

Abstract
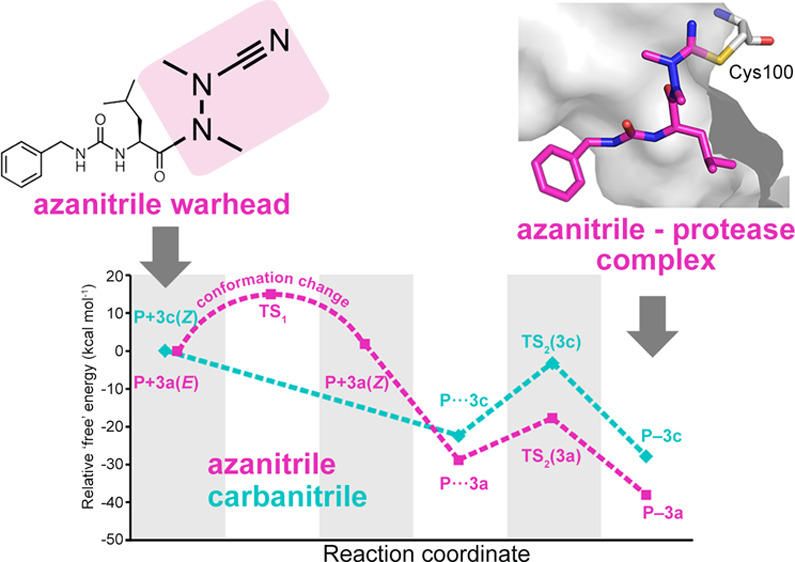
Azapeptide nitriles are postulated to reversibly covalently react with the active-site cysteine residue of cysteine proteases and form isothiosemicarbazide adducts. We investigated the interaction of azadipeptide nitriles with the cathepsin B1 drug target (SmCB1) from Schistosoma mansoni, a pathogen that causes the global neglected disease schistosomiasis. Azadipeptide nitriles were superior inhibitors of SmCB1 over their parent carba analogs. We determined the crystal structure of SmCB1 in complex with an azadipeptide nitrile and analyzed the reaction mechanism using quantum chemical calculations. The data demonstrate that azadipeptide nitriles, in contrast to their carba counterparts, undergo a change from E- to Z-configuration upon binding, which gives rise to a highly favorable energy profile of noncovalent and covalent complex formation. Finally, azadipeptide nitriles were considerably more lethal than their carba analogs against the schistosome pathogen in culture, supporting the further development of this chemotype as a treatment for schistosomiasis.
Keywords: azapeptide inhibitors, cysteine proteases, protein structures, structure−activity relationships, schistosomiasis
Azapeptides, peptides in which the CαH of at least one amino acid has been replaced with nitrogen, have emerged as particularly important peptidomimetic structures (Figure 1). Compared to their parent carbapeptide analogs, bioactive azapeptides can possess improved potency and target selectivity as well as superior pharmacokinetics.1−5 Azadipeptide nitriles were introduced as a class of efficient inhibitors of human cysteine cathepsins.6−9 This chemotype supported the successful development of activity-based probes, modified for organelle-specific delivery to lysosomal cysteine proteases and applied as PET-imaging agents for tumor-associated cathepsin activity.10−12 These reports have highlighted that azadipeptide nitriles can enrich the portfolio of inhibitors of cysteine proteases suitable as activity-based probes13,14 and potential therapeutics against parasitic and protozoal infections.15−18 Similar to the well-established dipeptide nitriles, azadipeptide nitriles are thought to undergo a covalent, reversible interaction with the target proteases by forming a stabilized isothiosemicarbazide adduct (Figure 1).6−12
Figure 1.
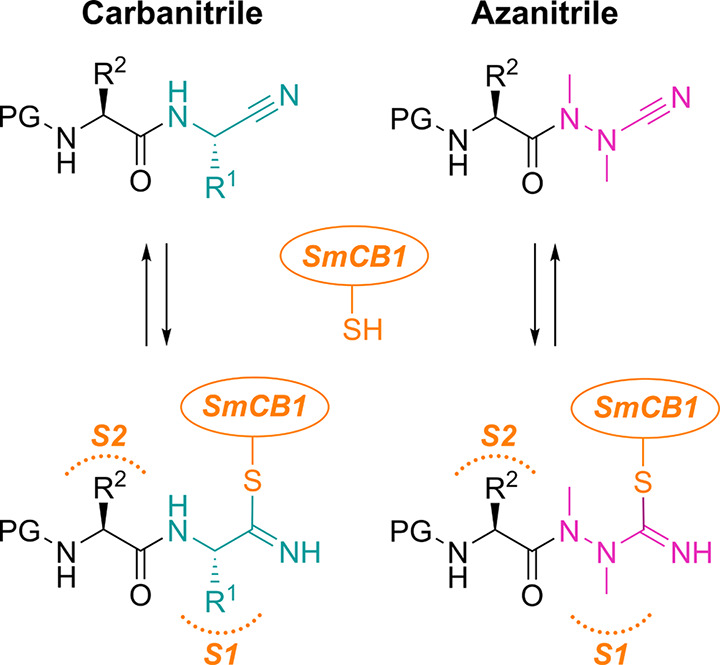
Carba- and azadipeptide nitriles and their reaction with cysteine proteases. Isoelectronic CαH/N exchange in the warhead (cyan/magenta) of dipeptide nitriles (left) leads to azadipeptide nitriles (right). Two carbohydrazide nitrogens in azanitriles need to be alkylated to circumvent spontaneous heterocyclization.6 R1 and R2 are substituents in amino acid residues at the P1 and P2 positions (binding in the enzyme subsites S1 and S2), respectively; PG is a protecting group. The depicted azadipeptide bears an aza-alanine nitrile at the P1 position. Reactive warheads of both chemotypes form a covalent, reversible bond with the thiol of the catalytic cysteine residue (orange) of papain-family cysteine proteases (represented by SmCB1).
Here, we investigated the interaction of aza- and carbadipeptide nitriles with cathepsin B1 (SmCB1) from the human blood fluke, Schistosoma mansoni.19,20 Trematode flatworms of the genus Schistosoma cause schistosomiasis (bilharzia), a chronic disease that infects over 200 million people in tropical and subtropical areas.21 As treatment and control of schistosomiasis rely on just one drug, praziquantel, there is impetus to identify new antischistosomals.22−25 The cysteine protease SmCB1 is a central digestive enzyme of the parasite and has been validated as a chemotherapeutic target for the cure of schistosomiasis.20,26 Previously, we determined the crystal structure of SmCB1 as a footing for rational drug development.20,27,28
In this study, we compared the functional properties of dipeptides with azanitrile and carbanitrile warheads and found azanitriles to be superior. The azadipeptide nitriles were potent inhibitors of SmCB1 with antischistosomal activity and, thus, represent a new class of potential drug leads. We present the first crystallographic analysis of a protease–azanitrile inhibitor complex and quantum chemical calculations to provide mechanistic insight into the phenomenon of azanitrile warhead reactivity.
Results and Discussion
SAR Analysis of Aza- and Carbadipeptide Nitriles Reveals a High Potency of SmCB1 Inhibitors with an Azanitrile Warhead
We have evaluated a set of 18 azadipeptide and 50 carbadipeptide nitriles in vitro as potential inhibitors of the SmCB1 protease. The compound scaffold is defined by positions P3 through P1 (Schechter and Berger nomenclature),29 and the substitutions were selected to provide a high diversity in all positions. The compounds were screened against recombinant SmCB1, and their Ki values were determined using a kinetic inhibition assay with the fluorogenic substrate Cbz-Phe-Arg-AMC. The data for seven representative pairs of both chemotypes are shown in Table 1. Analogs with the azanitrile warhead (1a–7a) were more potent than those with the carbanitrile warhead (1c–7c). This general trend was confirmed for the entire set of test compounds (Tables S1 and S2). Most of the aza compounds were effective in nanomolar concentrations with slow-binding kinetics, whereas only 10% of the carba compounds had Ki values <1 μM and had fast-binding behavior with linear progress curves (Figure 2), a typical feature of peptidic (carba)nitrile inhibitors of cysteine proteases.9,30,31 Using the example of the Cbz-capped phenylalanine pair (2a and 2c), additional variations in the carbanitrile warhead by the stepwise introduction of methyl groups at the aminoacetonitrile unit (i.e., the incorporation of alanine, sarcosine, or N-methylalanine-derived nitrile building blocks in 8c–10c) did not improve the inhibition of SmCB1 (Table 2).
Table 1. Pairs of Peptidomimetics with Aza- or Carbanitrile Warheads and Their Inhibition Potency against SmCB1.

The Ki values were determined using a kinetic activity assay with the fluorogenic peptide substrate Cbz-Phe-Arg-AMC.
Figure 2.

Different binding kinetics of SmCB1 inhibitors with azanitrile and carbanitrile warheads represented by 2a and 2c, respectively. Progress curves show the hydrolysis of the fluorogenic substrate Cbz-Phe-Arg-AMC by SmCB1 in the presence of increasing inhibitor concentrations. (A) The azanitrile exhibited a time-dependent inhibition characterized by nonlinear progress curves typical of slow-binding kinetics. (B) Linear progress curves obtained for the carbanitrile are characteristic of fast-binding inhibitors. In dose–response plots, the derived steady-state reaction velocities were plotted against inhibitor concentration, and the inhibition constants Ki were obtained. In the kobs versus [I] plot, the first-order rate constants kobs from the time-dependent progress curves were plotted against inhibitor concentrations to show a linear dependence. For details on data fitting and kinetic parameters, see the Methods.
Table 2. Effect of Variations in the Carbanitrile Warhead on SmCB1 Inhibition.

The Ki values were determined using a kinetic activity assay with the fluorogenic peptide substrate Cbz-Phe-Arg-AMC.
Table 3 shows the SAR analysis of the P1 to P3 positions of the azanitrile scaffold. The presence of the azanitrile warhead was not sufficient for strong potency, as compound 1a, which lacks a residue side chain for the P2–S2 interaction, was not active. Larger hydrophobic and aromatic residues at the P2 position strengthen binding to the protease (1a versus 2a, 11a). Also, modification of the α-nitrogen substituent in the aza-amino nitrile unit of 2a by extended hydrophobic P1 substituents could improve inhibition (2a versus 8a, 9a). In the P3 position, the introduction of a benzyl urea moiety as present in 3a markedly increased the inhibitory potency. A comparison of P3 triaryl compounds, each with leucine in the P2 position (4a, 5a, 17a), showed that the amide linkage and the associated triaryl group (5a) was preferred over the urea and methylurea linkages (4a and 17a, respectively). The amide bond contributes to the binding affinity because its methylation decreased potency 300-fold (5a versus 6a). When the entire structure of 5a was maintained but leucine was replaced with a less appropriate P2 amino acid, i.e., homocycloleucine in 7a, a 40-fold decrease in potency was measured. Also, we analyzed the second-order rate constants of azadipeptide nitriles (k2nd, Table S1). The k2nd values of the five most potent compounds with single-digit nM Ki values (from 4.6 to 6.2 nM) were particularly high for 3a, 5a, and 11a (20.4, 29.0, and 32.7 × 103 M–1 s–1, respectively) and somewhat lower for 8a and 9a (5.2 and 2.1 × 103 M–1 s–1), indicating a slower association of the enzyme–inhibitor complex, presumably due to the presence of the large P1 substituents in 8a and 9a (R1 = Bn–CH2– and CH3–(CH2)4–, respectively).
Table 3. Inhibition of SmCB1 and Antischistosomal Activity of Azadipeptide Nitriles.

The abbreviations
used are as follows:
The Ki values were determined using a kinetic activity assay with the fluorogenic peptide substrate Cbz-Phe-Arg-AMC.
Phenotypic changes in the parasite in the presence of 10 μM inhibitor are indicated for 24 and 48 h (see Table S3) and converted to a severity score on a scale from 0 (no effect) to 4 (severe).
2a is listed in both P1 and P2 sections.
N-Methylated leucine.
Phenotypic Effects of Azanitriles on Schistosoma mansoni
A panel of 35 azanitrile and carbanitrile inhibitors that were effective against SmCB1 (Tables 1–3 and S2) was phenotypically screened in vitro at 10 μM against S. mansoni newly transformed schistosomula (NTS), the postinvasive stage of the parasite that feeds on host blood. The resulting phenotypic responses were graded from 0 through 4, i.e., from the least to the most severe. Figure 3 compares the severity scores for the azanitrile and carbanitrile analog pairs (for complete phenotyping data for the panel, see Table S3). We found that both inhibitor chemotypes substantially differed in their bioactivities.
Figure 3.
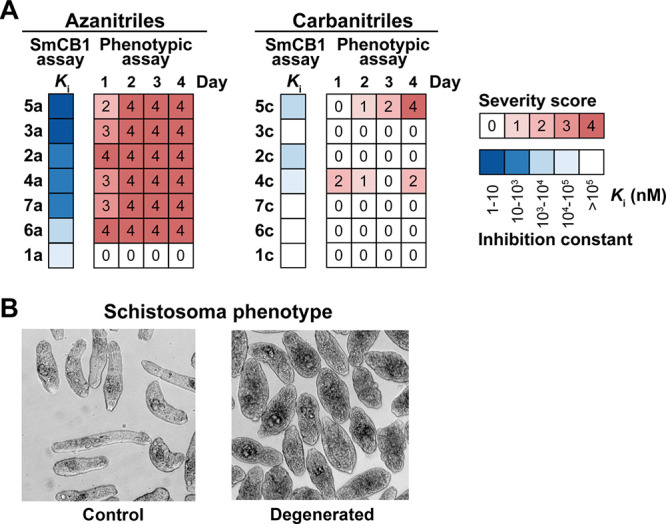
Correlation of the antischistosomal activity with the inhibition of SmCB1 by azanitrile and carbanitrile analogs. (A) Pairs of analogs with azanitrile and carbanitrile warheads were phenotypically screened against S. mansoni newly transformed schistosomula (NTS) and the data arising was compared to those for the inhibition of SmCB1. Phenotypic changes in the parasite (Table S3) were observed every day for 4 days in the presence of 10 μM inhibitor. Changes were converted to a severity score on a scale from 0 (no effect) to 4 (severe; red heat map). Ki values for SmCB1 inhibition (from Table 1) are shown in the blue heat map. (B) An example of an inhibitor-induced degenerated phenotype in NTS of S. mansoni versus untreated controls (for details, see Table S3).
The azanitrile inhibitors caused rapid and severe phenotypes (scores of 3 and 4) except for 1a, which was ineffective against NTS and SmCB1. In contrast, the incubation of NTS with the carbanitrile analogs resulted in more occasional and lower severity scores. The correlation between the severity scores generated by the azanitriles against the parasite and their potency of inhibition of SmCB1 was highly significant (Table S3, Spearman correlation test with nonzero value coefficients, 20 000 permutations, p < 0.001). The most inhibitory azanitriles (with Ki values in the single-digit nM range) were also tested for their cytotoxicity against two human cell lines. They displayed low cytotoxicity at the same concentrations used in the NTS assay (Table S4), indicating that the observed phenotypic changes were specific to the parasite. In conclusion, azadipeptide nitriles that target SmCB1 were demonstrated to be effective antischistosomal compounds.
Crystallographic Analysis Identifies the Binding Mode of Azanitriles to SmCB1
The crystal structure of SmCB1 in complex with the single-digit nanomolar inhibitor 3a was determined at a resolution of 1.3 Å (PDB ID: 6YI7; Table S5). The binding mode of 3a in the active site is presented in Figure 4. The inhibitor’s P1 to P3 residues occupy the S1 to S3 binding subsites of SmCB1. Through the nucleophilic attack of the thiol group of the catalytic Cys100 residue, an isothiosemicarbazide adduct is formed that incorporates the CAB atom of the nitrile moiety (Figures 4D and S1). The NAA atom of the nitrile moiety is stabilized by two hydrogen bonds to the backbone amide of Cys100 and the side chain amide of Gln94 (Figure 4D). Analogous interactions were observed in the structure of rat cathepsin B complexed with a carbanitrile inhibitor.32
Figure 4.

Binding mode of the azadipeptide nitrile inhibitor 3a in the SmCB1 active site. (A) Overall crystal structure of the SmCB1–3a complex in surface (enzyme) and stick (inhibitor) representation. In the SmCB1 active site (boxed), the catalytic residues Cys100 (yellow) and His270 (pink) and major subsite residues (cyan) are highlighted. (B) Chemical structure of 3a forming a covalent bond with the S atom of the catalytic Cys100. The azanitrile warhead is boxed in gray, and atom labeling is indicated (hydrogen atoms are omitted). (C) Zoomed view of (A) showing the active site residues that form nonpolar interactions (green) with 3a (in stick representation with carbon atoms in magenta); the catalytic residues are also indicated. (D) The P1 to P3 positions of the inhibitor bind the corresponding S1 to S3 subsites of the SmCB1 active site. Dashed lines indicate hydrogen bonds formed between SmCB1 residues (gray) and 3a (magenta); heteroatoms have standard color coding (O, red; N, blue; S, yellow). Coordinates are deposited under PDB code 6YI7.
Azadipeptides are atropochiral molecules due to the restricted rotation around the methylated N–N axis. The induction of chirality was demonstrated by methylation of the hydrazine fragment in model azapeptides, which led to the E-configuration of the respective CO–N bond and, hence, to atropisomerism.33 However, E-configured peptidomimetic ligands should possess a weaker affinity for the active site of a protease than the Z-configured isomers. This is also supported by data for cathepsin inhibition involving E- and Z-locked azadipeptide nitriles.34 The configuration of nonlocked azadipeptide nitriles bound to the protease active site is so far unknown. Noteworthy, in this study, we were able to demonstrate the Z-configuration of the C(O)–NAE(NAC) fragment for the enzyme-bound prototypical inhibitor 3a (Figure 4).
Insights were also gained into the binding with the S2 and S3 subsites. At the P2 position, 3a contains a leucine substituent making nonpolar interactions with residues Ala271 and Leu146 of SmCB1 (Figure 4C). Residue Glu316 in the S2 subsite has been shown to rotate out of the binding pocket to avoid steric clash with bulky ligand substituents.20 In the SmCB1–3a complex, the flexibility of Glu316 was demonstrated by its dual conformation with one of the conformations making contact with the P2 leucine. The P3 position of the inhibitor is formed by a benzylaminocarbonyl group. The lower value of the electron density of this substituent suggests its static or dynamic disorder (Figure S2). In the crystallographic model, one conformation of the benzylaminocarbonyl moiety was modeled into the electron density map, representing the major position. Nevertheless, it is obvious that the P3 substituent can acquire several alternative positions within the S3 subsite, which is rather wide and less engaged in ligand binding.20
Quantum Chemical Calculations Demonstrate the Highly Favorable Energetics of the Azanitrile Reaction Mechanism
Computational approaches based on quantum mechanics have proven powerful in exploring catalytic mechanisms of cysteine proteases and designing their covalent inhibitors.20,35−39 In the present study, quantum chemical calculations were employed to analyze the “free” energy profile of the binding reaction of 3a to SmCB1, proceeding via a noncovalent intermediate to the final covalent complex (Figure 5A; “free” energies refer to the sums of gas phase energies and solvation free energies). We observed different configurations of the planar amide group C(O)–N(N) at the warhead (Figure 5B). The bound 3a adopts the Z-configuration as seen in the SmCB1–3a crystallographic complex; however, the modeling of unbound 3a reveals that the E-configuration is more stable in solvent (by 1.9 kcal mol–1) (Table S6 and Figure S3). This is in line with the preferred E-conformation determined experimentally for other free azanitriles and trisubstituted hydrazides in general.33,34,40,41 Thus, 3a undergoes a conformational change upon binding to the enzyme. The activation energy for the E- to Z-conversion calculated using the implicit solvent model was approximately 15 kcal mol–1 (Table S6). This rotational barrier is a possible explanation for the slow binding of azanitriles to SmCB1 observed in the inhibition kinetics (Figure 2). On the contrary, for the carbanitrile, 3c, the Z-configuration of the C(O)–N(C) moiety was more stable than the E-configuration for both the modeled SmCB1–3c complex and the uncomplexed 3c (by 3.1 and 2.0 kcal mol–1, respectively) (Figure S3 and the Supplementary Results). This agrees with the Z-configuration reported for various protease-bound and free carbanitriles.32,33 The fact that no conformational change is required for the interaction of 3c with the enzyme determines the fast-binding kinetics of carbanitriles to SmCB1 (Figure 2).
Figure 5.

Computational analysis of the binding reaction of azanitrile and carbanitrile inhibitors to the active site of SmCB1. (A) The “free” energy profile of the binding of azanitrile 3a and carbanitrile 3c was determined using quantum chemical calculations. Individual states along the reaction pathway (indicated by numbers) are defined by their relative “free” energies (Table S6). (B) The unbound azanitrile inhibitor has the E-configuration in solution (with minimum “free” energy) and undergoes a conformational change to the Z-configuration that was also demonstrated crystallographically in the SmCB1–3a complex. (C) Modeled states upon binding of the azanitrile inhibitor to the active site include an initial noncovalent complex (4), a transition state with proton transfer from His270 to H2O (5), and a final covalent complex after proton transfer to the nitrile group (6). The distance (sulfur–carbon) of the catalytic Cys100 and the inhibitor’s CAB atom is 3.2, 2.3, and 1.8 Å, respectively.
We next investigated the intermediate noncovalent complexes of 3a and 3c with SmCB1. In optimized noncovalent models, the catalytic residue His270 is positively charged, and the thiolate sulfur atom of Cys100 is separated by about 3.2 Å from the CAB atom of the inhibitor (Figure 5C). The formation of the noncovalent complex is energetically favorable for both inhibitors with a higher “free” energy gain for 3a (Figure 5A, Table S6). The stronger noncovalent interaction of 3a might be the result of a distinct electrostatic pattern of the azanitrile warhead (Figure S4 and the Supplementary Results). Conversion to the covalent complex is associated with the formation of a covalent bond between the S atom of Cys100 and CAB atom of the inhibitor and a concomitant water-mediated proton transfer from His270 to the NAA atom of the nitrile group (Figure 5C). The transformation proceeds via a transition state with the S···CAB distance of about 2.3 Å and a proton localized on H3O+ (crystallographic water molecule 628). The transition barrier is 11.1 and 19.2 kcal mol–1 for 3a and 3c, respectively, and the transition state has lower “free” energy than the initial separated reactants (Table S6). The final covalent complexes with a S–CAB separation of 1.8 Å are more stable by 9.2 and 5.4 kcal mol–1 for 3a and 3c, respectively, compared to the noncovalent complexes, thus indicating that the “free” energy gain is higher for 3a also in the last step of the reaction (Table S6).
To conclude, the azanitrile 3a has more favorable thermodynamics of its reaction with SmCB1 by 10.2 kcal mol–1 compared to its carbanitrile analog (Table S6), which corresponds to their inhibitory potencies differing by several orders of magnitude (Table 1). The computational analysis showed that the carbanitrile inhibitor has the Z-configuration both in solution and in covalent complex with SmCB1, whereas the azanitrile inhibitor undergoes the E- to Z-transformation. Further, the Z-configuration of the azanitrile in the SmCB1 noncovalent complex is more stable than the E-configuration (by 3.5 kcal mol–1, Supplementary Results), suggesting that azanitrile binding involves a “configurational selection”42 determined by the topology of the SmCB1 active site. The linear dependence of the kobs values on the inhibitor concentrations (Figure 2) indicates a one-step kinetic reaction mechanism of the slow-binding azanitrile inhibitor,43 which is, however, composed of several distinct mechanistic events in the physicochemical reaction scheme. On the basis of computational analysis, E- to Z-conversion is the kinetic controlling step, while the covalent bond formation is not supposed to be attributed to the slow binding because the transition barrier TS2 of azanitrile is even lower than that of the fast-binding carbanitrile. The slow binding observed for azanitriles is a known kinetic feature of several other chemotypes of covalent inhibitors of cysteine proteases.35,44−46
Conclusions
We investigated the inhibition potency of dipeptidomimetics with azanitrile and carbanitrile warheads against the cysteine protease SmCB1, a drug target from the human blood fluke S. mansoni. A screen of 68 compounds, including analog pairs of both chemotypes, demonstrated the nanomolar inhibition potency of the azanitriles and their general superior potency over their carbanitrile counterparts. Furthermore, the azanitriles were more quickly lethal to the schistosome parasite in vitro and, accordingly, represent a new class of compounds for the development of schistosomiasis drugs. The study provides a platform for further improvement of the azanitrile scaffold based on, for example, the introduction of a P3 triaryl moiety that is connected via an amide bond to a hydrophobic P2 amino acid and that proved particularly advantageous for SmCB1 inhibition. Further research will also address the selectivity of azanitrile inhibitors that are, in general, highly potent against human cysteine cathepsins with endopeptidase activity.6,7
The atomic-level azanitrile–target interaction has not been elucidated up to now. In this study, we solved the first crystal structure of SmCB1 in complex with an azadipeptide nitrile and structurally analyzed the azanitrile-binding mode. By then analyzing the quantum chemical “free” energy profiles of the azanitrile- and carbanitrile-binding reactions with SmCB1, we demonstrated that the azanitriles alone undergo an E- to Z-conformational change upon binding to the enzyme, which we propose as an explanation for the different inhibition kinetic behavior of both chemotypes. Furthermore, azanitriles have a significantly higher “free” energy gain in three consecutive steps along the reaction coordinate, including formation of the initial noncovalent inhibitor–enzyme complex, the transition state, and the final covalent complex with the inhibitor warhead linked to the enzyme catalytic site. The data on the distinct conformational dynamics and binding energetics of the azanitrile warhead provide a mechanistic insight into the reactivity of azanitrile peptidomimetics.
Methods
Synthesis of Inhibitors
General Conditions
Except for compound 3c, the inhibitors used in this study have been synthesized as described.6−8,47 Thin-layer chromatography was carried out on Merck (Darmstadt, Germany) aluminum sheets, silica gel 60 F254. Detection was performed with a UV light at 254 nm. Preparative column chromatography was performed on Merck silica gel (0.063–0.200 mm, 60 Å). Melting points were determined on a Büchi (Essen, Germany) 510 oil bath apparatus. 1H NMR (500 MHz) and 13C NMR (125 MHz) spectra were recorded on a Bruker Avance DRX 500 spectrometer and 1H NMR (600 MHz) and 13C NMR (150 MHz) spectra, on a Bruker Avance III 600 NMR spectrometer. Chemical shifts δ are given in ppm referring to the signal center using the solvent peaks for reference: DMSO-d6 2.49/39.7 ppm. LC-MS analyses were carried out on an API2000 (Applied Biosystems, Darmstadt, Germany) mass spectrometer coupled to an Agilent (Santa Clara, CA, USA) 1100 LC system using a Phenomenex Luna C18 column (Phenomenex, Aschaffenburg, Germany; 50 × 2.0 mm, particle size 3 μm). The purity of the compounds was determined using the diode array detector (DAD) of the LC-MS instrument between 196 and 400 nm. HRMS spectra were recorded on a microTOF-Q (Bruker, Köln, Germany) mass spectrometer connected to a Dionex (Thermo Scientific, Braunschweig, Germany) Ultimate 3000 LC via an ESI interface using a Nucleodur C18 Gravity column (50 × 2.0 mm I.D., 3 μm, Macherey-Nagel, Düren, Germany). The compounds were of at least 95% purity. All compounds passed the PAINS filter using a false positive remover.48
(S)-tert-Butyl 1-(Cyanomethylamino)-4-methyl-1-oxopentan-2-ylcarbamate (II)

(S)-2-(tert-Butoxycarbonylamino)-4-methylpentanoic acid hydrate (I, 5.39 g, 21.6 mmol) was dissolved in anhydrous THF (30 mL) and cooled to −25 °C. N-Methylmorpholine (2.61 mL, 2.41 g, 23.8 mmol) and isobutyl chloroformate (3.10 mL, 3.25 g, 23.8 mmol) were consecutively added to the stirred solution. Aminoacetonitrile monosulfate (6.65 g, 43.1 mmol) was suspended in H2O (3 mL). The resulting suspension was treated with 5 N NaOH (17.0 mL) under ice-cooling and added to the reaction mixture when the precipitation of N-methylmorpholine hydrochloride occurred. It was allowed to warm to room temperature within 0.5 h and stirred for a further 1.5 h. After evaporation of the solvent, the resulting aqueous residue was extracted with EtOAc (3 × 30 mL). The combined organic layer was washed with aq. 10% KHSO4 (30 mL), H2O (30 mL), a sat. aq. NaHCO3 solution (30 mL), H2O (30 mL), and brine (30 mL). The solvent was dried over Na2SO4, filtered, and evaporated to dryness. The crude residue was purified by recrystallization from EtOAc/n-hexane to obtain II as a white solid (4.01 g, 69%); mp 118–120 °C (lit.49 mp 114–116 °C) (Scheme 1).
Scheme 1. Synthesis of Compound 3c.

Reagents and conditions: (a) (1) ClCO2i-Bu, NMM, THF, −25 °C and (2) H2NCH2CN × H2SO4, NaOH, H2O, THF, −25 °C to rt, 2 h; (b) TFA, CH2Cl2, rt, 2 h; (c) benzyl isocyanate, Et3N, CH2Cl2, 0 °C for 15 min to rt for 16 h.
1H NMR (600 MHz, DMSO-d6) δ 0.83 (d, 3J = 6.5 Hz, 3H) and 0.86 (d, 3J = 6.6 Hz, 3H, CH(CH3)2), 1.33–1.39 (m, 1H, CH–CHH), 1.36 (s, 9H, C(CH3)3), 1.43 (ddd, 2J = 13.6 Hz, 3J = 10.1 Hz, 3J = 5.2 Hz, 1H, CH–CHH), 1.52–1.63 (m, 1H, CH(CH3)2), 3.95 (td, 3J = 9.1 Hz, 3J = 4.8 Hz, 1H, NH–CH), 4.08–4.12 (m, 2H, CH2–C≡N), 6.96 (d, 3J = 8.2 Hz, 1H, O–CO–NH), 8.52 (t, 3J = 5.7 Hz, 1H, CH–CO–NH). 13C NMR (150 MHz, DMSO-d6) δ 21.56, 23.06 (CH(CH3)2), 24.41 (CH(CH3)2), 27.25 (NH–CH2), 28.35 (C(CH3)3), 40.58 (CH–CH2), 52.65 (NH–CH), 78.27 (C(CH3)3), 117.74 (C≡N), 155.55 (O–CO–NH), 173.49 (CH–CO–NH). LC-MS (ESI) (90% H2O to 100% MeOH in 10 min, then 100% MeOH for 10 min, DAD 196–200 nm), 100% purity, m/z = 270.0 ([M + H]+), 287.0 ([M + NH4]+).
(S)-2-(3-Benzylureido)-N-(cyanomethyl)-4-methylpentanamide (3c)

(S)-tert-Butyl 1-(cyanomethylamino)-4-methyl-1-oxopentan-2-ylcarbamate (II, 2.02 g, 7.50 mmol) was dissolved in anhydrous CH2Cl2 (200 mL) and treated with TFA (50 mL). The reaction mixture was stirred at room temperature for 2 h. The solvent was then evaporated, and the residue was diluted with CH2Cl2 (4 × 50 mL) and evaporated to remove the excess TFA. The crude product was dissolved in anhydrous CH2Cl2 (30 mL) and cooled to 0 °C. Subsequently, benzyl isocyanate (2.01 mL, 2.18 g, 16.3 mmol) was added, followed by triethylamine (5.15 mL, 3.76 g, 37.1 mmol). Stirring at 0 °C was prolonged for 15 min, and it then continued at room temperature overnight. After evaporation of the solvent, the residue was suspended in H2O (50 mL) and extracted with EtOAc (3 × 75 mL). The organic layer was washed with aq. 10% KHSO4 (50 mL), H2O (50 mL), a sat. aq. NaHCO3 solution (50 mL), H2O (50 mL), and brine (50 mL), dried over Na2SO4, filtered, and evaporated to dryness. The crude residue was purified by preparative column chromatography using a gradient of petroleum ether/EtOAc (1:1) to EtOAc (100%), followed by recrystallization from EtOAc of the product containing fractions to give a white solid (0.34 g, 15%); mp 192–193 °C (Scheme 1).
1H NMR (500 MHz, DMSO-d6) δ 0.85 (d, 3J = 6.6 Hz, 3H) and 0.88 (d, 3J = 6.7 Hz, 3H, CH(CH3)2), 1.33–1.45 (m, 2H, CH–CH2), 1.53–1.63 (m, 1H, CH(CH3)2), 4.07–4.11 (m, 2H, CH2–C≡N), 4.18–4.22 (m, 2H, CH2–NH–CO–NH), 6.17 (d, 3J = 8.4 Hz, 1H, NH–CH), 6.42 (t, 3J = 6.0 Hz, 1H, CH2–NH–CO–NH), 7.17–7.25 (m, 3H, 2H, 4H), 7.26–7.33 (m, 2H, 3H), 8.66 (t, 3J = 5.6 Hz, 1H, CH–CO–NH). 13C NMR (125 MHz, DMSO-d6) δ 21.96, 23.09 (CH(CH3)2), 24.42 (CH(CH3)2), 27.19 (NH–CH2), 41.91 (CH–CH2), 43.06 (CH2–NH–CO–NH), 51.59 (CH–CH2), 117.73 (C≡N), 126.77 (C-4), 127.13 (C-2), 128.39 (C-3), 140.75 (C-1), 157.76 (NH–CO–NH), 174.07 (CH–CO–NH). LC-MS (ESI) (90% H2O to 100% MeOH in 10 min, then 100% MeOH for 10 min, DAD 210–400 nm), 99% purity, m/z = 302.9 ([M + H]+). HRMS, calcd. for C16H22N4O2: [M + H]+m/z 303.1814; found: 303.1816.
Production of Recombinant SmCB1
A nonglycosylated mutant of the SmCB1 zymogen (Uniprot accession Q8MNY2) was expressed using the pPICZαA vector in the yeast Pichia pastoris, activated by S. mansoni legumain and purified as described previously.27,50 All purification steps were performed under reducing conditions in the presence of 2 mM dithiothreitol and 1 mM EDTA under an argon atmosphere to prevent the active site cysteine residue from oxidation.
Preparation of the SmCB1–Inhibitor Complex
The freshly activated SmCB1 (0.5 mg mL–1) was incubated with a 4-fold molar excess of the inhibitor 3a in 10 mM sodium acetate, pH 5.5, containing 20 mM cysteine and 1 mM EDTA, for 3 h at room temperature under an argon atmosphere. The enzyme inhibition was monitored using a kinetic assay with the fluorogenic substrate Cbz-Phe-Arg-AMC.20 The complex was buffer-exchanged into 5 mM sodium acetate, pH 5.5, and concentrated to the final concentration of 4 mg mL–1 using Amicon Ultracel-10k centrifugal units (Millipore, Burlington, USA); the inhibitor 3a was maintained in a 4-fold molar excess to SmCB1 in the mixture.
Protein Crystallization and Data Collection
Crystals of the SmCB1–3a complex were obtained by vapor diffusion in a hanging drop. The drop consisting of 1 μL of protein solution and 1 μL of reservoir solution was equilibrated over 1 mL of reservoir solution at 20 °C. The reservoir solution contained 180 mM ammonium acetate, 80 mM sodium citrate, and 27% PEG 1500, pH 6.1. The protein concentration of the stock solution of the complex was 4 mg mL–1 (in 5 mM sodium acetate, pH 5.5). The obtained needle-shaped crystal was flash-cooled by plunging into liquid nitrogen without cryoprotection. Diffraction data were collected at 100 K on the beamline MX14.1 operated by the Helmholtz-Zentrum Berlin at the BESSY II electron storage ring (Berlin-Adlershof, Germany).51 Diffraction data were processed using the XDS suite of programs.52 Crystal parameters and data collection statistics are given in Table S5.
Crystal Structure Determination
The SmCB1–3a complex crystallized in the orthorhombic space group P212121 containing one molecule in the asymmetric unit and a solvent content of ∼41% (Table S5). The structure of the SmCB1–3a complex was determined by molecular replacement with the program Molrep53 from the CCP4 package54 using the structure of the mature SmCB1 (PDB code: 3S3Q)20 as the search model. Model refinement was carried out using the program REFMAC 5.2 from the CCP4 package,54 interspersed with manual adjustments using Coot.55 The structure was refined using data to a resolution of 1.3 Å. The final crystallographic model contains residues 70–323 (the SmCB1 zymogen numbering is used throughout the paper). Anisotropic refinement of all atomic displacement parameters (ADPs; B-factors) was included in the refinement protocol. The geometric restraints for ligand 3a were constructed by the program Libcheck54 using 3a optimized by the DFT-D3/B3LYP/DZVP method combined with the COSMO56 implicit solvent model. The optimization was performed by the Turbomole7.057 and Cuby458 programs. The 3a molecule was modeled with an occupancy factor of 1 into generally well-defined electron density. The final refinement statistics are given in Table S5. The quality of the final model was validated with Molprobity.59 Atomic coordinates and structure factors have been deposited in the Protein Data Bank with the accession code 6YI7. The structure was analyzed using the program CONTACT.54 All figures showing structural presentations were prepared with the program PyMOL 1.4 (Schrödinger, New York, USA).
Inhibition Assays
Inhibition measurements were performed in triplicates in 96-well microplates (100 μL assay volume) at 37 °C. SmCB1 (20–40 pM) was added to a mixture of the fluorogenic substrate Cbz-Phe-Arg-AMC (20 μM) and an inhibitor (0–100 μM) in 0.1 M sodium acetate, pH 5.5, containing 2.5 mM dithiothreitol and 0.1% PEG 6000. The substrate hydrolysis was monitored in an Infinite M1000 microplate reader (Tecan, Männedorf, Switzerland) at excitation and emission wavelengths of 360 and 465 nm, respectively, for up to 15 and 60 min for fast- and slow-binding inhibitors, respectively. Fast-binding inhibitors showed linear progress curves, and the apparent inhibition constant Ki′ was determined by nonlinear regression using the equation vs/v0 = 1/(1 + [I]/Ki′) by the GraFit software (Erithacus Software, East Grinstead, UK), where vs is the steady-state velocity, v0 is the velocity in the absence of an inhibitor, and [I] is the inhibitor concentration. For slow-binding inhibitors, an observed first-order rate constant kobs was calculated at each inhibitor concentration by fitting the progress curve to the equation P = vst + (vi – vs)(1 – exp(kobst))/kobs + d, where P is the product formation, vs is the steady-state velocity, t is the reaction time, vi is the initial velocity, and d is the offset. The inhibition constant Ki′ was determined by nonlinear regression using the equation vs/v0 = 1/(1 + [I]/Ki′). The true inhibition constants Ki were then calculated using the equation Ki = Ki′/(1 + [S]/Km), where [S] is the substrate concentration and Km is the Michaelis constant. The apparent second-order rate constant k2nd′ was determined by fitting to the linear equation kobs = k2nd′[I] + koff, where koff is the first-order rate constants for the dissociation of the enzyme–inhibitor complex and the true constant k2nd was calculated by the correction k2nd = k2nd′(1 + [S]/Km). The Km value determined for Cbz-Phe-Arg-AMC and SmCB1 was 25 μM. In none of the assay systems did the final concentration of DMSO exceed 1.5%.
Interaction Energy Calculations
The crystal structure of SmCB1 in complex with 3a was used for molecular modeling. Hydrogen atoms were added to the protein by the Reduce and Leap programs in AMBER 1460 and to the ligand using PyMOL 1.7.6. Aspartates, glutamates, lysines, arginines, and histidines were charged (except for the neutral His270 in covalent complexes). Hydrogen atoms were relaxed by annealing from 1000 to 0 K at the MM level in AMBER 14. The FF14SB force field was used for the protein while the GAFF force field was used for the ligand. The cooling runs were 4 ps long with a 1 fs step and Berendsen thermostat used. Fourteen crystallographic water molecules were considered for the modeled complex. The 3c ligand was built manually using the PyMOL 1.7.6.
The QM part comprised residues within 2.5 Å of 3a (i.e., 300 atoms). The QM part was treated at the DFT-D3/BLYP/TZVPP level for single-point energy calculations. For gradient optimizations, we used the DFT-D3/BLYP/DZVP level.61 The rest of the system was treated at the PM6-D3H4X level.62,63 The environment was described by the COSMO implicit solvent model.56 The coupling between QM and SQM was done by Cuby4,58 which calls Turbomole 7.057 and Mopac64 for QM and SQM, respectively. Residues further than 6 Å of 3a in the crystal structure were frozen during the optimization.
To generate the noncovalent complexes of 3a and 3b with SmCB1, it was necessary to break the covalent bond between the inhibitor and the SG atom of Cys100 and transfer the hydrogen atom of the C=N–H group of the inhibitor to His270.35 The bond was broken by employing harmonic restraints and relaxed scans. The obtained noncovalent complexes were reoptimized without any restraint and scored using the standard QM-based scoring function.35,65,66 The relative “free” energy of the noncovalent complex was computed as a sum of gas phase interaction energy (ΔEint), interaction desolvation free energy (ΔΔGsolv), and the change of the conformational “free” energy of the ligand (ΔGconf′).35,65,66 The ligand structures were optimized in solution by the same methodology as the complex. The solvation free energy of the studied ligands was computed by the SMD/HF/6-31G* method67 implemented in Gaussian09.68
Life Cycle of Schistosoma mansoni
S. mansoni (NMRI strain) is routinely maintained in the CDIPD by cycling between Biomphalaria glabrata snails and Golden Syrian hamsters.26,69,70 Vertebrate animal use is supported under a protocol approved by UC San Diego’s Institutional Animal Care and Use Committee (IACUC). The protocol complies with United States federal regulations regarding the care and use of laboratory animals: Public Law 99-158, the Health Research Extension Act, and Public Law 99-198, the Animal Welfare Act, which is regulated by USDA, APHIS, CFR, Title 9, Parts 1, 2, and 3. Informed consent of all participating subjects was obtained.
Schistosome Phenotypic Assay
Newly transformed schistosomula (NTS) of S. mansoni were prepared by mechanically transforming infective larvae (cercariae) as described previously.70,71 NTS (200–300 parasites) were incubated in 200 μL of Basch Medium 169 containing 5% FBS, 100 U mL–1 penicillin, and 100 μg mL–1 streptomycin at 5% CO2 and 37 °C.70,72,73 Inhibitors were added at the final concentration of 10 μM,20,69 and changes in phenotypes were observed every 24 h for up to 4 days. A constrained nomenclature of “descriptors” is used to record the multiple and dynamic changes in movement, shape, and translucence of which the schistosome parasite is capable (Table S3).70 These descriptors are then converted into an ordinal “severity score” system from 0 (no effect) to 4 (maximum effect), which allows for the relative comparison of compound effects, as described.74,75 Images of NTS were captured using a Zeiss Axiovert 40 C inverted microscope (10× objective) and a Zeiss AxioCam MRc digital camera controlled by AxioVision 40 (v. 4.8.1.0) software.
Acknowledgments
The authors thank Brian M. Suzuki, Jiří Brynda, and Vojtěch Spiwok for technical assistance with the phenotypic assays involving the schistosome parasite, the crystallography, and the statistics analysis, respectively. We also thank Helena Mertlíková-Kaiserová for cytotoxicity data. This work was supported by grant LTAUSA19109 from the Ministry of Education, Youth and Sports of the Czech Republic (MEYS), grant NV18-05-00345 from the Ministry of Health of the Czech Republic, project ChemBioDrug CZ.02.1.01/0.0/0.0/16_019/0000729 from ERDF/OPRDE, Gilead Sciences & IOCB Research Center, and institutional projects RVO 61388963 and PROGRES Q25. J.F. and M.L. were supported by project LM2015070 from MEYS. Phenotypic screening at the CDIPD was supported in part by R21AI126296 from NIH-NIAID and OPP1171488 from the Bill and Melinda Gates Foundation. Diffraction data were collected on MX14.1 at the BESSY II electron storage ring operated by the Helmholtz-Zentrum Berlin.
Glossary
Abbreviations
- AMC
aminomethylcoumarin
- Bn
benzyl
- Cbz
benzyloxycarbonyl
- Cha
cyclohexylalanine
- Hph
homophenylalanine
- NTS
newly transformed schistosomula
- RMSD
root-mean-square deviation
- SmCB1
cathepsin B1 from Schistosoma mansoni
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00644.
Supplementary methods, results, and six tables: (S1) inhibition of SmCB1 by azadipeptide nitriles, (S2) inhibition of SmCB1 by carbadipeptide nitriles, (S3) antischistosomal activity of azanitrile and carbanitrile inhibitors, (S4) cytotoxicity of the most potent azanitrile inhibitors of SmCB1, (S5) X-ray data collection and refinement statistics, and (S6) energy calculation of the binding reaction of the inhibitors 3a and 3c to SmCB1; four figures: (S1) atom labeling scheme for the inhibitor 3a, (S2) electron density maps for the inhibitor 3a, (S3) energy calculation of the conformation change of the inhibitors 3a and 3c, and (S4) the molecular surface of the electrostatic potential (ESP) of the inhibitors 3a and 3c (PDF)
Accession Codes
Atomic coordinates and experimental structure factors for the SmCB1–3a complex have been deposited in the Protein Data Bank with accession code 6YI7.
Author Contributions
‡ A.J. and M.H. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Chingle R.; Proulx C.; Lubell W. D. (2017) Azapeptide Synthesis Methods for Expanding Side-Chain Diversity for Biomedical Applications. Acc. Chem. Res. 50 (7), 1541–1556. 10.1021/acs.accounts.7b00114. [DOI] [PubMed] [Google Scholar]
- Proulx C.; Sabatino D.; Hopewell R.; Spiegel J.; Garcia Ramos Y.; Lubell W. D. (2011) Azapeptides and their therapeutic potential. Future Med. Chem. 3 (9), 1139–64. 10.4155/fmc.11.74. [DOI] [PubMed] [Google Scholar]
- Verhelst S. H.; Witte M. D.; Arastu-Kapur S.; Fonovic M.; Bogyo M. (2006) Novel aza peptide inhibitors and active-site probes of papain-family cysteine proteases. ChemBioChem 7 (6), 943–950. 10.1002/cbic.200600001. [DOI] [PubMed] [Google Scholar]
- Mir F. M.; Atmuri N. D. P.; Bourguet C. B.; Fores J. R.; Hou X.; Chemtob S.; Lubell W. D. (2019) Paired Utility of Aza-Amino Acyl Proline and Indolizidinone Amino Acid Residues for Peptide Mimicry: Conception of Prostaglandin F2alpha Receptor Allosteric Modulators That Delay Preterm Birth. J. Med. Chem. 62 (9), 4500–4525. 10.1021/acs.jmedchem.9b00056. [DOI] [PubMed] [Google Scholar]
- Sexton K. B.; Kato D.; Berger A. B.; Fonovic M.; Verhelst S. H.; Bogyo M. (2007) Specificity of aza-peptide electrophile activity-based probes of caspases. Cell Death Differ. 14 (4), 727–32. 10.1038/sj.cdd.4402074. [DOI] [PubMed] [Google Scholar]
- Löser R.; Frizler M.; Schilling K.; Gütschow M. (2008) Azadipeptide nitriles: highly potent and proteolytically stable inhibitors of papain-like cysteine proteases. Angew. Chem., Int. Ed. 47 (23), 4331–4334. 10.1002/anie.200705858. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Lohr F.; Furtmann N.; Kläs J.; Gütschow M. (2011) Structural optimization of azadipeptide nitriles strongly increases association rates and allows the development of selective cathepsin inhibitors. J. Med. Chem. 54 (1), 396–400. 10.1021/jm101272p. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Lohr F.; Lülsdorff M.; Gütschow M. (2011) Facing the gem-dialkyl effect in enzyme inhibitor design: preparation of homocycloleucine-based azadipeptide nitriles. Chem. - Eur. J. 17 (41), 11419–23. 10.1002/chem.201101350. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Schmitz J.; Schulz-Fincke A. C.; Gütschow M. (2012) Selective nitrile inhibitors to modulate the proteolytic synergism of cathepsins S and F. J. Med. Chem. 55 (12), 5982–6. 10.1021/jm300734k. [DOI] [PubMed] [Google Scholar]
- Yang P. Y.; Wang M.; Li L.; Wu H.; He C. Y.; Yao S. Q. (2012) Design, synthesis and biological evaluation of potent azadipeptide nitrile inhibitors and activity-based probes as promising anti-Trypanosoma brucei agents. Chem. - Eur. J. 18 (21), 6528–41. 10.1002/chem.201103322. [DOI] [PubMed] [Google Scholar]
- Loh Y.; Shi H.; Hu M.; Yao S. Q. (2010) Click” synthesis of small molecule-peptide conjugates for organelle-specific delivery and inhibition of lysosomal cysteine proteases. Chem. Commun. (Cambridge, U. K.) 46 (44), 8407–9. 10.1039/c0cc03738a. [DOI] [PubMed] [Google Scholar]
- Löser R.; Bergmann R.; Frizler M.; Mosch B.; Dombrowski L.; Kuchar M.; Steinbach J.; Gütschow M.; Pietzsch J. (2013) Synthesis and radiopharmacological characterisation of a fluorine-18-labelled azadipeptide nitrile as a potential PET tracer for in vivo imaging of cysteine cathepsins. ChemMedChem 8 (8), 1330–44. 10.1002/cmdc.201300135. [DOI] [PubMed] [Google Scholar]
- Van Kersavond T.; Nguyen M. T. N.; Verhelst S. H. L. (2017) Synthesis and Application of Activity-Based Probes for Proteases. Methods Mol. Biol. 1574, 255–266. 10.1007/978-1-4939-6850-3_19. [DOI] [PubMed] [Google Scholar]
- Tan M. S. Y.; Davison D.; Sanchez M. I.; Anderson B. M.; Howell S.; Snijders A.; Edgington-Mitchell L. E.; Deu E. (2020) Novel broad-spectrum activity-based probes to profile malarial cysteine proteases. PLoS One 15 (1), e0227341 10.1371/journal.pone.0227341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deu E. (2017) Proteases as antimalarial targets: strategies for genetic, chemical, and therapeutic validation. FEBS J. 284 (16), 2604–2628. 10.1111/febs.14130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau P. D.; Miller B. W.; McCall L. I.; Almaliti J.; Reher R.; Hirata K.; Le T.; Siqueira-Neto J. L.; Hook V.; Gerwick W. H. (2019) Design of Gallinamide A Analogs as Potent Inhibitors of the Cysteine Proteases Human Cathepsin L and Trypanosoma cruzi Cruzain. J. Med. Chem. 62 (20), 9026–9044. 10.1021/acs.jmedchem.9b00294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettari R.; Previti S.; Maiorana S.; Amendola G.; Wagner A.; Cosconati S.; Schirmeister T.; Hellmich U. A.; Zappala M. (2019) Optimization Strategy of Novel Peptide-Based Michael Acceptors for the Treatment of Human African Trypanosomiasis. J. Med. Chem. 62 (23), 10617–10629. 10.1021/acs.jmedchem.9b00908. [DOI] [PubMed] [Google Scholar]
- Ettari R., Previti S., Di Chio C., and Zappala M. (2020) Falcipain-2 and falcipain-3 inhibitors as promising antimalarial agents. Curr. Med. Chem., 10.2174/0929867327666200730215316. [DOI] [PubMed] [Google Scholar]
- Sajid M.; McKerrow J. H.; Hansell E.; Mathieu M. A.; Lucas K. D.; Hsieh I.; Greenbaum D.; Bogyo M.; Salter J. P.; Lim K. C.; Franklin C.; Kim J. H.; Caffrey C. R. (2003) Functional expression and characterization of Schistosoma mansoni cathepsin B and its trans-activation by an endogenous asparaginyl endopeptidase. Mol. Biochem. Parasitol. 131 (1), 65–75. 10.1016/S0166-6851(03)00194-4. [DOI] [PubMed] [Google Scholar]
- Jílková A.; Řezáčová P.; Lepšík M.; Horn M.; Váchová J.; Fanfrlík J.; Brynda J.; McKerrow J. H.; Caffrey C. R.; Mareš M. (2011) Structural Basis for Inhibition of Cathepsin B Drug Target from the Human Blood Fluke, Schistosoma mansoni. J. Biol. Chem. 286 (41), 35770–35781. 10.1074/jbc.M111.271304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colley D. G.; Bustinduy A. L.; Secor W. E.; King C. H. (2014) Human schistosomiasis. Lancet 383 (9936), 2253–2264. 10.1016/S0140-6736(13)61949-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caffrey C. R. (2007) Chemotherapy of schistosomiasis: present and future. Curr. Opin. Chem. Biol. 11 (4), 433–439. 10.1016/j.cbpa.2007.05.031. [DOI] [PubMed] [Google Scholar]
- Caffrey C. R.; Secor W. E. (2011) Schistosomiasis: from drug deployment to drug development. Curr. Opin. Infect. Dis. 24 (5), 410–417. 10.1097/QCO.0b013e328349156f. [DOI] [PubMed] [Google Scholar]
- Thetiot-Laurent S. A.; Boissier J.; Robert A.; Meunier B. (2013) Schistosomiasis chemotherapy. Angew. Chem., Int. Ed. 52 (31), 7936–7956. 10.1002/anie.201208390. [DOI] [PubMed] [Google Scholar]
- Caffrey C. R., El-Sakkary N., Mäder P., Krieg R., Becker K., Schlitzer M., Drewry D. H., Vennerstrom J. L., and Grevelding C. G. (2019) Drug Discovery and Development for Schistosomiasis. In Neglected Tropical Diseases (Swinney D., Pollastri M., Mannhold R., Buschmann H., and Holenz J., Eds.), Wiley, Weinheim, Germany. [Google Scholar]
- Abdulla M. H.; Lim K. C.; Sajid M.; McKerrow J. H.; Caffrey C. R. (2007) Schistosomiasis mansoni: novel chemotherapy using a cysteine protease inhibitor. PLoS Med. 4 (1), e14 10.1371/journal.pmed.0040014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn M.; Jílková A.; Vondrášek J.; Marešová L.; Caffrey C. R.; Mareš M. (2011) Mapping the Pro-Peptide of the Schistosoma mansoni Cathepsin B1 Drug Target: Modulation of Inhibition by Heparin and Design of Mimetic Inhibitors. ACS Chem. Biol. 6 (6), 609–617. 10.1021/cb100411v. [DOI] [PubMed] [Google Scholar]
- Jílková A.; Horn M.; Řezáčová P.; Marešová L.; Fajtova P.; Brynda J.; Vondrášek J.; McKerrow J. H.; Caffrey C. R.; Mareš M. (2014) Activation Route of the Schistosoma mansoni Cathepsin B1 Drug Target: Structural Map with a Glycosaminoglycan Switch. Structure 22 (12), 1786–1798. 10.1016/j.str.2014.09.015. [DOI] [PubMed] [Google Scholar]
- Schechter I.; Berger A. (1967) On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 27 (2), 157–62. 10.1016/S0006-291X(67)80055-X. [DOI] [PubMed] [Google Scholar]
- Thompson S. A.; Andrews P. R.; Hanzlik R. P. (1986) Carboxyl-modified amino acids and peptides as protease inhibitors. J. Med. Chem. 29 (1), 104–11. 10.1021/jm00151a018. [DOI] [PubMed] [Google Scholar]
- Schmitz J.; Gilberg E.; Löser R.; Bajorath J.; Bartz U.; Gütschow M. (2019) Cathepsin B: Active site mapping with peptidic substrates and inhibitors. Bioorg. Med. Chem. 27 (1), 1–15. 10.1016/j.bmc.2018.10.017. [DOI] [PubMed] [Google Scholar]
- Greenspan P. D.; Clark K. L.; Tommasi R. A.; Cowen S. D.; McQuire L. W.; Farley D. L.; van Duzer J. H.; Goldberg R. L.; Zhou H.; Du Z.; Fitt J. J.; Coppa D. E.; Fang Z.; Macchia W.; Zhu L.; Capparelli M. P.; Goldstein R.; Wigg A. M.; Doughty J. R.; Bohacek R. S.; Knap A. K. (2001) Identification of dipeptidyl nitriles as potent and selective inhibitors of cathepsin B through structure-based drug design. J. Med. Chem. 44 (26), 4524–4534. 10.1021/jm010206q. [DOI] [PubMed] [Google Scholar]
- Ottersbach P. A.; Schnakenburg G.; Gütschow M. (2012) Induction of chirality: experimental evidence of atropisomerism in azapeptides. Chem. Commun. (Cambridge, U. K.) 48 (46), 5772–4. 10.1039/c2cc31161e. [DOI] [PubMed] [Google Scholar]
- Ottersbach P. A.; Schmitz J.; Schnakenburg G.; Gütschow M. (2013) An access to aza-Freidinger lactams and E-locked analogs. Org. Lett. 15 (3), 448–51. 10.1021/ol3030583. [DOI] [PubMed] [Google Scholar]
- Fanfrlík J.; Brahmkshatriya P. S.; Řezáč J.; Jílková A.; Horn M.; Mareš M.; Hobza P.; Lepšík M. (2013) Quantum Mechanics-Based Scoring Rationalizes the Irreversible Inactivation of Parasitic Schistosoma mansoni Cysteine Peptidase by Vinyl Sulfone Inhibitors. J. Phys. Chem. B 117 (48), 14973–14982. 10.1021/jp409604n. [DOI] [PubMed] [Google Scholar]
- Mladenovic M.; Fink R. F.; Thiel W.; Schirmeister T.; Engels B. (2008) On the origin of the stabilization of the zwitterionic resting state of cysteine proteases: a theoretical study. J. Am. Chem. Soc. 130 (27), 8696–705. 10.1021/ja711043x. [DOI] [PubMed] [Google Scholar]
- Paasche A.; Schirmeister T.; Engels B. (2013) Benchmark Study for the Cysteine-Histidine Proton Transfer Reaction in a Protein Environment: Gas Phase, COSMO, QM/MM Approaches. J. Chem. Theory Comput. 9 (3), 1765–77. 10.1021/ct301082y. [DOI] [PubMed] [Google Scholar]
- Schirmeister T.; Kesselring J.; Jung S.; Schneider T. H.; Weickert A.; Becker J.; Lee W.; Bamberger D.; Wich P. R.; Distler U.; Tenzer S.; Johe P.; Hellmich U. A.; Engels B. (2016) Quantum Chemical-Based Protocol for the Rational Design of Covalent Inhibitors. J. Am. Chem. Soc. 138 (27), 8332–5. 10.1021/jacs.6b03052. [DOI] [PubMed] [Google Scholar]
- Quesne M. G.; Ward R. A.; de Visser S. P. (2013) Cysteine protease inhibition by nitrile-based inhibitors: a computational study. Front. Chem. 1, 39. 10.3389/fchem.2013.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löser R.; Pitzschler R.; Köckerling M. (2017) Synthesis and X-ray Crystal Structure of N’-Cyano-N,N’-dimethyl-4-nitrobenzohydrazide. Crystals 7 (10), 290. 10.3390/cryst7100290. [DOI] [Google Scholar]
- Licandro E.; Perdicchia D. (2004) N-acylhydrazines: Future perspectives offered by new syntheses and chemistry. Eur. J. Org. Chem. 2004 (4), 665–675. 10.1002/ejoc.200300416. [DOI] [Google Scholar]
- Vogt A. D.; Di Cera E. (2012) Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry 51 (30), 5894–902. 10.1021/bi3006913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland R. A. (2013) Slow Binding Inhibitors. In Evaluation of Enzyme Inhibitors in Drug Discovery: A Guide for Medicinal Chemists and Pharmacologists, pp 203–244, John Wiley. [PubMed] [Google Scholar]
- Miller B.; Friedman A. J.; Choi H.; Hogan J.; McCammon J. A.; Hook V.; Gerwick W. H. (2014) The marine cyanobacterial metabolite gallinamide A is a potent and selective inhibitor of human cathepsin L. J. Nat. Prod. 77 (1), 92–9. 10.1021/np400727r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokhen M.; Khazanov N.; Albeck A. (2011) The mechanism of papain inhibition by peptidyl aldehydes. Proteins: Struct., Funct., Genet. 79 (3), 975–85. 10.1002/prot.22939. [DOI] [PubMed] [Google Scholar]
- Brady K. D. (1998) Bimodal inhibition of caspase-1 by aryloxymethyl and acyloxymethyl ketones. Biochemistry 37 (23), 8508–15. 10.1021/bi9803325. [DOI] [PubMed] [Google Scholar]
- Laube M.; Frizler M.; Wodtke R.; Neuber C.; Belter B.; Kniess T.; Bachmann M.; Gütschow M.; Pietzsch J.; Löser R. (2019) Synthesis and preliminary radiopharmacological characterisation of an 11C-labelled azadipeptide nitrile as potential PET tracer for imaging of cysteine cathepsins. J. Labelled Compd. Radiopharm. 62 (8), 448–459. 10.1002/jlcr.3729. [DOI] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. (2010) New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 53 (7), 2719–40. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Löser R.; Schilling K.; Dimmig E.; Gütschow M. (2005) Interaction of papain-like cysteine proteases with dipeptide-derived nitriles. J. Med. Chem. 48 (24), 7688–707. 10.1021/jm050686b. [DOI] [PubMed] [Google Scholar]
- Jílková A.; Horn M.; Mareš M. (2020) Structural and Functional Characterization of Schistosoma mansoni Cathepsin B1. Methods Mol. Biol. 2151, 145–158. 10.1007/978-1-0716-0635-3_12. [DOI] [PubMed] [Google Scholar]
- Mueller U.; Forster R.; Hellmig M.; Huschmann F. U.; Kastner A.; Malecki P.; Puhringer S.; Rower M.; Sparta K.; Steffien M.; Uhlein M.; Wilk P.; Weiss M. S. (2015) The macromolecular crystallography beamlines at BESSY II of the Helmholtz-Zentrum Berlin: Current status and perspectives. Eur. Phys. J. Plus 130 (7), 141. 10.1140/epjp/i2015-15141-2. [DOI] [Google Scholar]
- Kabsch W. (2010) Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66 (2), 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. (2000) An approach to multi-copy search in molecular replacement. Acta Crystallogr., Sect. D: Biol. Crystallogr. 56 (12), 1622–1624. 10.1107/S0907444900013780. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Ballard C. C.; Cowtan K. D.; Dodson E. J.; Emsley P.; Evans P. R.; Keegan R. M.; Krissinel E. B.; Leslie A. G.; McCoy A.; McNicholas S. J.; Murshudov G. N.; Pannu N. S.; Potterton E. A.; Powell H. R.; Read R. J.; Vagin A.; Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67 (4), 235–242. 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60 (12), 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Klamt A.; Schuurmann G. (1993) Cosmo - a New Approach to Dielectric Screening in Solvents with Explicit Expressions for the Screening Energy and Its Gradient. J. Chem. Soc., Perkin Trans. 2 (5), 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- Ahlrichs R.; Bar M.; Haser M.; Horn H.; Kolmel C. (1989) Electronic-Structure Calculations on Workstation Computers - the Program System Turbomole. Chem. Phys. Lett. 162 (3), 165–169. 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]
- Řezáč J. (2016) Cuby: An integrative framework for computational chemistry. J. Comput. Chem. 37 (13), 1230–7. 10.1002/jcc.24312. [DOI] [PubMed] [Google Scholar]
- Lovell S. C.; Davis I. W.; Arendall W. B. III; de Bakker P. I.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. (2003) Structure validation by Cα geometry: φ, ψ and Cβ deviation. Proteins: Struct., Funct., Genet. 50 (3), 437–450. 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Case D. A., Babin V., Berryman J. T., Betz R. M., Cai Q., Cerutti D. S., Cheatham T. E., Darden T. A., Duke R. E., Gohlke H., Goetz A. W., Gusarov S., Homeyer N., Janowski P., Kaus J., Kolossváry I., Kovalenko A., Lee T. S., LeGrand S., Luchko T., Luo R., Madej B., Merz K. M., Paesani F., Roe D. R., Roitberg A., Sagui C., Salomon-Ferrer R., Seabra G., Simmerling C. L., Smith W., Swails J., Walker R. C., Wang J., Wolf R. M., Wu X., and Kollman P. A. (2014) AMBER 14, University of California San Francisco, San Francisco (USA).
- Hostaš J.; Řezáč J. (2017) Accurate DFT-D3 Calculations in a Small Basis Set. J. Chem. Theory Comput. 13 (8), 3575–3585. 10.1021/acs.jctc.7b00365. [DOI] [PubMed] [Google Scholar]
- Stewart J. J. (2007) Optimization of parameters for semiempirical methods V: modification of NDDO approximations and application to 70 elements. J. Mol. Model. 13 (12), 1173–213. 10.1007/s00894-007-0233-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Řezáč J.; Hobza P. (2012) Advanced Corrections of Hydrogen Bonding and Dispersion for Semiempirical Quantum Mechanical Methods. J. Chem. Theory Comput. 8 (1), 141–51. 10.1021/ct200751e. [DOI] [PubMed] [Google Scholar]
- Stewart J. J. P. (2004) Optimization of parameters for semiempirical methods IV: extension of MNDO, AM1, and PM3 to more main group elements. J. Mol. Model. 10 (2), 155–164. 10.1007/s00894-004-0183-z. [DOI] [PubMed] [Google Scholar]
- Fanfrlík J.; Bronowska A. K.; Řezáč J.; Přenosil O.; Konvalinka J.; Hobza P. (2010) A reliable docking/scoring scheme based on the semiempirical quantum mechanical PM6-DH2 method accurately covering dispersion and H-bonding: HIV-1 protease with 22 ligands. J. Phys. Chem. B 114 (39), 12666–78. 10.1021/jp1032965. [DOI] [PubMed] [Google Scholar]
- Lepšík M.; Řezáč J.; Kolář M.; Pecina A.; Hobza P.; Fanfrlík J. (2013) The Semiempirical Quantum Mechanical Scoring Function for In Silico Drug Design. ChemPlusChem 78 (9), 921–931. 10.1002/cplu.201300199. [DOI] [PubMed] [Google Scholar]
- Marenich A. V.; Cramer C. J.; Truhlar D. G. (2009) Performance of SM6, SM8, and SMD on the SAMPL1 test set for the prediction of small-molecule solvation free energies. J. Phys. Chem. B 113 (14), 4538–43. 10.1021/jp809094y. [DOI] [PubMed] [Google Scholar]
- Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Petersson G. A., Nakatsuji H., Li X., Caricato M., Marenich A. V., Bloino J., Janesko B. G., Gomperts R., Mennucci B., Hratchian H. P., Ortiz J. V., Izmaylov A. F., Sonnenberg J. L., Williams Ding F., Lipparini F., Egidi F., Goings J., Peng B., Petrone A., Henderson T., Ranasinghe D., Zakrzewski V. G., Gao J., Rega N., Zheng G., Liang W., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Throssell K., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M. J., Heyd J. J., Brothers E. N., Kudin K. N., Staroverov V. N., Keith T. A., Kobayashi R., Normand J., Raghavachari K., Rendell A. P., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Millam J. M., Klene M., Adamo C., Cammi R., Ochterski J. W., Martin R. L., Morokuma K., Farkas O., Foresman J. B., and Fox D. J. (2016) Gaussian 16, Rev. C.01, Gaussian, Inc., Wallingford (USA).
- Fajtová P.; Štefanić S.; Hradilek M.; Dvořák J.; Vondrášek J.; Jílková A.; Ulrychová L.; McKerrow J. H.; Caffrey C. R.; Mareš M.; Horn M. (2015) Prolyl Oligopeptidase from the Blood Fluke Schistosoma mansoni: From Functional Analysis to Anti-schistosomal Inhibitors. PLoS Neglected Trop. Dis. 9 (6), e0003827 10.1371/journal.pntd.0003827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdulla M. H.; Ruelas D. S.; Wolff B.; Snedecor J.; Lim K. C.; Xu F.; Renslo A. R.; Williams J.; McKerrow J. H.; Caffrey C. R. (2009) Drug discovery for schistosomiasis: hit and lead compounds identified in a library of known drugs by medium-throughput phenotypic screening. PLoS Neglected Trop. Dis. 3 (7), e478 10.1371/journal.pntd.0000478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Štefanić S.; Dvořák J.; Horn M.; Braschi S.; Sojka D.; Ruelas D. S.; Suzuki B.; Lim K.-C.; Hopkins S. D.; McKerrow J. H.; Caffrey C. R. (2010) RNA Interference in Schistosoma mansoni Schistosomula: Selectivity, Sensitivity and Operation for Larger-Scale Screening. PLoS Neglected Trop. Dis. 4 (10), e850 10.1371/journal.pntd.0000850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontovyč A.; Ulrychová L.; Horn M.; Dvořák J. (2020) Collection of Excretory/Secretory Products from Individual Developmental Stages of the Blood Fluke Schistosoma mansoni. Methods Mol. Biol. 2151, 55–63. 10.1007/978-1-0716-0635-3_5. [DOI] [PubMed] [Google Scholar]
- Dvořák J.; Fajtová P.; Ulrychová L.; Leontovyč A.; Rojo-Arreola L.; Suzuki B. M.; Horn M.; Mareš M.; Craik C. S.; Caffrey C. R.; O’Donoghue A. J. (2016) Excretion/secretion products from Schistosoma mansoni adults, eggs and schistosomula have unique peptidase specificity profiles. Biochimie 122, 99–109. 10.1016/j.biochi.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser J.; Schurigt U.; Suzuki B. M.; Caffrey C. R.; Holzgrabe U. (2015) Anti-Schistosomal Activity of Cinnamic Acid Esters: Eugenyl and Thymyl Cinnamate Induce Cytoplasmic Vacuoles and Death in Schistosomula of Schistosoma mansoni. Molecules 20 (6), 10873–83. 10.3390/molecules200610873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long T.; Neitz R. J.; Beasley R.; Kalyanaraman C.; Suzuki B. M.; Jacobson M. P.; Dissous C.; McKerrow J. H.; Drewry D. H.; Zuercher W. J.; Singh R.; Caffrey C. R. (2016) Structure-Bioactivity Relationship for Benzimidazole Thiophene Inhibitors of Polo-Like Kinase 1 (PLK1), a Potential Drug Target in Schistosoma mansoni. PLoS Neglected Trop. Dis. 10 (1), e0004356 10.1371/journal.pntd.0004356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.