

SUMMARY
Protein synthesis inhibitors (e.g., cycloheximide) block mitotic entry, suggesting that cell cycle progression requires protein synthesis until right before mitosis. However, cycloheximide is also known to activate p38 mitogen-activated protein kinase (MAPK), which can delay mitotic entry through a G2/M checkpoint. Here, we ask whether checkpoint activation or a requirement for protein synthesis is responsible for the cycloheximide effect. We find that p38 inhibitors prevent cycloheximide-treated cells from arresting in G2 phase and that G2 duration is normal in approximately half of these cells. The Wee1 inhibitor MK-1775 and Wee1/Myt1 inhibitor PD0166285 also prevent cycloheximide from blocking mitotic entry, raising the possibility that Wee1 and/or Myt1 mediate the cycloheximide-induced G2 arrest. Thus, protein synthesis during G2 phase is not required for mitotic entry, at least when the p38 checkpoint pathway is abrogated. However, M phase progression is delayed in cycloheximide-plus-kinase-inhibitor-treated cells, emphasizing the different requirements of protein synthesis for timely entry and completion of mitosis.
Graphical Abstract
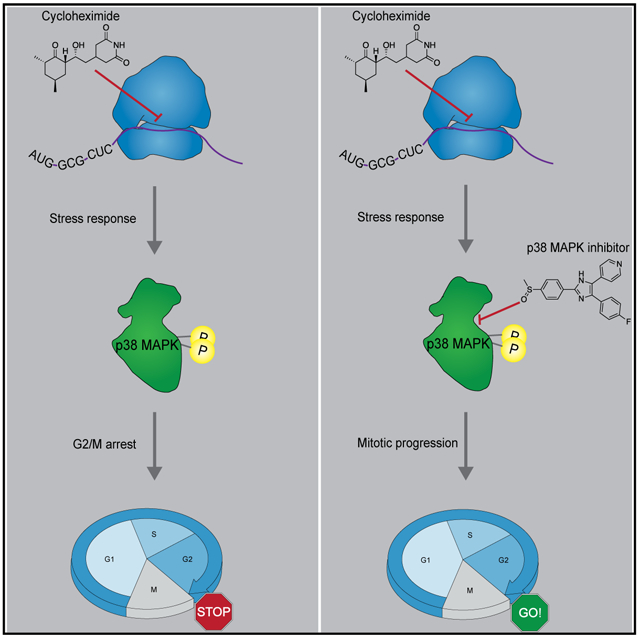
In Brief
Protein synthesis inhibitors have long been known to prevent G2 phase cells from entering mitosis. Lockhead et al. demonstrate that this G2 arrest is due to the activation of p38 MAPK, not insufficient protein synthesis, arguing that protein synthesis in G2 phase is not absolutely required for mitotic entry.
INTRODUCTION
Early studies on human cells in tissue culture as well as cells in the intestinal crypt of rats demonstrated that protein synthesis inhibitors, like cycloheximide and puromycin, prevent cells from entering mitosis, unless the cells were already in late G2 phase at the time of treatment (Donnelly and Sisken, 1967; Verbin and Farber, 1967). The discovery of mitotic cyclins, activators of the cyclin-dependent kinases (Cdks), which accumulate prior to mitosis, provided a plausible explanation for these observations (Evans et al., 1983; Moreno et al., 1989; Morgan, 2007). Indeed, supplementing a cycloheximide-arrested Xenopus laevis egg extract with exogenous cyclin B is sufficient to promote mitotic progression (Murray et al., 1989), as is supplementing an RNase-treated extract with cyclin B mRNA (Murray and Kirschner, 1989), and blocking the synthesis of cyclin B1 and B2 prevents mitotic entry (Minshull et al., 1989). This argues that the synthesis of this particular protein is of singular importance for M phase initiation.
In human cells, mitotic cyclins, mainly cyclins A2, B1, and B2, start to accumulate around the time of the G1/S transition as a result of the activation of cyclin transcription by E2F-family transcription factors (Dyson, 1998) and stabilization of the cyclin proteins via antigen-presenting cell (APC)/CCdh1 inactivation (Reimann et al., 2001). At the end of S phase, the ATR-mediated DNA replication checkpoint is turned off and a FOXM1-mediated transcriptional circuit is activated (Lemmens et al., 2018; Saldivar et al., 2018). At about the same time, the pace of cyclin B1 accumulation (Akopyan et al., 2014; Deibler and Kirschner, 2010; Frisa and Jacobberger, 2009; Jacobberger et al., 2012; Pines and Hunter, 1991), as well as the accumulation of other pro-mitotic regulators, including Plk1, Bora, and Aurora A, increases (Akopyan et al., 2014; Macůrek et al., 2008; Seki et al., 2008). These changes in transcription and protein abundances are thought to culminate in the activation of mitotic kinases, especially Cdk1, and the inactivation of the counteracting phosphatases PP1 and PP2A-B55 (Crncec and Hochegger, 2019; Heim et al., 2017). Cdk1 activity—judged by substrate phosphorylation—rises throughout G2 phase (Akopyan et al., 2014; Lindqvist et al., 2007) and sharply increases toward the end of G2 phase (Akopyan et al., 2014; Gavet and Pines, 2010b). Cdk1-cyclin B1 then translocates from the cytoplasm to the nucleus just prior to nuclear envelope breakdown (Hagting et al., 1999; Jin et al., 1998; Li et al., 1997; Pines and Hunter, 1991; Santos et al., 2012).
The final increase in cyclin B1-Cdk1 activity, and decrease in PP2A-B55 activity, is thought to be due to the flipping of two bistable switches. Two feedback loops, a double-negative feedback loop involving the Cdk1-inhibitory kinases Wee1/Myt1 and a positive feedback loop involving the Cdk1-activating phosphatase Cdc25, keep Cdk1 activity low until cyclin B1 has reached a threshold concentration, beyond which the system switches from low to high Cdk1 activity and high to low Wee1/ Myt1 activity (Figure 1A; Novak and Tyson, 1993; Pomerening et al., 2003; Sha et al., 2003). At the same time, a double-negative feedback loop centered on PP2A-B55 flips and leads to an abrupt decrease of PP2A-B55 activity (Gharbi-Ayachi et al., 2010; Mochida et al., 2010, 2016; Rata et al., 2018; Vinod and Novak, 2015).
Figure 1. Measuring the Duration of Cell Cycle Phases Using Fluorescently Labeled PCNA and Histone H2B in MCF10A Cells.

(A) Schematic of the regulation of Cdk1 activity at the G2/M transition by cyclins and multiple feedback loops. The protein synthesis inhibitor cycloheximide (CHX) can block cyclin accumulation; it also activates p38 MAPK, which can delay G2/M progression by inhibiting Cdc25 and/or potentially activating Wee1/Myt1 (Reinhardt and Yaffe, 2009). The small-molecule inhibitors SB202190 and SB203580 and PD0166285 and MK-1775 have been used in this study to inhibit p38 MAPK or Wee1/Myt1 activity, respectively.
(B) eYFP-PCNA can be used to determine the onset of S phase, the completion of S phase, and the onset of mitosis (nuclear envelope breakdown); histone H2B-mTurquoise (used here) or histone H2B-mCherry can be used to determine anaphase onset. Scale bars: 10 μm.
(C) Three examples of cells showing the disappearance of eYFP-PCNA foci (yellow arrows) at the end of S phase. Times (in the format h:min) were aligned to the time of entry into G2 phase. Scale bars: 10 μm.
(D) Frequency distributions of G2 phase duration measured in MCF10A cells expressing H2B-mCherry and eYFP-PCNA either in the absence (“medium,” gray; n = 104) or presence of 0.1% DMSO (blue; n = 100). Means and standard deviations are indicated.
The cyclin B1 threshold concentration is determined by the amounts of Cdc25 and Wee1/Myt1 activity present (Tsai et al., 2014). In somatic cells, several signaling pathways impinge upon Cdc25 and/or Wee1 to delay the G2-to-M transition in the face of stresses (Reinhardt and Yaffe, 2009). These include the ATM/ATR kinases, which activate Chk1 and Chk2, which in turn can inactivate Cdc25 and activate Wee1 by phosphorylating 14-3-3 binding sites in the two Cdk1 regulators. These pathways play a role in delaying mitosis in the presence of DNA damage and may also help prevent premature mitosis in cells undergoing normal DNA replication (Blackford and Jackson, 2017; Lemmens et al., 2018; Reinhardt and Yaffe, 2009; Saldivar et al., 2017). In addition, a protein kinase cascade that includes the MKK3 and MKK6 mitogen-activated protein (MAP) kinase kinases, p38 MAP kinase (MAPK), and the downstream kinase MAPKAP kinase 2 (MK2) has been implicated in restricting the activity of mitotic kinases during DNA replication and blocking mitotic entry in response to cellular stress by phosphorylating and therefore inhibiting Cdc25 via the same 14-3-3 binding site (Lemmens et al., 2018; Manke et al., 2005; Matsusaka and Pines, 2004). Interestingly, p38 activation has been observed in response to protein synthesis stresses, including cycloheximide (Kyriakis et al., 1994); indeed, cycloheximide is often used as a positive control for maximal activation of p38. These findings raise the question of whether the cycloheximide-dependent G2 delay is caused by blocking the synthesis of proteins required for mitotic entry or rather activation of the p38-dependent G2/M checkpoint.
Here, we used live-cell markers of cell cycle progression combined with small-molecule inhibitors to dissect the contribution of protein synthesis to G2 and mitotic progression. We show that inhibition of Wee1/Myt1 shortens the duration of G2 phase in a dose-dependent manner and allows cells to progress into mitosis in the presence of cycloheximide. Moreover, p38 inhibition overcomes a cycloheximide-induced G2 arrest, arguing that p38-mediated checkpoint activation causes the arrest and not insufficient protein synthesis. However, although G2 protein synthesis is not required for mitotic entry, it is required for normal mitotic progression. These findings suggest that the burst of cyclin synthesis that normally occurs during G2 phase serves as a “just-in-time” preparation for mitotic progression but does not trigger mitotic entry.
RESULTS
We chose MCF10A cells, a spontaneously immortalized human mammary epithelial cell line, for these studies, because they are euploid, non-tumorigenic, and have been studied extensively (Debnath et al., 2003; Soule et al., 1990). To determine when S phase ends and G2 phase begins, we stably expressed an eYFP-PCNA (proliferating cell nuclear antigen) fusion protein, a live-cell marker of DNA replication (Hahn et al., 2009; Leonhardt et al., 2000; Zerjatke et al., 2017). eYFP-PCNA forms bright foci within the nucleus during S phase, which become brighter and less numerous as S phase progresses (Figures 1B and 1C). At the end of S phase, eYFP-PCNA foci dissolve and fluorescence becomes diffuse, marking the S/G2 transition (Figure 1C). Upon nuclear envelope breakdown (NEB), nuclear eYFP-PCNA disperses throughout the cell; this can be taken as a marker for the G2/M transition (or, more precisely, of the prophase/prometaphase transition; Figure 1B). Thus, eYFP-PCNA proved to be well suited to measure G2 duration, from the time of G2 onset (the disappearance of foci) to the time of G2 termination (taken as the time when eYFP-PCNA exited the nucleus due to NEB; Zerjatke et al., 2017). In addition, we stably expressed histone H2B fused to a fluorescent protein in order to monitor the different stages of mitotic progression (Figure 1B).
Typical mean G2 durations were found to be 4 h with a standard deviation of 42 min (Figure 1D). This is within the range of previously reported durations for G2 phase in a variety of cell lines (Akopyan et al., 2014; Araujo et al., 2016; Baserga, 1985; Essers et al., 2005; Gerlich et al., 2006; Hahn et al., 2009). Because many of our subsequent experiments required the addition of DMSO-solubilized drugs to a final DMSO concentration of 0.1%, we ensured that this concentration of DMSO does not measurably impact G2 progression (Figure 1D).
Live-Cell Imaging Confirms that Cycloheximide Blocks Entry into Mitosis
Early studies on fixed cells showed that the protein synthesis inhibitors puromycin and cycloheximide cause cells to arrest in G2 phase (Donnelly and Sisken, 1967; Verbin and Farber, 1967). We confirmed this finding by live-cell microscopy using the PCNA probe to demarcate G2 phase. We followed asynchronously growing cells in cell culture for 4–6 h and then added cycloheximide (10 μg/mL) and continued to follow the cells for another 6–10 h (Figure 2A). This allowed us to identify cells that had exited S phase during the initial imaging period, determine accurately how much time these cells had spent in G2 phase prior to drug addition, and finally determine the fate of these cells in response to the drug treatment.
Figure 2. Cycloheximide Blocks Entry into Mitosis.

(A) Schematic of the experimental setup: asynchronously grown cells were imaged for 4–6 h in order to determine the time when cells exited S phase. After this period, DMSO or small-molecule inhibitors were added, and cells were followed for another 6–12 h to determine whether and when cells entered and progressed through mitosis.
(B and C) Montages of MCF10A cells expressing H2B-mCherry and eYFP-PCNA followed over the time course of the experiment described in (A). Times (in the format h:min) were aligned to the point of DMSO or cycloheximide addition. Four cells are shown that had spent different amounts of time in G2 phase at the time of cycloheximide addition and subsequently either arrested in G2 phase (B) or entered mitosis (C).
(D) Cell cycle progression in MCF10A cells expressing H2B-Turquoise and eYFP-PCNA and treated with DMSO (left; n = 130) or CHX (right; n = 165). Each row represents timing data from a single cell. The majority of cells treated with cycloheximide arrested in G2 phase.
(E) Logistic regression analysis. This estimates the probability of a cell entering mitosis as a function of how much time the cell had spent in G2 phase at the time of drug addition, for the experiment shown in (D). Circles indicate the fraction of cells that entered mitosis by 5.5 h after entry into G2 phase; this cutoff was the time at which 95% of the DMSO-treated control cells had entered mitosis. The solid lines show the logistic fit for the data, and the lightly colored areas indicate the 95% confidence intervals.
(F) Mitotic indices for MCF10A cells expressing H2B-Turquoise and eYFP-PCNA cells treated with DMSO or CHX. Shown are two independent experiments (circles and triangles, respectively). At least 3,605 cells were counted for each time point.
Cells treated with DMSO alone progressed into mitosis (130 out of 130 cells), but cycloheximide addition arrested the large majority of cells (153 cells out of 165) in G2 phase (Figures 2B-2D). Cycloheximide-treated cells were more likely to progress into mitosis if the drug was added late in G2 phase. Of the 12 cycloheximide-treated cells that did enter mitosis, 10 had spent more than 3 h in G2 phase (>75% of the duration of a normal G2 phase) at the time of cycloheximide addition (Figures 2C and 2D). Based on logistic regression analysis, the probability that a cycloheximide-treated cell will enter mitosis if the cycloheximide is added 2 h after the start of G2 phase is 1% (with a 95% confidence interval [CI] of 0%–7%); if added 3 h after the start of G2 phase, it rises to 4% (95% CI 1%–11%), and if it is added 4 h after the start of G2 phase, the duration of a typical normal G2 phase, the probability is 19% (95% CI 6%–45%; Figure 2E). The fraction of mitotic cells in the cell population (mitotic index) remained approximately constant throughout the experiment for the DMSO-treated population but decreased to near-zero within 60 min after cycloheximide treatment (Figure 2F). Together, these findings confirm that cycloheximide-treated G2 cells do arrest, as previously noted (Donnelly and Sisken, 1967; Verbin and Farber, 1967), and imply that cells remain sensitive to cycloheximide treatment until late in G2 phase.
Wee1/Myt1 Inhibition Shortens G2 Phase and Restores Mitotic Entry in Cycloheximide-Treated G2 Phase Cells
The Wee1/Myt1 kinases are key regulators of the G2/M transition that, when active, restrain Cdk1-cyclin B activity. Fluorescence resonance energy transfer (FRET) studies have indicated that Cdk1 activation begins just prior to the nuclear translocation of Cdk1-cyclin B1—thus very late in G2 phase—which suggests that Wee1 and Myt1 may be active during almost all of G2 phase (Gavet and Pines, 2010a, 2010b). However, other studies have suggested that some Cdk1 activation can be detected early in G2 phase (Akopyan et al., 2014), which could mean that Wee1/Myt1 is switched off earlier. These possibilities can be distinguished by determining how much the duration of G2 phase can be shortened by Wee1/Myt1 inhibition. In the former case, the minimal duration of G2 phase would be near zero; in the latter case, it would be longer, with the minimal duration of G2 phase corresponding to how long the interval normally is between the inactivation of Wee1/Myt1 and the onset of M phase.
We used two small-molecule inhibitors, MK-1775 and PD0166285, to inhibit Wee1 and Wee1/Myt1 activity, respectively. MK-1775 inhibits Wee1 with a half maximal inhibitory concentration (IC50) of 5.2 nM and 100-fold selectivity over Myt1 (Hirai et al., 2009). PD0166285 is an inhibitor of both Wee1 and Myt1 and so may inhibit Cdk1 Tyr 15 more completely than MK-1775 does. However, it also inhibits the upstream kinase Chk1, although with a higher IC50 value (reported IC50 values of 24 nM for Wee1, 72 nM for Myt1, and 3.4 μM for Chk1; Wang et al., 2001). Treating an asynchronously growing cell culture with different concentrations of MK-1775 or PD0166285 reduced the Wee1-mediated phosphorylation of Cdk1 at Tyr 15 in a dose-dependent manner (Figure 3A), with a greater effect obtained with the Wee1/Myt1 inhibitor PD0166285 than with the more potent Wee1-only inhibitor MK-1775.
Figure 3. Wee1 Inhibition by MK-1775 Shortens G2 Phase and Restores Mitotic Entry in a Fraction of Cycloheximide-Treated G2 Phase Cells.

(A) Asynchronously growing MCF10A cells were treated with DMSO, different concentrations of the Wee1 inhibitor MK-1775 or the Wee1/Myt1-inhibitor PD0166285 for 1 h. The phosphorylation state of tyrosine 15 of Cdk1 as a measure of Wee1/Myt1 activity was analyzed by immunoblotting. α-tubulin was used as loading control. Uncropped immunoblots are shown in Figure S6A.
(B) G2 duration as a function of MK-1775 concentration. The duration of G2 phase of cells expressing H2B-mCherry and eYFP-PCNA was measured by live-cell fluorescence microscopy in the presence of DMSO or different concentrations of MK-1775. Only cells that had not entered G2 phase at the time of treatment and showed a distinct G2 phase were included in this analysis (n > 52 cells for all conditions).
(C) Montages of MCF10A cells expressing H2B-mCherry and eYFP-PCNA followed over the time course of the experiment described in Figure 2A. Times (in the format h:min) were aligned to the point of DMSO/drug addition (10 μg/mL CHX and/or 1 μM MK-1775).
(D) Cell cycle progression for MCF10A cells expressing H2B-mCherry and eYFP-PCNA and treated with DMSO (n = 100), cycloheximide (n = 103), MK-1775 (n = 105), or cycloheximide plus MK-1775 (n = 105). Each row represents timing data from a single cell. The majority of cells treated with cycloheximide arrested in G2 phase, whereas more of the cells treated with 1 μM MK-1775 in addition to cycloheximide entered mitosis. Rows marked with a purple square denote cells that underwent abnormal mitoses, often lacking proper metaphase and cytokinesis. A biological replicate is shown in Figure S1A.
(E) Logistic regression analysis. Probability of a cell entering mitosis as a function of how long the cell had been in G2 phase at the time of drug addition for the experiment shown in (D). Circles indicate the fraction of cells that entered mitosis by 5.3 h after entry into G2 phase; this cutoff was the time at which 95% of the DMSO-treated control cells had entered mitosis. The solid lines show the logistic fits for the data, and the lightly colored areas indicate the 95% confidence intervals. Note that the green (MK-1775) data points corresponding to a probability of 1.0 have been shifted upward to make them more visible.
(F) Mitotic indices for MCF10A cells expressing H2B-mCherry and eYFP-PCNA treated with DMSO, CHX, 1 μM MK-1775, or CHX plus 1 μM MK-1775. At least 4,547 cells were counted for each time point.
We first investigated the impact of the Wee1-only inhibitor MK-1775 on the duration of G2 phase by live-cell imaging. Cells were treated with different concentrations ranging from 15 nM to 2 μM MK-1775, and 52–100 cells that entered and completed G2 phase over a 10-h imaging period were tracked and their G2 durations determined. MK-1775 shortened G2 phase in a graded fashion. The highest concentration of MK-1775 (2 μM) resulted in a G2 duration of 116 ± 27 min compared to 242 ± 41 min in the DMSO-treated cells (mean ± SD; Figure 3B). The finding that Wee1 inhibition by MK-1775 gradually shortens but does not abolish G2 phase in MCF10A cells suggests that Wee1 is required for keeping cells from entering M phase, at least in the latter half of G2 phase.
Considering the central role of Wee1 in controlling G2/M, we asked whether Wee1 inhibition was able to overcome the cycloheximide-induced G2 arrest. We used the same experimental setup as described in Figure 2A but, after the initial imaging period, added DMSO, cycloheximide, 1 μM MK-1775, or cycloheximide plus 1 μM MK-1775 to the cells. Again (cf. Figure 2D), cells treated with DMSO progressed into mitosis with normal G2 duration, whereas cycloheximide prevented most cells from entering mitosis (Figures 3C-3E and S1A). Consistent with the idea that Wee1 inhibition shortens G2 phase, a majority of cells (72/105) treated with MK-1775 entered mitosis within 1 h of drug treatment (compared to 26/100 for the DMSO-treated cells). Cells that were treated with MK-1775 late in G2 phase tended to enter mitosis more quickly than those treated early in G2 phase (Figure S1B). The fraction of cells in mitosis spiked about 3-fold within the first hour of MK-1775 treatment (Figure 3F) but returned to baseline around 3 h after drug addition. Strikingly, whereas cycloheximide blocked mitotic entry in most cells (92/103), about half of the cells (55/105) treated with cycloheximide plus 1 μM MK-1775 entered mitosis. The probability of a cell entering mitosis in the presence of cycloheximide depended on the time the cell had spent in G2 phase prior to drug addition and increased sharply if cells had spent at least 1.5 to 2 h in G2 phase (Figure 3E). As we had observed for cells treated with the Wee1 inhibitor alone, the mitotic index for cells treated with MK-1775 plus cycloheximide showed a pronounced spike within the first hour of treatment; however, the spike decayed to zero within the next hour. Higher concentrations of MK-1775 (2 μM and 4 μM) mildly increased the percentage of cells entering mitosis in the presence of cycloheximide but caused severe mitotic delays in the presence or absence of cycloheximide (Figure S1C). Taken together, these data indicate that MK-1775 can partially override a cycloheximide-induced G2 phase arrest.
We next asked how the Wee1/Myt1 inhibitor PD0166285 compared to the Wee1-only inhibitor MK-1775. Similarly to MK-1775, PD0166285 shortened G2 phase gradually as the inhibitor concentration was increased (Figure 4A), but the shortening was more pronounced in the case of PD0166285. A concentration of 1 μM PD0166285 resulted in a G2 duration of 38 ± 17 min compared to 255 ± 31 min in the DMSO-treated control (mean ± SD; Figure 4A). The stronger effect of PD0166285 compared to MK-1775 could be due to its inhibition of Myt1, although it is also possible that inhibition of Chk1 or some other kinase could contribute.
Figure 4. Wee1/Myt1 Inhibition by PD0166285 Shortens G2 Phase and Restores Mitotic Entry in Cycloheximide-Treated G2 Phase Cells.

(A) The duration of G2 phase was measured by live-cell fluorescence microscopy of cells expressing H2B-mCherry and eYFP-PCNA in the presence of DMSO or different concentrations of PD0166285. Only cells that had not entered G2 phase at the time of treatment and showed a distinct G2 phase were included in this analysis (n > 28 cells for all conditions).
(B) Montages of MCF10A cells expressing H2B-mCherry and eYFP-PCNA followed over the time course of the experiment described in Figure 2A. Times (in the format h:min) were aligned to the point of DMSO/drug addition (10 μg/mL cycloheximide [CHX] and/or 1 μM PD0166285).
(C) Cell cycle progression for MCF10A cells expressing H2B-mCherry and eYFP-PCNA and treated with DMSO (n = 113), cycloheximide (n = 116), PD0166285 (n = 100), or cycloheximide plus PD0166285 (n = 111). Each row represents timing data from a single cell. The majority of cells treated with cycloheximide arrested in G2 phase, although cells treated with 1 μM PD0166285 alone or cycloheximide plus 1 μM PD0166285 progressed into mitosis shortly after treatment with the drug. A biological replicate is shown in Figure S2A.
(D) Logistic regression analysis. Probability of a cell entering mitosis as a function of how long the cell had been in G2 phase at the time of drug addition for the experiment shown in (C). Circles indicate the fraction of cells that entered mitosis by 7.8 h after entry into G2 phase; this cutoff was the time at which 95% of the DMSO-treated control cells had entered mitosis. The solid lines show the logistic fits for the data, and the lightly colored areas indicate the 95% confidence intervals. Note that the green (PD0166285) and purple (PD0166285+CHX) data points corresponding to a probability of 1.0 have been shifted upward to make them visible.
(E) Mitotic indices for MCF10A cells expressing H2B-mCherry and eYFP-PCNA treated with DMSO, CHX, PD0166285, or CHX plus PD0166285. At least 3,226 cells were counted for each time point.
Likewise, we found that Wee1/Myt inhibition can overcome the cycloheximide-induced G2 arrest, and the rescue was more complete than that afforded by MK1775 treatment. Essentially all of the cycloheximide-treated cells were able to enter mitosis in the presence of PD0166285 (Figures 4B, 4C, and S2A), and cells progressed into mitosis with similar dynamics as cells treated with PD0166285 alone (Figures 4E and S2B). In contrast to cycloheximide treatment alone, the probability of a cell entering mitosis was ~100% and was independent of the time the cell had spent in G2 phase at the time of drug addition (Figure 4D). The override was also observed when cells were treated with cycloheximide for 2 h prior to the addition of PD0166285 (Figure S2C).
The MK-1775 and PD0166285 data suggest that the Wee1/Myt1 switch is normally thrown very late in G2 phase. Moreover, they indicate that there is sufficient cyclin (and any other proteins essential for M phase entry) present even early in G2 phase to allow rapid mitotic entry, provided that Wee1/Myt1 activity is low. Moreover, the level of Wee1/Myt1 activity determines the length of G2 phase. These results are consistent with and extend the findings of previous studies on the effects of Wee1/Myt1 inhibition (Araujo et al., 2016; Chang et al., 2016; Heijink et al., 2015; Krek and Nigg, 1991; Wang et al., 2001).
Cycloheximide Treatment in S Phase Blocks Cell Cycle Progression, Even in the Absence of Wee1/Myt1 Activity
Previous studies of Wee1 inhibitors have shown that they can drive chemotherapy-treated and p53 mutant cell lines into mitosis without completing DNA replication (Aarts et al., 2012; Chang et al., 2016). Similarly, overexpression of Cdc25B (but not Cdc25C) has also been reported to drive S phase HeLa cells into mitosis (Karlsson et al., 1999). To further explore the connection between protein synthesis and M phase entry, we analyzed the cell cycle progression of cells treated in S phase with DMSO, cycloheximide, Wee1 or Wee1/Myt1 inhibitor, or Wee1 or Wee1/Myt1 inhibitor plus cycloheximide. After being treated with DMSO in S phase, 100% of cells (99/99) entered G2 phase, and 93% (92/99) of these cells progressed into mitosis during the 12-h imaging period (Figure 5A). In contrast, only 29% of cells (29/100) treated with cycloheximide during S phase progressed into G2 phase (Figure 5A), whereas most other cells (60/100) continued to display PCNA foci (albeit dimmer foci than those seen in control cells), suggesting that S phase was never completed (Figure 5C). None of the cells treated with cycloheximide alone progressed into mitosis (Figures 5A and 5B).
Figure 5. Cycloheximide Treatment in S Phase Blocks Cell Cycle Progression, Even in the Absence of Wee1/Myt1 Activity.

(A) Cell cycle progression for MCF10A cells expressing H2B-mCherry and eYFP-PCNA treated during S phase with either DMSO (n = 99), cycloheximide (n = 100), 1 μM MK-1775 (n = 100), cycloheximide plus 1 μM MK-1775 (n = 100), or 4 μM MK-1775 alone (n = 99). Each row represents a single cell. Rows marked with a purple square denote cells that underwent abnormal mitoses, often lacking proper metaphase and cytokinesis.
(B) Cell cycle progression for MCF10A cells expressing H2B-mCherry and eYFP-PCNA treated during S phase with either DMSO (n = 99), cycloheximide (n = 98), 1 μM PD0166285 (n = 99), or cycloheximide plus 1 μM PD0166285 (n = 105). Each row represents timing data from a single cell. Rows marked with a purple square denote cells that underwent abnormal mitoses, often lacking proper metaphase and cytokinesis.
(C) Montage of an MCF10A cell expressing H2B-mCherry and eYFP-PCNA treated with cycloheximide during S phase. In this cell (and most cells), the PCNA foci became weaker after cycloheximide treatment, yet they never completely disappeared, suggesting that these cells remained in S phase. Time (in the format h:min) was aligned to the point of cycloheximide addition.
(D) Montage of an MCF10A cell expressing H2B-mCherry and eYFP-PCNA treated with 1 μM PD0166285 during S phase that progressed into mitosis in the presence of PCNA foci (suggesting that the cell never completed S phase). Time (in the format h:min) was aligned to the point of drug addition. Note the abnormal mitotic progression without a proper metaphase and cytokinesis.
(E) Mitotic duration, measured as the time from nuclear envelope breakdown (NEB) to anaphase, for cells treated either in G2 phase or S phase with DMSO, PD0166285, or cycloheximide plus PD0166285. Whereas treatment with PD0166285 or treatment with PD0166285 plus cycloheximide in G2 phase only slightly extended mitosis (see also Figure 7), treatment with these drugs in S phase dramatically increased the duration of mitosis.
All cells (100/100) treated with 1 μM MK-1775 during S phase entered G2 phase and progressed into mitosis. We rarely (0/100 for the experiment shown in Figure 5A and 4/102 for a replicate experiment) observed S phase cells prematurely entering mitosis after treatment with 1 μM MK-1775, but the number of these premature transitions into mitosis increased significantly (24/99 cells) if cells were treated with 4 μM MK-1775 (Figure 5A). Similarly, all cells (99/99) treated with 1 μM PD0166285 during S phase entered mitosis; 38 of them progressed into mitosis in the presence of PCNA foci and without displaying a detectable G2 phase (Figures 5B and 5D). This suggests that, at some time during S phase, there is enough pro-mitotic activity to drive cells into mitosis if Wee1 and Myt1 are inhibited. However, most of the cells that had directly progressed from S phase into mitosis after PD0166285 addition failed to undergo proper chromosome segregation and cytokinesis (28/38 failures compared to 3/61 cells which displayed a G2 phase; Figure 5B), and even those cells that carried out some duration of G2 phase prior to mitotic entry required more time to progress through mitosis (Figure 5E). Presumably, this is at least partially due to the cells’ incomplete DNA replication.
Only 35% of cells (35/100) treated with cycloheximide plus 1 μM MK-1775 during S phase entered G2 phase, and none of these cells entered mitosis, as might be expected given that cells treated with cycloheximide plus MK-1775 early in G2 phase generally failed to progress into M phase. On the other hand, the Wee1/Myt1 inhibitor PD0166285 was able to completely overcome a cycloheximide-induced arrest when cells had entered G2 at the time of drug addition (Figure 4C). However, only 32% of cells (34/105) treated in S phase with cycloheximide plus PD0166285 entered mitosis, 9 of them directly from S phase. About one-third of the cells (38/105) never completed S phase, and 19% (20/105) entered G2 phase, but not mitosis. Frequently, cells that managed to enter mitosis exhibited extended and qualitatively abnormal mitotic progression (Figures 5B and 5E). These findings support the hypothesis that some S phase protein synthesis is required for mitotic entry, even in the absence of the Cdk1-inhibiting activity of Wee1 and Myt1.
Wee1/Myt1 Counteract Pro-mitotic Activities that Accumulate during G2 Phase
To further investigate the relationship between Wee1/Myt1 activity, G2 duration, and a cell’s ability to enter mitosis, we followed untreated cells for 6 h, treated cells with cycloheximide for 2 h, and then added different Wee1/Myt1 inhibitor concentrations and assessed the cell’s fate (Figures S2C and S2D). At the lowest concentration of PD0166285 (0.125 μM), only a fraction of cells (31%; 32/104) progressed into mitosis, and the probability for a cell to enter mitosis increased as the time the cell had spent in G2 phase prior to cycloheximide addition increased (Figure S2D). With increasing PD0166285 concentrations, more cells were able to enter mitosis, even if they had spent less time in G2 phase prior to drug addition (Figures S2C and S2D). These results are again consistent with the idea that pro-mitotic activities accumulate throughout G2 phase and are opposed by Wee1/Myt1 activity during this time; the later in G2 phase, the more pro-mitotic activities have accumulated and the less completely Wee1/Myt1 needs to be inhibited in order to flip the mitotic switch.
p38 Inhibition Allows Cells to Enter Mitosis in the Presence of Cycloheximide
The data presented so far are consistent with the hypothesis that, in G2 phase, cyclin synthesis triggers mitotic entry: cycloheximide blocks mitotic entry, and the effects of MK-1775 and PD0166285 suggest that some pro-mitotic activity, possibly cyclin, gradually accumulates throughout G2 phase. However, it also remains possible that the ability of cycloheximide to block mitotic entry is due to its activation of p38 MAPK and MK2, rather than to any effect on cyclin accumulation (Kyriakis et al., 1994; Reinhardt and Yaffe, 2009).
To address this issue directly, we treated cells with cycloheximide plus one of two p38 MAPK inhibitors (SB202190 and SB203580) that act as high-affinity inhibitors of p38α and p38β (MAPK14 and MAPK11) and as lower affinity inhibitors of other protein kinases (Davies et al., 2000). We verified that, as previously reported, cycloheximide stimulated the phosphorylation of p38 and its downstream target Hsp27 and that the inhibitors decreased cycloheximide-induced Hsp27 phosphorylation in a dose-dependent fashion (Figure 6A). We sometimes also observed a reduction of p38 phosphorylation itself when using higher inhibitor concentrations (10 and 50 μM), which could be either an off-target effect or a consequence of feedback in the regulation of p38 activity (Figure S3A). Tyrosine 15 phosphorylation of Cdk1 was not impacted by cycloheximide or p38 inhibition (Figure S3A). The Wee1 inhibitor MK-1775 did not impact the phosphorylation state of p38 or Hsp27 (Figure S3B). Treating cells with 1 μM PD0166285, on the other hand, reduced both the cycloheximide-induced phosphorylation of p38 and Hsp27. The observed reduction in phosphorylation could be a consequence of either cross-talk between Wee1/Myt1 and p38, Chk1 and p38, or an off-target effect.
Figure 6. p38 MAPK Inhibition Allows Cells to Enter Mitosis in the Presence of Cycloheximide.

(A) Asynchronously growing cells were treated for 6 h with DMSO, cycloheximide, or cycloheximide plus either of the p38 MAPK inhibitors SB202190 or SB203580. The phosphorylation state of p38 as well as the phosphorylation state of the p38 substrate Hsp27 was analyzed by immunoblotting to assess the activation state of p38. Uncropped immunoblots are shown in Figure S6B.
(B) Montages of MCF10A cells expressing H2B-mCherry and eYFP-PCNA followed over the time course of the experiment described in Figure 3A treated with cycloheximide or cycloheximide plus either SB202190 or SB203580. Times (in the format h:min) were aligned to the point of drug addition.
(C) Cell cycle progression for MCF10A cells expressing H2B-mCherry and eYFP-PCNA treated with DMSO (n = 99), CHX (n = 100), SB202190 (n = 92), SB203580 (n = 90), CHX+SB202190 (n = 95), or CHX+SB203580 (n = 91). Each row represents timing data from a single cell. The majority of cells treated with cycloheximide arrested in G2 phase, whereas cells treated with CHX plus SB202190 or SB203580 (50 μM) progressed into mitosis in most cases. Rows marked with a purple square denote cells that underwent abnormal mitoses, often lacking proper metaphase and cytokinesis.
(D) Logistic regression analysis. Probability of a cell to enter mitosis as a function of how long the cell has already been in G2 phase at the time of drug addition for the experiment shown in (C). Circles indicate the fraction of cells that entered mitosis by 5 h after entry into G2 phase; this cutoff was the time at which 95% of the DMSO-treated control cells had entered mitosis. The solid lines show the logistic fit for the data, and the lightly colored areas indicate the 95% confidence intervals.
(E) Mitotic indices for MCF10A cells expressing H2B-mCherry and eYFP-PCNA cells treated with DMSO, CHX, SB202190, SB203580, CHX+SB202190, or CHX+SB203580. At least 3,672 cells were counted for each time point.
An additional similar experiment is shown in Figures S4A and S4B.
Both p38 inhibitors almost completely prevented cycloheximide from blocking the progression of G2 phase cells into M phase (Figures 6B and 6C). Whereas only 4 out of 100 cells treated with cycloheximide alone entered M phase, 83 out of 100 SB202190- and 77 out of 92 SB203580-treated cells did enter M phase in the presence of cycloheximide (Figure 6C). For cells treated with cycloheximide alone, the probability of entering M phase was near zero unless the cells were treated late in G2 phase (Figures 6C and 6D), as shown earlier (Figures 2E, 3D, and 4C); however, the probability of entering M phase for cells treated with cycloheximide plus either SB202190 or SB203580 was 10%–20% for cells treated at the start of G2 phase and rose to near 100% for cells treated 2 h after the start of G2 phase (Figures 6C and 6D). Overall, 45 out of 95 cells treated with SB202190 plus cycloheximide and 60 out of 91 cells treated with SB203580 plus cycloheximide exhibited a normal G2 phase duration (between the 5th and 95th percentiles of the G2 durations of DMSO-treated control cells; Figure S3C). However, a significant fraction of the cells treated early during G2 phase had prolonged G2 durations, whereas cells treated later in G2 phase mostly exhibited a normal G2 duration (Figure S3D). The combination of cycloheximide plus SB202190 or SB203580 caused a small increase in the mitotic index followed by a slow decline to lower levels but rescued the sharp decline in mitotic cells observed in cycloheximide-treated cultures (Figures 6E and S4B). The structurally similar but inactive compound SB202474 did not prevent cycloheximide from blocking M phase entry (Figure S4). Similar results were obtained in HeLa and hTERT-RPE1 cells (Figures S4C and S4D).
Thus, the p38 MAPK inhibitors SB202190 and SB203580 allow the majority of the cycloheximide-treated G2 phase cells to progress into M phase. This suggests that p38-mediated checkpoint effects, rather than a lack of protein synthesis per se, are principally responsible for the arrest of cycloheximide-treated G2 phase cells.
Cells treated with SB202190 or SB203580 alone generally progressed through G2 phase normally, although about 12% of cells showed a prolonged G2 phase (Figure S3C) and a few cells (4/92 and 2/90) failed to enter M phase (Figure 6C). Thus, in normal, unperturbed cells, p38 has relatively little effect on G2 duration and M phase.
Protein Synthesis during G2 Phase Is Required for Normal Mitotic Progression
So far, we have shown that inhibition of either Wee1/Myt1 or p38 can overcome a cycloheximide-induced arrest and allow G2 phase cells to progress into mitosis in the absence of protein synthesis. However, the requirements to enter mitosis and to successfully progress through mitosis might differ (see, e.g., Gavet and Pines, 2010b). Consistent with this notion, we had already observed that cells treated with the Wee1/Myt1 inhibitor during S phase could enter mitosis (with little or no G2 phase) but then exhibited pronounced mitotic errors (Figure 5). In addition, a few cells treated with cycloheximide plus PD0166285 during G2 phase exhibited very long mitoses (Figure 4D), as did some cells treated with cycloheximide plus either of the p38 inhibitors (Figure 6C).
Accordingly, we examined the duration of mitosis in cells treated in G2 phase with cycloheximide plus either the Wee1 inhibitor, the Wee1/Myt1 inhibitor, or one of the p38 inhibitors. Cells treated with MK-1775 showed some delay in mitotic progression, with 42.9% of cells exhibiting protracted mitoses, taken here as mitoses longer than the 95th percentile of mitotic durations in DMSO-treated cells (Figure 7A). Treatment with MK-1775 plus cycloheximide also caused some cells to progress through mitosis more slowly, though the effect was somewhat less pronounced than that seen with MK-1775 alone (Figure 7A). The milder effect might be due to a smaller fraction of the cycloheximide plus MK-1775-treated early G2 phase cells entering mitosis compared with the cells treated with MK-1775 alone (Figure 3E). A substantial fraction of cells treated with PD0166285 with or without cycloheximide exhibited a protracted mitosis (47% for PD-treated cells and 58% for cells treated with PD plus cycloheximide; Figure 7B). Thus, cells that successfully entered mitosis after Wee1 or Wee1/Myt1 inhibition in the presence or absence of cycloheximide were, nevertheless, delayed in their progression through M phase. These results confirm previous findings that Wee1/Myt1 inhibition extends the mitotic duration (Araujo et al., 2016). The duration of mitosis was a dose-dependent function of both MK-1775 and PD0166285 concentration (Figure 7C). Moreover, the greater the shortening of G2 phase, the greater the delay in M phase (compare Figure 7C with Figures 3B, 4A, and S5).
Figure 7. Protein Synthesis during G2 Phase Is Required for Normal Mitotic Progression.

(A) Frequency distribution of mitotic durations (measured from NEB to anaphase onset) of cells treated with DMSO (n = 100), 1 μM MK-1775 (n = 105), or CHX plus 1 μM MK-1775 (n = 54) during G2 phase. Cells were considered to exhibit a protracted mitosis if the mitotic duration exceeded the 95th percentile of the mitotic duration of DMSO-treated cells (>39.5 min). p values were calculated using a nonparametric Mann-Whitney test (****p < 0.0001).
(B) Frequency distribution of mitotic durations (measured from NEB to anaphase onset) of cells treated with DMSO (n = 113), 1 μM PD0166285 (n = 100), or CHX plus 1 μM PD0166285 (n = 110) during G2 phase. Cells were considered to exhibit a protracted mitosis if the mitotic duration exceeded the 95th percentile of the mitotic duration of DMSO-treated cells (>30 min). p values were calculated using a nonparametric Mann-Whitney test (*p < 0.05 and ****p < 0.0001).
(C) Mitotic duration was measured for cells that progressed through G2 phase and entered mitosis in the presence of DMSO or different concentrations of MK-1775 or PD0166285. Mitotic duration increased with higher concentrations of MK-1775 or PD0166285 (and shorter G2 duration; see Figures 3B and 4A).
(D) Frequency distribution of mitotic durations of cells treated with DMSO (n = 99), SB202190 (n = 88), SB203580 (n = 88), CHX+SB202190 (n = 76), or CHX+SB203580 (n = 77) during G2 phase. Cells were considered to exhibit a protracted mitosis if the mitotic duration exceeded the 95th percentile of the mitotic duration of DMSO-treated cells (>30 min). p values were calculated using a nonparametric Mann-Whitney test.
For the p38 inhibitors SB202190 and SB203580, the drugs had some effect on the duration of mitosis, even in the absence of cycloheximide; 17% (SB202190) and 4.5% (SB203580) of the drug-treated cells exhibited a protracted mitosis versus 0% for the DMSO-treated controls (Figure 7D). This suggests that p38 function may contribute to M phase progression in at least a sub-set of cells. A greater proportion of cells treated with either of the inhibitors plus cycloheximide exhibited a protracted mitosis (50% and 34%, respectively), suggesting that the protein synthesis that normally occurs during G2 phase helps cells to progress through M phase in a timely fashion.
DISCUSSION
Here, we have used live-cell imaging to confirm the decades-old observation (Donnelly and Sisken, 1967; Verbin and Farber, 1967) that the protein synthesis inhibitor cycloheximide prevents G2 phase cells from entering mitosis (Figure 2). Based on logistic regression analysis, the point of no return—the time at which the cell becomes refractory to cycloheximide treatment—occurs only at the end of G2 phase (Figure 2). On a cell biological level, this timing approximately corresponds to when the antephase checkpoint is silenced (Pines and Rieder, 2001; Rieder and Cole, 1998, 2000). On a biochemical level, this is about when the activity of cyclin B1-Cdk1 rises to maximal levels (Akopyan et al., 2014; Gavet and Pines, 2010b; Jacobberger et al., 2012). It is possible that all three of these phenomena are manifestations of the flipping of the bistable Cdk1/PP2A switch from its interphase to its M phase state.
Both the Wee1 inhibitor MK-1775 and the Wee1/Myt1 inhibitor PD0166285 shortened G2 phase in a gradual, concentration-dependent manner (Figures 3B and 4A). The effects of MK-1775 and PD0166285 on cell cycle progression have been investigated previously (Araujo et al., 2016; Chang et al., 2016; Heijink et al., 2015; Krek and Nigg, 1991; Wang et al., 2001), yet the combination of acute inhibition using small-molecule inhibitors and live cell imaging allows us now to understand the dynamic nature of these effects in more detail. Acute inhibition of Wee1 or Wee1/Myt1 causes cells that have already spent some time in G2 phase to quickly enter mitosis. This results in a transient sharp increase in the mitotic cell population (Figures 3F and 4E). Consistent with previous studies, we find that treatment with 4 μM MK-1775 results in long mitotic delays and an increased mitotic population (Figure S1C), though we did not observe delays of a similar magnitude using more moderate concentrations of MK-1775 (1 μM) or any concentration of PD0166285 (Figures 3 and 4). Note that PD0166285 treatment also resulted in an increased mitotic population, which appeared to be mainly caused by cells delayed in mitosis because they had prematurely entered mitosis without completing DNA replication.
The cycloheximide effect appears not to be due to the inhibition of protein synthesis per se but rather to the activation of p38 MAPKs. Indeed, activation of p38 MAPK in response to protein synthesis inhibition is well known (Kyriakis et al., 1994), although the mechanism leading to this activation is not well understood but might involve induced conformational changes in the 28S ribosomal subunit and might to some extent be decoupled from the inhibition of protein synthesis (Iordanov et al., 1997; Shifrin and Anderson, 1999). Consistent with this notion, concentrations of the protein synthesis inhibitor anisomycin that do only mildly inhibit protein synthesis and other activators of p38, like H2O2, have been shown to decrease the fraction of cells in prophase and prometaphase (Matsusaka and Pines, 2004).
Accordingly, the p38 inhibitors SB202190 and SB203580 largely restored mitotic entry in cycloheximide-treated cells (Figures 6, S2, and S3). Likewise, the Wee1 inhibitor allowed about half of the cycloheximide-treated cells to progress into mitosis, consistent with the hypothesis that the effects of the p38 inhibitors are ultimately mediated by the Cdc25 and/or Wee1/Myt1 proteins (Figure 4). The override was even more pronounced using the Wee1/Myt1 inhibitor PD0166285, possibly because of the additional effect of this inhibitor on Myt1 and/or p38 activation. Taken together, these results suggest that G2 phase cyclin synthesis, and G2 phase protein synthesis in general, is not strictly required for timely progression into M phase.
Note, however, that, even though G2 phase protein synthesis is not required when p38 or Wee1 and Myt1 are inhibited, it is theoretically possible that protein synthesis could still be required when these kinases are not pharmacologically inhibited. For example, if there was some p38-inhibitory protein that was normally synthesized in G2 phase and whose synthesis was normally required for progression into M phase, this protein synthesis requirement would be expected to be abrogated in cells treated with p38 or Wee1/Myt1 inhibitors. However, there is no affirmative evidence for such a protein, and p38 inhibition alone, in contrast to Wee1/Myt1 inhibition, does not alter the duration of G2 phase in the absence of cycloheximide. Therefore, the simplest interpretation of the present findings is that the effect of cycloheximide on G2/M progression is due to checkpoint activation rather than protein synthesis inhibition.
These findings also suggest that the accelerating accumulation of cyclin B1 that normally begins at about the onset of G2 phase is not the trigger for mitosis, or at least not the only trigger, because normal G2 durations can be seen in the absence of such protein synthesis. This conclusion fits well with loss-of-function studies that show that, even though cyclin B1 is essential in mouse embryos (Brandeis et al., 1998; Strauss et al., 2018), substantial inhibition of cyclin B1 synthesis yields relatively subtle delays in the timing of mitotic entry (Gong and Ferrell, 2010; Gong et al., 2007; Hégarat et al., 2020). Likewise, the current findings fit well with the observation that cyclin B1 overexpression has little or no effect on cell cycle dynamics (Resnitzky et al., 1994). Note that the current findings also suggest that cell size is probably not the ultimate trigger of mitotic entry, because cell growth would be expected to be inhibited by protein synthesis inhibitors.
What then is the trigger for mitosis? One possibility is that the cessation of some low but non-zero levels of ATR- or ATM-mediated checkpoint signaling (Gong and Ferrell, 2010; Lemmens et al., 2018; Saldivar et al., 2018) at the S/G2 boundary might set into motion a signal transduction process that leads to inactivation or degradation of Wee1/Myt1 (Ayad et al., 2003; Michael and Newport, 1998; Watanabe et al., 2004) and activation of Cdc25. Consistent with this hypothesis, we observed that the duration of G2 phase is a sensitive function of the basal level of Wee1/Myt1 activity (Figures 3B and 4A). Another possibility is that the translocation of cyclin A2 from the nucleus to the cytoplasm, an event that occurs during late S phase, may initiate the events that lead to mitotic entry (Cascales et al., 2017; Jackman et al., 2002). A third possibility is that the Bora-Aurora A-Plk1 pathway is the critical trigger (Seki et al., 2008; Vigneron et al., 2018). How exactly cells integrate the different signaling pathways in order to decide whether or not to enter mitosis remains an important, open question in somatic cell cycle regulation.
Cycloheximide-treated cells rescued by p38 MAPK inhibition, and cells entering mitosis precociously due to Wee1/Myt1 inhibition, did take longer to progress through and exit mitosis (Figure 7). In both cases, these cells would be expected to enter mitosis with lower cyclin B1 levels than normal. The lower cyclin B1 levels could result in all or some mitotic substrates being phosphorylated more slowly, resulting in the observed mitotic delays.
G2 phase protein synthesis, or cyclin B1 synthesis more specifically, appears to represent “just-in-time” preparation for the next phase of the cell cycle. This concept, borrowed from supply chain management, has been proposed to apply to protein synthesis and complex assembly in the bacterial cell cycle (McAdams and Shapiro, 2003). Even though the proteins involved in the bacterial cell cycle bear little resemblance to those that regulate the eukaryotic cell cycle, perhaps this concept applies to both regulatory systems.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Enquiries on reagents and resources should be directed to, and will be fulfilled by the lead contact, James Ferrell (james.ferrell@stanford.edu).
Material Availability
Plasmids and cell lines generated in this study will be made freely available upon request.
Data and Code Availability
The datasets and code supporting the current study are available from the corresponding author on request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture methods
MCF10A human mammary epithelial cells were a kind gift from Sabrina Spencer and were cultured in growth medium DMEM:F12 (GIBCO, #11320-033) containing 5% horse serum (GIBCO, catalog number 16050114), 20 ng/ml EGF (PreproTech, AF-100-15), 0.5 mg/mL hydrocortisone (Sigma-Aldrich, H0888-1g), 100 ng/ml cholera toxin (Sigma-Aldrich, C8052-2mg) 10 μg/ml insulin (Sigma-Aldrich, I1882-100mg), 1% penicillin, and 1% streptomycin (both from Life Technologies, catalog number 15140-122) as described previously (Debnath et al., 2003; Soule et al., 1990). HeLa cells (CCL-2) were purchased from the ATCC and cultured in Dulbecco’s modified Eagle medium containing high glucose and pyruvate (Invitrogen, #11995-073) supplemented with 10% fetal bovine serum (Axenia Biologix, #F001), 1% penicillin, 1% streptomycin, and 4 mM L-glutamine (all from Gemini Bio-Products, #400-110). HEK293T cells were obtained from ATCC (CRL-3216) and cultured in the same medium as HeLa. hTERT RPE-1 cells were obtained from ATCC (CRF-4000) and cultured in DMEM:F12 (GIBCO, catalog number 11320-033), 10% fetal bovine serum (Axenia Biologix, #F001), 0.01 mg/ml hygromycin B (Invitrogen, 10687-010), 1% penicillin, and 1% streptomycin. All cells were maintained at 37°C and 5% CO2 and discarded after passage 25.
Stable cell lines
To obtain MCF10A cells stably expressing eYFP-PCNA, we sub-cloned eYFP-PCNA from the eYFP-PCNA construct (Hahn et al., 2009) into the pTRIP-EF1α lentiviral transfer vector (Dardalhon et al., 2001), kindly provided by Ed Grow at Stanford University, by using the XbaI and BamHI restriction sites. To make lentivirus, we incubated 1 mL Opti-MEM and 36 μL FuGENE6 for 5 min at room temperature, then added 10 μg pTRIP-EF1α-eYFP-PCNA, and 6.6 μg pCMVΔR8.74, 3.3 μg pMD.G-VSVG, and 3.3 μg pRev (all kindly provided by Ed Grow), and incubated for 30 min. We used this to transfect HEK293T cells in Opti-MEM for 6 h at 37°C and 5% CO2 and then exchanged with fresh Opti-MEM. We harvested medium containing virus 48 h, 72 h, and 96 h later, filtered out cell debris with a sterile 0.45-μm filter (Millipore), concentrated by centrifuging for 20 min at 3600 rpm in Amicon-Ultra 15 Filter Units with a 100,000 kDa MW cutoff (Millipore), and froze down at −80°C. To transduce MCF10A cells, we added concentrated virus and 5 μg/ml polybrene (Sigma-Aldrich) to MCF10A cells in growth media, incubated for 24 h, and replaced with growth media. After culturing cells for 5 more days, we sorted for eYFP-positive cells by fluorescence-activated cell sorting. To obtain MCF10A cells stably expressing eYFP-PCNA and histone H2B-mCherry or histone H2B-mTurquoise we made lentivirus as above with histone H2B-mCherry or histone H2B-mTurquoise sub-cloned into the CSII-EF lentiviral transfer vector (Spencer et al., 2013). We used it to transduce MCF10A cells stably expressing eYFP-PCNA and then sorted for cells positive for both fluorescent proteins using fluorescence-activated cell sorting. The hTERT RPE-1 cells stably expressing eYFP-PCNA and histone H2B-mTurquoise cells were produced in the same manner.
To obtain HeLa cells stably expressing eYFP-PCNA, we linearized the eYFP-PCNA construct (Hahn et al., 2009) by incubating with FspI (New England Biolabs) and purifying with ethanol precipitation. We co-transfected linearized eYFP-PCNA and linearized hygromycin marker (Clontech) with FuGENE6 (Promega) at a ratio of 1 μg eYFP-PCNA to 0.1 mg hygromycin marker to 6 μL FuGENE6 according to the manufacturer’s instructions except that we washed cells with Opti-MEM (Invitrogen), transfected cells in Opti-MEM, and incubated in Opti-MEM for 5 h at 37°C and 5% CO2 before replacing with growth medium. We split cells 48 h later and after 25 more h added 400 μg/ml hygromycin B (Invitrogen). We picked colonies 11 days later using cloning rings and expanded a clone that had correct PCNA localization in the nucleus and could form PCNA foci.
METHOD DETAILS
Chemical inhibitors
PD0166285 was generously provided by Pfizer and later purchased from EMD Millipore (#513028) and stored frozen as a 25 mM stock in DMSO (Sigma-Aldrich) and used at a final concentration of 1 μM if not specified differently. Cycloheximide was purchased from Sigma-Aldrich, stored frozen as a 10 mg/ml stock in water and used at a final concentration of 10 μg/ml. SB202474 (EMD Millipore, #559387) and SB202190 (Sigma, S7067) were stored frozen as 50 mM or 10 mM stock solutions in DMSO. SB203580 (EMD Millipore, #559387) was stored frozen as a 50 mM stock solution. SB202474, SB202190 and SB203580 were used at a final concentration of 50 μM if not specified differently. MK-1775 was purchased as a 10 mM stock solution in DMSO (Selleckchem, S1525).
Live-cell time-lapse microscopy and image analysis
Cells were seeded into 96-well plates (Costar) or collagen-coated (PureCol, Advanced BioMatrix, #5005-100ML) 96-well glass bottom plate (Cellvis, P96-1.5H-N) the day before microscopy at such a density that they were sub-confluent even at the end of the experiment. To prevent drying, each well of the plate contained between 100 and 200 μL of growth medium. Images were taken in 10 min or 15 min intervals, depending on the needs of the experiment, on the ImageXpress Micro System Standard Model (Molecular Devices) controlled by the MetaXpress 5.1 software (Molecular Devices) using the 10X objective (NA = 0.3, Plan Fluor) or the 20X objective (NA = 0.45, Plan Fluor ELWD). Cells were kept alive inside the microscope in a humidified chamber at 37°C and 5% CO2. We used the YFP-LIVE filter cube for imaging eYFP-PCNA, the CFP-LIVE filter cube for imaging histone H2B-mTurquoise, the HcRED-LIVE filter cube for imaging histone H2B-mCherry. Combined exposure through all the filter cubes did not exceed 700 msec per frame. We used 4x gain and 1x1 or 2x2 binning.
Raw TIFF images were exported using the MetaXpress 5.1 software (Molecular Devices) and collated into time series by well and site using a script written in MATLAB (MathWorks) or Python. Cells were tracked manually and each relevant change in a fluorescent reporter (PCNA focus disappearance, etc.) was recorded in Excel (Microsoft). For some analyses we used a graphical user interface written in LabView (National Instruments) that recorded the frame number and cell coordinates by responding to a mouse click and exported results to Excel (Microsoft). To allow features like PCNA foci to be easily perceived, images were typically min-max adjusted, and we sometimes allowed the H2B-mCherry image to be saturated in the mitotic stages in order to allow the low intensity H2B-mCherry signal in interphase to be perceivable. A custom-written MATLAB script provided by Tobias Meyer’s laboratory was used to count the total number of cells in every time frame in order to calculate mitotic indices. False color and merged-channel images were generated using Fiji (Schindelin et al., 2012).
Immunoblotting and antibodies
2 mL of MCF10A cell suspension at a cell concentration of 1.5 × 105 cells/ml was seeded into a 6-well plate (Falcon, #353046) and grown for 48 h at 37°C. The medium was exchanged and the cells were grown for another 6 h. An equal volume of medium containing the prediluted inhibitors was then added to the cells, and the cells were incubated for 30 min (for PD0166285) or 6 h (for cycloheximide and cycloheximide plus either SB202190 or SB203580). The medium was then removed, cells were washed twice with cold PBS and lysed with lysis buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, 1x Phosstop #4906845001, 1x cOmplete #11873580001). Samples were boiled in SDS-PAGE sample buffer and analyzed by SDS-PAGE and immunoblotting. The following antibodies were used: rabbit α-Cdk1 phospho-Tyr15 (Cell Signaling Technology, #9111L), mouse α-Cdk1 (Santa Cruz Biotechnology, #SC-54), mouse α-tubulin (Santa Cruz Biotechnology, #SC-32293), rabbit α-HSP27 phospho-Ser82 (Cell Signaling Technology, #2401), mouse α-HSP27 (Cell Signaling Technology, #2402), rabbit α-p38 MAPK (Cell Signaling Technology, #9212) and rabbit α-p38 MAPK phospho-Thr180/Tyr182 (Cell Signaling Technology, #9211).
QUANTIFICATION AND STATISTICAL ANALYSIS
Logistic regression analysis
Logistic regression analysis is a method for estimating how the probability of a binary outcome—in our case, whether a cell does or does not ultimately progress into mitosis—varies as a function of time and treatment conditions. The underlying assumption is that the odds of progressing into mitosis scale multiplicatively with time, which means that the time course data should be approximated by a logistic function, with parameters for the steepness and time of the transition from low to high probability. We binarized cell outcomes (i.e., the cell either did or did not progress into mitosis within the average G2 phase duration plus two standard deviations of the DMSO-treated control), plotted the fraction of cells that attained the outcome as a function of the time of drug addition, and fitted the data to a logistic function using the LogitModelFit command in Mathematica 10. The 95% confidence bands were calculated using code deposited in the Mathematica Stack Exchange (https://mathematica.stackexchange.com/questions/26616/how-can-i-compute-and-plot-the-95-confidence-bands-for-a-fitted-logistic-regres).
Other statistical analyses
Further statistical analyses (e.g., mean, percentiles, nonparametric Mann-Whitney test) were performed using Prism 8.0.2 (Graphpad). Statistical parameters, e.g., the number of cells analyzed (n), are described in the figure legends of the respective figure. All experiments except the immunoblot analyses have been performed in at least two biological replicates – meaning that cells were freshly plated, imaged, and independently treated with the respective drugs – of which usually one representative experiment is shown (often – and for all main conclusions - we show a replicate in the Supplemental Information).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-Cdk1 phospho-Tyr15 | Cell Signaling Technology | #9111L; RRID:AB_331460 |
| mouse anti-Cdk1 | Santa Cruz Biotechnology | #SC-54; RRID:AB_627224 |
| mouse anti-tubulin | Santa Cruz Biotechnology | #SC-32293; RRID:AB_628412 |
| rabbit anti-HSP27 phospho-Ser82 | Cell Signaling Technology | #2401; RRID:AB_331644 |
| mouse anti-HSP27 | Cell Signaling Technology | #2402; RRID:AB_331761 |
| rabbit anti-p38 MAPK | Cell Signaling Technology | #9212; RRID:AB_330713 |
| rabbit anti-p38 MAPK phospho-Thr180/Tyr182 | Cell Signaling Technology | #9211; RRID:AB_331641 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PD0166285 | EMD Millipore | #513028 |
| Cycloheximide | Sigma-Aldrich | 01810-5G |
| SB202474 | EMD Millipore | #559387 |
| SB202190 | Sigma | S7067 |
| SB203580 | EMD Millipore, | #559387 |
| MK-1775 | Selleckchem | S1525 |
| horse serum | GIBCO | #16050-114 |
| fetal bovine serum | Axenia Biologix | #F001 |
| choleratoxin | Sigma-Aldrich | C8052-2mg |
| insulin | Sigma-Aldrich | I1882-100mg |
| hygromycin B | Invitrogen | 10687-010 |
| FuGENE6 | Promega | E2691 |
| Collagen (PureCol) | Advanced BioMatrix | #5005-100ML |
| 96-well glass bottom plate | Cellvis | P96-1.5H-N |
| Experimental Models: Cell Lines | ||
| MCF10A | Laboratory of Tobias Meyer | |
| HEK293T | ATCC | CRL-3216 |
| HeLa | ATCC | CCL-2 |
| hTERT RPE-1 | ATCC | CRL-3216 |
| MCF10A eYFP-PCNA H2B-mTurquoise | This study | N/A |
| MCF10A eYFP-PCNA H2B-mCherry | This study | N/A |
| hTERT RPE-1 eYFP-PCNA H2B-mTurquoise | This study | N/A |
| HeLa eYFP-PCNA H2B-mTurquoise | This study | N/A |
| Recombinant DNA | ||
| pTRIP-EF1α | (Dardalhon et al., 2001) | N/A |
| pTRIP-EF1α-eYFP-PCNA | This study | N/A |
| CSII-EF | (Spencer et al., 2013) | N/A |
| CSII-EF- H2B-mTurquoise | This study | N/A |
| CSII-EF- H2B-mCherry | This study | N/A |
| Software and Algorithms | ||
| Mathematica 10 and 12 | Wolfram Research | https://www.wolfram.com/mathematica/ |
| Graphpad Prism 8.0.2 and 8.4 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Fiji (ImageJ, version 2.00-rc-49/1.51e) | (Schindelin et al., 2012) | https://imagej.net/Fiji |
| MetaXpress 5.1 software | Molecular Devices | https://www.moleculardevices.com/ |
| Excel | MIcrosoft | N/A |
| Python 3.7.3 | (Van Rossum and Drake, 2009) | https://www.python.org/ |
| MATLAB R2015b | MathWorks | https://www.mathworks.com |
| Other | ||
| ImageXpress Micro System Standard Model | Molecular Devices | https://www.moleculardevices.com/ |
Highlights.
Cycloheximide (CHX) prevents mitotic entry right up until the G2/M transition
Inhibition of Wee1 or Wee1/Myt1 can overcome a cycloheximide-induced G2 arrest
Activation of p38 MAPK, not lack of protein synthesis, accounts for the CHX arrest
Mitotic entry in the absence of protein synthesis causes delays in mitotic progression
ACKNOWLEDGMENTS
We thank Mary Teruel, Michael Zhao, and the Stanford High Throughput Biosciences Facility for help with the ImageExpress microscopy. We thank Arne Lindqvist, Tim Stearns, Tobias Meyer, KarleneA. Cimprich, Aaron F. Straight, Frederick G. Westhorpe, Whitney L. Johnson, and Bradley T. French for helpful discussions. We are indebted to Sabrina L. Spencer, Ed Grow, Tony Yu-Chen Tsai, Paul G. Rack, M. Cristina Cardoso, and Michael W. Davidson for sharing reagents. We acknowledge help with LabView from Taru Roy; help with image analysis from Colin J. Fuller; and help with MATLAB from Feng-Chiao Tsai, Sabrina Spencer, Jeremy B. Chang, Graham A. Anderson, and Tony Yu-Chen Tsai. We are grateful to Pfizer for providing PD0166285. We thank the Stanford FACS Facility and the Stanford High-Throughput Biosciences Facility for technical assistance and Silke Hauf, Oshri Afanzar, Xianrui Cheng, Yuping Chen, William Huang, Shixuan Liu, and Connie Phong for comments on the manuscript. The work was supported by grants from the National Institutes of Health (R01 GM046383 and R35 GM131792; J.E.F.), a Stanford Graduate Fellowship (S.L.), the Stanford Training Grant in Chemical and Molecular Pharmacology (T32 GM067586; S.L.), a postdoctoral fellowship from the German Research Foundation (KA 4476/1-1; J.K.), Cancer Research UK (FC001596; S.S.), the UK Medical Research Council (FC001596; S.S.), and the Wellcome Trust (FC001596; S.S.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.Org/10.1016/j.celrep.2020.107901.
REFERENCES
- Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, Toniatti C, Ashworth A, and Turner NC (2012). Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer Discov. 2, 524–539. [DOI] [PubMed] [Google Scholar]
- Akopyan K, Silva Cascales H, Hukasova E, Saurin AT, Müllers E, Jaiswal H, Hollman DA, Kops GJ, Medema RH, and Lindqvist A (2014). Assessing kinetics from fixed cells reveals activation of the mitotic entry network at the S/G2 transition. Mol. Cell 53, 843–853. [DOI] [PubMed] [Google Scholar]
- Araujo AR, Gelens L, Sheriff RS, and Santos SD (2016). Positive feedback keeps duration of mitosis temporally insulated from upstream cell-cycle events. Mol. Cell 64, 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayad NG, Rankin S, Murakami M, Jebanathirajah J, Gygi S, and Kirschner MW (2003). Tome-1, a trigger of mitotic entry, is degraded during G1 via the APC. Cell 113, 101–113. [DOI] [PubMed] [Google Scholar]
- Baserga R (1985). The Biology of Cell Reproduction (Harvard University). [Google Scholar]
- Blackford AN, and Jackson SP (2017). ATM, ATR, and DNA-PK: the trinity at the heart of the DNA damage response. Mol. Cell 66, 801–817. [DOI] [PubMed] [Google Scholar]
- Brandeis M, Rosewell I, Carrington M, Crompton T, Jacobs MA, Kirk J, Gannon J, and Hunt T (1998). Cyclin B2-null mice develop normally and are fertile whereas cyclin B1-null mice die in utero. Proc. Natl. Acad. Sci. USA 95, 4344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascales HS, Burdova K, Müllers E, Stoy H,von Morgen P, Macurek L, and Lindqvist A (2017). Cyclin A2 localises in the cytoplasm at the S/G2 transition to activate Plk1. bioRxiv. 10.1101/191437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Q, Chandrashekhar M, Ketela T, Fedyshyn Y, Moffat J, and Hedley D (2016). Cytokinetic effects of Wee1 disruption in pancreatic cancer. Cell Cycle 15, 593–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crncec A, and Hochegger H (2019). Triggering mitosis. FEBS Lett. 593, 2868–2888. [DOI] [PubMed] [Google Scholar]
- Dardalhon V, Herpers B, Noraz N, Pflumio F, Guetard D, Leveau C, Dubart-Kupperschmitt A, Charneau P, and Taylor N (2001). Lentivirus-mediated gene transfer in primary T cells is enhanced by a central DNA flap. Gene Ther. 8, 190–198. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, and Cohen P (2000). Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem. J 351, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, and Brugge JS (2003). Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods 30, 256–268. [DOI] [PubMed] [Google Scholar]
- Deibler RW, and Kirschner MW (2010). Quantitative reconstitution of mitotic CDK1 activation in somatic cell extracts. Mol. Cell 37, 753–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly GM, and Sisken JE (1967). RNA and protein synthesis required for entry of cells into mitosis and during the mitotic cycle. Exp. Cell Res. 46, 93–105. [DOI] [PubMed] [Google Scholar]
- Dyson N (1998). The regulation of E2F by pRB-family proteins. Genes Dev. 12, 2245–2262. [DOI] [PubMed] [Google Scholar]
- Essers J, Theil AF, Baldeyron C, van Cappellen WA, Houtsmuller AB, Kanaar R, and Vermeulen W (2005). Nuclear dynamics of PCNA in DNA replication and repair. Mol. Cell. Biol 25, 9350–9359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans T, Rosenthal ET, Youngblom J, Distel D, and Hunt T (1983). Cyclin: a protein specified by maternal mRNA in sea urchin eggs that is destroyed at each cleavage division. Cell 33, 389–396. [DOI] [PubMed] [Google Scholar]
- Frisa PS, and Jacobberger JW (2009). Cell cycle-related cyclin b1 quantification. PLoS ONE 4, e7064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O, and Pines J (2010a). Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J. Cell Biol 189, 247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavet O, and Pines J (2010b). Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev. Cell 18, 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlich D, Koch B, Dupeux F, Peters JM, and Ellenberg J (2006). Live-cell imaging reveals a stable cohesin-chromatin interaction after but not before DNA replication. Curr. Biol 16, 1571–1578. [DOI] [PubMed] [Google Scholar]
- Gharbi-Ayachi A, Labbé JC, Burgess A, Vigneron S, Strub JM, Brioudes E, Van-Dorsselaer A, Castro A, and Lorca T (2010). The substrate of Greatwall kinase, Arpp19, controls mitosis by inhibiting protein phosphatase 2A. Science 330, 1673–1677. [DOI] [PubMed] [Google Scholar]
- Gong D, and Ferrell JE Jr. (2010). The roles of cyclin A2, B1, and B2 in early and late mitotic events. Mol. Biol. Cell 21, 3149–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong D, Pomerening JR, Myers JW, Gustavsson C, Jones JT, Hahn AT, Meyer T, and Ferrell JE Jr. (2007). Cyclin A2 regulates nuclear-envelope breakdown and the nuclear accumulation of cyclin B1. Curr. Biol 17, 85–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagting A, Jackman M, Simpson K, and Pines J (1999). Translocation of cyclin B1 to the nucleus at prophase requires a phosphorylation-dependent nuclear import signal. Curr. Biol 9, 680–689. [DOI] [PubMed] [Google Scholar]
- Hahn AT, Jones JT, and Meyer T (2009). Quantitative analysis of cell cycle phase durations and PC12 differentiation using fluorescent biosensors. Cell Cycle 8, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hégarat N, Crncec A, Suarez Peredo Rodriguez MF, Echegaray Iturra F, Gu Y, Busby O, Lang PF, Barr AR, Bakal C, Kanemaki MT, et al. (2020). Cyclin A triggers mitosis either via the Greatwall kinase pathway or cyclin B. EMBO J. 39, e104419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijink AM, Blomen VA, Bisteau X, Degener F, Matsushita FY, Kaldis P, Foijer F, and van Vugt MA (2015). A haploid genetic screen identifies the G1/S regulatory machinery as a determinant of Wee1 inhibitor sensitivity. Proc. Natl. Acad. Sci. USA 112, 15160–15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim A, Rymarczyk B, and Mayer TU (2017). Regulation of cell division. Adv. Exp. Med. Biol 953, 83–116. [DOI] [PubMed] [Google Scholar]
- Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, et al. (2009). Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther 8, 2992–3000. [DOI] [PubMed] [Google Scholar]
- Iordanov MS, Pribnow D, Magun JL, Dinh TH, Pearson JA, Chen SL, and Magun BE (1997). Ribotoxic stress response: activation of the stress-activated protein kinase JNK1 by inhibitors of the peptidyl transferase reaction and by sequence-specific RNA damage to the alpha-sarcin/ricin loop in the 28S rRNA. Mol. Cell. Biol 17, 3373–3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackman M, Kubota Y, den Elzen N, Hagting A, and Pines J (2002). Cyclin A- and cyclin E-Cdk complexes shuttle between the nucleus and the cytoplasm. Mol. Biol. Cell 13, 1030–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobberger JW, Avva J, Sreenath SN, Weis MC, and Stefan T (2012). Dynamic epitope expression from static cytometry data: principles and reproducibility. PLoS ONE 7, e30870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Hardy S, and Morgan DO (1998). Nuclear localization of cyclin B1 controls mitotic entry after DNA damage. J. Cell Biol 141, 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson C, Katich S, Hagting A, Hoffmann I, and Pines J (1999). Cdc25B and Cdc25C differ markedly in their properties as initiators of mitosis. J. Cell Biol 146, 573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krek W, and Nigg EA (1991). Mutations of p34cdc2 phosphorylation sites induce premature mitotic events in HeLa cells: evidence for a double block to p34cdc2 kinase activation in vertebrates. EMBO J. 10, 3331–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyriakis JM, Banerjee P, Nikolakaki E, Dai T, Rubie EA, Ahmad MF, Avruch J, and Woodgett JR (1994). The stress-activated protein kinase subfamily of c-Jun kinases. Nature 369, 156–160. [DOI] [PubMed] [Google Scholar]
- Lemmens B, Hegarat N, Akopyan K, Sala-Gaston J, Bartek J, Hochegger H, and Lindqvist A (2018). DNA replication determines timing of mitosis by restricting CDK1 and PLK1 activation. Mol. Cell 71, 117–128.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, Zink D, and Cardoso MC (2000). Dynamics of DNA replication factories in living cells. J. Cell Biol 149, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Meyer AN, and Donoghue DJ (1997). Nuclear localization of cyclin B1 mediates its biological activity and is regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 94, 502–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindqvist A, van Zon W, Karlsson Rosenthal C, and Wolthuis RM (2007). Cyclin B1-Cdk1 activation continues after centrosome separation to control mitotic progression. PLoS Biol. 5, e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, and Medema RH (2008). Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 455, 119–123. [DOI] [PubMed] [Google Scholar]
- Manke IA, Nguyen A, Lim D, Stewart MQ, Elia AE, and Yaffe MB (2005). MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 17, 37–48. [DOI] [PubMed] [Google Scholar]
- Matsusaka T, and Pines J (2004). Chfr acts with the p38 stress kinases to block entry to mitosis in mammalian cells. J. Cell Biol 166, 507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAdams HH, and Shapiro L (2003). A bacterial cell-cycle regulatory network operating in time and space. Science 301, 1874–1877. [DOI] [PubMed] [Google Scholar]
- Michael WM, and Newport J (1998). Coupling of mitosis to the completion of S phase through Cdc34-mediated degradation of Wee1. Science 282, 1886–1889. [DOI] [PubMed] [Google Scholar]
- Minshull J, Blow JJ, and Hunt T (1989). Translation of cyclin mRNA is necessary for extracts of activated xenopus eggs to enter mitosis. Cell 56, 947–956. [DOI] [PubMed] [Google Scholar]
- Mochida S, Maslen SL, Skehel M, and Hunt T (2010). Greatwall phosphorylates an inhibitor of protein phosphatase 2A that is essential for mitosis. Science 330, 1670–1673. [DOI] [PubMed] [Google Scholar]
- Mochida S, Rata S, Hino H, Nagai T, and Novák B (2016). Two bistable switches govern M phase entry. Curr. Biol 26, 3361–3367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Hayles J, and Nurse P (1989). Regulation of p34cdc2 protein kinase during mitosis. Cell 58, 361–372. [DOI] [PubMed] [Google Scholar]
- Morgan DO (2007). The Cell Cycle: Principles of Control (New Science). [Google Scholar]
- Murray AW, and Kirschner MW (1989). Cyclin synthesis drives the early embryonic cell cycle. Nature 339, 275–280. [DOI] [PubMed] [Google Scholar]
- Murray AW, Solomon MJ, and Kirschner MW (1989). The role of cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature 339, 280–286. [DOI] [PubMed] [Google Scholar]
- Novak B, and Tyson JJ (1993). Modeling the cell division cycle: M-phase trigger, oscillations, and size control. J. Theor. Biol 165, 101–134. [Google Scholar]
- Pines J, and Hunter T (1991). Human cyclins A and B1 are differentially located in the cell and undergo cell cycle-dependent nuclear transport. J. Cell Biol 115, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pines J, and Rieder CL (2001). Re-staging mitosis: a contemporary view of mitotic progression. Nat. Cell Biol 3, E3–E6. [DOI] [PubMed] [Google Scholar]
- Pomerening JR, Sontag ED, and Ferrell JE Jr. (2003). Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol 5, 346–351. [DOI] [PubMed] [Google Scholar]
- Rata S, Suarez Peredo Rodriguez MF, Joseph S, Peter N, Echegaray Iturra F, Yang F, Madzvamuse A, Ruppert JG, Samejima K, Platani M, et al. (2018). Two interlinked bistable switches govern mitotic control in mammalian cells. Curr. Biol 28, 3824–3832.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann JD, Gardner BE, Margottin-Goguet F, and Jackson PK (2001). Emi1 regulates the anaphase-promoting complex by a different mechanism than Mad2 proteins. Genes Dev. 15, 3278–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt HC, and Yaffe MB (2009). Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol 21, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resnitzky D, Gossen M, Bujard H, and Reed SI (1994). Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Mol. Cell. Biol 14, 1669–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, and Cole RW (1998). Entry into mitosis in vertebrate somatic cells is guarded by a chromosome damage checkpoint that reverses the cell cycle when triggered during early but not late prophase. J. Cell Biol 142, 1013–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, and Cole R (2000). Microtubule disassembly delays the G2-M transition in vertebrates. Curr. Biol 10, 1067–1070. [DOI] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Hamperl S, Bocek MJ, Chung M, Bass TE, Cisneros-Soberanis F, Samejima K, Xie L, Paulson JR, Earnshaw WC, et al. (2018). An intrinsic S/G2 checkpoint enforced by ATR. Science 361, 806–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos SD, Wollman R, Meyer T, and Ferrell JE Jr. (2012). Spatial positive feedback at the onset of mitosis. Cell 149, 1500–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki A, Coppinger JA, Jang CY, Yates JR, and Fang G (2008). Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 320, 1655–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha W, Moore J, Chen K, Lassaletta AD, Yi CS, Tyson JJ, and Sible JC (2003). Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts. Proc. Natl. Acad. Sci. USA 100, 975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shifrin VI, and Anderson P (1999). Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J. Biol. Chem 274, 13985–13992. [DOI] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD Jr., Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, and Brooks SC (1990). Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 50, 6075–6086. [PubMed] [Google Scholar]
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, and Meyer T (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss B, Harrison A, Coelho PA, Yata K, Zernicka-Goetz M, and Pines J (2018). Cyclin B1 is essential for mitosis in mouse embryos, and its nuclear export sets the time for mitosis. J. Cell Biol 217, 179–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai TY, Theriot JA, and Ferrell JE Jr. (2014). Changes in oscillatory dynamics in the cell cycle of early Xenopus laevis embryos. PLoS Biol. 12, e1001788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Rossum G, and Drake FL (2009). Python 3 Reference Manual (Create-Space). [Google Scholar]
- Verbin RS, and Farber E (1967). Effect of cycloheximide on the cell cycle of the crypts of the small intestine of the rat. J. Cell Biol 35, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneron S, Sundermann L, Labbé J-C, Pintard L, Radulescu O, Castro A, and Lorca T (2018). Cyclin A-cdk1-dependent phosphorylation of bora is the triggering factor promoting mitotic entry. Dev. Cell 45, 637–650.e7. [DOI] [PubMed] [Google Scholar]
- Vinod PK, and Novak B (2015). Model scenarios for switch-like mitotic transitions. FEBS Lett. 589, 667–671. [DOI] [PubMed] [Google Scholar]
- Wang Y, Li J, Booher RN, Kraker A, Lawrence T, Leopold WR, and Sun Y (2001). Radiosensitization of p53 mutant cells by PD0166285, a novel G(2) checkpoint abrogator. Cancer Res. 61, 8211–8217. [PubMed] [Google Scholar]
- Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, and Osada H (2004). M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc. Natl. Acad. Sci. USA 101, 4419–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerjatke T, Gak IA, Kirova D, Fuhrmann M, Daniel K, Gonciarz M, Müller D, Glauche I, and Mansfeld J (2017). Quantitative cell cycle analysis based on an endogenous all-in-one reporter for cell tracking and classification. Cell Rep. 19, 1953–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets and code supporting the current study are available from the corresponding author on request.