

Abstract
A better understanding of the molecular mechanisms underlying disease is key for expediting the development of novel therapeutic interventions. Disease mechanisms are often mediated by interactions between proteins. Insights into the physical rewiring of protein–protein interactions in response to mutations, pathological conditions, or pathogen infection can advance our understanding of disease etiology, progression, and pathogenesis and can lead to the identification of potential druggable targets. Advances in quantitative mass spectrometry (MS)‐based approaches have allowed unbiased mapping of these disease‐mediated changes in protein–protein interactions on a global scale. Here, we review MS techniques that have been instrumental for the identification of protein–protein interactions at a system‐level, and we discuss the challenges associated with these methodologies as well as novel MS advancements that aim to address these challenges. An overview of examples from diverse disease contexts illustrates the potential of MS‐based protein–protein interaction mapping approaches for revealing disease mechanisms, pinpointing new therapeutic targets, and eventually moving toward personalized applications.
Keywords: affinity purification, mass spectrometry, networks, protein–protein interactions, proximity labeling
Subject Categories: Molecular Biology of Disease,
This Review discusses mass spectrometry techniques that have been instrumental for identifying protein‐protein interactions. Examples from diverse disease contexts illustrate the potential of these approaches for revealing disease mechanisms and therapeutic targets.
Introduction
Identifying the principal molecular basis of human diseases is crucial for successful prevention, diagnosis, and treatment. In the past two decades, scientists have placed a lot of hope on large genomic studies for deciphering disease mechanisms. Nevertheless, despite the wealth of genomic information gathered, the molecular mechanism of most diseases remains unknown. This can be explained at least in part by the fact that many human diseases are complex and do not follow a classical genotype to phenotype model. They may result from multiple genetic changes, epigenetic modifications, or infection by a pathogen. The fallacy of expecting simple genetic changes to explain complex disease phenotypes has been demonstrated especially well in the case of cancer, where a distinct collection of mutations is often not exclusive to a given cancer type (Junttila & de Sauvage, 2013; Leiserson et al, 2015). Additionally, mutations of a single gene can lead to multiple different diseases, with the corresponding proteins having several functions in different cellular contexts (Nadeau, 2001). Consequently, extracting useful diagnostic or prognostic information from genetics alone can be difficult.
Considering genetic information in the context of disrupted cellular processes and networks can help overcome this challenge. Systems biology approaches, which aim to provide a comprehensive picture of a biological process by quantifying all observable components and their relationships, are well‐suited to understand the influence of disease mutations on a complex network of interconnected pathways. Proteins are the key components of these networks. Often, individual proteins do not perform any of their functions in isolation but accomplish the task through direct interactions with other proteins. As such, studying protein–protein interaction (PPI) networks has become a powerful tool for identifying the functional consequences of genetic variation. In this approach, disease‐related gene mutations are mapped to vital PPIs of cellular processes. Comparison of disease states with the wild‐type reference map—either through the introduction of proteins carrying mutations or exogenous expression of pathogen proteins—promises to reveal how networks change during disease pathogenesis (Krogan et al, 2015; Willsey et al, 2018).
Cellular proteins are directly responsible for adaptation to disease‐mediated changes. Because of the connectivity between proteins, the impact of a disease‐related mutation is not restricted to a specific gene product. Instead, it affects the entire network and can accordingly impact the activity of a whole subset of proteins. Instead of focusing on individual genes or loci implicated in human disease, PPI‐based analyses study the parts of pathway connections that are most changed by the disease state, thus offering an alternative to identify a mutation’s impact on cellular function. Interacting proteins can be visualized using a network‐based approach, with nodes representing the “bait” proteins of interest of a PPI study. Nodes are connected by edges to the interacting proteins identified by Affinity Purification Mass Spectrometry (AP‐MS), proximity labeling, Cross‐Linking Mass Spectrometry (XL‐MS), or other types of experiments. This mapping is performed in both the diseased state and non‐diseased or WT states, and variations between the global regulation of PPIs in the networks are monitored. The introduction of disease‐related mutations can lead to perturbations in these networks, including a complete loss of interactions, partial loss of specific interactions, or a rewiring or gain in new interactions (Fig 1). This connectivity suggests that small changes to a PPI network, such as the introduction of mutations to a particular gene, can cause significant changes at multiple nodes across the system. Changes in the interaction partners of the disease‐related protein, either during disease progression or following an infection, might contribute to a specific disease state, potentially linking genotype and phenotype. Applying a network‐based approach to study human diseases has multiple clinical and therapeutic advantages. The finding that a gene or protein is implicated in a given biochemical process or disease suggests that its interacting proteins may also play a role in the same processes, thus providing potential mechanistic explanations and therapeutic implications beyond a single gene or protein.
Figure 1. A systems‐level approach for converting genetic information into a pathway‐level understanding of data.
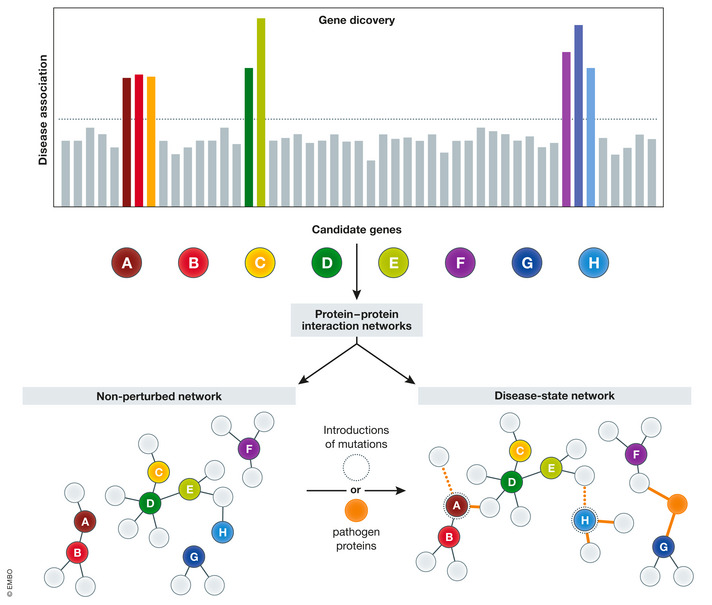
Genetic variants, which may occur rarely across individuals with a specific disease, can be used as the basis of PPI networks. Comparisons of WT PPI networks and PPI networks with disease‐related mutations introduced can aid in determining the functional significance of these mutations. Similarly, the introduction of pathogenic proteins can determine which host pathways are hijacked over the course of an infection.
Here, we review the current state of research using mass spectrometry (MS)‐based global and unbiased PPI networks to study human disease. Throughout, we will highlight current challenges of the field, and how new advances in the mapping of PPI networks address some of them. For a detailed examination of other PPI identification tools not relying on MS for detection, we refer the reader to other reviews (e.g., Snider et al, 2015; Beltran et al, 2017).
MS‐based methods for global PPI studies
Liquid chromatography‐MS (LC‐MS) is a sensitive, accurate, and selective method to quantify proteins (Richards et al, 2015; Aebersold & Mann, 2016). One of its major benefits to identify PPIs is the global and unbiased nature of MS proteomics. This is in contrast to other methods for identifying PPIs, including yeast‐2‐hybrid (Y2H), which maps physical, binary interactions of a predetermined set of proteins of interest (Walhout & Vidal, 2001). The general workflow of utilizing discovery MS to develop PPI networks is outlined in Box 1 and illustrated in Fig 2. Below, we summarize a variety of methods that, when combined with quantitative MS, allow the proteome‐level analysis of complex biological systems.
Box 1.
The general workflow of discovery MS starts with digesting a mixture of proteins into peptides with defined cleavage sites (e.g., using trypsin), which are separated using liquid chromatography and their mass‐to‐charge (m/z) is measured in a mass spectrometer. In standard tandem MS/MS experiments, the sequence of individual peptides will be determined by collecting a second MS spectrum after induced fragmentation. Taken together, the m/z data of fragments and full peptides are then used to computationally search large databases specific to the organism of interest and thus identify proteins in the original mixture (Fig 2A). To identify candidate interactors in protein–protein interaction studies, data will be “scored” to determine the accuracy of the identified interaction. This is oftentimes done by combining several parameters such as reproducibility, specificity, and abundance of each detected protein. A variety of scoring algorithms exists for this purpose, including MiST, CompPASS, and SAINT (Sowa et al, 2009; Choi et al, 2011; Teo et al, 2014, 2016; Morris et al, 2014; Verschueren et al, 2015). The general methodology of each algorithm differs—for example, SAINT incorporates quality controls and quantitative data for a given prey to determine the probability that an interaction between the prey and bait protein is a true positive, while CompPASS utilizes several scoring parameters that ultimately focus on abundance, uniqueness, and reproducibility to distinguish between true interactors and contaminant background proteins (Christianson et al, 2011). The output of these programs is a table of filtered, scored data that can be imported into network visualization tools such as Cytoscape (Shannon et al, 2003).
In addition to computational approaches assessing the specificity of PPIs by comparing to appropriate controls, a variety of different MS methods exists for quantifying changes between different conditions (Fig 2B–E). Label‐free quantitation allows comparing the relative abundances of identified proteins in an unlimited number of samples (Fig 2B). However, there are limitations with this approach, one of them being that for comparison purposes, identical amounts of each sample should be injected on the column for analysis. When this is not possible, normalization of the data may be required. Additionally, to reduce instrumental bias, samples being compared should be analyzed in a single acquisition batch on the mass spectrometer. Randomization of run order can also help avoid systematic errors. Metabolic or isobaric labeling approaches such as Stable Isotope Labeling with Amino Acids in Cell Culture (SILAC) and tandem mass tag (TMT) or other isobaric labels allow the user to multiplex multiple samples together, increasing experimental throughput. SILAC metabolically incorporates stable heavy amino acids at the protein level (Fig 2C; Ong et al, 2002; Szklarczyk et al, 2019), while isobaric tagging methods utilize NHS‐activated molecules that label free amines with chemical tags in vitro following digestion (Fig 2D). All labeling methods rely on the inclusion of additional control samples to which a mass label is added, so that in a mixture of control and experimental sample the origin of a respective protein interactor can be traced (Ong et al, 2002; Thompson et al, 2003; Mann, 2014). Together, these methods allow comparison across different conditions or timepoints or to discriminate between specific and non‐specific interactions (Wiese et al, 2007; Virreira Winter et al, 2018). Additionally, targeted MS strategies, such as parallel reaction monitoring (PRM) or multiple/selective reaction monitoring (MRM/SRM), can also be used to validate interactions with greater consistency, sensitivity, and accuracy (Lange et al, 2008; Gallien et al, 2012; Peterson et al, 2012). Briefly, unique peptides of the target protein are selected during assay development. These are then monitored through their signature fragment ions for precise quantitation in the final experiment (Fig 2E).
Among the identified proteins in MS‐based interaction studies, numerous non‐specific interactors or contaminants are copurified together with the protein of interest. Therefore, it is necessary to analyze PPI studies in a way that separates true interactors from artifacts. This can be done, in part, through careful experimental design and suitable controls. Importantly, appropriate controls such as an unrelated protein carrying the same tag, or the tag alone, need to be included to determine the specificity of interaction (Jäger et al, 2011b). For example, GFP can be used as a bait in control experiments. It is unlikely for GFP to form interactions with many proteins, and identified interactors are presumably false positives due to the epitope tags or the affinity capture method (Morris et al, 2014). Additionally, each type of affinity tag can capture specific background contaminations. These contaminants can be accessed via the CRAPome database (Mellacheruvu et al, 2013), a public repository of interactions generated from negative control data, and filtered out of experiments. Contamination can also result from carryover of overexpressed proteins, with residual amount of protein identified in subsequent MS experiments despite not actually being present as an interactor. Strict wash steps between experimental conditions may be required to alleviate this problem.
Figure 2. Overview of different mass spectrometry techniques.
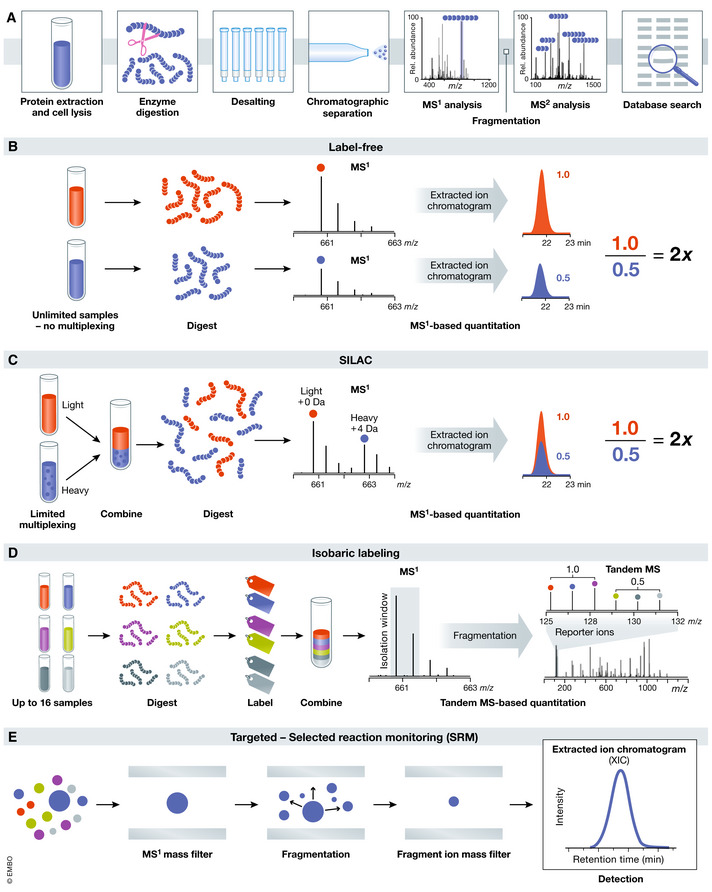
(A) Workflow for bottom‐up proteomics. Preparing proteomic samples for LC‐MS/MS analysis requires protein extraction, proteolysis, and, optionally, peptide‐level fractionation. Online LC separation of complex peptide mixtures introduces analytes into the mass spectrometer for precursor and fragment ion mass analysis. Tandem mass spectra are matched to theoretical spectra generated in silico to garner peptide sequences that are used for protein inference. (B) Label‐free quantitation. Following protein digestion, for each sample, an equal amount of peptides is separately loaded on the column. Relative quantitation is performed by comparing the extracted peak intensity of a given peptide across runs in the dataset. (C) SILAC. During cell culture, “light” or “heavy” versions of specific amino acids are metabolically incorporated into samples. Following sample preparation, cell lysates are mixed in equal total protein ratios and digested into peptides. Intensities of peptide extracted ion chromatograms from the MS1 scan can be used to quantify relative protein abundances between samples. (D) Isobaric labeling. Each sample is digested into peptides, labeled with a unique isobaric label, and mixed in equal ratios. During MS/MS analysis, each tag yields a fragment with a unique mass that can be used for relative quantitation. (E) Targeted MS. In SRM, each fragment of a protein of interest is individually monitored and quantified. The peptide of interest is first isolated, and its characteristic fragments can be monitored for quantitation. Only the specific peptide and fragment masses selected by the user are monitored over the analysis.
Affinity purification mass spectrometry (AP‐MS)
AP‐MS experiments (Fig 3A) utilize epitope tagging, where short peptide or protein tags (for example, FLAG‐, TAP‐, Strep‐Tag, or c‐myc (Chang, 2006)) are fused to the protein of interest—either in the context of an exogenous expression construct or under the gene’s endogenous promoter using gene editing technologies like CRISPR‐Cas9. The resulting bait protein functions as an affinity capture probe for interacting, or “prey” proteins, eliminating the need for specific antibodies to proteins of interest, as would be the case in lower throughput immunoprecipitation (IP) experiments. The affinity tag can easily be purified on a matrix recognizing the epitope. After washing steps to eliminate non‐specific interactors, interacting proteins can be identified via MS.
Figure 3. Overview of MS‐based methods to determine protein–protein interaction networks.
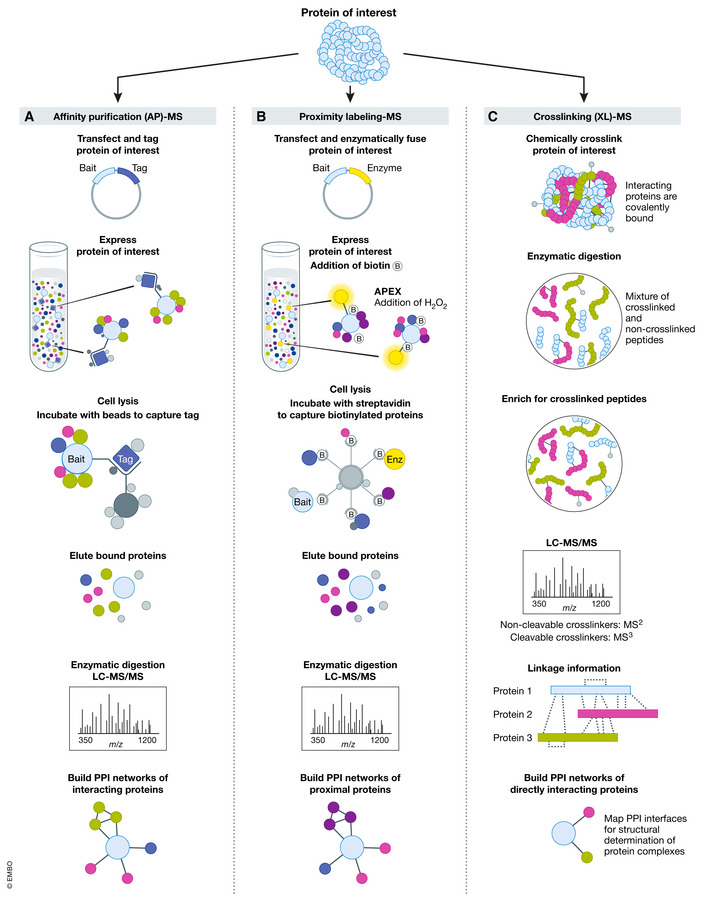
(A) General workflow for identifying interacting proteins using AP‐MS. Bait proteins are endogenously tagged and expressed in cells, followed by cell lysis and affinity purification of bait proteins and interacting prey proteins. The mixture is digested and analyzed by LC‐MS/MS. Following data processing to determine true interactors (BOX), bait and prey proteins can be incorporated into PPI networks. (B) Identification of proximal proteins using proximity labeling. The protein of interest is fused with a promiscuous ligase and expressed in cells. Following the addition of biotin, proteins interacting within the fusion protein’s labeling radius are tagged and can be subsequently lysed and captured using an affinity matrix. The mixture is digested and analyzed by LC‐MS/MS. Following data processing to determine true proximal proteins (BOX), bait and proximal proteins can be incorporated into PPI networks. (C) Direct interactions via cross‐linked peptides using XL‐MS. Following cross‐linking with the appropriate reagent, cells are lysed and digested, and the mixture is enriched for peptides tagged with the cross‐linker. Following LC‐MS/MS, data interpretation is performed to identify cross‐linked peptides and build PPI networks of directly interacting proteins.
Advances in high‐throughput AP‐MS methodologies have enabled the identification of 1,000s of protein complexes and PPIs in large‐scale interaction networks, both in models of healthy and disease states. The largest assembly of such PPI networks is the BioPlex database, which has, to date, compiled over 56,533 interactions with 10,961 proteins in HEK293T cells (Huttlin et al, 2015, 2017). Publicly available data sets like these, including hu.MAP 2.0 (Drew et al, 2017; preprint: Drew et al, 2020), represent important resources for biomedical research efforts and have spurred a multitude of discoveries of molecular mechanisms underlying disease, some of which we discuss further below.
A limitation of AP‐MS is the need for milder lysis conditions than those typically employed in MS experiments. Membrane proteins can be hard to capture using this approach due to problems in protein extraction (Sastry et al, 2009; Pankow et al, 2016). Weaker or more transient interactions are also prone to loss during extraction or washing steps. Tandem affinity purification (TAP) tagging affixes two separate proteins or peptide tags to a fusion protein of interest (Rigaut et al, 1999), and using one tag that can endure harsher lysis or washing conditions (e.g., His‐tag) can increase the recovery rate of proteins that are lost in regular AP‐MS experiments (Puig et al, 2001). However, this comes at the disadvantage of more laborious sample preparation and purification, as well as potential artifacts due to the addition of large tags to the protein of interest. Irrespective of the number of tags employed, non‐specific interactors that remain after washing can cause background issues, requiring careful selection of negative controls. Another limitation of AP‐MS is the lysis‐induced mixing of cellular compartments that do not normally interact, which can result in false positive PPI identifications. Possible solutions to deconvolute the effects of compartment mixing are currently being explored and will be discussed in the section New Methodology. It is possible that introducing a tag to the N‐ or C‐terminus may disrupt normal protein function, making it advantageous to test tagging both termini. It is also important to note that AP‐MS does not readily differentiate direct interactors from indirect interactors. On the other hand, AP‐MS offers many advantages over earlier strategies for determining interactions (e.g. Y2H), including high sensitivity and the quantification of multiple interactors at the same time (non‐binary). AP‐MS also allows detecting post‐translational modifications (PTMs) on interacting proteins (Matsuura et al, 2008). Following data generation, label‐free quantification can provide an intensity value for a given protein. This quantitative information can be used to perform comparative analyses and can thus help determine whether an interaction is specific to the protein of interest.
Proximity labeling
Proximity labeling represents a complementary strategy to traditional AP‐MS experiments (Han et al, 2018). In this case, proximal proteins are monitored by expressing in cells a bait protein of interest fused to a promiscuous labeling enzyme (Fig 3B). The addition of a small molecule substrate, such as biotin, allows the covalent tagging of endogenous proteins within a 10–20 nm range, capturing the protein’s surrounding environment, including potential interactors. After cell lysis, proteins are denatured and solubilized, followed by selective enrichment of biotinylated proteins, commonly through streptavidin binding, and identification by MS. Because of the strong binding affinity between biotin and streptavidin, proximity labeling permits more efficient protein extraction, lysis methods and harsher washing conditions than AP‐MS, allowing the identification of weak or transient interactions that might be lost with other methodologies. The procedure includes the use of detergents during lysis, as complexes are not required to remain intact during lysis and purification.
Various proximity labeling methodologies have been established. BioID utilizes BirA, a biotin ligase with specific mutations rendering the enzyme promiscuous. BirA catalyzes the transformation of biotin to a more reactive form, and the resultant biotin cloud reacts with primary amines of proteins in its vicinity, resulting in their covalent biotinylation (Roux et al, 2018). Subcellular compartments that have been targeted by BioID include the nuclear envelope (Kim et al, 2016b), centrosome (Antonicka et al, 2020), nucleus (preprint: Go et al, 2019), cytoplasm (Redwine et al, 2017), Golgi apparatus (Liu et al, 2018), ER (Hoffman et al, 2019), endosome, lysosome, mitochondrial matrix (Antonicka et al, 2020), cell–cell junctions (Fredriksson et al, 2015), and flagella (Kelly et al, 2020), with labeling efficiency limited in the ER (Roux et al, 2018; preprint: Go et al, 2019). Due to slow reaction kinetics, BioID requires labeling for 18–24 h to produce sufficient material for identification by MS, which can lead to off‐target labeling and high background, and somewhat restricts the type of experiments amenable to BioID. Additionally, due to its timescale, BioID experiments are limited to the generation of static interaction maps. An alternative to BioID, BioID2, was developed by introducing mutations to the biotin ligase of Aquifex aeolicus. This significantly smaller enzyme decreases the disruption to the fusion protein, allowing improved targeting and localization to subcellular compartments (Kim et al, 2016a). However, it still requires over 16 h of labeling. To improve labeling efficiency and speed, Branon et al (2018) performed directed evolution on BirA, which resulted in two faster‐acting enzymatic variations: TurboID carrying 15 mutations and miniTurbo carrying 13 mutations and a deletion of the N‐terminal domain. The high affinity of these enzymes for biotin allows comparable labeling to BioID in under ten minutes.
Another class of proximity labels arose from modifications to peroxidases, enzymes responsible for catalyzing redox reactions. Horseradish peroxidase (HRP) is the best‐studied peroxidase and has been employed for proximity labeling. However, it suffers from poor labeling efficiency in reducing environments (Trinkle‐Mulcahy, 2019). Engineered ascorbic acid peroxidase (APEX) does not have this drawback, and can be genetically introduced as a tag on bait proteins of interest (Rhee et al, 2013; Hung et al, 2016). Following the timed addition of H2O2, APEX oxidizes phenol derivatives to biotin‐phenoxyl radicals that covalently react with electron rich amino acids, providing biotin labeling kinetics on the order of minutes (Martell et al, 2012). The rapid labeling capabilities of APEX offer speed comparable to that of many biological processes and thus make this approach well‐suited to investigate transient or dynamically changing protein interactions. APEX labeling can be performed in most subcellular environments, as it retains activity in reducing environments, including the cytosol (Martell et al, 2012). Nevertheless, the need for peroxide has been criticized due to its potentially harmful effect on cells and prevents APEX labeling in living organisms. Newer iterations of proximity labeling methodology seek to avoid potential toxicity issues while requiring short labeling times.
The recently introduced, contact‐specific SplitID divides the TurboID enzyme in separate, inactive fragments (Cho et al, 2020). These two fragments recombine when in close proximity, as with interacting proteins. This method is well suited for organelle contact sites, where each fragment is targeted to a specific organelle, and subsequently, biotinylation is restricted to their contact sites, eliminating off‐target labeling. Similarly, the N‐ and C‐terminal fragments of split APEX are inactive when separated, but when joined through molecular interactions promote peroxidase activity (Han et al, 2019).
Experimental design should be carefully considered before undertaking a proximity labeling experiment. With all proximity labeling techniques, proteins neighboring the bait are captured throughout the experiment. Proteins that are not direct interactors but colocalize during the labeling period, simply due to diffusion through the enzymatic labeling region, can lead to high background, making it difficult to distinguish proteins that really reside in the immediate environment (Lobingier et al, 2017). The parallel analysis of the expressed ligase without an attached bait protein can help identify proteins not expected to be interactors. A protein’s presence in this control sample can arise from natural interactions with the ligase (Roux et al, 2018) and proteins that attach to the streptavidin used for enrichment. Similar to AP‐MS, it is possible that insertion of an enzyme at the N‐ or C‐ terminus may alter protein function. Prior to generating an enzyme‐expressing stable cell line, enzymatic fusion on both the N‐ and C‐termini of the protein of interest should be tested to ensure there is no disruption to normal localization (Sears et al, 2019). Another possibility is that proteins that are in proximity to the non‐labeled terminus fall outside the labeling radius and will therefore not be detected. As such, separate experiments where the N‐terminus and C‐terminus are labeled may be advantageous.
Cross‐linking mass spectrometry (XL‐MS)
Although AP‐MS can identify which proteins are within the same complex, it does not provide information on which members of the complex are actually in direct physical contact. XL‐MS is an approach that can fill this gap (Fig 3C). It provides structural information by identifying proximate amino acid pairs—including weak or transient interactions—covalently linked by a chemical cross‐linker of a specific length. The obtained distance restraint information can be used to determine the topology and orientation of subunits in the complex and PPI binding interfaces (Yu & Huang, 2018). The cross‐linking reaction is performed at near‐native conditions. Cross‐linked peptides are generated through enzymatic digestion, and the resultant cross‐linked peptides are enriched, followed by MS analysis and identification via database searching. Data analysis provides information on the sequence assignment of cross‐linked peptides and localization of specific cross‐linked amino acid residues. When paired with integrative modeling techniques, the physical interaction data derived from XL‐MS can be used to inform structural biology and computational modeling studies. Restraint information is obtained for both inter‐ and intra‐linked proteins and has been used to determine the structure of various protein complexes (Shi et al, 2014) and proteome‐wide interactions (Weisbrod et al, 2013). A limitation of XL‐MS is related to the data analysis step. Spectra contain a mix of various types of cross‐linked peptides, and complexities arise as all possible combinations of these peptides must be considered. However, rapid progress has been made in developing software to assist with this task, including software specific for cleavable and non‐cleavable cross‐linkers (Liu et al, 2017; Lu et al, 2018).
A variety of different cross‐linkers is available, targeting various amino acid side chains and distances between binding interfaces (Kao et al, 2011; Gutierrez et al, 2016). To increase the sensitivity and accuracy of XL‐MS, many different methods have been developed for enriching cross‐linked peptides (Tang et al, 2005; Rinner et al, 2008; Leitner et al, 2012; Kaake et al, 2014), utilizing the advantages of varying cross‐linker chemistry (Kaake et al, 2014), as well as optimizing data acquisition and analysis workflows (Liu et al, 2015; O’Reilly & Rappsilber, 2018). When cross‐linking is performed prior to cell lysis, in vivo interactions are stabilized and able to survive harsh denaturing and wash conditions, removing background contamination while preserving weak or transient interactions (Kaake et al, 2014).
The methods described above provide complementary approaches for interactome mapping. Integration of these techniques in MS workflows can aid the characterization of disease‐associated molecules, as we detail in the following sections.
Disease Network Analysis
Implementation of disease networks
An early study used AP‐MS to identify binding partners for 338 bait proteins specifically selected for their roles in various human diseases, including cancer, diabetes, and obesity (Ewing et al, 2007). Following stringent data filtering, the authors reported 6,463 interactions between 2,235 proteins and demonstrated the tendency of bait proteins to pull‐down functionally related interaction partners. Since then, AP‐MS and proximity labeling have been used in several small‐ and large‐scale studies to identify the interaction partners of proteins implicated in a variety of diseases (Table 1).
Table 1.
Selection of disease focused MS‐based PPI studies
| Disease area | Specific disease | Method | Citation |
|---|---|---|---|
| Autosomal | Neurofibromatosis type 1 | AP‐MS | Kobayashi et al (2019) |
| Cancer | Breast Cancer | BioID | Kazazian et al (2017) |
| Cancer | Breast Cancer | BioID | Luo et al (2018) |
| Cancer | Breast Cancer | BioID | Zhu et al (2019) |
| Cancer | Breast Cancer | BioID | Thuault et al (2020) |
| Cancer | Breast Cancer | BioID | Kothari et al (2020) |
| Cancer | Leukemia | BioID | Okuyama et al (2019) |
| Cancer | Lung Cancer | BioID | Kim et al (2017) |
| Cancer | Non‐Small‐cell Lung Cancer | BioID | Manshouri et al (2019) |
| Cancer | Prostate Cancer | BioID | Reina‐Campos et al (2019) |
| Cancer | AP‐MS | Hauri et al (2013) | |
| Cancer | AP‐MS | O’Connor et al (2020) | |
| Cardiac | Cardiac Disease | AP‐MS | Waldron et al (2016) |
| Cardiac | Cardiac Disease | BioID | Chu et al (2018) |
| Cardiac | Heart Failure | AP‐MS | Chiang et al (2018) |
| Metabolic | Diabetes Mellitus | BioID | Zhang et al (2018) |
| Neurological | Autism Spectrum Disorder | IP‐MS | Li et al (2015) |
| Neurological | Huntington’s Disease | AP/IP‐MS | Shirasaki et al (2012) |
| Neurological | Amyotrophic Lateral Sclerosis and Frontotemporal Dementia | BioID | Chou et al (2018) |
| Neurological | Amyotrophic Lateral Sclerosis, Autism Spectrum Disorder | AP‐MS | Malty et al (2017) |
| Neurological | Huntington's Disease, Parkinson's Disease, Alzheimer's Disease | AP‐MS | Hosp et al (2015) |
Directly comparing interaction networks and their changes in response to disease‐related perturbations can be used to identify the disease mechanisms of specific mutations. For example, Nissim et al (2019) used this approach to validate the causal role of an inherited mutation in the RAS oncogene family‐like 3 (RABL3), which they found to be significantly associated with familial pancreatic cancer. The biological function of RABL3 was previously largely unknown. Comparison of WT and mutated RABL3 interactomes revealed differential interactions that implicated RABL3 as a KRAS regulator and possible biomarker of pancreatic cancer. The power of comparing disease‐related PPI networks with their healthy counterparts was also highlighted in a study by the Yates lab (Pankow et al, 2015), who used IP‐MS to determine interactions driving the phenotype of cystic fibrosis (CF). CF is a Mendelian disorder predominantly caused by protein instability resulting from an in‐frame deletion of phenylalanine 508 in the CFTR gene. The resulting misfolding of ∆F508 CFTR recruits alternate chaperones, creating an altered PPI network defined by the addition of novel interaction partners not present in the WT CFTR network. In total, 209 interacting proteins significantly differed in either relative abundance or addition or removal of nodes between WT and mutant CFTR. As glycosylation of ∆F508 CFTR can partially restore its function, changes in interacting partners occurring between WT and ∆F508 CFTR were analyzed under a shift to lower temperature and inhibition of histone deacetylase, conditions known to promote CFTR glycosylation. Both conditions yielded significant changes between their respective WT and ∆F508 CFTR interactomes. For example, incubation at 30°C results in the removal of 89% of interactions unique to ∆F508 CFTR, including interactions involved with degradation proteins of ubiquitin‐mediated pathways and ERAD, heat‐shock proteins involved in protein folding, and RNA‐processing proteins, and rescued proteins involved in ER quality control. Surprisingly, many of the proteins showing differential interactions between WT and ∆F508 CFTR were associated with other diseases caused by protein misfolding or aggregation, such as neurodegenerative diseases, suggesting common mechanisms and pathways for these otherwise unrelated diseases (Pankow et al, 2015).
Host–pathogen interactions
Comparative network analysis can go beyond individual genes and mutations. As viruses and many intracellular bacteria require the host protein machinery to propagate, host–pathogen interactions represent an obvious choice for the application of disease network analysis (Shah et al, 2015). Unbiased AP‐MS approaches have been used to identify these interactions for a wide range of viruses and bacteria (Table 2). As mentioned above, proximity labeling can help resolve even short‐lived PPIs, such as those expected during the replication of pathogens. As an example, BioID was used to identify proteins associated with the replication/transcription complex (RTC) of the coronavirus mouse hepatitis virus (V’kovski et al, 2019). Over 500 proteins proximal to the RTC were identified, uncovering a spatial link between translational and replicational complexes. More global approaches aimed at creating networks across complete pathogen proteomes have been reported, including human immunodeficiency virus (Jäger et al, 2011a), Hepatitis C Virus (Ramage et al, 2015), Kaposi’s Sarcoma‐associated Herpesvirus (Davis et al, 2015), and many more, outlined in Table 2. AP‐MS and gene perturbation techniques were combined to map and compare host–pathogen PPI networks for the related flaviviruses Dengue and Zika virus, in both human and mosquito hosts (Shah et al, 2018). Twenty‐eight host proteins common to both humans and mosquitoes were found to interact with Zika and dengue viruses, including the SEC61 translocon complex. Chemical inhibition of SEC61 halted replication of both viruses in human and mosquito hosts, demonstrating the power of comparative analyses of PPI networks in identifying shared pathways that can be exploited to develop powerful broad‐acting antiviral strategies. Using large‐scale host–pathogen interaction studies to develop host‐directed strategies promises to reveal more broadly acting antivirals with reduced potential for viral escape (Batra et al, 2018; Shah et al, 2018). This approach can also be used for exploring drug repurposing, where drugs already approved for treatment of a specific disease may target the same proteins implicated in another disease. This was recently applied to find treatment options for coronavirus disease 2019 (COVID‐19), the disease caused by the pandemic coronavirus strain severe acute respiratory syndrome coronavirus 2 (SARS‐CoV‐2). Currently, no antiviral drugs or vaccines are available for the treatment of SARS‐CoV‐2 or the related coronaviruses SARS‐CoV or Middle Eastern respiratory syndrome (MERS). AP‐MS PPI networking for 26 of the 29 SARS‐CoV‐2 proteins generated a map of 332 interactions between host and viral proteins. Identified host proteins included 66 druggable human proteins known to be targeted by 69 compounds approved for use in humans or in advanced clinical investigations. A subset of these was tested for antiviral activity, revealing several candidates for expedited drug development (Gordon et al, 2020b). Similar PPI maps were generated for SARS‐CoV‐1 and MERS‐CoV, enabling comparisons between these less transmittable but more lethal coronaviruses (Gordon et al, 2020a). To facilitate these types of studies, a large amount of available data for virus interactomes has been made available in several dedicated databases, including VirusHostNet and VirusMentha (Calderone et al, 2015; Guirimand et al, 2015).
Table 2.
Selection of host–pathogen focused MS‐based PPI studies
| Pathogen | Method | Citation |
|---|---|---|
| Chlamydia trachomatis | AP‐MS | Mirrashidi et al (2015) |
| Cytomegalovirus | IP‐MS | Moorman et al (2010) |
| Epstein–Barr Virus | AP‐MS | Georges and Frappier (2017) |
| Epstein–Barr Virus | BioID | Rider et al (2018) |
| Herpesvirus | BioID | Cheerathodi and Meckes (2020) |
| HIV‐1 | BioID | Le Sage et al (2015) |
| Human bocavirus 1 | BioID | Wang et al (2020) |
| Human Papillomavirus | AP‐MS | White et al (2012a, 2012b) |
| Mycobacterium tuberculosis | AP‐MS | Penn et al (2018) |
| Respiratory Syncytial Virus | IP‐MS | Wu et al (2012) |
| West Nile Virus | AP‐MS | Li et al (2019) |
| Zika Virus | BioID | Coyaud et al (2018) |
Cancer‐associated protein interaction networks
The importance of studying disease networks was recently highlighted in a meta‐analysis showing that, for example, in cancer, proteins encoded by cancer driver genes interact with other cancer driver proteins more than randomly expected (Bouhaddou et al, 2019). Additionally, cancer‐associated mutations are often located on protein interfaces responsible for protein and ligand binding (Buljan et al, 2018). Although these somatic mutations may drive important cellular changes, determining the impact of cancer‐associated mutations on protein function is difficult, as these mutations usually occur only rarely across the human population and, consequently and statistically, appear to have a minimum impact on disease phenotypes (Fragoza et al, 2019). Large‐scale interaction studies using the most significantly disease‐associated genes as baits, especially when there are no or few hits deemed significant on a genome‐wide scale, can help identify biologically relevant mutations. This systems‐level view offers a powerful method to study interactions, where individual genes are knocked out or mutated, and perturbed interactions can be linked to specific pathways (Krogan et al, 2015; Willsey et al, 2018). In this way, mutations affecting similar pathways can be identified to develop new drug targets. For example, the serine/threonine protein phosphatase 2A (PP2A) usually acts as a tumor suppressor through the negative regulation of several oncogenic signaling pathways. Mutations in PPP2R1A, the gene encoding a PP2A subunit responsible for binding additional catalytic and regulatory subunits, are present in various cancer types, but their exact mechanisms of tumorigenesis are not fully understood. Using AP‐MS to study the effects of PP2A’s most frequently mutated residue, Arg‐183, on global protein interactions across multiple cell types, Narla et al demonstrated that its role in tumorigenesis might be due to reduced binding of several tumor suppressive regulatory subunit family members (O’Connor et al, 2020).
RAS family genes are GTPases whose activation or deactivation is dependent on whether they are bound to GTP or GDP, respectively. Activating RAS mutations are present in approximately 20% of human cancers, particularly lung, colorectal, and pancreatic cancers (Gimple and Wang, 2019). Gaps in our knowledge of RAS biology, including how RAS genes activate downstream pathway members and a poor understanding of RAS effectors and regulators, have contributed to a lack of therapeutic approaches (Stephen et al, 2014), with alterations in RAS family genes associated with poor patient prognosis in pan‐cancer studies (Gao et al, 2013). Understanding the molecular functions of RAS interactors may help develop effective therapeutics. Kennedy et al (2020) investigated how a common KRAS mutation, KRASG13D, affects EGFR signaling in colorectal cancer (CRC) cells by utilizing AP‐MS to compare the network of 95 bait proteins involved in this pathway in WT and KRAS‐mutated cells. Significant differences in PPIs suggest that the majority of rewiring in the EGFR network results from the gain or loss of interacting proteins, linking KRAS activity to a myriad of adaptive network alterations spanning from core interactions throughout the network periphery. These vast rearrangements might offer an explanation for the failure of single‐pathway inhibitors to treat KRAS‐mutated cancers, and point towards the need for combinatorial therapies. To capture transient and dynamic RAS signaling targets, Kovalski et al utilized BioID to identify neighboring proteins of WT and mutant RAS isoforms H‐, K‐, and N‐RAS. These mutations represent the most frequently altered genes for each cancer type, and BioID experiments were performed in cell lines relevant to the specific RAS mutant—bladder cancer cells for H‐RAS, colon cancer cells for KRAS, and melanoma cells for N‐RAS. Of the 690 proteins identified as proximal to RAS, 150 were common across all mutations. These proteins were enriched for known RAS functions, including cytoskeleton formation and cell junction integrity. To help identify the proteins essential for RAS‐mediated tumor growth, CRISPR‐Cas9 knockout screening of the identified proximal proteins was performed. Seventeen of these proteins were found to negatively impact cancer cell proliferation, representing novel RAS‐related proteins required for cancer cell growth. Among these, the mTORC2 complex was identified as a target of oncogenic RAS, particularly mTOR and MAPKAP1, which support mTORC2 kinases signaling at the plasma membrane (Kovalski et al, 2019).
As protein kinases modulate cellular signaling and are frequently mutated in various cancers, kinase inhibitors represent the largest category of anticancer drug targets. DYRK2, a 26S proteasome regulating kinase (Banerjee et al, 2019), has been implicated in cancer progression, both as an oncogene and as a tumor suppressor (Mimoto et al, 2017). Mehnert et al combined AP‐MS, BioID, and XL‐MS to establish the molecular response to five cancer‐associated DYRK2 mutations. These results were compared with WT DYRK2 and a catalytically inactive, non‐cancerous DYRK2 mutation to determine the effect of each mutation on cancer‐relevant interactions and biochemical pathways (Mehnert et al, 2020). The different mutations resulted in varied perturbations of the interactome, with a C‐terminal truncated mutant and the catalytically inactive mutant causing the strongest interactome changes, including a loss of interactions with the DYRK2 kinase core complex. Of note, the interaction networks generated from AP‐MS and BioID were highly complementary, suggesting good coverage of both stable and transient interactions. These mutations also altered the interactions with the Y‐complex, changing the phosphorylation status of DYRK2. In addition to interactome remodeling, differential phosphorylation was observed for known cancer driver proteins following DYRK2 mutation, further implicating the selected mutations in cancer progression. Significant topological changes between the mutants were detected by XL‐MS, suggesting these changes may steer the observed interactome alterations.
Integrating orthogonal data
Integrating MS‐based PPI interactomes with complementary data can further boost the discovery potential. For example, genetic interaction data can aid in determining functional relationships between genes. Genetic interaction studies map the phenotypic readout between pairs of depleted genes, allowing thousands of comparisons between genes linked to specific biological processes (Roguev et al, 2013). This strategy provides information on which genes are working in concert, and this can be used to develop novel therapeutic strategies targeting synthetic lethal partners of inactivated tumor suppressors (Helleday, 2011). Importantly, the information about functional relationships is not limited to the identification of direct physical interactions and can reveal the molecular cross‐talk between pathways. For example, coupling GI studies with PPI networks identified novel regulators of β‐catenin and helped define the functional networks required for the survival of β‐catenin‐active cancers (Rosenbluh et al, 2016). This approach was recently adapted to investigate the genetic interactions fundamental to HIV‐1 infection. 356 human genes linked to HIV‐1, including host‐dependency factors identified in a previous AP‐MS study (Jäger et al, 2011a), were depleted in a pairwise manner, resulting in over 63,000 comparisons. This study identified the RNA deadenylase complex CNOT, which had not previously been implicated in this process, as a mediator of HIV‐1 infection through the innate immune response (Gordon et al, 2020c).
Integrating data across networks of different diseases offers yet another powerful avenue. For example, network analysis has been used to study the role of viral infection in cancer development. Several cancers can develop as a result of viral infection, suggesting that tumors and viruses may affect the same pathways even if the genes they directly alter are different. To determine the role of viral proteins in this process, studies have used AP‐MS to determine the PPI networks across tumor‐associated proteins from different viral species. A more focused analysis was performed for all viral proteins of human papillomavirus (HPV). Identifying cancer genes in these networks highlighted the rewiring of Notch signaling across different cancer‐associated viruses (Rozenblatt‐Rosen et al, 2012). Integrating differential mutation patterns of HPV‐associated cancer samples using network propagation, on the other hand, identified oncogenic events phenocopied by a virus infection, including increased tumor cell invasion depending on both viral and human proteins that were found to interact (Eckhardt et al, 2018).
Pairing MS‐based interaction data with structural biology techniques can provide powerful insights into structural changes occurring during disease progression. Disease‐related protein complexes can be mapped using XL‐MS, X‐ray crystallography, and cryo‐EM to guide the development of therapeutic compounds. Such a strategy was used to study the progression of the Ebola virus (EBOV) infection, a well‐studied virus that results in recurrent deadly outbreaks. The virus–host interactome for six of the seven EBOV proteins revealed 194 high‐confidence interactions, including one between the viral VP30 protein and the human ubiquitin ligase RBBP6. X‐ray crystallography revealed that this interaction mimics the EBOV nucleoprotein (NP) binding to VP30 at the same interface, blocking this virus–virus PPI required for viral transcription. RBBP6 thus represents a host restriction factor, and peptide mimics successfully inhibited EBOV replication in cell culture. These findings suggest that the binding interface of VP30‐RBBP6 represents a potential therapeutic target for inhibitors (Batra et al, 2018). Other groups have used similar strategies to identify subunit orientation and binding sites implicated in disease pathogenesis. Cryo‐EM was used in combination with AP‐MS identified interactions between the intracellular pathogen Chlamydia trachomatis and the human proteome to determine the crystal structure of the Chlamydia complex, providing novel insight into retromer assembly (Elwell et al, 2017). Although cryo‐EM provides near‐atomic resolution for protein structures, certain arrangements are prone to areas of low electron density, making it difficult to resolve individual subunits without orthogonal structural information (Yu & Huang, 2018). Possible crystal structures can be fitted within the cryo‐EM volume, with the distance restraints from XL‐MS experiments providing the location and orientation of subunits. Henry et al used this approach to identify the active structure and binding mechanisms of apolipoprotein E4 (ApoE4). ApoE4 is involved in lipid transport and is linked to Alzheimer’s disease. Two conformations of ApoE4 were resolved, revealing an activation mechanism of ApoE4 based on the accessibility of the receptor‐binding region (Henry et al, 2018).
Taken together, disease networks have the power to reveal underlying biological mechanisms not only for the disease model under investigation, but can also increase our understanding of basic biological mechanisms more broadly.
New developments
Since protein–protein interactions are dynamic and can differ between tissues or cell lines or in response to stress, time, environmental stimulus, and disease state, creating a complete interactome remains a challenge (Ideker & Krogan, 2012). To mitigate this and our understanding of disease networks, we rely on continuous innovations in high‐throughput technologies, MS methods and adaptations, and new combinations of existing approaches.
Adapting advanced proteomics techniques can bring us closer to a complete fundamental understanding of a given biological system and can consequently allow its manipulation for disease treatment. Especially, more quantitative MS methods, such as targeted approaches and new acquisition techniques, are driving the field forward (Fig 4). Of these, data‐independent analysis (DIA) has shown particular promise in the analysis of AP‐MS samples. DIA is a powerful MS/MS data acquisition technique that attempts to quantify all peptides expressed in a given proteome (Gillet et al, 2012; Collins et al, 2013). Rather than sampling the most intense precursor ions present in an MS1 scan, as done in traditional data‐dependent approaches, all peptides within a defined m/z range are fragmented, allowing data to be collected for all peptides in a mixture. This process is repeated until the entire m/z range is fragmented (Fig 4A). As the same subset of peptides is always fragmented between DIA experiments, this can lead to improved sample reproducibility and fewer missing values compared to traditional data‐dependent acquisition (DDA). Additionally, DIA methods provide highly accurate and reproducible quantitation, similar to that achieved with targeted proteomics methods, while offering the advantage of unbiased analysis (Gillet et al, 2012). The increased dynamic range and sensitivity of these new MS methods promise more complete PPI network descriptions, by decreasing false negatives when probing for the interaction between two given proteins. They also allow for quantitative detection of changes in interactions. For instance, Lambert et al used DIA to develop a pipeline to score altered interactors following drug exposure or related to disease state. This pipeline was used to quantify differences in interactors between WT and melanoma‐associated sequence variants in the human kinase CDK4. DIA captured known interacting partners and discovered new interactors, revealing specificity in the recruitment of HSP90 to CDK4 mutants at Arg24 (Lambert et al, 2013).
Figure 4. New MS methodologies and preparation developments for PPI analysis.
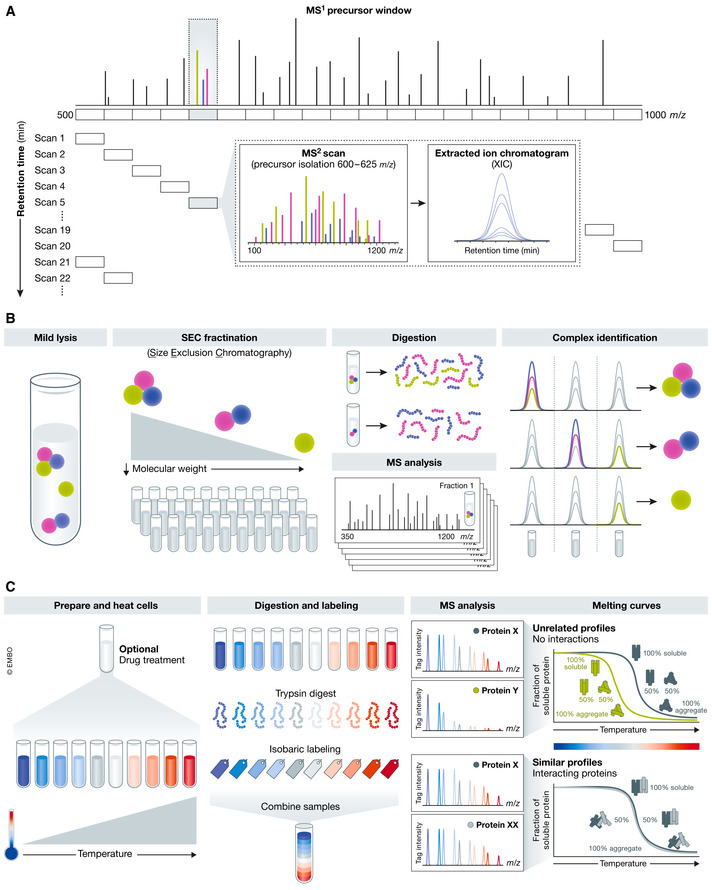
(A) DIA. Rather than individually isolating and fragmenting a given peak in an MS1 spectrum, DIA collectively fragments all precursors present in a specified m/z window. (B) Complex identification utilizing SEC. Native protein complexes are fractionated by SEC, each fraction is digested, and LC‐MS/MS is employed to identify the proteins present in each fraction. This information can be utilized to identify complexed proteins. (C) Following exposure to a temperature gradient and TMT‐labeling, thermal proteome profiling allows identification of complexed proteins.
In addition to the sensitivity issues of traditional methods, another limitation is that sample sets can easily reach sizes that are difficult to manage. For instance, it can be difficult to individually tag and analyze all disease‐related point mutations belonging to a single gene. Extending the analysis across several genes or conditions can become laborious. To alleviate this problem, strategies to identify PPIs on a global scale without requiring affinity purification have been explored. These techniques can be complementary to traditional AP‐MS workflows, as the goal is to characterize changes across the entire interactome in a single experiment (Smits & Vermeulen, 2016). For example, Aebersold and colleagues have advanced the concept of protein correlation profiling using complex‐centric analysis to systematically detect protein complexes (Fig 4B). Here, protein complexes are fractionated by size exclusion chromatography (SEC; Kristensen et al, 2012; Hu et al, 2019). Each fraction is then proteolytically digested and analyzed by DIA MS, and explored using a newly developed software package, CCprofiler. Interactions can be inferred based on proteins present in the same fraction. In a proof‐of‐principle experiment, the authors found 462 protein complexes comprising 2,127 protein subunits in the HEK293 cell line. This technique shows great promise for large‐scale PPI identification without the need for epitope tagging (Heusel et al, 2019). On the other hand, Nordlund et al adapted their cellular thermal shift assay (Savitski et al, 2014) to develop thermal proximity coaggregation (TPCA), a high‐throughput method to monitor PPIs across the proteome (Dai et al, 2018). The basis of TPCA is that different proteins will become denatured at certain temperatures. By analyzing this denaturation‐mediated loss of solubility across specific timepoints, a melting curve can be generated. As individual proteins denature, any interacting proteins will have similar solubility profiles and can be analyzed by MS. An advantage of this method is that interactions are studied in vivo. Intact cells are heated to the desired temperature prior to cell lysis, preserving existing interactions and eliminating false interactions occurring after lysis. This approach makes use of sample multiplexing with TMT, combining up to sixteen samples that are heated to different temperatures (Fig 4C). Comparison of melting curves obtained for 7,693 proteins identified from K562 cells with 111,776 publicly available PPIs showed that known interacting protein pairs were statistically more likely to have equivalent melting curves than random pairs of proteins. Melting curves were then generated across multiple cell lines, identifying a core set of common complexes and complexes unique to each cell line (Tan et al, 2018). This approach was used to study cells treated with cancer drugs, identifying the expected binding partners and several novel interactors and downstream effectors (Savitski et al, 2014).
A common critique of MS‐based approaches is the mixing of cellular compartments during lysis and the resulting loss of important information on the subcellular localization of the proteins of interest. To address this, a variety of MS methods has been adapted in recent technological developments under the umbrella term of spatial proteomics (Trotter et al, 2010; Geladaki et al, 2019; Lundberg & Borner, 2019). These new strategies provide quantitative, high‐throughput means for assessing global protein translocation in health and disease states. For example, APEX proximity labeling can be used to extract subcellular localization information from “bystander proteins” that are within the labeling environment of the protein of interest, but are not interactors. A proof‐of‐principle study showed that this approach can be used to provide both spatial and temporal resolution during the rapid signaling cascade of G protein‐coupled receptor signaling (Lobingier et al, 2017). In addition to new MS methodologies, existing approaches are evolving to capture a wider picture of disease networks. For example, APEX was recently adapted to label functional, intact mouse hearts, determining the mechanism behind adrenaline stimulated cardiac function (Liu et al, 2020). New technologies as well as novel combinations of existing approaches for purification and detection of proteins and their interaction partners will continue to enable advances in the study of PPI networks and informing novel therapeutic approaches.
Conclusions and outlook
Improvements in MS instrumentation, workflows, and data analysis have enabled the collection of very large datasets and greatly increased our ability to place disease‐related mutations and alterations in a biological context. Because of the connectivity between proteins, the impact of a genetic mutation is not restricted to a specific gene product. Instead, it affects an entire network, impacting the activity of whole subsets of proteins. The unbiased study of these interactions can provide clues about the spatial and temporal locations of molecules and allows us to identify specific pathways affected by different disease states. These data can be further integrated with structural information obtained from cryo‐EM or XL‐MS or functionally analyzed using genetic interaction maps, ideally placing complexes into pathways and providing clues on their mechanisms.
Disease networks should, however, not be viewed in isolation. Instead, it is critical to evaluate the biological significance of the mechanisms indicated by an interactome, just like with all systems biology approaches (Eckhardt et al, 2020). For example, immortalized cell lines are often used for PPI studies as they are easily scalable for large experiments. Although easy to manipulate, these cell lines do not necessarily capture biologically relevant relationships in more complex tissues and organisms. Newer genetic tools, including CRISPR/Cas9‐based genome engineering of primary cells, can lead to the development of more physiologically accurate and functionally relevant disease models to study interactions. As technology continues to advance and the availability and throughput of these methods increases, we are moving closer to integrating these approaches into personalized medicine applications. Their utility does not end with elucidating biological mechanisms, but also reveal the network disruption points for a particular patient and inform physicians about the most promising interventions.
Conflict of interest
The Krogan Laboratory has received research support from Vir Biotechnology and F. Hoffmann‐La Roche.
Acknowledgements
This research was funded by grants from the National Institutes of Health (P50AI150476, U19AI135990, U19AI135972, R01AI143292, U54CA209891, R01AG059751, U54NS100717, P01HL146366 and U01MH115747 to NJK); from the Defense Advanced Research Projects Agency (HR0011‐19‐2‐0020, HR00111‐9‐S0092‐FP‐FP‐002 and HR00111‐92‐0021 to NJK) and funding from F. Hoffmann‐La Roche and Vir Biotechnology. We are grateful to Qiongyu Li, Monita Muralidharan, Robyn Kaake, and Ruth Hüttenhain for their input on this manuscript.
Mol Syst Biol. (2021) 17: e8792
References
- Aebersold R, Mann M (2016) Mass‐spectrometric exploration of proteome structure and function. Nature 537: 347–355 [DOI] [PubMed] [Google Scholar]
- Antonicka H, Lin Z‐Y, Janer A, Aaltonen MJ, Weraarpachai W, Gingras A‐C, Shoubridge EA (2020) A high‐density human mitochondrial proximity interaction network. Cell Metab 32: 479–497.e9 [DOI] [PubMed] [Google Scholar]
- Banerjee S, Wei T, Wang J, Lee JJ, Gutierrez HL, Chapman O, Wiley SE, Mayfield JE, Tandon V, Juarez EF et al (2019) Inhibition of dual‐specificity tyrosine phosphorylation‐regulated kinase 2 perturbs 26S proteasome‐addicted neoplastic progression. Proc Natl Acad Sci USA 116: 24881–24891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra J, Hultquist JF, Liu D, Shtanko O, Von Dollen J, Satkamp L, Jang GM, Luthra P, Schwarz TM, Small GI et al (2018) Protein interaction mapping identifies RBBP6 as a negative regulator of Ebola virus replication. Cell 175: 1917–1930.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran PMJ, Jean Beltran PM, Federspiel JD, Sheng X, Cristea IM (2017) Proteomics and integrative omic approaches for understanding host–pathogen interactions and infectious diseases. Mol Syst Biol 13: 922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhaddou M, Eckhardt M, Chi Naing ZZ, Kim M, Ideker T, Krogan NJ (2019) Mapping the protein‐protein and genetic interactions of cancer to guide precision medicine. Curr Opin Genet Dev 54: 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branon TC, Bosch JA, Sanchez AD, Udeshi ND, Svinkina T, Carr SA, Feldman JL, Perrimon N, Ting AY (2018) Efficient proximity labeling in living cells and organisms with TurboID. Nat Biotechnol 36: 880–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buljan M, Blattmann P, Aebersold R, Boutros M (2018) Systematic characterization of pan‐cancer mutation clusters. Mol Syst Biol 14: e7974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone A, Licata L, Cesareni G (2015) VirusMentha: a new resource for virus‐host protein interactions. Nucleic Acids Res. 43: D588–D592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang I‐F (2006) Mass spectrometry‐based proteomic analysis of the epitope‐tag affinity purified protein complexes in eukaryotes. Proteomics 6: 6158–6166 [DOI] [PubMed] [Google Scholar]
- Cheerathodi MR, Meckes DG Jr (2020) BioID combined with mass spectrometry to study herpesvirus protein‐protein interaction networks. Methods Mol Biol 2060: 327–341 [DOI] [PubMed] [Google Scholar]
- Chiang DY, Alsina KM, Corradini E, Fitzpatrick M, Ni L, Lahiri SK, Reynolds JO, Pan X, Scott L Jr, Heck AJR et al (2018) Rearrangement of the protein phosphatase 1 interactome during heart failure progression. Circulation 138: 1569–1581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KF, Branon TC, Rajeev S, Svinkina T, Udeshi ND, Thoudam T, Kwak C, Rhee H‐W, Lee I‐K, Carr SA et al (2020) Split‐TurboID enables contact‐dependent proximity labeling in cells. Proc Natl Acad Sci USA 117: 12143–12154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H, Larsen B, Lin Z‐Y, Breitkreutz A, Mellacheruvu D, Fermin D, Qin ZS, Tyers M, Gingras A‐C, Nesvizhskii AI (2011) SAINT: probabilistic scoring of affinity purification‐mass spectrometry data. Nat Methods 8: 70–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C‐C, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, Sayegh M, Donlin‐Asp PG, Chen YH, Duong DM et al (2018) TDP‐43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci 21: 228–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson JC, Olzmann JA, Shaler TA, Sowa ME, Bennett EJ, Richter CM, Tyler RE, Greenblatt EJ, Harper JW, Kopito RR (2011) Defining human ERAD networks through an integrative mapping strategy. Nat Cell Biol 14: 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu M, Novak SM, Cover C, Wang AA, Chinyere IR, Juneman EB, Zarnescu DC, Wong PK, Gregorio CC (2018) Increased cardiac arrhythmogenesis associated with gap junction remodeling with upregulation of RNA‐binding protein FXR1. Circulation 137: 605–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins BC, Gillet LC, Rosenberger G, Röst HL, Vichalkovski A, Gstaiger M, Aebersold R (2013) Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14‐3‐3 system. Nat Methods 10: 1246–1253 [DOI] [PubMed] [Google Scholar]
- Coyaud E, Ranadheera C, Cheng D, Gonçalves J, Dyakov BJA, Laurent EMN, St‐Germain J, Pelletier L, Gingras A‐C, Brumell JH et al (2018) Global interactomics uncovers extensive organellar targeting by Zika virus. Mol Cell Proteomics 17: 2242–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai L, Zhao T, Bisteau X, Sun W, Prabhu N, Lim YT, Sobota RM, Kaldis P, Nordlund P (2018) Modulation of protein‐interaction states through the cell cycle. Cell 173: 1481–1494.e13 [DOI] [PubMed] [Google Scholar]
- Davis ZH, Verschueren E, Jang GM, Kleffman K, Johnson JR, Park J, Von Dollen J, Maher MC, Johnson T, Newton W et al (2015) Global mapping of herpesvirus‐host protein complexes reveals a transcription strategy for late genes. Mol Cell 57: 349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew K, Lee C, Huizar RL, Tu F, Borgeson B, McWhite CD, Ma Y, Wallingford JB, Marcotte EM (2017) Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Mol Syst Biol 13: 932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew K, Wallingford JB, Marcotte EM (2020) hu.MAP 2.0: Integration of over 15,000 proteomic experiments builds a global compendium of human multiprotein assemblies. bioRxiv 10.1101/2020.09.15.298216 [PREPRINT] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt M, Zhang W, Gross AM, Von Dollen J, Johnson JR, Franks‐Skiba KE, Swaney DL, Johnson TL, Jang GM, Shah PS et al (2018) Multiple routes to oncogenesis are promoted by the human papillomavirus‐host protein network. Cancer Discov 8: 1474–1489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt M, Hultquist JF, Kaake RM, Hüttenhain R, Krogan NJ (2020) A systems approach to infectious disease. Nat Rev Genet 21: 339–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elwell CA, Czudnochowski N, von Dollen J, Johnson JR, Nakagawa R, Mirrashidi K, Krogan NJ, Engel JN, Rosenberg OS (2017) Chlamydia interfere with an interaction between the mannose‐6‐phosphate receptor and sorting nexins to counteract host restriction. eLife 6: e22709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom‐Cerajewski L, Robinson MD, O’Connor L, Li M et al (2007) Large‐scale mapping of human protein‐protein interactions by mass spectrometry. Mol Syst Biol 3: 89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragoza R, Das J, Wierbowski SD, Liang J, Tran TN, Liang S, Beltran JF, Rivera‐Erick CA, Ye K, Wang T‐Y et al (2019) Extensive disruption of protein interactions by genetic variants across the allele frequency spectrum in human populations. Nat Commun 10: 4141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson K, Van Itallie CM, Aponte A, Gucek M, Tietgens AJ, Anderson JM (2015) Proteomic analysis of proteins surrounding occludin and claudin‐4 reveals their proximity to signaling and trafficking networks. PLoS One 10: e0117074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B (2012) Targeted proteomic quantification on quadrupole‐orbitrap mass spectrometer. Mol Cell Proteomics 11: 1709–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6: l1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geladaki A, Kočevar Britovšek N, Breckels LM, Smith TS, Vennard OL, Mulvey CM, Crook OM, Gatto L, Lilley KS (2019) Combining LOPIT with differential ultracentrifugation for high‐resolution spatial proteomics. Nat Commun 10: 331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges AA, Frappier L (2017) Affinity purification‐mass spectroscopy methods for identifying Epstein‐Barr virus‐host interactions. Methods Mol Biol 1532: 79–92 [DOI] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Reiter L, Bonner R, Aebersold R (2012a) Targeted data extraction of the MS/MS spectra generated by data‐independent acquisition: a new concept for consistent and accurate proteome analysis. Mol Cell Proteomics 11: O111.016717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gimple RC, Wang X (2019) RAS: striking at the core of the oncogenic circuitry. Front Oncol 9: 965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Go CD, Knight JDR, Rajasekharan A, Rathod B, Hesketh GG, Abe KT, Youn J‐Y, Samavarchi‐Tehrani P, Zhang H, Zhu LY et al (2019) A proximity biotinylation map of a human cell. bioRxiv 166 10.1101/796391 [PREPRINT] [DOI] [Google Scholar]
- Gordon DE, Hiatt J, Bouhaddou M, Rezelj VV, Ulferts S, Braberg H, Jureka AS, Obernier K, Guo JZ, Batra J et al (2020a) Comparative host‐coronavirus protein interaction networks reveal pan‐viral disease mechanisms. Science 370: eabe9403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL et al (2020b) A SARS‐CoV‐2 protein interaction map reveals targets for drug repurposing. Nature 583: 459–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon DE, Watson A, Roguev A, Zheng S, Jang GM, Kane J, Xu J, Guo JZ, Stevenson E, Swaney DL et al (2020c) A quantitative genetic interaction map of HIV infection. Mol Cell 78: 197–209.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guirimand T, Delmotte S, Navratil V (2015) VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res 43: D583–D587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez CB, Yu C, Novitsky EJ, Huszagh AS, Rychnovsky SD, Huang L (2016) Developing an acidic residue reactive and sulfoxide‐containing ms‐cleavable homobifunctional cross‐linker for probing protein‐protein interactions. Anal Chem 88: 8315–8322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Li J, Ting AY (2018) Proximity labeling: spatially resolved proteomic mapping for neurobiology. Curr Opin Neurobiol 50: 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y, Branon TC, Martell JD, Boassa D, Shechner D, Ellisman MH, Ting A (2019) Directed evolution of split APEX2 peroxidase. ACS Chem Biol 14: 619–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauri S, Wepf A, van Drogen A, Varjosalo M, Tapon N, Aebersold R, Gstaiger M (2013) Interaction proteome of human Hippo signaling: modular control of the co‐activator YAP1. Mol Syst Biol 9: 713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T (2011) The underlying mechanism for the PARP and BRCA synthetic lethality: clearing up the misunderstandings. Mol Oncol 5: 387–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry N, Krammer E‐M, Stengel F, Adams Q, Van Liefferinge F, Hubin E, Chaves R, Efremov R, Aebersold R, Vandenbussche G et al (2018) Lipidated apolipoprotein E4 structure and its receptor binding mechanism determined by a combined cross‐linking coupled to mass spectrometry and molecular dynamics approach. PLoS Comput Biol 14: e1006165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heusel M, Bludau I, Rosenberger G, Hafen R, Frank M, Banaei‐Esfahani A, van Drogen A, Collins BC, Gstaiger M, Aebersold R (2019) Complex‐centric proteome profiling by SEC‐SWATH‐MS. Mol Syst Biol 15: e8438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AM, Chen Q, Zheng T, Nicchitta CV (2019) Heterogeneous translational landscape of the endoplasmic reticulum revealed by ribosome proximity labeling and transcriptome analysis. J Biol Chem 294: 8942–8958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosp F, Vossfeldt H, Heinig M, Vasiljevic D, Arumughan A, Wyler E, Genetic and Environmental Risk for Alzheimer’s Disease GERAD1 Consortium , Landthaler M, Hubner N, Wanker EE et al (2015) Quantitative interaction proteomics of neurodegenerative disease proteins. Cell Rep 11: 1134–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu LZ, Goebels F, Tan JH, Wolf E, Kuzmanov U, Wan C, Phanse S, Xu C, Schertzberg M, Fraser AG et al (2019) EPIC: software toolkit for elution profile‐based inference of protein complexes. Nat Methods 16: 737–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung V, Udeshi ND, Lam SS, Loh KH, Cox KJ, Pedram K, Carr SA, Ting AY (2016) Spatially resolved proteomic mapping in living cells with the engineered peroxidase APEX2. Nat Protoc 11: 456–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K et al (2015) The BioPlex network: a systematic exploration of the human interactome. Cell 162: 425–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H et al (2017) Architecture of the human interactome defines protein communities and disease networks. Nature 545: 505–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ideker T, Krogan NJ (2012) Differential network biology. Mol Syst Biol 8: 565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K et al (2011a) Global landscape of HIV‐human protein complexes. Nature 481: 365–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S, Gulbahce N, Cimermancic P, Kane J, He N, Chou S, D’Orso I, Fernandes J, Jang G, Frankel AD et al (2011b) Purification and characterization of HIV‐human protein complexes. Methods 53: 13–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junttila MR, de Sauvage FJ (2013) Influence of tumour micro‐environment heterogeneity on therapeutic response. Nature 501: 346–354 [DOI] [PubMed] [Google Scholar]
- Kaake RM, Wang X, Burke A, Yu C, Kandur W, Yang Y, Novtisky EJ, Second T, Duan J, Kao A et al (2014) A new in vivo cross‐linking mass spectrometry platform to define protein‐protein interactions in living cells. Mol Cell Proteomics 13: 3533–3543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao A, Chiu C‐L, Vellucci D, Yang Y, Patel VR, Guan S, Randall A, Baldi P, Rychnovsky SD, Huang L (2011) Development of a novel cross‐linking strategy for fast and accurate identification of cross‐linked peptides of protein complexes. Mol Cell Proteomics 10: M110.002212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazazian K, Go C, Wu H, Brashavitskaya O, Xu R, Dennis JW, Gingras A‐C, Swallow CJ (2017) Plk4 Promotes Cancer Invasion and Metastasis through Arp2/3 Complex Regulation of the Actin Cytoskeleton. Cancer Res. 77: 434–447 [DOI] [PubMed] [Google Scholar]
- Kelly FD, Tran KD, Hatfield J, Schmidt K, Sanchez MA, Landfear SM (2020) A cytoskeletal protein complex is essential for division of intracellular amastigotes of Leishmania mexicana . J Biol Chem 295: 13106–13122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy SA, Jarboui M‐A, Srihari S, Raso C, Bryan K, Dernayka L, Charitou T, Bernal‐Llinares M, Herrera‐Montavez C, Krstic A et al (2020) Extensive rewiring of the EGFR network in colorectal cancer cells expressing transforming levels of KRAS. Nat Commun 11: 499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DI, Jensen SC, Noble KA, Kc B, Roux KH, Motamedchaboki K, Roux KJ (2016a) An improved smaller biotin ligase for BioID proximity labeling. Mol Biol Cell 27: 1188–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DI, Jensen SC, Roux KJ (2016b) Identifying protein‐protein associations at the nuclear envelope with BioID. Methods Mol Biol 1411: 133–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim BR, Coyaud E, Laurent EMN, St‐Germain J, Van de Laar E, Tsao M‐S, Raught B, Moghal N (2017) Identification of the SOX2 interactome by BioID reveals EP300 as a mediator of SOX2‐dependent squamous differentiation and lung squamous cell carcinoma growth. Mol Cell Proteomics 16: 1864–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi D, Tokuda T, Sato K, Okanishi H, Nagayama M, Hirayama‐Kurogi M, Ohtsuki S, Araki N (2019) Identification of a specific translational machinery via TCTP‐EF1A2 interaction regulating NF1‐associated tumor growth by affinity purification and data‐independent mass spectrometry acquisition (AP‐DIA). Mol Cell Proteomics 18: 245–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothari C, Osseni MA, Agbo L, Ouellette G, Déraspe M, Laviolette F, Corbeil J, Lambert J‐P, Diorio C, Durocher F (2020) Machine learning analysis identifies genes differentiating triple negative breast cancers. Sci Rep 10: 10464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalski JR, Bhaduri A, Zehnder AM, Neela PH, Che Y, Wozniak GG, Khavari PA (2019) The functional proximal proteome of oncogenic ras includes mTORC2. Mol Cell 73: 830–844.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen AR, Gsponer J, Foster LJ (2012) A high‐throughput approach for measuring temporal changes in the interactome. Nat Methods 9: 907–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan NJ, Lippman S, Agard DA, Ashworth A, Ideker T (2015) The cancer cell map initiative: defining the hallmark networks of cancer. Mol Cell 58: 690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert J‐P, Ivosev G, Couzens AL, Larsen B, Taipale M, Lin Z‐Y, Zhong Q, Lindquist S, Vidal M, Aebersold R et al (2013) Mapping differential interactomes by affinity purification coupled with data‐independent mass spectrometry acquisition. Nat Methods 10: 1239–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange V, Picotti P, Domon B, Aebersold R (2008) Selected reaction monitoring for quantitative proteomics: a tutorial. Mol Syst Biol 4: 222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiserson MDM, Vandin F, Wu H‐T, Dobson JR, Eldridge JV, Thomas JL, Papoutsaki A, Kim Y, Niu B, McLellan M et al (2015) Pan‐cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet 47: 106–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A, Reischl R, Walzthoeni T, Herzog F, Bohn S, Förster F, Aebersold R (2012) Expanding the chemical cross‐linking toolbox by the use of multiple proteases and enrichment by size exclusion chromatography. Mol Cell Proteomics 11: M111.014126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Sage V, Cinti A, Valiente‐Echeverría F, Mouland AJ (2015) Proteomic analysis of HIV‐1 Gag interacting partners using proximity‐dependent biotinylation. Virol J. 12: 138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Ma Z, Shi M, Malty RH, Aoki H, Minic Z, Phanse S, Jin K, Wall DP, Zhang Z et al (2015) Identification of human neuronal protein complexes reveals biochemical activities and convergent mechanisms of action in autism spectrum disorders. Cell Syst 1: 361–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Johnson JR, Truong B, Kim G, Weinbren N, Dittmar M, Shah PS, Von Dollen J, Newton BW, Jang GM et al (2019) Identification of antiviral roles for the exon‐junction complex and nonsense‐mediated decay in flaviviral infection. Nat Microbiol 4: 985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Rijkers DTS, Post H, Heck AJR (2015) Proteome‐wide profiling of protein assemblies by cross‐linking mass spectrometry. Nat Methods 12: 1179–1184 [DOI] [PubMed] [Google Scholar]
- Liu F, Lössl P, Scheltema R, Viner R, Heck AJR (2017) Optimized fragmentation schemes and data analysis strategies for proteome‐wide cross‐link identification. Nat Commun 8: 15473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, Kushner J, Yang L, Chen B‐X, Kushnir A et al (2020) Mechanism of adrenergic CaV1.2 stimulation revealed by proximity proteomics. Nature 577: 695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Doray B, Kornfeld S (2018) Recycling of Golgi glycosyltransferases requires direct binding to coatomer. Proc Natl Acad Sci USA 115: 8984–8989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobingier BT, Hüttenhain R, Eichel K, Miller KB, Ting AY, von Zastrow M, Krogan NJ (2017) An approach to spatiotemporally resolve protein interaction networks in living cells. Cell 169: 350–360.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Millikin RJ, Solntsev SK, Rolfs Z, Scalf M, Shortreed MR, Smith LM (2018) Identification of MS‐cleavable and noncleavable chemically cross‐linked peptides with metamorpheus. J Proteome Res 17: 2370–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg E, Borner GHH (2019) Spatial proteomics: a powerful discovery tool for cell biology. Nat Rev Mol Cell Biol 20: 285–302 [DOI] [PubMed] [Google Scholar]
- Luo Y, He J, Yang C, Orange M, Ren X, Blair N, Tan T, Yang J‐M, Zhu H (2018) UCH‐L1 promotes invasion of breast cancer cells through activating Akt signaling pathway. J Cell Biochem 119: 691–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malty RH, Aoki H, Kumar A, Phanse S, Amin S, Zhang Q, Minic Z, Goebels F, Musso G, Wu Z et al (2017) A map of human mitochondrial protein interactions linked to neurodegeneration reveals new mechanisms of redox homeostasis and NF‐κB signaling. Cell Systems 5: 564–577.e12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann M (2014) Fifteen years of stable isotope labeling by amino acids in cell culture (SILAC). Methods Mol Biol 1188: 1–7 [DOI] [PubMed] [Google Scholar]
- Manshouri R, Coyaud E, Kundu ST, Peng DH, Stratton SA, Alton K, Bajaj R, Fradette JJ, Minelli R, Peoples MD et al (2019) ZEB1/NuRD complex suppresses TBC1D2b to stimulate E‐cadherin internalization and promote metastasis in lung cancer. Nat Commun 10: 5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY (2012) Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol 30: 1143–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura A, Ito M, Sakaidani Y, Kondo T, Murakami K, Furukawa K, Nadano D, Matsuda T, Okajima T (2008) O‐linked N‐acetylglucosamine is present on the extracellular domain of notch receptors. J Biol Chem 283: 35486–35495 [DOI] [PubMed] [Google Scholar]
- Mehnert M, Ciuffa R, Frommelt F, Uliana F, van Drogen A, Ruminski K, Gstaiger M, Aebersold R (2020) Multi‐layered proteomic analyses decode compositional and functional effects of cancer mutations on kinase complexes. Nat Commun 11: 3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D, Wright Z, Couzens AL, Lambert J‐P, St‐Denis NA, Li T, Miteva YV, Hauri S, Sardiu ME, Low TY et al (2013) The CRAPome: a contaminant repository for affinity purification‐mass spectrometry data. Nat Methods 10: 730–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimoto R, Imawari Y, Hirooka S, Takeyama H, Yoshida K (2017) Impairment of DYRK2 augments stem‐like traits by promoting KLF4 expression in breast cancer. Oncogene 36: 1862–1872 [DOI] [PubMed] [Google Scholar]
- Mirrashidi KM, Elwell CA, Verschueren E, Johnson JR, Frando A, Von Dollen J, Rosenberg O, Gulbahce N, Jang G, Johnson T et al (2015) Global mapping of the inc‐human interactome reveals that retromer restricts chlamydia infection. Cell Host Microbe 18: 109–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman NJ, Sharon‐Friling R, Shenk T, Cristea IM (2010) A targeted spatial‐temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol Cell Proteomics 9: 851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JH, Knudsen GM, Verschueren E, Johnson JR, Cimermancic P, Greninger AL, Pico AR (2014) Affinity purification‐mass spectrometry and network analysis to understand protein‐protein interactions. Nat Protoc 9: 2539–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau JH (2001) Modifier genes in mice and humans. Nat Rev Genet 2: 165–174 [DOI] [PubMed] [Google Scholar]
- Nissim S, Leshchiner I, Mancias JD, Greenblatt MB, Maertens O, Cassa CA, Rosenfeld JA, Cox AG, Hedgepeth J, Wucherpfennig JI et al (2019) Mutations in RABL3 alter KRAS prenylation and are associated with hereditary pancreatic cancer. Nat Genet 51: 1308–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor CM, Leonard D, Wiredja D, Avelar RA, Wang Z, Schlatzer D, Bryson B, Tokala E, Taylor SE, Upadhyay A et al (2020) Inactivation of PP2A by a recurrent mutation drives resistance to MEK inhibitors. Oncogene 39: 703–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuyama K, Strid T, Kuruvilla J, Somasundaram R, Cristobal S, Smith E, Prasad M, Fioretos T, Lilljebjörn H, Soneji S et al (2019) PAX5 is part of a functional transcription factor network targeted in lymphoid leukemia. PLoS Genet 15: e1008280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong S‐E, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics 1: 376–386 [DOI] [PubMed] [Google Scholar]
- O’Reilly FJ, Rappsilber J (2018) Cross‐linking mass spectrometry: methods and applications in structural, molecular and systems biology. Nat Struct Mol Biol 25: 1000–1008 [DOI] [PubMed] [Google Scholar]
- Pankow S, Bamberger C, Calzolari D, Martínez‐Bartolomé S, Lavallée‐Adam M, Balch WE, Yates JR 3rd (2015) ∆F508 CFTR interactome remodelling promotes rescue of cystic fibrosis. Nature 528: 510–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankow S, Bamberger C, Calzolari D, Bamberger A, Yates JR 3rd (2016) Deep interactome profiling of membrane proteins by co‐interacting protein identification technology. Nat Protoc 11: 2515–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn BH, Netter Z, Johnson JR, Von Dollen J, Jang GM, Johnson T, Ohol YM, Maher C, Bell SL, Geiger K et al (2018) An Mtb‐human protein‐protein interaction map identifies a switch between host antiviral and antibacterial responses. Mol Cell 71: 637–648.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ (2012) Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Mol Cell Proteomics 11: 1475–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado‐Nilsson E, Wilm M, Séraphin B (2001) The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24: 218–229 [DOI] [PubMed] [Google Scholar]
- Ramage HR, Kumar GR, Verschueren E, Johnson JR, Von Dollen J, Johnson T, Newton B, Shah P, Horner J, Krogan NJ et al (2015) A combined proteomics/genomics approach links hepatitis C virus infection with nonsense‐mediated mRNA decay. Mol Cell 57: 329–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redwine WB, DeSantis ME, Hollyer I, Htet ZM, Tran PT, Swanson SK, Florens L, Washburn MP, Reck‐Peterson SL (2017) The human cytoplasmic dynein interactome reveals novel activators of motility. eLife 6: e28257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina‐Campos M, Linares JF, Duran A, Cordes T, L’Hermitte A, Badur MG, Bhangoo MS, Thorson PK, Richards A, Rooslid T et al (2019) Increased serine and one‐carbon pathway metabolism by PKCλ/ι deficiency promotes neuroendocrine prostate cancer. Cancer Cell 35: 385–400.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee H‐W, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, Ting AY (2013) Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339: 1328–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards AL, Merrill AE, Coon JJ (2015) Proteome sequencing goes deep. Curr Opin Chem Biol 24: 11–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider MA, Cheerathodi MR, Hurwitz SN, Nkosi D, Howell LA, Tremblay DC, Liu X, Zhu F, Meckes DG Jr (2018) The interactome of EBV LMP1 evaluated by proximity‐based BioID approach. Virology 516: 55–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Séraphin B (1999) A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Rinner O, Seebacher J, Walzthoeni T, Mueller LN, Beck M, Schmidt A, Mueller M, Aebersold R (2008) Identification of cross‐linked peptides from large sequence databases. Nat Methods 5: 315–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguev A, Talbot D, Negri GL, Shales M, Cagney G, Bandyopadhyay S, Panning B, Krogan NJ (2013) Quantitative genetic‐interaction mapping in mammalian cells. Nat Methods 10: 432–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbluh J, Mercer J, Shrestha Y, Oliver R, Tamayo P, Doench JG, Tirosh I, Piccioni F, Hartenian E, Horn H et al (2016) Genetic and proteomic interrogation of lower confidence candidate genes reveals signaling networks in β‐catenin‐active cancers. Cell Syst 3: 302–316.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Burke B, May DG (2018) BioID: a screen for protein‐protein interactions. Curr. Protoc. Protein Sci. 91: 19.23.1–19.23.15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt‐Rosen O, Deo RC, Padi M, Adelmant G, Calderwood MA, Rolland T, Grace M, Dricot A, Askenazi M, Tavares M et al (2012) Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 487: 491–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry MSR, Zhou W, Baneyx F (2009) Integrity of N‐ and C‐termini is important for E. coli Hsp31 chaperone activity. Protein Sci 18: 1439–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitski MM, Reinhard FBM, Franken H, Werner T, Savitski MF, Eberhard D, Martinez Molina D, Jafari R, Dovega RB, Klaeger S et al (2014) Tracking cancer drugs in living cells by thermal profiling of the proteome. Science 346: 1255784 [DOI] [PubMed] [Google Scholar]
- Sears RM, May DG, Roux KJ (2019) BioID as a tool for protein‐proximity labeling in living cells. Methods Mol Biol 2012: 299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PS, Wojcechowskyj JA, Eckhardt M, Krogan NJ (2015) Comparative mapping of host–pathogen protein–protein interactions. Curr Opin Microbiol 27: 62–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PS, Link N, Jang GM, Sharp PP, Zhu T, Swaney DL, Johnson JR, Von Dollen J, Ramage HR, Satkamp L et al (2018) Comparative flavivirus‐host protein interaction mapping reveals mechanisms of dengue and Zika virus pathogenesis. Cell 175: 1931–1945.e18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13: 2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaki DI, Greiner ER, Al‐Ramahi I, Gray M, Boontheung P, Geschwind DH, Botas J, Coppola G, Horvath S, Loo JA et al (2012) Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 75: 41–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Fernandez‐Martinez J, Tjioe E, Pellarin R, Kim SJ, Williams R, Schneidman‐Duhovny D, Sali A, Rout MP, Chait BT (2014) Structural characterization by cross‐linking reveals the detailed architecture of a coatomer‐related heptameric module from the nuclear pore complex. Mol Cell Proteomics 13: 2927–2943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits AH, Vermeulen M (2016) Characterizing protein‐protein interactions using mass spectrometry: challenges and opportunities. Trends Biotechnol 34: 825–834 [DOI] [PubMed] [Google Scholar]
- Snider J, Kotlyar M, Saraon P, Yao Z, Jurisica I, Stagljar I (2015) Fundamentals of protein interaction network mapping. Mol Syst Biol 11: 848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa ME, Bennett EJ, Gygi SP, Harper JW (2009) Defining the human deubiquitinating enzyme interaction landscape. Cell 138: 389–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen AG, Esposito D, Bagni RK, McCormick F (2014) Dragging ras back in the ring. Cancer Cell 25: 272–281 [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta‐Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P et al (2019) STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Res 47: D607–D613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan CSH, Go KD, Bisteau X, Dai L, Yong CH, Prabhu N, Ozturk MB, Lim YT, Sreekumar L, Lengqvist J et al (2018) Thermal proximity coaggregation for system‐wide profiling of protein complex dynamics in cells. Science 359: 1170–1177 [DOI] [PubMed] [Google Scholar]
- Tang X, Munske GR, Siems WF, Bruce JE (2005) Mass spectrometry identifiable cross‐linking strategy for studying protein−protein interactions. Anal Chem 77: 311–318 [DOI] [PubMed] [Google Scholar]
- Teo G, Koh H, Fermin D, Lambert J‐P, Knight JDR, Gingras A‐C, Choi H (2016) SAINTq: Scoring protein‐protein interactions in affinity purification ‐ mass spectrometry experiments with fragment or peptide intensity data. Proteomics 16: 2238–2245 [DOI] [PubMed] [Google Scholar]
- Teo G, Liu G, Zhang J, Nesvizhskii AI, Gingras A‐C, Choi H (2014) SAINTexpress: improvements and additional features in Significance Analysis of INTeractome software. J Proteomics 100: 37–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AKA, Hamon C (2003) Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem 75: 1895–1904 [DOI] [PubMed] [Google Scholar]
- Thuault S, Mamelonet C, Salameh J, Ostacolo K, Chanez B, Salaün D, Baudelet E, Audebert S, Camoin L, Badache A (2020) A proximity‐labeling proteomic approach to investigate invadopodia molecular landscape in breast cancer cells. Sci Rep 10: 6787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle‐Mulcahy L (2019) Recent advances in proximity‐based labeling methods for interactome mapping. F1000Res 8: 135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trotter MWB, Sadowski PG, Dunkley TPJ, Groen AJ, Lilley KS (2010) Improved sub‐cellular resolution via simultaneous analysis of organelle proteomics data across varied experimental conditions. Proteomics 10: 4213–4219 [DOI] [PubMed] [Google Scholar]
- Verschueren E, Von Dollen J, Cimermancic P, Gulbahce N, Sali A, Krogan NJ (2015) Scoring large‐scale affinity purification mass spectrometry datasets with MiST. Curr Protoc Bioinformatics 49: 8.19.1–8.19.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virreira Winter S, Meier F, Wichmann C, Cox J, Mann M, Meissner F (2018) EASI‐tag enables accurate multiplexed and interference‐free MS2‐based proteome quantification. Nat Methods 15: 527–530 [DOI] [PubMed] [Google Scholar]
- V’kovski P, Gerber M, Kelly J, Pfaender S, Ebert N, Braga Lagache S, Simillion C, Portmann J, Stalder H, Gaschen V et al (2019) Determination of host proteins composing the microenvironment of coronavirus replicase complexes by proximity‐labeling. eLife 8: e42037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walhout AJM, Vidal M (2001) High‐throughput yeast two‐hybrid assays for large‐scale protein interaction mapping. Methods 24: 297–306 [DOI] [PubMed] [Google Scholar]
- Waldron L, Steimle JD, Greco TM, Gomez NC, Dorr KM, Kweon J, Temple B, Yang XH, Wilczewski CM, Davis IJ et al (2016) The cardiac TBX5 interactome reveals a chromatin remodeling network essential for cardiac septation. Dev Cell 36: 262–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xu P, Cheng F, Li Y, Wang Z, Hao S, Wang J, Ning K, Ganaie SS, Engelhardt JF et al (2020) Cellular cleavage and polyadenylation specificity factor 6 (CPSF6) mediates nuclear import of human bocavirus 1 NP1 protein and modulates viral capsid protein expression. J Virol 94: e01444‐19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod CR, Chavez JD, Eng JK, Yang L, Zheng C, Bruce JE (2013) In vivo protein interaction network identified with a novel real‐time cross‐linked peptide identification strategy. J Proteome Res 12: 1569–1579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EA, Kramer RE, Tan MJA, Hayes SD, Harper JW, Howley PM (2012a) Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J Virol 86: 13174–13186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EA, Sowa ME, Tan MJA, Jeudy S, Hayes SD, Santha S, Münger K, Harper JW, Howley PM (2012b) Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc Natl Acad Sci USA 109: E260–E267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiese S, Reidegeld KA, Meyer HE, Warscheid B (2007) Protein labeling by iTRAQ: a new tool for quantitative mass spectrometry in proteome research. Proteomics 7: 340–350 [DOI] [PubMed] [Google Scholar]
- Willsey AJ, Jeremy Willsey A, Morris MT, Wang S, Willsey HR, Sun N, Teerikorpi N, Baum TB, Cagney G, Bender KJ et al (2018) The psychiatric cell map initiative: a convergent systems biological approach to illuminating key molecular pathways in neuropsychiatric disorders. Cell 174: 505–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W, Tran KC, Teng MN, Heesom KJ, Matthews DA, Barr JN, Hiscox JA (2012) The interactome of the human respiratory syncytial virus NS1 protein highlights multiple effects on host cell biology. J Virol 86: 7777–7789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C, Huang L (2018) Cross‐linking mass spectrometry: an emerging technology for interactomics and structural biology. Anal Chem 90: 144–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C, Li L, Zhang Z, Bi M, Wang H, Su W, Hernandez K, Liu P, Chen J, Chen M et al (2019) A non‐canonical role of YAP/tead is required for activation of estrogen‐regulated enhancers in breast cancer. Mol Cell 75: 791–806.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]