

Summary
SEC14p is the yeast phosphatidylinositol (PI)/phosphatidylcholine (PC) transfer protein, and it effects an essential stimulation of yeast Golgi secretory function. We now report that the SEC14p localizes to the yeast Golgi and that the SEC14p requirement can be specifically and efficiently bypassed by mutations in any one of at least six genes. One of these suppressor genes was the structural gene for yeast choline kinase (CKI), disruption of which rendered the cell independent of the normally essential SEC14p requirement. The antagonistic action of the CKI gene product on SEC14p function revealed a previously unsuspected influence of biosynthetic activities of the CDP-choline pathway for PC biosynthesis on yeast Golgi function and indicated that SEC14p controls the phospholipid content of yeast Golgi membranes in vivo.
Introduction
Our efforts have been directed at determining the role of the Saccharomyces cerevisiae SEC14 gene product in stimulating yeast Golgi secretory function. We have shown that SEC14p is a cytosolic factor that stimulates essential Golgi secretory functions and is conserved across wide phylogenetic boundaries (Bankaitis et al., 1989). Furthermore, we have established that SEC14p is the phosphatidylinositol (PI)/phosphatidylcholine (PC) transfer protein of yeast and that the PI/PC transfer activities exhibit a marked in vitro lability when assayed in cell-free extracts prepared from sec14ts mutant yeast (Bankaitis et al., 1990). Those findings established an in vivo function for a phospholipid transfer protein, even though these proteins have been the subject of intense biochemical and physical characterization (Helmkamp, 1986). Such characterization notwithstanding, a number of fundamental questions regarding phospholipid transfer protein function remain. The most basic of these include: do the in vitro phospholipid transfer activities of these proteins accurately reflect catalysis of phospholipid equilibration between membranes in vivo; what organelle membranes do these proteins interact with in vivo; and what is the precise role of these proteins in vivo? We have used the power of the yeast system to address these questions in the case of SEC14p.
In this report we demonstrate that SEC14p exhibits a colocalization with the yeast Golgi complex, the organelle that is dysfunctional in sec14 mutant strains. Moreover, we show that mutations in any one of at least six genes render the yeast cell independent of the normally essential SEC14p requirement. On the basis of our demonstration that bypass suppression of sec14 defects could be achieved by loss-of-function mutations in structural genes for enzymes that participate in the CDP-choline pathway for PC biosynthesis, we propose that SEC14p functions in phospholipid equilibration in vivo—a function that is directly consistent with the biochemical activity of SEC14p in vitro. These results reveal a previously unsuspected relationship between yeast Golgi function and phospholipid biosynthesis. Based upon our demonstration that SEC14p colocalizes with yeast Golgi bodies in vivo, SEC-14p-mediated phospholipid equilibration is proposed to be critical to the secretory competence of Golgi membranes in yeast.
Results
SEC14p Colocalizes with the Yeast Golgi Marker KEX2p
Previous experiments had demonstrated that, while SEC14p distributed primarily to the yeast cytosol, significant amounts of SEC14p were observed to be associated with particulate fractions (Bankaitis et al., 1989). It was of interest to determine whether the particulate fraction represented a Golgi-associated SEC14p population. This possibility was investigated both by immunolocalization and cofractionation of SEC14p and KEX2p, an integral membrane protein of the yeast Golgi (Franzusoff et al., 1991), in wild-type yeast cells. The immunofluorescence data are shown in Figure 1.
Figure 1.
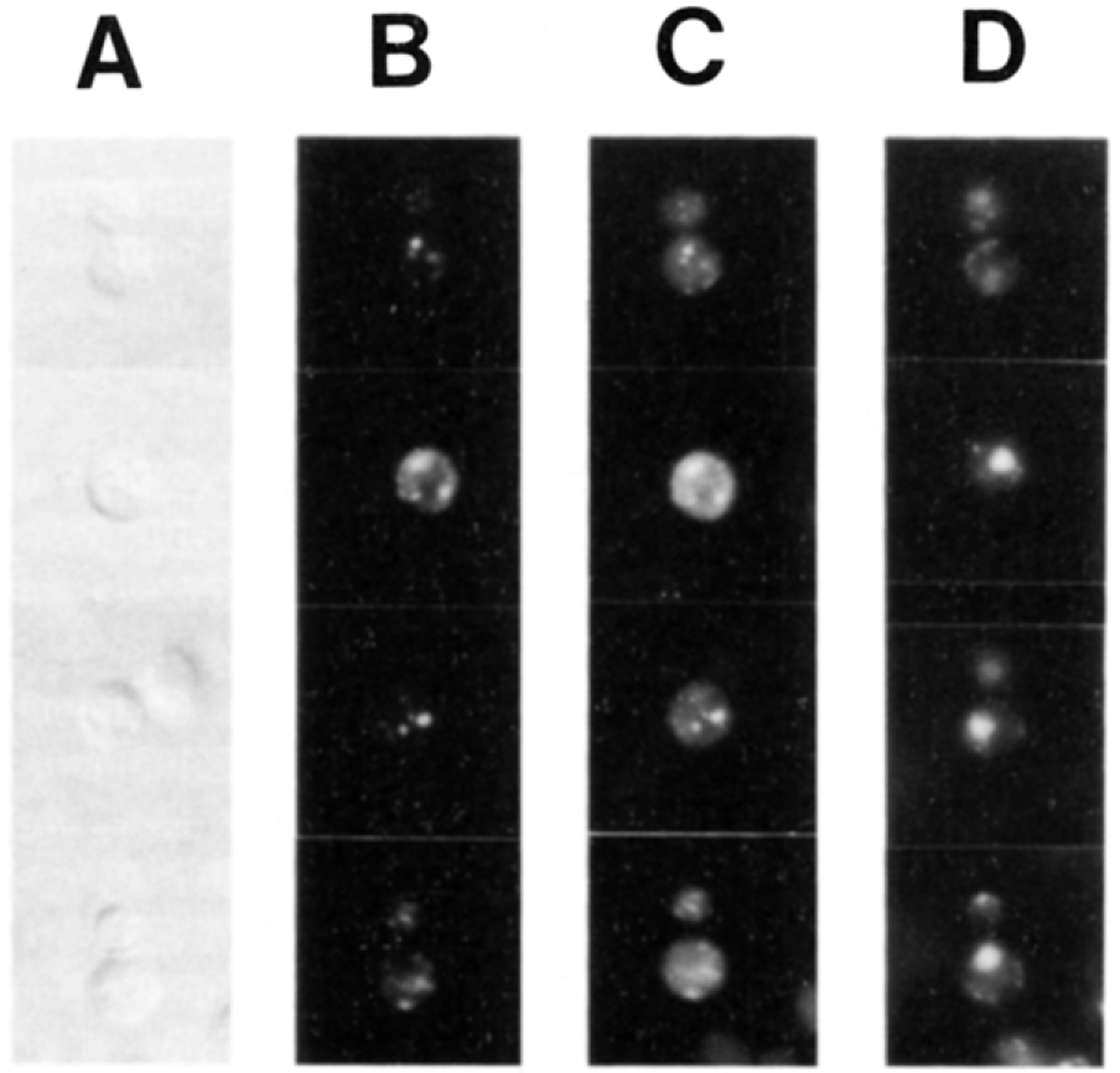
SECl4p Colocalizes with the Yeast Golgi Complex In Vivo Wild-type yeast cells were grown at 25°C and double-stained for fluorescence microscopy with TRITC-conjugated antibodies directed against antibodies bound to KEX2p, a Golgi marker, and FITC-conjugated antibodies directed against SEC14p. Cell outlines are revealed by Nomarski (A). The KEX2p (B) and SEC14p (C) staining profiles are also shown. Nuclear fluorescence is shown in (D) as determined by DAPI (4′, 6′-diamidino-2-phenylindole) staining.
Yeast cells were fixed, permeabilized, and incubated with affinity-purified KEX2p primary antibodies (from R. Fuller) and tetramethyl rhodamine isothiocyanate (TRITC)-conjugated goat anti-rabbit IgG secondary serum. After blocking with bulk rabbit IgG and extensive washing, the cells were finally treated with fluorescein isothiocyanate (FITC)-conjugated SEC14p antibodies. Visualization of the rhodamine staining profile of these yeast cells identified a punctate staining in the cytoplasm that was characteristic of KEX2p localization, as rigorously established by Franzusoff et al. (1991), and revealed the distribution of the yeast Golgi bodies (Figure 1). The authenticity of this signal was confirmed by its absence when cells were stained with TRITC-conjugated secondary antibodies alone, or when Δkex2 yeast cells were tested (data not shown). Treatment of the wild-type yeast cells with the FITC-conjugated SEC14p antibodies also revealed a punctate staining pattern in the cytoplasm that was superimposed upon a diffuse cytoplasmic staining (Figure 1). We generally observed between four and eight brightly stained ovoid bodies per cell. Both the punctate and cytoplasmic fluorescein staining were SEC14p specific, as confirmed by loss of signal upon preincubation of the FITC-conjugated SEC14p antibodies with purified SEC14p antigen, use of preimmune serum to stain the cells, and use of FITC-conjugated SEC14p antibodies to stain yeast sec14° mutant strains (such as those sec14–88::URA3 strains described below). Moreover, the SEC14p staining pattern was independent of whether the cells were costained for KEX2p or not (data not shown). The punctate KEX2p and SEC14p staining patterns appeared to be coincident (compare Figures 1B and 1C). This apparent coincidence of staining was observed for all 150 cells that were so analyzed. Finally, the punctate KEX2p and SEC14p profiles were distinct from nuclear and mitochondrial fluorescence as determined by DAPI staining (Figure 1D). These data suggested that the punctate SEC14p staining revealed a SEC14p population that was associated with the yeast Golgi complex in vivo.
To obtain an independent confirmation of the conclusions reached from the immunofluorescence data, we determined whether the membrane-associated SEC14p codistributed with the KEX2p in subcellular fractionation experiments. A representative experiment is shown in Table 1. The bulk of KEX2p (68%) was recovered in the P100 fraction, in agreement with previous reports (Fuller et al., 1989). A significant amount of KEX2p (i.e., 17% of total) was measured in the P12 fraction as well. SEC14p exhibited a considerable association with sedimentable fractions. About 9% and 20% of total SEC14p was measured in the P12 and P100 fractions, respectively, while about 53% of the total SEC14p was detected in the 100,000 × g supernatant fraction. This relative distribution of SEC14p between cytosolic (53%) and membrane (29%) fractions was in agreement with that previously reported (Bankaitis et al., 1989). We noted that the bulk of the plasma membrane ATPase, the vacuolar dipeptidylaminopeptidase (DPAP-B), the endoplasmic reticulum (ER) marker NADPH cytochrome c reductase, and the mitochondrial cytochrome c oxidase were recovered in either the P1 or P12 fractions. Nevertheless, significant amounts of ATPase (13%), DPAP-B (27%), and reductase (17%) were also detected in the P100 fraction.
Table 1.
Distribution of Marker Proteins in Supernatant and Pellet Fractions
| KEX2p | SEC14p | PM ATPase | DPAP-B | Cyt Reductase | Cyt Oxidase | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Act | % | Act | % | Act | % | Act | % | Act | % | Act | % | |
| Lysate | 2394 | 100 | 2280 | 100 | 30 | 100 | 815 | 100 | 3.11 | 100 | 3.48 | 100 |
| S1 | 2018 | 84 | 1783 | 78 | 10 | 33 | 405 | 50 | 1.6 | 51 | 2.79 | 80 |
| P1 | 321 | 13 | 405 | 18 | 20 | 67 | 417 | 51 | 1.7 | 56 | 0.65 | 19 |
| S12 | 1716 | 72 | 1656 | 73 | 6 | 20 | 257 | 31 | 0.74 | 24 | 0.06 | 2 |
| P12 | 398 | 17 | 209 | 9 | 5 | 17 | 131 | 16 | 0.75 | 24 | 2.72 | 78 |
| S100 | 57 | 2 | 1217 | 53 | 2 | 7 | 28 | 4 | 0.09 | 3 | ND | ND |
| P100 | 1616 | 68 | 445 | 20 | 4 | 13 | 216 | 27 | 0.53 | 17 | 0.03 | 1 |
Wild-type (CTY182) cells were grown in YPD medium at 25°C; the cells were converted to spheroplasts; and osmotic lysates were generated. The lysates were centrifuged at 1000 × g to generate P1 and S1. S1 was recentrifuged at 12,000 × g to generate P12 and S12. The latter was spun at 100,000 × g to yield the P100 and S100 fractions. Aliquots of each fraction were quantitated for KEX2p, SEC14p, and DPAP-B by ELISA. NADPH cytochrome c (Cyt c) reductase, cytochrome c oxidase, and plasma membrane (PM) ATPase were assayed by established methods (see Experimental Procedures). ELISA units are relative values that were calculated from a mean A492 signal that was determined to fall within the linear portion of a standard curve that was obtained by titrating yeast lysate for relative concentration of a particular protein ligand (see Mimms et al., 1990). Quantitative ELISAs were run at 5000 x , 1000 x , and 200 x dilutions of lysate for KEX2p, SEC14p, and DPAP-B, respectively, and are expressed in units per fraction. Unit values for NADPH cytochrome c reductase (μmol of cytochrome c reduced per min), cytochrpme c oxidase (μmol of cytochrome c oxidized per min), and plasma membrane ATPase (μmol of phosphate produced pej min) have been described (see Experimental Procedures). These are expressed on a per fraction basis as well. The activity (Act) of each fraction is given in the appropriate units. The percent of the total relative activity (%) for each fraction with respect to each activity measured is also given. ND = none detectable.
To further resolve Golgi membranes from other contaminating membranes, the P100 fraction was loaded under a self-forming 30%–60% sorbitol flotation gradient and centrifuged to equilibrium (see Experimental Procedures). Fractions were subsequently collected and assayed for all six markers indicated above. As shown in Figure 2A, KEX2p and SEC14p cofractionated in a single peak that corresponded to the 58% sorbitol region of the gradient. The ATPase and reductase activities were apparent only at the very bottom of the gradient, whereas DPAP-B was observed to reside in lighter membranes that were confined to a peak in the 45%–46% sorbitol region of the gradient. The KEX2p-enriched fractions were pooled and determined to contain 61% of total KEX2p and 16% of total SEC14p. We note that these numbers signified nearly quantitative recoveries of KEX2p and SEC14p that were loaded onto the flotation gradient (67% and 20% of the total, respectively). These pooled fractions contained only 1% (or less) of the total ATPase, reductase, and DPAP-B measured in the original lysate (Figure 2A).
Figure 2. Distribution of Marker Proteins across Gradient Fractions.
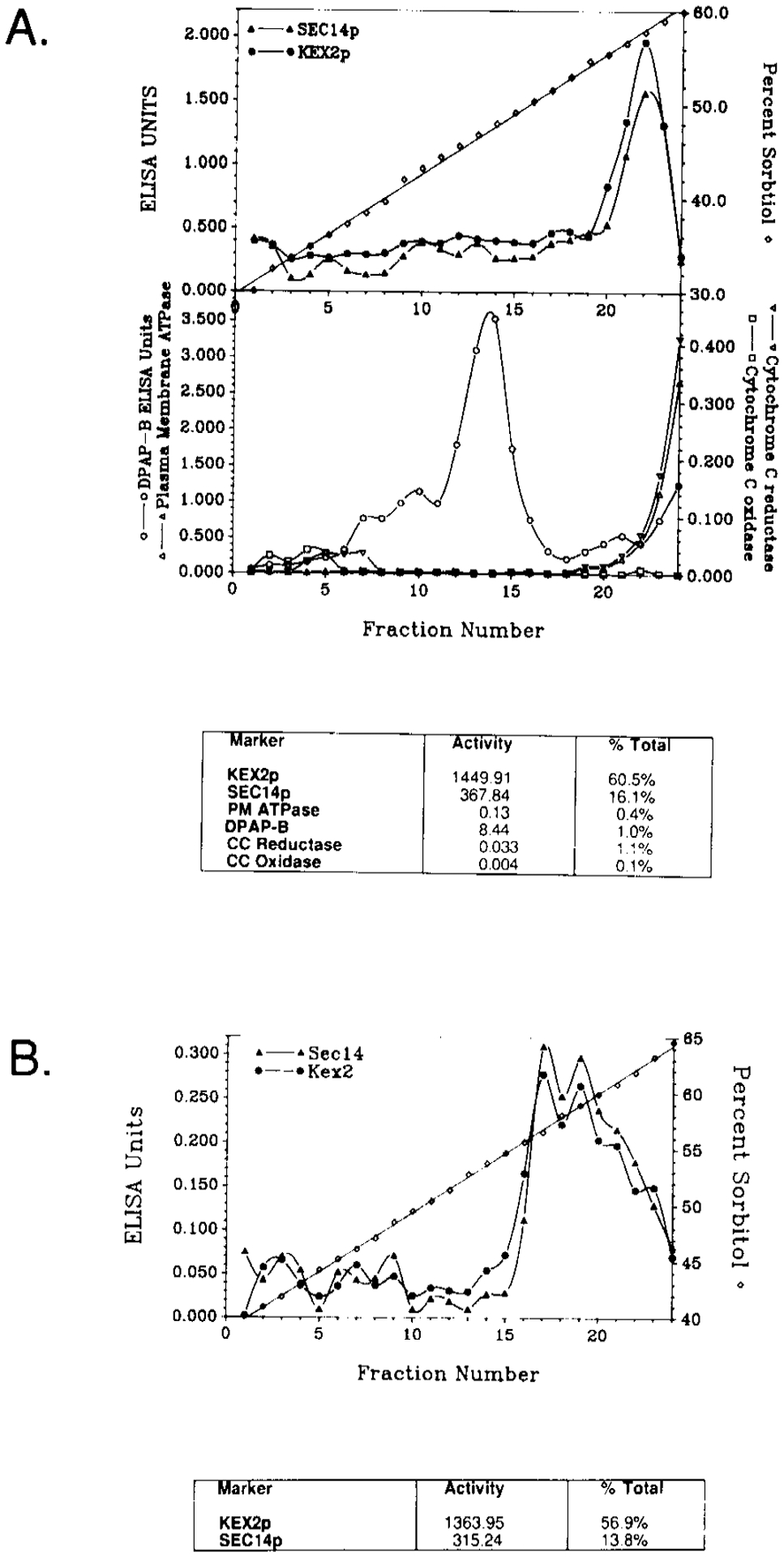
(A) The P100 fraction was collected and loaded under a 30%–60% self-forming sorbitol flotation gradient. After centrifugation to equilibrium, 0.5 ml fractions were collected and assayed for the indicated markers as described in the legend to Table 1 and the Experimental Procedures. Unit values are expressed on a per fraction basis. The relative quantities of each marker in the peak KEX2p fraction pool are tabulated both in terms of units per fraction and percent of total.
(B) The KEX2p fraction pool was layered upon a 40%–65% sorbitol sedimentation gradient. The gradient was fractionated and assayed for KEX2p and SEC14p by quantitative ELISA. Again, the unit values for KEX2p and SEC14p are tabulated in terms of units per fraction and percent of total.
The pooled fractions enriched for KEX2p were layered onto a 40%–65% sorbitol gradient and sedimented to equilibrium by a final round of centrifugation (see Experimental Procedures). Fractions were again collected and analyzed for KEX2p and SEC14p by enzyme-linked immunosorbant assay (ELISA). KEX2p and SEC14p cofractionated in this gradient as well (Figure 2B). Both species were recovered in a single, rather broad peak located in the 56–60% sorbitol region of the gradient. The peak fractions were pooled and determined to contain 57% of total KEX2p and 14% of total SEC14p, respectively (Figure 2B). Again, these data signified essentially quantitative recoveries of the input KEX2p (61%) and SEC14p (16%).
Taken together, the apparent colocalization of KEX2p and SEC14p by immunofluorescence, coupled with the near quantitative cofractionation of P100 SEC14p with purified Golgi membranes, argued strongly that SEC14p associated with the yeast Golgi complex in vivo.
Identification of Six Genes That Suppress sec14 Defects
The sec14–1ts allele is a transition mutation that converts GLY266 (GGT) of SEC14p to ASP (GAT), and renders both the PI and PC transfer activities of SEC14p labile in vitro (Cleves et al., 1989; Bankaitis et al., 1990). The conditional-lethal phenotype of sec14–1ts provided a powerful selection for Ts+ revertants, and we noted that sec14–1ts was a readily revertible allele (~10−6 Ts+ revertants per cell per generation). We isolated and characterized a total of 107 independent and spontaneously arising revertants from the sec14–1ts haploid strains CTY1–1A and CTY2–1C (see Experimental Procedures and Table 4). Standard dominance tests and meiotic segregation analyses showed that 26 of the mutants exhibited a recessive suppression of sec14–1ts (the remaining 81 exhibited a dominant suppressor phenotype), and that each suppressor mutation segregated in the manner expected for a single nuclear gene. Moreover, backcrosses of the suppressor mutants to SEC14 strains indicated independent assortment of each suppressor allele with respect to sec14–1ts. These data led us to conclude that all 107 suppressor mutations were unlinked to sec14–1ts. Complementation analysis demonstrated that the 26 recessive suppressor mutations cleanly fell into four complementation groups, designated sac1 and bsr2-bsr4 (bsr:bypass SEC14p recessive; see below). These were represented by 14, 7, 3, and 2 mutants, respectively. The sac1 mutants uniformly acquired a new cs phenotype and identified a locus whose function also modulates the actin cytoskeleton (Cleves et al., 1989; Novick et al., 1989). We have previously reported a characterization of the sac1 mutants (Cleves et al., 1989), and these shall not be treated in detail here. None of the other bsr mutants exhibited any obvious conditional phenotypes.
Table 4.
Yeast Strains
| Strain | Genotype | Origin |
|---|---|---|
| CTY182 | MATa, ura3–52, Δhis3–200, Iys2–801 | Bankaitis et al. (1989) |
| CTY1–1A | MATa, ura3–52, Δhis3–200, Iys2–801, sec14–1ts | Bankaitis et al. (1989) |
| CTY2–1C | MATα, ade2–101, sec14–1ts | Cleves et al. (1989) |
| CTY68 | MATa, ade2–101, ura3–52, Δhis3–200, Δtrpl, sec14–1ts | This study |
| CTY69 | MATα, ade2–101, ura3–52, Δhis3–200, Δtrpl, sec14–1ts | This study |
| CTY70 | MATa, ade2–101, ura3–52, Δhis3–200, Iys2–801, Δtrpl, sec14–1ts | This study |
| CTY76 | MATα, ade2–101, ura3–52, Δhis3–200, Iys2–801, seel4–1ts | This study |
| CTY110 | CTY1–1A bsd1–110 | This study |
| CTY114 | CTY1–1A bsd1–114 | This study |
| CTY124 | CTY1–1A bsdl-124 | This study |
| CTY160 | MATa, ura3–52, Δhis3–200, sec14–1ts, bsr4-l | This study |
| CTY214 | MATa, ura3–52, ade2–101, leu2–3, 112, his4–519, sec14–ts | This study |
| CTY215 | MATα, ade2–101och, leu2–3, 112, sec14–1ts | This study |
| CTY217 | MATα, ade2–101, ura3–52, Ahis3–200, bsd2–1, sec14–129::HIS3 | This study |
| CTY218 | MATα, ade2–101, ura3–52, Ahis3–200, bsd2–1, sec14A1::HIS3 | This study |
| CTY221 | MATa, ura3–52, Δhis3–200, Iys2–801, bsdl-124, seel4–129::HIS3 | This study |
| CTY222 | MATa, ura3–52, Δhis3–200, Iys2–801, bsdl-124, sec14Δ1::HIS3 | This study |
| CTY225 | MATa, ura3–52, Δhis3–200, Iys2–801, bsr2–2, sec14Δ1::HIS3 | This study |
| CTY226 | MATa, ura3–52, Δhis3–200, Iys2–801, bsr3–1, sec14Δ1::HIS3 | This study |
| CTY227 | MATa, ura3–52, Δhis3–200, bsr4–1, sec14Δ1::HIS3 | This study |
| CTY228 | MATa, ura3–52, Δhis3–200, Iys2–801, bsr2–2, sed4–129::HIS3 | This study |
| CTY229 | MATa, ura3–52, Δhis3–200, Iys2–801, bsr3–1, sec14–129::HIS3 | This study |
| CTY230 | MATa, ura3–52, Δhis3–200, bsr4–1, sec14–129::HIS3 | This study |
| CTY410 | MATa, Δhis3–200, leu2Δ9, cho2::LEU2 | S. Henry |
| CTY411 | MATα, ade2–101, Ahis3–200, Ieu2, trp1, can1, opi3::URA3 | S. Henry |
| CTY416 | MATα, ade5, Ieu2, cdg1 | S. Henry |
| HJ051 | MATa, ura3–52, his−, leu−, trp−, ept1::URA3 | Hjelmstad and Bell (1988) |
| CTYD1 | This study | |
| CTYD43 | Cleves et al. (1989) | |
| CTYD100 | This study | |
| CTYD100.1 | This study | |
| CTYD110.1 | This study | |
| CTYD114.1 | This study | |
| CTYD124.1 | This study |
To determine the number of genes represented by the dominant suppressor mutants, we crossed representative mutants derived from CTY2–1C to the complete set of such mutants derived from CTY1–1A, and vice versa. These diploids were then subjected to meiotic analysis. We found that 80 of the 81 dominant suppressor mutations were tightly linked (composite tetrad data of 816 PD:0 NPD:5 TT). These comprise the bsd1 (bsd: bypass SEC-14p dominant; see below) linkage group and, owing to the collective tightness of the linkage, were presumed to be allelic. One dominant suppressor mutation was unlinked to the bsd1 group and defined the bsd2 gene.
Taken together, the 107 suppressors of sec14–1ts identified six genes. Four (SAC1, BSR2, BSR3, and BSR4) were defined by recessive mutations, whereas two (BSD1 and BSD2) were defined by dominant mutations. Data demonstrating nontrival mechanisms for suppression of sec14 defects by these mutations are provided below.
Suppression Is Effected via Bypass of SEC14p Function
Three general models for extragenic suppression of sec14–1ts were entertained: alteration of gene products that interact with SEC14p or otherwise regulate its activity, upregulation of Sec14pts synthesis, and a bypass of SEC14p function. These models can be distinguished on the basis of the ability of any particular suppressor to overcome the lethality associated with sec14 disruption alleles. In the first two models, the lethality of sec14 null mutations is. predicted to be epistatic to suppression. The bypass model, however, predicts that suppression will be epistatic to lethality of the sec14 null allele.
We have previously reported the construction of. two sec14 disruption alleles that represent recessive-lethal mutations, sec14–129::H1S3 and sec14W1::H1S3 (Bankaitis et al., 1989). The former lesion encodes a truncated SEC14p that exhibits no measureable PI/PC transfer activity (see below), thereby identifying sec14–129::HIS3 as a reasonable null allele. This disruption was recombined into sec14–1ts, Δhis3–200 homozygous diploids that were heterozygous for the suppressor locus in question, and these diploids were subjected to meiotic analysis. In Table 2, we present the data obtained for three different alleles of bsd1 with respect to suppression of sec14–129::H1S3. Asci exhibiting 4:0 and 3:1 segregations of viable to non-viable spores were recovered in each of the crosses involving the bsd1–110, bsd1–114, and bsd1–124 alleles. As expected, each ascus of the 4:0 class exhibited two His+ progeny and each 3:1 class ascus exhibited only one His+ segregant. These data indicated a bypass mode of suppression for bsd1 mutations. The same conclusion was reached for the bsr2, bsr3, bsr4, and bsd2 mutants (data not shown). In all cases but for bsd2 (defined by a single allele) we tested several alleles for suppression of sec14–129::H1S3 and always observed the occurrence of viable His+ meiotic progeny.
Table 2.
Bypass Suppression of a sec14 Disruption by bsd Alleles
| Ascus Types | |||||
|---|---|---|---|---|---|
| (Viable/Nonviable) | |||||
| Strain | Relevant Genotype | 4:0 | 3:1 | 2:2 | |
| CTYD110.1 | 2 | 2 | 2 | ||
| CTYD114.1 | 2 | 11 | 2 | ||
| CTYD124.1 | 6 | 7 | 3 | ||
Suppression of a sec14 disruption allele. The indicated diploid strains were sporulated and subjected to standard meiotic analysis (Sherman et al., 1983). Four-spore asci were dissected and classified with respect to the ratio of viable to nonviable meiotic progeny observed for each ascus. An interpretation of the data is presented in the text.
To confirm that the bsr and bsd mutants were rendered independent of the normally essential SEC14p requirement, the sec14–129::H1S3 lesion was recombined directly into bsr and bsd haploid yeast strains by transformation with the appropriate linear DNA, followed by selection for His+ prototrophs (see Experimental Procedures). His+ transformants were readily obtained and showed no obvious growth defects. These sec14–129::H1S3 derivative strains were then tested for the expected replacement events by probing radiolabeled cell-free extracts prepared from such mutants for SEC14p antigen. Treatment of extracts prepared from SEC14/sec14–129::H1S3 diploid strains with SEC14p antiserum precipitated two radiolabeled species (Figure 3). The major species was represented by the 35 kd SEC14p. The second SEC14p species was a stable and rather prominent polypeptide of 16–17 kd in apparent molecular mass. The size of that sec14–129::H1S3 gene product was consistent with the fact that the N-terminal 129 residues of SEC14p were represented in the primary translation product (Bankaitis et al., 1989; Cleves et al., 1989).
Figure 3. Suppression of a sec14 Disruption Allele.
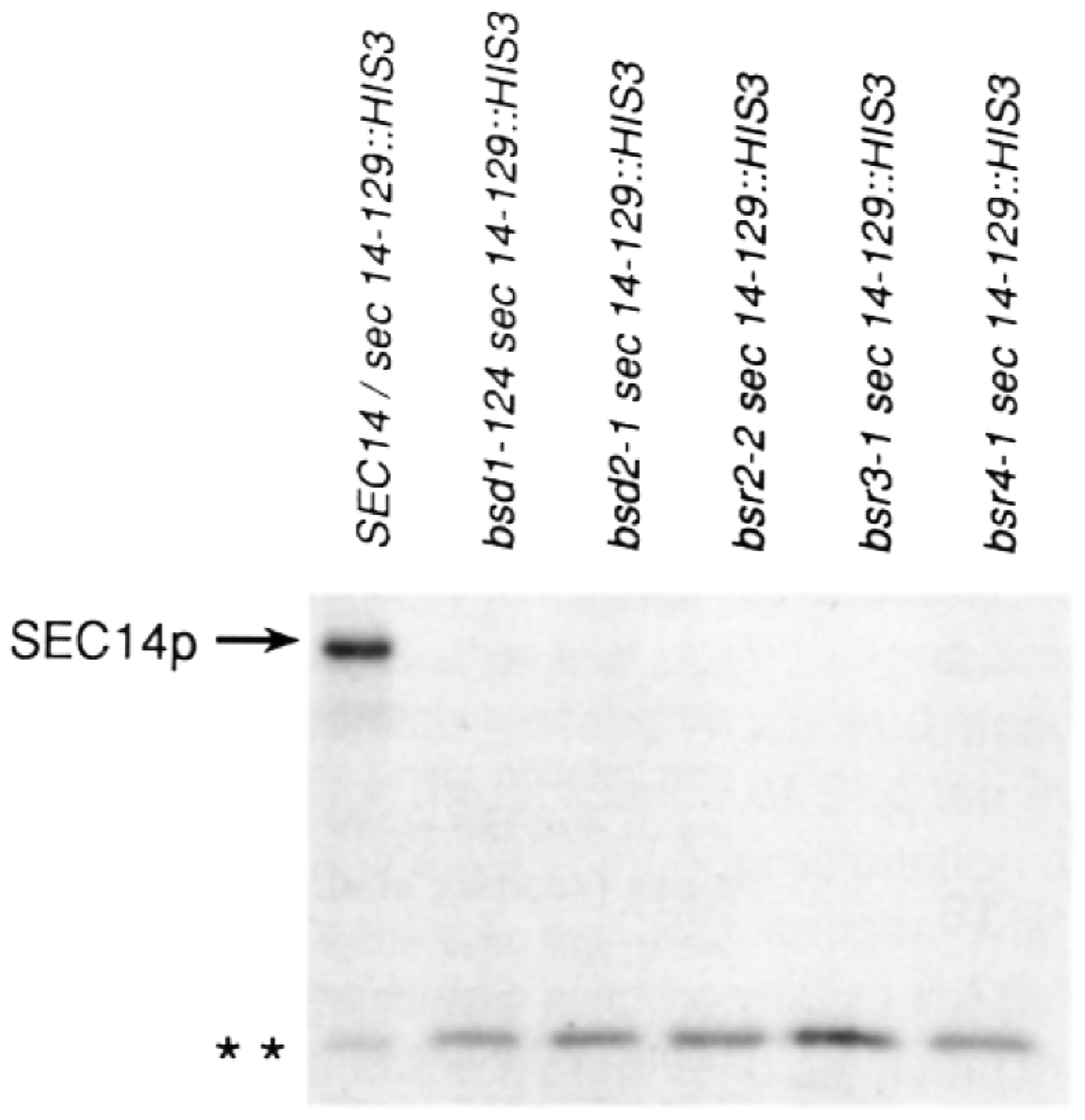
The appropriate yeast strains were grown to early logarithmic growth phase (OD600 = 0.5) in glucose (2%) minimal medium at 30°C. Yeast cells (0.5 OD600 units) were radiolabeled for 20 min with 100 μCi of Trans-Label (>1000 Ci/mmol; ICN Radiochemicals, Irvine, CA), and radiolabeling was terminated by addition of trichloroacetic acid (5% final). SEC14p immunoreactive species were specifically precipitated and resolved by SDS-PAGE and autoradiography as previously described (Bankaitis et al., 1989; Salama et al., 1990). The relevant genotypes of the corresponding yeast strains are indicated above (strains CTYD43, CTY221, CTY217, CTY228, CTY229, and CTY230, respectively). Complete genotypes for these strains are given in Table 4. The positions of the full-length SEC14p and the truncated sec14–129::HIS3 (* *) gene product are indicated at left. The immunoprecipitates were normalized on the basis of total incorporated cpm in the clarified extracts. The samples obtained from the suppressor strains were severalfold overloaded with respect to those obtained from the diploid strains.
Treatment of extracts prepared from haploid suppressor strains transformed to His+ with linear sec14–129::H1S3 DNA yielded only one.radiolabeled SEC14p species, and it represented the truncated form of SEC14p (Figure 3). No full-length SEC14p was detected, even though the suppressor mutant samples were some 3- to 5-fold overloaded with respect to the diploid control samples. These data proved that the bsr arid bsd suppressor mutations were epistatic to the lethality associated with sec14 disruption alleles. Since no PI/PC transfer protein activity was detected in bsr and bsd strains carrying the sec14–129::H1S3 lesion (data not shown), the data eliminated a number of trivial modes for suppression such as upregulation of Sec14pts, informational Suppression, some nonspecific stabilization of the thermolabile Sec14pts, and activation of some cryptic PI/PC transfer protein function.
Bypass Suppression Is Efficient
The ability of the bsr and bsd mutations to restore wild-type growth properties to yeast strains carrying sec14 disruption alleles suggested that the bypass suppression was efficient. We measured the secretory capacities of bsr and bsd strains carrying the normally lethal sec14–129::H1S3 mutation and expressed these values as the secretion index, the ratio of extracellular invertase to total invertase measured for a particular strain.
Representative data are presented in Table 2. In the case of the wild-type strain, essentially all of the invertase activity was extracellularly disposed (secretion index = 0.98). The sec14–1ts strain, however, exhibited most of its invertase activity in a latent form (secretion index = 0.17) (Table 3). The distribution of invertase activity in the bsr and bsd strains carrying sec14–129::H1S3 was very similar to that of the SEC14 strain. Secretion indices of greater than 0.90 were measured in all cases (Table 3). The kinetics of vacuolar protein biogenesis in these suppressor strains were normal as well (unpublished data). These data demonstrated that suppression of sec14–129::H1S3 was efficient.
Table 3.
Invertase Secretion in Suppressor Strains
| Strain | Relevant Genotype | Total Invertase | Extracellular Invertase | Secretion Index |
|---|---|---|---|---|
| CTY182 | SEC14 | 343 | 335 | 0.98 |
| CTY1–1A | sec14–1ts | 385 | 65 | 0.17 |
| CTY228 | bsr2–2, sec14–129::HIS3 | 342 | 312 | 0.91 |
| CTY229 | bsr3–1, sec14–129::HIS3 | 389 | 357 | 0.92 |
| CTY230 | bsr4–1, sec14–129::HIS3 | 357 | 343 | 0.96 |
| CTY221 | bsd1–124, sec14–129::HIS3 | 391 | 372 | 0.95 |
| CTY217 | bsd2–1, sec14–129::HIS3 | 292 | 285 | 0.98 |
Invertase secretion in supressor strains that lack the SEC14p. Yeast cells were cultured to mid-logarithmic growth phase in YPD at 30°C and shifted to YP (0.1% glucose) for 30 min and finally shifted to 37°C for 2 hr. Total and extracellular invertase were determined and are expressed in units of activity as described in the Experimental Procedures. The secretion index, a relative measure of secretion competence calculated from these values (Salama et al., 1990; Experimental Procedures), is also given.
Bypass Suppression Requires the Late Stages of the Secretory Pathway
One possible mechanism for bypass of SEC14p may require that a segment of the normal secretory pathway itself be bypassed in the suppressor mutants. Such an alternative mode of secretion might exhibit two distinguishing characteristics: the failure of secretory proteins to be fully modified during transit to the cell surface, and an uncoupling of secretion from some of the late sec functions that are essential for protein transport to the cell surface.
To determine whether the invertase secreted by sec14–129::H1S3 suppressor strains contained fully matured outer chain carbohydrate, we employed an immunological assay for acquisition of mannose in terminal α-1→3 linkage (Franzusoff and Schekman, 1989). Radiolabeled invertase was quantitatively immunoprecipitated with anti-invertase serum in the first round, the samples split, and the second round of quantitative immunoprecipitation with either control invertase or test α-1→3 mannose linkage-specific antisera followed. The precipitates were subsequently resolved by SDS-PAGE and autoradiography. Representative data are shown in Figure 4. The invertase precipitated from the control wild-type strain migrated as a characteristically heterogeneous smear in the 95–135 kd molecular mass range. As expected, this invertase was immunoprecipitated by both the control invertase and the test α-1→3 mannose linkage-specific antisera. The data for the sec14–129::H1S3, bsr3–1 and sec14–129::H1S3, bsd1–124 double mutants are also presented. We note that the results for these mutants were the same as those observed for the wild-type strain. The radiolabeled invertase was, in each case, precipitated by both the control invertase and the test α-1→3 mannose antisera (Figure 4). These data were representative of the results obtained for the other bsr and bsd mutants.
Figure 4. Invertase Glycosylation in the Suppressor Strains.
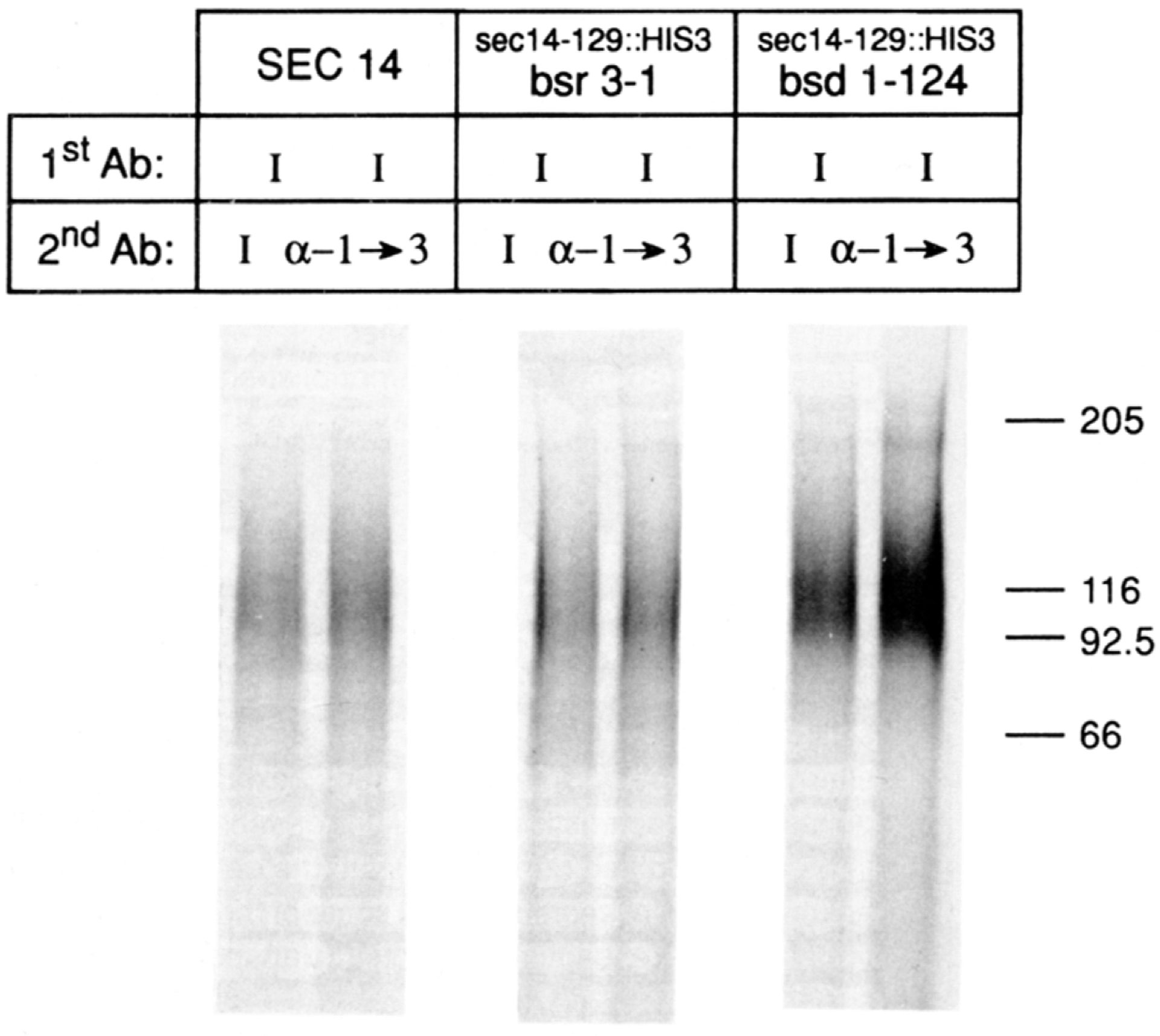
The appropriate yeast strains were grown to early logarithmic growth phase (OD600 = 0.5) in glucose (2%) minimal medium, washed two times with distilled water, and resuspended in glucose (0.1%) minimal medium. After a 2 hr incubation at 25°C to allow derepression of invertase synthesis, the cells (2 OD600 units) were shifted to 37°C and allowed to incorporate radiolabel (250 μCi of Trans-Label) for 1 hr. Invertase antigen was precipitated from clarified extracts by an initial round (1stAb) of immunoprecipitation using invertase-specific antibodies (I) directed solely against polypeptide epitopes (Klionsky et al., 1988). The precipitates were split and subjected to a second round (2ndAb) of immunoprecipitation with either the control invertase sera (I) or the test antibodies (α-1→3) raised specifically against α-1→3 mannose linkages of outer chain carbohydrate (Franzusoff and Schekman, 1989). The relevant genotypes of the yeast strains employed (i.e., CTY182, CTY229, and CTY221, respectively) are given at the top of the corresponding lanes, as are the designations for the 2nd Ab used. Precipitates were resolved on SDS gels that were 7.5% in acrylamide.
To determine whether the range of suppression by bsr and bsd mutations extended beyond sec14 defects, we used standard genetic methods to construct the appropriate bsr, sects and bsd, sects double mutants. These were subsequently tested for growth at 37°C, a nonpermissive temperature for all sects mutants (Novick et al., 1980). We restricted our analysis to the late-acting (i.e., secretory vesicle-accumulating) sec mutants because of the late Golgi execution point for SEC14p (Franzusoff and Schekman, 1989; our unpublished data) and previous analyses of the range of suppression by sac1CS that suggested restriction of suppression to late secretory pathway defects (Cleves et al., 1989). Our data indicated that suppression by the bsr and bsd mutations was sec14 specific. None of the defects in the 10 late-acting sec genes tested (i.e., sec1-sec6, sec8, sec9, sec10, sec15; Novick et al., 1980) were suppressed at 37°C by either bsr or bsd.
These data showed that the course of secretory protein biogenesis was normal in the sec14 bypass suppressor strains. Moreover, the results suggested that the late stages of the secretory pathway were required for secretion in these strains. We concluded that major segments of the secretory pathway were not being bypassed in the suppressor mutants, and that secretion involved the participation of all of the proper compartments.
BSR4 Is the Structural Gene for the Yeast Choline Kinase
We sought to isolate clones of the BSR genes. Recovery of such clones was guided by the phenotypic suppression of sec14–1ts by the bsr mutations. The recessive nature of this suppression permitted the recognition of candidate BSR clones on the basis of screening the appropriate sec14–1ts, bsr double mutants for rescue of the sec14–1ts phenotype. These efforts were most immediately successful in the case of BSR4. We isolated a genomic clone of BSR4 and confirmed its authenticity by standard integrative genetic mapping experiments (see Experimental Procedures).
The sequence of a 2683 bp EcoRI–HindIII restriction fragment that carried the intact BSR4 gene was determined. The largest open reading frame spanned 582 codons and could encode a primary translation product of 66,323 daltons in size. Comparison of the BSR4p primary sequence with those in an updated yeast protein data base compiled by one of us (M. G.) indicated that the BSR4p sequence was identical to that of yeast choline kinase described by Hosaka et al. (1989). These data proved that BSR4 was the structural gene for choline kinase. We shall henceforth refer to BSR4 within the context of the established CKI nomenclature.
cki Disruption Mutations Are Bypass Suppressors of sec14 Defects
The recessive nature of cki-mediated suppression of sec-14 mutations suggested that these suppressor alleles represented simple cki loss-of-function mutations. We constructed a cki-284::HIS3 allele that resulted in a truncation of the C-terminal 298 residues of choline kinase and recombined it into the diploid yeast strain CTYD100.1 (MATa/MATα, ura3/ura3, Δhis3/Δhis3, SEC14/sec14–88::URA3, CKI/CKI), thereby generating the heterozygous CKI/cki-284::HIS3 configuration The corresponding diploid was then subjected to meiotic analysis to test the ability of cki-284::HIS3 to suppress the haploid-lethal sec14–88::URA3 defect. In a total of 20 tetrads analyzed, our ability to obtain haploid Ura+His+ progeny, but no Ura+His− progeny, was indicative of suppression of sec14–88::URA3 by cki-284::HIS3. The His+ progeny were uniformly unable to incorporate [14C]choline into PC in vivo, as expected for mutants deficient in choline kinase activity (data not shown).
To confirm that the Ura+ His+ meiotic progeny had inherited sec14–88::URA3, the four sister segregants derived from an ascus that yielded 4 viable:0 nonviable spores were tested for the presence of SEC14p in cell-free lysates by a quantitative immunoprecipitation assay. Vacuolar carboxypeptidase Y (CPY) antigen was also monitored in these lysates so as to normalize the data. As the data in Figure 5 show, the parental diploid and the two ura−his− segregants contained the unadulterated SEC14p. In contrast, the Ura+His+ segregants lacked SEC14p immunoreactive material as measured by the immunoprecipitation assay. This was the result expected for sec14–88::URA3 strains, as the SEC14p antiserum employed was directed at epitopes residing between amino acids 111 and 284, inclusive, of SEC14p (Bankaitis et al., 1989). This disruption precluded expression of that region of SEC14p. Equivalent amounts of radiolabeled CPY were recovered from these five lysates, indicating that the data were properly normalized. We concluded that one mechanism by which the normally essential SEC14p requirement can be bypassed involved the loss of choline kinase activity.
Figure 5. Suppression of sec14–88::URA3 by a cki Disruption Allele .
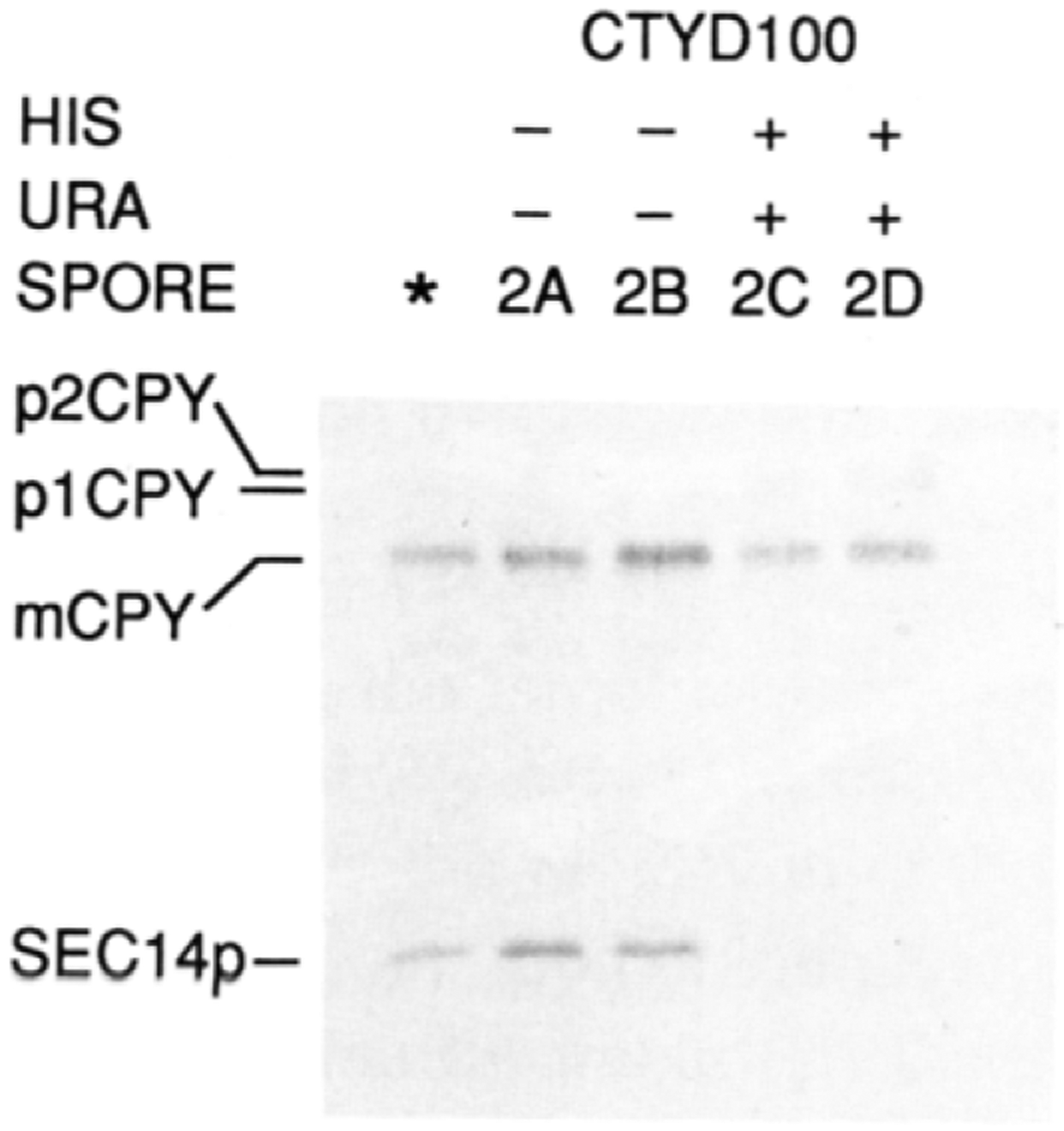
The radiolabeled CPY and SEC14p profiles from meiotic progeny derived from an ascus that exhibited four viable spores, in a cross where sec14–88::URA3 and cki-284::HIS3 were segregating, are shown. Also included are the profiles obtained for the parental diploid strain CTYD100 (*). The details of the cross are given in the text. The meiotic progeny were cultured in glucose (2%) minimal medium, to an OD600 of approximately 0.5, at 37°C. A 30 min labeling period (100 μCi of Trans-Label) followed and was terminated with trichloroacetic acid. SEC14p and CPY immunoreactive materials were recovered and visualized after SDS-PAGE and autoradiography. The positions of the SEC14p, CPY, and p1 and p2 proCPY forms are indicated at the left. The Ura and His phenotypes of the meiotic progeny are indicated above the corresponding lanes. Further details are provided in the text.
Suppression of sec14 Defects Is a General Consequence of Mutations in the CDP-Choline Pathway for PC Biosynthesis
Yeast are able to synthesize PC by either one of two mechanisms: via methylation of phosphatidylethanolamine (PE) (the methylation pathway), and via utilization of exogenously supplied choline (the CDP-choline pathway). The choline kinase is an enzyme that catalyzes the first step of the latter pathway (for a review see Carman and Henry, 1989), and these alternative routes for PC biosynthesis are depicted in Figure 6A. To test if bypass of SEC14 was a general consequence of defects in PC biosynthesis, we constructed a series of sec14–1ts mutants that were blocked at defined steps in either the methylation or CDP-choline pathways for PC biosynthesis. The ability of these strains to grow and secrete protein at the restrictive temperature (37°C) was then determined.
Figure 6. Suppression of sec14 Defects by Mutations in the CDP-Choline Pathway for PC Biosynthesis.
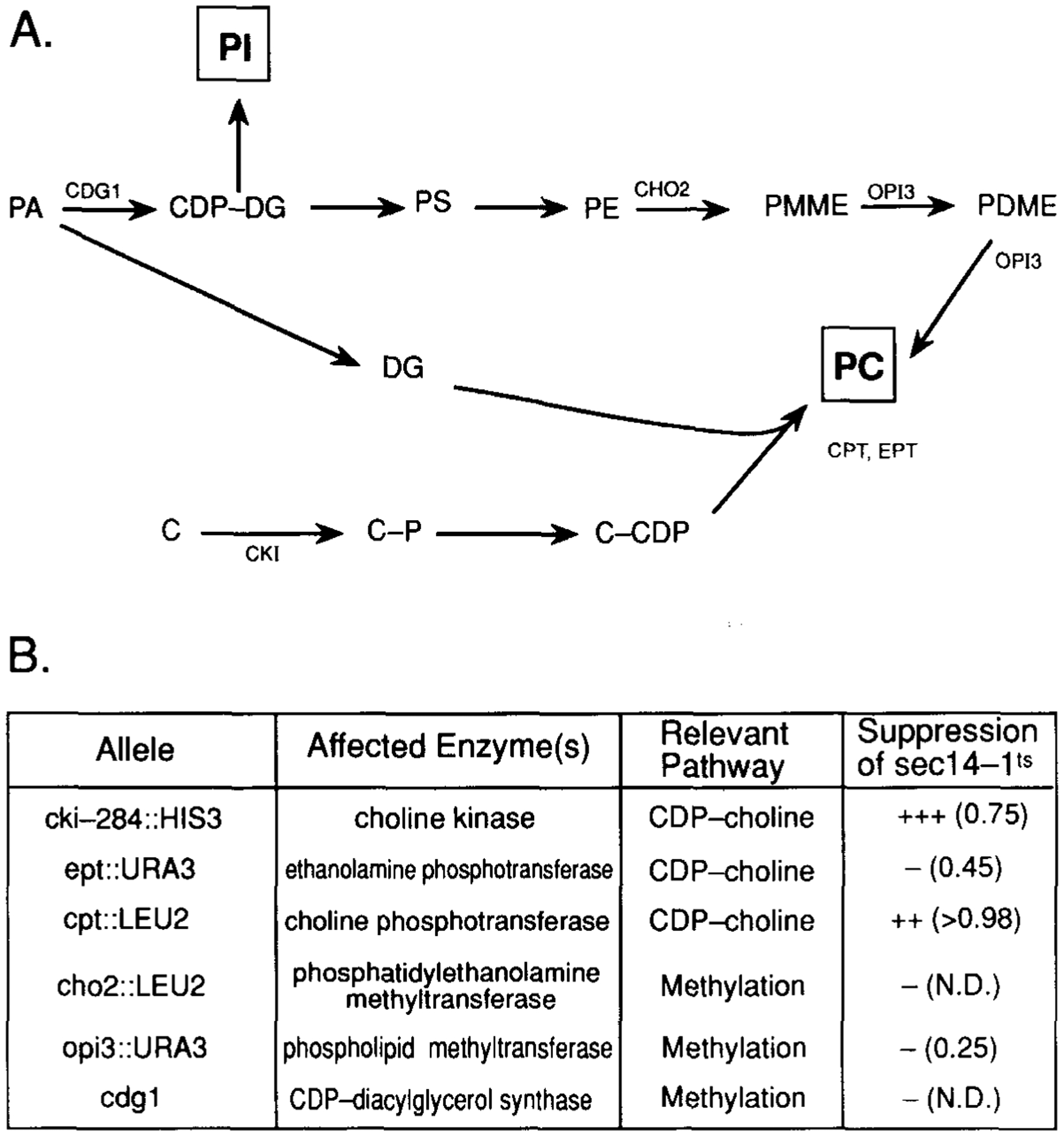
(A) The two pathways for PC biosynthesis in yeast are diagrammed, and the gene designation for structural enzymes of each pathway are indicated at the appropriate site(s) of action (for a review see Carman and Henry, 1989). Abbreviations: PA, phosphatidic acid; CDP-DG, CDP-diacylglycerol; DG, diacylglycerol; PI, phosphatidylinositol; PS, phosphatidylserine; PE, phosphatidylethanolamine; PMME, phosphatidylmonomethylethanolamine; PDME, phosphatidyldimethylethanolamine; PC, phosphatidylcholine; C, choline; C-P, choline-phosphate; C-CDP, CDP-choline.
(B) The mutations in PC biosynthesis that were tested for suppression of sec14–1ts are listed. Also given are the structural enzyme and PC biosynthetic pathway affected by each mutation. Suppression was determined by constructing sec14–1ts haploid yeast strains carrying each designated mutant allele. The relevant crosses involved: meiotic analysis of CTYD100, HJ051 × CTY76, CTY214 transformed with cpt1::LEU2 as described (Hjelmstad and Bell, 1987), CTY215 × CTY410, CTY68 × CTY411, and CTY2–1C × CTY416, respectively. Complete genotypes of these strains are presented in Table 4. Phenotypic suppression was followed by testing for growth and formation of single colonies on YPD agar at 37°C, and is qualitatively indicated. The secretion index, where determined, is given in parentheses and was calculated from the ratio of extracellular invertase:total invertase as described in the legend to Table 3. These values are to be compared with those measured in these experiments for wild-type yeast (>0.98) and sec14-1ts yeast (0.20) incubated at 37°C (data not shown). The experimental conditions were exactly as those described in the legend to Table 3. N.D. = not determined.
A compilation of the data is provided in Figure 6B. In addition to suppression of sec14 defects by cki-disruption alleles, disruption of the CPT1 gene (which encodes the ultimate enzyme of the CDP-choline pathway, the choline phosphotransferase) resulted in a suppression of sec14–1ts with respect to growth and secretion at 37°C. The cpt1::LEU2 lesion also suppressed sec14–129::HIS3. It had previously been established by Hjelmstad and Bell (1987, 1988) that both the CPT1 and the EPT1 gene products participate in the ultimate step in PC synthesis via the CDP-choline pathway, although the CPT1 gene product represents the major activity with respect to PC synthesis and the EPT1 gene product (the ethanolamine phosphotransferase) is primarily associated with PE synthesis. In this regard, we noted that, while ept1::URA3 failed to phenotypically suppress sec14-1ts, some partial suppression was detected by biochemical criteria. That is, the ept1::URA3 allele improved the secretion index of sec14–1ts yeast from approximately 0.20 to approximately 0.45 (Figure 6B).
The demonstration that independent blocks at the first (cki) and last (cpt1) steps of PC biosynthesis via the CDP-choline pathway suppressed the growth and secretory defects associated with sec14–1ts led us to conclude that suppression was a general consequence of CDP-choline pathway dysfunction. None of the three defects associated with the methylation pathway for PC biosynthesis tested (i.e., cho2::LEU2, opi3::URA3, and cdg1) had any apparent compensatory effect on sec14–1ts (Figure 6B). Thus, suppression of sec14–1ts was limited to defects in the CDP-choline pathway for PC biosynthesis. We tested whether the normally essential SEC14p requirement could be bypassed by culturing sec14–1ts yeast in choline-free media, a condition under which the CDP-choline pathway should not generate net PC synthesis within the yeast cell. Choline deprivation failed to elicit any detectable suppression of sec14–1ts. It should be noted, however, that these nutritional conditions did not guarantee the absence of net PC synthesis in the Golgi via a salvage mechanism that utilizes the CDP-choline pathway.
Discussion
Phospholipid transfer proteins are a class of cytosolic proteins that are ubiquitous among eukaryotic cells and were initially recognized by their ability to catalyze exchange of phospholipids between membranes in vitro. The PI transfer proteins (PI-TPs) in particular have recently commanded a great deal of attention because of their conservation of structural and catalytic properties and their ability to effect a net transfer of PI from a donor membrane enriched in this phospholipid to an acceptor membrane that is PI deficient. Although PI-TPs exhibit significant preference for PI, they do mobilize PC, and net movement of PI in one direction is dependent on countermovement of PC. Our demonstration that SEC14p is the PI-TP of yeast provided an assignment of an in vivo function for a phospholipid transfer protein, namely, an essential involvement of the SEC14p in stimulating yeast Golgi secretory function (Bankaitis et al., 1990). Our findings that SEC14p exhibited colocalization with the yeast Golgi marker KEX2p in double-label immunofluorescence experiments (Figure 1) and that the sedimentable SEC14p codistributed with the KEX2p in subcellular fractions that were highly enriched in yeast Golgi membranes (Table 1, Figure 2) indicated that SEC14p exhibited a physical association with the yeast Golgi complex in vivo. On the basis of the quantitative fractionation experiments (Table 4, Figure 2), we estimate that at least 20% of the total cellular SEC14p was Golgi associated. Since it is the Golgi that is dysfunctional in sec14 strains (Novick et al., 1980), and the Golgi defect is manifested quite rapidly upon imposition of restrictive conditions dysfunction (Bankaitis et al., 1989), the cumulative data argue strongly for a direct role for SECl4p in stimulating yeast Golgi secretory function. Moreover, the enrichment of SEC14p to a minor organelle membrane is rather surprising, as the in vitro activity of this protein does not provide any clear basis for such a specificity.
Two extreme models for SEC14p function have been proposed, and these are distinguished by the in vivo relevance of the PI/PC transfer activity of SEC14p (for a discussion see Bankaitis et al., 1990). First, the phospholipid transfer activity measured for the SEC14p in vitro may accurately reflect SECl4p function in vivo. In this case, the SEC14p would be considered to function in the mobilization of PI and/or PC between intracellular membranes. Such an activity could maintain an appropriate phospholipid balance between organelle membranes, generate an appropriate phospholipid environment in a particular membrane (i.e., Golgi), or both. Alternatively, the in vitro phospholipid transfer activity of SEC14p might not accurately reflect SEC14p function in vivo. For example, SEC14p may exhibit some other undefined function that is regulated by binding of PI or PC to SEC14p. In this case, the in vitro phospholipid transfer activity Would be entirely artifactual.
In this study, we present data that are most readily incorporated within the framework of the phospholipid equilibration model for SEC14p function. The experimental evidence is summarized as follows: First, characterization of 107 independently obtained mutants that exhibited suppression of the sec14–1ts missense mutation revealed only extragenic suppressor mutations. These identified six genes (SAC1, BSR2, BSR3, BSR4, BSD1, and BSD2). Second, in all cases, suppression was via bypass of SEC14p (Table 2, Figure 3), and suppression was efficient (Table 3). In no case did suppression involve detectable activation of some cryptic PI/PC transfer protein activity. Third, the bypass suppression appeared to circumvent only the SEC14p requirement. That is, secretory protein traffic in the suppressor strains was considered to occur via the normal route on the basis of proper maturation of secretory invertase outer chain carbohydrate (Figure 4) and the requirement for the 10 late-acting SEC gene products in protein secretion from the suppressor strains. Fourth, molecular characterization of BSR4 revealed its identity with the structural gene for yeast choline kinase (CKI), the enzyme that catalyzes the first step in the CDP-choline pathway for PC biosynthesis. Moreover, cki disruption mutations were efficient bypass suppressors (Figure 5). Finally, disruptions in CPT1, the structural gene for the enzyme that catalyzes the ultimate step in PC biosynthesis via the CDP-choline pathway, also suppressed sec14 defects (Figure 6). Our findings that the SEC14p requirement could be specifically and efficiently bypassed by mutations that directly affect PC biosynthesis are consistent with a phospholipid equilibration function for SEC-14p. These data are not easily reconciled with the hypothesis that some essential, but as yet undefined, SEC14p function is regulated by PI or PC binding to SEC14p.
Our observation that cki and cpt1 mutations effectively bypass the SEC14p requirement provides the first link between biosynthetic protein transport, intracellular phospholipid transport, and phospholipid biosynthesis via the CDP-choline pathway. It will be of interest to determine whether BSR2 and BSR3 are allelic to CPT1 or CCT, the structural gene for the penultimate enzyme of this pathway (Tsukagoshi et al., 1987). Our preliminary data show that bsr2 and bsr3 strains are unable to incorporate [14C] choline into PC in vivo, indicating defects in the CDP-choline pathway in these strains (T. P. M. and V. A. B., unpublished data). Bypass suppression of sec14 was not reproduced by inactivation of the methylation pathway for PC synthesis, however. Mutational blocks in either the methylation of PE (cho2::LEU2), subsequent methylations of PE derivatives to PC (opi3::URA3), or reductions in the capacity to synthesize CDP-DG (CDP-diacylglycerol) (cdg1) failed to phenotypically suppress sec14–1ts (Figure 6).
Why do mutations in the CDP-choline pathway for PC synthesis result in a bypass of SEC14p, whereas mutations in the methylation pathway have no such effect? We offer two general possibilities. First, PC biosynthesis via the CDP-choline pathway might be compartmentalized to the yeast Golgi, whereas synthesis via the methylation pathway might not. In this case, the SEC14p would function to remove PC from the Golgi. Bypass of SEC14p would then be a logical consequence of inactivation of the CDP-choline pathway. This model requires that yeast localize the membrane-bound enzymes of the CDP-choline system to the Golgi. In mammalian cells, these enzymes are located in the ER and the Golgi (Van Meer, 1989). Our observation that sec14 defects were not suppressed by exclusion of choline from media is not immediately consistent with the idea that synthesis of PC via the CDP-choline route is the target for suppression.
An alternative possibility is that inactivation of the CDP-choline pathway alters the regulation of the methylation pathway. In this case, an appropriate phospholipid composition (perhaps some favorable PI/PC ratio) might be generated by bulk phospholipid synthesis in the ER and subsequently imposed upon the Golgi by bulk membrane flow. This would also obviate the need for a PI/PC equilibrating activity such as that we suggest is provided by SEC14p. In any case, these suppression data provide the first indication of a more general physiological involvement for CDP-choline pathway function in yeast—one that is phenotypically distinguishable from its recognized role in net PC synthesis from choline.
Our finding that SEC14p represents the sole measurable PI or PC transfer acivity in yeast (Aitken et al., 1990; Bankaitis et al., 1990), coupled with the data in this report indicating that SEC14p function can be efficiently and specifically bypassed without activation of some cryptic PI/PC transfer activity, allows us to comment on a previous model for phospholipid transfer protein function in vivo. Wieland et al. (1987) pointed out that the rate of phospholipid egress from the mammalian ER due to bulk membrane flow far outstrips the rate at which ER phospholipid can be regenerated by synthesis, and it is likely that all eukaryotic cells are faced with this incongruity. These workers argued that phospholipid retrieval back to the ER must occur to correct the imbalance between the ER phospholipid egress and synthesis rates. It was suggested that phospholipid transfer proteins could play a determining role in such a retrieval mechanism (Wieland et al., 1987), and a proposal as to how phospholipid transfer proteins might drive such a process has been offered (Rothman, 1990). Our bypass data indicate that SEC14p cannot play a determining role in such phospholipid retrieval in yeast, even though the SEC14p ligands (PI and PC) represent the two most abundant phospholipids in this organism. One alternative possibility is that the determining mechanism for such phospholipid retrieval to the yeast ER will involve an as yet undemonstrated retrograde vesicular pathway, like the one perturbed by brefeldin A in mammalian cells (Lippincott-Schwartz et al., 1989).
In conclusion, we have presented evidence that supports a phospholipid equilibration function for SEC14p and have shown that SEC14p colocalizes with yeast Golgi bodies in vivo. The data provide evidence that directly links phospholipid biosynthesis and intracellular phospholipid transport to secretory pathway function, specifically at the level of Golgi function in yeast. The idea that SEC14p is involved in controlling the secretory competence of yeast Golgi membranes by modulating their phospholipid content underscores the value of genetic approaches in the study of intercompartmental protein transport. The elegant in vitro systems designed to study this problem (such as those pioneered by Rothman and colleagues; see Rothman and Orci, 1990) uniformly rely on transport-competent membranes to be provided by living cells. This raises the likelihood that the execution points of functions such as those performed by SEC14p precede those events measured by current in vitro assays. Now that a biochemical basis for SEC14p function has been suggested, combined biochemical and genetic approaches can be directed toward a detailed mechanistic appreciation of how SEC14p drives yeast Golgi function.
Experimental Procedures
Strains, Media, and Reagents
Complete genotypes of haploid and diploid yeast strains used in this work are provided in Table 4. Yeast complex media (YPD) and Wickerham’s minimal media have been described (Sherman et al., 1983). Standard Escherichia coli media were employed (Silhavy et al., 1984). E. coli strains MC1061 (Casadaban and Cohen, 1980), KK2186 (Bankaitis et al., 1989), and DH5 (Hanahan, 1983) were used for plasmid propagation and purification. The reagents employed in this study have been described (Bankaitis et al., 1989; Cleves et al., 1989). Additional reagents included horseradish peroxidase-conjugated antibodies (Bio-Rad Laboratories, Richmond, CA). Bovine serum albumin (fraction V), cycloheximide, yeast cytochrome c (type III), NADPH, o-dianisidine, glucose oxidase, horseradish peroxidase, pyruvate kinase type II, ATP, phosphoenolpyruvate, p-nitrophenol, sorbitol, Triton X-100, triethanolamine, and Tween 20 were from Sigma (St. Louis, MO). immunolon-2 ELISA plates and o-phenylenediamine were obtained from Dynatech Laboratories (Chantilly, VA) and Abbott Laboratories (North Chicago, IL), respectively.
Immunofluorescence Microscopy
The immunofluorescence microscopy techniques we employed were essentially as those previously described (Kilmartin and Adams, 1984). Yeast strain CTY182 (see Table 4), carrying a multicopy plasmid into which we engineered KEX2 (YEp KEX2), was grown to an OD600 of 1.0 in minimal medium. These cells overproduced KEX2p, which facilitated visualization of yeast Golgi. KEX2p-overproducer yeast strains preserve the native KEX2p localization and are not prone to artifacts of dosage-induced mislocalization (Franzusoff et al, 1991). Following fixation in 3.7% formaldehydes, the cells were converted to spheroplasts with lyticase (Enzogenetics; Corvallis, OR) (Bankaitis et al., 1989), permeabilized with 1.0% Triton X-100 in 1x phosphate-buffered saline (PBS), and incubated at 22°C in PBS for 3 hr with affinity-purified rabbit anti-KEX2p polyclonal serum (1:100 dilution). After extensive washing with PBS, the samples were incubated for 3 hr with TRITC-conjugated goat anti-rabbit IgG secondary serum (Sigma Chemical Co., St. Louis, MO). Finally, after incubation with an excess of bulk rabbit IgG (Sigma) and another extensive washing regimen, the cells were similarly incubated with FITC-conjugated rabbit anti-SEC14p polyclonal serum, or the corresponding preimmune serum, that had been exhaustively cross-adsorbed against sec14–88::URA3 yeast strains that fail to make any relevant SEC14p antigen. DAPI (0.05 μg/ml) was added 5 min prior to the final PBS wash regimen. The fixed cells were suspended in mounting solution (1 mg/ml p-phenylenediamine in 90% glycerol) and mounted under a glass coverslip immediately prior to viewing. Samples were visualized with a Zeiss Axioplan microscope equipped for Nomarski and fluorescence microscopy. Micrographs were generated from 4 s exposures of TMAX 400 film.
Subcellular Fractionation
Yeast strain CTY182 was grown to early logarithmic growth phase in a 1 liter culture in YPD medium at 25°C with shaking. Cycloheximide (100 μg/ml) was added and, after a 15 min incubation, cells were poisoned with NaN3 (10 mM). After a wash with cold 10 mM NaN3, the cells were resuspended in 100 ml of spheroplast buffer (0.8 M sorbitol, 50 mM potassium phosphate [pH 7.5], 10 mM NaN3, 40 mM 2-mercaptoethanol), and lyticase (Enzogenetics, Corvallis, OR) was added to 0.5 μg/ml. The cells were incubated for 45 min at 30°C with occasional gentle agitation. Spheroplasts were pelleted by centrifugation at 1000 × g for 6 min, washed with 0.8 M sorbitol, 50 mM potassium phosphate (pH 7.5) buffer that was 10 mM in NaN3, and resuspended in 30 ml of ice-cold lysis buffer (0.3 M sorbitol, 10 mM triethanolamine [pH 7.2], 1 mM EDTA). The cells were then incubated on ice for 20 min with occasional gentle agitation. This lysis procedure results in efficient spheroplast lysis but preserves the integrity of intracellular organelles (Bankaitis et al., 1989; data not shown).
Lysates were adjusted to a final 1.0 M sorbitol concentration and centrifuged at 1000 × g for 6 min to remove unlysed cells (P1). The supernatant (S1) was centrifuged at 12,000 × g for 15 min to yield the P12 and S12 fractions. The S12 fraction was finally layered onto a 1 ml step of a 1.8 M sorbitol, 0.1 mM PMSF, 10 mM triethanolamine (pH 7.2) solution that was floating on a 4.28 M sorbitol, 0.1 mM PMSF, 10 mM triethanolamine (pH 7.2) cushion in an SW28 rotor tube (Beckman Instruments Inc) and centrifuged at 100,000 × g for 3 hr at 8°C. The resulting S100 was carefully removed from the bottom cushion onto which the sedimentable material (P100 fraction) had pelleted. This P100 was subsequently loaded under a self-forming 30–60% sorbitol gradient (stepped in increments of 10%) in 10 mM triethanolamine (pH 7.2) and centrifuged to equilibrium at 200,000 × g in an SW41 rotor (Beckman Instruments, Inc) for 40 hr. The linear gradient (gradient 1) was fractionated from the top to the bottom (0.5 ml fractions). The peak KEX2p fractions were pooled, diluted 1:1 with 10 mM triethanolamine (pH 7.2), and layered onto a self-forming 40%–65% sorbitol gradient (gradient 2) that was stepped in increments of 6.25%. After centrifugation at 200,000 × g for 40 hr, the gradient was fractionated from the top to the bottom in 0.5 ml fractions.
The presence of various marker polypeptides in the collected fractions was determined either by enzyme assay or ELISA. NADPH cytochrome c reductase and cytochrome c oxidase were assayed by the methods of Kreibich et al. (1973) and Mason et al. (1973), respectively. Vanadate-sensitive ATPase was assayed according to the method of Bowman and Slayman (1979). KEX2p, SEC14p, and DPAP-B were followed by a quantitative ELISA method that was modified from the procedure described by Mimms et al. (1990). ELISA activity was determined using a direct sandwich assay in which fractions were diluted 1000-fold, dried onto microtiter wells at 60°C, washed with 0.5% Tween 20 in PBS, and blocked with 3% bovine serum albumin, 0.05% Tween 20 in PBS (blocking buffer). Wells were incubated with primary antisera that had either been affinity-purified or had been extensively preadsorbed against the appropriate yeast deletion strains. After extensive washing with PBS, the wells were incubated with horseradish peroxidase-antibody conjugate in blocking buffer, incubated with o-phenylenediamine substrate, quenched, and the A492 was determined on an MR600 plate reader (Dynatech Instruments, Torrance, CA).
Genetic Techniques and Suppressor Isolation
Standard genetic methods for mating of haploid yeast strains, complementation analysis, and tetrad analysis were employed (Sherman et al., 1983). Plasmid DNA was purified using the alkaline lysis method (Silhavy et al., 1984). Procedures for gene disruption in yeast (Rothstein, 1983) and transformation of DNA into yeast (Ito et al., 1983) and E. coli (Silhavy et al., 1984) have been described previously. The isolation of the sec14–1ts suppressor mutants and the criteria for assessing Ts+ and ts phenotypes were described by Cleves et al. (1989). The sec14–129::HIS3 disruption was recombined into the appropriate yeast strains as described by Cleves et al. (1989). General DNA sequencing protocols are given in Cleves et al. (1989).
Immunoprecipitations and Invertase Assays
Radiolabeling of yeast cells with 35S-labeled amino acids and preparation of clarified extracts were performed essentially as described by Bankaitis et al. (1989). Specific conditions of radiolabeling are given in the corresponding figure legends. Immunoprecipitation of SEC14p, invertase, α-1→3 mannose linkage-containing material, and CPY from clarified extracts was accomplished by our standard immunoprecipitation regimen (Bankaitis et al., 1989). Rabbit polyclonal antisera raised against the SECl4p (Bankaitis et al., 1989), invertase (Klionsky et al., 1988), α-1→3 mannose linkages (Franzusoff and Schekman, 1989), and CPY (Klionsky et al., 1988) were employed at a 1:500 dilution to insure quantitative recovery of cognate antigen. Immunoprecipitates were resolved by SDS-PAGE and autoradiography as described by Schauer et al. (1985).
Invertase assays were performed exactly as reported by Bankaitis et al. (1989) using the method of Goldstein and Lampen (1979). Details of culture conditions are given in the legend to Table 2. Invertase units were defined as nmol of glucose produced per min at 30°C. The secretion index related secreted invertase to total invertase (i.e., units of secreted invertase/units of total invertase) as described by Salama et al. (1990).
Cloning of BSR4
The scheme for isolating genomic clones of BSR4 was based on the recessive nature of sec14–1ts suppression by bsr4 alleles. Yeast strain CTY160 was transformed to Ura+ at 30°C with a yeast genomic library propagated in YCp50 (Rose et al., 1987). The Ura+ transformants were patched onto selective medium, then replica plated onto duplicate YPD plates incubated at 30°C and 37°C, respectively. Of the 939 Ura+ transformants so analyzed, 2 were found to have acquired an unselected ts phenotype. In one case, the ts phenotype exhibited plasmid linkage and that plasmid was designated pCTY300. This plasmid was considered to likely carry BSR4 because it exhibited the expected property of specific complementation of bsr4. That is, pCTY300 failed to complement bsr2 or bsr3 mutations.
Integrative genetic mapping was used to prove that pCTY300 carried the authentic BSR4 gene. A 9 kb BamHI fragment that was excised from the pCTY300 insert and carried the 3′ end of BSR4 was subcloned into Ylp5 (Botstein et al., 1979). The resultant plasmid was gapped within the insert by digestion with endonuclease Kpnl, and the linearized construct was recombined into the genome of yeast strain CTY70 via the homology presented by the insert DNA. The Ylp5-bearing strain was mated to strain CTY160, and the resultant diploid was subjected to meiotic analysis. Twenty-six tetrads were analyzed for segregation of Ts+ (marking bsr4), ts− (marking BSR4), and Ura+ (marking the integrated Ylp5 construct). For every tetrad analyzed, the meiotic progeny exhibited the expected 2:2 segregation of Ts+:ts− and Ura+:ura−. Moreover, all Ts+ progeny were Ura− and all ts spores were Ura+. These data demonstrated a tight linkage of BSR4 and the Ylp5-borne URA3 (26PD:ONPD:OTT), thereby proving that pCTY300 carried BSR4.
Construction of cki-284::HIS3 and sac14–88::URA3
Plasmid pCTY307 is a YCp50 derivative that carries CKI on a 2.7 kb HindIII–CIal restriction fragment. A 1.8 kb BamHI restriction fragment carrying HIS3 was subcloned into the unique BamHI site of that fragment. This BamHI site resides essentially in the center of the CKI coding sequence (Hosaka et al., 1989). The resulting disruption truncates CKI at codon 284. The cki-284::HIS3 allele was recovered on a 4.0 kb CIal-Hpal restriction fragment for linear transformation into the appropriate yeast strains.
The sec14–88::URA3 allele was constructed by linearizing plasmid pRE26 at its unique Kpnl site and inserting a 1.2 kb Kpnl fragment that carries the yeast URA3 gene. pRE26 is a derivative of pUC9 that carries the entire SEC14 gene, extending from a genomic MIuI site at −381 to a genomic BgIII site at approximately +2000 (Bankaitis et al., 1989), subcloned into the blunted EcoRI and SaII polylinker half-sites, respectively. This disruption truncated SEC14 after codon 88 and was excised as a 3.7 kb EcoRI–BgIII fragment for linear transformation into yeast.
Acknowledgments
We wish to thank Bob Bell, Bob Fuller, Susan Henry, Phil Bassford, and John Cronan for plasmids, strains, antisera, and helpful discussions. We are particularly indebted to Jim Rothman for sharing his insightful comments and ideas. Finally, we wish to thank Kathy Beal and Sandy Henson for expert preparation of the manuscript. These studies were supported by grants to V. A. B. from the National Science Foundation (DCB-8620076) and the American Heart Association Illinois Affiliate (880752). J. R. A. and W. D. were supported by a grant from the NIH (GM35143) to W. D.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC Section 1734 solely to indicate this fact.
References
- Aitken JF, van Heusden GPH, Temkin M, and Dowhan W (1990). The gene encoding the phosphatidylinositol transfer protein is essential for cell growth. J. Biol. Chem 265, 4711–4717. [PubMed] [Google Scholar]
- Bankaitis VA, Malehorn DE, Emr SD, and Greene R (1989). The Saccharomyces cerevisiae SEC14 gene encodes a cytosolic factor that is required for transport of secretory proteins from the yeast Golgi complex. J. Cell Biol 108, 1271–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis VA, Aitken JF, Cleves AE, and Dowhan W (1990). An essential role for a phospholipid transfer protein in yeast Golgi function. Nature 347, 561–562. [DOI] [PubMed] [Google Scholar]
- Botstein D, Falco SC, Stewart S, Brennan M, Scherer S, Stinch-comb DI, Struhl K, and Davis R (1979). Sterile host yeast (SHY): a eukaryotic system of biological containment for recombinant DNA experiments. Gene 8, 17–24. [DOI] [PubMed] [Google Scholar]
- Bowman B, and Slayman CW (1979). The effects of vanadate on the plasma membrane ATPase of Neurospora crassa. J. Biol. Chem 254, 2928–2934. [PubMed] [Google Scholar]
- Carman GM, and Henry SA (1989). Phospholipid biosynthesis in yeast. Annu. Rev. Biochem 58, 635–669. [DOI] [PubMed] [Google Scholar]
- Casadaban MJ, and Cohen SN (1980). Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol 138, 179–207. [DOI] [PubMed] [Google Scholar]
- Cleves AE, Novick PJ, and Bankaitis VA (1989). Mutations in the SAC1 gene suppress defects in yeast Golgi and yeast actin function. J. Cell Biol 109, 2939–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzusoff A, and Schekman R (1989). Functional compartments of the yeast Golgi apparatus are defined by the sec7 mutation. EMBO J. 8, 2695–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzusoff A, Redding K, Crosby J, Fuller RS, and Schekman R (1991). Localization components involved in protein transport and through the yeast Golgi apparatus. J. Cell Biol 112, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller RS, Brake A, and Thorner J (1989). Yeast prohormone processing enzyme (KEX2 gene product) is a Ca2+-dependent serine protease. Proc. Natl. Acad. Sci. USA 86, 1434–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein A, and Lampen JL (1979). β-d-fructofuranoside fructohydrolase from yeast. Meth. Enzymol 42, 504–511. [DOI] [PubMed] [Google Scholar]
- Hanahan D (1983). Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol 166, 557–580. [DOI] [PubMed] [Google Scholar]
- Helmkamp GM Jr. (1986). Phospholipid transfer proteins: mechanisms of action. J. Bioenerg. Biomembr 18, 71–90. [DOI] [PubMed] [Google Scholar]
- Hjelmstad RH, and Bell RM (1987). Mutants of Saccharomyces cerevisiae defective in sn-1,2-diacylglycerol cholinephosphotransferase: isolation, characterization, and cloning of the CPT1 gene. J. Biol. Chem 262, 3909–3917. [PubMed] [Google Scholar]
- Hjelmstad RH, and Bell RM (1988). The sn-1,2-diacylglycerol ethanolamine-phosphotransferase activity of Saccharomyces cerevisiae: isolation of mutants and cloning of the EPT1 gene. J. Biol. Chem 263, 19748–19757. [PubMed] [Google Scholar]
- Hosaka K, Kodaki T, and Yamashita S (1989). Cloning and characterization of the yeast CKI gene encoding choline kinase and its expression in Escherichia coli. J. Biol. Chem 264, 2053–2059. [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, and Kimura A (1983). Transformation of intact yeast cells treated with alkaline cations. J. Bacteriol 153, 163–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilmartin JV, and Adams AEM (1984). Structural rearrangements of tubulin and actin during the cell cycle of the yeast Saccharomyces. J. Cell Biol 98, 922–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ, Banta LM, and Emr SD (1988). Intracellular sorting and processing of a yeast vacuolar hydrolase: proteinase A propeptide contains vacuolar targeting information. Mol. Cell. Biol 8, 2105–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreibich G, Debey P, and Sabatini D (1973). Selective release of content from microsomal vesicles without membrane disassembly. I. Permeability changes induced by low detergent concentrations. J. Cell Biol 58, 436–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippincott-Schwartz J, Yuan LC, Bonifacino JS, and Klausner RD (1989). Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane recycling from Golgi to ER. Cell 56, 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason T, Poyton R, Wharton D, and Schatz G (1973). Cytochrome c oxidase from baker’s yeast. I. Isolation and properties. J. Biol. Chem 248, 1346–1354. [PubMed] [Google Scholar]
- Mimms L, Floreani M, Tyner J, Whitters E, Rosenlof R, Wray L, Goetze A, Sarin V, and Eble K (1990). Discrimination of hepatitis B virus subtypes using monoclonal antibodies to the pre-S1 and pre-S2 domains of the viral envelopes. Virology 176, 604–619. [DOI] [PubMed] [Google Scholar]
- Novick P, Field C, and Schekman R (1980). Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway. Cell 21, 205–215. [DOI] [PubMed] [Google Scholar]
- Novick P, Osmond BC, and Botstein D (1989). Suppressors of yeast actin mutations. Genetics 121, 659–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose M, Novick P, Thomas J, Botstein D, and Fink G (1987). A Saccharomyces cerevisiae genomic plasmid bank based on a centromere-containing shuttle vector. Gene 60, 237–243. [DOI] [PubMed] [Google Scholar]
- Rothman JE (1990). Phospholipid transfer market. Nature 347, 519–520. [DOI] [PubMed] [Google Scholar]
- Rothman JE, and Orci L (1990). Movement of proteins through the Golgi stack: a molecular dissection of vesicular transport. FASEB J. 4, 1460–1468. [DOI] [PubMed] [Google Scholar]
- Rothstein RJ (1983). One-step gene disruption in yeast. Meth. Enzymol 101, 202–211. [DOI] [PubMed] [Google Scholar]
- Salama SR, Cleves AE, Malehorn DE, Whitters EA, and Bankaitis VA (1990). Cloning and characterization of the Kluyvero-myces lactis SEC14: a gene whose product stimulates Golgi secretory function in S. cerevisiae. J. Bacteriol 172, 4510–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman F, Fink GR, and Hicks JB (1983). Methods in Yeast Genetics (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; ). [Google Scholar]
- Silhavy TJ, Berman ML and Enquist LW (1984). Experiments with Gene Fusions (Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; ). [Google Scholar]
- Tsukagoshi Y, Nikawa J-I, and Yamashita S (1987). Molecular cloning and characterization of the gene encoding cholinephosphate cytidylyltransferase in Saccharomyces cerevisiae. Eur. J. Biochem 169, 477–486. [DOI] [PubMed] [Google Scholar]
- Van Meer G (1989). Lipid traffic in animal cells. Annu. Rev. Cell Biol 5, 247–275. [DOI] [PubMed] [Google Scholar]
- Wieland FT, Gleason ML, Serafini TA, and Rothman JE (1987). The rate of bulk flow from the endoplasmic reticulum to the cell surface. Cell 50, 289–300. [DOI] [PubMed] [Google Scholar]