

Abstract
Gastrointestinal stromal tumors (GIST) are the most common mesenchymal tumor of the gut. GIST are thought to arise solely from interstitial cells of Cajal (ICC), a KIT-positive population that controls gut motility. Activating gain-of-function mutations in KIT and PDGFRA are the most frequent driver events, and most of these tumors are responsive to the tyrosine kinase inhibitor imatinib. Less common drivers include mutant BRAFV600E and these tumors are resistant to imatinib. A mouse model of GIST was recently reported using Etv1, the master transcriptional regulator of ICC-intramuscular (IM) and ICC-myenteric (MY), to induce mutant Braf expression. ICC hyperplasia was observed in Etv1CreERT2;BrafLSL-V600E/+ mice but loss of Trp53 was required for development of GIST. We identified previously expression of the pan-ErbB negative regulator, LRIG1, in two distinct subclasses of ICC (ICC-deep muscular plexus [DMP] in small intestine and ICC-submucosal plexus [SMP] in colon) and that LRIG1 regulated their development from smooth muscle cell progenitors. Using Lrig1CreERT2 to induce BrafV600E, we observed ICC hyperplasia beyond the confines of ICC-DMP and ICC-SMP expression, suggesting smooth muscle cells as the cell-of-origin. To examine this possibility, we selectively activated BrafV600E in smooth muscle cells. Myh11CreERT2; BrafLSL-V600E/+ mice developed not only ICC hyperplasia but also GIST and in the absence of Trp53 disruption. In addition to providing a simpler model for mutant Braf GIST, these results provide conclusive evidence for smooth muscle cells as an alternative cell-of-origin for GIST.
Keywords: Animal model, Stomach, Smooth muscle, Neoplasia
Introduction
Interstitial cells of Cajal (ICC) are thought to be the sole cell-of-origin of gastrointestinal stromal tumors (GIST). In the normal gut wall, ICC are a KIT-positive population, which forms networks within the musculature of the gastrointestinal tract and serves as pacemakers and transducers of neural inputs that control contraction of smooth muscle to regulate gut motility [1,2]. GIST are found most often in the stomach but can occur throughout the gastrointestinal tract. Activating gain-of-function mutations in KIT and PDGFRA are the most frequent driver events. These tumors have attracted considerable interest in the oncology community since they are highly responsive to imatinib, the tyrosine kinase inhibitor that targets KIT as well as ABL. Less common driver events include activating mutations in BRAF; mutant BRAF GIST account for 3-13%[3–6] of wild-type KIT and PDGFR GIST, and these tumors are much less responsive to imatinib. Common markers of GIST are KIT and ANO1 (present in nearly all cases), CD34 (70% of cases), and ETV1 (50–60% of cases).
Mouse models of GIST include mice with a germline mutation in Kit, which exhibit ICC hyperplasia, a GIST precursor, followed by full-blown GIST formation, further supporting ICC as the GIST cell-of-origin [7]. Recently, Ping Chi and co-workers used Etv1, the master transcriptional regulator of ICC-intramuscular (IM) and ICC-myenteric (MY), as an inducible Cre driver to activate BrafV600E. Administration of tamoxifen to activate Cre in Etv1CreERT2; BrafLSL-V600E/+mice resulted in ICC hyperplasia and GIST, providing direct support for ICC as GIST cell-of-origin. In this model, loss of Trp53 was required for development of GIST [8]
We previously reported that LRIG1, a type 1 transmembrane pan-ErbB negative regulator, is expressed in gastrointestinal epithelial cells with stem/progenitor properties [9–11]. In addition, LRIG1 null mice in a mixed C57BL/6 x 129 background spontaneously develop duodenal adenomas [9,12]. In separate studies, we showed that LRIG1 is also expressed in a subset of ICC (ICC-deep muscular plexus [DMP] in small intestine and ICC-submucosal plexus [SMP] in colon). These two populations of ICC reside beneath the submucosa within or juxtaposed to the inner circular muscle layer. We did not detect expression of LRIG1 in ICC-MY present between the inner circular and the outer longitudinal muscle or in ICC within the musculature of the stomach [13]. In that report, we also found that LRIG1 regulates postnatal development of ICC-DMP and ICC-SMP from smooth muscle progenitors [13].
Prior to publication of the report by Ping Chi and co-workers, we had set out to use Lrig1CreERT2 mice to inducibly-activate mutant BrafV600E in ICC-DMP and ICC-SMP. Although Lrig1CreERT2; BrafLSL-V600E/+ mice developed ICC hyperplasia and GIST, they occurred beyond the confines of ICC-DMP and ICC-SMP expression, most notably in the stomach and myenteric plexus of the small intestine and colon. Based on the link between Lrig1 and smooth muscle [13], we speculated that Braf V600E GIST may have arisen from smooth muscle. Using an inducible smooth muscle Cre driver (Myh11CreERT2) to activate mutant Braf, we show that ICC hyperplasia and GIST can arise from smooth muscle, thus identifying smooth muscle as an alternate GIST cell of origin.
Materials and methods
Animals
The generation of Lrig1<tm1.(cre/ERT)Rjc> (Lrig1CreERT2) mice has been reported previously [9]. BrafLSL-V600E [14], Myh11CreERT2 mice [15] and RosaLSL-YFP mice [16] were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). C57BL/6-Gt(ROSA)26Sortm1(KIT)Geno (RosaLSL-Kitc/+) mice were generated in collaboration with GenOway Transgenic Service (Lyons, France, see supplementary material, Figure S1). In brif, the targeting vector was obtained by introducing human mutated KIT (exon 11 codons 557–558 deletion mutation [17,18]) cDNA cassette, polyA (pA) sequences of the human growth hormone (hGH pA) and a Kozak sequence into GenOway’s Rosa26 “Quick Knock-in™” targeting vector, already containing the ubiquitous CAG promoter and a floxed STOP cassette. The targeting construct was transfected into ES cells by electroporation, followed by G418 selection and subsequent injection into BalbC blastocysts. Genotyping was performed by PCR using the following three primers: 89089-AAG CAC TTG CTC TCC CAA AGT CG, Rosa-GCA GTG AGA AGA GTA CCA CCA TG, and 89091 -ACC TTT TGA TAA GGC TGC AGA AG, yielding bands of 641 and 256 bp for wild-type and inducible alleles, respectively. To assess Cre-inducible recombination, PCR using the following three primers was performed: 89082-GAT GGT TGA GAA GAG CCT GTC TGG A, 89083-TAC AGC TCC TGG GCA ACG TGC TG, and 89084-CCT CGT GCT TTA CGG TAT CGC CG, yielding bands of 524 and 247 bp for wild-type and induced alleles, respectively. To induce recombination, mice were given a single, intraperitoneal (i.p.) injection of tamoxifen (Sigma-Aldrich, St. Louis, MO, USA) at the concentrations and time points indicated in the text and figure legends. The ages of mice at sacrifice are indicated in figure legends. Unless otherwise stated, the greater curvature of the corpus was used for gastric examinations, ileum for the small intestine and the proximal part for the colon. All mouse experiments were approved by Institutional Animal Care and Use Committee at Vanderbilt University Medical Center.
Tissue processing and immunofluorescence
For formalin-fixed paraffin-embedded blocks, tissues were fixed with either 10% neutral buffered formalin or 4% paraformaldehyde (PFA) before embedding. Sections were deparaffinized, rehydrated and H&E stained (Sigma). Immunostaining for ANO1 (rabbit anti-TMEM16A 1:100, ab53212, Abcam, Cambridge, UK) was performed on FFPE sections with antigen retrieval using Trilogy (Cell Marque, Rocklin, CA, USA). All other immunostaining was performed on frozen sections. Tissues were washed with PBS and fixed with 4% PFA (Thermo Fischer Scientific, Waltham, MA, USA) for 1 h at 4 °C, followed by consecutive 15% and 30% sucrose immersions before freezing in Optimal Cutting Temperature (OCT) compound (Sakura Finetek, Torrance, CA, USA). Cryosections cut at 6 μm for single focus images and 15 μm for Z-stacked images, were mounted onto glass slides and incubated at room temperature for 30 min in PBS containing 0.1% Triton 100-X (PBST) and 2.5% normal donkey serum to reduce nonspecific immunostaining. Sections were counterstained with 4’,6-diamidino-2-phenylindole (DAPI) for detection of nuclei. Primary and secondary antibodies used were: Rat anti-mouse KIT (1:250, #CBL1360, Millipore, Burlington, MA, USA); rat anti-mouse CD34 (1: 200, 14-0341-81, eBioscience, San Diego, CA, USA); rat anti-mouse PDGFRA (1:100, 16-1401-82, eBioscience); mouse anti-smooth muscle actin (SMA) conjugated with Cy3 (1:2000, C6198, Sigma); mouse anti-SMA (1:100, sc-53015, Santa Cruz, Dallas, TX, USA); goat anti-SM22 (1:200, ab10135, Abeam); donkey anti-rat IgG conjugated with Alexa 488 (1:500, A21208, Life Technologies, Carlsbad, CA) and goat anti-rat IgG conjugated with Alexa 568 (1:500, A11077, Life Technologies). Micrographic images were obtained using an Olympus IX-71 (Olympus, Center Valley, PA) and Nikon A1R (Nikon, Tokyo, Japan). Unless stated otherwise, images presented are representatives of no less than 3 mouse samples. All tissue images are oriented with mucosal side up except for overt tumor tissue.
Western blotting
Jejunal walls were stripped from the mucosal layer of the tissue and lysed in RIPA buffer supplemented with complete protease inhibitor cocktail and phosSTOP (Roche, Basel, Switzerland) followed by sonication. Aliquots of proteins (10 μg) were loaded onto an SDS-PAGE gel after 10 min high-speed centrifugation at 4 °C. The following antibodies were used for western blotting: phosphorylated ERK1/2 (1:1000, #9101, Cell Signaling Technology, Danvers, MA, USA), total ERK1/2 (1:1000, #9102 ,Cell Signaling Technology). Densitometry was performed using ImageJ (NIH, Bethesda, MD).
Results
Activation of mutant BRAF in Lrig1-expressing cells results in ICC hyperplasia
As noted in the introduction, LRIG1 is expressed in two ICC populations, one present within the inner circular smooth muscle layer of the small intestine (ICC-DMP) and the other at the boundary of the submucosa and the inner circular muscle layers of the colon (ICC-SMP). We also found LRIG1 was not expressed in ICC-MY located in the outer longitudinal smooth muscle layer or in gastric ICC. A limitation to using Lrig1 as a Cre driver is that it is expressed in many epithelial stem/progenitor cells. In fact, 12 wk after a single injection of tamoxifen (2 mg per mouse) to 8-wk old Lrig1CreERT2/+; BrafLSL-V600E/+ mice, they developed oral tumors and were unable to eat (supplementary material, Figure S2A); histologically, the tumors were squamous papillomas similar to those reported in Kras mutant mice [19]. Prior to the development of these obstructing oral lesions, mice were sacrificed 8–10 wk after tamoxifen administration. Activation of mutant BRAF in the intestinal wall was confirmed using western blotting analysis by upregulation of phosphorylated ERK, a downstream signaling molecule of BRAF (supplementary material, Figure S3). Upon gross inspection of the abdomen, the most striking finding was a markedly thickened gastric wall. Histologically, the outer half of the muscularis propria was replaced by hyperplastic spindle-shaped cells with dysplastic features (Figure 1A) and squamous hyperplasia of the forestomach was also observed (supplementary material, Figure S4). By immunofluorescence, these hyperplastic regions were devoid of smooth muscle actin (SMA) and partially positive for KIT (Figure 1B), resembling ICC hyperplasia in the gastric wall of Kit mutant mice [7]. The outer longitudinal muscle layer was almost completely replaced by lesions with diffuse staining for CD34 (Figure 1B,C), an additional marker for GIST. Active proliferation of the hyperplastic lesions was confirmed by detection of Ki67 in these CD34-positive cells (Figure 1C and supplementary material, Figure S5). Similar lesions, chiefly in the longitudinal muscle and also marked by KIT and CD34 staining, were observed in small intestine and colon, although they were less prominent than those in the stomach (Figure 1D).
Figure 1.
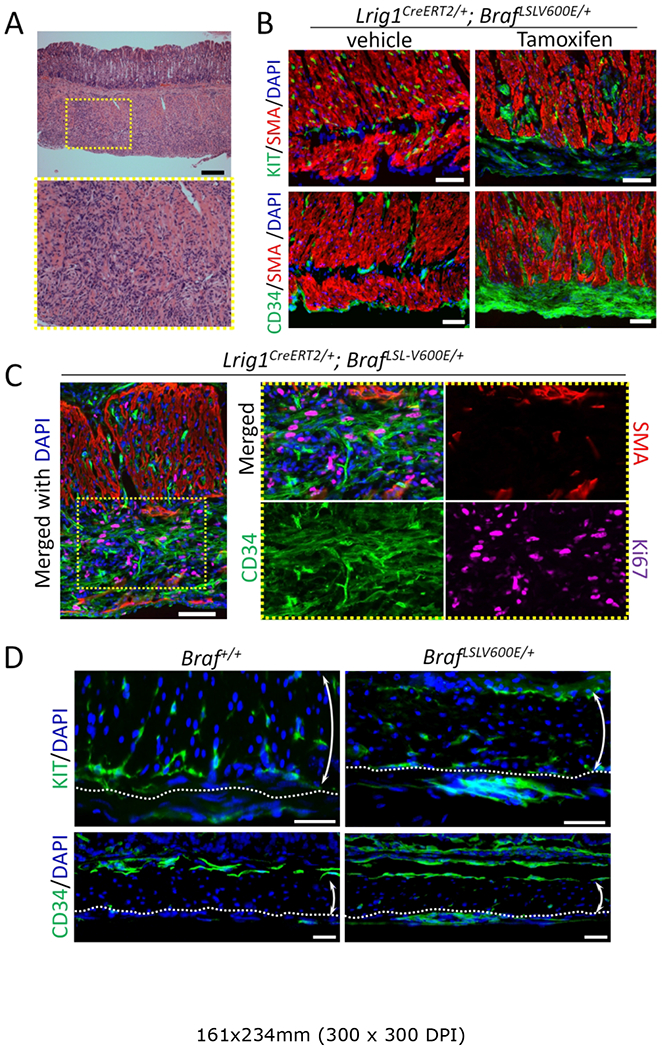
Activation of mutant BRAF in LRIG1-expressing cells results in ICC hyperplasia in mouse stomach and colon. (A) H&E staining of the stomach of an adult Lrig1CreERT2/+; BrafLSL-V600E/+ mouse 10 wk after a single injection of 0.5 mg per mouse tamoxifen. Lower panel is a higher magnification image of the yellow dotted box in the upper panel. Scale bars, 500 μm. (B) Immunofluorescence images of the gastric muscle layer of Lrig1CreERT2/+; BrafLSL-V600E/+ treated as above or vehicle control stained for KIT (upper panels, green), CD34 (lower panels, green) and SMA (red). Nuclei were stained with DAPI. Scale bars, 50 μm. (C) Immunofluorescence images of Lrig1CreERT2/+; BrafLSL-V600E/+ tissue sections 3 wks after a single 0.5 mg per mouse tamoxifen injection stained for CD34 (green), SMA (red) and Ki67 (magenta). Images of the greater curvature of the corpus were captured. The area enclosed in the yellow dotted box of the left most panels is enlarged in the right four panels. Nuclei were stained with DAPI. Scale bars, 50 μm. (D) Immunofluorescence images of colon sections from Lrig1CreERT2/+; BrafLSL-V600E/+ treated as described in B and stained for KIT (upper panels, green) and CD34 (lower panels, green). Dotted lines indicate the myenteric side of the circular muscular layer. Double-headed arrows indicate the circular muscular layer. Nuclei were stained with DAPI. Scale bars, 50 μm.
There are several lines of evidence that these lesions did not arise from Lrig1-expressing ICC. First, the most striking phenotype was in the stomach, and as noted earlier, LRIG1 is not expressed in gastric ICC. Secondly, the outer longitudinal smooth muscle layer of the stomach was replaced by CD34-positive hyperplastic cells, a finding that is unlikely to be due solely to hyperplasia of ICC. Finally, the lesions in the small intestine and colon were most prominent in the outer longitudinal muscular layer distinct from the location of LRIG1-expressing ICC-DMP and SMP. Nor is ICC-MY likely to be the origin of these lesions because colonic ICC-MY is negative for LRIG1 expression (Figure 2A) and expression of LRIG1 in small intestinal ICC-MY is weak [13].
Figure 2.
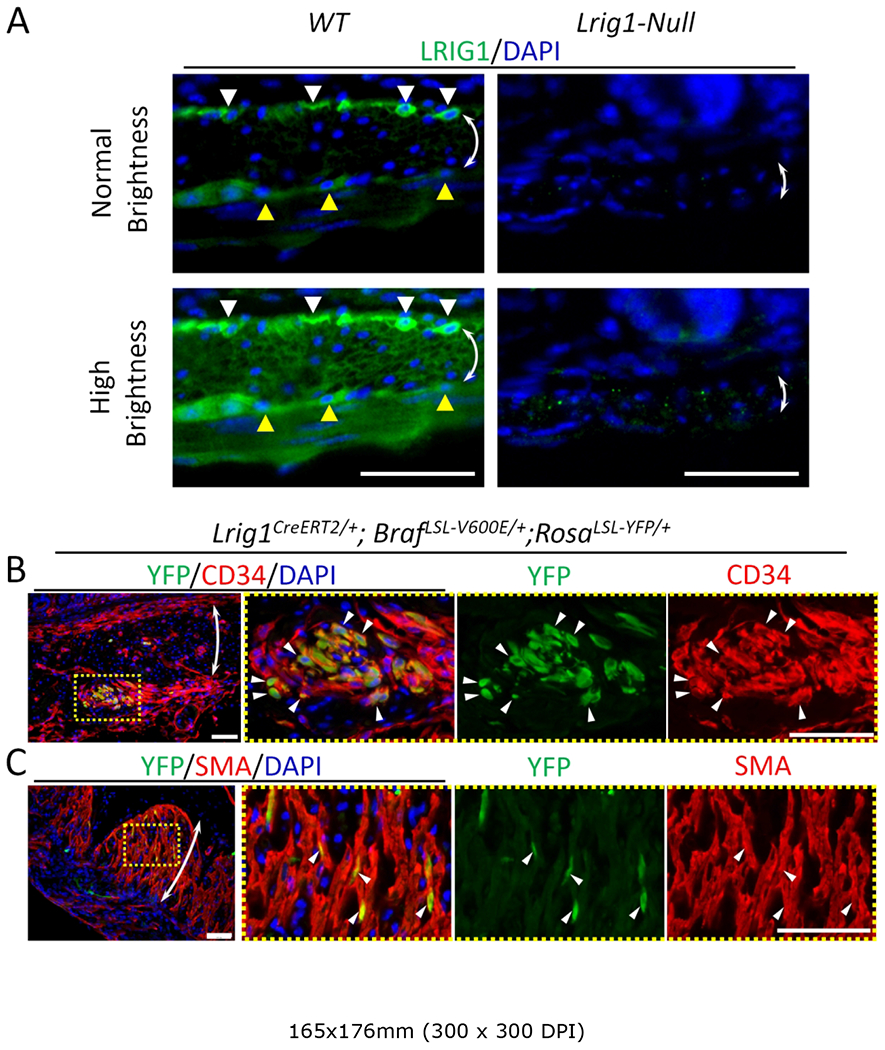
Evidence that ICC hyperplasia in Lrig1CreERT2/+; BrafLSL-V600E/+ mouse is driven from LRIG1 expression in smooth muscle. (A) Immunofluorescence images of wild-type (WT, left panels) and Lrig1CreERT2/CreERT2 (Lrig1-null, right panels) small intestinal tissue sections stained for LRIG1 (green). Lower panels are modified from upper panel images by adjusting brightness and contrast. White and yellow arrowheads indicate ICC-DMP and ICC-MY, respectively. Double-headed arrows indicate the circular muscular layer. Nuclei were stained with DAPI. Scale bars, 50 μm. (B,C) Immunofluorescence images of Lrig1CreERT2/+; BrafLSL-V600E/+; RosaLSL-YFP/+ gastric tissue sections 2 wks after a 0.5 mg per mouse tamoxifen injection stained for YFP (green), CD34 (B, red) and SMA (C, red). Areas enclosed in yellow dotted boxes of the left most panels are enlarged in the right three panels. Representative cells positive for both markers are indicated by white arrowheads. Double-headed arrows indicate the circular muscular layer. Nuclei were stained with DAPI. Scale bars, 50 μm.
These observations led us to more carefully evaluate the expression of LRIG1 in the smooth muscle layer. Indeed, we found that both the inner circular and outer longitudinal smooth muscle cells do express LRIG1, although at much lower levels than in ICC-DMP, ICC-SMP or overlying epithelial cells (Figure 2A). The specificity of LRIG1 staining was confirmed by its absence in Lrig1 null mice (right panel Figure 2A). To further evaluate whether recombination mediated by the Lrig1CreERT2 driver occurs in smooth muscle, Lrig1CreERT2/+; BrafLSL-V600E/+; RosaLSL-YFP/+ mice were generated to lineage trace Lrig1-expressing cells. Two weeks after a single injection of tamoxifen, there were YFP-positive clusters in CD34-positive lesions in the stomach (Figure 2B), as well as YFP-positive cells in unaffected smooth muscle cells (Figure 2C). Taken together, these results suggest that the lesions detected in the Lrig1CreERT2/+; BrafLSL-V600E/+ gut wall likely originate from smooth muscle cells rather than LRIG1-expressing ICC.
Smooth muscle-specific expression of BrafV600E results in GIST-like lesions
In order to activate mutant BRAF exclusively in smooth muscle cells, we turned to a smooth muscle-specific gene, Myh11, as a Cre-driver. As noted above, we previously reported ICC-DMP and ICC-SMP originate from smooth muscle cells during postnatal development of the ICC network [13]. Thus, a constitutively expressed Myh11Cre driver will not allow tracing of smooth muscle cell lineages without also marking these ICC populations. Instead, we used a conditional Myh11CreERT2 driver to induce recombination after postnatal day 7 since ICC-DMP and ICC-SMP no longer express smooth muscle markers after this time [13]. To assess how tightly regulated CreERT2-mediated recombination is in this model, we first examined spatiotemporal expression of YFP in Myh11CreERT2; RosaLSL-YFP/+ mice. In the absence of tamoxifen, YFP expression was observed after postnatal day 14 (Figure 3A and supplementary material, Figure S6), indicating some leakiness in this promoter; however, YFP expression was restricted to smooth muscle cells.
Figure 3.
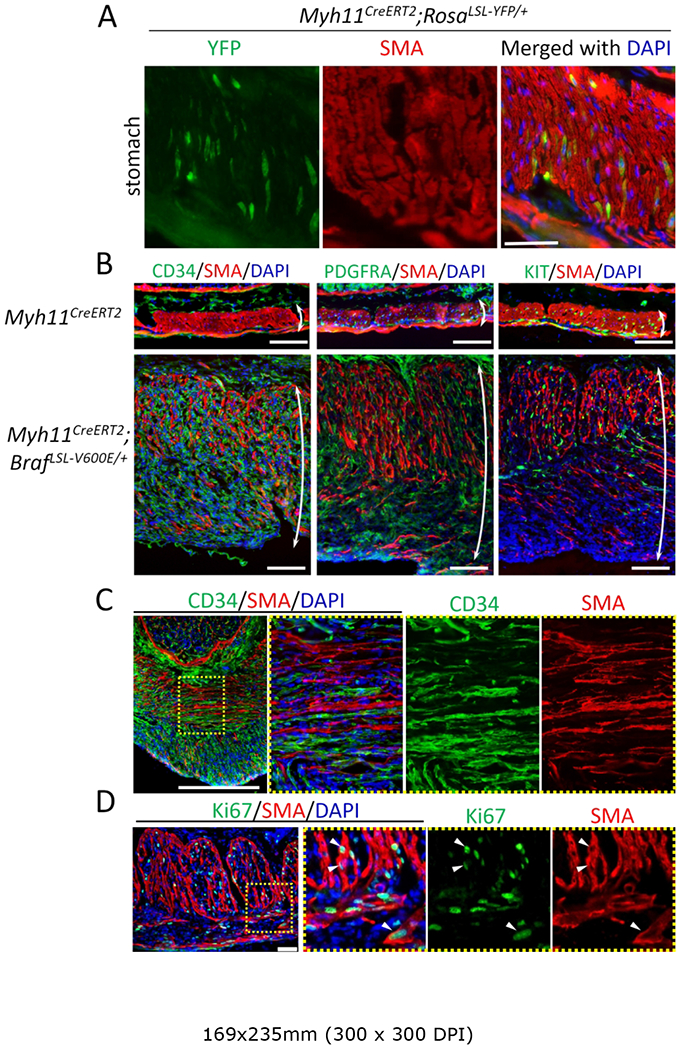
Activation of mutant BRAF in gastric smooth muscle resulted in rapid onset of ICC hyperplasia. (A) Immunofluorescence image of the gastric muscle layer in a Myh11CreERT2; RosaLSL-YFP/+ mouse prior to tamoxifen injection. Gastric section of a 6-wk-old mouse stained for YFP (green) and SMA (red). Nuclei were stained with DAPI. Scale bar, 100 μm. (B,C) Immunofluorescence images of gastric muscle layer from Myh11CreERT2 and Myh11CreERT2; BrafLSL-V600E/+ mice given a single 0.1 mg per mouse tamoxifen injection at 6-wks of age and sacrificed two weeks later. Longitudinal (B) and cross (C) sections were stained for CD34 (left panel of B and C, green), PDGFRA (middle panel of B, green), KIT (right panel of B, green) and SMA (red). Images of the greater curvature of the corpus were captured. Double-headed arrows indicate the full thickness of the smooth muscle layer. Area enclosed in a yellow dotted box of the left-most panel of C is enlarged in the right three panels. Nuclei were stained with DAPI. Scale bars, 100 μm. (D) Immunofluorescence image of the gastric muscle layer from a Myh11CreERT2; BrafLSL-V600E/+ mouse given single 0.1 mg per mouse tamoxifen injection at 6-wks of age and sacrificed two weeks later. Staining was performed for Ki67 (green) and SMA (red). Images of the greater curvature of the corpus were captured. The area enclosed in the yellow dotted box of the left-most panel is enlarged in the right three panels. Nuclei were stained with DAPI. Scale bars, 50 μm.
To further examine whether smooth muscle cells may act as a cell-of-origin for GIST, Myh11CreERT2; BrafLSL-V600E/+ mice were generated. At a standard dose of tamoxifen (2 mg per mouse), mice became moribund within 1–2 wk, possibly due to impaired gut motility. A single small dose of tamoxifen (0.1 mg per mouse) was given to Myh11CreERT2;BrafLSL-V600E/+ mice at 6-wk of age and mice were sacrificed two wk later. These mice largely recapitulated the phenotype exhibited by Lrig1CreERT2/+;BrafLSL-V600E/+ mice, except for the absence of the oral and forestomach squamous hyperplasia.
Once again, the phenotype was most pronounced in the stomach, which was grossly thickened. Histologically, the gastric wall was largely composed of smooth muscle with hyperplastic stromal cells evident (Figure 3B). Similar to Lrig1CreERT2/+;BrafLSL-V600E/+ mice, the gastric lesions were negative for both SMA and SM22, differentiated smooth muscle markers, (supplementary material, Figure S7) and were diffusely positive for CD34 (Figure 3B, left panel). The lesions were also positive for PDGFRA (Figure 3B, middle panel), an additional marker for GIST. Immunofluorescence staining for KIT was patchy, and most KIT-positive cells appeared to be preexisting ICC (Figure 3B right). Of note, residual SMA-positive cells in the smooth muscle layer exhibited ICC-like morphology with cell bodies exhibiting long processes (Figure 3C and supplementary material Figure S8). Ki67-positive proliferating cells, which are rarely seen in smooth muscle, were found in both SMA-positive and −negative cells (Figure 3D). As noted in Lrig1CreERT2/+; BrafLSL-V600E/+ mice, similar, but less prominent, lesions were observed in the small and large intestinal wall (supplementary material, Figure S9).
Combining the results from Lrig1CreERT2/+; BrafLSL-V600E/+ mice and Myh11CreERT2;BrafLSL-V600E/+ mice, we have shown that BrafV600E expression in smooth muscle cells results in GIST-like lesions in stomach, small intestine and colon.
Myh11CreERT2; BrafLSL-V600E/+ mice generate overt GIST
We then sought to determine whether Myh11CreERT2; BrafLSL-V600E/+ mice might develop overt GIST over time. One month after receiving a single dose of 0.1 mg per mouse tamoxifen, these mice appeared moribund. By decreasing the dose of tamoxifen to 0.005 mg per mouse, we were able to maintain the mice for longer periods of time. Four months after administration of tamoxifen to 6-wk-old-mice, KIT-positive foci emerged, although not in every CD34-positive cell (Figure 4A). At this time, 4 of 11 mice had tumors in the smooth muscle layer of the gut and bladder (Table 1). These tumors were solid and devoid of a lumen (Figure 4B). Microscopically, the tumors had a spindle-shaped cellular morphology with an appearance of dedifferentiated GIST (Figure 4C). These tumors expressed CD34, PDGFRA, KIT and ANO1 (Figure 4D), supporting the diagnosis of GIST. Taken together, our results show that ICC hyperplasia and overt GIST may arise from smooth muscle cells.
Figure 4.
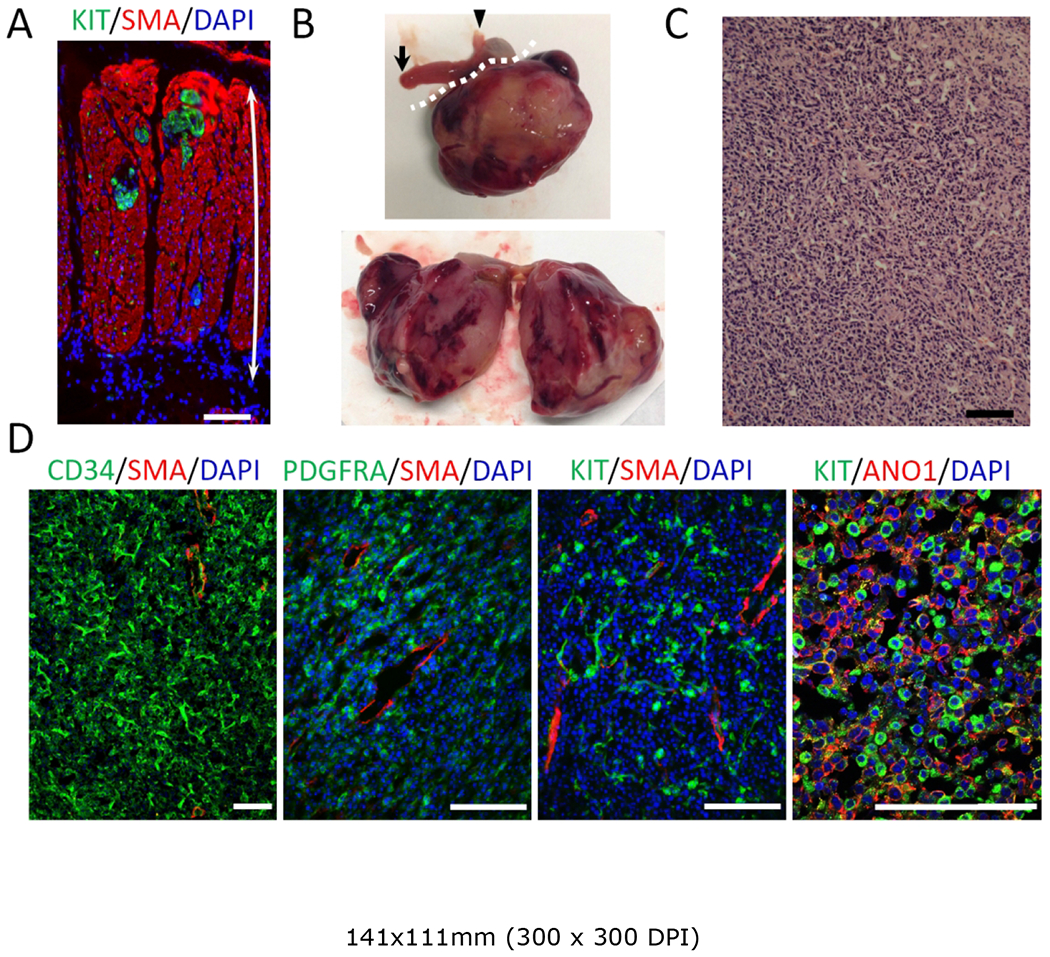
Activation of mutant BRAF in smooth muscle results in overt GIST. (A) Immunofluorescence image of the gastric muscle layer from a Myh11CreERT2; BrafLSL-V600E/+ mouse given a single injection of 0.1 mg per mouse tamoxifen at 6-wks of age and sacrificed 4 months later. Staining was performed for KIT (green) and SMA (red). Double-headed arrows indicate the full thickness of the smooth muscle layer. Nuclei were stained with DAPI. Scale bars, 100 μm. See also Table 1 for number and location of overt GIST detected in eleven Myh11CreERT2; BrafLSL-V600E/+ mice examined. (B) Macroscopic appearance of an overt GIST formed in a gastric wall. The white dotted line in the upper panel demarcates the tumor. Arrowhead indicates the esophagus and arrow indicates the duodenum. The right panel shows a bisected tumor. (C) H&E staining of GIST. Scale bar, 100 μm. (D) Immunofluorescence images of tumor stained for CD34 (green), PDGFR (green), KIT (green), SMA (red) and AN01 (red). Nuclei were stained with DAPI. Scale bars, 100 μm. Histology shown is representative of the three overt GIST tissues in gastrointestinal tract.
Table 1.
Overt GIST detected in Myh11CreERT2; BrafLSL-V600E/+ mice.
| Location | Number of mice |
|---|---|
| Stomach | 2 |
| Cecum | 1 |
| Bladder | 1 |
| Total | 4 out of 11 (36.4%) |
Lrig1CreERT2/+; RosaLSL-Kitc/+ mice also exhibit GIST-like lesions
Since mutant KIT GIST are much more common than mutant BRAF GIST, we also examined the consequences of inducing gain-of-function mutant KIT in LRIG1-expressing cells. A single dose of tamoxifen (2 mg per mouse) was given to adult Lrig1CreERT2/+; RosaLSL-Kitc/+ mice and the mice were sacrificed one month later. GIST-like small lesions were observed in the muscularis propria of the stomach and intestine, although the lesions were less prominent than those seen in Lrig1CreERT2/+; BrafLSL-V600E/+ mice (Figure 5A,B). There was diffuse expression of CD34 and SMA in the outer longitudinal smooth muscle of these Lrig1CreERT2/+; RosaLSL-Kitc/+ mice; as noted above (Figure 1B), CD34 expression was also diffuse in the outer longitudinal muscle of Lrig1CreERT2/+; BrafLSL-V600E/+ mice whereas SMA staining was absent. Cell clusters positive for CD34 and KIT were observed in the smooth muscle layer (Figure 5A,B). Thus, GIST can arise from smooth muscle cells driven not only by mutant Braf but also by mutant Kit.
Figure 5.
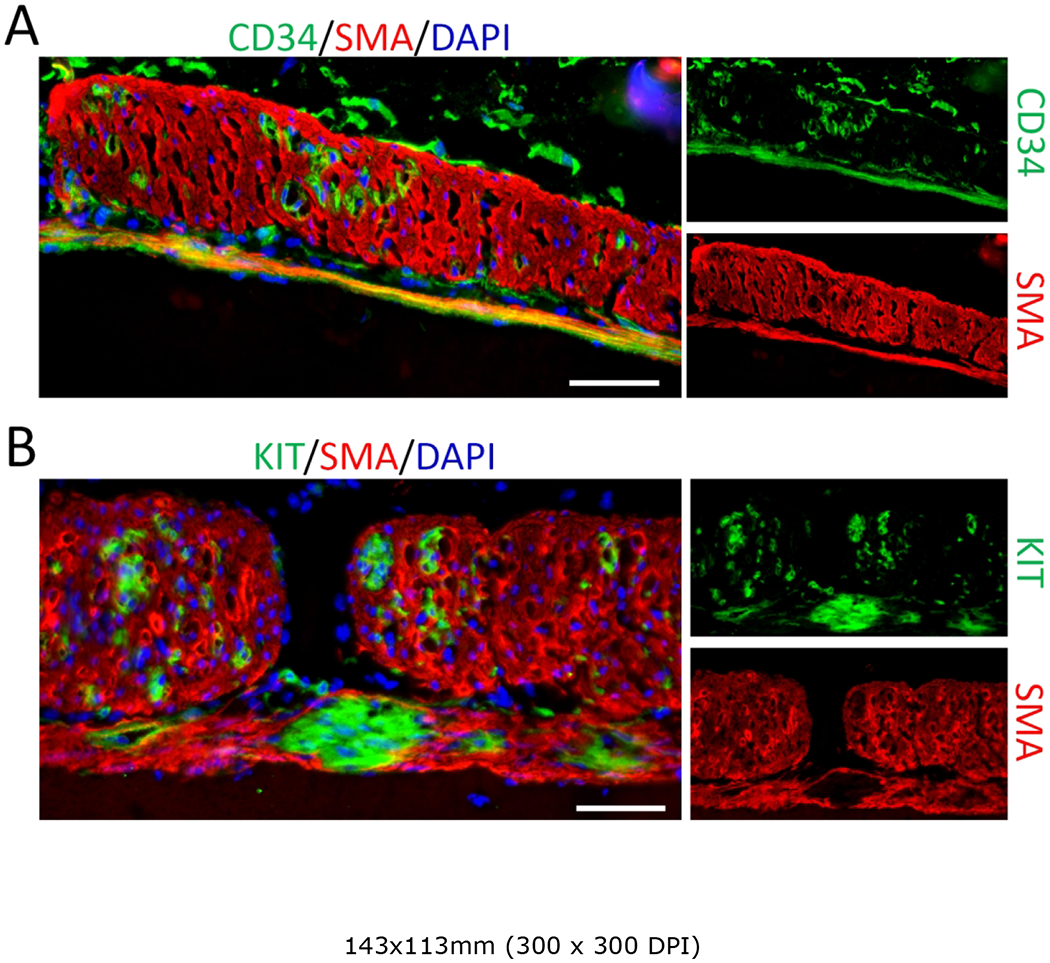
Activation of mutant KIT in Lrig1-expressing cells results in GIST-like lesions. (A,B) Immunofluorescence images of the gastric muscle layer of Lrig1CreERT2/+; RosaLSL-Kitc/+ mice given a single injection of 2 mg tamoxifen per mouse at 6-wks of age and sacrificed one month later. Tissue sections were stained for CD34 (A, green), KIT (B, green) and SMA (red). Images of the greater curvature of the corpus were captured. Nuclei were stained with DAPI. Scale bars, 50 μm.
Discussion
These studies were initiated using an inducible Lrig1CreERT2 to activate BrafV600E in two subpopulations of ICC, ICC-DMP and ICC-SMP, which we previously showed express the pan-ErbB negative regulator and tumor suppressor, LRIG1. In contrast to ICC-MY that are found in the outer longitudinal muscle layer, ICC-DMP and ICC-SMP are found within or juxtaposed to the inner circular muscle in the small intestine and colon, respectively. Upon activation of BrafV600E in LRIGI-expressing cells, we found GIST-like lesions in the gastric smooth muscle layer and in the intestinal longitudinal muscle layer, both of which are distinct from the location of LRIG1-expressing ICC populations. Based on our prior observation that LRIG1 regulates development of submucosal ICC-DMP and ICC-SMP from smooth muscle, we examined LRIG1 expression in smooth muscle cells more closely and identified low but detectable levels of LRIG1 expression in smooth muscle cells. With this information in hand, we turned to an inducible smooth muscle Cre driver to see if this cell population could be a source of GIST. Activation of mutant BRAF in Myh11CreERT2; BrafLSL-V600E/+ mice led to full blown GIST, demonstrating that smooth muscle cells may act as an alternate GIST cell-of-origin.
Our model complements the Etv1CreERT2; BrafLSL-V600E/+mouse model developed by Chi and co-workers [8]. Etv1 is the master transcriptional regulator of two subclasses of ICC distinct from ICC-DMP and ICC-SMP; these are termed ICC-MY and ICC-IM [8,20]. in their model, BRAFV600E expression alone was insufficient to generate overt GIST but required loss of Trp53. In a similar vein, additional mutations are required for mutant BRAF serrated adenomas to progress to colon cancer [21–23] and benign nevi to melanoma [24,25]. Interestingly, our result shows that BRAF mutation alone, in the absence of Trp53 disruption, was sufficient to generate GIST when mutant BRAF was expressed in smooth muscle cells, suggesting a differential dependency of driver mutations for GIST that arise from different cells-of-origin.
Further investigation of the Etv1- and Myh11-driven mutant Braf models might uncover distinct features of the GIST tumors when they originate from ICC versus smooth muscle. In contrast to the introduction of mutant Braf, mutant Kit was not as efficient in inducing multiple lesions, ICC-hyperplasia, or overt GIST. These findings may reflect the presence of a differential cellular context in smooth muscle cells that is favorable for the Braf mutation to induce progression to GIST. Alternatively, differences in recombination efficiency may explain differences in severity between the Braf mutant and Kit mutant models. Further studies should be conducted to clarify detailed mechanisms of differences of how different oncogenic drivers result in different spectrums of ICC hyperplasia and GIST formation.
In patients, most GIST with mutant KIT and PDGFRA are highly responsive to imatinib, a small molecule tyrosine kinase inhibitor (TKI), that targets KIT, as well as BCR-ABL and FIP1L1-PDFGRA, with about 80% of patients with metastatic disease achieving a partial response or stable disease [26]. Importantly, the response to therapeutics is highly correlated with the genotype of GIST. Patients without KIT or PDGFRA mutation show primary resistance to imatinib, which is caused by alternative mutations such as in BRAF, RAS, NF1 or SDHB deficiency [27,28]. While a report suggests a possibility for the use of dabrafenib, a BRAF inhibitor, in BRAF mutant GIST [29], more effective treatment regimens are needed for GIST due to these less commonly occurring events. In this regard, our Braf mutant GIST model may be a useful tool.
In summary, we report a new mouse model of BRAF mutant GIST. Under control of the smooth muscle Myh11 driver, conditional expression of constitutively active BrafV600E was sufficient to generate GIST. In addition to providing a unique genetic model for Braf mutant GIST, this is the first report to show that smooth muscle can be the origin of GIST. There might exist a portion of clinical GIST that originate from smooth muscle but not ICC. Such differences in the GIST cell-of-origin may have important implications for the treatment and prognosis of the disease.
Supplementary Material
Acknowledgements
The authors thank Nicholas Markham and Hiroaki Niitsu for technical and editorial assistance. This work was supported in part by funding from GIST Cancer Research Fund (MCH) ,VA Merit Review Grant (MCH, 2I01BX000338-05) and MEXT KAKENHI (JP18008400).
JK is a member of the Department of Clinical Bio-resource Research and Development in Kyoto University, which is sponsored by KBBM, Inc. MCH is a consultant for Novartis, Deciphera Pharmaceuticals, Blueprint Medicines, and Molecular MD. MCH has provided expert testimony to Novartis. MCH receives research support from Deciphera Pharmaceuticals, Blueprint Medicines and has equity interest in Molecular MD. BPR is an Associate Editor of The Journal of Pathology. No other conflicts of interest were declared.
References
- 1.Thomson L, Robinson TL, Lee JCF, Farraway LA, Hughes MJG, Andrews DW, et al. Interstitial cells of Cajal generate a rhythmic pacemaker current. Nat Med. 1998;4:848–51. [DOI] [PubMed] [Google Scholar]
- 2.lino S, Ward SM, Sanders KM. Interstitial cells of Cajal are functionally innervated by excitatory motor neurones in the murine intestine. J Physiol. 2004;556:521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Huss S, Pasternack H, Ihle MA, Merkelbach-Bruse S, Heitkötter B, Hartmann W, et al. Clinicopathological and molecular features of a large cohort of gastrointestinal stromal tumors (GISTs) and review of the literature: BRAF mutations in KIT/PDGFRA wild-type GISTs are rare events. Hum Pathol. 2017;62:206–14. [DOI] [PubMed] [Google Scholar]
- 4.Agaram NP, Wong GC, Guo T, Maki RG, Singer S, Dematteo RP, et al. Novel V600E BRAF mutations in imatinib-naive and imatinib-resistant gastrointestinal stromal tumors. Genes Chromosomes Cancer. 2008;47:853–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agaimy A, Terracciano LM, Dirnhofer S, Tornillo L, Foerster A, Hartmann A, et al. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin Pathol. 2009;62:613–6. [DOI] [PubMed] [Google Scholar]
- 6.Hostein I, Faur N, Primois C, Boury F, Denard J, Emile J-F, et al. BRAF Mutation Status in Gastrointestinal Stromal Tumors. Am J Clin Pathol. 2010;133:141–8. [DOI] [PubMed] [Google Scholar]
- 7.Rubin BP, Antonescu CR, Scott-Browne JP, Comstock ML, Gu Y, Tanas MR, et al. A Knock-In Mouse Model of Gastrointestinal Stromal Tumor Harboring Kit K641E. Cancer Res. 2005;65:6631–9. [DOI] [PubMed] [Google Scholar]
- 8.Ran L, Murphy D, Sher J, Cao Z, Wang S, Walczak E, et al. ETV1-Positive Cells Give Rise to BRAFV600E -Mutant Gastrointestinal Stromal Tumors. Cancer Res. 2017;77:3758–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Powell AE, Wang Y, Li Y, Poulin EJ, Means AL, Washington MK, et al. The Pan-ErbB Negative Regulator Lrig1 Is an Intestinal Stem Cell Marker that Functions as a Tumor Suppressor. Cell. 2012;149:146–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Poulin EJ, Powell AE, Wang Y, Li Y, Franklin JL, Coffey RJ. Using a new Lrig1 reporter mouse to assess differences between two Lrig1 antibodies in the intestine. Stem Cell Res. 2014;13:422–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Choi E, Lantz TL, Vlacich G, Keeley TM, Samuelson LC, Coffey RJ, et al. Lrig1+ gastric isthmal progenitor cells restore normal gastric lineage cells during damage recovery in adult mouse stomach. Gut. 2018;67:1595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Shi C, Lu Y, Poulin EJ, Franklin JL, Coffey RJ. Loss of Lrig1 Leads to Expansion of Brunner Glands Followed by Duodenal Adenomas with Gastric Metaplasia. Am J Pathol. 2015;185:1123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kondo J, Powell AE, Wang Y, Musser MA, Southard-Smith EM, Franklin JL, et al. LRIG1 Regulates Ontogeny of Smooth Muscle-Derived Subsets of Interstitial Cells of Cajal in Mice. Gastroenterology. 2015;149:407–419.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dankort D, Filenova E, Collado M, Serrano M, Jones K, McMahon M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007;21:379–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wirth A, Benyó Z, Lukasova M, Leutgeb B, Wettschureck N, Gorbey S, et al. G12–G13–LARG-mediated signaling in vascular smooth muscle is required for salt-induced hypertension. Nat Med. 2008;14:64–8. [DOI] [PubMed] [Google Scholar]
- 16.Srinivas S, Watanabe T, Lin C-S, William CM, Tanabe Y, Jessell TM, et al. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001; 1:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martín J, Poveda A, Llombart-Bosch A, Ramos R, López-Guerrero JA, del Muro JG, et al. Deletions Affecting Codons 557-558 of the c-KIT Gene Indicate a Poor Prognosis in Patients With Completely Resected Gastrointestinal Stromal Tumors: A Study by the Spanish Group for Sarcoma Research (GEIS). J Clin Oncol. 2005;23:6190–8. [DOI] [PubMed] [Google Scholar]
- 18.Yan L, Zou L, Zhao W, Wang Y, Liu B, Yao H, et al. Clinicopathological significance of c-KIT mutation in gastrointestinal stromal tumors: a systematic review and meta-analysis. Sci Rep. 2015;5:13718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Caulin C, Nguyen T, Longley MA, Zhou Z, Wang X-J, Roop DR. Inducible Activation of Oncogenic K-ras Results in Tumor Formation in the Oral Cavity. Cancer Res. 2004;64:5054–8. [DOI] [PubMed] [Google Scholar]
- 20.Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, et al. ETV1 is a lineage-specific survival factor in GIST and cooperates with KIT in oncogenesis. Nature. 2010;467:849–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rad R, Cadinanos J, Rad L, Varela I, Strong A, Kriegl L, et al. A Genetic Progression Model of BrafV600E-Induced Intestinal Tumorigenesis Reveals Targets for Therapeutic Intervention. Cancer Cell. 2013;24:15–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakamoto N, Feng Y, Stolfi C, Kurosu Y, Green M, Lin J, et al. BRAFV600E cooperates with CDX2 inactivation to promote serrated colorectal tumorigenesis. Dang CV, editor. eLife. 2017;6:e20331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tong K, Pellón-Cárdenas O, Sirihorachai VR, Warder BN, Kothari OA, Perekatt AO, et al. Degree of Tissue Differentiation Dictates Susceptibility to BRAF-Driven Colorectal Cancer. Cell Rep. 2017;21:3833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. [DOI] [PubMed] [Google Scholar]
- 25.Yeh I Recent advances in molecular genetics of melanoma progression: implications for diagnosis and treatment. F1000Research [Internet]. 2016. [cited 2018 Jun 20];5 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4926755/ [DOI] [PMC free article] [PubMed]
- 26.Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80. [DOI] [PubMed] [Google Scholar]
- 27.Szucs Z, Thway K, Fisher C, Bulusu R, Constantinidou A, Benson C, et al. Molecular subtypes of gastrointestinal stromal tumors and their prognostic and therapeutic implications. Future Oncol. 2016;13:93–107. [DOI] [PubMed] [Google Scholar]
- 28.Antonescu CR, DeMatteo RP. CCR 20th Anniversary Commentary: A Genetic Mechanism of Imatinib Resistance in Gastrointestinal Stromal Tumor—Where Are We a Decade Later? Clin Cancer Res. 2015;21:3363–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Falchook GS, Trent JC, Heinrich MC, Beadling C, Patterson J, Bastida CC, et al. BRAF Mutant Gastrointestinal Stromal Tumor: First report of regression with BRAF inhibitor dabrafenib (GSK2118436) and whole exomic sequencing for analysis of acquired resistance. Oncotarget. 2013;4:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.