, Lu Y, Sugi K, et al. (2017). KLF2 and KLF4 control endothelial identity and vascular integrity. JCI Insight 2, e91700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santambrogio L, Berendam SJ, and Engelhard VH (2019). The Antigen Processing and Presentation Machinery in Lymphatic Endothelial Cells. Front. Immunol 10, 1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnittler HJ (1998). Structural and functional aspects of intercellular junctions in vascular endothelium. Basic Res. Cardiol 93 (Suppl 3), 30–39. [DOI] [PubMed] [Google Scholar]
- Seebach J, Dieterich P, Luo F, Schillers H, Vestweber D, Oberleithner H, Galla HJ, and Schnittler HJ (2000). Endothelial barrier function under laminar fluid shear stress. Lab. Invest 80, 1819–1831. [DOI] [PubMed] [Google Scholar]
- Simmons RD, Kumar S, Thabet SR, Sur S, and Jo H (2016). Omics-based approaches to understand mechanosensitive endothelial biology and atherosclerosis. Wiley Interdiscip. Rev. Syst. Biol. Med 8, 378–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son DJ, Kumar S, Takabe W, Kim CW, Ni CW, Alberts-Grill N, Jang IH, Kim S, Kim W, Won Kang S, et al. (2013). The atypical mechanosensitive microRNA-712 derived from pre-ribosomal RNA induces endothelial inflammation and atherosclerosis. Nat. Commun 4, 3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, and Satija R (2019). Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarbell JM, Shi ZD, Dunn J, and Jo H (2014). Fluid Mechanics, Arterial Disease, and Gene Expression. Annu. Rev. Fluid Mech 46, 591–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ten Dijke P, Egorova AD, Goumans MJ, Poelmann RE, and Hierck BP (2012). TGF-β signaling in endothelial-to-mesenchymal transition: the role of shear stress and primary cilia. Sci. Signal 5, pt2. [DOI] [PubMed] [Google Scholar]
- Ueno H, Kanellakis P, Agrotis A, and Bobik A (2000). Blood flow regulates the development of vascular hypertrophy, smooth muscle cell proliferation, and endothelial cell nitric oxide synthase in hypertension. Hypertension 36, 89–96. [DOI] [PubMed] [Google Scholar]
- Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, et al. (2017). Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356, eaah4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi T, Naito H, Suehiro JI, Lin Y, Kawaji H, Iba T, Kouno T, Ishikawa-Kato S, Furuno M, Takara K, et al. (2018). CD157 Marks Tissue-Resident Endothelial Stem Cells with Homeostatic and Regenerative Properties. Cell Stem Cell 22, 384–397.e6. [DOI] [PubMed] [Google Scholar]
- Wang N, Miao H, Li YS, Zhang P, Haga JH, Hu Y, Young A, Yuan S, Nguyen P, Wu CC, and Chien S (2006). Shear stress regulation of Krüppel-like factor 2 expression is flow pattern-specific. Biochem. Biophys. Res. Commun 341, 1244–1251. [DOI] [PubMed] [Google Scholar]
- Wang KC, Yeh YT, Nguyen P, Limqueco E, Lopez J, Thorossian S, Guan KL, Li YJ, and Chien S (2016). Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. USA 113, 11525–11530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, Hamers AAJ, Cochain C, Vafadarnejad E, Saliba AE, et al. (2018). Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res 122, 1675–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Q, Zeng L, Zhang Z, Margariti A, Ali ZA, Channon KM, Xu Q, and Hu Y (2006). Sca-1+ progenitors derived from embryonic stem cells differentiate into endothelial cells capable of vascular repair after arterial injury. Arterioscler. Thromb. Vasc. Biol 26, 2244–2251. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Takahashi T, Asahara T, Ohura N, Sokabe T, Kamiya A, and Ando J (2003). Proliferation, differentiation, and tube formation by endothelial progenitor cells in response to shear stress. J. Appl. Physiol. (1985) 95, 2081–2088. [DOI] [PubMed] [Google Scholar]
- Zhao J, Zhang S, Liu Y, He X, Qu M, Xu G, Wang H, Huang M, Pan J, Liu Z, et al. (2020). Single-cell RNA sequencing reveals the heterogeneity of liver-resident immune cells in human. Cell Discov. 6, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The FastQ files generated from the scRNaseq and scATACseq workflow are available at the NCBI BioProject repository (accession number PRJNA646233). The accession number for the scRNA-seq and scATAC-seq data is https://www.ncbi.nlm.nih.gov/bioproject: PRJNA646233. The code generated during this study are available at GitHub (https://github.com/JoLab-Emory/SingleCell).