

Abstract
Purpose of Review
Fetuses of diabetic mothers are at increased risk for congenital malformations. Research in recent decades using animal and embryonic stem cell models has revealed many embryonic developmental processes that are disturbed by maternal diabetes. The aim of this review is to give clinicians a better understanding of the reasons for rigorous glycemic control in early pregnancy, and to provide background to guide future research.
Recent Findings
Mouse models of diabetic pregnancy have revealed mechanisms for altered expression of tissue-specific genes that lead to malformations that are more common in diabetic pregnancies, such as neural tube defects (NTDs) and congenital heart defects (CHDs), and how altered gene expression causes apoptosis that leads to malformations. Embryos express the glucose transporter, GLUT2, which confers susceptibility to malformation, due to high rates of glucose uptake during maternal hyperglycemia and subsequent oxidative stress; however, the teleological function of GLUT2 for mammalian embryos may be to transport the amino sugar glucosamine (GlcN) from maternal circulation to be used as substrate for glycosylation reactions and to promote embryo cell growth. Malformations in diabetic pregnancy may be not only due to excess glucose uptake but also due to insufficient GlcN uptake.
Summary
Avoiding maternal hyperglycemia during early pregnancy should prevent excess glucose uptake via GLUT2 into embryo cells, and also permit sufficient GLUT2-mediated GlcN uptake.
Keywords: Diabetic pregnancy, Neural tube defects, Diabetic embryopathy, Hyperglycemia in pregnancy, Oxidative stress in pregnancy, Pax3
Introduction
Maternal diabetes existing prior to pregnancy, either type 1 or type 2, increases risk of congenital malformations, a diabetic complication referred to as “diabetic embryopathy” [1–6]. Even in recent years, congenital malformations are increased up to 5-fold in diabetic pregnancies [7–14]. Malformations occur within the first 10 weeks of pregnancy during early organogenesis; almost any organ system can be affected, but NTDs, including anencephaly and spina bifida, and CHDs, are among the most common that occur [6, 10, 13–15]. Maternal obesity also increases risk for the same birth defects as occur in diabetic pregnancies, which could be due to undiagnosed type 2 diabetes or impaired glucose tolerance [16–20]. Currently, there are no strategies to reduce risk for diabetic embryopathy other than prepregnancy counseling to institute rigorous glycemic control prior to conception, and to administer folic acid as advised for all women of childbearing age [11, 21, 22]. Even in planned, well-controlled pregnancies of diabetic women, tight glycemic control comes at a risk, because the incidence of severe hypoglycemia is increased in the first trimester [23–26]. Thus, understanding how maternal diabetes causes congenital malformations is crucial in order to design strategies to prevent them.
Development of Mouse Models to Study Mechanisms Causes of Diabetic Embryopathy
Until the early 1990s, most studies using animal models of diabetic embryopathy employed the rat, in which diabetes was induced with the pancreatic beta cell toxins alloxan or streptozotocin (STZ) prior to, or at the beginning of pregnancy, or yolk sac-enclosed postimplantation rat or mouse embryos were cultured in diabetic rat serum or in high glucose media. These embryos displayed dysmorphogenesis, including NTDs, and underwent growth and developmental delay [27–34]. Some studies indicated that high glucose alone was teratogenic, while others suggested that ketones or altered lipid metabolism were also involved. By the early 1990s, several genes that control mammalian embryogenesis had been identified, and recent development of transgenic and targeted genome mutagenesis technologies made it possible to study regulatory pathways in mutant mouse lines. My laboratory hypothesized that maternal diabetes disturbs expression of genes that regulate early embryonic organogenesis. We developed a mouse model of diabetic pregnancy to test this hypothesis. In our model, diabetes is induced in female mice with STZ and then treated with subcutaneously implanted, sustained-release insulin pellets before mating with nondiabetic males. The insulin pellets maintain euglycemia prior to pregnancy, and on embryonic day (E) 4.5 (equivalent to week 3 of human pregnancy, dated by last menstrual period (LMP)), the insulin released by the pellets is no longer sufficient and diabetic mice become hyperglycemic (> 17 mmol/L); control mice remain euglycemic with fed blood glucose levels of 8–9.5 mmol/L (compared with human nondiabetic blood glucose < 7.8 mmol/L 2 h after meals). There was no growth or developmental delay, but NTDs, primarily exencephaly, were significantly increased [35]. The NTDs were morphologically very similar to the NTDs of Pax3Sp/Sp (Splotch) mouse embryos that are caused by a loss-of-function mutation in Pax3 [36], a developmental regulator that is expressed in embryonic neuroepithelium and somitic mesoderm. We found that Pax3 expression was significantly reduced in whole embryos of diabetic mice as determined by reverse transcription-polymerase chain reaction (RT-PCR), and specifically in neuroepithelium, which gives rise to the closed neural tube (which forms the scaffolding of the central neural system) and the neural crest cells, as determined by in situ hybridization. Furthermore, TUNEL apoptosis assays demonstrated that the neuroepithelium in unfused regions of neural tubes of embryos of diabetic mice and Pax3Sp/Sp embryos was undergoing apoptosis, which provided a cellular mechanism for failure of the neural tube to close in affected embryos [35]. While there could be additional genes or signaling pathways involved in neural tube closure that are disturbed by maternal diabetes, because Pax3Sp/Sp embryos develop NTDs and CHDs with 100% penetrance [37], simply reducing Pax3 expression in neuroepithelium below a critical threshold is sufficient to cause a NTD or CHD. We then set about asking two main questions: (a) How does maternal diabetes inhibit Pax3 expression? and (b) How does insufficient PAX3 protein production lead to NTDs and CHDs? Findings from experiments addressing these questions are described below and diagrammed schematically in Fig. 1a.
Fig. 1.
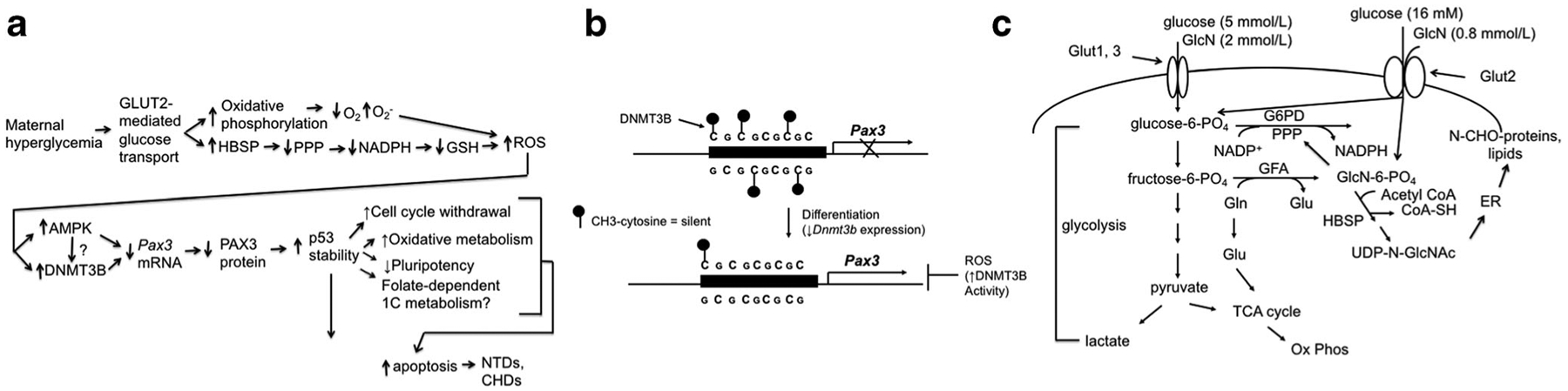
a—schematic diagram of pathways by which maternal diabetes causes NTDs and CHDs. Maternal hyperglycemia causes increased glucose transport into embryo cells via GLUT2. Among the pathways by which increased glucose uptake causes oxidative stress (increased reactive oxygen species (ROS)) are as follows: increased glucose metabolism, including increased oxidative phosphorylation, which consumes oxygen faster than it can be replenished, leading to increased superoxide production; increased substrate for the hexosamine biosynthetic pathway (HBSP), which inhibits generation of NADPH from the pentose phosphate pathway (PPP), which decreases the reducing equivalents needed to generate reduced glutathione (GSH). Increased ROS stimulate activities of AMP-activated protein kinase (AMPK) and DNA methyltransferase 3B (DNMT3B). Both increased AMPK and DNMT3B activities inhibit production of Pax3 mRNA, but it is not known if the effects of ROS on DNMT3B are mediated by phosphorylation by AMPK. Decreased Pax3 mRNA leads to decreased PAX3 protein levels. Because PAX3 stimulates p53 degradation, decreased PAX3 increases p53 stability. Increased p53 protein leads to cell cycle withdrawal, increased oxidative metabolism, decreased pluripotency, and potential changes in folate-dependent 1-Carbon (1C) metabolism. It is not known which individual, or combination, of these processes leads to apoptosis, but increased p53-dependent apoptosis, causes NTDs and CHDs. b—diagram of regulation of the Pax3 CpG island. In undifferentiated embryo cells, methylation of cytosine residues within a CpG island within the Pax3 transcription regulatory element by DNMT3B prevents Pax3 transcription. Upon differentiation, decreased Dnmt3b expression leads to demethylation of the Pax3 CpG island, thereby permitting Pax3 transcription. However, oxidative stress (ROS) stimulates DNMT3B activity, which maintains the Pax3 CpG island methylation pattern of undifferentiated cells, thereby inhibiting Pax3 expression. c—biochemical pathways influenced by GLUT2-transported GlcN metabolism. Glucosamine (GlcN) is transported into embryo cells by GLUT2, which has a KM of 0.8 mmol/L for GlcN. During maternal hyperglycemia, glucose is transported by GLUT2, which has a KM of 16 mmol/L for glucose, and glucose inhibits transport of GlcN. At physiological glucose concentrations, glucose concentrations, glucose is transported by GLUT1 and GLUT3, which have KMs of 5 mmol/L for glucose. GLUT1 can also transport GlcN, with a KM of 2 mmol/L (although circulating GlcN concentrations this high are unlikely to be achieved even when ingesting GlcN supplements). GLUT2-transported GlcN becomes phosphorylated (GlcN-6-P) and enters the HBSP, providing uridine diphosphate N-acetylglucosamine (UDP-N-GlcNAc). UDP-N-GlcNAc is transferred onto lipid carrier molecules in the endoplasmic reticulum (ER) membrane, where it is used for the initiation of N-glycosylation (N-CHO) reactions. Fructose-6-P plus the amino acid Glutamine (Gln) can be converted to GlcN-6-P plus Glutamate (Glu) by the enzyme glutamine fructose-6-phosphate amidotransferase (GFA). However, exogenous GlcN spares fructose-6-P and Gln, providing more glycolytic metabolites, increasing lactate production, and maintaining substrates for the tricarboxylic acid cycle (TCA) and oxidative phosphorylation (Ox Phos). Among the glycolytic metabolites that are increased by exogenous GlcN is glucose-6-P, which stimulates activity of the rate-limiting enzyme of the pentose phosphate pathway (PPP), glucose-6-phosphate dehydrogenase (G6PD), thereby increasing NADPH production. Stimulation of glycolysis and the PPP increase substrates for growth and biomass accumulation
How Does Maternal Diabetes Inhibit Pax3 Expression in Neuroepithelium?
Because the incidence of congenital malformations in pregnancies of women with either type 1 and type 2 diabetes is related to poor glycemic control [2–4], we investigated whether hyperglycemia is responsible for the adverse effects of maternal diabetes on neuroepithelial Pax3 expression, apoptosis, and NTDs. To test this, we induced hyperglycemia for 10 h on E7.5 (equivalent to approximately week 4.5 of human pregnancy), the day prior to the onset of Pax3 expression and the beginning of neural tube closure, with hourly subcutaneous glucose injection of pregnant nondiabetic mice, and we selectively lowered blood glucose levels by injection of pregnant diabetic mice with phlorizin (an SGLT (sodium/glucose cotransporter) −1 and − 2 inhibitor, to normalize blood glucose levels by inhibiting renal reabsorption of glucose) on E6.5–9.5. Transient hyperglycemia on E7.5 was sufficient to inhibit Pax3 expression, increase neural tube apoptosis, and increase NTDs, as long as blood glucose levels on E7.5 were elevated to ≥ 13.9 mmol/L (compared with normal blood glucose levels in nondiabetic pregnant and nonpregnant mice of 8–9.5 mmol/L, and human < 7.8 mmol/L). Phlorizin significantly reduced NTDs in embryos of diabetic mice, as long as blood glucose levels were lowered to < 13.9 mmol/L [38]. This demonstrated that hyperglycemia ≥ 13.9 mmol/L is necessary and sufficient to cause the gene expression and cellular changes leading to NTDs in diabetic pregnancy.
Interestingly, mouse embryos express Slc2a2, the gene encoding the high KM (~ 16 mmol/L) glucose transporter, GLUT2, beginning at the 8-cell stage, and at least through E9.5 (equivalent to week 5 of human pregnancy), as determined from RNA extracted from whole embryos [39]. Because embryos also express the low KM (~ 5 mmol/L) glucose transporters, GLUT1 and GLUT3, encoded by Slc2a1 and Slc2a3, respectively [39–41], which would transport glucose more efficiently than GLUT2 during euglycemia, and embryos would not normally be exposed to glucose concentrations from maternal circulation that are near the GLUT2 KM for glucose, this suggests that embryos may express Slc2a2 to transport a solute other than glucose. Nevertheless, because the adverse effects of hyperglycemia occurred when maternal glucose levels were near the GLUT2 KM for glucose [38], we hypothesized that GLUT2, not GLUT1 or GLUT3, would cause high rates of glucose transport into the embryo during maternal hyperglycemia, leading to malformations. We showed that expression of Slc2a2 does not differ between embryos of diabetic and nondiabetic mice, and that when Slc2a2+/− female and male mice were crossed, in support of our hypothesis, Slc2a2+/− and Slc2a2−/− embryos are protected from NTDs induced by maternal hyperglycemia compared with wild type embryos [42].
Several groups that have used rodent models have provided evidence that oxidative stress mediates the molecular and cellular abnormalities leading to diabetic embryopathy [43–48]. Administration of antioxidants such as vitamin E or the reduced glutathione (GSH) precursor, N-acetylcysteine, or transgenic expression of superoxide dismutases (Cu/Zn-SOD and Mn-SOD), supported involvement of increased free radical production and/or decreased free radical scavenging [43–46, 48–50]. We showed that maternal hyperglycemia induces markers of oxidative stress and that vitamin E and the GSH precursor, glutathione-ethyl ester (GSH-EE), suppressed the effects of hyperglycemia to inhibit Pax3 expression and to increase NTDs; furthermore, the mitochondrial complex III inhibitor, antimycin A, which increases superoxide production, replicated the effects of hyperglycemia to inhibit Pax3 expression in neuroepithelium and to increase NTDs [51]. Importantly, the effects of oxidative stress appear to be due to transcriptional or post-transcriptional effects, not DNA damage, because concentrations of antimycin A that inhibited Pax3 expression by P19 embryonal carcinoma cells in vitro did not induce DNA strand breaks [51]. The processes by which increased rates of glucose uptake induce oxidative stress in embryo cells are complex and appear to involve both increased free radical production and decreased free radical scavenging. We have identified two mechanisms: hypoxia, and increased flux through the hexosamine biosynthetic pathway (HBSP).
Pre- and early postimplantation embryos exist in a relatively hypoxic environment, and exhibit high rates of glycolytic metabolism until coordinated provision of O2 from maternal circulation, development of the circulatory system, and cellular differentiation shift fuel metabolism toward increased mitochondrial biogenesis and aerobic metabolism [52–56]. However, excess glucose uptake may generate higher than normal amounts of substrate for oxidative phosphorylation, potentially consuming oxygen faster than it can be delivered, which can increase superoxide production [57]. We found that 3 h of hyperglycemia of pregnant nondiabetic mice on E7.5 induced embryo hypoxia, as determined by O2-sensing microelectrodes [58]. Furthermore, housing pregnant nondiabetic mice in a hypoxic (12% O2) chamber on E7.5 replicated the effects of maternal hyperglycemia on markers of oxidative stress, Pax3 expression, and NTDs, and the effects of hypoxia on Pax3 and NTDs were inhibited by vitamin E and GSH-EE; conversely, housing pregnant diabetic mice in a hyperoxic (30% O2) chamber on E7.5 suppressed the effects of maternal diabetes on oxidative stress, Pax3 expression, and NTDs [58]. Thus, increased glucose uptake during maternal hyperglycemia appears to create an excessive hypoxic condition, thereby increasing superoxide production.
We also considered that increased glycolytic flux would increase activity of the HBSP, in which fructose-6-PO4, and the amino acid, glutamine (Gln) are converted into glucosamine (GlcN)-6-PO4. GlcN-6-PO4 competes with glucose-6-PO4 for binding to glucose-6-PO4 dehydrogenase (G6PD) [59], the rate limiting enzyme of the pentose phosphate pathway (PPP). The PPP is an important source of NADPH, which is needed to generate GSH from oxidized glutathione (GSSG). We showed that maternal hyperglycemia increases HBSP flux, inhibits G6PD activity, and decreases GSH/GSSG. Administration of GlcN (at doses that would be taken up via GLUT1 [60]) replicated the effects of high glucose on oxidative stress, Pax3 expression, and NTDs, and the effects of exogenous GlcN were inhibited by GSH-EE [61]. This suggests that “glucose loading” of embryo cells by maternal hyperglycemia can decrease free radical scavenging through inhibition of the PPP.
Recent evidence indicates that glycolytic, oxidative, and one-carbon (1C) metabolism, through changes in ratios of NAD+/NADH, NADP+/NADPH, and substrates for epigenetic modifications of DNA and histones by methylation, regulate the differentiated state of embryonic stem cells (ESCs) through changes in gene expression [62, 63••, 64, 65••]. Notably, it has recently been recognized that both cytoplasmic and mitochondrial folate-dependent 1C metabolism are major sources of NADPH in cancer cells and regulate cellular redox status through increasing GSH/GSSG ratios [62, 65••]. What is not yet known is whether 1C metabolism, and consequently, NADP+/NADPH and GSH/GSSG ratios, is altered in embryos of hyperglycemic mothers, or if decreased production of NADPH, due to inhibition of the PPP, depletes substrates for epigenetic modifications of DNA and histones generated by 1C metabolism. There is some evidence that NTDs are decreased when pregnant diabetic rats or mice are administered supplemental folic acid [66, 67], but it has not been determined whether this is due to buffering of NADP+/NADPH and GSH/GSSG ratios. There is also evidence that Pax3Sp/Sp embryos may be protected from NTDs with supplemental folic acid [68]. This suggests that there are downstream effects of PAX3 that are mediated by folic acid. Because the only required PAX3 activity for neural tube closure and cardiac outflow tract septation is mediated by p53 (see below), it is possible that p53 regulates 1C metabolism, and that the effects of supplemental folic acid to reduce NTDs in embryos of diabetic rats and mice are due to p53-regulated 1C metabolism instead of, or in addition to, blocking oxidative stress-mediated inhibition of Pax3 expression.
We considered that AMP-activated protein kinase (AMPK) might sense increased oxidative stress and directly or indirectly inhibit Pax3 expression. AMPK is activated by hypoxia, the hydroxyl radical, and other cellular stresses in which the AMP/ATP ratio is increased [69, 70]. Activated AMPK can regulate transcription by phosphorylating co-activators and transcription factors [71–74]. We showed that maternal hyperglycemia stimulates AMPK activity, and that stimulating AMPK inhibits Pax3 expression in neuroepithelium and induces NTDs [75]. Therefore, while oxidative stress may occur throughout the embryo in response to maternal hyperglycemia, direct or indirect regulation of a tissue specifically expressed gene by an enzyme that is regulated by redox status explains how focused malformations occur.
Differentiation-regulated and tissue-specific genes are kept transcriptionally silent by methylation of CpG islands within their transcriptional regulatory elements, and demethylation of these CpG islands during differentiation is associated with their transcriptional induction [76–80]. We investigated whether oxidative stress inhibits Pax3 expression by regulating methylation of a CpG island within the Pax3 neuroepithelial transcription control region. We found that maternal hyperglycemia and oxidative stress inhibit the differentiation-induced hypomethylation of this CpG island, and that failure to hypomethylate the Pax3 CpG island contributes to inhibition of Pax3 expression. By knocking down expression of each of the DNA methyltransferases (DNMTs), it appears that inhibition of hypomethylation is due to stimulation of DNMT3B activity, with no effect on DNMT1 or DNMT3A [81] (Fig. 1b). A CpG island upstream of the human PAX3 gene has 79% identity with the mouse CpG island, suggesting that they may be under similar transcriptional regulation. It still remains to be determined if DNMT3B activity is regulated by phosphorylation, or if it is phosphorylated by AMPK. It also remains to be determined if any histone-modifying enzymes are regulated by oxidative stress, and if so, whether AMPK mediates these effects.
How Does Insufficient PAX3 Protein Production Lead to NTDs?
Pax3 is one of nine members of the Pax gene family that encode DNA-binding transcription factors that play important roles in Metazoan development [82]. PAX proteins contain a DNA-binding paired domain, a partial or complete DNA-binding homeodomain, and some PAX proteins contain a conserved octapeptide [82]. We and others have identified several direct or indirect targets of PAX3 in tissues where it is expressed, including genes encoding N-CAM, myelin basic protein, the c-Met receptor, MyoD, Myf-5, PAX7, Msx2, Dep-1, and cdc46 [83–92]. It has been generally thought that PAX3 controls neural tube closure, neural crest migration, and skeletal muscle development by serving as a transcription factor of genes that regulate these processes.
Expression of Pax genes, including Pax3, causes oncogenic transformation of cell lines and tumor formation in nude mice, and PAX gene expression is reactivated in many human malignancies [82, 93]. These observations, and that the growth of neural folds of embryos of diabetic mice and Pax3Sp/Sp embryos toward each other appeared to have simply aborted during NTD formation, were what prompted us to examine neuroepithelial apoptosis in these embryos. That neuroepithelial apoptosis was observed in embryos of diabetic mice and Pax3Sp/Sp embryos but not in embryos of nondiabetic mice or Pax3+/+ embryos [35] suggested that, either due to failure of a PAX3-dependent developmental program, or a requirement for PAX3 to promote growth or inhibit apoptosis, PAX3-deficient cells die, thereby leading to NTDs.
To test whether PAX3 inhibits apoptosis, specifically due to a p53-mediated process, we generated Pax3+/Spp53+/− double heterozygous mice and crossed them with each other. We recovered their embryos on E10.5 (equivalent to week 6 of human pregnancy, based on LMP), scored them for NTDs, and assayed them for apoptosis by TUNEL staining [94]. As expected, 100% of Pax3Sp/Spp53+/+ embryos displayed spina bifida and/or exencephaly, and apoptosis at sites of NTDs. Remarkably, homozygous p53 knockout rescued not only apoptosis in Pax3Sp/Spp53−/− embryos but also neural tube closure, as none of the Pax3Sp/Spp53−/− embryos displayed NTDs. These results altered our understanding of how PAX3 directs neural tube closure. They indicated that, while PAX3 might regulate expression of genes that control neural tube closure, it is not required to do so, but PAX3 is required to inhibit p53-dependent apoptosis. We assayed p53 mRNA and p53 protein in wild type, Pax3+/Sp, and Pax3Sp/Sp embryos and found that there was no effect of Pax3 genotype on p53 mRNA, but there was less p53 protein in wild type compared with Pax3Sp/Sp embryos. This suggests that PAX3 has no effect on p53 gene expression, but that it suppresses p53 translation or stability, so that when PAX3 protein is insufficient in neuroepithelial cells, p53 protein increases. Consequently, p53-dependent neuroepithelial cell death occurs.
As noted above, CHDs, cardiac outflow tract defects (COTDs) in particular, are among the most common malformations that occur in human diabetic pregnancy [7, 95]. Defective PAX3-dependent cardiac neural crest cell (CNCC) migration from neuroepithelium is responsible for COTD that occur with 100% penetrance in Pax3Sp/Sp embryos [96, 97]. Based on our findings of hyperglycemia-induced oxidative stress-mediated inhibition of Pax3 expression, and dependency on p53 for NTDs in PAX3-deficient embryos, we investigated whether COTDs occur in our mouse model of diabetic pregnancy and if they occur by the same molecular mechanism. We used two mouse lines, in which LacZ or Green Fluorescent Protein (GFP) were inserted into the Pax3 gene. LacZ interrupted the Pax3 coding sequence and functioned as a Pax3 knockout, so served to report cells that encoded a nonfunctional PAX3, whereas GFP was inserted downstream of the Pax3 coding sequence and had its own internal ribosome entry site (IRES), so served as a reporter of Pax3 expression. We also crossed these mice with p53+/− mice. We found that maternal hyperglycemia inhibited Pax3-expressing CNCC migration and increased COTDs, due to loss of these cells to apoptosis; furthermore, these effects on CNCC migration were due to oxidative stress and were inhibited by vitamin E and GSH-EE [98]. Moreover, the effects of PAX3 deficiency on CNCC migration, apoptosis, and COTDs were prevented by p53 knockout [99]. This indicates that the same cellular and molecular mechanisms may be responsible for congenital malformations in diabetic pregnancy of tissues that are dependent on Pax3.
To investigate how PAX3 regulates p53 synthesis or stability, we used mouse ESCs that can be induced to form Pax3-expressing neuronal precursors. We induced Pax3 in neuronal precursors, transfected with or without Pax3 shRNA expression plasmids to knock down PAX3 protein levels, or transfection of a Pax3 expression plasmid into undifferentiated ESCs. We found that PAX3 has no effect on p53 mRNA levels, but reduces p53 steady state protein levels through decreased p53 stability [100••]. Interestingly, PAX3 physically associated with p53 and the ubiquitin ligase, MDM2, and stimulated ubiquitination of p53 by MDM2, marking p53 for degradation. Surprisingly, when we generated full length PAX3 protein, or only some of the structural domains, and p53 and MDM2, using bacterial expression vectors, and then assayed p53 ubiquitination in vitro, we found that each of the DNA-binding domains (the paired domain and the homeodomain), alone or together, associated with p53 and MDM2 and stimulated MDM2-mediated p53 ubiquitination. Moreover, expressing only the DNA-binding domains, alone or together, even in the absence of the PAX3 transcription regulatory domains, decreased steady state levels of p53 in ESCs [100••]. Taken together with our findings on neural tube and cardiac outflow tract development in embryos lacking both PAX3 and p53 [94, 99], this indicates that PAX3 is not needed to function as a transcription factor to direct neural tube closure or cardiac neural crest migration, but its DNA-binding domains are needed to bind to p53 and stimulate its degradation, thereby inhibiting some p53-dependent process(es), in order to allow neural tube closure and cardiac neural crest migration.
If PAX3 is not needed to regulate neural tube closure and neural crest migration, why is it needed to inhibit a p53-mediated process, and what might that process be? There is recent evidence from the cancer and stem cell fields that p53 is transcriptionally activated by Ser and Thr phosphorylation; in stem cells, this occurs during differentiation of pluripotent cells; in cancer cells, these sites are unphosphorylated or absent. Transcriptionally active p53 promotes differentiation and loss of pluripotency, senescence, or cell cycle withdrawal, and increased oxidative/decreased glycolytic metabolism [101–113]. We can speculate that PAX3 is necessary to suppress p53 activity once neuroepithelial (or neural crest) commitment has initiated; neuroepithelium and neural crest progenitors continuously proliferate as the embryo elongates more than 2-fold and as the neural folds undergo convergent extension, and neural crest cells migrate from the neuroepithelium, all the while blocking further differentiation. Thus, induction of Pax3 expression may be regulated by transition from predominantly glycolytic metabolism to increasing oxidative metabolism that accompanies differentiation [52, 53, 114–116], and PAX3, in turn, may be needed to “brake” oxidative metabolism (and therefore, cell cycle withdrawal and further differentiation) through titrating p53 steady state levels.
There are other functions of PAX3 on the neural tube or neural crest that have been learned from study of mouse strains carrying various mutant Splotch alleles. Others have observed decreased proliferation, but not necessarily apoptosis, in embryos carrying different mutant Pax3 alleles; some of these defects can be complemented with specific 1C donors and may involve epigenetic mechanisms, such as DNA and histone methylation [117–121]. Whether any of these processes are dependent or independent of PAX3 regulation of p53 has not been investigated. However, because the only essential function of PAX3 to control neural tube closure and CNCC migration appears to be to inhibit p53 stability, it is possible that p53 regulates neural tube closure and CNCC migration through regulation of 1C metabolism.
What Is the Normal GLUT2 Function in Embryos?
As noted above, GLUT2 expressed by mouse embryos confers high rates of glucose uptake during maternal hyperglycemia and is required for hyperglycemia-induced NTDs [42]. Surprisingly, Slc2a2+/− and Slc2a2−/− embryos were recovered on E10.5 (equivalent to week 6 of human pregnancy) at fewer than predicted Mendelian frequencies regardless of whether the mothers were hyperglycemic [42], indicating that GLUT2 plays an important function for pre- or early postimplantation embryo survival at physiological glucose concentrations. As explained previously, this function is probably not to transport glucose, but may be to transport another solute. GLUT2 has been shown to be a low KM (0.8 mmol/L) GlcN transporter (whereas GLUT1 is a high KM (2 mmol/L) GlcN transporter) [60]. GlcN in maternal circulation is derived primarily from de novo synthesis in the liver [122, 123]. As described above, GlcN-6-PO4 can be formed from fructose-6-PO4 + Gln to enter the HBSP. The end product of the HBSP is uridine diphosphate N-acetylglucosamine (UDP-N-GlcNAc), which is used in glycosylation reactions in the synthesis of glycoproteins, proteoglycans, and glycolipids [124]. GlcN-6-PO4 can also be synthesized upon phosphorylation of exogenously transported GlcN. We hypothesized that GLUT2 may be important for early embryos to transport GlcN from maternal circulation in order to ensure sufficient substrate for the HBSP. Testing this hypothesis is not possible in vivo because, while it is possible to raise maternal GlcN concentrations (which would increase GLUT1- rather than GLUT2-mediated transport), it is not possible to lower or eliminate maternal GlcN. However, it can be tested in vitro using ESCs that expresses functional GLUT2 transporters.
Most ESC lines do not express functional GLUT2 transporters, probably because they historically have been established from blastocysts in conventional high glucose (25 mmol/L) media [125]. Because blastocysts express GLUT2, culture in high glucose would drive high rates of glucose uptake, which induces oxidative stress [126]. Therefore, ESC lines that survive in high glucose media apparently lose expression of functional GLUT2 transporters and are unresponsive to changes in glucose concentrations in media that correspond to physiological nondiabetic glucose levels (7.7 mmol/L) and those that mimic “hyperglycemic” (17.5 mmol/L) conditions [127••]. The experiments described above to study Pax3 CpG island methylation were performed using the D3 ESC line that was established in media containing 25 mmol/L glucose and is unresponsive to changes in glucose concentrations. Oxidative stress was induced downstream of glucose uptake and metabolism using antimycin A [75]). We recently established an ESC line in low glucose (5.5 mmol/L) media that we named “LG-ESC” [127••]. Unlike D3 ESCs, which express only low KM (4.2 mmol/L) glucose transporters and in which changes in glucose concentrations did not induce markers of oxidative stress or inhibit Pax3 expression, LG-ESCs express high KM (15.8 mmol/L) glucose Z in glucose concentrations did induce markers of oxidative stress and inhibit Pax3 expression [127••]. Therefore, LG-ESCs could be used to test the hypothesis that GLUT2 functions as a GlcN transporter and that exogenous GlcN is beneficial to embryo cells.
GlcN at 0.8 mmol/L is transported by LG-ESCs, and is dependent on GLUT2, as transport was significantly inhibited by knockdown of Glut2 mRNA with shRNA [128••]. GlcN stimulated proliferation and is dependent on GLUT2. Metabolic profiling and study of flux through glycolysis and oxidative phosphorylation indicated that GlcN stimulates proliferation by increasing substrate for the HBSP, thereby decreasing dependence on fructose-6-PO4 and Gln for the HBSP, and increasing substrates for glycolysis and Gln metabolism. Increased glycolytic metabolites included glucose-6-PO4, which increased activity and products of the PPP [128••]. Therefore, exogenous GLUT2-transported GlcN increases the supply of glycolytic, PPP-, and Gln-derived substrates for biomass accumulation and energy production, and also ensures sufficient substrate for glycosylation reactions. GlcN may be an essential nutrient for the embryo (that is, embryo cells can synthesize GlcN-6-PO4 from fructose-6-PO4 + Gln, but not in sufficient amounts) and may explain the survival disadvantage of Slc2a2+/− and Slc2a2−/− embryos. Pathways influenced by GLUT2-transported GlcN are diagrammed in Fig. 1c.
Interestingly, 16 mmol/L glucose inhibited GLUT2-mediated transport of GlcN by 40%, whereas 0.8 mmol/L GlcN inhibited GLUT2-mediated transport of glucose by only 14%. Thus, the recognition that GLUT2 expression may be necessary for normal embryo growth and development to transport GlcN from maternal circulation raises the possibility that some of the adverse effects of maternal hyperglycemia are due to inhibition of GLUT2-mediated GlcN transport. For example, impaired GlcN uptake would divert more fructose-6-PO4 and Gln into the HBSP, thereby decreasing glycolytic intermediates, including glucose-6-PO4, and Gln-derived substrates for growth. However, it is difficult to explain how growth could be restricted to structures that develop malformations, rather than affect the entire embryo. Moreover, while rat embryos of diabetic mothers often display growth and developmental delay, mouse embryos of diabetic mothers, even those displaying malformations, are of normal size and developmental stage for gestational age. Another possibility is that synthesis of HBSP-derived UDP-N-GlcNAc may be insufficient for necessary glycosylation reactions, and insufficient protein glycosylation induces the unfolded protein response (UPR) and endoplasmic reticulum (ER) stress [129]. Others have observed maternal diabetes-induced UPR and ER stress, leading to caspase-8-dependent apoptosis, in a mouse model of diabetic embryopathy [130–132]. The authors attributed the induction of ER stress to oxidative stress [47, 130], but whether the ER stress is due, at least in part, to an UPR resulting insufficient substrate for glycosylation has not been determined. Again, how an UPR leading to apoptosis could be restricted to structures that develop malformations is not known.
Of note, analyses of polymorphisms in parental and off-spring DNA from the National Birth Defects Prevention Study found interactions between a variant of ENPP1 (which encodes a regulator of insulin signaling) carried by mothers and two variants of SLC2A2 carried by fetuses to increase NTD risk, and between two maternal common polymorphic variants of LEP (which encodes the adipocyte hormone Leptin) and the same 2 fetal SLC2A2 variants to decrease NTD risk [133]. The SLC2A2 polymorphisms were within coding regions of the gene, suggesting effects on protein function or stability. In another study, novel gene variants in coding and noncoding variants of SLC2A2 were found to be associated with NTDs in Mexican American subjects [134]. Whether these variants affect GlcN transport and/or maternal circulating GlcN concentrations is not known.
Conclusions
There has been much progress in the past few decades in the understanding of the mechanisms by which maternal diabetes causes congenital malformations. Some findings are clinically translatable. For example, while it has long been known that increased congenital malformations are associated with hyperglycemia in early pregnancy [11, 21, 22], these recent findings indicate that avoiding maternal hyperglycemia would prevent not only excess GLUT2-mediated glucose uptake by embryo cells but also insufficient GlcN uptake. Other findings are mechanistically informative, but not immediately translatable. For example, whereas experimental use of antioxidants has revealed some of the processes by which hyperglycemia induces oxidative stress, and some of the mechanisms by which the adverse effects of oxidative stress occur; it would not be advisable to prophylactically administer supplemental antioxidants to diabetic women who could become pregnant. We found that doses of vitamin E that were effective in mice to prevent diabetic embryopathy were at the upper end of nontoxic doses [51], and because transition to increasingly aerobic fuel metabolism is linked to cellular differentiation [52, 53, 114–116], excess reducing agents may interfere with metabolic cues that regulate cell fate determination, as well as induction of free radical scavenging enzymes. GlcN supplements, to compete with high glucose concentrations for GLUT2-mediated transport, would also not be advisable because high concentrations of GlcN can be transported by GLUT1, with a KM of 2 mmol/L, which would compete with glucose for transport during euglycemia [60]. We have shown that excess GlcN transport during euglycemia inhibits the PPP and decreases GSH, resulting in oxidative stress [61]. As described above, further investigation may determine whether diabetes disturbs 1C-derived NADPH production, and whether normal NADPH levels can be restored by higher doses of folic acid. Functional analyses of human SLC2A2 (GLUT2) variants may provide further information about how this transporter affects neural tube development. Future research is still necessary to improve support of women with diabetes to reduce congenital malformations in their pregnancies.
Funding
This work was supported by the National Institutes of Health (R01 DK052865, R01 DK058300, R01 DK104649 to MRL, and P30 DK036836 to the Joslin Diabetes Center).
Abbreviations
- CNNC
Cardiac neural crest cells
- COHDs
Cardiac outflow tract defects
- CHDs
Congenital heart defects
- DNMT
DNA methyltransferase
- ESC
Embryonic stem cell
- GlcN
Glucosamine
- G6PD
Glucose-6-PO4 dehydrogenase
- Gln
Glutamine
- GSH-EE
Glutathione-ethyl ester
- GFP
Green fluorescent protein
- HBSP
Hexosamine biosynthetic pathway
- IRES
Internal ribosome entry site
- LMP
Last menstrual period
- NTDs
Neural tube defects
- 1C
One carbon
- GSSG
Oxidized glutathione
- PPP
Pentose phosphate pathway
- GSH
Reduced glutathione
- SGLT
Sodium/glucose cotransporter
- SOD
Superoxide dismutase
Footnotes
Conflict of Interest Mary R. Loeken declares that she has no conflicts of interest.
References
Papers of particular interest, published recently, have been highlighted as:
•• Of major importance
- 1.Mills JL. Malformations in infants of diabetic mothers. Teratology. 1982;25(3):385–94. 10.1002/tera.1420250316. [DOI] [PubMed] [Google Scholar]
- 2.Miodovnik M, Mimouni F, Dignan PSJ, Berk MA, Ballard JL, Siddiqi TA, et al. Major malformations in infants of IDDM women: vasculopathy and early first-trimester poor glycemic control. Diabetes Care. 1988;11:713–8. [DOI] [PubMed] [Google Scholar]
- 3.Schaefer-Graf UM, Buchanan TA, Xiang A, Songster G, Montoro M, Kjos SL. Patterns of congenital anomalies and relationship to initial maternal fasting glucose levels in pregnancies complicated by type 2 and gestational diabetes. Am J Obstet Gynecol. 2000;182(2):313–20. [DOI] [PubMed] [Google Scholar]
- 4.Ylinen K, Aula P, Stenman UH, Kesaniemi-Kuokkanen T, Teramo K. Risk of minor and major fetal malformations in diabetics with high haemoglobin A1c values in early pregnancy. Br Med J (Clin Res Ed). 1984;289(6441):345–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.White P. Pregnancy complicating diabetes. Am J Med. 1949;7(5): 609–16. [DOI] [PubMed] [Google Scholar]
- 6.Kucera J. Rate and type of congenital anomalies among offspring of diabetic women. J Reprod Med. 1971;7:61–70. [PubMed] [Google Scholar]
- 7.Loffredo CA, Wilson PD, Ferencz C. Maternal diabetes: an independent risk factor for major cardiovascular malformations with increased mortality of affected infants. Teratology. 2001;64(2): 98–106. [DOI] [PubMed] [Google Scholar]
- 8.Sheffield JS, Butler-Koster EL, Casey BM, McIntire DD, Leveno KJ. Maternal diabetes mellitus and infant malformations. Obstet Gynecol. 2002;100(5 Pt 1):925–30. [DOI] [PubMed] [Google Scholar]
- 9.Wren C, Birrell G, Hawthorne G. Cardiovascular malformations in infants of diabetic mothers. Heart. 2003;89(10):1217–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Correa A, Gilboa SM, Besser LM, Botto LD, Moore CA, Hobbs CA, et al. Diabetes mellitus and birth defects. Am J Obstet Gynecol. 2008;199(3):237 e1–9. 10.1016/j.ajog.2008.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evers IM, de Valk HW, Visser GH. Risk of complications of pregnancy in women with type 1 diabetes: nationwide prospective study in the Netherlands. BMJ. 2004;328(7445):915–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cea-Soriano L, Garcia-Rodriguez LA, Brodovicz KG, Masso Gonzalez E, Bartels DB, Hernandez-Diaz S. Safety of noninsulin glucose-lowering drugs in pregnant women with pregestational diabetes: a cohort study. Diabetes Obes Metab. 2018;20:1642–51. 10.1111/dom.13275. [DOI] [PubMed] [Google Scholar]
- 13.Ludvigsson JF, Neovius M, Soderling J, Gudbjornsdottir S, Svensson AM, Franzen S, et al. Periconception glycaemic control in women with type 1 diabetes and risk of major birth defects: population based cohort study in Sweden. BMJ. 2018;362:k2638 10.1136/bmj.k2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oyen N, Diaz LJ, Leirgul E, Boyd HA, Priest J, Mathiesen ER, et al. Pre-pregnancy diabetes and offspring risk of congenital heart disease: a nation-wide cohort study. Circulation. 2016;133:2243–53. 10.1161/CIRCULATIONAHA.115.017465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Becerra JE, Khoury MJ, Cordero JF, Erickson JD. Diabetes mellitus during pregnancy and the risks for specific birth defects: a population-based case-control study. Pediatrics. 1990;85(1):1–9. [PubMed] [Google Scholar]
- 16.Gilboa SM, Correa A, Botto LD, Rasmussen SA, Waller DK, Hobbs CA, et al. Association between prepregnancy body mass index and congenital heart defects. Am J Obstet Gynecol. 2010;202(1):51 e1–e10. 10.1016/j.ajog.2009.08.005. [DOI] [PubMed] [Google Scholar]
- 17.Watkins ML, Rasmussen SA, Honein MA, Botto LD, Moore CA. Maternal obesity and risk for birth defects. Pediatrics. 2003;111(5 Part 2):1152–8. [PubMed] [Google Scholar]
- 18.Shaw GM, Velie EM, Schaffer D. Risk of neural tube defect-affected pregnancies among obese women. JAMA. 1996;275: 1093–6. [DOI] [PubMed] [Google Scholar]
- 19.Werler MM, Louik C, Shapiro S, Mitchell AA. Prepregnant weight in relation to risk of neural tube defects. JAMA. 1996;275(14):1089–92. 10.1001/jama.1996.03530380031027. [DOI] [PubMed] [Google Scholar]
- 20.Helle EIT, Biegley P, Knowles JW, Leader JB, Pendergrass S, Yang W, et al. First trimester plasma glucose values in women without diabetes are associated with risk for congenital heart disease in offspring. J Pediatr. 2017;195:275–8. 10.1016/j.jpeds.2017.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bell R, Glinianaia SV, Tennant PW, Bilous RW, Rankin J. Periconception hyperglycaemia and nephropathy are associated with risk of congenital anomaly in women with pre-existing diabetes: a population-based cohort study. Diabetologia. 2012;55(4):936–47. 10.1007/s00125-012-2455-y. [DOI] [PubMed] [Google Scholar]
- 22.Kitzmiller JL, Wallerstein R, Correa A, Kwan S. Preconception care for women with diabetes and prevention of major congenital malformations. Birth Defects Res A Clin Mol Teratol. 2010;88(10):791–803. 10.1002/bdra.20734. [DOI] [PubMed] [Google Scholar]
- 23.Robertson H, Pearson DW, Gold AE. Severe hypoglycaemia during pregnancy in women with type 1 diabetes is common and planning pregnancy does not decrease the risk. Diabet Med. 2009;26(8):824–6. 10.1111/j.1464-5491.2009.02769.x. [DOI] [PubMed] [Google Scholar]
- 24.Evers IM, ter Braak EW, de Valk HW, van Der Schoot B, Janssen N, Visser GH. Risk indicators predictive for severe hypoglycemia during the first trimester of type 1 diabetic pregnancy. Diabetes Care. 2002;25(3):554–9. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen LR, Pedersen-Bjergaard U, Thorsteinsson B, Johansen M, Damm P, Mathiesen ER. Hypoglycemia in pregnant women with type 1 diabetes: predictors and role of metabolic control. Diabetes Care. 2008;31(1):9–14. 10.2337/dc07-1066. [DOI] [PubMed] [Google Scholar]
- 26.Temple RC, Aldridge VJ, Murphy HR. Prepregnancy care and pregnancy outcomes in women with type 1 diabetes. Diabetes Care. 2006;29(8):1744–9. 10.2337/dc05-2265. [DOI] [PubMed] [Google Scholar]
- 27.Horii KI, Watanabe GI, Ingalls TH. Experimental diabetes in pregnant mice. Prevention of congenital malformations in offspring by insulin. Diabetes. 1966;15(3):194–204. [DOI] [PubMed] [Google Scholar]
- 28.Sadler T. Effects of maternal diabetes on early embryogenesis: II. Hyperglycemia-induced exencephaly. Teratology. 1980;21(3): 349–56. [DOI] [PubMed] [Google Scholar]
- 29.Sadler TW, et al. Teratology. 1980;21(3):339–47. [DOI] [PubMed] [Google Scholar]
- 30.Cockroft DL, Coppola PT. Teratogenic effects of excess glucose on head-fold rat embryos in culture. Teratology. 1977;16:141–6. [DOI] [PubMed] [Google Scholar]
- 31.Buchanan TA, Denno KM, Sipos GF, Sadler TW. Diabetic teratogenesis. In vitro evidence for a multifactorial etiology with little contribution from glucose per se. Diabetes. 1994;43(5):656–60. [DOI] [PubMed] [Google Scholar]
- 32.Wentzel P, Eriksson UJ. Insulin treatment fails to abolish the teratogenic potential of serum from diabetic rats. Eur J Endocrinol. 1996;134:459–66. [DOI] [PubMed] [Google Scholar]
- 33.Eriksson UJ, Dahlstrom E, Larsson KS, Hellerstrom C. Increased incidence of congenital malformations in the offspring of diabetic rats and their prevention by maternal insulin therapy. Diabetes. 1982;31:1–6. [DOI] [PubMed] [Google Scholar]
- 34.Strieleman PJ, Metzger BE. Glucose and scyllo-inositol impair phosphoinositide hydrolysis in the 10.5-day cultured rat conceptus–a role in dysmorphogenesis? Teratology. 1993;48: 267–78. [DOI] [PubMed] [Google Scholar]
- 35.Phelan SA, Ito M, Loeken MR. Neural tube defects in embryos of diabetic mice: role of the Pax-3 gene and apoptosis. Diabetes. 1997;46(7):1189–97. [DOI] [PubMed] [Google Scholar]
- 36.Chalepakis G, Goulding M, Read A, Strachan T, Gruss P. Molecular basis of splotch and Waardenburg Pax-3 mutations. Proc Natl Acad Sci U S A. 1994;91:3685–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Auerbach R. Analysis of the developmental effects of a lethal mutation in the house mouse. J Exp Zool. 1954;127:305–29. [Google Scholar]
- 38.Fine E, Horal M, Chang T, Fortin G, Loeken M. Evidence that hyperglycemia causes altered gene expression, apoptosis, and neural tube defects in a mouse model of diabetic pregnancy. Diabetes. 1999;48:2454–62. [DOI] [PubMed] [Google Scholar]
- 39.Hogan A, Heyner S, Charron MJ, Copeland NG, Gilbert DJ, Jenkins NA, et al. Glucose transporter gene expression in early mouse embryos. Development. 1991;113(1):363–72. [DOI] [PubMed] [Google Scholar]
- 40.Trocino RA, Akazawa S, Takino H, Takao Y, Matsumoto K, Maeda Y, et al. Cellular-tissue localization and regulation of the GLUT-1 protein in both the embryo and the visceral yolk sac from normal and experimental diabetic rats during the early postimplantation period. Endocrinology. 1994;134(2):869–78. [DOI] [PubMed] [Google Scholar]
- 41.Matsumoto K, Akazawa S, Ishibashi M, Trocino RA, Matsuo H, Yamasaki H, et al. Abundant expression of GLUT1 and GLUT3 in rat embryo during the early organogenesis period. Biochem Biophys Res Commun. 1995;209(1):95–102. [DOI] [PubMed] [Google Scholar]
- 42.Li R, Thorens B, Loeken MR. Expression of the gene encoding the high Km glucose transporter 2 by the early postimplantation mouse embryo is essential for neural tube defects associated with diabetic embryopathy. Diabetologia. 2007;50(3):682–9. 10.1007/s00125-006-0579-7. [DOI] [PubMed] [Google Scholar]
- 43.Siman CM, Eriksson UJ. Vitamin E decreases the occurrence of malformations in the offspring of diabetic rats. Diabetes. 1997;46: 1054–61. [DOI] [PubMed] [Google Scholar]
- 44.Sivan E, Reece EA, Wu Y-K, Homko CJ, Polansky M, Borenstein M. Dietary vitamin E prophylaxis and diabetic embryopathy: morphologic and biochemical analysis. Am J Obstet Gynecol. 1996;175:793–9. [DOI] [PubMed] [Google Scholar]
- 45.Viana M, Herrera E, Bonet B. Terotogenic effects of diabetes mellitus in the rat. Prevention by vitamin E. Diabetologia. 1996;39:1041–6. [DOI] [PubMed] [Google Scholar]
- 46.Hagay ZJ, Weiss Y, Zusman I, Peled-Kamar M, Reece EA, Eriksson UJ, et al. Prevention of diabetes-associated embryopathy by overexpression of the free radical scavenger copper zinc superoxide dismutase in transgenic mouse embryos. Am J Obstet Gynecol. 1995;173:1036–41. [DOI] [PubMed] [Google Scholar]
- 47.Yang P, Zhao Z, Reece EA. Activation of oxidative stress signaling that is implicated in apoptosis with a mouse model of diabetic embryopathy. Am J Obstet Gynecol. 2008;198(1):130 e1–7. 10.1016/j.ajog.2007.06.070. [DOI] [PubMed] [Google Scholar]
- 48.Trocino RA, Akazawa S, Ishibashi M, Matsumoto K, Matsuo H, Yamamoto H, et al. Significance of glutathione depletion and oxidative stress in early embryogenesis in glucose-induced rat embryo culture. Diabetes. 1995;44:992–8. [DOI] [PubMed] [Google Scholar]
- 49.Eriksson UJ, Borg LAH. Protection by free oxygen radical scavenging enzymes against glucose-induced embryonic malformations in vitro. Diabetologia. 1991;34:325–31. [DOI] [PubMed] [Google Scholar]
- 50.Forsberg H, Borg LA, Cagliero E, Eriksson UJ. Altered levels of scavenging enzymes in embryos subjected to a diabetic environment. Free Radic Res. 1996;24:451–9. [DOI] [PubMed] [Google Scholar]
- 51.Chang TI, Horal M, Jain S, Wang F, Patel R, Loeken MR. Oxidant regulation of gene expression and neural tube development: insights gained from diabetic pregnancy on molecular causes of neural tube defects. Diabetologia. 2003;46:538–45. [DOI] [PubMed] [Google Scholar]
- 52.Akazawa S, Unterman T, Metzger BE. Glucose metabolism in separated embryos and investing membranes during organogenesis in the rat. Metabolism. 1994;43(7):830–5. [DOI] [PubMed] [Google Scholar]
- 53.Shepard TH, Tanimura T, Robkin MA. Energy metabolism in early mammalian embryos. Symp Soc Dev Biol. 1970;29:42–58. [PubMed] [Google Scholar]
- 54.Fischer B, Bavister BD. Oxygen tension in the oviduct and uterus of rhesus monkeys, hamsters and rabbits. J Reprod Fertil. 1993;99(2):673–9. [DOI] [PubMed] [Google Scholar]
- 55.Rodesch F, Simon P, Donner C, Jauniaux E. Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet Gynecol. 1992;80(2):283–5. [PubMed] [Google Scholar]
- 56.Quinn P, Harlow GM. The effect of oxygen on the development of preimplantation mouse embryos in vitro. J Exp Zool. 1978;206(1): 73–80. [DOI] [PubMed] [Google Scholar]
- 57.Paddenberg R, Ishaq B, Goldenberg A, Faulhammer P, Rose F, Weissmann N, et al. Essential role of complex II of the respiratory chain in hypoxia-induced ROS generation in the pulmonary vasculature. Am J Phys Lung Cell Mol Phys. 2003;284(5):L710–9. [DOI] [PubMed] [Google Scholar]
- 58.Li R, Chase M, Jung SK, Smith PJS, Loeken MR. Hypoxic stress in diabetic pregnancy contributes to impaired embryo gene expression and defective development by inducing oxidative stress. Am J Physiol Endocrinol Metab. 2005;289(4):E591–9. [DOI] [PubMed] [Google Scholar]
- 59.Kanji MI, Toews ML, Carper WR. A kinetic study of glucose-6-phosphate dehydrogenase. J Biol Chem. 1976;251(8):2258–62. [PubMed] [Google Scholar]
- 60.Uldry M, Ibberson M, Hosokawa M, Thorens B. GLUT2 is a high affinity glucosamine transporter. FEBS Lett. 2002;524(1–3):199–203. [DOI] [PubMed] [Google Scholar]
- 61.Horal M, Zhang Z, Virkamaki A, Stanton R, Loeken MR. Activation of the hexosamine pathway causes oxidative stress and abnormal embryo gene expression: involvement in diabetic teratogenesis. Birth Defects Res Part A Clin Mol Teratol. 2004;70:519–27. [DOI] [PubMed] [Google Scholar]
- 62.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510(7504):298–302. 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.••.Ryall JG, Cliff T, Dalton S, Sartorelli V. Metabolic reprogramming of stem cell epigenetics. Cell Stem Cell. 2015;17(6):651–62. 10.1016/j.stem.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reviews how metabolites generated during glycolytic and oxidative processes of stem cells regulated enzymes that stimulate epigenetic modifications affecting transcriptional regulation during differentiation and lineage commitment.
- 64.Sperber H, Mathieu J, Wang Y, Ferreccio A, Hesson J, Xu Z, et al. The metabolome regulates the epigenetic landscape during naive-to-primed human embryonic stem cell transition. Nat Cell Biol. 2015;17(12):1523–35. 10.1038/ncb3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.••.Lewis CA, Parker SJ, Fiske BP, McCloskey D, Gui DY, Green CR, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol Cell. 2014;55(2):253–63. 10.1016/j.molcel.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper shows how folate-mediated 1 Carbon metabolism in cytosol and mitochondria compartments regulates NADPH production.
- 66.Oyama K, Sugimura Y, Murase T, Uchida A, Hayasaka S, Oiso Y, et al. Folic acid prevents congenital malformations in the offspring of diabetic mice. Endocr J. 2008;56(1):29–37. [DOI] [PubMed] [Google Scholar]
- 67.Wentzel P, Eriksson UJ. A diabetes-like environment increases malformation rate and diminishes prostaglandin E(2) in rat embryos: reversal by administration of vitamin E and folic acid. Birth Defects Res A Clin Mol Teratol. 2005;73(7):506–11. 10.1002/bdra.20145. [DOI] [PubMed] [Google Scholar]
- 68.Wlodarczyk BJ, Tang LS, Triplett A, Aleman F, Finnell RH. Spontaneous neural tube defects in splotch mice supplemented with selected micronutrients. Toxicol Appl Pharmacol. 2006;213(1):55–63. 10.1016/j.taap.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 69.Hardie DG. Roles of the AMP-activated/SNF1 protein kinase family in the response to cellular stress. Biochem Soc Symp. 1999;64:13–27. [PubMed] [Google Scholar]
- 70.Choi SL, Kim SJ, Lee KT, Kim J, Mu J, Birnbaum MJ, et al. The regulation of AMP-activated protein kinase by H(2)O(2). Biochem Biophys Res Commun. 2001;287(1):92–7. [DOI] [PubMed] [Google Scholar]
- 71.Yang W, Hong YH, Shen XQ, Frankowski C, Camp HS, Leff T. Regulation of transcription by AMP-activated protein kinase: phosphorylation of p300 blocks its interaction with nuclear receptors. J Biol Chem. 2001;276(42):38341–4. [DOI] [PubMed] [Google Scholar]
- 72.Hong YH, Varanasi US, Yang W, Leff T. AMP-activated protein kinase regulates HNF4alpha transcriptional activity by inhibiting dimer formation and decreasing protein stability. J Biol Chem. 2003;278(30):27495–501. [DOI] [PubMed] [Google Scholar]
- 73.Bergeron R, Ren JM, Cadman KS, Moore IK, Perret P, Pypaert M, et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am J Physiol Endocrinol Metab. 2001;281(6):E1340–6. [DOI] [PubMed] [Google Scholar]
- 74.Lee M, Hwang JT, Lee HJ, Jung SN, Kang I, Chi SG, et al. AMP-activated protein kinase activity is critical for hypoxia-inducible factor-1 transcriptional activity and its target gene expression under hypoxic conditions in DU145 cells. J Biol Chem. 2003;278(41):39653–61. [DOI] [PubMed] [Google Scholar]
- 75.Wu Y, Viana M, Thirumangalathu S, Loeken MR. AMP-activated protein kinase mediates effects of oxidative stress on embryo gene expression in a mouse model of diabetic embryopathy. Diabetologia. 2012;55(1):245–54. 10.1007/s00125-011-2326-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454(7205): 766–70. 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Borgel J, Guibert S, Li Y, Chiba H, Schubeler D, Sasaki H et al. Targets and dynamics of promoter DNA methylation during early mouse development. Nat Genet. 2010;42(12):1093–100. 10.1038/ng.708. [DOI] [PubMed] [Google Scholar]
- 78.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nature reviews Genetics. 2013;14(3):204–20. 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 79.Fouse SD, Shen Y, Pellegrini M, Cole S, Meissner A, Van Neste L et al. Promoter CpG methylation contributes to ES cell gene regulation in parallel with Oct4/Nanog, PcG complex, and histone H3 K4/K27 trimethylation. Cell Stem Cell. 2008;2(2):160–9. 10.1016/j.stem.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448(7153):553–60. 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wei D, Loeken MR. Increased DNA methyltransferase 3b (dnmt3b)-mediated CpG island methylation stimulated by oxidative stress inhibits expression of a gene required for neural tube and neural crest development in diabetic pregnancy. Diabetes. 2014;63(10):3512–22. 10.2337/db14-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robson EJ, He SJ, Eccles MR. A PANorama of PAX genes in cancer and development. Nat Rev Cancer. 2006;6(1):52–62. [DOI] [PubMed] [Google Scholar]
- 83.Kwang SJ, Brugger SM, Lazik A, Merrill AE, Wu LY, Liu YH, et al. Msx2 is an immediate downstream effector of Pax3 in the development of the murine cardiac neural crest. Development. 2002;129(2):527–38. [DOI] [PubMed] [Google Scholar]
- 84.Borycki AG, Li J, Jin F, Emerson CP, Epstein JA. Pax3 functions in cell survival and in pax7 regulation. Development. 1999;126(8):1665–74. [DOI] [PubMed] [Google Scholar]
- 85.Moase CE, Trasler DG. N-CAM alterations in splotch neural tube defect mouse embryos. Development. 1991;113(3):1049–58. [DOI] [PubMed] [Google Scholar]
- 86.Kioussi C, Gross MK, Gruss P. Pax3: a paired domain gene as a regulator in PNS myelination. Neuron. 1995;15:553–62. [DOI] [PubMed] [Google Scholar]
- 87.Tajbakhsh S, Rocancourt D, Cossu G, Buckingham M. Redefining the genetic heirarchies controlling skeletal myogenesis: Pax-3 and Myf-5 act upstream of MyoD. Cell. 1997;89:127–38. [DOI] [PubMed] [Google Scholar]
- 88.Maroto M, Reshef R, Munsterberg AE, Koester S, Goulding M, Lassar AB. Ectopic Pax-3 activates MyoD and Myf-5 expression in embryonic mesoderm and neural tissue. Cell. 1997;89:139–48. [DOI] [PubMed] [Google Scholar]
- 89.Epstein JA, Shapiro DN, Chang J, Lam PYP, Maas RL. Pax3 modulates expression of the c-Met receptor during limb muscle development. Proc Natl Acad Sci U S A. 1996;93:4213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Laborda J. 36B4 cDNA used as an estradiol-independent mRNA control is the cDNA for human acidic ribosomal phosphoprotein PO. Nucleic Acids Res. 1991;19:3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hill AL, Phelan SA, Loeken MR. Reduced expression of Pax-3 is associated with overexpression of cdc46 in the mouse embryo. Dev Genes Evol. 1998;208:128–34. [DOI] [PubMed] [Google Scholar]
- 92.Cai J, Phelan SA, Hill AL, Loeken MR. Identification of Dep-1, a new gene that is regulated by the transcription factor, Pax-3, as a marker for altered embryonic gene expression during diabetic pregnancy. Diabetes. 1998;47:1803–5. [DOI] [PubMed] [Google Scholar]
- 93.Maulbecker CC, Gruss P. The oncogenic potential of Pax genes. EMBO J. 1993;12:2361–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pani L, Horal M, Loeken MR. Rescue of neural tube defects in Pax-3-deficient embryos by p53 loss of function: implications for Pax-3-dependent development and tumorigenesis. Genes Dev. 2002;16(6):676–80. 10.1101/gad.969302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ferencz C, Rubin JD, McCarter RJ, Clark EB. Maternal diabetes and cardiovascular malformations: predominance of double outlet right ventricle and truncus arteriosus. Teratology. 1990;41(3): 319–26. 10.1002/tera.1420410309. [DOI] [PubMed] [Google Scholar]
- 96.Kirby ML, Turnage KL 3rd, Hays BM. Characterization of conotruncal malformations following ablation of “cardiac” neural crest. Anat Rec. 1985;213(1):87–93. [DOI] [PubMed] [Google Scholar]
- 97.Conway SJ, Henderson DJ, Copp AJ. Pax3 is required for cardiac neural crest migration in the mouse: evidence from the splotch (Sp2H) mutant. Development. 1997;124:505–14. [DOI] [PubMed] [Google Scholar]
- 98.Morgan SC, Relaix F, Sandell LL, Loeken MR. Oxidative stress during diabetic pregnancy disrupts cardiac neural crest migration and causes outflow tract defects. Birth Defects Res A Clin Mol Teratol. 2008;82(6):453–63. 10.1002/bdra.20457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Morgan SC, Lee HY, Relaix F, Sandell LL, Levorse JM, Loeken MR. Cardiac outflow tract septation failure in Pax3-deficient embryos is due to p53-dependent regulation of migrating cardiac neural crest. Mech Dev. 2008;125(9–10):757–67. 10.1016/j.mod.2008.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.••.Wang XD, Morgan SC, Loeken MR. Pax3 stimulates p53 ubiquitination and degradation independent of transcription. PLoS One. 2011;6(12):e29379 10.1371/journal.pone.0029379. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated that PAX3 DNA-binding domains associate with MDM2 and p53 and stimulate p53 degradation separate from PAX3’s activity as a transcription factor.
- 101.Ma W, Sung HJ, Park JY, Matoba S, Hwang PM. A pivotal role for p53: balancing aerobic respiration and glycolysis. J Bioenerg Biomembr. 2007;39(3):243–6. [DOI] [PubMed] [Google Scholar]
- 102.Lundberg AS, Hahn WC, Gupta P, Weinberg RA. Genes involved in senescence and immortalization. Curr Opin Cell Biol. 2000;12(6):705–9. [DOI] [PubMed] [Google Scholar]
- 103.Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, et al. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460(7259): 1132–5. 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kawamura T, Suzuki J, Wang YV, Menendez S, Morera LB, Raya A, et al. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature. 2009;460(7259):1140–4. 10.1038/nature08311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kondoh H, Lleonart ME, Gil J, Wang J, Degan P, Peters G, et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 2005;65(1):177–85. [PubMed] [Google Scholar]
- 106.Li M, He Y, Dubois W, Wu X, Shi J, Huang J. Distinct regulatory mechanisms and functions for p53-activated and p53-repressed DNA damage response genes in embryonic stem cells. Mol Cell. 2012;46(1):30–42. 10.1016/j.molcel.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lin T, Chao C, Saito S, Mazur SJ, Murphy ME, Appella E, et al. p53 induces differentiation of mouse embryonic stem cells by suppressing Nanog expression. Nat Cell Biol. 2005;7(2):165–71. [DOI] [PubMed] [Google Scholar]
- 108.Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, et al. p53 regulates mitochondrial respiration. Science. 2006;312(5780):1650–3. 10.1126/science.1126863. [DOI] [PubMed] [Google Scholar]
- 109.Pan G, Thomson JA. Nanog and transcriptional networks in embryonic stem cell pluripotency. Cell Res. 2007;17(1):42–9. 10.1038/sj.cr.7310125. [DOI] [PubMed] [Google Scholar]
- 110.Qin H, Yu T, Qing T, Liu Y, Zhao Y, Cai J, et al. Regulation of apoptosis and differentiation by p53 in human embryonic stem cells. J Biol Chem. 2007;282(8):5842–52. 10.1074/jbc.M610464200. [DOI] [PubMed] [Google Scholar]
- 111.Solozobova V, Blattner C. Regulation of p53 in embryonic stem cells. Exp Cell Res. 2010;316(15):2434–46. 10.1016/j.yexcr.2010.06.006. [DOI] [PubMed] [Google Scholar]
- 112.Utikal J, Polo JM, Stadtfeld M, Maherali N, Kulalert W, Walsh RM, et al. Immortalization eliminates a roadblock during cellular reprogramming into iPS cells. Nature. 2009;460(7259):1145–8. 10.1038/nature08285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yang A, Shi G, Zhou C, Lu R, Li H, Sun L, et al. Nucleolin maintains embryonic stem cell self-renewal by suppression of the p53-dependent pathway. J Biol Chem. 2011;286:43370–82. 10.1074/jbc.M111.225185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mandal S, Lindgren AG, Srivastava AS, Clark AT, Banerjee U. Mitochondrial function controls proliferation and early differentiation potential of embryonic stem cells. Stem Cells. 2011;29(3): 486–95. 10.1002/stem.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Facucho-Oliveira JM, St John JC. The relationship between pluripotency and mitochondrial DNA proliferation during early embryo development and embryonic stem cell differentiation. Stem Cell Rev. 2009;5(2):140–58. 10.1007/s12015-009-9058-0. [DOI] [PubMed] [Google Scholar]
- 116.Ellington SK. In vitro analysis of glucose metabolism and embryonic growth in postimplantation rat embryos. Development. 1987;100(3):431–9. [DOI] [PubMed] [Google Scholar]
- 117.Conway SJ, Bundy J, Chen J, Dickman E, Rogers R, Will BM. Decreased neural crest stem cell expansion is responsible for the conotruncal heart defects within the splotch (Sp(2H))/Pax3 mouse mutant. Cardiovasc Res. 2000;47(2):314–28. [DOI] [PubMed] [Google Scholar]
- 118.Fleming A, Copp AJ. Embryonic folate metabolism and mouse neural tube defects. Science. 1998;280(5372):2107–9. [DOI] [PubMed] [Google Scholar]
- 119.Sudiwala S, Palmer A, Massa V, Burns AJ, Dunlevy LPE, de Castro SCP, et al. Cellular mechanisms underlying Pax3-related neural tube defects and their prevention by folic acid. Dis Model Mech. 2019;12(11). 10.1242/dmm.042234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Burren KA, Savery D, Massa V, Kok RM, Scott JM, Blom HJ, et al. Gene-environment interactions in the causation of neural tube defects: folate deficiency increases susceptibility conferred by loss of Pax3 function. Hum Mol Genet. 2008;17(23):3675–85. 10.1093/hmg/ddn262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ichi S, Costa FF, Bischof JM, Nakazaki H, Shen YW, Boshnjaku V, et al. Folic acid remodels chromatin on Hes1 and Neurog2 promoters during caudal neural tube development. J Biol Chem. 2010;285(47):36922–32. 10.1074/jbc.M110.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Spiro RG. Role of insulin in two pathways of glucose metabolism: in vivo glucosamine and glycogen synthesis. Ann N Y Acad Sci. 1959;82:366–73. [DOI] [PubMed] [Google Scholar]
- 123.Setnikar I, Rovati LC. Absorption, distribution, metabolism and excretion of glucosamine sulfate. A review. Arzneimittel-Forschung. 2001;51(9):699–725. 10.1055/s-00311300105. [DOI] [PubMed] [Google Scholar]
- 124.Fleischer B. Mechanism of glycosylation in the Golgi apparatus. J Histochem Cytochem. 1983;31(8):1033–40. [DOI] [PubMed] [Google Scholar]
- 125.Abbondanzo S, Gadi I, Stewart C. Derivation of embryonic stem cell lines In: Wasserman PM, Pamphilis MLD, editors. Methods in enzymology. New York: Academic Press; 1993. p. 803–23. [DOI] [PubMed] [Google Scholar]
- 126.Wang F, Thirumangalathu S, Loeken MR. Establishment of new mouse embryonic stem cell lines is improved by physiological glucose and oxygen. Cloning Stem Cells. 2006;8(2):108–16. [DOI] [PubMed] [Google Scholar]
- 127.••.Jung JH, Wang XD, Loeken MR. Mouse embryonic stem cells established in physiological-glucose media express the high KM Glut2 glucose transporter expressed by normal embryos. Stem Cells Transl Med. 2013;2(12):929–34. 10.5966/sctm.2013-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes glucose transport kinetics and sensitivity to high glucose concentrations of LG-ESC lines that were islated in physiological glucose media.
- 128.••.Jung JH, Iwabuchi K, Yang Z, Loeken MR. Embryonic stem cell proliferation stimulated by altered anabolic metabolism from glucose transporter 2-transported glucosamine. Sci Rep. 2016;6: 28452 10.1038/srep28452. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper provides functional evidence that GLUT2 may function as a glucosamine transporter for embryo cells to stimulate growth.
- 129.Shang J, Korner C, Freeze H, Lehrman MA. Extension of lipidlinked oligosaccharides is a high-priority aspect of the unfolded protein response: endoplasmic reticulum stress in Type I congenital disorder of glycosylation fibroblasts. Glycobiology. 2002;12(5):307–17. [DOI] [PubMed] [Google Scholar]
- 130.Wang F, Reece EA, Yang P. Superoxide dismutase 1 overexpression in mice abolishes maternal diabetes-induced endoplasmic reticulum stress in diabetic embryopathy. Am J Obstet Gynecol. 2013;209(4):345 e1–7. 10.1016/j.ajog.2013.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wang F, Wu Y, Gu H, Reece EA, Fang S, Gabbay-Benziv R, et al. Ask1 gene deletion blocks maternal diabetes-induced endoplasmic reticulum stress in the developing embryo by disrupting the unfolded protein response signalosome. Diabetes. 2015;64(3): 973–88. 10.2337/db14-0409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhao Z, Yang P, Eckert RL, Reece EA. Caspase-8: a key role in the pathogenesis of diabetic embryopathy. Birth Defects Res B Dev Reprod Toxicol. 2009;86(1):72–7. 10.1002/bdrb.20185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lupo PJ, Mitchell LE, Canfield MA, Shaw GM, Olshan AF, Finnell RH, et al. Maternal-fetal metabolic gene-gene interactions and risk of neural tube defects. Mol Genet Metab. 2014;111(1): 46–51. 10.1016/j.ymgme.2013.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ruggiero JE, Northrup H, Au KS. Association of facilitated glucose transporter 2 gene variants with the myelomeningocele phenotype. Birth Defects Res A Clin Mol Teratol. 2015;103:479–87. 10.1002/bdra.23358. [DOI] [PMC free article] [PubMed] [Google Scholar]