

Abstract
The unforeseen emergence of coronavirus disease 2019 (COVID-19), a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) at the Wuhan province of China in December 2019, subsequently its abrupt spread across the world has severely affected human life. In a short span of time, COVID-19 has sacked more than one million human lives and marked as a severe global pandemic, which is drastically accountable for the adverse effect directly to the human society, particularly the health care system and the economy. The unavailability of approved and effective drugs or vaccines against COVID-19 further created conditions more adverse and terrifying. To win the war against this pandemic within time there is a desperate need for the most adequate therapeutic treatment, which can be achieved by the collaborative research work among scientists worldwide. In continuation of our efforts to support the scientific community, a review has been presented which discusses the structure and the activity of numerous molecules exhibiting promising SARS-CoV-2 and other CoVs inhibition activities. Furthermore, this review offers an overview of the structure, a plausible mechanism of action of SARS-CoV-2, and crucial structural features substantial to inhibit the primary virus-based and host-based targets involved in SARS-CoV-2 treatment. We anticipate optimistically that this perspective will provide the reader and researcher’s better understanding regarding COVID-19 and pave the path in the direction of COVID-19 drug discovery and development paradigm.
Keywords: Coronavirus, COVID-19, SARS-CoV-2, Spike glycoprotein, ACE-2 receptor, 3CL protease, Cathepsin L proteinase, RdRp, TMPRSS2, Inhibitors. contents
Graphical abstract

1. Introduction
The COVID-19 pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is continuously spreading its giant wings across the world, and as of Oct 20, 2020, more than 40 million confirmed cases were reported in more than 215 countries and territories and with 1,123,180 confirmed deaths. The top three countries reporting the maximum confirmed cases to date are the United States of America, India, and Brazil, with 8,456,653, 7,597,063, and 5,251,127 cases, respectively [1]. The most common symptoms of COVID-19 in humans are fever, dry cough, and tiredness, whereas severe symptoms include difficulty breathing or shortness of breath, chest pain, high blood pressure, and loss of speech or movement because of central nervous system conditions leading to multiple organ failure [2,3]. In severe instances, it has been observed that SARS-CoV-2 infection generates bilateral pneumonia, acute respiratory distress syndrome, kidney failure, and eventually, death [[4], [5], [6]]. Furthermore, the current data reveal the SARS-CoV-2 mortality rate between 3% and 5, which is much lower than other Coronaviridae family member’s mortality rate, such as SARS-CoV (9–15%) and MERS-CoV (34–37%) [7,8]. However, tragically, SARS-CoV-2 is considerably transmissible and causes more severe health effect on the elderly people or with comorbidities, which is the existence of more than one chronic disease such as diabetes, obesity, and cardiovascular at the same time, that weaken the immune system of the human body [9]. The risk for severe illness from SARS-CoV-2 increases with age; older adults above 65 years of age are at higher risk (23-fold) and account for 80% of hospitalizations [10]. In the middle of the crisis, the COVID-19 spreading rate and scale are much higher and more alarming than previous coronaviral epidemics. In many countries, the number of SARS-CoV-2 cases is rising continuously at an alarming rate, whereas the countries that succeeded in slowing their initial outbreak are now experiencing a second wave with the rise in COVID-19 cases [11]. Heretofore, scientists have been unsuccessful in identifying specific countermeasures, such as drugs, vaccines, and personal protection equipment (PPE), for the effective control or elimination of the SARS-CoV-2; therefore, to control the transmission of the virus, we have only basic protective measures recommended by the World Health Organization (WHO), i.e., “Infection prevention and control guidance-Covid 19” [12].
The COVID-19 outbreak not only has caused high infection and fatality rates but also responsible for the detrimental effect globally on the society, economy, and geopolitics. As this pandemic is encompassing the world, it is instigating a high level of concern within the society and particularly among the elderly people, health care providers, and people with underlying health conditions. With the introduction of forced quarantine and nationwide lockdown as measures to control virus transmission, it is expected that this kind of measures, in the long run, could have severe effects on humans, including acute panic, obsessive behaviors, hoarding, paranoia, depression, alcohol and drug use, self-harm or suicidal behavior, and post-traumatic stress disorder (PTSD) among people [13]. Considering the severity of the COVID-19 pandemic and the unavailability of any approved drugs or vaccines for its treatment and prevention has forced the researchers globally to emphasize the discovery and development of therapeutics targeting SARS-CoV-2. Both industry and academia are working together to appropriately characterize the virus and test in animals [[14], [15], [16], [17], [18], [19]]. The successful isolation and sequencing of SARS-CoV-2 genes have possibly revealed that this virus is a member of the Beta coronavirus genus and has positive-sense single-stranded RNA. The SARS-CoV-2 is closely comparable to other members of the genus, for example, acute respiratory syndrome CoV (SARS-CoV) and Middle East Respiratory Syndrome Cov (MERS-CoV). The SARS-CoV-2 exhibits a 79.5% similarity with SARS-CoV with a 94.6% resemblance in the amino acid sequence of seven conserved nonstructural proteins. Consequently, the developed understanding and relevant information obtained from the clinical data against SARS and MARS can increase the chance of success in the design and synthesis of efficacious therapeutic agents to treat COVID-19 infection.
The human civilization has grappled with the health and humanitarian crisis unleashed by the global coronavirus pandemic; there is a massive challenge and a great opportunity for the scientific community to centralize the efforts in the development of an effective drug/vaccine against contagious coronavirus. One of the relevant approaches to control the COVID-19 pandemic is to develop an effective vaccine targeting SARS-CoV-2 and in past vaccines showed a significant effect in controlling the preceding epidemics. The earnest efforts by scientists are in progress in this direction, and the results are encouraging. However, many vaccines are only partially effective or work better only for some age groups than others, and the immunity a vaccine provides can decrease with time. Even if one of the COVID-19 vaccines is proven safe and effective, its large-scale manufacture and global distribution in a short span of time is itself a big challenge, and it is obvious that it will remain unavailable to many people, particularly living in the developing countries. Therefore, the discovery of a vaccine will not be the absolute solution; we need treatment to fight COVID-19. Treatment will also be helpful to manage illness resulting from imperfections in vaccine effectiveness and uptake. In the case of COVID-19, an early-stage treatment involving a combination of antiviral compounds/drugs can be one of the effective treatments of COVID-19. People with severe COVID-19 are treated in the hospital with the repurposing drugs with debatable results; however, there is no treatment available for people with early signs or mild symptoms of COVID-19 or laboratory-confirmed infected persons with high risk. The discovery and development of COVID-19 drugs could protect patients from getting sicker and dying; moreover, it will support in slowing down the virus spread and help people to live longer. It can also be used as a prophylactic treatment.
Drug discovery and development is a complicated and time-consuming process that involves several stages to produce a safe and efficacious drug. The process broadly can be divided into the following stages: a) Drug discovery that includes target selection, lead finding, lead optimization, and pharmacological profiling; b) Preclinical research involve pharmacokinetics, pharmacodynamics, and short-term toxicology studies in in vitro and in vivo settings as well as formulation and synthesis scale-up. c) Clinical research encompasses testing of the drug in humans to check its safety and effectiveness. Clinical research is mainly divided into four phases: Phase 1, Phase II, Phase III, and Phase IV that involve the study of the drug in the respective areas of human pharmacology: safety, therapeutic exploration, dose-ranging, therapeutic confirmation or comparison, and post-marketing surveillance. d) The Food and Drug Administration (FDA) review stage includes a thorough review of submitted drug-related data by FDA review teams and deciding to approve or disapprove the drug. e) FDA post-market-safety monitoring process involve monitoring of marketed drug safety in public by the FDA. For a newly discovered drug to complete all mandatory clinical phases and regulatory approval to be available in the market takes at least 10 years and involves multidisciplinary (chemistry, biochemistry, physiology, microbiology, and pharmacology), multi-sector cooperation, and regulations of the FDA. The success of the drug discovery and development process not only depends on the industrial and academic collaboration between the specialists from the medicinal, chemistry, and biology disciplines, but also on the collaboration of pharmaceutical R and D specialist and clinical research teams, composed of doctors, pharmacists, nurses, chemists, and other health specialists. To win this war against COVID-19, we urgently require an effective and safe drug in the immediate future, and this could be achieved only by research collaboration across the globe. Researchers keeping their individual academic progress aside must rapidly coordinate nationally and internationally. Moreover, the countries on urgent priority must create an effective global scientific partnership, particularly for COVID-19 research, which will allow the universities and researchers to engage across borders and facilitate international collaboration. Appreciatively, the publishers, funders, and scientific societies around the globe started sharing research findings and making available the COVID-19 related data free of charge, that is helping the scientist across disciplines and worldwide to establish a more rapid connection. This sharing of data in the public domain must continue while maintaining the publications, grants, intellectual property rights, and patents. We are optimistic that collaborative research work among scientists worldwide can lead the world into a COVID-19 safe future.
We have compiled this review under the research objective focused on the discovery and development of a therapeutic drug to reduce the severity of COVID-19. Theoretically, all the enzymes and proteins responsible for viral replication and managing host cellular machinery are significant targets in the development of a potential agent for COVID-19 treatment [20,21]. This review briefly summarized small molecules reported in the literature with promising activity against various SARS-CoV-2 targets, including primary virus-based and host-based targets. This perspective is within the context of our ongoing drug design and discovery research work [[22], [23], [24], [25], [26], [27], [28], [29]], and here we deliver to the reader summary of the SARS-CoV-2 medicinal chemistry research outcomes, and we anticipate that it will update readers with the current progress in SARS-CoV-2 drug discovery. Being a medicinal chemist and keeping in mind our responsibility towards society and the scientific community, we mark this review as our small contribution towards the global war against COVID-19.
2. Structure and mechanism of action of SARS-CoV-2
The coronavirus genome encompasses 30000 nucleotides and made up of Spike (S) glycoprotein, Envelop (E) protein, Membrane (M) protein, Nucleocapsid (N) protein, Hemagglutenin esterase dimer (HE) and several non-structural proteins (nsp) (Fig. 1 ) [20,21,30,31].
Fig. 1.

Schematic representation of SARS-CoV-2 and its structural proteins.
The N-protein is located inside the capsid, protects the viral RNA genome by coating it into a ribonucleoprotein complex, and performs multiple functions, which include viral RNA replication and transcription. It contains an N-terminal RNA binding domain, an S-terminal dimerization domain, and a central Ser/Arg-rich linker for primary phosphorylation. The process of viral replication and transcription begins with the binding of the N-terminal of the N protein to the RNA genome; therefore, the development of molecules as inhibitors of RNA binding to the N terminal domain of N protein of coronavirus can stop the viral RNA replication and transcription and thus can be an interesting pharmacological target. The Spike (S) glycoprotein is a structural transmembrane protein present on the outer envelope of the virion, and it facilitates virus entry by contacting specific receptors located on the surface of the host cell. In functional form, it exists as a homotrimer, and each monomer is made up of approximately 1200–1400 amino acid residues and comprises a single peptide (1–13 amino acids) at the N-terminus, the S1 and S2 subunit bearing 14–685 and 686–1273 residues, respectively. The S1 subunit consists of the N - terminal domain (NTD, 14–305 residues) and the C-terminal domain (CTD), also known as the receptor-binding domain (RBD, 319–541 residues). It assists in the host-guest interaction and encourages virus entry by recognizing protein receptors of the host cells. Besides, the S2 subunit consists of the fusion peptide (FP) (788–806 residues), heptapeptide repeat sequence 1 (HR1) (912–984 residues), HR2 (1163–1213 residues), TM domain (1213–1237 residues), and a cytoplasmic domain (1237–1273 residues), and is responsible for fusion of viral and cellular membranes. The S protein homotrimers resemble the crown-like corona around the virus; furthermore, the bulbous head and stalk region in S protein monomers are shaped by S1 and S2 subunits. The E-protein is the smallest membrane protein, consisting of 76–109 amino acids, and is in charge of assembly and morphogenesis of virions and pathogenesis. The M-protein is ample on the viral surface and because of its membrane-bending properties responsible for virus fusion, assembly, and budding function. Hemagglutinin-esterase dimer (HE) is placed on the surface of the virus and is essential for host cell infection through virus entry but is expendable for replication. The non-structural proteins (nsps) of SARS-CoV-2 include highly conserved 16 nsps, responsible for different functions, encompassing the generation of replication-transcription complex (RTC). The nsps are an interesting druggable target; however, the success rate increases with the understanding of its role in viral infection as well as the availability of crystal and ligand structure. In the discovery of anti-SARS-CoV-2 therapies, some of the nsps are necessary for the virus, such as chymotrypsin-like protease (3CLpro, nsp 5), papain-like protease (PLpro, nsp 3), RNA-dependent RNA polymerase (RdRp, nsp12) in complex with cofactors nsp7, and nsp8 and helicase (nsp 13), and are considered as promising targets for the treatment of SARS-CoV-2.
The mechanism of action of SARS-CoV-2 of a human cell comprising viral entry, replication, and RNA packing are drawn in Fig. 2 [21,[30], [31], [32], [33]]. The spike (S) protein of coronavirus binds to angiotensin-converting enzyme 2 (ACE2) receptors positioned on the surface of several human cells, particularly in lung cells for viral entry. The membrane fusion is one of the significant phases in a virus infection cycle, and the spike (S) protein and different cellular proteases play a crucial role in this phase. The membrane fusion is mostly achieved through two pathways: the endosomal/clathrin-dependent and nonendosomal/clathrin-independent paths. Inside the endosomal/clathrin-dependent route, the internalized viral particles fuse with the host target membrane through the S2 subunit of S-protein at low pH, followed by the S-protein cleavage by the host protease cathepsin L. In the nonendosomal/clathrin-independent path, the fusion with cellular membrane leads to the host protease cleavage of the S protein via cell membrane-associated proteases, for example, TMPRSS2. The membrane fusion results in the discharge of viral genetic material into the cytoplasm, followed by replication/transcription complex (RTC)- mediated replication and transcription processes. The viral RNA attaches to the host ribosome and produces two polyproteins (pp1a and pp1ab) encoding 16 nsps, which is very vital in the making of new mature virions. These two significant polyproteins undergo proteolytic cleavage by the coronavirus main proteinase (3CLpro) and the papain-like protease (PLpro). Additionally, RNA-dependent RNA polymerase (RdRp) is responsible for replicating the RNA genome. The structural viral proteins comprising E, M, and S proteins are synthesized in the cytoplasm of the host cell and then implanted into the endoplasmic reticulum (ER) and transported to the endoplasmic reticulum-Golgi intermediate compartment (ERGIC). In addition, the encapsidation of replicated genomes by N protein in the cytoplasm leads to the formation of nucleocapsid, which is further merged within the ERGIC membrane. This enables the self-assembly and formation of new virions that are finally exported from infected cells. The new virions enclosed by smooth-walled vesicles are transported to the cell membrane and subsequently exported out of the infected cell via the exocytosis process to infect new cells. In the meantime, during viral production, the endoplasmic reticulum of the host cell experiences the accumulation of pressure and stress that accelerate its death. However, it is worth mentioning that the novel COVID-19 complete mechanism of action has been as yet unavailable.
Fig. 2.

The schematic diagram showing the SARS-CoV-2 entry mechanism, viral replication and viral RNA packing in the human cell.
3. Potential therapeutic agents for COVID-19 treatment
In the last few months, the race to discover novel treatments for COVID -19 has immensely accelerated, and numerous peptides and small molecules have been recognized as encouraging therapeutic agents against SARS-CoV-2 [[34], [35], [36], [37], [38], [39], [40], [41], [42], [43], [44], [45]]. Recently, the constrained peptides have received much attention in the area of drug development; however, a certain inherent admonition limits its use as a drug [[46], [47], [48]]. This perspective encompasses the relevant information specifically associated with the structure and the activity of the small molecules and peptidomimetics that belong to the category of potential candidates acting against the SARS-CoV-2. All the molecules are categorized in different groups depending upon their action against various therapeutic targets. The basic information provided in this section may lead to the novel drug design and discovery against COVID-19.
3.1. Angiotensin-converting enzyme-2 (ACE-2) inhibitors
ACE-2 is a homolog of ACE and encompasses a single zinc-binding catalytic domain, which exhibits a 42% resemblance to the human ACE active region [49]. SARS-CoV entry into the cell works through the interaction of the viral spike protein with different host receptors present in the cell surface, and in this respect, ACE-2 has emerged as a primary virus receptor. The recent research findings have recognized that SARS-CoV-2 also involves ACE-2 for infection, and therefore significant attention has been given to understand its mechanism to facilitate the development of potent ACE-2 inhibitors for the treatment of the virus [[50], [51], [52], [53]].
3.1.1. Chloroquine (1) and hydroxychloroquine (2)
Chloroquine (1) (Fig. 3 ) is a potent drug exclusively used in the treatment of malaria and amoebiasis. Chloroquine (1), under cell culture conditions, is effective in blocking the interaction of RBD of SARS-CoV to ACE-2 with an ED50 of 4.4 μM [54,55]. In 2020, M. Wang et al. discovered that 1 effectively inhibits the SARS-CoV-2 infection with an EC50 of 1.13 μM, CC50 > 100 μM, and SI > 88.50 in Vero E6 cells. Additionally, chloroquine (1) was also found to increase its antiviral effect with immune-modulating activity in vivo [56]. Hydroxychloroquine (2) (Fig. 3), an analog of chloroquine (1), and a well-recognized antimalarial and anti-autoimmune agent recently discovered as a potent agent for SARS-CoV-2 treatment. In 2020, X. Yao et al. demonstrated that in in vitro anti-SARS-CoV-2 activities, hydroxychloroquine (2) is more effective (EC50 = 0.72 μM) than chloroquine (1) (EC50 = 5.47 μM) [57]. These molecules possibly block virus infection by increasing the endosomal pH needed for membrane fusion between virus and host cell and also interferes with the glycosylation of cellular receptor ACE-2, therefore specifically inhibiting the SARS-CoV/SARSCoV-2. Interestingly, during in vitro testing, hydroxychloroquine (2) effectively reduced the viral copy number of SARS-CoV-2 [58]. The clinical trials conducted with a combination of the hydroxychloroquine (2) and azithromycin in 20 patients (EUCTR2020-000890-25) in France and with hydroxychloroquine (2) in 30 patients (15 each for the hydroxychloroquine and the control group) in China demonstrated a significant reduction in viral load and promising prospects in the hydroxychloroquine group, respectively; however, both the trials expressed the requirement for a larger sample size for further validating the results [59,60]. Next, to examine the efficacy of hydroxychloroquine (2) in COVID-19 treatment, another study was conducted on 62 patients in China. Hydroxychloroquine (2) treatment displayed promising results and significantly reduced the patients’ clinical recovery time, and encouraged the resolution of pneumonia (ChiCTR2000029559) [61]. Further, two clinical trials conducted with large-scale COVID-19 patients (368 and 1376) in the USA revealed the negative results associated with the efficacy and safety of hydroxychloroquine (2). In 368 COVID-19 patients, hydroxychloroquine (2) alone or in combination with azithromycin was ineffective in reducing the risk of death or mechanical ventilation over supportive care. Additionally, an increase in mortality rate with hydroxychloroquine (2) treatment alone was observed [62]. The clinical study conducted with 1376 patients, including moderate to severe COVID-19 infection, revealed the inefficiency of hydroxychloroquine (2) in COVID-19 treatment. Both hydroxychloroquine (2) receiving patients (811) and patients with no given drug (placebo group) showed a comparable risk from intubation or death [63]. Additionally, hydroxychloroquine (2) and chloroquine (1) are known to cause several side effects, including ventricular arrhythmias, QT prolongation, retinopathy, and other cardiac-related toxicity in patients [[64], [65], [66]]. In June 2020, the FDA revoked the emergency authorization for hydroxychloroquine (2) and chloroquine (1) as a treatment for COVID-19, and the WHO decided to suspend the SOLIDARITY trial of hydroxychloroquine (2) [67,68].
Fig. 3.

Chemical structure of Angiotensin-converting enzyme-2 (ACE-2) inhibitors 1–10.
3.1.2. NAAE [N-(2-aminoethyl)-1 aziridine-ethanamine] (3)
M. J. Huentelman et al. applied the structure-based approach to recognize novel human ACE-2 inhibitors and, in addition, with the ability to block SARS coronavirus spike protein-mediated cell fusion [69]. Approximately 14,0000 compounds with known 3D structures were screened through in silico molecular docking studies, and the molecules with the highest predicted binding scores were recognized. Subsequently, selected molecules were assayed for ACE2 enzymatic inhibitory activity as well as for SARS coronavirus spike protein-mediated cell fusion inhibition activity. This approach results in the identification of NAAE (3) (Fig. 3) as a novel ACE2 inhibitor and with an ability to block the SARS coronavirus spike protein-mediated cell fusion effectively. The NAAE exhibited very promising results in inhibiting a SARS-CoV pseudotyped virus with an IC50 value of 57 ± 7 μmol/L and a Ki of 459 μmol/L in a dose-dependent inhibition of ACE2 catalytic activity. The NAAE (3) ability to control ACE2 activity and inhibit SARS spike protein-mediated cell fusion is quite promising, and therefore, we endorse its application as a lead compound for further optimization in SARS-CoV-2 infection treatment.
3.1.3. GW280264X (4), TAPI-0 (5) and TAPI-2 (6)
D. W. Lambert et al. discovered that during viral admittance, the SARS-CoV receptor, ACE2, undergoes proteolytic shedding and furthermore identified a disintegrin and metalloproteinase (ADAM17) and TACE (TNF-α converting enzyme, a member of the ADAM) responsible for ACE2 shredding [70]. The GW280264X (4) (Fig. 3) was recognized as an ADAM-specific inhibitor with the ability to reduce ACE2 shredding at 1 nM against SARS-CoV [71]. In addition, two compounds, TAPI-0 (5) and TAPI-2 (6) (Fig. 3), were identified as promising TACE inhibitors. Both TAPI-0 (5) and TAPI-2 (6) effectively reduced ACE2 shredding against SARS-CoV (IC50 = 100 and 200 nM, respectively) [70].
3.1.4. MLN-4760 (7)
MLN-4760 (7) (Fig. 3) is one of the most potent ACE2 inhibitors with an IC50 of 440 pM. The results of the crystal structure studies of the MLN-4760 (7) bound ACE2 complex revealed the key binding interactions within the active site and provided knowledge about the residues engaged in catalysis and substrate specificity. The study discusses the importance of N-terminal and C-terminal subdomain movement of ACE-2 upon MLN-4760 (7) binding, which is supposed to be responsible for the positioning of critical residues in order to stabilize the bound inhibitor [72].
3.1.5. HTCC (8) and HMHTCC (9)
The human coronavirus NL63 (HCoV-NL63) is known as a common cold pathogen that may cause severe lower respiratory tract diseases, particularly in children and patients with underlying disease. It is also responsible for causing croup in children that is because of swelling inside the trachea due to infection. This condition affects normal breathing and results in a “barking” cough and hoarse voice symptoms. Considering the significance of the polymer as potent antimicrobial agent, A. Milewska et al. developed a series of polymer-based compounds with promising anticoronaviral activity. Both cationically-modified Chitosan derivative, N-(2-hydroxypropyl) -3-trimethylammonium Chitosan chloride (HTCC) (8) (Fig. 3), and hydrophobically modified HTCC (9) (Fig. 3) exhibited promising coronavirus HCoV-NL63 inhibition activity [73]. The polymer-based compound 8 showed IC50 and CC50 values of ∼50 nM and ∼0.8 μM, respectively, and compound 10 displayed IC50 and CC50 values of ∼230 nM and ∼1 μM, respectively. Additionally, the suggestion has been made that the HTCC (8) is capable of interfering with the viral admittance process by blocking the interaction of HCoV-NL63 with its ACE2 receptor [74].
3.1.6. Telmisartan (10)
A considerable degree of angiotensin II on the lung interstitium encourages apoptosis and further leads to acute respiratory distress syndrome (ARDS). The ACE-2 inhibitors or angiotensin blockers can be used effectively for the COVID-19 treatment; however, scientists must be cognizant of the fact that the effect of ACE inhibitors on COVID-19 is debatable [75,76]. Telmisartan (10) (Fig. 3) is a non-peptide angiotensin II receptor antagonist used for hypertension treatment and belongs to the benzimidazole class of molecules. Recently telmisartan is selected as an alternative for COVID-19 treatment in patients prior to the development of ARDS and went clinical trials (NCT04355936) [77]. The study with telmisartan included a total of 78 COVID-19 patients; 40 of them received telmisartan, and 38 were kept in the control group. At day 5 the C-reactive protein (CRP) serum levels was 51.1 ± 44.8 mg/L (mean ± SD; n = 28) and 24.2 ± 31.4 mg/L (mean ± SD; n = 32, p < 0.05) in the control and telmisartan group, respectively. The median discharge time for the telmisartan and control group was observed 9 days and 15 days, respectively. Insignificant adverse effects related to telmisartan were reported and no major differences were observed for ICU admissions and death. In the preliminary results, the telmisartan displayed anti-inflammatory effect and improvement in the hospitalized patients infected with SARS-CoV-2. In the present preliminary report, despite the small number of patients studied, telmisartan, a well-known inexpensive, safe antihypertensive drug administered in high doses, demonstrates anti-inflammatory effects and improved morbidity improvement in hospitalized patients infected with SARS-CoV-2 patients [78].
3.1.7. Recombinant human angiotensin-converting enzyme 2 (rhACE2)
The soluble recombinant human Angiotensin-converting Enzyme 2 (rhACE2) possibly responsible for blocking the SARS-CoV-2 entry by blocking the interaction of S protein with the cellular ACE2. In addition, it’s established that rhACE2 could inhibit SARS-CoV-2 replication in cellular and embryonic stem cell-derived organoids, and its monitored administration can reduce serum level of angiotensin II by directing the substrate away from the ACE [79,80]. A soluble recombinant human ACE2 (rhACE2; APN01) developed by Apeiron Biologics already submitted to phase II trial for ARDS treatment, and China (NCT04287686) is assessing the role of rhACE2 in COVID-19 pneumonia. APN01 mimics the human enzyme ACE2 that is accountable for virus admittance. The virus, instead of binding to ACE2 present on the cell surface, binds to the APN01 and therefore preventing the virus from infecting the cells. Furthermore, APN01 also responsible for reducing the harmful inflammatory reactions in the lungs and therefore provide protection against lung injury caused by acute respiratory distress syndrome (ARDS) [81]. The results of APN01 testing on a small group of patients suffering from ARDS are very positive and encouraging; however, to reach the final decision, further studies are mandatory in this direction [82,83].
3.2. SARS-CoV-2 3CL protease (Mpro) inhibitors
The 3-chymotrypsin-like cysteine (3CL) protease is the main protease found in coronaviruses and is responsible for cleavage and transformation of two large polyproteins (pp1a and pp1ab) into mature nsps [84,85]. Considering the significance of 3CL protease (3C-like protease or main protease) in processing the polyprotein that is translated from the viral RNA, it is considered as one of the key drug targets for coronavirus infection. Reports suggest the 96% similarity between the sequencing of 3CL pro in SARS-CoV and SARSCoV-2 and the very nominal differences on the surface of the proteins [86]. It is expected that the SARS-CoV 3CLpro inhibitors can also work as SARS-CoV-2 3CLpro inhibitors. In recent years, a large number of small molecules, peptides and peptidomimetics are developed as SARS-CoV 3CLPro inhibitors [[87], [88], [89], [90]]; however, this section comprises recently reported selected SARS-CoV-2 3CLPro inhibitors for discussion.
3.2.1. Lopinavir (11) and ritonavir (12)
Lopinavir (11) (Fig. 4 ) and ritonavir (12) (Fig. 4) are a class of protease inhibitors, manufactured by AbbVie Corporation and used in combination with other medications to treat human immunodeficiency virus (HIV) infection. The molecular interaction of lopinavir (11) and ritonavir (12) with 3CLpro of SARS-CoV were examined through theoretical studies and the results suggest the proper fitting of these two drugs at the substrate-binding pocket of SARS-CoV 3CLpro [91]. In addition, the binding interactions of lopinavir (11) and ritonavir (12) with the SARS-CoV-2 proteinase were studied using molecular modeling and quantum chemical methods [92]. The lopinavir (11) and ritonavir (12) in combination are selected for coronavirus pneumonia treatment; however, its efficacy and safety are still not clearly established. Reports revealed the antiviral effect of lopinavir (11) at EC50 value of 26.1 μM but not ritonavir (12) against SARS-CoV-2 in vitro [93]. The pharmacokinetic studies indicated that the metabolism of lopinavir (11) could be inhibited by ritonavir (12) and therefore these two drugs combination could extend the systemic exposure to 11 and maintain its concentration in the circulation for a longer time. In clinical trials, this drug combination was effective in lowering the body temperature and restoring homeostasis however, unable to considerably accelerate the clinical improvement and reduce mortality in patients with serious COVID-19 [94]. A clinical study conducted at China with lopinavir-ritonavir on 199 patients with severe COVID-19 infection showed no advantageous of lopinavir-ritonavir treatment except the standard care (ChiCTR2000029308) [95]. Another trail conducted on 127 COVID-19 patients in Hong Kong with triple combination therapy (lopinavir-ritonavir, ribavirin, and b-interferon) displayed that in comparison to only lopinavir-ritonavir treatment the triple-drug combination therapy was safer and effective in reducing the viral shredding duration in mild to moderate symptom patients [96]. Not enough evidence available to confirm the regular use of lopinavir and ritonavir in the COVID-19 treatment. In July 2020, WHO discontinued the SOLIDARITY trial of lopinavir-ritonavir treatment for COVID-19 [97].
Fig. 4.

Chemical structure of the SARS-CoV-2 3CL protease (Mpro) inhibitors 11–26.
3.2.2. N3 (13) and other COVID-19 virus Mpro inhibitors (14–20)
H. Yang and coworkers combined the structure-assisted drug design, virtual drug screening, and high-throughput screening method to identify the lead compound that targets the main protease (Mpro) of SARS-CoV-2 [98]. The research group identified the peptidomimetic N3 (13) (Fig. 4) as a potent irreversible inhibitor of COVID-19 virus Mpro by using computer-aided drug design and then determined the crystal structure of Mpro of SARS-CoV-2 in complex with N3 (13) (Fig. 5 A). The N3 inhibitor binds with the residues of 164–168 in the long strand 155–168 residues and with residues 189–191 of the loop connecting between the second and third region. Additionally, N3 forms several hydrogen bonds with the main chain of the residues in the substrate-binding pocket. These interactions of N3 and COVID-19 Mpro lock the inhibitor inside the substrate-binding pocket. Subsequently, by using fluorescence resonance energy transfer assay they screened <10,000 compounds, including approved drugs, clinical-trial drug candidates, and natural products. The seven compounds were identified as primary hits, including Ebselen (14) (IC50 = 0.67 μM), Tideglusib (15) (IC50 = 1.55 μM), Carmofur (16) (IC50 = 1.82 μM), TDZD-8 (17) (IC50 = 2.5 μM), Disulfiram (18) (IC50 = 9.35 μM), Shikonin (19) (IC50 = 15.75 μM), and PX-12 (20) (IC50 = 21.39 μM) (Fig. 4). Further, to confirm their inhibition activity in vitro the Ebselen (14) and N3 (13) were evaluated in a cell-based assay. Both Ebselen (14) and N3 (13) displayed promising SARS-CoV-2 inhibition activity with EC50 values of 4.67 μM and 16.77 μM, respectively.
Fig. 5.

A) 3-D complex of SARS-CoV-2 M pro (blue color) interacts with N3 (13) inhibitor (orange color) (PDB: 6LU7). B) 3-D complex of SARS-CoV-2 M pro (green color) interacts with compound 21 (orange color) (PDB: 6LZE). C) 3-D complex of SARS-CoV-2 M pro (purple color) interacts with compound 22 (green color) (PDB: 6M0K). D) 3-D complex of SARS-CoV-2 M pro (red color) interacts with compound 25 (green color) (PDB: 6Y2F).
3.2.3. Compound 21 and 22
W. Dai et al. analyzed the substrate-binding pocket of SARS-CoV-2 Mpro (PDB: 2H2Z) and designed and synthesized two compounds that is N-((S)-3-cyclohexyl-1-oxo-1-(((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)propan-2-yl)-1H-indole-2-carboxamide (21) (Fig. 4) and N-((S)-3-(3-fluorophenyl)-1-oxo-1-(((S)-1-oxo-3-((S)-2-oxopyrrolidin-3-yl)propan-2-yl)amino)propan-2-yl)-1H-indole-2-carboxamide (22) (Fig. 4) to target Mpro [99]. The compounds 21 and 22 effectively inhibited (100% and 86%, respectively) SARS-CoV-2 Mpro at 1 μM concentration. In addition, both compounds (21 and 22) exhibited promising IC50 values (0.053 ± 0.005 μM and 0.040 ± 0.002 μM, respectively) in fluorescence resonance energy transfer (FRET)–based cleavage assay. Next, to understand the mechanism of inhibition of SARS-CoV-2 by 21 and 22, the group determined the X-ray crystal structures of SARS-CoV-2 Mpro in complex with 21 and 22 (Fig. 5B and C). The result revealed that compounds 21 and 22 follow a similar inhibitory mechanism to occupy the substrate-binding pocket and block the enzyme activity of SARS-CoV-2 Mpro. In both inhibitors (21 and 22), the C and O atoms of the aldehyde group form covalent interaction and hydrogen bond, respectively, with the Cys145 in the S1’ site, and therefore stabilizes the inhibitor conformations. The (S)-γ-lactam ring of 21 fits well into the S1 site by forming a hydrogen bond with the side chain of His 163 through its oxygen atom and with the main chain of Phe 140 and the side chain of Glu166 by NH group. The amide bonds on the chain of 21 form hydrogen bonds with the main chains of His 164 and Glu166 residues. The cyclohexyl group present in the compound 21 fits well into the S2 site by stacking with the imidazole ring of His41 and by forming hydrophobic interactions with the side chains of Met49, Tyr54, Met165, Asp187, and Arg188. The indole group present on 21 stabilized by forming a hydrogen bond with Glu166 and interacting with chains of residues Pro168 and Gln189 through hydrophobic interactions. Additionally, the multiple water molecules play a significant role in binding and stabilizing compound 21 by forming serval hydrogen bonds. Compound 22 exhibited a similar inhibitor-binding mode as compound 21; however, the small difference in binding mode was possibly because of the presence of 3-fluorophenyl group in 22. The 3-fluorophenyl group experiences a downward rotation and form hydrophobic interactions with the side chains of residues His41, Met49, Met165, Val186, Asp187, and Arg188. The 3-fluorophenyl group of 22 is stabilized through a hydrogen bond with the side chain of Gln189. The compound 21 and 22 showed promising anti–SARS-CoV-2 infection activity in cell culture via plaque assay, with EC50 = 0.53 ± 0.01 μM and 0.72 ± 0.09 μM respects. Both 21 and 22 compounds showed encouraging cytotoxicity profiles with CC50 values of >100 μM and selectivity indices (SI) > 189 for 21 and > 139 for 22. Additionally, 21 and 22 displayed promising in vivo pharmacokinetic properties, and compound 21 exhibited low toxicity and thus suggesting the significance of further studies with these compounds.
3.2.4. α-ketoamides (23–26)
L. Zhang et al. reported the X-ray structures of the unliganded SARS-CoV-2 Mpro and its complex with an α-ketoamide inhibitor [100,101]. Bearing in mind the similarity between SARS-CoV and SARS-CoV-2 and antiviral activity of compound 23 (Fig. 4) against MERS-CoV, the group modified the structure of 23 and synthesized compounds 24, 25, and 26 (Fig. 4). Compound 23 inhibits the SARS-CoV-2 Mpro with IC50 = 0.18 ± 0.02 μM whereas its structural modification reduces the inhibitory activity (24, IC50 = 2.39 ± 0.63 and 25, IC50 = 0.67 ± 0.18 μM. The x-ray crystal structure of the complex between α-ketoamide 25 and the Mproof SARS-CoV-2 was determined (Fig. 5D). The result suggests the formation of thiohemiketal in a reversible reaction via nucleophilic attack of the catalytic Cys145 onto the α-keto group of the compound 25 and the stabilization of oxyanion or hydroxyl group though hydrogen bond from His41. In compound 25, the amide oxygen accepts a hydrogen bond from the main-chain amides of Gly143, Cys145, and partly Ser144 and results in the formation of canonical “oxyanion hole” of the cysteine protease. The compound 26 bearing NH2 in place of the Boc group showed almost no activity. These compounds were well tolerated in the mice, and in addition, 24 and 25 showed a tendency to concentrate more in the lungs; therefore, these compounds could be directly administered to the lungs.
3.2.5. N-substituted isatin compounds (27–35)
Taking into consideration the SARS-CoV 3CLpro inhibitory activity of a series of N-substituted 5-carboxamide-isatin compounds, L. H. Lai and his coworkers decided to utilize an isatin scaffold to design its derivatives compounds (29 compounds) and evaluated them for SARS-CoV-2 3CLpro inhibition, followed by molecular docking studies to identify potential bonding modes and established structure activity-relationship (SAR) [102,103].
In the enzyme assay, Tideglusib (15) (Fig. 4) was used as a positive control with a measured IC50 value (1.91 μM). The SARS-CoV-2 3CLpro inhibition activity results revealed that for a high inhibitory activity, the isatin scaffold must have a hydrophobic group of the nitrogen atom and the carboxamide group on the benzene ring. Three compounds, including 27 (Fig. 6 ) (IC50 = 0.053 ± 0.010 μM), 28 (Fig. 6) (IC50 = 0.045 ± 0.007 μM) and 29 (Fig. 6) (IC50 = 0.047 ± 0.007 μM) discovered as the most potent compounds with promising SARS-CoV-2 3CLpro inhibition activity. The substitution of 6-bromo group present on the naphthalene ring of compound 29 with 6-phenyl group resulted in compound 30 (Fig. 6) with a lower inhibitory effect (IC50 = 24.9 ± 4.6 μM). In addition, the substitution of the amide group in compound 28 (Fig. 6) with acetamide group afforded 31 (Fig. 6) with significantly reduced inhibition activity (IC50 = 39.2 ± 10.5 μM). Moreover, the introduction of 3 or 4 carbon alkyl chain on the nitrogen atom while sustaining the amide group on the benzene ring or replacing it with iodine in the isatin scaffold causes a decrease in the SARS-CoV-2 3CLpro inhibition activity (32 (Fig. 6) IC50 = 10.2 ± 1.0 μM, 33 (Fig. 5) IC50 = 17.8 ± 0.7 μM, and 34 (Fig. 6), IC50 = 41.8 ± 8.0 μM). In the isatin scaffold, the introduction of the benzyl group at nitrogen and carboxylic acid N-hydroxysuccinimide ester on the benzene ring exhibited a detrimental effect on the activity (35, IC50 = 15.5 ± 1.2 μM).
Fig. 6.

Chemical structure of SARS-CoV-2 3CL protease (Mpro) inhibitors 27–35.
To understand the binding mode of these compounds, the group utilized an induced-fit docking procedure to build the complex model of the most potent compound 28 and SARS-CoV-2 3CLpro. Compound 28 fits easily in the substrate-binding pocket by making H-bonds with the side-chain carboxyl groups of Asn 142 and Glu166 and the main-chain amino group of Cys 145 through its carboxamide at C-5 and oxygens at C-2 and C-3, respectively. The naphthyl ring of compound 28 fits into the hydrophobic groove formed by Met 49 and Met 165 and forms π-π stacking interaction with His 41.
3.3. Spike (S) glycoprotein inhibitors
The Spike (S) glycoprotein of SARS-CoV-2 is extremely conserved and responsible for receptor recognition, viral attachment, and admittance into host cells. Therefore, it is considered a crucial target for the development of COVID-19 prevention and treatment drug and vaccine for the prevention and treatment of [[104], [105], [106]]. In general, the S protein is present in a metastable, prefusion conformation, but after interaction with the host, the S protein experiences extensive structural rearrangements that facilitate fusion of the virus with the host cell membrane [107,108]. Certainly, the S protein sequence and structure of SARS-CoV and SARS-CoV-2 protein bear resemblances, but still, several SARS-CoV antibodies are unsuccessful in binding to the SARS-CoV-2 spike protein; therefore, it is essential to develop exceptional COVID-19 treatment strategies.
3.3.1. S1 subunit: RBD inhibitors
Both SARS-CoV and SARS-CoV-2 gain admittance into the host cell through binding of the S1 subunit situated RBD to the cell receptor ACE2 in the region of amino peptidase N. Approximately 73%–76% similarity is observed in the sequencing of SARS-CoV-2 and SARS-CoV RBD [109,110]. Recently, X Wang and coworkers resolved the crystal structure of the receptor-binding domain (RBD) of the spike protein of SARS-CoV-2 bound to the cell receptor ACE2 (Fig. 7 A) [111]. Detailed studies regarding the contact region for virus and ACE2 suggest different interaction patterns for SARS-CoV and SARS-CoV-2. In the SARS-CoV-2 case, more residues, i.e., 18 of the RBD are in direct interaction with 20 residues of the ACE2, whereas in SARS-CoV RBD, only 16 residues contact 20 residues of the ACE2. Additionally, substantial surface area is concealed with SARS-CoV-2 S CTD in complex with ACE2 than with SARS S RBD. The SARS-CoV-2 exhibit enhanced interaction with ACE2 because of the mutation of key residues, such as F486 and E484, a substitute of I472 and P470, respectively, in SARS RBD and responsible for forming strong π –π interaction (aromatic groups) with ACE2 Y83 and ionic interactions with K31 [108,112,113]. Because of the structural understanding of the ACE2 receptor bound to the SARS-CoV-2 spike receptor-binding domain, the S protein and its RBD are encouraging targets for the development of antiviral drugs against SARS-CoV and SARS-CoV-2. In this respect, several peptide analogs, monoclonal antibodies, and protein chimeras are recognized as RBD inhibitors; however, in literature, the availability of small molecules targeting the RBD is minimal.
Fig. 7.

(A) Crystal structure of SARS-CoV-2 spike receptor-binding domain (orange color) bound with ACE2 (green color) (PDB: 6M0J). (B) 3D structure of SARS-CoV-2 RNA-dependent RNA polymerase in complex with cofactors (PDB: 6M71).
3.3.1.1. SSAA09E2 (36)
Adedeji et al. screened approximately 14,000 compounds (following Lipinski’s rule of five) using a cell-based assay to identify SARS-CoV entry inhibitors. The group discovered the first small molecule, SSAA09E2 (36) (Fig. 8 ), capable of blocking SARS-CoV by interfering with the ACE2–SARS-S RBD interaction [114]. The immunoprecipitation and immunoblot assays were used to examine the inhibition mechanism of SARS-CoV entry, and results revealed that SSAA09E2 (36) interferes with the interaction of the RBD of SARS-CoV S protein and ACE2 and efficiently blocks their collaboration (IC50 = 3.1 μM and CC50 = >100 μM). Further study suggests that the SSAA09E2 (36) does not tamper with the surface expression of ACE2 and mostly affects the initial recognition of the virus by directly interfering with SARS-S RBD–ACE2 interactions.
Fig. 8.

Chemical structure of RBD inhibitors 36–38.
3.3.1.2. K22 (37)
A. Lundin et al. screened 16,671 compounds bearing different chemical structures for anti-human coronavirus 229E activity [115]. The small molecule K22 (37) (Fig. 8) was identified as a potent inhibitor of HCoV-229E with an IC50 = 0.7 μM and CC50 = 110 μM. The mechanism of action study of K22 confirms its significant role during the early steps of the viral life cycle, possibly by interacting with viral particles and therefore inactivating their binding. Additionally, K22 was found to be able to inhibit a wide range of coronavirus, including MERS–CoV in primary human epithelial cultures, which signify the human coronavirus infection admittance port.
3.3.1.3. Rilapladib (38)
A. V. Ravi and coworkers performed in silico analysis of 113 quinoline drugs to identify the plausible anti-SARS-CoV-2 therapeutics [116]. Among all the screened compounds, a hydroquinoline-based small molecule drug Rilapladib (38) (Fig. 8), developed by GlaxoSmithKline, was found to target the RBD of the Spike-ACE2 complex. The Rilapladib (38) is mostly used for the treatment of atherosclerotic plaques and Alzheimer’s disease and works as lipoprotein-associated phospholipase A2 (Lp-PLA2) inhibitor or 1-alkyl-2-acetylglycerophosphocholine esterase inhibitor. The results revealed that the appropriate fitting of Rilapladib (38) into the interface of the RBD of the Spike-ACE2 complex with −9.7 kcal/mol binding energy. The Rilapladib (38) showed many strong binding interactions with the amino acid residues present at the RBD interface of the Spike-ACE2 complex (His 34, Glu 35, Glu 37, Asp 38, Leu 39.) and therefore recommend Rilapladib (38) as inhibitors of ACE2-mediated viral admittance of SARS-CoV-2.
3.3.2. S2 subunit inhibitors
The S2 subunit particularly responsible for viral fusion and admittance consists of FP, HR1, HR2, TM domain, and cytoplasmic domain fusion (CT) [107,[117], [118], [119]]. The fusion peptide (FP) is composed of 15–20 amino acid residues, mostly hydrophobic, and during the prehairpin conformation of S protein, it anchors to the target membrane. HR1 and HR2 contain mainly hydrophobic, traditionally bulky, polar, hydrophilic, and charged residues and are situated at the C-terminus of a hydrophobic FP and at the N-terminus of the TM domain, respectively. They are responsible for forming the six-helical bundle (6-HB), which is significant for the viral fusion and the admittance function of the S2 subunit. S protein anchors to the viral membrane through the TM domain and the S2 subunit ends in a CT tail. Both SARS-CoV and SARS-CoV-2 S proteins, particularly in their HR region, share a high degree of conservation. Therefore, the development of small molecules mimicking broadly neutralizing antibodies (bnAbs) targeting the S2 domain of SARS-CoV S protein and with better oral delivery can be considered a promising approach in SARS-CoV-2 therapeutic research.
3.3.2.1. TGG (39), luteolin (40) and quercetin (41)
X. Xu and coworkers developed a screening method involving two steps, i.e., frontal affinity chromatography-mass spectrometry (FAC/MS) and pseudotyped virus infection assay to recognize molecules that inhibit the SARS-CoV entry into the host cell [120]. Through this approach, two small molecules, TGG (39) (1,2,3,6-Tetragalloyl-beta-d-glucopyranose) (Fig. 9 ) and luteolin (40) (Fig. 9), emerged as potential candidates. The inhibitory effect of TGG (39) and luteolin (40) against infection by a wild-type SARS-CoV was determined using an MTT assay, and the results revealed that both of these compounds significantly inhibit in a dose-dependent fashion (IC50 = 4.5 and 10.6 μM, respectively). Additionally, the group determined the cytotoxicity of these compounds against the Vero E6 cells by using MTT assay, and the results displayed CC50 of TGG (39) and luteolin (40) 1.08 and 0.155 mM, respectively, and with selective index values 240.0 and 14.62, respectively. Next, the acute toxicity assessment provided 50% lethal doses of TGG (39) and luteolin (40) as 456 and 232.2 mg/kg, respectively, indicating the applicability of these two molecules in mice at relatively high concentration. Later, bearing in mind the structural similarity of quercetin (41) (Fig. 9) with luteolin (40), it was also evaluated for SARS-CoV entry antagonizing effect. Quercetin (41) in assays with the HIV-luc/SARS pseudotyped virus exhibited antiviral activity with an EC50 of 83.4 μM and very low cytotoxicity (CC50 = 3.32 mM).
Fig. 9.

Chemical structure of S2 subunit inhibitors 39–43.
3.3.2.2. ADS-J1 (42)
ADS-J1 (42) (Fig. 9), is one of the most promising viral entry inhibitors with IC50 = 3.89 μM. The molecular docking analysis results revealed that ADS-J1 can fit and bind properly into the deep pocket of the SARS-CoV S protein HR region and, therefore, efficiently block the SARS-CoV entry into host cells [121]. Recent studies with ADS-J1 in a pseudovirus-based inhibition assay displayed its ability to inhibit MERS-CoV infection with IC50 = 0.6 μM, CC50 = 26.9 μM, and selectivity index = ∼45 [122].
3.3.2.3. Arbidol (43)
Arbidol (43) (Fig. 9), also known as umifenovir, is a broad-spectrum drug approved for human use in Russia and China against influenza A and influenza B viral infections [123]. Abidol is an indole analog containing an amine and hydroxyl group at the 4 and 5 positions, respectively, as essential moieties for its antiviral effect. Arbidol (43) blocks the viruses-cell fusion by binding to HA protein and significantly inhibit SARS-CoV-2 virus infection in vitro (IC50 = 4.11 μM, CC50 = 31.79 μM, and SI = 7.73) [124]. The protein sequence analysis of Arbidol suggests the similarity of its small region (aa947−aa1027) present in the S2 domain of SARS-CoV-2 spike glycoprotein with influenza virus H3N2 HA. Therefore, its mechanism of action is possibly through targeting the SARS-CoV-2 spike glycoprotein and by blocking its trimerization and thus inhibiting the host cell adhesion and takeover. Presently, Arbidol (43) is in phase IV clinical trials against COVID-19 (NCT04350684 and NCT04260594), the existing clinical trial data are not sufficient to consider this drug as an effective treatment for COVID-19 infection, and consequently, further study is mandatory in this direction [125].
3.4. Cathepsin L proteinase inhibitors
One of the key steps in viral infection is the activation of the SARS-CoV-2 spike protein by cleavage of proteases, and diverse lysosomal cathepsins are essential for coronavirus admittance in humans via endocytosis [126,127]. Recent reports suggest that particularly cathepsin L is involved in SARS-CoV-2 endocytosis entry, not cathepsin B or calpain [128]. Considering, the significant role of cathepsin L in the SARS CoV infection cycle and the capability to prevent the progression of pulmonary fibrosis, it could be considered as one of the significant therapeutic options for COVID-19 treatment [129].
3.4.1. CID 23631927 (44) and CID 16725315 (SID26681509) (45)
P. P. Shah et al. examined tetrahydroquinoline oxocarbazate (CID 23631927) (44) and related thiocarbazate (CID 16725315) (45) (Fig. 10 ) as a human cathepsin L inhibitor and viral entry inhibitor of the SARS-CoV and Ebola pseudotype virus [130]. In the cathepsin L inhibition assay, the CID 23631927 (44) displayed an IC50 value of 6.9 nM. Additionally, the CID 23631927 (44) exhibited activity in blocking both SARS-CoV (IC50 = 273 ± 49 nM) and Ebola virus (IC50 = 193 ± 39 nM) entry into human embryonic kidney 293T cells and was nontoxic to human aortic endothelial cells at 100 μM. In comparison, the CID 16725315 (45) was less potent as a cathepsin L inhibitor with IC50 = 56 nM. The oxocarbazate CID 23631927 (44) successfully blocked SARS-CoV and Ebola pseudotype virus entry into human cells and emerged as a slow-binding, reversible inhibitor of human cathepsin L with activity in the subnanomolar range.
Fig. 10.

Chemical structure of Cathepsin L Proteinase inhibitors 44–48.
X. Ou et al. studied the effect of cathepsin inhibitors on the SARS-CoV-2 entry [131]. To examine the requirement of cathepsin L for SARS-CoV-2 entry, HEK 293/hACE2 cells were treated with the cathepsin L selective inhibitor CID 16725315 (SID26681509) (45). This cathepsin L inhibition treatment reduced the entry of SARS-CoV-2 pseudovirus into 293/hACE2 above 76% and therefore revealing the significant role of cathepsin L for priming of SARS-CoV-2 S protein in the lysosome for cell entry.
3.4.2. SSAA09E1 (46)
A O. Adedeji et al. used a dose-dependence fluorescence inhibition assay to study the cathepsin L activity of the few compounds [114]. Among all the compounds, SSAA09E1 (46) (Fig. 10) emerged as a promising cathepsin L inhibitor. SSAA09E1 (46), a nonpeptidomimetic small molecule, is considered a novel antiviral agent for SARS-CoV infection, and it inhibits the cathepsin L with IC50 values of 5.33 ± 0.61 μM. In a pseudotype-based assay in 293T cells, SSAA09E1 (46) displayed the EC50 = ∼6.4 μM, and no cytotoxicity was noticed below 100 μM.
3.4.3. MDL28170 (47)
P. Bates et al. performed the high-throughput screening of a library of 1000 compounds and identified MDL28170 (47) (Fig. 10) as an inhibitor of cathepsin L [132]. In an antiviral activity assay, the MDL28170 (47) efficiently and specifically inhibited the cathepsin L-mediated substrate cleavage and blocked SARS-CoV viral entry (IC50 = 2.5 nM and EC50 = 100 nM). Indeed, MDL28170 (47) exhibited potent inhibitory activity and is a promising initial candidate for antiviral inhibitors of SARS-CoV viral admittance; on the other hand, its cytotoxicity data is unavailable.
3.4.4. K11777 (48)
Y. Zhou et al. used SARS-CoV assay to screen a library of cysteine protease inhibitors with established activity against human cathepsins, SARS-CoV, and filoviruses, including EBOV [133]. Among all the compounds, K11777 (48) (Fig. 10) was identified as potent inhibitors of SARS-CoV and Ebola virus entry. K11777 (48) efficiently inhibited SARS-CoV pseudovirus entry (IC50 value of 0.68 ± 0.09 nM) and displayed no toxicity (CC50 value > 10 μM). For inhibition of MERS-CoV and NL63 envelope higher concentration of K11777 (48) was required (IC50 = 46 nM for MERS-CoV and IC50 = <7 nM for NL63).
3.5. Transmembrane Serine Protease 2 (TMPRSS2) inhibitors
The SARS-CoV virus admittance into the host cells is facilitated, either by endosomal cysteine proteases cathepsin L or by the cell membrane-associated serine protease TMPRSS2 by cleavage of the viral S protein [52,134]. The research findings in the literature suggest that, specifically, TMPRSS2 facilitates viral activation and is therefore considered as one of the essential host factors for SARS-CoV-2 pathogenicity [135]. The maximum expression of human TMPRSS2 is found in the epithelia of the gastrointestinal, urogenital, and respiratory tracts [136]. In recent years, TMPRSS2 developed as a potential drug target for antiviral drug discovery, and therefore, we can anticipate that promising TMPRSS2 inhibitors can serve as a potential therapy for COVID 19.
3.5.1. Camostat (49)
S. Matsuyama et al. reported that Camostat (49) (Fig. 11 ), a commercially available serine protease inhibitor developed in 1980, efficiently inhibits the TMPRSS2 activity and consequently blocks SARS-CoV infection (at 10 μM) [137]. By increasing the concentration of Camostat (49) to 100 μM, the inhibition efficiency was not more than 65%, suggesting that 35% of virus entry was through the endosomal cathepsin pathway. Therefore, the treatment of the cells was performed with a combination of camostat (49) and EST [(23, 25) trans-epoxysuccinyl- L-leucylamindo-3-methyl butane ethyl ester], a cathepsin inhibitor, and the results revealed >95% infection blockage.
Fig. 11.

Chemical structure of Transmembrane Serine Protease 2 (TMPRSS2) inhibitor 49.
This simultaneous treatment successfully prohibited cell entry and the multistep growth of SARS-CoV in human Calu-3 airway epithelial cells, suggesting the dual blockade of viral entry from the cell surface and through the endosomal pathway. In a recent study (in vitro), Hoffmann et al. reported that camosatat (49) could block the SARSCoV-2 entry into primary lung cells and thus revealing its significance as TMPRSS2 inhibitors in SARS-CoV-2 treatment in the near future [52].
3.6. Furin cleavage site inhibitors
Remarkably, Furin is a type 1 membrane-bound protease enzyme expressed in multiple tissues belonging to the subtilisin-like proprotein convertase family. The unique furin-like cleavage site present in the spike protein (S) of SARS-CoV-2 but absent in other SARS-like CoVs, is responsible for high infectivity and transmissibility of SARS-CoV-2 [138,139]. The Furin is highly expressed in the lungs, and in addition, SARS-CoV-2 has multiple furin cleavage sites, which increases the probability of being cleaved by furin-like proteases and hence increases the infectivity of SARS-CoV-2 [[140], [141], [142]]. The potent furin inhibitors may be successfully utilized in blocking the viral entry process.
3.6.1. Decanoyl-RVKR-chloromethylketone (dec-RVKR-CMK) (50) and MI-1851 (51)
Decanoyl-Arg-Val-Lys-Arg-chloromethylketone (dec-RVKR-cmk) (50) (Fig. 12 ) is a small, synthetic, and clinically suitable furin inhibitor that is used as a reference inhibitor to examine the effects of furin and related proprotein convertases [143]. It irreversibly blocks furin and is significantly responsible for the inhibition of viral infection. G R Whittaker and coworkers reported that dec-RVKR-CMK (50) efficiently (IC50 = 75 μM) blocks the MERS-CoV S protein-mediated entry and viral infection in HEK-293T cells. In addition, dec-RVKR-CMK (50) in combination with camostat (49) (cathepsin inhibitor) significantly inhibits MERS-CoV infection [144]. Recently, Eva Böttcher-Friebertshäuser and coworkers reported MI-1851 (51) (Fig. 12), a synthetic and peptidomimetic furin inhibitor, as a strong inhibitor of SARS-CoV-2 replication. MI-1851 (51) successfully inhibited the proteolytic processing of the S protein from SARS-CoV-2 by endogenous furin in HEK293 cells. MI-1851 (51), in combination with different TMPRSS2 inhibitors, exhibited promising antiviral activity against SARS-CoV-2 than an equimolar amount of any single serine protease inhibitor [145]. Moreover, dec-RVKR-CMK) (50), being the first inhibitor of furin, was used for principal studies to introduce cell-target antiviral compounds; however, currently, dec-RVKR-CMK) (50) has been replaced by novel inhibitors that possess minimal side effects, such as MI-1851 (51).
Fig. 12.

Chemical structure of Furin cleavage site inhibitors 50–51.
3.7. RNA-dependent RNA polymerase (RdRp) inhibitors
RdRp is an enzyme that plays a significant role in the RNA virus life cycle, including coronavirus [[146], [147], [148]]. During infection, RdRp facilitates the transcription and replication of RNA. Non-structural proteins (nsps) accumulated into a multi-subunit polymerase complex, including nsp12-nsp7-nsp8 subcomplexes, mediate transcription and replication of the viral genome. Particularly, nsp12 is the catalytic unit with RdRp activity and conduct polymerase reaction effectively in the presence of nsp7 and nsp8 cofactors. For antiviral drug development, RdRp is a favorable drug target because it has no human counterpart and, in addition, is important for the virus life cycle. Recently, the structure of SARS-CoV-2 RNA-dependent RNA polymerase in complex with cofactors and is included in PDB database (Fig. 7 B), which will definitely facilitate the drug design and development process on RdRp target [149]. The RdRp of SARS-CoV-2 comprises a larger N-terminal extension and polymerase domain. The polymerase domain consists of three subdomains: a fingers subdomain (residues L366 to A581 and K621 to G679), a palm subdomain (residues T582 to P620 and T680 to Q815), and a thumb subdomain (residues H816 to E920). The active site domain of the SARS-CoV-2 RdRp is formed by the conserved polymerase motifs A-G in the palm domain and configured like other RNA polymerases. The SARS-CoV-2 RdRp homology modeling using SARS-CoV-1 RdRp as a template expressed 97.08% sequence identity to the template with 100% of the residues in the allowed regions and 97.5% in the most-favored regions [150].
3.7.1. Remdesivir (52)
Remdesivir (52) (GS-5734) (Fig. 13 ), an adenosine analog developed by Gilead Science, is an established antiviral drug against a wide range of infections with RNA viruses and is significantly effective against various CoVs in cell cultures and in a mouse model of SARS-CoV infection [151]. Remdesivir (52) is found to be highly active against MERS-CoV and SARS-CoV with EC50 = 0.074 ± 0.023 μM and 0.069 ± 0.036 μM, respectively. Recently, the Remdesivir (52) and other broad-spectrum antivirals targeting RdRp, such as ribavirin (53), Penciclovir (54), and Favipiravir (55) (Fig. 13) are examined against SARS-CoV-2 in Vero E6 cells. The results of this study are quite encouraging for Remdesivir (52) with EC50 = 0.77 μM and selectivity index = 129.87. The half-maximal effective concentration (EC50) and half-cytotoxic concentration (CC50) for Ribavirin (53) and Penciclovir (54) is as follows: EC50 = 109.50 μM, CC50 > 400 μM and EC50 = 95.96 μM, CC50 > 400 μM, respectively [56]. The mechanism of action of nucleoside prodrug Remdesivir (53) in CoVs is by inhibiting the RdRp and evade the action of the exoribonuclease (nsp 14) [153]. Remdesivir (53) underwent several clinical trials (NCT04323761 and NCT04292899) with the intention to examine its therapeutic potential in patients [[154], [155], [156], [157], [158]]. The results from remdesivir (53) clinical trials have been dispersed and broadly discussed [159]. In Nov 2020, the WHO recommended against the use of remdesivir in hospitalized patients, irrespective of disease severity, because currently there is no evidence to support that remdesivir improves survival and other outcomes in COVID-19 infected patients [160].
Fig. 13.

Chemical structure of RNA-Dependent RNA Polymerase (RdRp) inhibitors 52–56.
3.7.2. Favipiravir (55)
Favipiravir (55) (T-705), is developed by Toyama Chemical Co., Ltd as RdRp inhibitor for the treatment of influenza viruses in Japan [161]. Favipiravir is a pyrazine-derived antiviral drug molecule applied against RNA viruses. The active form of T-705 (55) is host enzymes converted Favipiravir ribofuranosyl-50-triphosphate (Favipiravir-RTP) (56) that selectively inhibits the RdRp. Favipiravir (55) exhibited activity by assimilating into the nascent viral RNA and thus terminating the viral replication processes. It has a wide range of antiviral activities, including inhibition of ZIKV in Vero cells with EC50 = 3.5–3.8 μg/mL and Zaire Ebolavirus in Vero E6 cells with EC50 = 10.5 μg/mL [162]. Recently, Favipiravir (55) emerged as a promising candidate for the SARS-CoV-2 treatment with EC50 = 61.88 μM, CC50 > 400 μM, and SI > 6.46 in Vero 6 cells [56]. At present, Favipiravir (55) is under Phase III clinical trials (NCT04336904). However, it is important to mention that Favipiravir use is associated with risks such as teratogenicity and embryo toxicity [163].
4. Conclusion and perspectives
The COVID-19 disease, triggered by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), in a short stretch of time has spread globally and is declared affirmed as a global pandemic by the World Health Organization (WHO). The human civilization is severely affected by the unprecedented COVID-19, the situation around the world remains complicated, and the entire human race has been gripped with anxiety. A continuous increase in the number of COVID-19 cases and the lack of an effective treatment is raising alarm in the scientific community and simultaneously encouraging scientists to accelerate the efforts in the field of COVID-19 drug discovery and development process. Remarkably, the SARS-CoV-2 shares a similar infection mechanism as other coronaviruses, which opened the window to investigate the appropriateness of well-known inhibitors against diverse CoVs to fight the war against SARS-CoV-2. Quite a few primary virus-based and host-based target inhibitors, including peptides, natural products, and small molecules, are discovered with anti-coronavirus activity. However, the lack of in vitro and in vivo results of the potential molecules and the poor safety, bioavailability, and pharmacokinetic profiles of the compounds in the preclinical trials make the situation more challenging. At present, the vaccine, monoclonal antibodies, convalescent sera from SARS-CoV-2 survivors, repurposing drugs, and small molecules inhibitor are encouraging approaches to develop effective COVID-19 treatment [164].
It is our firm belief that the discovery and development of small molecules as inhibitors of druggable targets of SARS-CoV-2 can facilitate the process of identifying the effective drug for the COVID-19 treatment. The chemical structure and activity of molecules with promising inhibition activity against the primary virus-based and host-based targets of SARS-CoV-2 including ACE-2 inhibitors, 3CL protease (Mpro) inhibitors, RBD inhibitors, S2 subunit inhibitors, Cathepsin L Proteinase inhibitors, TMPRSS2 inhibitors, Furin cleavage site inhibitors, and RdRp inhibitors are displayed in Table 1 . As evident from the structure of molecules acting as ACE-2 inhibitors (1–9), the hydrophilic functional groups (hydroxyl, amino, and carboxyl groups) are most common in peptidomimetics, chitosan, and small molecules. The chloroquine (1), hydroxychloroquine (2), and MLN-4760 (7) with ACE-2 inhibition activity in the micromolar range have chloro substituents on the aromatic ring. The peptidomimetics, including α-keto amides such as 11, 13, 21–25, showed promising SARS-CoV-2 3CL protease (Mpro) inhibition activity. Aromaic heterocyclic and cycloalkane rings are common structural features in these peptidomimetics. In contrast, the small molecule 14 having the Se atom in the ring also exhibited 3CL protease (Mpro) inhibition activity in the micromolar range. Based on the structure-activity relationship and docking analysis of isatin derivatives (27–35), a few significant structural features are recommended for SARS CoV-2 3CLpro inhibition activity. This includes the carboxamide group at C-5 and hydrophobic aromatic substituents at the N-1 position of the isatin ring. Any type of modification at C-5, including the introduction of a carbon atom between a carboxamide group and the isatin ring, as well as replacement of a carboxamide group with a carboxylic group or ester group, was incompatible for binding. The introduction of aromatic rings, such as naphthalene and benzothiophene, at the N-1 position of the isatin scaffold increases the rigidity, results in better fitting of the molecule in the pocket, and exhibits an increase in inhibitory activity. However, there is a limitation to the introduction of the larger substituent on the naphthalene ring, which showed an unfavorable effect on inhibition activity. Both molecule 36 and 37 bearing an amide bond, more than one aromatic ring, and a piperazine or piperadine ring emerged as a promising RBD inhibitor with activity in the micromolar range. Among the S2 subunit inhibitors (39–43), the most common functional groups are the hydroxyl groups. In the d-glucopyranose derivative (39) and 4-chomenone derivatives 40 and 41, the presence of multiple hydroxyl groups reveals the importance of small hydrophilic groups such as hydroxyl for the S2 subunit inhibition activity. A large molecule such as 42 with one triazine, two phenyl, and two naphthyl rings connected to each other through amino and diazine linkages and with four sulfonic acid groups fits well in the SARS-CoV S protein HR region and shows inhibition activity in the micromolar range. Arbidol (43), a small molecule and a derivative of indole-3-carboxylate, also exhibited S2 inhibition activity in the micromolar range. Except molecule 46, a thiourea derivative with thiophene ring, all the molecules included as Cathepsin L Proteinase and Furin cleavage site inhibitors are peptidomimetics (44–45, 47–48, and 50–51). The diaminomethylideneamino group is common in both in TMPRSS2 inhibitor 49 and Furin cleavage site inhibitors 50 and 51. In the structure of RNA-Dependent RNA Polymerase (RdRp) inhibitors, (52–55) monocyclic or bicyclic nitrogen-containing heterocyclic rings and small hydrophilic groups such as hydroxyl, amino, and carboxamide are common.
Table 1.
Molecules with inhibition activity against various main virus-based and host based coronavirus targets.
| Name | Structure | Targets | Virus | IC50//EC50 | Ref. |
|---|---|---|---|---|---|
| Chloroquine | 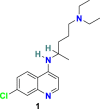 |
ACE2 | SARS-CoV-2 | EC50 = 1.13 μM | 56 |
|
Hydroxychlo -roquine |
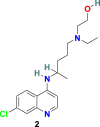 |
ACE2 | SARS-CoV-2 | EC50 = 0.72 μM | 57 |
| NAAE | 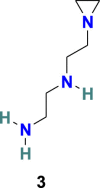 |
ACE2 | SARS-CoV | IC50 = 57 μM | 69 |
| GW280264X | 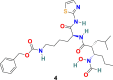 |
ACE2 | SARS-CoV | IC50 = 1 nM | 71 |
| TAPI-0 | 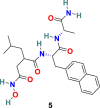 |
ACE2 | SARS-CoV | IC50 = 100 nM | 70 |
| TAPI-2 | 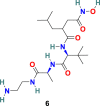 |
ACE2 | SARS-CoV | IC50 = 200 nM | 70 |
| MLN-4760 | 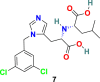 |
ACE2 | - | IC50 = 440 pM | 72 |
| HTCC | 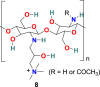 |
ACE2 | HCoV-NL63 | IC50 = ∼50 nM | 73, 74 |
| HMHTCC | 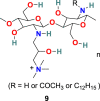 |
ACE2 | HCoV-NL63 | IC50 = ∼230 nM | 73, 74 |
| Lopinavir | 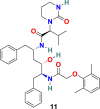 |
3CL protease | SARS-CoV-2 | EC50 = 26.1 μM | 93 |
| N3 | 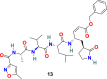 |
3CL protease | SARS-CoV-2 | EC50 = 16.77 μM | 98 |
| Ebselen | 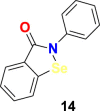 |
3CL protease | SARS-CoV-2 | EC50 = 4.67 μM | 98 |
| compound 21 | 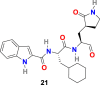 |
3CL protease | SARS-CoV-2 | EC50 = 0.53 μM | 99 |
| compound 22 | 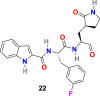 |
3CL protease | SARS-CoV-2 | EC50 = 0.72 μM | 99 |
| compound 23 | 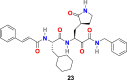 |
3CL protease | SARS-CoV-2 | IC50 = 0.18 μM | 100-101 |
| compound 24 | 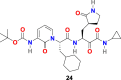 |
3CL protease | SARS-CoV-2 | IC50 = 2.39 μM | 100-101 |
| compound 25 | 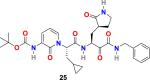 |
3CL protease | SARS-CoV-2 | IC50 = 0.67 μM | 100-101 |
| compound 27 | 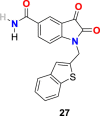 |
3CL protease | SARS-CoV-2 | IC50 = 0.053 μM | 103 |
| compound 28 | 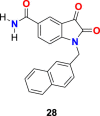 |
3CL protease | SARS-CoV-2 | IC50 = 0.045 μM) | 103 |
| compound 29 | 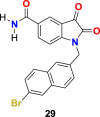 |
3CL protease | SARS-CoV-2 | IC50 = 0.047 μM | 103 |
| compound 30 | 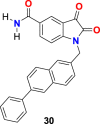 |
3CL protease | SARS-CoV-2 | IC50 = 24.9 μM | 103 |
| compound 31 | 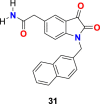 |
3CL protease | SARS-CoV-2 | IC50 = 39.2 μM | 103 |
| compound 32 | 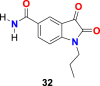 |
3CL protease | SARS-CoV-2 | IC50 = 10.2 μM | 103 |
| compound 33 | 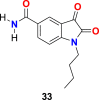 |
3CL protease | SARS-CoV-2 | IC50 = 17.8 μM | 103 |
| compound 34 | 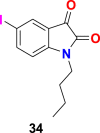 |
3CL protease | SARS-CoV-2 | IC50 = 41.8 μM | 103 |
| compound 35 | 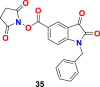 |
3CL protease | SARS-CoV-2 | IC50 = 15.5 μM | 103 |
| SSAA09E2 | 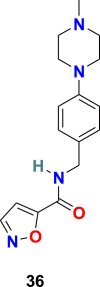 |
RBD | SARS-CoV | IC50 = 3.1 μM | 114 |
| K22 | 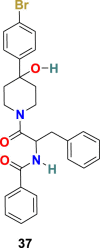 |
RBD | HCoV-229E | IC50 = 0.7 μM | 115 |
| TGG | 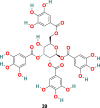 |
S2 subunit | SARS-CoV | IC50 = 4.5 μM | 120 |
| Luteolin | 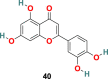 |
S2 subunit | SARS-CoV | IC50 = 10.6 μM | 120 |
| Quercetin | 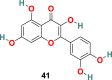 |
S2 subunit | SARS-CoV | IC50 = 83.4 μM | 120 |
| ADS-J1 | 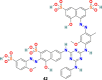 |
S2 subunit | SARS-CoV | IC50 = 3.89 μM | 121 |
| Arbidol | 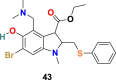 |
S2 subunit | SARS-CoV-2 | IC50 = 4.11 μM | 124 |
| CID 23631927 | 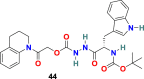 |
Cathepsin L Proteinase | SARS-CoV | IC50 = 6.9 nM | 130 |
| CID 16725315 | 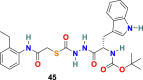 |
Cathepsin L Proteinase | SARS-CoV | IC50 = 56 nM | 130 |
| SSAA09E1 | 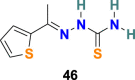 |
Cathepsin L Proteinase | SARS-CoV | IC50 = 5.33 μM | 114 |
| MDL28170 | 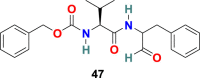 |
Cathepsin L Proteinase inhibitors | SARS-CoV | IC50 = 2.5 nM | 132 |
| K11777 | 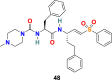 |
Cathepsin L Proteinase | SARS-CoV | IC50 = 0.68 nM | 133 |
| Camostat | 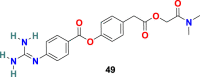 |
TMPRSS2 | SARS-CoV | IC50 = 10 μM | 137, 52 |
| dec-RVKR-CMK | 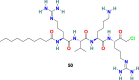 |
Furin cleavage site | MERS-CoV | IC50 = 75 μM | 144 |
| MI-1851 | 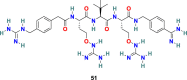 |
Furin cleavage site | SARS-CoV-2 | - | 145 |
| Remdesivir | 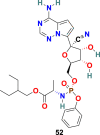 |
RdRp | SARS-CoV-2 | EC50 = 0.77 μM | 56 |
| Ribaviri | 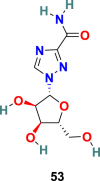 |
RdRp | SARS-CoV-2 | EC50 = 109.50 μM | 56 |
| Penciclovir | 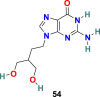 |
RdRp | SARS-CoV-2 | EC50 = 95.96 μM | 56 |
| Favipiravir | 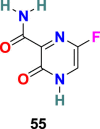 |
RdRp | SARS-CoV-2 | EC50 = 61.88 μM | 56 |
In conclusion, we anticipate that in the near future, inhibitors of virus- and host-based targets, particularly small molecules, would lead the way towards the development of FDA-approved drugs for the treatment of COVID-19. We have reviewed the structures of the most promising molecules effective against various targets significant in the SARS-CoV-2 treatment and found that the most common chemical structures are peptidomimetics and small molecules containing hydrophilic groups and heterocyclic aromatic/nonaromatic rings. Furthermore, to overcome the pharmacokinetic challenges, we can examine the effectiveness of small molecule inhibitors conjugated with biomolecules such as steroids, peptides, and sugars. Bearing in mind the limitations associated with the repurposing of approved drugs, which include issues related to therapeutic dosage, safety, physicochemical properties such as solubility, and permeability, among others, we must follow the traditional drug discovery process and focus on more rational approaches using virus-targeted and cell-targeted compounds which intervene in virus-cell interactions, primarily at the beginning of the infection. We can win this war against COVID-19 in a timely manner only through collective efforts from experts of different branches of chemistry, life sciences, pharmaceuticals sciences, social sciences, and industries. Prof. Yuval Noah Harari correctly said, “In this moment of crisis, the crucial struggle takes place within humanity itself. If this epidemic results in greater disunity and mistrust among humans, it will be the virus’s greatest victory. When humans squabble – viruses double. In contrast, if the epidemic results in closer global cooperation, it will be a victory not only against the coronavirus, but against all future pathogens” [165].
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
The authors, Dr. Shagufta and Dr. Irshad Ahmad are thankful to the School of Graduate Studies and Research, American University of Ras Al Khaimah, UAE for their support.
References
- 1.https://www.worldometers.info/coronavirus/.
- 2.https://www.who.int/emergencies/diseases/novel-coronavirus-2019/question-and-answers-hub/q-a-detail/q-a-coronaviruses.
- 3.https://www.aarp.org/health/conditions-treatments/info-2020/covid-19-brain-symptoms.html.
- 4.Lai C.-C., Shih T.-P., Ko W.-C., Tang H.-J., Hsueh P.-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int. J. Antimicrob. Agents. 2020;55:105924. doi: 10.1016/j.ijantimicag.2020.105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naicker S., Yang C.-W., Hwang S.-J., Liu B.-C., Chen J.-H., Jha V. The Novel Coronavirus 2019 epidemic and kidneys. Kidney Int. 2020;97:824–828. doi: 10.1016/j.kint.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huang C., Wang Y., Li X., Ren L., Zhao J., Hu Y., Zhang L., Fan G., Xu J., Gu X., Cheng Z., Yu T., Xia J., Wei Y., Wu W., Xie X., Yin W., Li H., Liu M., Xiao Y., Gao H., Guo L., Xie J., Wang G., Jiang R., Gao Z., Jin Q., Wang J., Cao B. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395:497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang C., W Horby P., G Hayden F., F Gao G. A novel coronavirus outbreak of global health concern. Lancet. 2020;395:470–473. doi: 10.1016/S0140-6736(20)30185-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toyoshima Y., Nemoto K., Matsumoto S., Nakamura Y., Kiyotani K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 2020;65:1075–1082. doi: 10.1038/s10038-020-0808-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ejaz H., Alsrhani A., Zafar A., Javed H., Junaid K., E Abdalla A., Abosalif K.O.A., Ahmed Z., Younas S. COVID-19 and comorbidities: deleterious impact on infected patients. J Infect PublicHealth. 2020;13(12):1833–1839. doi: 10.1016/j.jiph.2020.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mueller A.L., McNamara M.S., Sinclair D.A. Why does COVID-19 disproportionately affect older people? Aging (N Y) 2020;12(10):9959–9981. doi: 10.18632/aging.103344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.https://www.euronews.com/2020/09/25/is-europe-having-a-covid-19-second-wave-country-by-country-breakdown.
- 12.https://www.who.int/westernpacific/emergencies/covid-19/technical-guidance/infection-prevention-control.
- 13.Chakraborty I., Maity P. COVID-19 outbreak: migration, effects on society, global environment and prevention. Sci. Total Environ. 2020;728:138882. doi: 10.1016/j.scitotenv.2020.138882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou P., Yang X.L., Wang X.G., Hu B., Zhang L., Zhang W., H R Si, Zhu Y., Li B., Huang C.L., Chen H.D., Chen J., Luo Y., Guo H., Jiang R.D., Liu M.Q., Chen Y., Shen X.R., Wang X., Zheng X.S., Zhao K., Chen Q.J., Deng F., Liu L.L., Yan B., Zhan F.X., Wang Y.Y., Xiao G.F., Shi Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu F., Zhao S., Yu B., Chen Y.M., Wang W., G Song Z., Hu Y., W Tao Z., H Tian J., Y Pei Y., L Yuan M., L Zhang Y., H Dai F., Liu Y., M Wang Q., J Zheng J., Xu L., C Holmes E., Z Zhang Y. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chan J.F., H Kok K., Zhu Z., Chu H., K To K., Yuan S., Y Yuen K. Genomic characterization of the 2019 novel humanpathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microb. Infect. 2020;9(1):221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu R., Zhao X., Li J., Niu P., Yang B., Wu H., Wang W., Song H., Huang B., Zhu N., Bi Y., Ma X., Zhan F., Wang L., Hu T., Zhou H., Hu Z., Zhou W., Zhao L., Chen J., Meng Y., Wang J., Lin Y., Yuan J., Xie Z., Ma J., Liu W.J., Wang D., Xu W., Holmes E.C., Gao G.F., Wu G., Chen W., Shi W., Tan W. Genomic characterization and epidemiology of 2019 novel coronavirusimplications for virus origins and receptor binding. Lancet. 2020;395:565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H., Li X., Li T., Zhang S., Wang L., Wu X., Liu J. The genetic sequence, origin, and diagnosis of SARS-CoV-2. Eur. J. Clin. Microbiol. Infect. Dis. 2020;24:1–7. doi: 10.1007/s10096-020-03899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu A., Peng Y., Huang B., Ding X., Wang X., Niu P., Meng J., Zhu Z., Zhang Z., Wang J., Sheng J., Quan L., Xia Z., Tan W., Cheng G., Jiang T. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host Microbe. 2020;27(3):325–328. doi: 10.1016/j.chom.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xiu S., Dick A., Ju H., Mirzaie S., Abdi F., Cocklin S., Zhan P., Liu X. Inhibitors of SARS-CoV-2 entry: current and future opportunities. J. Med. Chem. 2020;63(21):12256–12274. doi: 10.1021/acs.jmedchem.0c00502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gil C., Ginex T., Maestro I., Nozal V., B Gil L., Geijo M.A.C., Urquiza J., Ramírez D., Alonso C., E Campillo N., Martinez A. COVID-19: drug targets and potential treatments. J. Med. Chem. 2020;63(21):12359–12386. doi: 10.1021/acs.jmedchem.0c00606. [DOI] [PubMed] [Google Scholar]
- 22.Shagufta, Ahmad I., Mathew S., Rehman S. 2020. Recent progress in selective estrogen receptor downregulators (SERDs) for the treatment of breast cancer. RSC Medicinal Chemistry. 2020;11:438–454. doi: 10.1039/c9md00570f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shagufta, Ahmad I. Transition metal complexes as proteasome inhibitors for cancer treatment. Inorg. Chim. Acta. 2020;506:119521. [Google Scholar]
- 24.Shagufta, Ahmad I. Tamoxifen a pioneering drug: an update on the therapeutic potential of tamoxifen derivatives. Eur. J. Med. Chem. 2018;143:515–531. doi: 10.1016/j.ejmech.2017.11.056. [DOI] [PubMed] [Google Scholar]
- 25.Shagufta, Ahmad I. An insight into the therapeutic potential of quinazoline derivatives as anticancer agents. MedChemComm. 2017;8:871–885. doi: 10.1039/c7md00097a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shagufta, Ahmad I., Panda G. Quest for steroidomimetics: amino acids derived steroidal and nonsteroidal architectures. Eur. J. Med. Chem. 2017;133:139–151. doi: 10.1016/j.ejmech.2017.03.054. [DOI] [PubMed] [Google Scholar]
- 27.Shagufta, Ahmad I. Recent insight into the biological activities of synthetic xanthone derivatives. Eur. J. Med. Chem. 2016;116:267–280. doi: 10.1016/j.ejmech.2016.03.058. [DOI] [PubMed] [Google Scholar]
- 28.Ahmad I., Shagufta Recent developments in steroidal and nonsteroidal aromatase inhibitors for the chemoprevention of estrogen-dependent breast cancer. Eur. J. Med. Chem. 2015;102:375–386. doi: 10.1016/j.ejmech.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 29.Ahmad I., Shagufta Sulfones: an important class of organic compounds with diverse biological activities. Int. J. Pharm. Pharmaceut. Sci. 2015;7(3):19–27. [Google Scholar]
- 30.Boopathi S., B Poma A., Kolandaivel P. Novel 2019 coronavirus structure, mechanism of action, antiviral drug promises and rule out against its treatment. J. Biomol. Struct. Dynam. 2020:1–10. doi: 10.1080/07391102.2020.1758788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Huang Y., Yang C., Xu X.-F., Xu W., S-W Liu Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020;41:1141–1149. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Duan Y., Yao Y., A Kumar S., H-L Zhu, Chang J. Current and future therapeutical approaches for COVID19. Drug Discov. Today. 2020;25(8):1545–1552. doi: 10.1016/j.drudis.2020.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng L., Zhang L., Huang J., S Nandakumar K., Liu S., Cheng K. Potential treatment methods targeting 2019-nCoV infection. Eur. J. Med. Chem. 2020;205:112687. doi: 10.1016/j.ejmech.2020.112687. 10.1016/j.ejmech.2020.112687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ghosh A.K., Brindisi M., Shahabi D., E Chapman M., D Mesecar A. Drug development and medicinal chemistry efforts toward SARS-coronavirus and covid-19 therapeutics. ChemMedChem. 2020;15:907–932. doi: 10.1002/cmdc.202000223. doi.org/10.1002/cmdc.202000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Antonio A.S., Wiedemann L.S.M., Junior V.F.V. Natural products’ role against COVID-19. RSC Adv. 2020;10:23379–23393. doi: 10.1039/d0ra03774e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kouznetsov V.V. COVID-19 treatment: much research and testing, but far, few magic bullets against SARS-CoV-2 coronavirus. Eur. J. Med. Chem. 2020;203:112647. doi: 10.1016/j.ejmech.2020.112647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sivasankarapillai V.S., M Pillai A., Rahdar A., P Sobha A., S Das S., C Mitropoulos A., H Mokarrar M., Z Kyzas G. On facing the SARS-CoV-2 (COVID-19) with combination of nanomaterials and medicine: possible strategies and first challenges. Nanomaterials. 2020;10:852. doi: 10.3390/nano10050852. doi:10.3390/nano10050852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salvi R., Patankar P. Emerging pharmacotherapies for COVID 19. Biomed. Pharmacother. 2020;128:110267. doi: 10.1016/j.biopha.2020.110267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amin S.A., Jha T. Fight against novel coronavirus: a perspective of medicinal chemists. Eur. J. Med. Chem. 2020;201:112559. doi: 10.1016/j.ejmech.2020.112559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Korkmaz B., Lesner A., M Adam S., Moss C., E Jenne D. Lung protection by cathepsin C inhibition: a new hope for COVID-19 and ARDS? J. Med. Chem. 2020;63(22):13258–13265. doi: 10.1021/acs.jmedchem.0c00776. [DOI] [PubMed] [Google Scholar]
- 41.Liang C., Tian L., Liu Y., Hui N., Qiao G., Li H., Shi Z., Tang Y., Zhang D., Xie X., Zhao X. A promising antiviral candidate drug for the COVID-19 pandemic: a mini-review of remdesivir. Eur. J. Med. Chem. 2020;201:112527. doi: 10.1016/j.ejmech.2020.112527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akhtar M.J. COVID19 inhibitors: a prospective therapeutics. Bioorg. Chem. 2020;101:104027. doi: 10.1016/j.bioorg.2020.104027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Catanzaro M., Fagiani F., Racchi M., Corsini E., Govoni S., Lanni C. Immune response in COVID-19: addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduction and Targeted Therapy. 2020;84:5. doi: 10.1038/s41392-020-0191-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saxena A. Drug targets for COVID-19 therapeutics: ongoing global efforts. J. Biosci. 2020;45:87. doi: 10.1007/s12038-020-00067-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morse J.S., Lalonde T., Xu S., R Liu W. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. Chembiochem. 2020;21(5):730–738. doi: 10.1002/cbic.202000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robertson N.S., R Spring D. Using peptidomimetics and constrained peptides as valuable tools for inhibiting protein−protein interactions. Molecules. 2018;23:959. doi: 10.3390/molecules23040959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gurevich E.V., V Gurevich V. Therapeutic potential of small molecules and engineered proteins. Handb. Exp. Pharmacol. 2014;219:1–12. doi: 10.1007/978-3-642-41199-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ngo H.X., Garneau-Tsodikova S. What are the drugs of the future? MedChemComm. 2018;9:757–758. doi: 10.1039/c8md90019a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Donoghue M., Hsieh F., Baronas E., Godbout K., Gosselin M., Stagliano N., Donovan M., Woolf B., Robison K., Jeyaseelan R., Breitbart B.E., Acton S. A novel angiotensin-converting enzyme−related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1−9. Circ. Res. 2000;87:e1–e9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 50.Zhang H., Penninger J.M., Li Y., Zhong N., Slutsky A.S. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46:586–590. doi: 10.1007/s00134-020-05985-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hoffmann M., Kleine-Weber H., Schroeder S., Kruger N., Herrler T., Erichsen S., S Schiergens T., Herrler G., H Wu N., Nitsche A., A Muller M., Drosten C., Pohlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yan R., Zhang Y., Li Y., Xia L., Guo Y., Zhou Q. Structural basis for the recognition of the SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448. doi: 10.1126/science.abb2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gao J., Tian Z., Yang X. Breakthrough: chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. BioSci. Trends. 2020;14:72–73. doi: 10.5582/bst.2020.01047. [DOI] [PubMed] [Google Scholar]
- 55.Vincent M.J., Bergeron E., Benjannet S., R Erickson B., E Rollin P., G Ksiazek T., G Seidah N., T Nichol S. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005;2:69. doi: 10.1186/1743-422X-2-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in Vitro. Cell Res. 2020;30:269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yao X., Ye F., Zhang M., Cui C., Huang B., Niu P., Liu X., Zhao L., Dong E., Song C., Zhan S., Lu R., Li H., Tan W., Liu D. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020. [DOI] [PMC free article] [PubMed]
- 58.Lan L., Xu D., Ye G., Xia C., Wang S., Li Y., Xu H. Positive RT-PCR test results in patients recovered from COVID-19. J. Am. Med. Assoc. 2020;323(15):1502–1503. doi: 10.1001/jama.2020.2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gautret P., Lagier J.-C., Parola P., Hoang V.T., Meddeb L., Mailhe M., Doudier B., Courjon J., Giordanengo V., Vieira V.E., Dupont H.T., Honore S., Colson P., Chabriere E., La Scola B., Rolain J.-M., Brouqui P., Raoult D. Hydroxychloroquine and azithromycin as a treatment of COVID-19: results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents. 2020 doi: 10.1016/j.ijantimicag.2020.105949. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60.Chen J., Liu D., Liu L., Liu P., Xu Q., Xia L., Ling Y., Huang D., Song S., Zhang D., Qian Z., Li T., Shen Y., Lu H. A pilot study of hydroxychloroquine in treatment of patients with moderate COVID-19. J. Zhejiang Univ. 2020;49:215–219. doi: 10.3785/j.issn.1008-9292.2020.03.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen Z., Hu J., Zhang Z., Jiang S., Han S., Yan D., Zhuang R., Hu B., Zhang Z. Efficacy of hydroxychloroquine in patients with COVID-19: results of a randomized clinical trial. MedRxiv. 2020 doi: 10.1101/2020.03.22.20040758. [DOI] [Google Scholar]
- 62.Magagnoli J., Narendran S., Pereira F., Cummings T.H., Hardin J.W., Sutton S.S., Ambati J. Outcomes of hydroxychloroquine usage in United States veterans hospitalized with COVID-19. Med. 2020;1(1):114–127. doi: 10.1016/j.medj.2020.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Geleris J., Sun Y., Platt J., Zucker J., Baldwin M., Hripcsak G., Labella A., Manson D.K., Kubin C., Barr R.G., Sobieszczyk M.E., Schluger N.W. Observational study of hydroxychloroquine in hospitalized patients with covid-19. N. Engl. J. Med. 2020;382(25):2411–2418. doi: 10.1056/nejmoa2012410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen C., Wang F., Lin C. Chronic hydroxychloroquine use associated with qt prolongation and refractory ventricular arrhythmia. Clin. Toxicol. 2006;44:173–175. doi: 10.1080/15563650500514558. [DOI] [PubMed] [Google Scholar]
- 65.Stas P., Faes D., Noyens P. Conduction disorder and qt prolongation secondary to long-term treatment with chloroquine. Int. J. Cardiol. 2008;127:e80–e82. doi: 10.1016/j.ijcard.2007.04.055. [DOI] [PubMed] [Google Scholar]
- 66.Yaylali S.A., Sadigov F., Erbil H., Ekinci A., A Akcakaya A. Chloroquine and hydroxychloroquine retinopathy-related risk factors in a turkish cohort. Int. Ophthalmol. 2013;33:627–634. doi: 10.1007/s10792-013-9748-0. [DOI] [PubMed] [Google Scholar]
- 67.Coronavirus (COVID-19) Update: FDA revokes emergency use authorization for chloroquine and hydroxychloroquine. 2020. https://www.fda.gov/news-events/press-announcements/coronavirus-covid-19-update-fda-revokes-emergency-use-authorization-chloroquine-and
- 68.Q&A : Hydroxychloroquine and COVID-19. 2020. https://www.who.int/news-room/q-a-detail/q-a-hydroxychloroquine-and-covid-19
- 69.Huentelman M.J., Zubcevic J., Prada J.A.H., Xiao X., Dimitrov D.S., Raizada M.K., Ostrov D.A. Structure-based discovery of a novel angiotensin-converting enzyme 2 inhibitor. Hypertension. 2004;44:903–906. doi: 10.1161/01.HYP.0000146120.29648.36. [DOI] [PubMed] [Google Scholar]
- 70.Lambert D.W., Yarski M., J Warner F., Thornhill P., T Parkin E., I Smith A., M Hooper N., J Turner A. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2) J. Biol. Chem. 2005;280:30113–30119. doi: 10.1074/jbc.M505111200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Haga S., Nagata N., Okamura T., Yamamoto N., Sata T., Yamamoto N., Sasazuki T., Ishizaka Y. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antivir. Res. 2010;85:551–555. doi: 10.1016/j.antiviral.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Towler P., Staker B., G Prasad S., Menon S., Tang J., Parsons T., Ryan D., Fisher M., Williams D., A Dales N., A Patane M., W Pantoliano M. ACE2 X-ray structures reveal a large hingebending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004;279:17996–18007. doi: 10.1074/jbc.M311191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Milewska A., Ciejka J., Kaminski K., Karewicz A., Bielska D., Zeglen S., Karolak W., Nowakowska M., Potempa J., J Bosch B., Pyrc K., Szczubialka K. Novel polymeric inhibitors of HCoV-NL63. Antivir. Res. 2013;97:112–121. doi: 10.1016/j.antiviral.2012.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Milewska A., Kaminski K., Ciejka J., Kosowicz K., Zeglen S., Wojarski J., Nowakowska M., Szczubiałka K., Pyrc K. HTCC: broad range inhibitor of coronavirus entry. PloS One. 2016:11. doi: 10.1371/journal.pone.0156552. e0156552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Diaz J.H. Hypothesis: angiotensin-converting enzyme inhibitors and angiotensin receptor blockers may increase the risk of severe COVID-19. J. Trav. Med. 2020;(3):27. doi: 10.1093/jtm/taaa041. No. taaa041. (doi: 10.1093/jtm/taaa041) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chary M.A., F Barbuto A., Izadmehr S., D Hayes B., M Burns M. COVID-19: therapeutics and their toxicities. J. Med. Toxicol. 2020;16(3):282–294. doi: 10.1007/s13181-020-00777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rothlin R.P., M Vetulli H., Duarte M., G Pelorosso F. Telmisartan as tentative angiotensin receptor blocker therapeutic for COVID-19. Drug Dev. Res. 2020 doi: 10.1002/ddr.21679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Duarte M., Pelorosso F.G., Nicolosi L., Salgado M.V., Vetulli H., Aquieri A., Azzato F., asconcel M.B., Castro M., Coyle J., Davolos I., Esparza E., Criado I.F., Gregori R., Mastrodonato P., Rubio M., Sarquis S., Wahlmann F., Rothlin R.P. Telmisartan for treatment of Covid-19 patients: an open randomized clinical trial. Preliminary report. medRxiv. 2020 doi: 10.1101/2020.08.04.20167205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.V Monteil H.K., Patricia P., Astrid H., Reiner A., S Wimmer M., Alexandra L., Elena G., P Carmen H., Felipe P., P Romero J., et al. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell Press. 2020;181(4):905–913. doi: 10.1016/j.cell.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang H., M Penninger J., Li Y., Zhong N., S Slutsky A. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med. 2020;46(4):586–590. doi: 10.1007/s00134-020-05985-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Khan A., Benthin C., Zeno B., E Albertson T., Boyd J., D Christie J., Hall R., Poirier G., J Ronco J., Tidswell M., et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care. 2017;21:234. doi: 10.1186/s13054-017-1823-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wösten-Van Asperen R.M., Lutter R., A Specht P., N Moll G., B van Woensel J., M van der Loos C., van Goor H., Kamilic J., Florquin S., P Bos A. Acute respiratory distress syndrome leads to reduced ratio of ACE/ACE2 activities and is prevented by angiotensin-(1−7) or an angiotensin II receptor antagonist. J. Pathol. 2011;225(4):618–627. doi: 10.1002/path.2987. [DOI] [PubMed] [Google Scholar]
- 83.Haschke M., Schuster M., Poglitsch M., Loibner H., Salzberg M., Bruggisser M., Penninger J., Krahenbuhl S. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin-converting enzyme 2 in healthy human subjects. Clin. Pharmacokinet. 2013;52(9):783–792. doi: 10.1007/s40262-013-0072-7. [DOI] [PubMed] [Google Scholar]
- 84.Thiel V., A Ivanov K., Putics A., Hertzig T., Schelle B., Bayer S., Weissbrich B., J Snijder E., Rabenau H., W Doerr H., E Gorbalenya A., Ziebuhr J. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84(9):2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 85.Hilgenfeld R. From SARS to MERS: crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014;281(18):4085–4096. doi: 10.1111/febs.12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee T.W., M Cherney M., Huitema C., Liu J., E James K., C Powers J., D Eltis L., James M.N. Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like azapeptide epoxide. J. Mol. Biol. 2005;353(5):1137–1151. doi: 10.1016/j.jmb.2005.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Morse J.S., Lalonde T., Xu S., R Liu W. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. Chembiochem. 2020;21(5):730–738. doi: 10.1002/cbic.202000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee H., Cao S., E Hevener K., Truong L., L Gatuz J., Patel K., K Ghosh A., E Johnson M. Synergistic inhibitor binding to the papain-like protease of human SARS coronavirus: mechanistic and inhibitor design implications. ChemMedChem. 2013;8(8):1361–1372. doi: 10.1002/cmdc.201300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ghosh A.K., Brindisi M., Shahabi D., Chapman M.E., D Mesecar A. Drug development and medicinal chemistry efforts toward SARS-coronavirus and Covid-19 therapeutics. ChemMedChem. 2020;15:907. doi: 10.1002/cmdc.202000223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pillaiyar T., Manickam M., Namasivayam V., Hayashi Y., Jung S.H. An overview of severe acute respiratory Syndrome−Coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J. Med. Chem. 2016;59:6595–6628. doi: 10.1021/acs.jmedchem.5b01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nukoolkarn V., S Lee V., Malaisree M., Aruksakulwong O., Hannongbua S. Molecular dynamic simulations analysis of ritronavir and lopinavir as SARS-CoV 3CLpro inhibitors. J. Theor. Biol. 2008;254(4):861–867. doi: 10.1016/j.jtbi.2008.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nutho B., Mahalapbutr P., Hengphasatporn K., C Pattaranggoon N., Simanon N., Shigeta Y., Hannongbua S., Rungrotmongkol T. Why are lopinavir and ritonavir effective against the NewlyEmerged coronavirus 2019? Atomistic insights into the inhibitory mechanisms. Biochemistry. 2020;59(18):1769–1779. doi: 10.1021/acs.biochem.0c00160. [DOI] [PubMed] [Google Scholar]
- 93.Choy K.T., Y Wong A., Kaewpreedee P., et al. Remdesivir, lopinavir, emetine,and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 178. 2020;104786 doi: 10.1016/j.antiviral.2020.104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ye X.T., L Luo Y., C Xia S., et al. Clinical efficacy of lopinavir/ritonavir in the treatment of Coronavirus disease. Eur. Rev. Med. Pharmacol. Sci. 2020;24(6):3390–3396. doi: 10.26355/eurrev_202003_20706. [DOI] [PubMed] [Google Scholar]
- 95.Cao B., Wang Y., Wen D., Liu W., Wang J., Fan G., Ruan L., Song B., Cai Y., Wei M., Li X., Xia J., Chen N., Xiang J., Yu T., Bai T., Xie X., Zhang L., Li C., Yuan Y., Chen H., Li H., Huang H., Tu S., Gong F., Liu Y., Wei Y., Dong C., Zhou F., Gu X., Xu J., Liu Z., Zhang Y., Li H., Shang L., Wang K., Li K., Zhou X., Dong X., Qu Z., Lu S., Hu X., Ruan S., Luo S., Wu J., Peng L., Cheng F., Pan L., Zou J., Jia C., Wang J., Liu X., Wang S., Wu X., Ge Q., He J., Zhan H., Qiu F., Guo L., Huang C., Jaki T., G Hayden F., W Horby P., Zhang D., Wang C. A trial of lopinavir-ritonavir in adults hospitalized with severe covid-19. N. Engl. J. Med. 2020;382(19):1787–1799. doi: 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.N Hung I.F., C Lung K., Tso E.Y.K., Liu R., Chung T.W.H., Y Chu M., Y Ng Y., Lo J., Chan J., R Tam A., P Shum H., Chan V., Wu A.K.L., M Sin K., S Leung W., L Law W., C Lung D., Sin S., Yeung P., Yip C.C.Y., R Zhang R., Fung A.Y.F., Yan E.Y.W., H Leung K., D Ip J., Chu A.W.H., M Chan W., Ng A.C.K., Lee R., Fung K., Yeung A., C Wu T., Chan J.W.M., W Yan W., M Chan W., Chan J.F.W., Lie A.K.W., Tsang O.T.Y., Cheng V.C.C., L Que T., S Lau C., H Chan K., To K.K.W., Y Yuen K. Triple combination of interferon beta-1b, lopinavireritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet. 2020:31042–31044. doi: 10.1016/S0140-6736(20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.WHO discontinues hydroxychloroquine and lopinavir/ritonavir treatment arms for Covid-19. 2020. https://www.who.int/news/item/04-07-2020-who-discontinues-hydroxychloroquine-and-lopinavir-ritonavir-treatment-arms-for-covid-19
- 98.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L.W., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of M(pro) from COVID-19 virus and discovery of its inhibitors. Nature. 2020;582:289. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- 99.Dai W., Zhang B., Su H., et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368(6497):1331–1335. doi: 10.1126/science.abb4489. eabb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. 2020;368(6489):409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang L., Lin D., Kusov Y., Nian Y., Ma Q., Wang J., von Brunn A., Leyssen P., Lanko K., Neyts J., de Wilde A., J Snijder E., Liu H., Hilgenfeld R. Alpha-Ketoamides as broad-spectruminhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J. Med. Chem. 2020;63(9):4562–4578. doi: 10.1021/acs.jmedchem.9b01828. [DOI] [PubMed] [Google Scholar]
- 102.Zhou L., Liu Y., Zhang W.L., Wei P., Huang C.K., Pei J.F., Yuan Y.X., Lai L.H. Isatin compounds as noncovalent SARS coronavirus 3C-like protease inhibitors. J. Med. Chem. 2006;49:3440–3443. doi: 10.1021/jm0602357. [DOI] [PubMed] [Google Scholar]
- 103.Liu P., Liu H., Sun Q., Liang H., Li C., Deng X., Liu Y., Lai L. Potent inhibitors of SARS-CoV-2 3C-like protease derived from N-substituted isatin compounds. Eur. J. Med. Chem. 2020;206:112702. doi: 10.1016/j.ejmech.2020.112702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gallagher T.M., J Buchmeier M. Coronavirus spike proteins in viral entry and pathogenesis. Virology. 2001;279(2):371–374. doi: 10.1006/viro.2000.0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., Xiang Z., Mu Z., Chen X., Chen J., Hu K., Jin Q., Wang J., Qian Z. Characterization of spike glycoprotein of SARSCoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020;11(1):1620. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Watanabe Y., D Allen J., Wrapp D., S McLellan J., Crispin M. Site specific glycan analysis of the SARS-CoV-2 spike. Science. 2020;369:330–333. doi: 10.1126/science.abb9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang Y., Yang C., Xu X.F., X W., Liu S.W. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020;41:1141–1149. doi: 10.1038/s41401-020-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang Q., Zhang Y., Wu L., Niu S., Song Chunli, Zhang Z., Lu G., Qiao C., Hu Y., Yuen K.-Y., Wang Q., Zhou H., Yan J., Qi J. Structural and Functional Basis Of SARS-CoV-2 entry by using human ACE2. Cell. 2020;181(4):894–904. doi: 10.1016/j.cell.2020.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gralinski L.E., D Menachery V. Return of the coronavirus: 2019-nCoV. Viruses. 2020;12(2):135. doi: 10.3390/v12020135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zhou P., L Yang X., G Wang X., Hu B., Zhang L., A Zhang W. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- 112.Wrapp D., Wang N., S Corbett K., A Goldsmith J., L Hsieh C., Abiona O., et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263. doi: 10.1126/science.abb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Walls A.C., J Park Y., A Tortorici M., Wall A., T McGuire A., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181:281–286. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Adedeji A.O., Severson W., Jonsson C., Singh K., R Weiss S., G Sarafianos S. Novel inhibitors of severe acute respiratory syndrome coronavirus entry that act by three distinct mechanisms. J. Virol. 2013;87:8017–8028. doi: 10.1128/JVI.00998-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lundin A., Dijkman R., Bergström T., Kann N., Adamiak B., Hannoun C., Kindler E., Jónsdóttir H.R., Muth D., Kint J., Forlenza M., Müller M.A., Drosten C., Thiel V., Trybala E. Targeting membrane-bound viral RNA synthesis reveals potent inhibition of diverse coronaviruses including the Middle East respiratory syndrome virus. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1004166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Alexpandi R., Mesquita J.F.D., K Pandian S., V Ravi A. Quinolines-based SARS-CoV-2 3CLpro and RdRp inhibitors and spike-RBD-ACE2 inhibitor for drug-repurposing against COVID-19: an in silico analysis. Front. Microbiol. 2020;1796:11. doi: 10.3389/fmicb.2020.01796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bosch B.J., Martina B.E.E., van der Zee R., Lepault J., J Haijema B., Versluis C., Heck A.J.R., de Groot R., E Osterhaus A.D.M., Rottier P.J.M. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl. Acad. Sci. USA. 2004;101:8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Deng Y., Liu J., Zheng Q., Yong W., Lu M. Structures and polymorphic interactions of two heptad-repeat regions of the SARS virus S2 protein. Structure. 2006;14:889–899. doi: 10.1016/j.str.2006.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ho M. Perspectives on the development of neutralizing antibodies against SARS-CoV-2. Antib Ther. 2020;3(2):109–114. doi: 10.1093/abt/tbaa009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Yi L., Li Z., Yuan K., Qu X., Chen J., Wang G., Zhang H., Luo H., Zhu L., Jiang P., Chen L., Shen Y., Luo M., Zuo G., Hu J., Duan D., Nie Y., Shi X., Wang W., Han Y., Li T., Liu Y., Ding M., Deng H., Xu X. Small molecules blocking the entry of severe acute respiratory syndrome coronavirus into host cells. J. Virol. 2004;78:11334–11339. doi: 10.1128/JVI.78.20.11334-11339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Greenough T.C., J Babcock G., Roberts A., J Hernandez H., Jr W.D., Thomas J., Coccia A., F Graziano R., Srinivasan M., Lowy I., W Finberg R., Subbarao K., Vogel L., Somasundaran M., Luzuriaga K., L Sullivan J., M Ambrosino D. Development and characterization of a severe acute respiratory syndrome-associated coronavirus-neutralizing human monoclonal antibody that provides effective immunoprophylaxis in mice. J. Infect. Dis. 2005;191:507–514. doi: 10.1086/427242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhao G., Du L., Ma C., Li Y., Li L., K Poon V., Wang L., Yu F., J Zheng B., Jiang S., Zhou Y. A safe and convenient pseudovirus-based inhibition assay to detect neutralizing antibodiesand screen for viral entry inhibitors against the novel human coronavirus MERS-CoV. Virol. J. 2013;10:266. doi: 10.1186/1743-422X-10-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kadam R.U., A Wilson I. Structural basis of influenza virus fusion inhibition by the antiviral drug Arbidol. Proc. Natl. Acad. Sci. U. S. A. 2017;114:206–214. doi: 10.1073/pnas.1617020114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wang X., Cao R., Zhang H., Liu J., Xu M., Hu H., Li Y., Zhao L., Li W., Sun X., Yang X., Shi Z., Deng F., Hu Z., Zhong W., Wang M. The anti-influenza virus drug, Arbidol is an efficientinhibitor of SARS-CoV-2 in vitro. Cell Discovery. 2020;6:28. doi: 10.1038/s41421-020-0169-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vankadari N. Arbidol: a Potential antiviral drug for the treatment of SARS-CoV-2 by blocking trimerization of the spike glycoprotein. Int. J. Antimicrob. Agents. 2020;56:105998. doi: 10.1016/j.ijantimicag.2020.105998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Dana D., K Pathak S. A review of small molecule inhibitors and functional probes of human cathepsin L. Molecules. 2020;25:698. doi: 10.3390/molecules25030698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhou Y., Vedantham P., Lu K., Agudelo J., Carrion R., Jr., Nunneley J.W., Barnard D., Pohlmann S., McKerrow J.H., Renslo A.R., Simmons G. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 2015;116:76–84. doi: 10.1016/j.antiviral.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., Xiang Z., Mu Z., Chen X., Chen J., Hu K., Jin Q., Wang J., Qian Z. Characterization of spike glycoprotein of SARSCoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020;11(1):1620. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu T., Luo S., Libby P., G-P Shi Cathepsin L-selective inhibitors: a potentially promising treatment for COVID-19 patients. Pharmacol. Ther. 2020;213:107587. doi: 10.1016/j.pharmthera.2020.107587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Shah P.P., Wang T., L Kaletsky R., C Myers M., E Purvis J., Jing H., M Huryn D., C Greenbaum D., B Smith A., Bates P., L Diamond S. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 2010;78:319–324. doi: 10.1124/mol.110.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Ou X., Liu Y., Lei X., Li P., Mi D., Ren L., Guo L., Guo R., Chen T., Hu J., Xiang Z., Mu Z., Chen X., Chen J., Hu K., Jin Q., Wang J., Qian Z. Characterization of spike glycoprotein ofSARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun. 2020;1620:11. doi: 10.1038/s41467-020-15562-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Simmons G., N Gosalia D., J Rennekamp A., D Reeves J., L Diamond S., Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. U. S. A. 2005;102:11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhou Y., Vedantham P., Lu K., Agudelo J., Carrion R., Jr., W Nunneley J., Barnard D. S Pöhlmann, J H McKerrow, A R Renslo, G Simmons. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res. 2015;116:76–84. doi: 10.1016/j.antiviral.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mollica V., Rizzo A., Massari F. The pivotal role of TMPRSS2 in coronavirus disease 2019 and prostate cancer. Future Oncol. 2020;16(27):2029–2033. doi: 10.2217/fon-2020-0571. doi: 10.2217/fon-2020-0571) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Iwata-Yoshikawa N., Okamura T., Shimizu Y., Hasegawa H., Takeda M., Nagata N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J. Virol. 2019:e01815–e01818. doi: 10.1128/JVI.01815-18. 93(6), No. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Vaarala M.H., S Porvari K., Kellokumpu S., P Kyllonen A., Vihko P.T. Expression of transmembrane serine protease TMPRSS2 in mouse and human tissues. J. Pathol. 2001:193. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH743>3.0.CO;2-T. (1),134−140. [DOI] [PubMed] [Google Scholar]
- 137.Kawase M., Shirato K., van der Hoek L., Taguchi F., Matsuyama S. Simultaneous treatmentof human bronchial epithelial cells with serine and cysteine protease inhibitors prevents severe acute respiratory syndrome coronavirus entry. J. Virol. 2012;86:6537–6545. doi: 10.1128/JVI.00094-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Walls A.C., J Park Y., A Tortorici M., Wall A., T McGuire A., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Coutard B., Valle C., de Lamballerie X., Canard B., G Seidah N., Decrol E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020;176:104742. doi: 10.1016/j.antiviral.2020.104742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Millet J.K., Whittaker G.R. Host cell proteases: critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015;202:120–134. doi: 10.1016/j.virusres.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hasan A., A Paray B., Hussain A., A Qadir F., Attar F., M Aziz F., Sharifi M., Derakhshankhah H., Rasti B., Mehrabi M., Shahpasand K., A Saboury A., Falahati M. A review on the cleavage priming of the spike protein on coronavirus by angiotensin-converting enzyme-2 and furin. J. Biomol. Struct. Dyn. 2020 doi: 10.1080/07391102.2020.1754293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Usul Afsar C. 2019-nCoV-SARS-CoV-2 (COVID-19) infection: Cruciality of Furin and relevance with cancer. Med. Hypotheses. 2020;140:109770. doi: 10.1016/j.mehy.2020.109770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Imran M., K Saleemi M., Chen Z., Wang X., Zhou D., Li Y., Zhao Z., Zheng B., Li Q., Cao S., Ye J. Decanoyl-arg-val-lys-arg-chloromethylketone: an antiviral compound that acts against flaviviruses through the inhibition of furin-mediated prM cleavage. Viruses. 2019;11(11):1011. doi: 10.3390/v11111011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Millet J.K., R Whittaker G. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. U. S. A. 2014;111:15214–15219. doi: 10.1073/pnas.1407087111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bestle D., R Heindl M., Limburg H., van T.V.L., Pilgram O., Moulton H., Stein D.A., Hardes K., Eickmann M., Dolnik O., Rohde C., Klenk H.D., Garten W., Steinmetzer T., Böttcher-Friebertshäuser E. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance. 2020 Jul 23;3(9) doi: 10.26508/lsa.202000786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.A J W te Velthuis, Arnold J.J., Cameron C.E., van den Worm S.H.E., Snijder E.J. The RNA polymerase activity of SARS-coronavirus nsp12 is primer dependent Nucleic Acids Res. 2010;38(1):203–214. doi: 10.1093/nar/gkp904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.McDonald S.M. RNA synthetic mechanisms employed by diverse families of RNA viruses. WIREs RNA. 2013;4(4):351–367. doi: 10.1002/wrna.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Chan J.F., H Kok K., Zhu Z., Chu H., K To K., Yuan S., Y Yuen K. Genomic characterization of the 2019 novel humanpathogenic coronavirus isolated from a patient with a typical pneumonia after visiting Wuhan. Emerg. Microb. Infect. 2020;9(1):221–236. doi: 10.1080/22221751.2020.1719902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L., Ge J., Zheng L., Zhang Y., Wang H., Zhu Y., Zhu C., Hu T., Hua T., Zhang B., Yang X., Li J., Yang H., Liu Z., Xu W., W Guddat L., Wang Q., Lou Z., Rao Z. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020;368(6492):779–782. doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Letko M., Marzi A., Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020;5:562e569. doi: 10.1038/s41564-020-0688-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sheahan T.P., C Sims A., L Graham R., D Menachery V., E Gralinski L., B Case J., R Leist S., Pyrc K., Y Feng J., Trantcheva I., Bannister R., Park Y., Babusis D., O Clarke M., L Mackman R., E Spahn J., A Palmiotti C., Siegel D., S Ray A., Cihlar T., Jordan R., R Denison M., S Baric R. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017:9. doi: 10.1126/scitranslmed.aal3653. 396), No. eaal3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Agostini M.L., L Andres E., C Sims A., L Graham R., P Sheahan T., Lu X., Smith E.C., B Case J., Y Feng J., Jordan R., S Ray A., Cihlar T., Siegel D., L Mackman R., O Clarke M., S Baric R., R Denison M. Coronavirus susceptibility to the antiviral Remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio. 2018;9(2):e00221–18. doi: 10.1128/mBio.00221-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Beigel J.H., M Tomashek K., E Dodd L., K Mehta A., S Zingman B., C Kalil A., Hohmann E., Y Chu H., Luetkemeyer A., Kline S., Lopez de Castilla D., W Finberg R., Dierberg K., Tapson V., Hsieh L., F Patterson T., Paredes R., A Sweeney D., R Short W., Touloumi G., C Lye D., Ohmagari N., Oh M., M Ruiz-Palacios G., Benfield T., Fatkenheuer G., G Kortepeter M., L Atmar R., B Creech C., Lundgren J., G Babiker A., Pett S., D Neaton J., H Burgess T., Bonnett T., Green M., Makowski M., Osinusi A., Nayak S., C Lane H. Remdesivir for the treatment of covid-19 d preliminary report, N. Engl. J. Med. 2020;383(19):1813–1826. doi: 10.1056/nejmoa2007764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Gilead Announces Results from Phase 3 Trial of Investigational Antiviral Remdesivir in Patients with Severe Covid-19 2020. https://www.gilead.com/news-and-press/press-room/press-releases/2020/4/gilead-announcesresults-from-phase-3-trial-of-investigational-antiviral-remdesivir-in-patients-with-severe-covid-19
- 156.Gilead’s Suspension of Covid-19 Trials in China Should Serve as a Bellwether for Studies in Other Countries. Clin. Trials Arena. 2020 https://www.clinicaltrialsarena.com/comment/gilead-remdesivir-covid-19-china-trials/ [Google Scholar]
- 157.Montefiore and Einstein Assess Remdesivir and Baricitinib for Covid-19. Clin. Trials Arena. 2020 https://www.clinicaltrialsarena.com/news/remdesivirplus-baricitinib-covid-19/ [Google Scholar]
- 158.Global Coronavirus COVID-19 Clinical Trial Tracker. 2020. https://www.covid-trials.org/
- 159.Singh A.K., Singh A., Singh R., Misra A. Remdesivir in COVID-19: a critical review of pharmacology, pre-clinical and clinical studies, Diabetes Metab. Syndr. Clin. Res. Rev. 2020;14(4):641–648. doi: 10.1016/j.dsx.2020.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.WHO recommends against the use of remdesivir in Covid-19 patients. 2020. https://www.who.int/news-room/feature-stories/detail/who-recommends-against-the-use-of-remdesivir-in-covid-19-patients
- 161.Oestereich L., Lüdtke A., Wurr S., Rieger T., Mu noz-Fontela C., Günther S. Successful treatment of advanced Ebola virus infection with T-705 (favipiravir) in a small animal model. Antivir. Res. 2014;105:17–21. doi: 10.1016/j.antiviral.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 162.Furuta Y., Komeno T., Nakamura T. Favipiravir (T-705), a broad spectrum inhibitor of viral RNA polymerase. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2017;93(7):449–463. doi: 10.2183/pjab.93.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Clinical Study To Evaluate The Performance And Safety Of Favipiravir in Covid-19. 2020. https://clinicaltrials.gov/ct2/show/NCT04336904
- 164.Ahamad S., Branch S., Harrelson S., K Hussain M., Saquib M., Khan S. Primed for global coronavirus pandemic: emerging research and clinical outcome. Eur. J. Med. Chem. 2021;209:112862. doi: 10.1016/j.ejmech.2020.112862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.In the Battle Against Coronavirus, Humanity Lacks Leadership. 2020. https://time.com/5803225/yuval-noah-harari-coronavirus-humanity-leadership/