

Abstract
The synaptic regulator, kalirin, plays a key role in synaptic plasticity and formation of dendritic arbors and spines. Dysregulation of the KALRN gene has been linked to various neurological disorders, including autism spectrum disorder, Alzheimer’s disease, schizophrenia, addiction and intellectual disabilities. Both genetic and molecular studies highlight the importance of normal KALRN expression for healthy neurodevelopment and function. This review aims to give an in-depth analysis of the structure and molecular mechanisms of kalirin function, particularly within the brain. These data are correlated to genetic evidence of patient mutations within KALRN and animal models of Kalrn that together give insight into the manner in which this gene may be involved in neurodevelopment and the etiology of disease. The emerging links to human disease from post-mortem, genome wide association (GWAS) and exome sequencing studies are examined to highlight the disease relevance of kalirin, particularly in neurodevelopmental diseases. Finally, we will discuss efforts to pharmacologically regulate kalirin protein activity and the implications of such endeavors for the treatment of human disease. As multiple disease states arise from deregulated synapse formation and altered KALRN expression and function, therapeutics may be developed to provide control over KALRN activity and thus synapse dysregulation. As such, a detailed understanding of how kalirin regulates neuronal development, and the manner in which kalirin dysfunction promotes neurological disease, may support KALRN as a valuable therapeutic avenue for future pharmacological intervention.
Keywords: KALRN, Kalirin, Dendritic spine, Synaptic plasticity, Schizophrenia, Autism spectrum disorder, Alzheimer’s disease, Developmental delay, Neurodevelopment, Neurodegeneration
1. Introduction
Since its discovery in 1996 (Alam et al., 1997; Kawai et al., 1999; Colomer et al., 1997), kalirin has emerged as a key player in the formation and stabilization of synaptic connections and neural circuits, as well as a factor in various psychiatric disorders. Kalirin (aka DUO/HAPIP (Colomer et al., 1997), encoded by the KALRN gene (aka ARH-GEF24), is a member of the Rho-guanosine nucleotide exchange factor (GEF) family. This review aims to outline the key discoveries that have propelled the study of kalirin to the forefront of synaptic neuroscience. We will discuss its role in disease, particularly diseases displaying altered synapse formation, emerging genetic relevance and the molecular pathways that mediate its regulation of neuronal biology. To our knowledge, no existing review on kalirin covers the broad scope of kalirin function, links to human disease and animal phenotypes in such depth and breadth. Special attention has been paid to the emerging links to human disease from post-mortem, genome wide association (GWAS) and exome sequencing studies. These resources provide both interesting case studies and highlight the disease relevance of kalirin, particularly in neurodevelopmental diseases. These observations will be supported by an in-depth summary of the functional roles of each protein domain, allowing correlation between point mutations found in patients and potential alterations in KALRN function that may underlie disease. Moreover, characterization of the available mouse models reveals disease related animal phenotypes that may allow the role of KALRN, and disease related alterations in protein function, to be linked to neurological dysfunction. Finally, we will address the attempts made to pharmacologically regulate KALRN, and provide arguments supporting this as a possible route to the treatment of human disease.
2. Discovery and characterization
Kalirin was isolated as a GEF-domain containing protein, and found to share considerable similarity to several members of the Dbl family of Rho-GEFs, with 60% homology to trio (Kawai et al., 1999; Alam et al., 1996). Originally designated as P-CIP10, kalirin was characterized as a Rac-GEF by its interaction with, and ability to induce actin and morphological rearrangements through, RAC1 (Alam et al., 1997). P-CIP10 was subsequently dubbed kalirin after the many-handed goddess, Kali, reflecting its multiple protein binding properties (see Section 2). Soon afterwards, another isoform, dubbed Δkal (also referred to as kalirin5) was isolated and expression profiling suggested complex transcriptional regulation of KALRN (Johnson et al., 2000). Further, a homologous human variant was isolated from skeletal muscle and dubbed duet (Kawai et al., 1999). Both Δkal and duet are distinct from kalirin in that they lack N-terminal structural regions (Kawai et al., 1999; Johnson et al., 2000). Duet was later found to be a splice variant of kalirin, present in both human and mouse and arising from a start codon between exons 34 and 35, whereas Δkal arose from transcriptional activation within the spectrin repeat domain (McPherson et al., 2002), see Fig. 1, Table1).
Fig. 1.
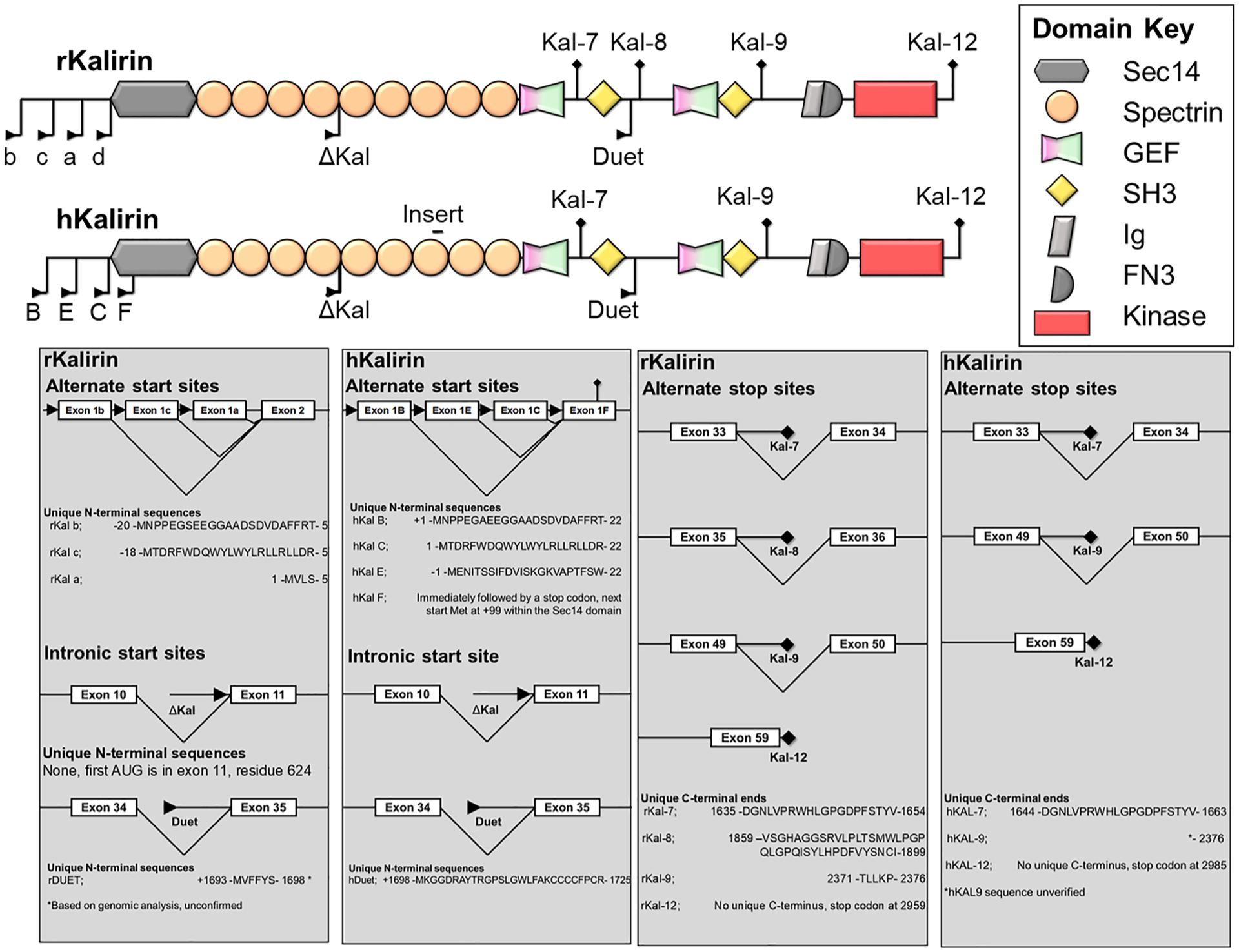
Alternate start sites and splicing generate a variety of human (h) and murine (r) kalirin isoforms. “insert” indicates a 9 amino acid insertion present in human kalirin, but not murine protein (Mandela and Ma, 2012). “b”, “c”, “a” and “d” indicate alternate start sites in murine KALRN, or “B”, “E” “C” and “F” in human KALRN, encoding the alternate N-terminal regions prior to the SEC14P domain. Delta-Kal indicates isoforms lacking the C-terminal region due to use of an internal start site within the spectrin repeat domains. Note - Kalirin7 (kal-7) is also referred to as Duo in some texts.
Table 1.
Genetic and protein properties and identifiers.
| DNA | Species | Gene Symbol | Chromosome | Ensembl Id | Gene position/length (GRCh38/GRCm38) |
| Human | KALRN | 3 | ENSG00000160145 | 124,080,023–124,726,325 (646,302 bp) | |
| Mouse | Kalrn | 16 | ENSMUSG00000061751 | 33,969,073–34,573,532 (604,459 bp) | |
| Protein | Species | Major Isoforms encoded | Uniprot Id | Length (a.a) | Mass (kDa) |
| Human | Kalirin-12 | O60229-1 | 2986 | 340 | |
| Kalirin-7 | O60229- | 1663 | 192 | ||
| Duet | O60229-4 | 1289 | 144 | ||
| Mouse | Kalirin-12 | A2CG49-1 | 2964 | 337 | |
| Kalirin-9 | A2CG49-2 | 2375 | 271 | ||
| Kalirin-7 | A2CG49-9 | 1654 | 191 |
Further transcriptional and expression profiles identified additional variants, arising from alternate promoters and splicing (Fig. 1), dubbed kalirin7, 8, 9 and 12 (Johnson et al., 2000; McPherson et al., 2002). This isoform complexity is compounded by the differential effects of alternate promoter-driven 5′ exons; A, E, C and F, and internal start sites resulting in N-terminally truncated kalirin isoforms (Δkal/duet) (Mains et al., 2011; Miller et al., 2015). Alternate promoters have been shown to be differentially expressed throughout the brain, and the resulting N-terminal extensions have been shown to alter lipid-binding properties and impart distinct cellular functions (Miller et al., 2015). For example, C-kalirin is expressed in all regions, whereas B-kalirin displays restricted expression within the prefrontal cortex (Mains et al., 2011).
Each isoform of kalirin exhibits distinct expression patterns within the brain throughout development, with kalirin9 (predominantly) and 12 expressed during embryonic neuronal development, whereas kalirin7 protein expression was observed only in adult (Hansel et al., 2001) cortex (Mandela and Ma, 2012). Indeed, in the adult cortex, expression appeared to switch almost exclusively to kalirin7, with the expression of other isoforms dramatically reduced (Hansel et al., 2001). During embryonic development kalirin8, 9 and 12 are present in multiple murine tissues, including intestines, tongue, kidney/adrenals, lungs, heart (Hansel et al., 2001) and smooth muscle cells (Wu et al., 2013). These studies highlight the restricted expression of kalirin7 within the mature cortex, and suggest a shift in KALRN gene function within later neuronal development to a role predominated by kalirin7.
3. Protein architecture and domains.
In order to understand the functional implications of each isoform generated by alternate splicing and promoter usage, it is necessary to assess the domains present and absent in each isoform. Each isoform expresses a distinct set of domains that together alter kalirin signaling. Here we will outline the role each domain and how they pertain to kalirin function.
3.1. SEC14P domain
Within the N-terminus and expressed in all isoforms (except those using intronic start sites, Δkal and Duet) lies a SEC14P domain (aka CRAL-TRIO domain), a well-characterized lipid-binding site (Schiller et al., 2008). Several SEC14P domains have been linked to intramolecular regulation of GEF activity of other related Rho-GEF proteins; for example, removal of the SEC14P domain from a kalirin homolog, Dbs, increased its RhoGEF activity (Kostenko et al., 2005). Indeed, several lines of evidence support an important role for kalirin’s SEC14P domain in regulating its activity, as detailed below.
The kalirin-SEC14P has been found to bind phosphatidylinositides (Schiller et al., 2008) but not phosphatidyl-ethanolamine, – choline or – serine (Ma et al., 2014). A kalirin7 construct lacking the SEC14P domain was deficient in driving spine size and formation (Ma et al., 2014). Studies indicate that phosphorylation of SEC14P is integral for synaptic plasticity; for example, phosphorylation of T95 in human within the SEC14P, in response to N-methyl-D-aspartate receptor (NMDAr) activation, was required for altered spine morphology, but not spine formation (Xie et al., 2007). Overexpression of a phosphomimetic kalirin7-T95E construct increased α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAr) amplitude, whereas T95A or wildtype kalirin7 was unable to achieve this (Xie et al., 2007). Moreover, T95 phosphorylation is required for LTP via the NMDAr-CAMKII pathway (Herring and Nicoll, 2016) and found to increase Rac-GEF activity and resulting PAK phosphorylation (Xie et al., 2007), suggesting T95 phosphorylation may induce kalirin activity. It has also been shown that the SEC14P and spectrin repeat domains alone can induce spine morphological change independently of the Rac-GEF domain (Schiller et al., 2008; Ma et al., 2014), suggesting that increased Rac-GEF activity may occur concurrently, yet be functionally distinct. Together, these results indicate a requirement of the SEC14P and post-translational modification for regulation of neuronal morphology and synaptic activity.
The role of the SEC14P in regulating kalirin activity may also be linked to neuroligin (nlgn) signaling; nlgn1 is known to increase spine density (reviewed in Bemben et al. (2015)), and knockdown of kalirin severely abrogates nlgn1-mediated spine formation (Paskus et al., 2019). The SEC14P domain was found to be the site of interaction, implicating SEC14P-nlgn1 binding in the de novo formation of spines (Paskus et al., 2019).
In summary, the SEC14P domain appears to play multiple roles; 1) it can drive de novo spine formation through nlgn-mediated mechanisms (Paskus et al., 2019). 2) It can act as a scaffold to alter spine morphology (Schiller et al., 2008; Ma et al., 2014), perhaps through known interactions with nlgns (Paskus et al., 2019) within the N-terminus. 3) It can drive Rac-GEF activity in response to NMDAr-CAMKII signaling (Xie et al., 2007) to regulate synaptic plasticity (Herring and Nicoll, 2016). Further work is required to delineate the overlap between these processes, but these studies highlight an integral role of the SEC14P domain in regulating kalirin function.
3.2. Spectrin repeat domains
Spectrin repeats are short helical bundles characterized as scaf-folding units imparting multiple protein–protein interactions on kalirin (Fig. 2). The majority of KALRN isoforms express spectrin repeat domains, with the exception of Δ-kalirin (that possesses only repeats 5–9) and duet (that lacks all repeats). Interestingly, despite a lack of catalytic activity, the spectrin repeat domains and SEC14P domain can together drive multiple phenotypic changes in neuronal morphology (Schiller et al., 2008; Ma et al., 2014), likely through targeting interacting partners to specific neuronal domains.
Fig. 2.
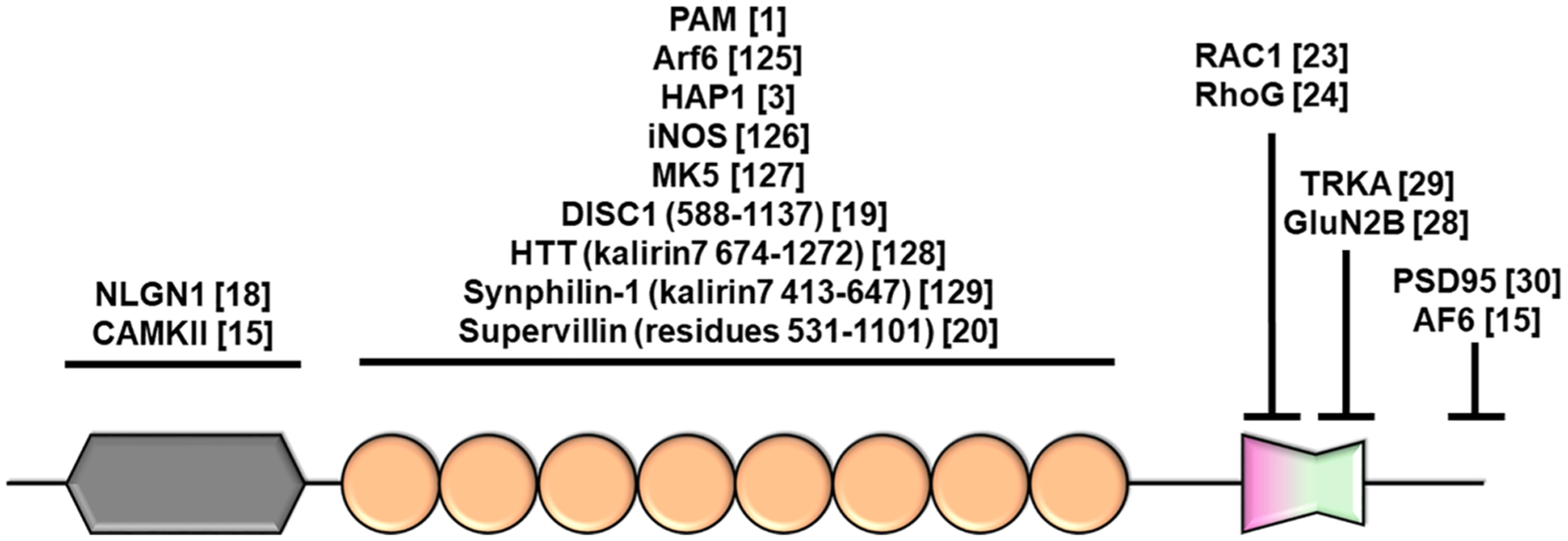
Kalirin7 interaction partners. Domain coloring/shape conform to Fig. 1.
Kalirin is found to bind DISC1 via the spectrin repeat domain (amino acids 588–1137 (Hayashi-Takagi et al., 2010). Interaction between kalirin7 and DISC1 was inhibits the interaction with, and activation of Rac (Hayashi-Takagi et al., 2010), suggesting that the spectrin repeat domain may provide interactions that can regulate kalirin catalytic activity. Interestingly, Supervillin was found to bind to both kalirin and its paralogue, trio, via spectrin repeats 4–7 (Son et al., 2015). Supervillin binding to trio increases GEF activity towards Rac. Although the effects of binding to kalirin were not assessed, the ability of DISC1 and Supervillin to regulate GEF activity by binding to the spectrin repeat domain suggests that this domain may play a key role in regulating activity. Interestingly, mutations within the spectrin8 region of trio result in the upregulation of the Rac-PAK pathway (PAK phosphorylation), and increased lamellipodial outgrowth and neurite outgrowth in cancer lines (Barbosa et al., 2020). As trio is highly homologous to kalirin, similar mechanisms may exist to regulate kalirin activity. Finally, motifs within the spectrin repeat domain can interact with the SRC homology 3 (SH3) domain (See 2.5), resulting in significantly lower activity compared to the isolated Rac-GEF domain (Schiller et al., 2006). Together, these results highlight the role of the spectrin repeats in regulating GEF activity (Fig. 3), likely by acting as a scaffold for a multitude of interacting proteins.
Fig. 3.
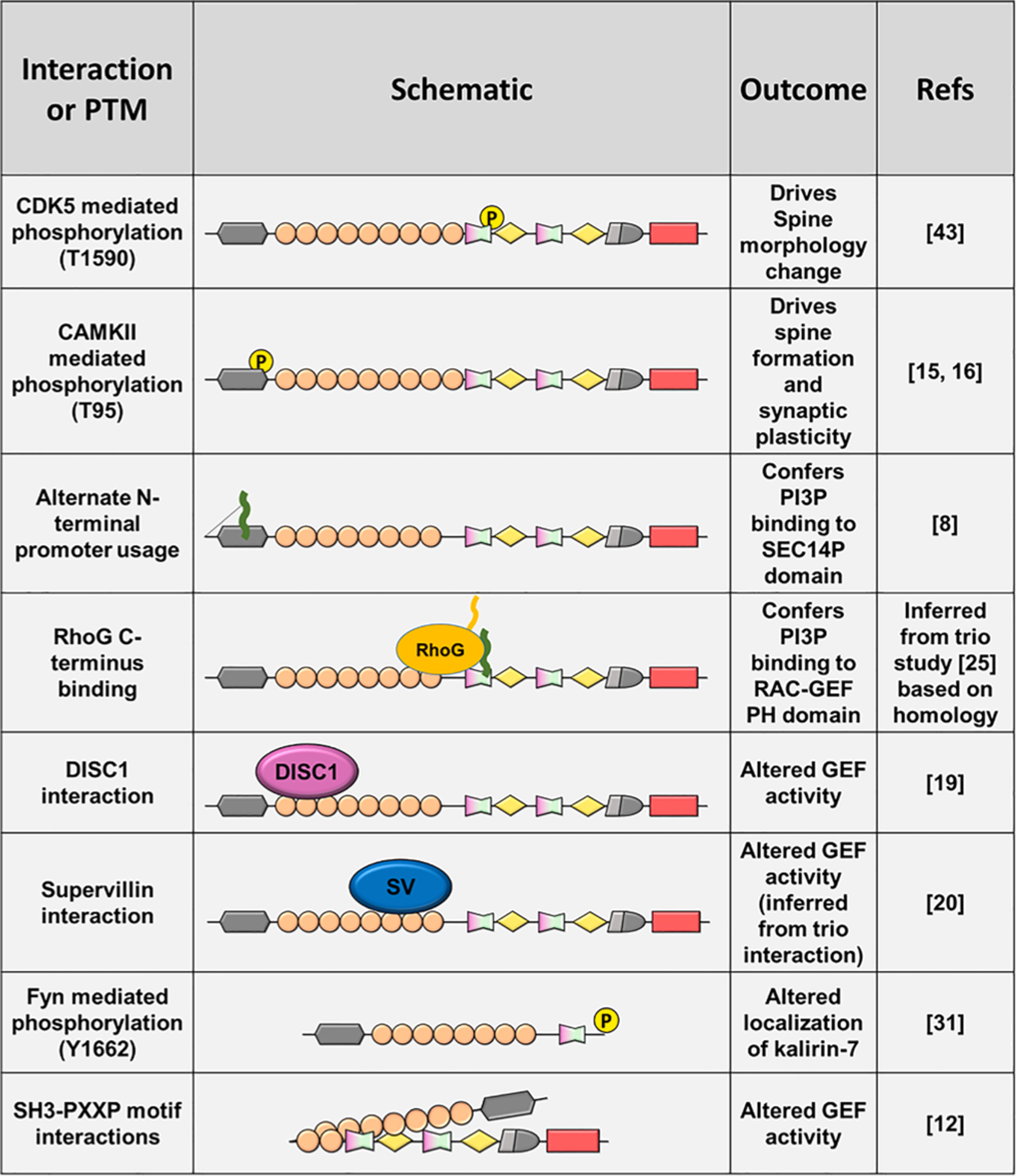
Multiple protein–protein interactions and post-translational (PTM) modifications alter kalirin function.
3.3. Rac-GEF domain
Within the cell, small GTPases remain in a guanosine-nucleotide diphosphate (GDP)-bound, inactive state. Guanosine nucleotide exchange factors (GEFs) interact with their cognate GTPase and stimulate the release of GDP. High intracellular concentrations of guanosine-nucleotide triphosphate (GTP) promotes its binding to and activation of GTPases. Each GTPase has intrinsic catalytic activity towards GTP and temporal control of activation is exerted through slow, GTPase-mediated hydrophilic cleavage of the gamma-phosphate of GTP. This process is accelerated by GTPase activating proteins (GAPs), returning the GTPase to its inactive GDP-bound form. The N-terminal GEF domain of kalirin has been shown extensively to act upon the Rho GTPase family, and in particular, Rac (Penzes et al., 2001a) and RhoG (May et al., 2002). This activity has been attributed to morphological change in neurons (Penzes et al., 2001a) highlighting an integral role of kalirin in regulating the Rac signaling pathway. Interestingly, the c-terminus of RhoG, but not Rac, can form a complex with the PH domain to alter lipid binding properties (Skowronek et al., 2004), suggesting alternate signaling mechanisms between substrate GTPases.
The kalirin Rac-GEF domain is comprised of a canonical dbl homology (DH), pleckstrin homology (PH) module, and is present in all kalirin isoforms, with the exception of duet. The DH domain directly interacts with Rac, driving the release of GDP and inducing activation of the GTPase. The PH domain of trio interacts with phosphatidyl inositol-4,5-bisphosphate (PI-4,5-P) and PI-4-P (Liu et al., 1998) and can directly alter DH catalytic activity (Bellanger et al., 2003), suggesting similar mechanisms for the kalirin PH domain, due to their high sequence homology. In addition, several studies have determined a role for the PH domain in mediating protein–protein interactions. The PH domain of kalirin was found to interact with GluN2B, a subunit of the NMDA receptor complex (Kiraly et al., 2011), and the receptor tyrosine kinase, TRKA (Chakrabarti et al., 2005). These interactions may allow Rac-GEF activity to be targeted to specific neuronal subdomains.
3.4. Kalirin7 unique C-terminus
Alternate splicing of the kalirin gene at exon 33 yields a variant with 20 unique C-terminal residues and a total size 1664 amino acids. This short isoform, expressed exclusively within the brain, encodes a PDZ binding domain (PBD) in its C-terminal region. This domain imparts PSD95-binding to kalirin (Penzes et al., 2001b), highlighting a specific role of kalirin7 within the post-synaptic density. Furthermore, this PBD is subject to post-translational modification: Y1662 phosphorylation within the PBD motif occurs in response to NRG1-ErbB4-Fyn pathway activation (Cahill et al., 2012).
3.5. SH3 domain
SRC Homology 3 (SH3) domains are around 60 amino acid domains that are involved in the formation of protein–protein interactions (reviewed in Mayer (2001)). SH3 domains, found in kalirin9 and 12, bind to the PxxP (P = proline, x = any residue) consensus motif and are present in 22 RhoGEFs (31% of family) suggesting a conserved role in regulation of GEF activity. Kalirin was found to interact with multiple PXXP peptides generated from internal sequences, suggesting the SH3 domain may regulate intramolecular interaction. Notably, the purified SH3 domain in complex with a PxxP peptide promoted SH3/Rac-GEF interaction and thereby limited GEF catalytic activity, and full-length kalirin showed dramatically reduced activity compared to the isolated Rac-GEF domain (Schiller et al., 2008). These results suggest that intramolecular SH3-PxxP interactions within kalirin may play a role in regulating kalirin function.
3.6. RhoA-GEF domain (isoforms 9, 12, duet)
Despite significant homology to the Rac-GEF domain, the RhoA-GEF domain exerts opposing effects in neurons via activation of RhoA (Penzes et al., 2001a). This domain is restricted to kalirin9 and 12 isoforms, which are expressed early in development. It is therefore logical to assume that RhoA signaling has some function in early, but not late neuronal development. Of note, introduction of kalirin9 or the isolated RhoA-GEF domain resulted in increased axonal growth in immature cultured cortical neurons (Penzes et al., 2001a).
Interestingly, RhoA activity is regulated by protein–protein interactions: Gαq is known to bind to the PH domain of trio (Rojas et al., 2007) to and promote activity. The region of binding is highly conserved between kalirin and trio, suggesting potential Gαq-mediated kalirin regulation. It has been postulated that Gαq binding favors the open, active form of trio, by restricting the position of the PH domain and revealing the RhoA binding site for GEF activity (Bandekar et al., 2019). Interestingly, a mutation within the RhoA-GEF domain can drive activity, and is associated with schizophrenia (Russell et al., 2018). Together, these results highlight a role of the RhoA-GEF in regulating early neuronal development, and reveal several mechanisms for the regulation of the activity of this domain.
3.7. Kalirin12 domains
3.7.1. Ig and FN3 domains.
The function of the Immunoglobilin-like C2-type (Ig) and Fibronectin type 3 (FN3) domains of kalirin are largely unknown. One potential mechanism of the kalirin Ig domain may be inferred from the ability of trio to bind to active RhoA via its Ig domain (Medley et al., 2000). This interaction resulted in relocalisation of trio, suggesting that following RhoA-GEF activity, RhoA may target trio to specific compartments. This may transduce distinct pathways, or sequester trio from a pool of inactive RhoA, thereby limiting activity. This feedback mechanism has not been shown for kalirin12 and it is notable that during evolution, the mammalian TRIO gene lost its FN3 domain, perhaps suggesting an alteration in kalirin12 regulation during evolution of the two genes (Kratzer et al., 2019). Further study is required to assess the binding partners and functional effects of the kalirin12 Ig and FN3 domains.
3.7.2. Kinase domain
Little is known about the function of the C-terminal regions unique to kalirin12. It is predicted to be a serine-kinase and has verified kinase activity; however its only characterized function is autophosphorylation of the skeletal muscle KALRN isoform, duet (Kawai et al., 1999). Comparative studies of kalirin9 vs kalirin12 suggest overlapping function (Penzes et al., 2001; May et al., 2002), however the ability of the isolated kinase domain to drive neurite outgrowth when overexpressed in DIV4 neurons suggests a non-redundant role of this domain in driving neurite extension (Yan et al., 2015). In addition, kalirin12 displays distinct subcellular localization, perhaps linked to the presence of the kinase domain. Whereas kalirin9 was observed in neurites, kalirin12 was present largely in the soma (Yan et al., 2015). Kalirin12 was also enriched in growth cones when transfected into hippocampal neurons (Mandela and Ma, 2012). Despite these observations, further study is required to define the role of the kalirin12 kinase domain in regulating kalirin activity and altered localization.
3.8. Isoforms, structure and function
As each domain plays a particular role in regulating kalirin function, it becomes increasingly evident that alternate isoform expression is likely to have profound effects on kalirin function. It is notable that with the exception of duet (identified only in muscle), all isoforms express the catalytic Rac-GEF domain, suggesting that this facet of kalirin function is central to its role in neurodevelopment. It seems that the expression of several domains, such as SEC14P and spectrin repeat domains, provide the substrate for regulation of kalirin localization and function in response to interacting partners. Thus, isoforms lacking these domains, such as Δ-isoforms, may play distinct cellular roles. For example, Δ-isoforms lack the SEC14p domain that is essential for interaction with nlgn1 and phospholipids within the membrane. This is likely to have an impact on the ability of Δ-isoforms to induce dendritic spine remodeling. In addition, several interacting partners (see Fig. 2) bind to kalirin with the spectrin repeat domain, and loss of these sites is likely to eliminate the regulatory effect of these partners. Kalirin-7 isoforms, on the other hand, lack the C-terminal RhoA-GEF domain. As RhoA and Rac functionally oppose each other, the presence of a single Rac-GEF domain in kalirin7 supports a strong preference for pro-synaptogenic Rac-signaling pathways. This is in line with the enhanced synaptogenic effects of kalirin7, as compared to other isoforms, no doubt enhanced by the kalirin7-specific PBD and localization to the dendritic spine. Moreover, kalirin7 is the only major isoform to lack a SH3 domain, suggesting altered SH3 mediated regulation of GEF activity. As kalirin7 is expressed later in development than kalirin9 and 12 (see 3.2, below), it suggests that Rac activation is dominant in the mature neuron at time points of synaptogenesis and synaptic plasticity, and that strong Rac activation is essential to these processes. In contract, kalirin9 and 12, expressed early in neuronal maturation, possess both a RhoA-GEF domain and regulatory SH3 domains, supporting RhoA regulation as a required function of early kalirin activity. The presence of this functionally apposed domain may facilitate stimulation of Rac and RhoA activity independently in response to varying neurite outgrowth cues. This combined with additional SH3 regulatory domains perhaps provides a more nuanced regulation of these early developmental processes.
Thus alternate promoter usage and splicing can generate kalirin isoforms that can shift kalirin function, either by altering the subcellular targeting, mechanisms of regulation, interacting partners, or by preferential GEF activity towards a subset of GTPases. In Section 3, we will discuss how these isoforms are expressed during development, and how changes in domain architecture may alter kalirin function to achieve precise control over key stages of neurodevelopment.
4. Kalirin Rac/RhoA signaling pathways
Kalirin regulates a host of neuronal properties, such as early axonal growth (Penzes et al., 2001a), dendritic arborization (Yan et al., 2016), dendritic spine formation (Penzes et al., 2001b, 2003), neuroendocrine secretion of hormones (Mandela et al., 2012, 2014) and AMPAr and NMDAr activity (Herring and Nicoll, 2016; Kiraly et al., 2011; Lemtiri-Chlieh et al., 2011; Li et al., 2019). The ability to regulate these effects has largely been attributed to kalirin’s role in initiating small GTPase signaling through Rac- and RhoA-specific GEF activity (Penzes et al., 2001a).
GTPases are ubiquitously expressed molecular switches that activate a host of cellular signaling pathways. GTPases are regulated spatially and temporally by GAPs (inhibitors of GTPase signaling), GEFs (activators of GTPase signaling) as well as post-translational modification, sequestration and membrane targeting. The GTPase superfamily is made up of multiple families based on homology to the archetypal Ras and together contribute to the regulation of almost all cellular processes. The Rho family of GTPases, made up of RhoA, RhoG, Rac and CDC42 are linked to cytoskeletal changes, translocation and cell morphology in non-neuronal cells (reviewed in Hall (1998)). Within these roles, each family member serves a distinct and often opposing function, with RhoA driving formation of actin stress fibers, focal adhesions and connections to the extracellular matrix while Rac and CDC42, promote actin polymerization to produce lamellipodial and filopodial protrusions (Hall, 1998). Within neurons, Rac and RhoA have key roles in maintaining and forming key neuronal architectures, through axon growth and guidance, dendritic arborization, spine growth and stabilization (reviewed in Stankiewicz and Linseman (2014)).
Neuropsychiatric diseases, such as intellectual disabilities (including syndromic genetic diseases, such as Rett and Fragile X), mood disorders (including bipolar and depression), epilepsy, psychosis and schizophrenia, have been extensively correlated to abnormal neuronal and synaptic development (reviewed in Forrest et al. (2018)). Neuronal structural alterations are critical for healthy brain maturation, and dysregulation of these processes can contribute to disease states. It is clear that the complexity in KALRN gene expression allows tissue, brain region and time-point specific control over key developmental processes. In this section we aim to define windows of isoform expression in order to correlate changes in kalirin function to neuronal development.
4.1. Early neuronal development
Maturing neurons alter their network connections through regulation of morphology and synaptic connectivity. The size and complexity of the dendritic arbor reflects the number of functional connections that can be made within a given circuit. In addition, axonal outgrowth is crucial to the development of neuronal circuits. Together these early morphological changes affect network formation within the prefontal cortex, critical to learning and memory processes. Deficits in either of these can result in severely limited synaptic connections and are extensively correlated to human neurodevelopmental disorders (Forrest et al., 2018). The RhoA family of GTPases are crucial in regulating the dendritic arbor (Nakayama et al., 2000) and axonal outgrowth (May et al., 2002), suggesting a role for kalirin in these early developmental processes.
Early development is predominated by the expression and action of kalirin9 and 12, which each possess both Rac and RhoA-GEF activity (Yan et al., 2015). One of most striking early discoveries on kalirin function was the observed increase in neurite length and growth cone formation in cortical neuronal cultures, following overexpression of these isoforms (DIV4) (Penzes et al., 2001a). These observations gave the first evidence supporting a role for kalirin in regulation of neuronal morphology. Conversely, shRNA mediated knockdown of all kalirin isoforms in DIV4 and DIV7 cultured hippocampal neurons (largely affecting expression of kalirin9 and 12) drastically impaired dendritic arborization (Yan et al., 2015). Indeed, knockdown of either kalirin9 or 12 yielded similar, though abrogated, decreases in dendritic complexity and neurite outgrowth compared to complete knockdown. These results highlight a central, overlapping role of kalirin9 and 12 in early dendritic arbor formation. In addition, kalirin has been implicated in axonal growth, with kalirin9 or 12 overexpression promoting fiber outgrowth in cervical ganglion neurons (May et al., 2002) and kalirin9 inducing axon extension in DIV2 cortical neurons (Penzes et al., 2001a). Rac and RhoA play crucial roles in axonal growth and dendritic arborization (Nakayama et al., 2000). It is therefore likely that Kalirin9 and 12 expression is intimately linked to early neuronal development by regulating these processes through the Rac- and RhoA-GEF domains present in these isoforms.
4.2. Synaptogenesis
Dendritic spines are the postsynaptic structures that regulate synaptic strength, and allow control over neuronal excitability. These structures are the site of synaptic input compartmentalization and provide regulation of synaptic signaling. By localizing critical proteins within a distinct sub-compartment, the dendritic spine can respond to changes in activity by enriching or reducing the abundance of neurotransmitter receptors and regulating the propagation of second messengers. Individual dendritic spines can also undergo structural growth, shrinkage, and are subject to experience dependent maintenance or elimination (Forrest et al., 2018).
Within the cortex, neurons undergo a striking and robust increase in dendritic complexity and dendritic spine density surges in a process dubbed synaptogenesis. This is followed by a period of pruning, where the number of dendritic spines and synapses declines, concurrently with cortical thinning throughout early childhood to adolescence (Rakic et al., 1994; Huttenlocher, 1984; Giedd et al., 1999). These developmental processes are correlated to the emergence of cognitive functions such as attention, working memory, cognitive control and response inhibition (reviewed in Penzes and Remmers (2012)). Alterations in dendritic spine number correlate to deficits in the formation of excitatory synapses, and to the progression of many neurological disorders (Forrest et al., 2018).
KALRN expression shifts from kalirin9 and 12 to kalirin7 during this period of spine formation (Hansel et al., 2001) suggestive of a role for kalirin7 in mediating these effects. Indeed, kalirin7 overexpression in cultured cortical neurons display remarkable increases in the formation and size of dendritic spines (Xie et al., 2007). Reciprocally, knockdown produces drastic loss of dendritic spines and altered spine morphology. Together, these results highlight a vital role for kalirin7 expression in maintaining dendritic spine size and number in mature neurons, together highlighting the role of kalirin in stabilizing synaptic connections.
Several key differences suggest a drastically altered role between kalirin9 and 12, and kalirin 7. Whereas kalirin9 and 12 activate both Rac and RhoA pathways to regulate axon and dendritic arbor growth, kalirin7 only has a single, Rac-GEF domain. As Rac and RhoA have opposing functions within the spine, with Rac driving spine growth and density and RhoA playing an opposing role, the expression of kalirin7 suggests a shift to pro-synaptic signaling, independent of RhoA. Moreover, kalirin7’s PBD strongly targets kalirin to the post-synaptic density through interaction with PSD95. This subcellular targeting is crucial for kalirin’s role in driving dendritic spine formation and growth (Penzes et al., 2001b). Thus, kalirin7 expression signals a shift in KALRN gene function to synapse formation and stabilization, concurrently with synaptogenesis.
4.3. Synaptic plasticity
In addition to regulating the formation of dendritic spines, kalirin plays an integral role in regulating synaptic plasticity; the activity dependent regulation of synapse strength by altered synaptic expression of ionotropic receptors, such as AMPAr (reviewed in Citri and Malenka (2008)). By altering the number or activity of synaptic receptors, neurons can regulate the strength of neurotransmitter response and this plasticity underlies network formation and, within the cortex, underlies the processes of learning and memory. Deficits in synaptic plasticity have been extensively linked to neurodevelopment disorders, including intellectual disability, developmental delay and schizophrenia (reviewed in Bliss et al. (2014)).
Kalirin is intrinsically linked to synaptic activity and plasticity; loss of Kalrn gene expression results in decreased AMPAr-mediated miniature excitatory postsynaptic currents (mEPSC) (Cahill et al., 2009) and knockdown of kalirin7 in CA1 pyramidal neurons causes a decrease in AMPAr and NMDAr mEPSC amplitude (Herring and Nicoll, 2016). Interestingly, dual knockdown of kalirin and its paralog, trio, resulted in an almost complete abrogation in mEPSC. Moreover, kalirin7 knockout animals display reduced NMDAr-mediated long term depression (Lemtiri-Chlieh et al., 2011) and potentiation (Kiraly et al., 2011; LaRese et al., 2017). This is perhaps linked to the ability of kalirin to directly interact with the NMDAr via the GluN2B subunit (Kiraly et al., 2011). Several correlates also exist between loss of Kalrn expression and loss of Wnt5 signaling; both result in impaired Rac activity and impairments in synaptic plasticity, spatial memory deficits and loss of dendritic spines and a reduced dendritic arbor (Chen et al., 2017) (see 6.1 for discussion on Kalrn knockout animal phenotypes). These observations suggest that Wnt5 signaling and kalirin work through a shared pathway likely involving Rac regulation. However, more work is required to confirm a mechanistic link between Wnt signaling and kalirin. These results highlight a strong requirement of kalirin in regulating synaptic activity, and the response to NMDAr-mediated synaptic plasticity.
Together, these studies reveal that early neuronal development of the dendritic arbor and axon extension are dependent on the activities of kalirin9 and 12. However, kalirin7 appears to have complex roles within later neurodevelopment, facilitating ionotropic receptor trafficking and activity, as well as dendritic branching and dendritic spine formation (Fig. 4). The expression of specific isoforms during development occurs concurrently with distinct stages of neurodevelopment, and are intrinsically linked to the formation and regulation of synaptic circuits that underlie cognitive function.
Fig. 4.
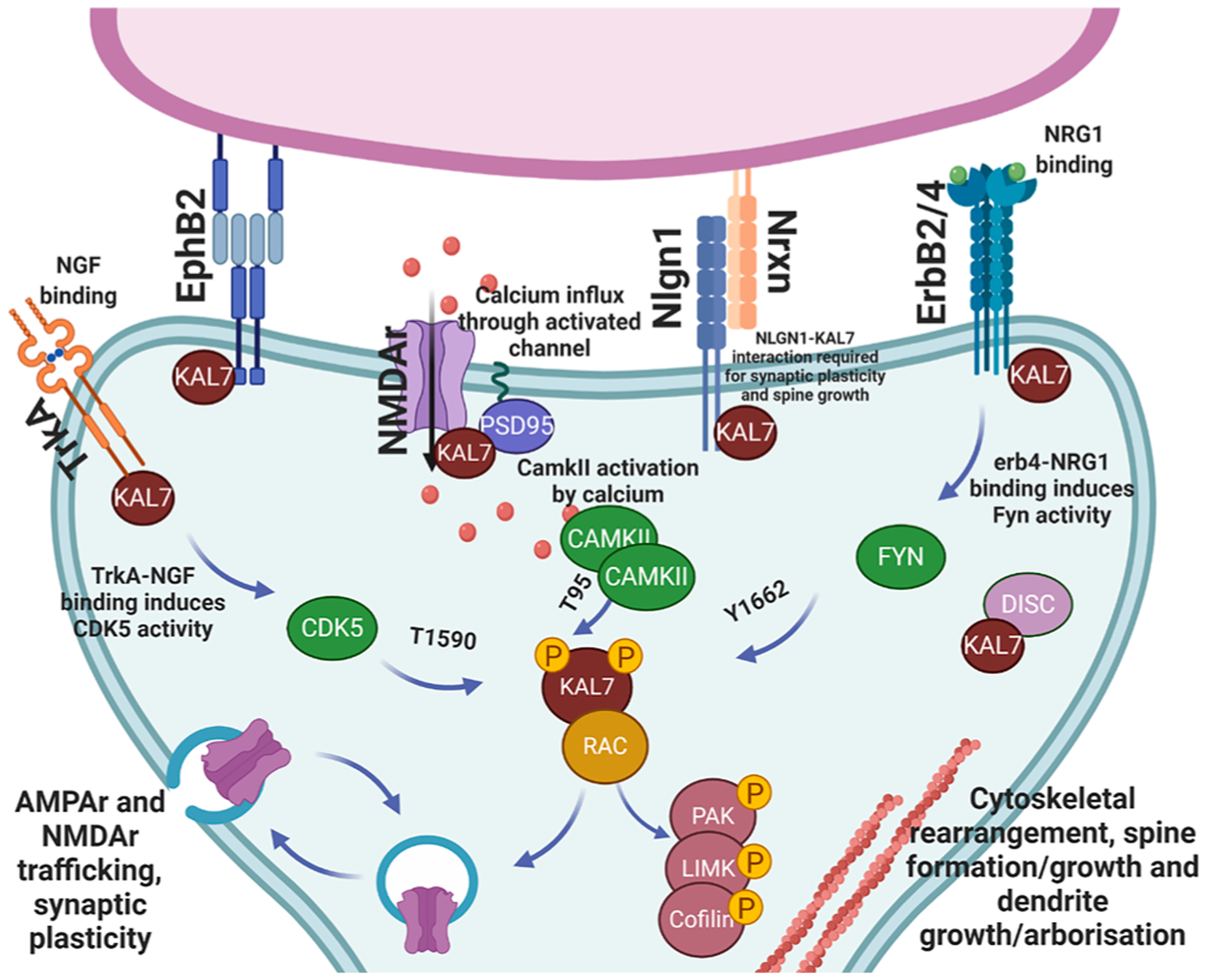
Post synaptic signaling pathways and interacting partners of kalirin7.
5. Genetics of KALRN in human disease
The KALRN gene, located on chromosome 3q21 (chr3:124,080,023–124,726,325) is made up of 66 exons that together produce multiple protein coding transcripts. KALRN and TRIO genes are 60% homologous, expressed in vertebrates, and result from the duplication of a common genetic ancestor (Kratzer et al., 2019). Key differences between vertebrate KALRN and TRIO paralogues are loss of the spectrin7 and FN3 expressing regions in TRIO (Kratzer et al., 2019), and differential promoter usage resulting in altered expression within the body. Both KALRN and TRIO display very low tolerance to loss of function (LOF) and truncating mutations (pLOF, 1) suggesting a high likelihood of haploinsufficiency (Karczewski et al., 2020), and LOF-intolerant gene mutations have been found to be highly enriched in neurodevelopmental disorders, such as schizophrenia (SCZ) (Pardinas et al., 2018). Interestingly, despite high homology and overlapping function, TRIO has been extensively linked to multiple neurological disorders (Ripke et al., 2013; Purcell et al., 2014; Fromer et al., 2014) with high penetrance, whereas the links between KALRN mutations and disease is less forthcoming. Despite this, KALRN mutations have been associated with multiple human diseases, particularly neuronal diseases associated with aberrant synapse formation (synaptopathies). In this section, KALRN mutations isolated in patient populations will be discussed, alongside the potential mechanistic effects of mutation, elucidated through post-mortem and molecular studies.
5.1. Autism spectrum disorder
Autism Spectrum Disorder (ASD) is a debilitating disorder defined by the central presentations of social communication deficits and repetitive behaviors. It is highly prevalent, and is estimated to affect 1 in 59 children in the USA (Baio et al., 2018) in line with global prevalence estimates of 1–2% (Kim et al., 2011). The age of onset is often before the age of three, implicating deficient prenatal or early postnatal neuronal development. Twin studies support a highly genetic component for the etiology of ASD, with a 50–95% chance of identical twins sharing a diagnosis (Colvert et al., 2015). Mutations in ASD genes have been linked to a wide range of neurological functions, from neurogenesis to neurite extension and synaptic function (reviewed in Gilbert and Man (2017)) and ASD patients have been observed to have increased dendritic spine density (Hutsler and Zhang, 2010).
A mutation within KALRN was found to be associated with ASD in a trial of 357 Faroese individuals, with 36 affected patients (Leblond et al., 2019). This de novo mutation in KALRN (c.6070A > G, N2024D) was predicted to be deleterious to protein function and lies within the second GEF domain, expressed in Kalirin9, and 12. A further protein truncating frameshift mutation, A367FS was isolated as a highly deleterious mutation linked to ASD (Satterstrom et al., 2020). Interestingly, although mutations in KALRN appear to be associated with ASD, trio has been reported to have many more ASD associated mutations (Sadybekov et al., 2017). This is perhaps surprising, considering the functional and structural similarities between these proteins. It is compelling to hypothesize that mutations within isoforms of KALRN, and TRIO, expressed early in development (kalirin9 and 12) are more linked to early onset diseases, such as ASD. Indeed, N2024D would affect the function of kalirin9 and 12 early in development, but may not impact the role of kalirin7 within synaptogenesis and synaptic plasticity, as this isoform fails to express the mutated domain. Thus, mutations that affect kalirin9 and 12, as well as trio, may confer higher risk for early developmental diseases, such as ASD.
5.2. Schizophrenia
SCZ affects ~1% of the population worldwide (Marco Piccinelli, 1997). The disorder is characterized by psychotic and mood deficits alongside cognitive impairment. SCZ has been found to be highly heritable with twin studies revealing a 50% chance of shared presentation (Sullivan et al., 2003) and risk to disease is often due to the inheritance of numerous rare, low effect size mutations that converge to produce altered neurodevelopment (Singh et al., 2017). Post-mortem studies of SCZ patients have been reported to display reduced dendritic spines and dendritic alterations (Glausier and Lewis, 2013). Kalirin lies within a synaptic SCZ signaling hub (Fig. 4) and genetic studies support a role for KALRN dysfunction in SCZ.
Compelling genetic data suggesting a role of KALRN in SCZ from GWAS or exome studies has been revealed due to the availability of large patient cohorts (Purcell et al., 2014; Howrigan et al., 2020). These studies have reported mutations within the spectrin repeat domain (R625C, Q862Q*, T1207M), GEF domains (D1338N, P2255T) and SH3 domains, (G1699V, E2373K) genetically associated with SCZ (Table 2, Fig. 5). Neuronal studies into the activity of GEF mutations reveals that abrogated Rac-GEF activity and increased RhoA-GEF activity may contribute to neuronal dysfunction and SCZ. This is in line with observed altered KALRN transcript expression which shows decreased total levels (likely representing the dominant isoform, kalirin7) (Narayan et al., 2008; Hill et al., 2006; Rubio et al., 2012). Thus, decreased kalirin levels, or kalirin LOF mutations, appear to be etiologically linked to SCZ. Interestingly, both D1338N (Russell et al., 2014) and the intronic variant Rs333332 (Cai et al., 2014) have been associated with altered cortical thickness, suggesting that mutations in KALRN may also impair cortical structural development, in line with observations of decreased brain tissue in SCZ patients (van Haren et al., 2008).
Table 2.
Non-synonymous mutations within KALRN associated to human disease.
| Mutants | Patient | Variant | Reference | Neuronal phenotype | Reference |
|---|---|---|---|---|---|
| R10* | DD | De novo | Deciphering Developmental Disorders (2017) | ||
| A357FS | ASD | De novo | Satterstrom (2020) | ||
| R625C | SCZ | Case(1)/Control(0) | Purcell (2014) | ||
| Q862Q* | SCZ | Case(1)/Control(0) | Purcell (2014) | ||
| V1015M | DD | De novo | Deciphering Developmental Disorders (2017) | ||
| T1207M | SCZ | case(2)-control(1) | Kushima (2012) | ||
| T1215K | ID, short stature | recessive variant (inherited) | Makrythanasis (2016) | ||
| D1338N | SCZ | Inherited | Russell (2018) | Reduced RAC-GEF Activity, Reduced spine density, size, Reduced cortical volume | Russell (2018) |
| E1577K | DD | De novo | Deciphering Developmental Disorders (2017) | ||
| G1699V | SCZ | De novo | Howrigan (2020) | ||
| N2024D | ASD | Case(1)/control (0) | Leblond (2019) | ||
| P2255T | SCZ | Case(15)-control(7) | Kushima (2012) | Increased Transcript stability, Increased RhoA-GEF activity, Reduced arborization, Reduced Spine size | Russell (2014) |
| E2373K | SCZ | Case(1)/Control(0) | Purcell (2014) |
FS, frameshift introduced;
premature stop codon.
Fig. 5.
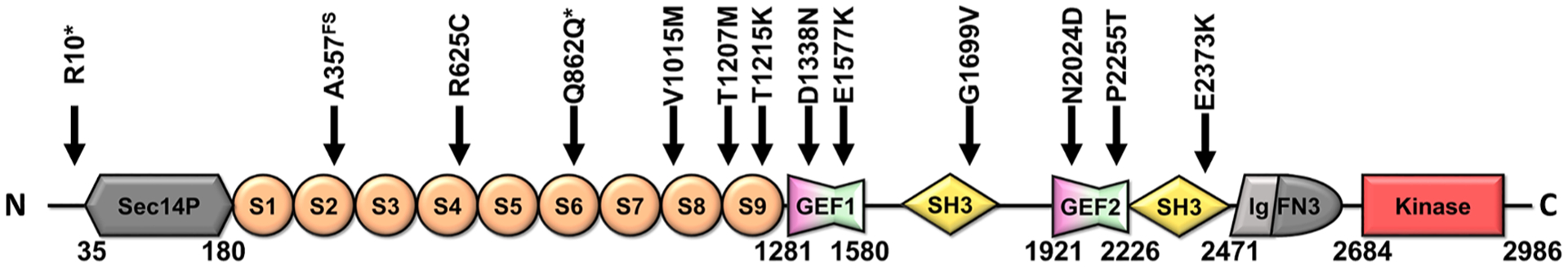
Kalirin12 protein architecture with positions of disease-associated mutations (Table 2),
Electroconvulsive therapy (ECT) can be an effective treatment for mood alterations, including those associated with schizophrenia, and has been shown in animal models to result in an upregulation of KALRN expression (Ma et al., 2002). Furthermore, ECT resulted in altered kalirin-PSD95 interaction, and increased Rac-GEF activity (Hayashi-Takagi et al., 2010). AS ECT has been linked to activation of NMDAr receptors, and NMDAr activity is known to both drive kalirin expression (Afroz et al., 2016) and activation (Herring and Nicoll, 2016), it is possible that ECT, via NMDAr activation, may play a role in the reduction of SCZ negative symptoms through kalirin. Another link between kalirin and SCZ is the observed effect of clozapine, a second-generation antipsychotic, in Kalrn knockout animals. Clozapine was found to be ineffective in reversing hyperactivity observed in Kalrn knockout mice, but was able to reverse altered locomotor activity in heterozygous mice (Cahill et al., 2009). These results suggest that kalirin expression is required for some sedative effects of clozapine. Interestingly, clozapine is known to target 5-HT2A serotonin receptors that are enriched within the cortex (Cardozo et al., 2017), and Kalirin7 is required for PAK activation and altered spine morphology in response to 5-HT2A activation (Jones et al., 2009). Thus, kalirin may play a role in transducing the response to clozapine, and in SCZ psychoses.
Of the above mutations, the neuronal effects of two missense mutations have been assessed in detail, revealing mechanisms that may underlie SCZ etiology. The first, D1338N, occurs within the Rac-GEF domain and was shown to impair GEF activity towards Rac and limit both dendritic arborization and spine density (Russell et al., 2014). Conversely, the P2255T mutation (Kushima et al., 2012) within the RhoA-GEF domain increased activity towards RhoA, again reducing dendritic arborization as well as spine size (Russell et al., 2018). Recent structural analyses (Bandekar et al., 2019) and biochemical analyses (Rojas et al., 2007; Lutz et al., 2007) reveal a regulatory role for Gαq in P63RhoGEF and trio activity. The proposed interaction site is highly conserved between kalirin and trio, suggesting this intermolecular regulation may also apply to kalirin. Analysis of crystal structure data (PDB-6DZ8, unpublished data) suggests that P2255T lies within a helical turn preceding the proposed PH-Gαq binding site (Rojas et al., 2007). It is compelling to hypothesize that P2255T may upregulate interaction with Gαq and thus drive catalytic activity above normal levels, although evidence of such a mechanism is lacking.
These findings highlight the opposing roles of the RhoA and Rac pathways within neurons, and support a shift in Rac to RhoA activation as a causative factor in SCZ. Indeed, the functional effect of RhoA overexpression in inhibiting spine formation (Tashiro et al., 2000) mimics post-mortem SCZ patient spine phenotypes (Glausier and Lewis, 2013). These results support tight control in the balance between RhoA and Rac activity as required for healthy neurodevelopment.
5.3. Intellectual disability and developmental delay
Intellectual disability usually presents in childhood, but is only formally diagnosed by standardized tests at maturity (Moeschler et al., 2014). Deficits in early childhood are characterized by delays in developmental benchmarks, such as age of first words and steps, and are often diagnosed as intellectual disability later in life. Intellectual disability has been associated with altered dendritic complexity (Kaufmann and Moser, 2000) and spine density (Purpura, 1974).
The first link between KALRN and intellectual disability was found in a consanguineous family harboring a homozygous mutation within the spectrin repeat domain (T1215K) (Makrythanasis et al., 2016). Further, heterozygous mutations observed in patients with developmental delay were linked to frameshift truncation (R10*), and protein coding variants (V1015M, E1577K, (Deciphering Developmental Disorders, 2017). These results suggest that KALRN contributes to normal neuronal development, and that altered expression or function may be a factor in the etiology of intellectual disability and developmental delay.
5.4. Addiction
Drug and alcohol addiction are common comorbidities of many psychiatric diseases. Mouse and human genetic analyses may reveal a potential role of KALRN in mediating a susceptibility to addiction. Mutations within KALRN have been associated with increased ventral striatal activation (rs6438839), a measure related to enhanced reward anticipation and a factor in addiction susceptibility (Pena-Oliver et al., 2016). Further, patients with rs4634050 within kalirin were recorded to partake in more binge drinking than those carrying the normal allele (Pena-Oliver et al., 2016). Perhaps in line with these genetic observations are the effects on cocaine self-administration in kalirin7 knockout mice. Kalirin7 knockout mice displayed robust deficiencies in behavior, neuronal morphology, cocaine-induced gene expression changes and spine plasticity (Kiraly et al., 2013, 2010; Wang et al., 2013), which may play a role in the observed increase in cocaine self-administration. These effects were blocked by ifenprodil (a GluN2B-selective inhibitor of the NMDAr) suggesting that kalirin7 contributes to the reinforcing effects of cocaine through glutamatergic signaling pathways (LaRese et al., 2017) and alterations in kalirin levels or function may play a role in the mechanisms of addiction susceptibility.
5.5. Alzheimer’s disease
Alzheimer’s disease (AD) is a neurodegenerative disease associated with profound deficits in working memory and cognitive function (DeTure and Dickson, 2019). Among its causes are the accumulation of amyloid plaques and neurofibrillary tangles that can impair neuronal health and synaptic function (reviewed in DeTure and Dickson (2019)).
A role for kalirin dysregulation has been proposed as a factor linked to AD progression. Downregulation of KALRN transcript expression was observed in a small cohort of AD patients (Youn et al., 2007), suggestive of a correlation between AD progression and kalirin dysregulation. Inducible nitric oxide synthase (iNOS) activity has been found to respond to inflammatory cues, resulting in oxidative damage and contributing to neuronal cell death (Asiimwe et al., 2016), and iNOS activity was found to be considerably higher in AD patients. Interestingly, kalirin is able to bind to iNOS and inhibit its activity (Youn et al., 2007). There appears, therefore, to be a potential link between downregulated kalirin levels, increased iNOS activity, and the progression of AD. These results have been recapitulated in animal models where kalirin protein expression was reduced in the Tau-P301S AD model (Dejanovic et al., 2018) and 3xTG-AD model (Cisse et al., 2017). Most compelling, however, is the ability of kalirin overexpression to reverse molecular and behavioral phenotypes in AD mouse models; kalirin overexpression was able to reverse soluble amyloid beta derived oligomer-induced dendritic spine density decreases in cultured neurons (Xie et al., 2019). AD patients with psychoses display an increase in beta amyloid (1–42)/(1–40) ratio as well as decreased kalirin that together may converge to limit dendritic spine formation and exacerbate AD symptoms (Murray et al., 2012). Interestingly, viral expression of the transcription factor XBP1 resulted in increased kalirin7 levels, and the reversal of spine density, synaptic plasticity and memory deficits in the 3xTG-AD mouse model (Cisse et al., 2017). Vitally, knockdown of kalirin7 abrogated these effects, suggesting that the effects of XBP1, at least in part, act through transcriptional activation of kalirin7. Together these results highlight that loss of KALRN expression is correlated to AD pathology, and that kalirin7 overexpression may be protective in AD.
5.6. Non-neuronal disease
Studies have also implicated kalirin in non-neuronal diseases. For example, KALRN has been identified as a genome wide significant risk factor in intracranial atherosclerotic stenosis (a causal factor in ischemic stroke) (Li et al., 2017; Dang et al., 2018, 2015). These effects have been correlated to three-fold overexpression of kalirin9 in stroke patients (Dang et al., 2018). The intronic KALRN variant rs9289231, has been identified as a risk factor in early onset coronary artery disease (Wang et al., 2007; Boroumand et al., 2014; Mofarrah et al., 2016). No alterations in serum kalirin transcript levels were observed in blood samples from CAD patients (Shafiei et al., 2018), however, kalirin has been shown to play a role in smooth muscle cell physiology. More specifically, Rac activation, through kalirin9, has been shown to promote smooth muscle cell proliferation and migration (Wu et al., 2013), a factor contributing to the progression of atherosclerosis. These results suggest that altered kalirin expression within smooth muscles cells may be affected by this genetic variant. One potential mechanism could be to upregulate expression of kalirin9 and/or 12 within smooth muscle cells, thereby driving pathological atherosclerosis.
In addition to intellectual disability, the homozygous T1215K (Makrythanasis et al., 2016) mutation was associated with short stature. Kalrn knockout animals show aberrant growth rates (male) and adult size (female), and cultured anterior pituitary cells from these animals displayed increased growth hormone and prolactin secretion (Mandela et al., 2012). These phenotypes were recapitulated by selective knockout in neuroendocrine cells (Mandela et al., 2014), suggesting that altered kalirin expression or function within these cells may affect hormonal regulation. These results suggest that normal KALRN expression may be required for development outside of the nervous system, perhaps linked to regulation of secreted hormone levels, and further confirm the varied function of non-kalirin7 isoforms throughout the body during development.
6. Animal models of KALRN
Multiple mouse models have substantially contributed to our understanding of kalirin’s role in neurodevelopment (see Table 3). The first total Kalrn knockout model (KalGEF1KO) introduced LoxP sites around exons 27, 28 and the intervening intron, that together code for the Rac-GEF domain (Cahill et al., 2009). Independently, another model was created through LoxP excision at exon13, within the spectrin repeat domain coding region, to generate the KalrnSR model (Mandela et al., 2012). Loss of these exons completely ablated Kalrn expression within the cortex and hippocampus, although it is likely that duet isoforms, whose promoter lies after the Rac-GEF domain, were unaffected. However, this isoform is largely only observed in skeletal muscle (Kawai et al., 1999).
Table 3.
Kalirin animal models with annotated morphological, anatomical and behavioral deficits. Arrows indicate increased or decreased phenotype.
| Animal model | Genetic details | Molecular phenotype | Structural/anatomical phenotype | Behavioral Phenotype |
|---|---|---|---|---|
| KalGEF1KO | Kalrn RAC-GEF domain knockout: exons 27 and 28 were eliminated | ↓ Rac activity in cortex but no impact on Rac levels in hypothalamus and hippocampus55
↓ AMPAr-mediated synaptic transmission in layer 5 pyramidal neurons55 No change in basal synaptic transmission on hippocampal Schaffer-collateral synapses116 ↓ LTP maintenance, but no effect on induction116 ↓ Activity-dependent spine plasticity as measured by a failure of spines on cultured cortical neurons to increase size and AMPAr expression following APV withdrawal113 |
↓ Dendritic spine density in frontal cortex (golgi staining) at 12 weeks55
↓ Hippocampal size (↓ hippocampal cell number)116 ↓ Dendrite arborization of layer V pyramidal neurons (both apical and basal dendrites) (golgi)113 ↓ Dendrite length (apical) of layer V pyramidal neurons (golgi)113 ↓ Neuron soma size in layer V frontal cortex113 ↑ Cell density in layer V frontal cortex113 ↓ Cortical thickness113 ↓ Dendritic spine density in cortex and hippocampus of aged mice (11–14 months; golgi116) |
↓ Morris water maze (spatial memory function)55 ↓ Y maze (working memory function at 4 months55 and 11–14 months116) ↓ Prepulse inhibition (sensory gating)55 ↓ Social approach assay (social behavior)55 ↑ Locomotor activity55,116 ↓ Social recognition task (11–14 months116) ↓ Acquisition and consolidation of contextual fear memories116 ↓ Acquisition and delayed consolidation of cue-induced fear memories116 |
| Kal7KO | Kalirin7 specific knockout mice: LoxP sites surround the unique kalirin7 exon, located between exons 33 and 34 to generate conditional knockout mice. Kal7cko/+ mice were crossed with Hprt-Cre mice (cre recombinase expressed in germ cells). | ↑ Input resistance115
↓ Membrane capacitance115 ↓ LTP (reduced EPSC amplitude following theta burst pairing)115 ↑ Kal12 and Kal9 protein in total cortical tissue lysates115 ↓ NR2B, Cdk5 protein in PSD samples115 ↓ Cocaine-induced increase in NR2B mRNA in NAc95 ↑ Cocaine-induced increase of Kal12 and Kal9 mRNA in NAc95 ↓ NMDA/AMPA receptor current ratio in layer 2/3 pyramidal neurons28 ↓ Cell surface expression of NR2B in cortical slices28 Kal7PH1 domain interacts with final juxtamembrane region of NR2B28 |
↓ Dendritic spine density in CA1 pyramidal neurons (apical dendrites) (golgi staining)115 but no effects on spine density were observed in the NAc97
↓ vGlut, PSD-95 in hippocampus (immunohistochemistry)115 ↓ Cocaine-induced increase in dendritic spine density in NAc (ballistic labeling)97*# ↓ Cocaine-induced increase in dendritic spine size97* *(20 mg/kg cocaine once daily for 8 days) #(20 mg/kg cocaine once daily for 4 days) |
↓ Anxiety-related behavior (elevated zero maze)115 ↓ Acquisition of contextual fear memories (passive avoidance)115 ↑ Development and persistence to cocaine-induced locomotor sensitization97 ↑ Reward sensitivity to cocaine (conditioned-place preference)97 ↑ Self-administration of low doses of cocaine (0.5 mg/kg/infusion and 0.25 mg/kg/infusion)95 ↑ Instrumental responding for food reinforcers95 |
| KalSR KO/KO | Kalirin spectrin repeat domain knockout mice: LoxP sites flanking exon 13 to generate conditional knockout mice.KalSRCKO/+ mice were crossed with Hprt-Cre mice to generate KalSRKO/KO mice |
↑ Basal secretion of growth hormone and prolactin from anterior pituitary cultures41
↓LTP in spinal neurons (reduced EPSC amplitude in spinal neurons following repeated stimulation of C-fibres)117 |
↓ Nicotinic acetylcholine receptor organization at the neuromuscular junction41 ↓ Skeletal muscle junctional folds (surface area)41 ↑ Sarcomere length41 ↓ Bone mass and remodeling119 ↓Osteoblasts in female mice119 ↑ Osteoblasts in male mice119 ↑ Osteoclasts in female mice119 ↑ Osteoclast activity and differentiation in vitro119 ↓Osteoprotegerin secretion in vtiro119 |
↓ Growth rate in both males and females41 ↓ Weight at weaning females only41 ↓ Anxiety-related behavior (elevated zero maze)41 ↓Acquisition of contextual fear memories (passive avoidance)41 ↓ Motor coordination (rotarod, wire hang test)41 ↑ Locomotor sensitization to cocaine56 |
| Cell Specific Animal Model | Genetic Details | Molecular Phenotype | Structural/Anatomical Phenotype | Behavioral Phenotype |
| SDH-KAL7−/− | Using Kal7 specific knockout mice, microinfusion of an adeno-associated virus 1/2 was used to deliver cre-recombinase directly to the spinal dorsal horn (SDH) of adult mice. | ↓ Dendritic spine density of SDH neurons (golgi)117 ↓ Inflammation-induced increase in spine density of SDH neurons following peripheral paw inflammation (golgi)117 ↓Thin dendritic spine density and stubby dendritic spine density of SDH neurons in vitro117 |
↓ Nociceptive response to formalin (phase II)117 ↓ Hypersensitivity to mechanical stimuli (complete Freund’s adjuvant)117 |
|
| KALSRDrd2-KO | KalSRCKO/+ mice were crossed with Drd2-Cre mice to generate KALSRDrd2-KO mice | ↓ Anxiety-related behavior (elevated zero maze) rescued with selective GluN2B antagonist ifenprodil.56 | ||
| KalSRPOMC-KO | KalSRCKO/+ mice were crossed with POMC-Cre mice to generate KALSRPOMC-KO mice | ↑ Serum corticosterone in females following restraint stress42 | ↓ Anxiety-related behavior in both males and females (elevated zero maze)42 ↓Acquisition of contextual fear memories in males and females (passive avoidance)42 |
6.1. KalGEF1KO behavioral and synaptic deficits
KalGEF1KO produced a deficit in cortico-specific Rac activity, consistent with a loss in kalirin-mediated GEF activity (Cahill et al., 2009), likely linked to the observed reduction in dendritic arborization (Xie et al., 2010). Furthermore, cortical spine density was reduced in the cortex, but not hippocampus, and this deficit emerged only in 12 week old mice. Furthermore, at 3 and 12 weeks, hippocampal neurons had normal dendritic spine density, but at 11 months showed a decrease in the presence of spines. Thus, despite loss of all isoforms, the functional effects of KalGEF1KO appears to selectively alter mature neuronal development at time points where kalirin7 expression dominates. These results indicate a vital role for kalirin7 in the adult brain, whereas the functions of kalirin9 and 12 display functional redundancy during early development; trio represents a potential candidate for this redundancy, due to structural and phenotypic overlap. Furthermore, KalGEF1KO animals displayed reduced prepulse inhibition, sociability and increased locomotor activity suggesting a wide range of behavioral deficits induced by loss of KALRN expression (Cahill et al., 2009; Vanleeuwen and Penzes, 2012). Interestingly, hyperactivity was observed at 12 weeks but not 3, suggesting a link to alterations in cortical spine density, which were only altered at similar developmental time points. It is notable that Kalrn knockout produces specific deficits in working memory, but not reference memory (Cahill et al., 2009; Vanleeuwen and Penzes, 2012), sharing a phenotype with GluR1, Nr2a and Disc1 knockout models. As kalirin lies within a signaling pathway involving each of these components (Fig. 4) there appears a specific functional, as well as mechanistic, link between these molecules.
6.2. A comparison of KalrnKO models
KalrnSR mice display similar deficits in behavior (Mandela et al., 2012). Although experimental paradigms differed, some correlates exist supporting shared developmental defects (see Table 3). For example, although hyperactivity was not directly addressed in the KalrnSR model, an increase in familiar and novel object exploration may correlate to hyperactive phenotypes (Mandela et al., 2012). As mature developmental processes appeared most drastically altered in both KalGEF1KO and KalrnSR knockout animals, selective knockout of the kalirin7 isoform (Kal7KO) allowed the role of this specific isoform to be assessed. By genetic excision of the kalirin7-specific exon, expression of this isoform was blocked (Ma et al., 2008), revealing reduced hippocampal spine density, similar to adult Kalrn knockout animals (Vanleeuwen and Penzes, 2012; Xie et al., 2011). KalGEF1KO (Xie et al., 2011); KalrnSR (Mandela et al., 2012) and Kal7KO (Ma et al., 2008) mice showed decreased anxiety-like behavior and impaired contextual fear learning, indicating functional kalirin7 is essential for anxiety and fear-related behaviors.
Several notable differences were observed between these knockout animals; whereas KalGEF1KO mice displayed hyperactivity (Cahill et al., 2009), none was observed in Kal7KO (Ma et al., 2008). Thus, despite the emergence of hyperactivity concurrently with cortical spine deficits in Kalrn knockout animals and periods of kalirin7 expression, hyperactivity appears to rely on kalirin7-independent mechanisms. Both full Kalrn knockout animals displayed deficits in working memory, whereas kalirin7 was unimpaired in this respect, though experimental paradigms may contribute somewhat to these differential findings. Both KalrnSR and Kal7KO mice displayed decreased anxiety-like behavior and hippocampal-dependent passive avoidance deficits (Mandela et al., 2012; Ma et al., 2008). Interestingly, knockout of Kalrn in neuroendocrine POMC-expressing cells specifically (KalrnSR-POMC) recapitulated these phenotypes, suggesting these may occur as a result of changes in secreted neuropeptides, such as ACTH, rather than altered neuronal connectivity (Mandela et al., 2014). Several phenotypes, were only evident in full knockout models; for example, Kal7KO did not show alterations in novel objection recognition, suggesting a role for loss of kalirin9 and 12 isoforms in these phenotypes (Mandela et al., 2012).
6.3. Conserved alterations in synaptic plasticity
Despite these differences, the role of kalirin in synaptic plasticity was highly conserved in full knockout and Kal7KO animals. Both displayed deficiencies in hippocampal LTP (Lemtiri-Chlieh et al., 2011; Xie et al., 2011) suggesting that regulation of the mature synapse is imparted by kalirin7 specifically. Extensive electrophysiological analysis of the Kal7KO model revealed the role of kalirin7 in regulating NMDAr-mediated synaptic plasticity. Indeed, both LTP (Kiraly et al., 2011; Ma et al., 2008) and LTD were strikingly impaired, but NMDAr independent synaptic plasticity mechanisms were unaffected (Lemtiri-Chlieh et al., 2011). To further strengthen the link to NMDAr dependent mechanisms, kalirin was found to interact with GluN2B and loss of kalirin impairs the sensitivity of neurons to the GluN2B selective inhibitor ifenprodil (Kiraly et al., 2011; LaRese et al., 2017). As NMDAr-mediated signaling is integral to synaptic mechanisms of cocaine addiction, it is perhaps not surprising that Kal7KO showed altered cocaine-mediated behavioral alterations and increased rates of self-administration (Kiraly et al., 2013). Kal7KO animals also showed reduced LTP response in spinal neurons, linked to the reduced ability of neurons to respond to inflammation and deficits in nociceptive sensitization in Kal7KO (Lu et al., 2015). Together, kalirin plays a vital role in the formation and regulation of synapses in both full Kalrn knockout and Kal7KO animals, further strengthening kalirin7 as a key regulator of neuronal connectivity.
6.4. Kalirin in neuropeptide secretion and hormonal regulation
Kalirin has been linked to the maturation of secretory granules, contributing to the secretion of neuropeptides and hormones (Ferraro et al., 2007) perhaps linked to its interaction with peptidylglycine alpha-amidating monooxygenase (PAM) within the trans-golgi network (Alam et al., 1996). Kalirin8, 12 and delta isoforms are expressed robustly in a range of secretory cells and overexpression of the kalirin GEF domain can induce secretion from neuroendocrine cells in the absence of stimulation, suggesting a role for kalirin in driving secretory granule maturation. Furthermore, KalrnSR animals have impaired skeletal bone mass, linked to impaired OPG secretion and osteoclastogenesis (Huang et al., 2014). These animals were also observed to have elevated growth hormone and prolactin secretion from pituitary cells (Mandela et al., 2012). In addition, loss of kalirin expression in POMC-expressing cells (largely neuroendocrine) results in altered basal secretion of various hormonal factors (Mandela et al., 2014). These results highlight an additional role of kalirin in regulating endocrine signaling and the secretory pathway.
These animal models reveal robust alterations in neuronal function that provide insight into the behavioral and physiological role of Kalrn; kalirin is required for normal synaptic plasticity, dendritic arborization and formation of spines, and alterations in these processes are linked to robust behavioral defects. In neuroendocrine cells, kalirin is involved in the secretory pathway, perhaps linked to anatomical and muscular deficits.
7. Pharmacological regulation of kalirin
Few pharmacological agents have been isolated to regulate kalirin activity, likely due to the difficulty in targeting the GEF-GTPase interface with small molecules. Due to the integral role of kalirin and trio in disease, however, various attempts have been made to develop pharmacological agents to regulate activity.
7.1. High throughput screening – aptamers and peptides
Proof of regulatory potential was generated when Schmidt and colleagues (Schmidt et al., 2002) developed an aptamer screen to identify peptides that interact with the RhoA-GEF domain of trio. One isolated aptamer (TRIAPα) and the resulting 36mer peptide (TRIPα) were shown to inhibit RhoA-GEF activity (IC50-4 μM) by competitive binding to the trio DH domain (Schmidt et al., 2002). Interestingly, TRIAPα was also found to inhibit kalirin RhoA-GEF activity in vitro. When expressed in COS cells, this interaction reduced RhoA-GEF activity and inhibited trio mediated neurite retraction in PC12 cells. This effect was not observed for kalirin-RhoA-GEF domain expressing cells, indicating selective inhibition of trio in a cellular context (Schmidt et al., 2002). There is potential, however, for the development of kalirin-selective peptides, in a manner similar to that highlighted through the development of a selective Tgat inhibitor (Bouquier et al., 2009). Tgat is an oncogenic splice variant of trio that encodes only the RhoA-GEF DH domain. TRIPα was unable to inhibit Tgat activity to the same level as the complete RhoA-GEF domain of trio, suggesting that the peptide made contacts with the PH domain, absent in Tgat. The altered binding surface of Tgat allowed the development of selective peptide inhibitors of Tgat over trio; TRIPαE32G and TRIPαT16ML17S. Both were able to inhibit Tgat activity in vitro (IC50-7.4 and 5.1 μM, respectively) and TRIPαE32G was shown to reduce Tgat signaling and inhibit Tgat-induced tumor formation (Bouquier et al., 2009).
Aptamer screening technologies therefore have the potential to yield inhibitors of kalirin, in a similar manner to trio and Tgat. However, the therapeutic benefit of such peptides may be limited by cell permeability issues. Indeed, TRIPα efficacy in a cellular context required coexpression by transfection, and the development and use of cell permeable TRIPα derivatives has not been forthcoming. These tools remain invaluable, however, for analyzing the RhoA mediated effects of kalirin and trio in vitro without the need for truncation mutants that may affect signaling independent of GEF activity. Furthermore, they provide proof of principle for selective inhibition of the GEF domains of trio and kalirin.
7.2. High throughput screening – small molecules
Subsequently, research focused on the generation of inhibitory agents with higher potential therapeutic benefit. The first small molecule with regulatory potential isolated, and the most widely characterized, is 1-(3-nitrophenyl)-1H-pyrrole-2,5-dione (NPPD) (Blangy et al., 2006). Isolated by yeast-3-hybrid studies searching for inhibitors of the trio-RhoG interaction, NPPD was found to inhibit GEF activity, and was selective for the trio Rac-GEF domain. Two structural analogues were also isolated, and all three were found to limit trio-RhoG activity, but not GEFs targeting RhoA or Arf1 (Blangy et al., 2006). Interestingly, sub-sequent analyses revealed that NPPD could also inhibit kalirin Rac-GEF activity and may act to stabilize the interaction between the Rac-GEF domain and Rac (Ferraro et al., 2007). Indeed, such a mechanism has observed for the Arf1-SEC7 interaction by BrefeldinA (Peyroche et al., 1999) suggesting that stabilizing the interaction between a GEF and its cognate GEF may limit activity by restricting turnover of substrates; i.e. converting the GEF into a dominant negative inhibitor through sequestration of its GTPase. However, NPPD was found to be toxic (Bouquier et al., 2009), perhaps limiting the usefulness of such a compound, and potentially confounding phenotypic effects in cellular contexts. As such, a less potent but more tolerated compound isolated in Blangey and colleagues’ initial screen was assessed; ITX1. Multiple analogues of this compound (notably ITX3) were shown to efficiently inhibit trio activity in vitro.
7.3. Cellular efficacy of small molecules
The utility of these compounds within a cellular context has been observed in multiple systems; ITX3 was able to reverse Rac-mediated neurite outgrowth in PC12 cells (Bouquier et al., 2009) and dendritic outgrowth and branching in hippocampal neuronal cultures (Yan et al., 2015). Notably, ITX3 had no effect on cultured neurons form KalrnSR knockout animals. As discussed (See 5.6), mutations within KALRN promote stroke and coronary heart disease. Kalirin9 and 12 are expressed in smooth muscle and can drive the proliferation and migration of these cells, factors that promote the progression of atherosclerosis. NPPD was found to limit kalirin Rac-GEF mediated phosphorylation of PAK1 in response to PDGF in smooth muscle cells (Wu et al., 2013). Furthermore, NPPD and ITX3 were able to limit PDGF-mediated smooth muscle cell migration, suggesting cellular efficacy and a potential route to inhibit kalirin within atherosclerosis. Within neuronal models, NPPD was also able to limit the ability of kalirin to drive synaptic plasticity mechanisms in hippocampal slices to a similar extent as observed in kalirin7 knockout animal slice (Lemtiri-Chlieh et al., 2011). Within neuroendocrine cells, kalirin’s GEF activity can drive the secretion of cellular hormones such as POMC and ACTH (Ferraro et al., 2007). NPPD was found to reverse the depletion of cellular hormones induced by overexpression of kalirin’s Rac-GEF domain in AtT-20 cells. Together, these results show that pharmacological inhibition of both the Rac-GEF and RhoA-GEF domains is possible and pave the way for the development of non-toxic, efficacious small molecules suitable for in vivo application.
7.4. Pharmacological promise of KALRN-regulatory molecules
Despite these exciting discoveries, problems arise over the suitability of pharmacological kalirin inhibition in reversing disease states. Indeed, the majority of kalirin and trio mutations in neurodevelopmental disorders are likely loss of function (such as frameshift truncations), and so further inhibition is likely to exacerbate patient symptoms. However, a subset of mutations have been characterized as gain of function; P2255T within KALRN (Russell et al., 2018) and spectrin-8 repeat domain mutations within TRIO (Barbosa et al., 2020) drive RhoA and Rac activity, respectively. Thus, NPPD, or similar small molecule inhibitors, may have benefit in neurological disorders, such as schizophrenia and developmental delay, where the GEF activity of kalirin is altered. Thus, kalirin remains a viable pharmacological target, although much work is required to isolate suitable in vivo and therapeutic tools.
8. Conclusions
The broad range of neurological processes affected by KALRN dysfunction and number of neurological disease-associated mutations within the KALRN gene highlight the importance of kalirin in neurodevelopment and disease. As a central regulator of dendritic structure, the formation dendritic of spines and synaptic plasticity, kalirin represents an exciting target for further basic research, as well as translational studies to assess its viability as a drug target in neurodevelopmental disease. (Koo et al., 2007; Ratovitski et al., 1999; Brand et al., 2012; McClory et al., 2018; Tsai et al., 2012)
Acknowledgements
This review and the corresponding Gene Wiki article are written as part of the Gene Wiki Review series–a series resulting from a collaboration between the journal GENE and the Gene Wiki Initiative. The Gene Wiki Initiative is supported by National Institutes of Health (GM089820). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. This work was funded by the NIMH grant R01MH071316.
This study makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from https://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by Wellcome.
The corresponding Gene Wiki entry for this review can be found here en.wikipedia.org/wiki/Kalirin.
Abbreviations:
- GEF
guanosine nucleotide exchange factor
- Nlgn
neuroligan
- SH3
SRC homology 3 domain
- GAP
GTPase activating proteins
- NMDAr
N-methyl-D-aspartate receptor
- AMPAr
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor
- DH
Dbl homology
- PH
Pleckstrin homology
- PI-4,5-P
phosphatidyl inositol-4,5-bisphosphate
- PBD
PDZ binding domain
- Ig
Immunoglobulin-like C2-type
- FN3
Fibronectin type 3
- mEPSC
Miniature excitatory postsynaptic current
- LOF
Loss of Function
- SCZ
Schizophrenia
- ASD
Autism Spectrum Disorder
- FS
Frameshift
- ECT
Electroconvulsive therapy
- AD
Alzheimer’s disease
- iNOS
Inducible nitric oxide synthase
- KalGEF1KO
Full knockout animal generated by excision of exon 27 and 28
- KalrnSR
Full knockout animal generated by excision of exon 13
- Kal7KO
Kalirin7 isoform knockout animal
- KalrnSR-POMC
knockout animal of Kalrn in neuroendocrine POMC-expressing cells
- NPPD
1-(3-nitrophenyl)-1H-pyrrole-2,5-dione
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Alam MR, et al. , 1997. Kalirin, a cytosolic protein with spectrin-like and GDP/GTP exchange factor-like domains that interacts with peptidylglycine alpha-amidating monooxygenase, an integral membrane peptide-processing enzyme. J. Biol. Chem 272 (19), 12667–12675. [DOI] [PubMed] [Google Scholar]
- Kawai T, Sanjo H, Akira S, 1999. Duet is a novel serine/threonine kinase with Dbl-Homology (DH) and Pleckstrin-Homology (PH) domains. Gene 227 (2), 249–255. [DOI] [PubMed] [Google Scholar]
- Colomer V, et al. , 1997. Huntingtin-associated protein 1 (HAP1) binds to a Trio-like polypeptide, with a rac1 guanine nucleotide exchange factor domain. Hum. Mol. Genet 6 (9), 1519–1525. [DOI] [PubMed] [Google Scholar]
- Alam MR, et al. , 1996. Novel proteins that interact with the COOH-terminal cytosolic routing determinants of an integral membrane peptide-processing enzyme. J. Biol. Chem 271 (45), 28636–28640. [DOI] [PubMed] [Google Scholar]
- Johnson RC, et al. , 2000. Isoforms of kalirin, a neuronal Dbl family member, generated through use of different 5’- and 3’-ends along with an internal translational initiation site. J. Biol. Chem 275 (25), 19324–19333. [DOI] [PubMed] [Google Scholar]
- McPherson CE, Eipper BA, Mains RE, 2002. Genomic organization and differential expression of Kalirin isoforms. Gene 284 (1–2), 41–51. [DOI] [PubMed] [Google Scholar]
- Mains RE, et al. , 2011. Kalrn promoter usage and isoform expression respond to chronic cocaine exposure. BMC Neurosci 12, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MB, et al. , 2015. An N-terminal amphipathic helix binds phosphoinositides and enhances kalirin Sec14 domain-mediated membrane interactions. J. Biol. Chem 290 (21), 13541–13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel DE, et al. , 2001. Kalirin, a GDP/GTP exchange factor of the Dbl family, is localized to nerve, muscle, and endocrine tissue during embryonic rat development. J. Histochem. Cytochem 49 (7), 833–844. [DOI] [PubMed] [Google Scholar]
- Mandela P, Ma XM, 2012. Kalirin, a key player in synapse formation, is implicated in human diseases. Neural Plast. 2012, 728161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JH, et al. , 2013. Kalirin promotes neointimal hyperplasia by activating Rac in smooth muscle cells. Arterioscler. Thromb. Vasc. Biol 33 (4), 702–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller MR, et al. , 2008. Autonomous functions for the Sec14p/spectrin-repeat region of Kalirin. Exp. Cell Res 314 (14), 2674–2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostenko EV, et al. , 2005. The Sec14 homology domain regulates the cellular distribution and transforming activity of the Rho-specific guanine nucleotide exchange factor Dbs. J. Biol. Chem 280 (4), 2807–2817. [DOI] [PubMed] [Google Scholar]
- Ma XM, et al. , 2014. Nonenzymatic domains of Kalirin7 contribute to spine morphogenesis through interactions with phosphoinositides and Abl. Mol. Biol. Cell 25 (9), 1458–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, et al. , 2007. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron 56 (4), 640–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring BE, Nicoll RA, 2016. Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc. Natl. Acad. Sci. USA 113 (8), 2264–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemben MA, et al. , 2015. The cellular and molecular landscape of neuroligins. Trends Neurosci. 38 (8), 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paskus JD, et al. , 2019. Synaptic Kalirin-7 and trio interactomes reveal a GEF protein-dependent neuroligin-1 mechanism of action. Cell Rep. 29 (10), 2944–2952 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi-Takagi A, et al. , 2010. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci 13 (3), 327–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son K, Smith TC, Luna EJ, 2015. Supervillin binds the Rac/Rho-GEF Trio and increases Trio-mediated Rac1 activation. Cytoskeleton (Hoboken) 72 (1), 47–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa S, et al. , 2020. Opposite modulation of RAC1 by mutations in TRIO is associated with distinct, domain-specific neurodevelopmental disorders. Am. J. Hum. Genet 106 (3), 338–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller MR, et al. , 2006. Regulation of RhoGEF activity by intramolecular and intermolecular SH3 domain interactions. J. Biol. Chem 281 (27), 18774–18786. [DOI] [PubMed] [Google Scholar]
- Penzes P, et al. , 2001a. Distinct roles for the two Rho GDP/GTP exchange factor domains of kalirin in regulation of neurite growth and neuronal morphology. J. Neurosci 21 (21), 8426–8434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May V, et al. , 2002. Kalirin Dbl-homology guanine nucleotide exchange factor 1 domain initiates new axon outgrowths via RhoG-mediated mechanisms. J. Neurosci 22 (16), 6980–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skowronek KR, et al. , 2004. The C-terminal basic tail of RhoG assists the guanine nucleotide exchange factor trio in binding to phospholipids. J. Biol. Chem 279 (36), 37895–37907. [DOI] [PubMed] [Google Scholar]
- Liu X, et al. , 1998. NMR structure and mutagenesis of the N-terminal Dbl homology domain of the nucleotide exchange factor Trio. Cell 95 (2), 269–277. [DOI] [PubMed] [Google Scholar]
- Bellanger JM, et al. , 2003. Different regulation of the Trio Dbl-Homology domains by their associated PH domains. Biol. Cell 95 (9), 625–634. [DOI] [PubMed] [Google Scholar]
- Kiraly DD, et al. , 2011. Kalirin binds the NR2B subunit of the NMDA receptor, altering its synaptic localization and function. J. Neurosci 31 (35), 12554–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti K, et al. , 2005. Critical role for Kalirin in nerve growth factor signaling through TrkA. Mol. Cell. Biol 25 (12), 5106–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, et al. , 2001b. The neuronal Rho-GEF Kalirin-7 interacts with PDZ domain-containing proteins and regulates dendritic morphogenesis. Neuron 29 (1), 229–242. [DOI] [PubMed] [Google Scholar]
- Cahill ME, et al. Control of interneuron dendritic growth through NRG1/erbB4-mediated kalirin-7 disinhibition. Mol. Psychiatry 17(1); 2012: 1, 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer BJ, 2001. SH3 domains: complexity in moderation. J. Cell Sci 114 (Pt 7), 1253–1263. [DOI] [PubMed] [Google Scholar]
- Rojas RJ, et al. , 2007. Galphaq directly activates p63RhoGEF and trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J. Biol. Chem 282 (40), 29201–29210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandekar SJ, et al. , 2019. Structure of the C-terminal guanine nucleotide exchange factor module of Trio in an autoinhibited conformation reveals its oncogenic potential. Sci. Signal 12 (569). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell TA, et al. , 2018. A Schizophrenia-Linked KALRN coding variant alters neuron morphology, protein function, and transcript stability. Biol. Psychiatry 83 (6), 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medley QG, et al. , 2000. The trio guanine nucleotide exchange factor is a RhoA target. Binding of RhoA to the trio immunoglobulin-like domain. J. Biol. Chem 275 (46), 36116–36123. [DOI] [PubMed] [Google Scholar]
- Kratzer MC, et al. , 2019. Evolution of the Rho guanine nucleotide exchange factors Kalirin and Trio and their gene expression in Xenopus development. Gene Expr. Patterns 32, 18–27. [DOI] [PubMed] [Google Scholar]
- Yan Y, Eipper BA, Mains RE, 2015. Kalirin-9 and Kalirin-12 play essential roles in dendritic outgrowth and branching. Cereb. Cortex 25 (10), 3487–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y, Eipper BA, Mains RE, 2016. Kalirin is required for BDNF-TrkB stimulated neurite outgrowth and branching. Neuropharmacology 107, 227–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, et al. , 2003. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron 37 (2), 263–274. [DOI] [PubMed] [Google Scholar]
- Mandela P, et al. , 2012. Kalrn plays key roles within and outside of the nervous system. BMC Neurosci. 13, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandela P, et al. , 2014. Elimination of Kalrn expression in POMC cells reduces anxiety-like behavior and contextual fear learning. Horm. Behav 66 (2), 430–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemtiri-Chlieh F, et al. , 2011. Kalirin-7 is necessary for normal NMDA receptor-dependent synaptic plasticity. BMC Neurosci. 12, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MX, et al. , 2019. Role of Cdk5 in Kalirin7-mediated formation of dendritic spines. Neurochem. Res 44 (5), 1243–1251. [DOI] [PubMed] [Google Scholar]
- Hall A, 1998. Rho GTPases and the actin cytoskeleton. Science 279 (5350), 509–514. [DOI] [PubMed] [Google Scholar]
- Stankiewicz TR, Linseman DA, 2014. Rho family GTPases: key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci 8, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest MP, Parnell E, Penzes P, 2018. Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci 19 (4), 215–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L, 2000. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J. Neurosci 20 (14), 5329–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P, Bourgeois JP, Goldman-Rakic PS, 1994. Synaptic development of the cerebral cortex: implications for learning, memory, and mental illness. Prog. Brain Res 102, 227–243. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR, 1984. Synapse elimination and plasticity in developing human cerebral cortex. Am. J. Ment. Defic 88 (5), 488–496. [PubMed] [Google Scholar]
- Giedd JN, et al. , 1999. Brain development during childhood and adolescence: a longitudinal MRI study. Nat. Neurosci 2 (10), 861–863. [DOI] [PubMed] [Google Scholar]
- Penzes P, Remmers C, 2012. Kalirin signaling: implications for synaptic pathology. Mol. Neurobiol 45 (1), 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citri A, Malenka RC, 2008. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology 33 (1), 18–41. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL, Morris RG, 2014. Synaptic plasticity in health and disease: introduction and overview. Philos. Trans. R. Soc. Lond. B Biol. Sci 369 (1633), 20130129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill ME, et al. , 2009. Kalirin regulates cortical spine morphogenesis and disease-related behavioral phenotypes. Proc. Natl. Acad. Sci. USA 106 (31), 13058–13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRese TP, et al. , 2017. Using Kalirin conditional knockout mice to distinguish its role in dopamine receptor mediated behaviors. BMC Neurosci. 18 (1), 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CM, et al. , 2017. Wnt5a is essential for hippocampal dendritic maintenance and spatial learning and memory in adult mice. Proc. Natl. Acad. Sci. USA 114 (4), E619–E628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karczewski KJ, et al. , 2020. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 581 (7809), 434–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardinas AF, et al. , 2018. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet 50 (3), 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, et al. , 2013. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat. Genet 45 (10), 1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell SM, et al. , 2014. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506 (7487), 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer M, et al. , 2014. De novo mutations in schizophrenia implicate synaptic networks. Nature 506 (7487), 179–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders S., 2017. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542 (7642), 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baio J, et al. , 2018. Prevalence of autism spectrum disorder among children aged 8 years – autism and developmental disabilities monitoring network, 11 Sites, United States, 2014. MMWR Surveill Summ. 67 (6), 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, et al. , 2011. Prevalence of autism spectrum disorders in a total population sample. Am. J. Psychiatry 168 (9), 904–912. [DOI] [PubMed] [Google Scholar]
- Colvert E, et al. , 2015. Heritability of autism spectrum disorder in a UK population-based twin sample. JAMA Psychiatry 72 (5), 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert J, Man HY, 2017. Fundamental elements in autism: from neurogenesis and neurite growth to synaptic plasticity. Front. Cell. Neurosci 11, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler JJ, Zhang H, 2010. Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94. [DOI] [PubMed] [Google Scholar]
- Leblond CS, et al. , 2019. Both rare and common genetic variants contribute to autism in the Faroe Islands. NPJ Genom. Med 4, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterstrom FK, et al. , 2020. Large-scale exome sequencing study implicates both developmental and functional changes in the neurobiology of autism. Cell 180 (3), 568–584 e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadybekov A, et al. , 2017. An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun 8 (1), 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco Piccinelli FGH, 1997. Gender Differences in the Epidemiology of Affective Disorders and Schizophrenia. World Health Organisation. [Google Scholar]
- Sullivan PF, Kendler KS, Neale MC, 2003. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 60 (12), 1187–1192. [DOI] [PubMed] [Google Scholar]
- Singh T, et al. , 2017. The contribution of rare variants to risk of schizophrenia in individuals with and without intellectual disability. Nat. Genet 49 (8), 1167–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glausier JR, Lewis DA, 2013. Dendritic spine pathology in schizophrenia. Neuroscience 251, 90–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howrigan DP, et al. , 2020. Exome sequencing in schizophrenia-affected parent-offspring trios reveals risk conferred by protein-coding de novo mutations. Nat. Neurosci 23 (2), 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayan S, et al. , 2008. Molecular profiles of schizophrenia in the CNS at different stages of illness. Brain Res. 1239, 235–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JJ, Hashimoto T, Lewis DA, 2006. Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 11 (6), 557–566. [DOI] [PubMed] [Google Scholar]
- Rubio MD, Haroutunian V, Meador-Woodruff JH, 2012. Abnormalities of the Duo/Ras-related C3 botulinum toxin substrate 1/p21-activated kinase 1 pathway drive myosin light chain phosphorylation in frontal cortex in schizophrenia. Biol. Psychiatry 71 (10), 906–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell TA, et al. , 2014. A sequence variant in human KALRN impairs protein function and coincides with reduced cortical thickness. Nat. Commun 5, 4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai DC, et al. , 2014. A genome-wide search for quantitative trait loci affecting the cortical surface area and thickness of Heschl’s gyrus. Genes Brain Behav. 13 (7), 675–685. [DOI] [PubMed] [Google Scholar]
- van Haren NE, et al. , 2008. Progressive brain volume loss in schizophrenia over the course of the illness: evidence of maturational abnormalities in early adulthood. Biol. Psychiatry 63 (1), 106–113. [DOI] [PubMed] [Google Scholar]
- Ma XM, Mains RE, Eipper BA, 2002. Plasticity in hippocampal peptidergic systems induced by repeated electroconvulsive shock. Neuropsychopharmacology 27 (1), 55–71. [DOI] [PubMed] [Google Scholar]
- Afroz S, et al. , 2016. Synaptic pruning in the female hippocampus is triggered at puberty by extrasynaptic GABAA receptors on dendritic spines. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardozo T, et al. , 2017. Chemistry-based molecular signature underlying the atypia of clozapine. Transl. Psychiatry 7 (2), e1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, et al. , 2009. Rapid modulation of spine morphology by the 5-HT2A serotonin receptor through kalirin-7 signaling. Proc. Natl. Acad. Sci. USA 106 (46), 19575–19580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushima I, et al. , 2012. Resequencing and association analysis of the KALRN and EPHB1 genes and their contribution to schizophrenia susceptibility. Schizophr. Bull 38 (3), 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz S, et al. , 2007. Structure of Galphaq-p63RhoGEF-RhoA complex reveals a pathway for the activation of RhoA by GPCRs. Science 318 (5858), 1923–1927. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Minden A, Yuste R, 2000. Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb. Cortex 10 (10), 927–938. [DOI] [PubMed] [Google Scholar]
- Moeschler JB, Shevell M, Committee on G, 2014. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics 134 (3), e903–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann WE, Moser HW, 2000. Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex 10 (10), 981–991. [DOI] [PubMed] [Google Scholar]
- Purpura DP, 1974. Dendritic spine “dysgenesis” and mental retardation. Science 186 (4169), 1126–1128. [DOI] [PubMed] [Google Scholar]
- Makrythanasis P, et al. , 2016. Exome sequencing discloses KALRN homozygous variant as likely cause of intellectual disability and short stature in a consanguineous pedigree. Hum Genom. 10 (1), 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena-Oliver Y, et al. , 2016. Mouse and human genetic analyses associate kalirin with ventral striatal activation during impulsivity and with alcohol misuse. Front. Genet 7, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraly DD, et al. , 2013. Constitutive knockout of kalirin-7 leads to increased rates of cocaine self-administration. Mol. Pharmacol 84 (4), 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. , 2013. Kalirin-7 mediates cocaine-induced AMPA receptor and spine plasticity, enabling incentive sensitization. J. Neurosci 33 (27), 11012–11022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraly DD, et al. , 2010. Behavioral and morphological responses to cocaine require kalirin7. Biol. Psychiatry 68 (3), 249–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeTure MA, Dickson DW, 2019. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener 14 (1), 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn H, et al. , 2007. Kalirin is under-expressed in Alzheimer’s disease hippocampus. J. Alzheimers Dis 11 (3), 385–397. [DOI] [PubMed] [Google Scholar]
- Asiimwe N, et al. , 2016. Nitric oxide: exploring the contextual Link with Alzheimer’s disease. Oxid. Med. Cell Longev 2016, 7205747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn H, et al. , 2007. Under-expression of Kalirin-7 Increases iNOS activity in cultured cells and correlates to elevated iNOS activity in Alzheimer’s disease hippocampus. J. Alzheimers Dis 12 (3), 271–281. [DOI] [PubMed] [Google Scholar]
- Dejanovic B, et al. , 2018. Changes in the synaptic proteome in tauopathy and rescue of tau-induced synapse loss by C1q antibodies. Neuron 100 (6), 1322–1336 e7. [DOI] [PubMed] [Google Scholar]
- Cisse M, et al. , 2017. The transcription factor XBP1s restores hippocampal synaptic plasticity and memory by control of the Kalirin-7 pathway in Alzheimer model. Mol. Psychiatry 22 (11), 1562–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, et al. , 2019. Kalirin-7 prevents dendritic spine dysgenesis induced by amyloid beta-derived oligomers. Eur. J. Neurosci 49 (9), 1091–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray PS, et al. , 2012. beta-Amyloid 42/40 ratio and kalirin expression in Alzheimer disease with psychosis. Neurobiol. Aging 33 (12), 2807–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, et al. , 2017. Genetic variant of kalirin gene is associated with ischemic stroke in a Chinese han population. Biomed. Res. Int 2017, 6594271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang M, et al. , 2018. Genetic variation of the kalirin gene is associated with ICAS in the Chinese population. J. Mol. Neurosci 66 (2), 157–162. [DOI] [PubMed] [Google Scholar]
- Dang M, et al. , 2015. KALRN rare and common variants and susceptibility to ischemic stroke in Chinese Han population. Neuromol. Med 17 (3), 241–250. [DOI] [PubMed] [Google Scholar]
- Wang L, et al. , 2007. Peakwide mapping on chromosome 3q13 identifies the kalirin gene as a novel candidate gene for coronary artery disease. Am. J. Hum. Genet 80 (4), 650–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boroumand M, et al. , 2014. The Kalirin gene rs9289231 polymorphism as a novel predisposing marker for coronary artery disease. Lab. Med 45 (4), 302–308. [DOI] [PubMed] [Google Scholar]
- Mofarrah M, et al. , 2016. Association of KALRN, ADIPOQ, and FTO gene polymorphism in type 2 diabetic patients with coronary artery disease: possible predisposing markers. Coron. Artery Dis 27 (6), 490–496. [DOI] [PubMed] [Google Scholar]
- Shafiei A, et al. , 2018. Association between serum kalirin levels and the KALRN gene rs9289231 polymorphism in early-onset coronary artery disease. J. Tehran Heart Cent 13 (2), 58–64. [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Cahill ME, Penzes P, 2010. Kalirin loss results in cortical morphological alterations. Mol. Cell. Neurosci 43 (1), 81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanleeuwen JE, Penzes P, 2012. Long-term perturbation of spine plasticity results in distinct impairments of cognitive function. J. Neurochem 123 (5), 781–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XM, et al. , 2008. Kalirin-7 is required for synaptic structure and function. J. Neurosci 28 (47), 12368–12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, et al. , 2011. Hippocampal phenotypes in kalirin-deficient mice. Mol. Cell. Neurosci 46 (1), 45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, et al. , 2015. A role for Kalirin-7 in nociceptive sensitization via activity-dependent modulation of spinal synapses. Nat. Commun 6, 6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro F, et al. , 2007. Kalirin/Trio Rho guanine nucleotide exchange factors regulate a novel step in secretory granule maturation. Mol. Biol. Cell 18 (12), 4813–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, et al. , 2014. The Rho-GEF Kalirin regulates bone mass and the function of osteoblasts and osteoclasts. Bone 60, 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, et al. , 2002. Identification of the first Rho-GEF inhibitor, TRIPalpha, which targets the RhoA-specific GEF domain of Trio. FEBS Lett. 523 (1–3), 35–42. [DOI] [PubMed] [Google Scholar]
- Bouquier N, et al. , 2009. Aptamer-derived peptides as potent inhibitors of the oncogenic RhoGEF Tgat. Chem. Biol 16 (4), 391–400. [DOI] [PubMed] [Google Scholar]
- Blangy A, et al. , 2006. Identification of TRIO-GEFD1 chemical inhibitors using the yeast exchange assay. Biol. Cell 98 (9), 511–522. [DOI] [PubMed] [Google Scholar]
- Peyroche A, et al. , 1999. Brefeldin A acts to stabilize an abortive ARF-GDP-Sec7 domain protein complex: involvement of specific residues of the Sec7 domain. Mol. Cell 3 (3), 275–285. [DOI] [PubMed] [Google Scholar]
- Bouquier N, et al. , 2009. A cell active chemical GEF inhibitor selectively targets the Trio/RhoG/Rac1 signaling pathway. Chem. Biol 16 (6), 657–666. [DOI] [PubMed] [Google Scholar]
- Koo TH, Eipper BA, Donaldson JG, 2007. Arf6 recruits the Rac GEF Kalirin to the plasma membrane facilitating Rac activation. BMC Cell Biol. 8, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratovitski EA, et al. , 1999. Kalirin inhibition of inducible nitric-oxide synthase. J. Biol. Chem 274 (2), 993–999. [DOI] [PubMed] [Google Scholar]
- Brand F, et al. , 2012. The extracellular signal-regulated kinase 3 (mitogen-activated protein kinase 6 [MAPK6])-MAPK-activated protein kinase 5 signaling complex regulates septin function and dendrite morphology. Mol. Cell. Biol 32 (13), 2467–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClory H, et al. , 2018. The COOH-terminal domain of huntingtin interacts with RhoGEF kalirin and modulates cell survival. Sci. Rep 8 (1), 8000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, et al. , 2012. The Guanine nucleotide exchange factor kalirin-7 is a novel synphilin-1 interacting protein and modifies synphilin-1 aggregate transport and formation. PLoS One 7 (12), e51999. [DOI] [PMC free article] [PubMed] [Google Scholar]